 Eleonóra Gál
Eleonóra Gál Jurij Dolenšek
Jurij Dolenšek Andraž Stožer2
Andraž Stožer2 László Czakó
László Czakó Attila Ébert
Attila Ébert Viktória Venglovecz
Viktória Venglovecz- 1Department of Pharmacology and Pharmacotherapy, University of Szeged, Szeged, Hungary
- 2Faculty of Medicine, University of Maribor, Maribor, Slovenia
- 3Faculty of Natural Sciences and Mathematics, University of Maribor, Maribor, Slovenia
- 4First Department of Medicine, University of Szeged, Szeged, Hungary
Anatomical proximity and functional correlations between the exocrine and endocrine pancreas warrant reciprocal effects between the two parts. Inflammatory diseases of the exocrine pancreas, such as acute or chronic pancreatitis, or the presence of cystic fibrosis disrupt endocrine function, resulting in diabetes of the exocrine pancreas. Although novel mechanisms are being increasingly identified, the intra- and intercellular pathways regulating exocrine–endocrine interactions are still not fully understood, making the development of new and more effective therapies difficult. Therefore, this review sought to accumulate current knowledge regarding the pathogenesis of diabetes in acute and chronic pancreatitis, as well as cystic fibrosis.
Introduction
The pancreas is a unique organ, having both the endocrine and exocrine functions. The exocrine pancreas is composed of acini, which are dome-shaped clusters of acinar cells that produce and secrete enzymes involved in the digestion of food. Enzymes are secreted into the ductal tree that is formed by another cell type, the -producing ductal cells that neutralise low pH from the stomach and provide an optimal environment for the enzymes to operate. Unlike the exocrine component, the endocrine pancreas produces hormones that enter the bloodstream and regulate carbohydrate metabolism. There is ever-accumulating evidence supporting the existence of a strong functional interrelationship between the exocrine and endocrine pancreas, and the repercussion of endocrine dysfunction in the exocrine function and vice versa (1–5). Therefore, understanding exocrine–endocrine interactions is crucial for the diagnosis and treatment of pancreatic diseases. Diabetes of the exocrine pancreas (DEP) develops secondary to exocrine pancreatic disorder. One of the most common diseases of the exocrine pancreas is acute and chronic pancreatitis (AP and CP, respectively). AP is a sudden inflammation of the pancreas, whereas CP is a persistent condition that arises from repeated damage of the pancreas and is associated with fibrosis, calcification and destruction of the gland. In both forms of pancreatitis, the risk of developing diabetes is high (6, 7); however, the underlying mechanism is not completely known. In addition to pancreatitis, cystic fibrosis (CF) is also often associated with diabetes, especially at advanced age (8–10). Although CF is not specifically an inflammatory disease, the secretory defect due to the dysfunction of the cystic fibrosis transmembrane conductance regulator (CFTR) Cl- channel causes inflammation and fibrosis of the pancreas that can also affect the endocrine functions.
Recently, DEP has been increasingly emphasized in clinical practice (11–14), although concerns remain regarding its treatment. Compared to type 1 (T1DM) and type 2 (T2DM) diabetes mellitus (DM), the pathogenesis of DEP has been relatively less researched, with most experimental data focusing on CF. Considering the lower incidence of DEP compared to the other two types of diabetes and the limited data available, diagnosing DEP remains difficult. In addition, no specific guidelines have been established for the treatment of DEP, although more and more recommendations have recently emerged (3, 15–18). Two recent reviews provided a detailed and comprehensive analysis of the current diagnostic and treatment guidelines for DEP and recommended the use of a novel nomenclature (12, 14) Accordingly, the classification of DEP includes new-onset diabetes after pancreatitis, i.e., post-pancreatitis diabetes mellitus (PPDM); pancreatic cancer-related diabetes; and CF-related diabetes (CFRD). Based on this, diabetes after AP should be termed PPDM-A, whereas diabetes after CP should be termed PPDM-C.
This review sought to accumulate current experimental knowledge regarding the endocrine–exocrine interactions, focusing on PPDM-A, PPDM-C, and CFRD.
Effect of the Exocrine Pancreas on Endocrine Function Under Physiological Conditions
Since the discovery of the “insulo-acinar axis” in early 1960s (19), the impact of insulin on pancreatic exocrine function has been extensively studied both in vivo and in vitro and these studies indicated that insulin has a significant effect on exocrine function (20–27). In contrast, much less information is available regarding the effect of exocrine cells on the endocrine function. In pigs, oral pancreatic enzyme pretreatment was shown to decrease plasma insulin levels for intravenous glucose tolerance test (GTT) and test meal (28). The same workgroup demonstrated that in exocrine pancreatic insufficient pigs, supplementation of exocrine enzymes reduced plasma insulin levels after both starvation and oral or intravenous GTT (29). Similarly, alpha-amylase supplementation also reduced plasma insulin levels after both intravenous and duodenal GTT, while it increased glucagon levels several folds in pigs with T1DM and T2DM (30). The inhibitory effect of amylase on insulin secretion has also been demonstrated in the insulin-producing cell line, BRIN-BD11 (30), and in hamsters with induced peripheral insulin resistance (IR) (31–33). In addition to amylase, lipase also plays an important role in insulin secretion by reducing the formation of long-chain fatty acids, which regulate glucose-induced insulin secretion through the activation of G-protein-coupled receptor, GPR40 (32, 34). These data indicate that amylase and lipase not only play a role in the breakdown of carbohydrates and fats but also directly or indirectly inhibit insulin release after ingestion of large amounts of carbohydrates or fats. The purpose of the inhibitory effect of amylase and lipase may be to keep insulin at a normal level; however, the exact significance of this is not entirely clear. One possible explanation is a regulatory mechanism that acts against insulin overproduction and thus exhaustion of beta cells. Among the proteases, chymotrypsin does not affect either insulin secretion, islet size, or the number of beta cells; however, this has only been demonstrated in hamsters and no other studies have been conducted to confirm these results in humans (32). In contrast, intracellular trypsin significantly increases the activity of KATP and reactivates the channel after complete rundown by modifying the regulatory protein of KATP or the channel itself (35, 36). Opening of the channel causes K+ efflux and, as a result, hyperpolarization of the membrane, which in turn inhibits voltage-gated Ca2+ channels and exocytosis of insulin granules. The physiological significance of the inhibitory effect of trypsin on insulin secretion is not fully understood, but it is likely to play an important role in normalizing serum insulin levels after a meal, such as amylase and lipase. Beside the regulation of insulin secretion, trypsin also plays a role in islet formation and differentiation through PAR-2 receptor activation and calcium signalling (37).
Effect of the Exocrine Pancreas on Endocrine Function Under Pathophysiological Conditions
Under certain pathological conditions, the secretion of digestive enzymes is altered. Inflammation of the pancreas destroys the parenchyma, and prolonged inflammation leads to the development of exocrine pancreatic insufficiency, a condition in which the production of pancreatic enzymes is greatly reduced. Due to decreased enzyme production, insulin secretion is released from the inhibitory effect of digestive enzymes that leads to hyperinsulinemia. Clinical studies have demonstrated that serum insulin level increases after AP and CP, although in both cases, hyperinsulinemia is explained by the decreased insulin clearance, and not by the overproduction of insulin (38, 39). Regardless of the cause of hyperinsulinemia, it is unclear whether this transiently high insulin level plays a role in DEP. Previous studies have demonstrated that hyperinsulinemia may be responsible for the development of IR (40–42), a condition in which cells become less sensitive to insulin. Because IR is considered as a precursor of diabetes, hyperinsulinemia during AP and CP may be a sign of subsequent DEP development. The following chapters describe the mechanisms that are thought to play a role in the development of DEP in AP, CP, and CF.
Post-Acute Pancreatitis Diabetes Mellitus
AP is an inflammatory disorder of the pancreas and one of the most frequent reasons for hospitalisation related to a gastrointestinal condition (43). AP can progress rapidly and cause severe symptoms like systemic inflammatory response, which may lead to multi-outcome and predisposes patients to diabetes. Research regarding the incidence of post-acute pancreatitis diabetes mellitus (PPDM-A), as well as factors associated therewith, has remained controversial. A recently published systematic review and meta-analysis that examined the incidence of new-onset DM after AP based on various criteria, including the severity of pancreatitis, aetiological factors, presence of necrosis and follow-up duration (7), showed a 23% overall incidence of DM after AP. Although similar results had been found by Das et al. (1), other workgroups showed much higher (44–47) or lower incidences (48, 49). Severity of pancreatitis, aetiology and the presence of necrosis have also been considered the most important risk factors for the development of diabetes (7). With regard to severity, some studies found no correlation between the development of diabetes and severity of pancreatitis (1, 44, 50–53), whereas others found a strong association between them, with the extent of necrosis being a decisive factor (44, 47, 51, 54, 55). Generally, the number of functionally active beta cells decreases as the extent of necrosis increases, leading to altered insulin secretion. Research regarding aetiological factors has been inconsistent. Majority of the studies indicate that diabetes is most often associated with alcohol-induced pancreatitis (7, 55), which may be partly explained by the increased incidence of diabetes among patients with alcoholic and biliary AP in such studies. With regard to other aetiological factors, hyperlipidaemia-induced pancreatitis has been found to cause diabetes in more patients (86%) compared to the other aetiologies (53). In addition, prior to the onset of diabetes, patients with AP often develop IR. Obesity is a risk factor in the development of IR (56, 57). Accumulated adipose tissue releases a number of inflammatory mediators that can affect insulin receptor or insulin binding to the receptor through various signalling pathways. Consistent with this, it has been reported that obesity increases the risk of IR development in patients with AP (58). Furthermore, in non-obese AP patients, increased intra-pancreatic fat deposition was associated with an increased HOMA-IR (homeostasis model assessment-IR) index (59). Among the inflammatory mediators, the adipocytokine IL-6 has been shown to be associated with elevated levels of chronic hyperglycaemia and IR after AP (60). This proinflammatory cytokine is released from adipose tissue and can inhibit both the insulin receptor and the action of insulin (61). Upregulation of IL-6 has been also shown in experimental models of AP where it is associated with enhanced local and systemic response. The association between IR and AP severity was investigated in a prospective clinical study, where IR was shown to be an independent factor in predicting AP severity (62). These data suggest that in addition to increasing the risk of diabetes development, IR also exacerbates the outcome of pancreatitis.
Experimental studies on rats have shown that sodium taurocholate-induced pancreatitis did not alter islet morphology or GLUT-2 expression but reduced insulin secretion in response to glucose stimulation, indicating functional deterioration of beta cells (63). Similar results were obtained in an L-arginine-induced rat model wherein no change in alpha and beta cell counts but a significant decrease in insulin secretion was observed 1 month after the induction of pancreatitis (64). Kinami et al. also found no morphological abnormalities in islet cells after the induction of AP but did observe a significant decrease in the serum levels of insulin and glucagon (65). Interestingly, they found that damage occurred earlier in alpha cells than beta cells. The exact mechanism that leads to a decrease in hormone production after AP is not completely known. Experiments in rats with acute necrotizing pancreatitis have shown that endoplasmic reticulum stress and nitric oxide production play significant roles in beta cell dysfunction (66–68), although other factors are presumably also involved. Our workgroup had also investigated the morphology and function of the endocrine pancreas after caerulein-induced AP in mice (unpublished data). Accordingly, we found that mice with caerulein-induced AP showed lower fasting insulin levels in the acute inflammation phase but higher fasting glucagon levels compared to the untreated control group. Fasting blood glucose levels were nonetheless normal. Intraperitoneal glucose tolerance tests in the same animals showed normal response to glucose by both hormones but significantly lower insulin levels in the caerulein-treated group. Blood glucose levels were normal and did not differ in caerulein-treated versus control mice. Immunofluorescent staining of whole pancreas tissue sections against insulin and glucagon did not reveal significant differences in islet morphology between the caerulein-treated and control groups. The aforementioned results suggest that the high levels of tissue necrosis observed in AP do not significantly affect islet morphology or islet cell counts but significantly alter serum hormone levels, particularly the level of insulin, which presumably leads to the deterioration of metabolism observed in patients with AP.
Furthermore, there is growing evidence that the exocrine pancreas and endocrine pancreas interact with each other not only through their secretions but also indirectly, through dysbiosis, resulting from inflammation. Sun et al. have shown that the level of an antimicrobial peptide, cathelicidin-related antimicrobial peptide (CRAMP), changes during inflammation (69). This peptide is produced by beta cells and plays an important role in shaping the immune environment of the pancreas. Production of CRAMP is regulated by short-chain fatty acids (SCFAs) which are produced by the gut microbiota. Since AP is associated with dysbiosis, the production of SCFAs and thus the level of CRAMP decreases during AP, which may lead to the development of T1D through the formation of an unfavourable immune environment (69). These data suggest that the development of diabetes in AP is a highly complex process, in which—besides the direct effect of the inflammatory environment in the pancreas—other indirect factors also play a role.
Post-Chronic Pancreatitis Diabetes Mellitus
CP is a progressive disease characterised by parenchymal destruction, as well as the presence of inflammation and fibrosis. Heavy alcohol consumption has been the most common cause of CP worldwide, although other factors, such as genetic mutations, hypertriglyceridemia, hypercalcaemia and pancreatic duct obstruction, may also play a role. According to the necrosis–fibrosis hypothesis, recurrent acute attacks on the pancreas cause irreversible damage of the exocrine pancreas and lead to the development of CP (70). Loss of islet cells and the development of hepatic IR are often associated with long-standing CP, which results in diabetes among such patients (71). CP is typically considered to be one of the major causes of DEP (72). Around 30% of CP patients develop diabetes, although such rates can reach as high as 90% depending on the follow-up duration and region (6, 73, 74). Alcoholism and distal pancreatectomy have been considered the most common independent risk factors for DEP in CP (75–77), while male sex, steatorrhea, and biliary stricture have also emerged as risk factors for diabetes development, with divided opinions regarding smoking (73, 74). Only a limited number of guidelines are available for the diagnosis and treatment of DEP in CP. The most accepted and currently used guideline was developed by the HaPanEU/United European Gastroenterology, which contains specific diagnostic and therapeutic recommendations (78). According to current recommendations, therapy should include treatment of hyperglycaemia, steatorrhea and malnutrition. Although recent efforts have been made to improve treatment, such patients still have higher mortality rates compared to those with CP alone, with the presence of diabetes considerably impairing their quality of life. Therefore, comprehensive knowledge regarding the pathomechanism of PPDM-C would definitely promote substantial improvements in therapy.
A number of experiments have been conducted to identify intra- and extracellular mechanisms related to the development of diabetes in CP. Most of these studies have used the long-term partial pancreatic duct ligation (PDL) technique for the induction of CP that causes extensive acinar cell damage and tissue fibrosis. According to the most widely accepted view, the major cause of impaired metabolism or diabetes in CP is defective insulin secretion (79–81). As the disease progresses, the pancreas becomes more fibrotic, which impedes proper blood supply to the islets, causing progressive loss of beta cells and impaired islet function. However, alterations in glucose metabolism have also been observed not only during disease progression but also during the early stages, which predisposes patients to the development of diabetes (82). In a clinical cohort, patients with CP who had no diabetes and pancreatic calcification showed higher fasting and mixed-meal glucose levels and lower insulin sensitivity compared to healthy controls (82). However, those with advanced CP without diabetes exhibited reduced beta cell mass, and the expression of pancreatic and duodenal homeobox gene as well as insulin gene decreased in these patients (83). Recent research has shown that the decrease in beta cell number among patients with CP was not due to cell death, but rather the epithelial–mesenchymal transition (EMT) of beta cells (84). Xiao et al. demonstrated that high levels of transforming growth factor β1 (TGF-β1) regulates EMT through the SMAD3/Stat3 signalling pathway. This hypothesis is supported by previous studies showing that TGF-β1 overexpression promotes massive fibrosis, abnormal islet distribution and the appearance of fibroblast-like cells and macrophages (85). Furthermore, the Stat3 antagonist forkhead box protein O1 has been shown to prevent the development of EMT, as well as diabetes. Furthermore, the inflammatory milieu resulting from pancreatitis also contributes to beta cell dysfunction. Several inflammatory mediators have been shown to inhibit insulin release (IL-1β, IFN-γ) or to be associated with IR (IL-6) (86–88). The accumulation of IFN-γ within the islets is presumably due to the differentiation of Th17 cells into IFN-γ-producing Th1 cells by the inflammatory environment (89). Under physiological conditions, insulin inhibits gluconeogenesis and glycogenolysis in the liver and promotes glycogen synthesis (90). Andersen et al. had shown that the inhibitory effect of insulin on hepatic glucose production is mediated by the reduction in hepatic GLUT-2 receptors (91). However, given the lack of sufficient insulin in cases with CP, no such reduction was observed, while GLUT-2 internalisation was inadequate (91, 92). In addition, they showed that insulin receptors interact with GLUT-2 transporters, a mechanism that allows insulin to regulate hepatic glucose transport (93). Apart from insulin, other islet hormones, such as PP, also have decreased secretion in CP, which can play a significant role in altered glucose metabolism (94). Several studies have revealed that PP cells on the periphery of the islets protect beta cells in the centre, while decreased PP production may be a sign of reduced insulin production and can be used to predict the development of diabetes (95–97). Bastidas et al. who characterised the effect of PP infusion on glucose tolerance and insulin response in CP dogs found that intravenous administration of PP did not improve glucose tolerance or insulin response (98) but did increase hepatic sensitivity to insulin. CP decreases the number of insulin receptors on hepatocytes, which probably plays role in the development of hepatic IR (99). Exogenous administration of PP increases the expression of hepatic insulin receptors, as well as the binding of insulin to its receptor (99, 100), thereby enhancing the sensitivity of hepatocytes to insulin (100). Studies have shown that intravenous administration of PP restored hepatic IR in rats, dogs and humans with CP (101–103), which could suggest the therapeutic use of PP. Banerjee et al. showed that packaging of PP into micelles increases its half-life and thus its efficacy (104). A randomised clinical trial investigating the effects of PP on insulin requirements in 10 patients with T1DM or DEP found that subcutaneous administration of PP reduced insulin requirements in these patients (105). In contrast to decreased hepatic insulin sensitivity, peripheral insulin sensitivity increases in CP, which may be explained by an increase in the number of insulin receptors on blood cells and an increase in insulin binding to its receptors (106, 107). However, activated inflammatory cells and cytokines present systemically in CP may play a role in the development of IR. Immune cells in the peripheral blood of CP patients show elevated expression of the cytokines, IL-2, IL-6, IL-12 and IFN-γ, and decreased expression of IL-4 and IL-10, indicating an increased inflammatory state (108). Similarly, plasma levels of IL-6, TNF-α and adiponectin are significantly elevated in CP (109). Chronic low-grade inflammation has long been considered to promote the development of IR (110). TNF-α is an adipose tissue-derived proinflammatory cytokine that causes IR by enhancing adipocyte lipolysis and increasing the serine/threonine phosphorylation of insulin receptor substrate-1 (IRS-1) through the JNK and IKKβ/NF-κB pathways (110–113). IFN-γ is suggested to promote IR by inhibiting insulin action and adiponectin secretion in adipocytes (114) and was shown to inhibit glucose-stimulated insulin response in CP (87). IL-6 is also recognised as an inflammatory mediator that causes IR by reducing the expression of GLUT4 and IRS-1 by activating the Janus kinase-signal transducer and activator of transcription (JAK-STAT) signalling pathway and increasing suppressor of cytokine signalling 3 (SOCS3) expression (115, 116). Through activation of STAT3, IL-6 can also lead to IR in skeletal muscle by inducing the expression of toll-like receptor-4 (TLR-4), which is suggested to be a major upstream molecule in the activation of NF-κB. Furthermore, IL-6 is also found to induce IR by impairing the synthesis of glycogen through downregulating the expression of miR-200s and upregulating that of FOG-2 (56, 117–119). Cytokines released from adipose tissue may therefore damage the insulin responsiveness also of skeletal muscle and further exacerbate IR in CP. Intramuscular adipose tissue content has been shown to affect the disease severity and survival rate in patients with pancreatic diseases (120, 121). IR in CP is exacerbated by obesity, which is a common concomitant condition in CP patients. In fact, obesity can be characterised as a state of chronic low-grade inflammation, promoting IR (122, 123). Clinical studies show that the duration of DM in CP patients correlates positively with BMI and obese patients are more likely to develop severe AP with a more intense systemic inflammatory response (124–126).
Serum glucagon levels can also change during CP. A previous study by Donowitz et al. involving 10 patients with CP found that CP reduced basal glucagon levels, while the infusion of L-alanine, an endogenous stimulator of glucagon secretion, was unable to enhance glucagon response (127). Moreover, basal blood glucose levels in these patients were higher compared to controls, indicating that glycaemic control is disturbed in CP. Similar results were found in another study that compared patients with T2DM to those who developed PPDM-C (128). This study found that both the diabetes and CP groups had higher serum glucose levels after OGTT compared to controls and the serum glucagon level also showed an initial increase in these groups, which is presumably due to the fact that postprandial glucagon release is not inhibited due to decreased endogenous insulin production. Interestingly, this increase in glucagon levels was not observed during intravenous GTT, suggesting that other intestinal hormones, such as incretin hormones, may also play a role in abnormal glucagon levels (129). In contrast, glucagon levels decreased under hypoglycaemic conditions. Previous studies have also observed low levels of glucagon during hypoglycaemia in patients with pancreatitis, with the rate of decrease being directly proportional to the stage of the disease (130, 131). Defective alpha cell function in diabetes and CP can be partly explained by damage to most of the beta cells, which render them unable to properly control the function of alpha cells (128). In addition, a recent study on a patient with advanced CP who had previously undergone partial pancreatectomy found that the number of alpha cells significantly increased mostly around middle-size ducts and in the lumen of the ducts, suggesting that newly formed alpha cells during regeneration may have ductal origin or that ductal cells play a substantial role in islet neogenesis (132). Figure 1 shows a possible mechanism for the development of DEP in CP.
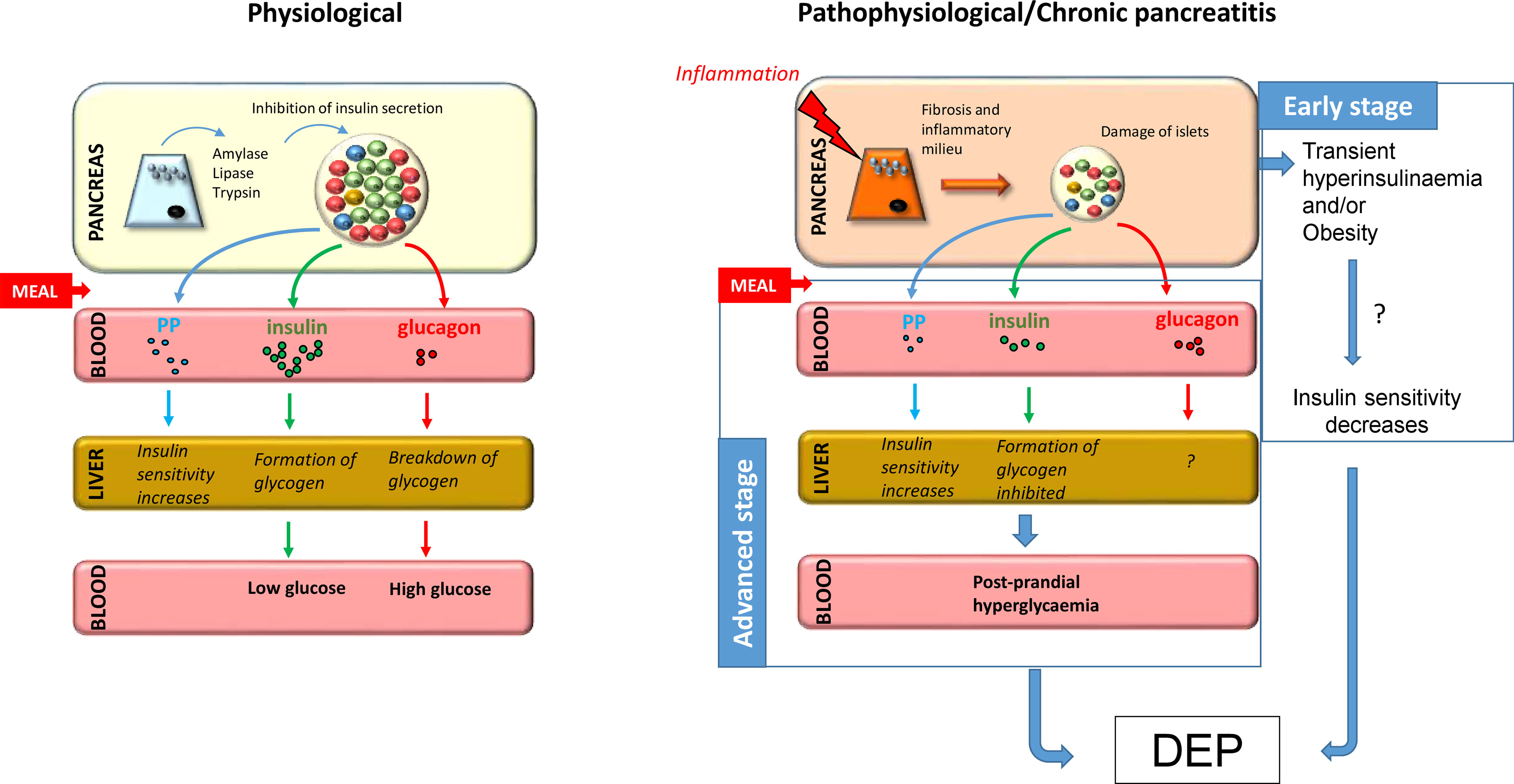
Figure 1 Putative mechanism for the development of DEP in CP. Under normal conditions, food ingestion triggers secretion of enzymes from acinar tissue, insulin from beta cells, and pancreatic polypeptide from PP cells and inhibits secretion of glucagon from alpha cells. In chronic pancreatitis, fibrosis and inflammatory milieu lead to destruction of islets and decreased hormone production, especially insulin and pancreatic polypeptide, resulting in decreased glycogen formation and increased hepatic IR, respectively. Taken together, this leads to post-prandial hyperglycaemia and predisposes patients to the development of diabetes at advanced stages. Furthermore, the early stage of the disease is often associated with hyperinsulinemia, which together with obesity, may play a role in the development of IR, which is a known precursor to diabetes.
Cystic Fibrosis-Related Diabetes
CF, the most common genetic disorder in Caucasian populations with a prevalence of 1 out of 3,500 individuals in the United States (133), is caused by mutations in the CFTR gene (134). Approximately, 2,000 CFTR gene mutations have been identified and classified into six groups depending on their biological effects (135). F508del, the most common mutation, causes an abnormal folding of the CFTR protein, which consequently degrades immediately after synthesis or does not function as a Cl− channel even when it reaches the cell membrane. The CFTR channel is located at the apical membrane of secretory epithelial cells and mediates the transport of Cl− and ions (136). The lungs and pancreas are the most affected organs in patients with CF. In the lungs, CFTR plays an essential role in the formation of airway surface liquid (ASL) (137) that lines the airway epithelium and ensures mucociliary clearance (138). Owing to the decreased Cl− secretion and increased Na+ absorption in CF, the volume and composition of ASL change, leading to airway surface dehydration and decreased mucociliary transport that favours airway infections (139). Persistent infections induce consecutive inflammation and greatly impairs respiratory function, ultimately leading to the death of patients with CF (140). The pancreas of patients with CF displays extensive fibrosis, fat infiltration and significant loss of islets (50%) (141). In the pancreas, CFTR is expressed on ductal cells and with the exchanger, responsible for the secretion of -rich pancreatic fluid (136). Defects in secretion increase the viscosity of the pancreatic fluid, which favours the formation of mucus plugs in the ductal tree and leads to premature activation of digestive enzymes, resulting in the destruction of the pancreas (142).
The introduction of novel therapies has considerably improved the survival of patients with CF in recent years. Currently, the median predicted survival age of patients with CF is 41–46 years, depending on the type of mutations and the gender of the patients (143, 144). A retrospective cohort analysis had shown that females have worse survival than males (144), for which the female hormone, oestrogen, is at least partly responsible. Oestrogen increases the incidence of the mucoid form of Pseudomonas aeruginosa in the lungs, which shows greater resistance to antibiotics and thus induces more severe inflammation (145). Nevertheless, the increased age of patients with CF has also been associated with an increased incidence of comorbidities. The most common comorbidity associated with CF is CF-related diabetes (CFRD) (8), a multifactorial disease that, although present more frequently in advanced age, can develop at any age (146). Over the age of 40, 83% of women and 64% of men with CF have been found to develop diabetes (147). Moreover, a strong correlation exists between the type of CFTR mutation and the development of diabetes. Patients with mild, class IV or class V mutations are less likely to develop diabetes than those with more severe, class I or class II mutations (147). Those with class I, II and III mutations exhibited significantly greater impairment in pancreatic exocrine functions compared to those with class IV and V mutations (148), which explains why diabetes is more common among class I and II mutations (147). The presence of diabetes substantially reduces the life expectancy of patients with CF (149), especially women (147, 150). Lewis et al. had shown that patients with CFRD had a 3.5 times higher mortality rate compared to those with CF (147). The shorter life expectancy among patients with CFRD can be mainly attributed to accelerated deterioration of lung function due to continuous bacterial infections. Hyperglycaemia in these patients provides energy for bacteria and promotes their growth (151). Furthermore, patients with CFRD present with poor nutritional status and microvascular complications (nephropathy, retinopathy and neuropathy) (9).
Diabetes in patients with CF is not considered T1DM or T2DM. Basically, severe damage to the exocrine pancreas destroys beta cells, resulting in decreased insulin secretion and therefore the development of CFRD. However, the exact molecular background for the development of diabetes is not fully elucidated in CF, partly due to the controversial role of CFTR on beta cells. One of the main reasons for this controversy is that CFTR is differentially expressed in different species and has various endocrine functions. Using isolated islets or a pancreatic beta cell line, mouse islets have been shown to express functionally active CFTR (152–156). Studies using genetically (F508del) or pharmacologically (CFTRinh172) developed CF mice have demonstrated characteristic changes in the morphology of the islets in the absence of CFTR (155, 156). Accordingly, although the size and insulin content of the islets decreased significantly, the cell number remained unchanged. In addition, centralisation of alpha cells can be observed. In both CF models, a non-hyperglycaemia-associated decrease in pancreatic and serum levels of insulin had been noted. Pharmacological inhibition of CFTR did not significantly affect serum glucagon levels but did increase it several folds in F508del mice (155, 156). The importance of CFTR in insulin secretion has also been confirmed in a mouse insulinoma cell line, MIN6, where genetic or pharmacological inhibition of CFTR caused a reduction in insulin secretion that was further decreased in the presence of oxidative stress (154). Guo et al. had shown that CFTR activation in beta cells is required for membrane depolarisation and calcium mobilisation associated with insulin secretion. They also demonstrated that incubation of F508del beta cells with the VX-809 corrector (Lumacaftor) dose-dependently increased insulin secretion (153). Nonetheless, oral administration of the CFTR corrector and activator (Lumacaftor and Ivacaftor) to patients with CF who have the F508del mutation did not improve glucose tolerance or insulin secretion (157, 158). In contrast to mice, Boom et al. found that CFTR was mainly localised to alpha cells in rat pancreatic islets (159). In the ferret CF model, the size and composition of islets show a dynamic change as the disease progresses (160). A comparison of CF ferrets at different stages showed that islet sizes and the number of alpha, beta and gamma cells increased significantly at more advanced stages. One study also demonstrated that the increased islet cell count presumably results from the transdifferentiation of ductal cells into islet cells, in which matrix metalloproteinase-7, a remodelling factor, plays an essential role (160).
Regarding the human pancreas, White et al. had recently shown that less than 1% of normal adult beta cells express CFTR (161). Further studies have indicated that insulin secretion defects in CFRD are not due to the lack of intrinsic CFTR function (162, 163). Hart et al. had shown that the absence of CFTR did not affect alpha and beta cell function and CFRD development much more related to beta cell loss and inflammatory cell infiltration (162). Moreover, Sun et al. found that CFTR regulates beta cells through pro-inflammatory factors released from exocrine cells (163). However, regardless of species, it is generally accepted that exocrine inflammation in CF damages the entire pancreas to such an extent that the number of beta cells and thus insulin secretion are considerably reduced (162). However, this hypothesis somewhat contradicts the fact that the remaining beta cells must produce enough insulin to prevent the development of diabetes, which suggests that CFTR intrinsically regulates insulin secretion and that the functional defect in residual beta cells causes the development of CFRD. Nevertheless, the development of CFRD with age suggests that the pancreas suffers progressive damage wherein a continuous decline of islet cells can be observed. The role of age in the pathomechanism of CFRD is also shown by the fact that insulin sensitivity also decreases over time in CF patients with glucose abnormalities (164). Unlike beta cells, however, much less information is available on the role of CFTR in alpha cells. In pancreatic alpha cells, CFTR inhibits glucagon secretion (165–167), presumably by stimulating KATP channels (167). Patients with CF exhibit dysregulated glucagon secretion, which probably also contributes to abnormalities in their glucose tolerance.
Conclusion
Considering that the exocrine pancreas and endocrine pancreas affect each other’s function through a number of pathways, it is not surprising to observe endocrine dysfunction in cases with exocrine insufficiency. During acute or chronic inflammation of the exocrine pancreas, the development of diabetes is highly dependent on the severity of the disease, whereas in CF, the development of diabetes has been found to increase with the length of the disease. However, all tree diseases are characterized by dysregulated hormone secretion. Given that early detection of disturbances in glucose homeostasis can prevent the development of more serious complications, monitoring carbohydrate metabolism in these patients is strongly recommended, as well as establishing appropriate diagnostic criteria of prime importance considering that DEP is often misdiagnosed as T2DM. After proper diagnosis of DEP, appropriate treatment of the disease constitutes another problem. Owing to the yet fully elucidated pathomechanism of DEP, developing specific therapies has remained difficult. Currently, treatment of DEP has been based on guidelines specific for the treatment of T2DM. Therefore, establishing guidelines that differentiate DEP according to not only other types of diabetes but also the different aetiologies thereof is greatly needed. Furthermore, there needs to be a strong emphasis on basic research considering that better understanding of the pathomechanism of the disease can substantially contribute to the identification of novel therapeutic targets.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This study was supported by the CFRD-SRC Grant (No. SRC 007), the National Research, Development and Innovation Office (SNN134497 to VV and K128222 to LC) grants, the New National Excellence Program of the Ministry of Human Capacities (UNKP-18-4), and the Slovenian Research Agency (research core funding nos. P3-0396 and I0-0029, as well as research projects nos. J3-9289, J4-9302, J1-9112, N3-0048, N3-0133, and N3-0170).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Das SL, Singh PP, Phillips AR, Murphy R, Windsor JA, Petrov MS. Newly Diagnosed Diabetes Mellitus After Acute Pancreatitis: A Systematic Review and Meta-Analysis. Gut (2014) 63(5):818–31. doi: 10.1136/gutjnl-2013-305062
2. Ewald N, Bretzel RG. Diabetes Mellitus Secondary to Pancreatic Diseases (Type 3c)–Are We Neglecting an Important Disease? Eur J Intern Med (2013) 24(3):203–6. doi: 10.1016/j.ejim.2012.12.017
3. Ewald N, Hardt PD. Diagnosis and Treatment of Diabetes Mellitus in Chronic Pancreatitis. World J Gastroenterol (2013) 19(42):7276–81. doi: 10.3748/wjg.v19.i42.7276
4. Gonzalez-Perez A, Schlienger RG, Rodriguez LA. Acute Pancreatitis in Association With Type 2 Diabetes and Antidiabetic Drugs: A Population-Based Cohort Study. Diabetes Care (2010) 33(12):2580–5. doi: 10.2337/dc10-0842
5. Noel RA, Braun DK, Patterson RE, Bloomgren GL. Increased Risk of Acute Pancreatitis and Biliary Disease Observed in Patients With Type 2 Diabetes: A Retrospective Cohort Study. Diabetes Care (2009) 32(5):834–8. doi: 10.2337/dc08-1755
6. Zhu X, Liu D, Wei Q, Lin H, Zhi M, Chen Y, et al. New-Onset Diabetes Mellitus After Chronic Pancreatitis Diagnosis: A Systematic Review and Meta-Analysis. Pancreas (2019) 48(7):868–75. doi: 10.1097/MPA.0000000000001359
7. Zhi M, Zhu X, Lugea A, Waldron RT, Pandol SJ, Li L. Incidence of New Onset Diabetes Mellitus Secondary to Acute Pancreatitis: A Systematic Review and Meta-Analysis. Front Physiol (2019) 10:637. doi: 10.3389/fphys.2019.00637
8. Doan LV, Madison LD. Cystic Fibrosis Related Diabetes (CFRD). Treasure Island (FL: StatPearls (2020).
9. Granados A, Chan CL, Ode KL, Moheet A, Moran A, Holl R. Cystic Fibrosis Related Diabetes: Pathophysiology, Screening and Diagnosis. J Cyst Fibros (2019) 18(Suppl 2):S3–9. doi: 10.1016/j.jcf.2019.08.016
10. Gilmour JA, Sykes J, Etchells E, Tullis E. Cystic Fibrosis-Related Diabetes Screening in Adults: A Gap Analysis and Evaluation of Accuracy of Glycated Hemoglobin Levels. Can J Diabetes (2019) 43(1):13–8. doi: 10.1016/j.jcjd.2018.04.008
11. Bhattamisra SK, Siang TC, Rong CY, Annan NC, Sean EHY, Xi LW, et al. Type-3c Diabetes Mellitus, Diabetes of Exocrine Pancreas - An Update. Curr Diabetes Rev (2019) 15(5):382–94. doi: 10.2174/1573399815666190115145702
12. Petrov MS, Basina M. DIAGNOSIS OF ENDOCRINE DISEASE: Diagnosing and Classifying Diabetes in Diseases of the Exocrine Pancreas. Eur J Endocrinol (2021) 184(4):R151–R63. doi: 10.1530/EJE-20-0974
13. Wynne K, Devereaux B, Dornhorst A. Diabetes of the Exocrine Pancreas. J Gastroenterol Hepatol (2019) 34(2):346–54. doi: 10.1111/jgh.14451
14. Petrov MS. DIAGNOSIS OF ENDOCRINE DISEASE: Post-Pancreatitis Diabetes Mellitus: Prime Time for Secondary Disease. Eur J Endocrinol (2021) 184(4):R137–R49. doi: 10.1530/EJE-20-0468
15. Johnston PC, Thompson J, McKee A, Hamill C, Wallace I. Diabetes and Chronic Pancreatitis: Considerations in the Holistic Management of an Often Neglected Disease. J Diabetes Res (2019) 2019:2487804. doi: 10.1155/2019/2487804
16. Moran A, Brunzell C, Cohen RC, Katz M, Marshall BC, Onady G, et al. Clinical Care Guidelines for Cystic Fibrosis-Related Diabetes: A Position Statement of the American Diabetes Association and a Clinical Practice Guideline of the Cystic Fibrosis Foundation, Endorsed by the Pediatric Endocrine Society. Diabetes Care (2010) 33(12):2697–708. doi: 10.2337/dc10-1768
17. Moran A, Pillay K, Becker D, Granados A, Hameed S, Acerini CL. ISPAD Clinical Practice Consensus Guidelines 2018: Management of Cystic Fibrosis-Related Diabetes in Children and Adolescents. Pediatr Diabetes (2018) 19(Suppl 27):64–74. doi: 10.1111/pedi.12732
18. Terzin V, Takacs R, Lengyel C, Varkonyi T, Wittmann T, Palinkas A, et al. Improved Glycemic Control in Pancreatic Diabetes Through Intensive Conservative Insulin Therapy. Pancreatology (2012) 12(2):100–3. doi: 10.1016/j.pan.2012.01.004
19. Hellman B, Wallgren A, Petersson B. Cytological Characteristics of the Exocrine Pancreatic Cells With Regard to Their Position in Relation to the Islets of Langerhans. A Study in Normal and Obese-Hyperglycaemic Mice. Acta Endocrinol (Copenh) (1962) 39:465–73. doi: 10.1530/acta.0.0390465
20. Campbell-Thompson ML, Kaddis JS, Wasserfall C, Haller MJ, Pugliese A, Schatz DA, et al. The Influence of Type 1 Diabetes on Pancreatic Weight. Diabetologia (2016) 59(1):217–21. doi: 10.1007/s00125-015-3752-z
21. Ferrer R, Medrano J, Diego M, Calpena R, Graells L, Molto M, et al. Effect of Exogenous Insulin and Glucagon on Exocrine Pancreatic Secretion in Rats In Vivo. Int J Pancreatol (2000) 28(1):67–75. doi: 10.1385/IJGC:28:1:67
22. Foulis AK, Stewart JA. The Pancreas in Recent-Onset Type 1 (Insulin-Dependent) Diabetes Mellitus: Insulin Content of Islets, Insulitis and Associated Changes in the Exocrine Acinar Tissue. Diabetologia (1984) 26(6):456–61. doi: 10.1007/BF00262221
23. Korc M, Owerbach D, Quinto C, Rutter WJ. Pancreatic Islet-Acinar Cell Interaction: Amylase Messenger RNA Levels Ar Determined by Insulin. Science (1981) 213(4505):351–3. doi: 10.1126/science.6166044
24. Otsuki M, Williams JA. Effect of Diabetes Mellitus on the Regulation of Enzyme Secretion by Isolated Rat Pancreatic Acini. J Clin Invest (1982) 70(1):148–56. doi: 10.1172/jci110588
25. Patel R, Singh J, Yago MD, Vilchez JR, Martinez-Victoria E, Manas M. Effect of Insulin on Exocrine Pancreatic Secretion in Healthy and Diabetic Anaesthetised Rats. Mol Cell Biochem (2004) 261(1-2):105–10. doi: 10.1023/b:mcbi.0000028744.13307.73
26. Saito A, Williams JA, Kanno T. Potentiation of Cholecystokinin-Induced Exocrine Secretion by Both Exogenous and Endogenous Insulin in Isolated and Perfused Rat Pancreata. J Clin Invest (1980) 65(4):777–82. doi: 10.1172/JCI109727
27. Soling HD, Unger KO. The Role of Insulin in the Regulation of -Amylase Synthesis in the Rat Pancreas. Eur J Clin Invest (1972) 2(4):199–212. doi: 10.1111/j.1365-2362.1972.tb00645.x
28. Pierzynowski SG, Goncharova K, Gregory PC, Westrom B, Podpryatov SE, Podpriatov SS, et al. Experiments Suggesting Extra-Digestive Effects of Enteral Pancreatic Amylase and Its Peptides on Glucose Homeostasis in a Pig Model. Sci Rep (2017) 7(1):8628. doi: 10.1038/s41598-017-07387-2
29. Lozinska L, Westrom B, Prykhodko O, Lindqvist A, Wierup N, Ahren B, et al. Decreased Insulin Secretion and Glucose Clearance in Exocrine Pancreas-Insufficient Pigs. Exp Physiol (2016) 101(1):100–12. doi: 10.1113/EP085431
30. Pierzynowska K, Oredsson S, Pierzynowski S. Amylase-Dependent Regulation of Glucose Metabolism and Insulin/Glucagon Secretion in the Streptozotocin-Induced Diabetic Pig Model and in a Rat Pancreatic Beta-Cell Line, BRIN-Bd11. J Diabetes Res (2020) 2020:2148740. doi: 10.1155/2020/2148740
31. Nozawa F, Yalniz M, Saruc M, Standop J, Egami H, Pour PM. Effects of Porcine Pancreatic Enzymes on the Pancreas of Hamsters. Part 2: Carcinogenesis Studies. JOP (2012) 13(5):482–7. doi: 10.6092/1590-8577/663
32. Nozawa F, Yalniz M, Saruc M, Standop J, Egami H, Pour PM. Effects of Fungal Pancreatic Enzymes on the Function of Islet Cells in Syrian Golden Hamsters. JOP (2013) 14(3):228–336. doi: 10.6092/1590-8577/903
33. Saruc M, Nozawa F, Yalniz M, Itami A, Pour PM. Effects of Porcine Pancreatic Enzymes on the Pancreas of Hamsters. Part 1: Basic Studies. JOP (2012) 13(5):476–81. doi: 10.6092/1590-8577/522
34. Kebede M, Alquier T, Latour MG, Semache M, Tremblay C, Poitout V. The Fatty Acid Receptor GPR40 Plays a Role in Insulin Secretion In Vivo After High-Fat Feeding. Diabetes (2008) 57(9):2432–7. doi: 10.2337/db08-0553
35. Proks P, Ashcroft FM. Modification of K-ATP Channels in Pancreatic Beta-Cells by Trypsin. Pflugers Arch (1993) 424(1):63–72. doi: 10.1007/BF00375103
36. Lee K, Ozanne SE, Rowe IC, Hales CN, Ashford ML. The Effects of Trypsin on ATP-Sensitive Potassium Channel Properties and Sulfonylurea Receptors in the CRI-G1 Insulin-Secreting Cell Line. Mol Pharmacol (1994) 46(1):176–85.
37. Wei C, Geras-Raaka E, Marcus-Samuels B, Oron Y, Gershengorn MC. Trypsin and Thrombin Accelerate Aggregation of Human Endocrine Pancreas Precursor Cells. J Cell Physiol (2006) 206(2):322–8. doi: 10.1002/jcp.20459
38. Bonora E, Rizzi C, Lesi C, Berra P, Coscelli C, Butturini U. Insulin and C-Peptide Plasma Levels in Patients With Severe Chronic Pancreatitis and Fasting Normoglycemia. Dig Dis Sci (1988) 33(6):732–6. doi: 10.1007/BF01540438
39. Pendharkar SA, Asrani VM, Xiao AY, Yoon HD, Murphy R, Windsor JA, et al. Relationship Between Pancreatic Hormones and Glucose Metabolism: A Cross-Sectional Study in Patients After Acute Pancreatitis. Am J Physiol Gastrointest Liver Physiol (2016) 311(1):G50–8. doi: 10.1152/ajpgi.00074.2016
40. Gray SL, Donald C, Jetha A, Covey SD, Kieffer TJ. Hyperinsulinemia Precedes Insulin Resistance in Mice Lacking Pancreatic Beta-Cell Leptin Signaling. Endocrinology (2010) 151(9):4178–86. doi: 10.1210/en.2010-0102
41. Petrides AS, Groop LC, Riely CA, DeFronzo RA. Effect of Physiologic Hyperinsulinemia on Glucose and Lipid Metabolism in Cirrhosis. J Clin Invest (1991) 88(2):561–70. doi: 10.1172/JCI115340
42. Zhang S, Wei M, Yue M, Wang P, Yin X, Wang L, et al. Hyperinsulinemia Precedes Insulin Resistance in Offspring Rats Exposed to Angiotensin II Type 1 Autoantibody In Utero. Endocrine (2018) 62(3):588–601. doi: 10.1007/s12020-018-1700-7
43. Lankisch PG, Apte M, Banks PA. Acute Pancreatitis. Lancet (2015) 386(9988):85–96. doi: 10.1016/S0140-6736(14)60649-8
44. Umapathy C, Raina A, Saligram S, Tang G, Papachristou GI, Rabinovitz M, et al. Natural History After Acute Necrotizing Pancreatitis: A Large US Tertiary Care Experience. J Gastrointest Surg (2016) 20(11):1844–53. doi: 10.1007/s11605-016-3264-2
45. Pelli H, Lappalainen-Lehto R, Piironen A, Jarvinen S, Sand J, Nordback I. Pancreatic Damage After the First Episode of Acute Alcoholic Pancreatitis and Its Association With the Later Recurrence Rate. Pancreatology (2009) 9(3):245–51. doi: 10.1159/000212089
46. Halonen KI, Pettila V, Leppaniemi AK, Kemppainen EA, Puolakkainen PA, Haapiainen RK. Long-Term Health-Related Quality of Life in Survivors of Severe Acute Pancreatitis. Intensive Care Med (2003) 29(5):782–6. doi: 10.1007/s00134-003-1700-8
47. Tu J, Yang Y, Zhang J, Yang Q, Lu G, Li B, et al. Effect of the Disease Severity on the Risk of Developing New-Onset Diabetes After Acute Pancreatitis. Med (Baltimore) (2018) 97(22):e10713. doi: 10.1097/MD.0000000000010713
48. Ibars EP, Sanchez de Rojas EA, Quereda LA, Ramis RF, Sanjuan VM, Peris RT. Pancreatic Function After Acute Biliary Pancreatitis: Does it Change? World J Surg (2002) 26(4):479–86. doi: 10.1007/s00268-001-0253-7
49. Shen HN, Yang CC, Chang YH, Lu CL, Li CY. Risk of Diabetes Mellitus After First-Attack Acute Pancreatitis: A National Population-Based Study. Am J Gastroenterol (2015) 110(12):1698–706. doi: 10.1038/ajg.2015.356
50. Lee YK, Huang MY, Hsu CY, Su YC. Bidirectional Relationship Between Diabetes and Acute Pancreatitis: A Population-Based Cohort Study in Taiwan. Med (Baltimore) (2016) 95(2):e2448. doi: 10.1097/MD.0000000000002448
51. Tu J, Zhang J, Ke L, Yang Y, Yang Q, Lu G, et al. Endocrine and Exocrine Pancreatic Insufficiency After Acute Pancreatitis: Long-Term Follow-Up Study. BMC Gastroenterol (2017) 17(1):114. doi: 10.1186/s12876-017-0663-0
52. Ho TW, Wu JM, Kuo TC, Yang CY, Lai HS, Hsieh SH, et al. Change of Both Endocrine and Exocrine Insufficiencies After Acute Pancreatitis in Non-Diabetic Patients: A Nationwide Population-Based Study. Med (Baltimore) (2015) 94(27):e1123. doi: 10.1097/MD.0000000000001123
53. Wu D, Xu Y, Zeng Y, Wang X. Endocrine Pancreatic Function Changes After Acute Pancreatitis. Pancreas (2011) 40(7):1006–11. doi: 10.1097/MPA.0b013e31821fde3f
54. Vipperla K, Papachristou GI, Slivka A, Whitcomb DC, Yadav D. Risk of New-Onset Diabetes Is Determined by Severity of Acute Pancreatitis. Pancreas (2016) 45(1):e14–5. doi: 10.1097/MPA.0000000000000536
55. Malecka-Panas E, Gasiorowska A, Kropiwnicka A, Zlobinska A, Drzewoski J. Endocrine Pancreatic Function in Patients After Acute Pancreatitis. Hepatogastroenterology (2002) 49(48):1707–12. doi: 10.1097/mpa.0b013e31821fde3f
56. Chen L, Chen R, Wang H, Liang F. Mechanisms Linking Inflammation to Insulin Resistance. Int J Endocrinol (2015) 2015:508409. doi: 10.1155/2015/508409
57. Khodabandehloo H, Gorgani-Firuzjaee S, Panahi G, Meshkani R. Molecular and Cellular Mechanisms Linking Inflammation to Insulin Resistance and Beta-Cell Dysfunction. Transl Res (2016) 167(1):228–56. doi: 10.1016/j.trsl.2015.08.011
58. Singh RG, Pendharkar SA, Cervantes A, Cho J, Miranda-Soberanis V, Petrov MS. Abdominal Obesity and Insulin Resistance After an Episode of Acute Pancreatitis. Dig Liver Dis (2018) 50(10):1081–7. doi: 10.1016/j.dld.2018.04.023
59. Ko J, Skudder-Hill L, Cho J, Bharmal SH, Petrov MS. The Relationship Between Abdominal Fat Phenotypes and Insulin Resistance in Non-Obese Individuals After Acute Pancreatitis. Nutrients (2020) 12(9):568–78. doi: 10.3390/nu12092883
60. Gillies N, Pendharkar SA, Asrani VM, Mathew J, Windsor JA, Petrov MS. Interleukin-6 is Associated With Chronic Hyperglycemia and Insulin Resistance in Patients After Acute Pancreatitis. Pancreatology (2016) 16(5):748–55. doi: 10.1016/j.pan.2016.06.661
61. Senn JJ, Klover PJ, Nowak IA, Mooney RA. Interleukin-6 Induces Cellular Insulin Resistance in Hepatocytes. Diabetes (2002) 51(12):3391–9. doi: 10.2337/diabetes.51.12.3391
62. Cho SK, Huh JH, Yoo JS, Kim JW, Lee KJ. HOMA-Estimated Insulin Resistance as an Independent Prognostic Factor in Patients With Acute Pancreatitis. Sci Rep (2019) 9(1):14894. doi: 10.1038/s41598-019-51466-5
63. Abe N, Watanabe T, Ozawa S, Masaki T, Mori T, Sugiyama M, et al. Pancreatic Endocrine Function and Glucose Transporter (GLUT)-2 Expression in Rat Acute Pancreatitis. Pancreas (2002) 25(2):149–53. doi: 10.1097/00006676-200208000-00006
64. Takacs T, Hegyi P, Jarmay K, Czako L, Gog C, Rakonczay Z Jr., et al. Cholecystokinin Fails to Promote Pancreatic Regeneration in Diabetic Rats Following the Induction of Experimental Pancreatitis. Pharmacol Res (2001) 44(5):363–72. doi: 10.1006/phrs.2001.0843
65. Kinami Y, Mura T, Sugii M, Miyazaki I. Function of Pancreatic Endocrine Cells in Experimental Acute Pancreatitis. World J Surg (1982) 6(4):471–7. doi: 10.1007/BF01657685
66. Deng WH, Chen C, Wang WX, Yu J, Li JY, Liu L. Effects of ORP150 on Appearance and Function of Pancreatic Beta Cells Following Acute Necrotizing Pancreatitis. Pathol Res Pract (2011) 207(6):370–6. doi: 10.1016/j.prp.2011.03.006
67. Hong YP, Guo WY, Wang WX, Zhao L, Xiang MW, Mei FC, et al. 4-Phenylbutyric Acid Attenuates Pancreatic Beta-Cell Injury in Rats With Experimental Severe Acute Pancreatitis. Int J Endocrinol (2016) 2016:4592346. doi: 10.1155/2016/4592346
68. Qader SS, Ekelund M, Andersson R, Obermuller S, Salehi A. Acute Pancreatitis, Expression of Inducible Nitric Oxide Synthase and Defective Insulin Secretion. Cell Tissue Res (2003) 313(3):271–9. doi: 10.1007/s00441-003-0764-7
69. Sun J, Furio L, Mecheri R, van der Does AM, Lundeberg E, Saveanu L, et al. Pancreatic Beta-Cells Limit Autoimmune Diabetes via an Immunoregulatory Antimicrobial Peptide Expressed Under the Influence of the Gut Microbiota. Immunity (2015) 43(2):304–17. doi: 10.1016/j.immuni.2015.07.013
70. Witt H, Apte MV, Keim V, Wilson JS. Chronic Pancreatitis: Challenges and Advances in Pathogenesis, Genetics, Diagnosis, and Therapy. Gastroenterology (2007) 132(4):1557–73. doi: 10.1053/j.gastro.2007.03.001
71. Czako L, Hegyi P, Rakonczay Z Jr., Wittmann T, Otsuki M. Interactions Between the Endocrine and Exocrine Pancreas and Their Clinical Relevance. Pancreatology (2009) 9(4):351–9. doi: 10.1159/000181169
72. Ewald N, Kaufmann C, Raspe A, Kloer HU, Bretzel RG, Hardt PD. Prevalence of Diabetes Mellitus Secondary to Pancreatic Diseases (Type 3c). Diabetes Metab Res Rev (2012) 28(4):338–42. doi: 10.1002/dmrr.2260
73. Pan J, Xin L, Wang D, Liao Z, Lin JH, Li BR, et al. Risk Factors for Diabetes Mellitus in Chronic Pancreatitis: A Cohort of 2,011 Patients. Med (Baltimore) (2016) 95(14):e3251. doi: 10.1097/MD.0000000000003251
74. Wang W, Guo Y, Liao Z, Zou DW, Jin ZD, Zou DJ, et al. Occurrence of and Risk Factors for Diabetes Mellitus in Chinese Patients With Chronic Pancreatitis. Pancreas (2011) 40(2):206–12. doi: 10.1097/mpa.0b013e31820032ae
75. Hwang HK, Park J, Choi SH, Kang CM, Lee WJ. Predicting New-Onset Diabetes After Minimally Invasive Subtotal Distal Pancreatectomy in Benign and Borderline Malignant Lesions of the Pancreas. Med (Baltimore) (2017) 96(51):e9404. doi: 10.1097/MD.0000000000009404
76. Yu J, Sun R, Han X, Liu Z. New-Onset Diabetes Mellitus After Distal Pancreatectomy: A Systematic Review and Meta-Analysis. J Laparoendosc Adv Surg Tech A (2020) 30(11):1215–22. doi: 10.1089/lap.2020.0090
77. Malka D, Hammel P, Sauvanet A, Rufat P, O'Toole D, Bardet P, et al. Risk Factors for Diabetes Mellitus in Chronic Pancreatitis. Gastroenterology (2000) 119(5):1324–32. doi: 10.1053/gast.2000.19286
78. Dominguez-Munoz JE, Drewes AM, Lindkvist B, Ewald N, Czako L, Rosendahl J, et al. Recommendations From the United European Gastroenterology Evidence-Based Guidelines for the Diagnosis and Therapy of Chronic Pancreatitis. Pancreatology (2018) 18(8):847–54. doi: 10.1016/j.pan.2018.09.016
79. Nealon WH, Townsend CM Jr., Thompson JC. The Time Course of Beta Cell Dysfunction in Chronic Ethanol-Induced Pancreatitis: A Prospective Analysis. Surgery (1988) 104(6):1074–9. doi: 10.5555/uri:pii:0039606088901705
80. Roy A, Sahoo J, Kamalanathan S, Naik D, Mohan P, Pottakkat B. Islet Cell Dysfunction in Patients With Chronic Pancreatitis. World J Diabetes (2020) 11(7):280–92. doi: 10.4239/wjd.v11.i7.280
81. Sasikala M, Talukdar R, Pavan kumar P, Radhika G, Rao GV, Pradeep R, et al. Beta-Cell Dysfunction in Chronic Pancreatitis. Dig Dis Sci (2012) 57(7):1764–72. doi: 10.1007/s10620-012-2086-7
82. Lundberg R, Beilman GJ, Dunn TB, Pruett TL, Freeman ML, Ptacek PE, et al. Early Alterations in Glycemic Control and Pancreatic Endocrine Function in Nondiabetic Patients With Chronic Pancreatitis. Pancreas (2016) 45(4):565–71. doi: 10.1097/MPA.0000000000000491
83. Mitnala S, Pondugala PK, Guduru VR, Rabella P, Thiyyari J, Chivukula S, et al. Reduced Expression of PDX-1 is Associated With Decreased Beta Cell Function in Chronic Pancreatitis. Pancreas (2010) 39(6):856–62. doi: 10.1097/MPA.0b013e3181d6bc69
84. Xiao X, Fischbach S, Zhang T, Chen C, Sheng Q, Zimmerman R, et al. SMAD3/Stat3 Signaling Mediates Beta-Cell Epithelial-Mesenchymal Transition in Chronic Pancreatitis-Related Diabetes. Diabetes (2017) 66(10):2646–58. doi: 10.2337/db17-0537
85. Sanvito F, Nichols A, Herrera PL, Huarte J, Wohlwend A, Vassalli JD, et al. TGF-Beta 1 Overexpression in Murine Pancreas Induces Chronic Pancreatitis and, Together With TNF-Alpha, Triggers Insulin-Dependent Diabetes. Biochem Biophys Res Commun (1995) 217(3):1279–86. doi: 10.1006/bbrc.1995.2906
86. Andersson AK, Flodstrom M, Sandler S. Cytokine-Induced Inhibition of Insulin Release From Mouse Pancreatic Beta-Cells Deficient in Inducible Nitric Oxide Synthase. Biochem Biophys Res Commun (2001) 281(2):396–403. doi: 10.1006/bbrc.2001.4361
87. Pavan Kumar P, Radhika G, Rao GV, Pradeep R, Subramanyam C, Talukdar R, et al. Interferon Gamma and Glycemic Status in Diabetes Associated With Chronic Pancreatitis. Pancreatology (2012) 12(1):65–70. doi: 10.1016/j.pan.2011.12.005
88. Petrov MS. Panorama of Mediators in Postpancreatitis Diabetes Mellitus. Curr Opin Gastroenterol (2020) 36(5):443–51. doi: 10.1097/MOG.0000000000000654
89. Talukdar R, Sasikala M, Pavan Kumar P, Rao GV, Pradeep R, Reddy DN. T-Helper Cell-Mediated Islet Inflammation Contributes to Beta-Cell Dysfunction in Chronic Pancreatitis. Pancreas (2016) 45(3):434–42. doi: 10.1097/MPA.0000000000000479
90. Klover PJ, Mooney RA. Hepatocytes: Critical for Glucose Homeostasis. Int J Biochem Cell Biol (2004) 36(5):753–8. doi: 10.1016/j.biocel.2003.10.002
91. Andersen DK, Ruiz CL, Burant CF. Insulin Regulation of Hepatic Glucose Transporter Protein is Impaired in Chronic Pancreatitis. Ann Surg (1994) 219(6):679–86. doi: 10.1097/00000658-199406000-00011
92. Nathan JD, Zdankiewicz PD, Wang J, Spector SA, Aspelund G, Jena BP, et al. Impaired Hepatocyte Glucose Transport Protein (GLUT2) Internalization in Chronic Pancreatitis. Pancreas (2001) 22(2):172–8. doi: 10.1097/00006676-200103000-00010
93. Eisenberg ML, Maker AV, Slezak LA, Nathan JD, Sritharan KC, Jena BP, et al. Insulin Receptor (IR) and Glucose Transporter 2 (GLUT2) Proteins Form a Complex on the Rat Hepatocyte Membrane. Cell Physiol Biochem (2005) 15(1-4):51–8. doi: 10.1159/000083638
94. Aslam M, Vijayasarathy K, Talukdar R, Sasikala M, Nageshwar Reddy D. Reduced Pancreatic Polypeptide Response is Associated With Early Alteration of Glycemic Control in Chronic Pancreatitis. Diabetes Res Clin Pract (2020) 160:107993. doi: 10.1016/j.diabres.2019.107993
95. Sliwinska-Mosson M, Marek G, Milnerowicz H. The Role of Pancreatic Polypeptide in Pancreatic Diseases. Adv Clin Exp Med (2017) 26(9):1447–55. doi: 10.17219/acem/65094
96. Sliwinska-Mosson M, Milnerowicz H. Distribution of Pancreatic Polypeptide-Secreting Endocrine Cells in Nondiabetic and Diabetic Cases. Appl Immunohistochem Mol Morphol (2017) 25(6):422–31. doi: 10.1097/PAI.0000000000000310
97. Valenzuela JE, Taylor IL, Walsh JH. Pancreatic Polypeptide Response in Patients With Chronic Pancreatitis. Dig Dis Sci (1979) 24(11):862–4. doi: 10.1007/BF01324903
98. Bastidas JA, Couse NF, Yeo CJ, Schmieg RE Jr., Andersen DK, Gingerich RL, et al. The Effect of Pancreatic Polypeptide Infusion on Glucose Tolerance and Insulin Response in Longitudinally Studied Pancreatitis-Induced Diabetes. Surgery (1990) 107(6):661–8.
99. Seymour NE, Volpert AR, Lee EL, Andersen DK, Hernandez C. Alterations in Hepatocyte Insulin Binding in Chronic Pancreatitis: Effects of Pancreatic Polypeptide. Am J Surg (1995) 169(1):105–9; discussion 10. doi: 10.1016/s0002-9610(99)80117-2
100. Seymour NE, Volpert AR, Andersen DK. Regulation of Hepatic Insulin Receptors by Pancreatic Polypeptide in Fasting and Feeding. J Surg Res (1996) 65(1):1–4. doi: 10.1006/jsre.1996.9999
101. Brunicardi FC, Chaiken RL, Ryan AS, Seymour NE, Hoffmann JA, Lebovitz HE, et al. Pancreatic Polypeptide Administration Improves Abnormal Glucose Metabolism in Patients With Chronic Pancreatitis. J Clin Endocrinol Metab (1996) 81(10):3566–72. doi: 10.1210/jcem.81.10.8855802
102. Goldstein JA, Kirwin JD, Seymour NE, Trachtenberg JE, Rademaker EA, Andersen DK. Reversal of In Vitro Hepatic Insulin Resistance in Chronic Pancreatitis by Pancreatic Polypeptide in the Rat. Surgery (1989) 106(6):1128–32; discussion 32-3. doi: 10.5555/uri:pii:0039606089903176
103. Sun YS, Brunicardi FC, Druck P, Walfisch S, Berlin SA, Chance RE, et al. Reversal of Abnormal Glucose Metabolism in Chronic Pancreatitis by Administration of Pancreatic Polypeptide. Am J Surg (1986) 151(1):130–40. doi: 10.1016/0002-9610(86)90023-1
104. Banerjee A, Onyuksel H. A Novel Peptide Nanomedicine for Treatment of Pancreatogenic Diabetes. Nanomedicine (2013) 9(6):722–8. doi: 10.1016/j.nano.2012.12.005
105. Rabiee A, Galiatsatos P, Salas-Carrillo R, Thompson MJ, Andersen DK, Elahi D. Pancreatic Polypeptide Administration Enhances Insulin Sensitivity and Reduces the Insulin Requirement of Patients on Insulin Pump Therapy. J Diabetes Sci Technol (2011) 5(6):1521–8. doi: 10.1177/193229681100500629
106. Muggeo M, Moghetti P, Faronato PP, Valerio A, Tiengo A, Del Prato S, et al. Insulin Receptors on Circulating Blood Cells From Patients With Pancreatogenic Diabetes: A Comparison With Type I Diabetes and Normal Subjects. J Endocrinol Invest (1987) 10(3):311–9. doi: 10.1007/BF03348136
107. Nosadini R, del Prato S, Tiengo A, Duner E, Toffolo G, Cobelli C, et al. Insulin Sensitivity, Binding, and Kinetics in Pancreatogenic and Type I Diabetes. Diabetes (1982) 31(4 Pt 1):346–55. doi: 10.2337/diab.31.4.346
108. Bhatnagar A, Wig JD, Majumdar S. Immunological Findings in Acute and Chronic Pancreatitis. ANZ J Surg (2003) 73(1–2):59–64. doi: 10.1046/j.1445-2197.2003.02621.x
109. Gasiorowska A, Talar-Wojnarowska R, Kaczka A, Borkowska A, Czupryniak L, Małecka-Panas E. Subclinical Inflammation and Endothelial Dysfunction in Patients With Chronic Pancreatitis and Newly Diagnosed Pancreatic Cancer. Digestive Dis Sci (2016) 61(4):1121–9. doi: 10.1007/s10620-015-3972-6
110. Shoelson SE, Lee J, Goldfine AB. Inflammation and Insulin Resistance. J Clin Invest (2006) 116(7):1793–801. doi: 10.1172/JCI29069
111. Hotamisligil GS. Molecular Mechanisms of Insulin Resistance and the Role of the Adipocyte. Int J Obes related Metab Disord J Int Assoc Study Obes (2000) 24(Suppl 4):S23–7. doi: 10.1038/sj.ijo.0801497
112. Kahn SE, Hull RL, Utzschneider KM. Mechanisms Linking Obesity to Insulin Resistance and Type 2 Diabetes. Nature (2006) 444(7121):840–6. doi: 10.1038/nature05482
113. Zhou X, You S. Rosiglitazone Inhibits Hepatic Insulin Resistance Induced by Chronic Pancreatitis and IKK-β/NF-κb Expression in Liver. Pancreas (2014) 43(8):1291–8. doi: 10.1097/mpa.0000000000000173
114. Wentworth JM, Zhang JG, Bandala-Sanchez E, Naselli G, Liu R, Ritchie M, et al. Interferon-Gamma Released From Omental Adipose Tissue of Insulin-Resistant Humans Alters Adipocyte Phenotype and Impairs Response to Insulin and Adiponectin Release. Int J Obes (2017) 41(12):1782–9. doi: 10.1038/ijo.2017.180
115. Lukic L, Lalic NM, Rajkovic N, Jotic A, Lalic K, Milicic T, et al. Hypertension in Obese Type 2 Diabetes Patients is Associated With Increases in Insulin Resistance and IL-6 Cytokine Levels: Potential Targets for an Efficient Preventive Intervention. Int J Environ Res Public Health (2014) 11(4):3586–98. doi: 10.3390/ijerph110403586
116. Serrano-Marco L, Barroso E, El Kochairi I, Palomer X, Michalik L, Wahli W, et al. The Peroxisome Proliferator-Activated Receptor (PPAR) β/δ Agonist GW501516 Inhibits IL-6-Induced Signal Transducer and Activator of Transcription 3 (STAT3) Activation and Insulin Resistance in Human Liver Cells. Diabetologia (2012) 55(3):743–51. doi: 10.1007/s00125-011-2401-4
117. Dou L, Zhao T, Wang L, Huang X, Jiao J, Gao D, et al. miR-200s Contribute to Interleukin-6 (IL-6)-Induced Insulin Resistance in Hepatocytes. J Biol Chem (2013) 288(31):22596–606. doi: 10.1074/jbc.M112.423145
118. Kim TH, Choi SE, Ha ES, Jung JG, Han SJ, Kim HJ, et al. IL-6 Induction of TLR-4 Gene Expression via STAT3 has an Effect on Insulin Resistance in Human Skeletal Muscle. Acta diabetologica (2013) 50(2):189–200. doi: 10.1007/s00592-011-0259-z
119. Yin J, Hao Z, Ma Y, Liao S, Li X, Fu J, et al. Concomitant Activation of the PI3K/Akt and ERK1/2 Signalling is Involved in Cyclic Compressive Force-Induced IL-6 Secretion in MLO-Y4 Cells. Cell Biol Int (2014) 38(5):591–8. doi: 10.1002/cbin.10235
120. Okumura S, Kaido T, Hamaguchi Y, Fujimoto Y, Masui T, Mizumoto M, et al. Impact of Preoperative Quality as Well as Quantity of Skeletal Muscle on Survival After Resection of Pancreatic Cancer. Surgery (2015) 157(6):1088–98. doi: 10.1016/j.surg.2015.02.002
121. Sternby H, Mahle M, Linder N, Erichson-Kirst L, Verdonk RC, Dimova A, et al. Mean Muscle Attenuation Correlates With Severe Acute Pancreatitis Unlike Visceral Adipose Tissue and Subcutaneous Adipose Tissue. United Eur Gastroenterol J (2019) 7(10):1312–20. doi: 10.1177/2050640619882520
122. Daniele G, Guardado Mendoza R, Winnier D, Fiorentino TV, Pengou Z, Cornell J, et al. The Inflammatory Status Score Including IL-6, TNF-α, Osteopontin, Fractalkine, MCP-1 and Adiponectin Underlies Whole-Body Insulin Resistance and Hyperglycemia in Type 2 Diabetes Mellitus. Acta Diabetologica (2014) 51(1):123–31. doi: 10.1007/s00592-013-0543-1
123. Osborn O, Olefsky JM. The Cellular and Signaling Networks Linking the Immune System and Metabolism in Disease. Nat Med (2012) 18(3):363–74. doi: 10.1038/nm.2627
124. Kumar KVSH, Manrai M, Sood AK, Sharma R. A Clinical Study of Insulin Resistance in Patients With Chronic Pancreatitis. Diabetes Metab Syndrome: Clin Res Rev (2017) 11:S283–6. doi: 10.1016/j.dsx.2017.03.003
125. Schrader H, Menge BA, Zeidler C, Ritter PR, Tannapfel A, Uhl W, et al. Determinants of Glucose Control in Patients With Chronic Pancreatitis. Diabetologia (2010) 53(6):1062–9. doi: 10.1007/s00125-010-1705-0
126. Sempere L, Martinez J, de Madaria E, Lozano B, Sanchez-Paya J, Jover R, et al. Obesity and Fat Distribution Imply a Greater Systemic Inflammatory Response and a Worse Prognosis in Acute Pancreatitis. Pancreatology (2008) 8(3):257–64. doi: 10.1159/000134273
127. Donowitz M, Hendler R, Spiro HM, Binder HJ, Felig P. Glucagon Secretion in Acute and Chronic Pancreatitis. Ann Intern Med (1975) 83(6):778–81. doi: 10.7326/0003-4819-83-6-778
128. Mumme L, Breuer TGK, Rohrer S, Schenker N, Menge BA, Holst JJ, et al. Defects in Alpha-Cell Function in Patients With Diabetes Due to Chronic Pancreatitis Compared With Patients With Type 2 Diabetes and Healthy Individuals. Diabetes Care (2017) 40(10):1314–22. doi: 10.2337/dc17-0792
129. Knop FK, Vilsboll T, Larsen S, Madsbad S, Holst JJ, Krarup T. Glucagon Suppression During OGTT Worsens While Suppression During IVGTT Sustains Alongside Development of Glucose Intolerance in Patients With Chronic Pancreatitis. Regul Pept (2010) 164(2–3):144–50. doi: 10.1016/j.regpep.2010.05.011
130. Larsen S, Hilsted J, Philipsen EK, Tronier B, Christensen NJ, Damkjaer Nielsen M, et al. Glucose Counterregulation in Diabetes Secondary to Chronic Pancreatitis. Metabolism (1990) 39(2):138–43. doi: 10.1016/0026-0495(90)90066-l
131. Linde J, Nilsson LH, Barany FR. Diabetes and Hypoglycemia in Chronic Pancreatitis. Scand J Gastroenterol (1977) 12(3):369–73. doi: 10.3109/00365527709180943
132. Webb MA, Chen JJ, James RFL, Davies MJ, Dennison AR. Elevated Levels of Alpha Cells Emanating From the Pancreatic Ducts of a Patient With a Low BMI and Chronic Pancreatitis. Cell Transplant (2018) 27(6):902–6. doi: 10.1177/0963689718755707
133. Mehta G, Macek M Jr., Mehta A, European Registry Working G. Cystic Fibrosis Across Europe: EuroCareCF Analysis of Demographic Data From 35 Countries. J Cyst Fibros (2010) 9(Suppl 2):S5–21. doi: 10.1016/j.jcf.2010.08.002
134. Kerem BS, Zielenski J, Markiewicz D, Bozon D, Gazit E, Yahav J, et al. Identification of Mutations in Regions Corresponding to the Two Putative Nucleotide (ATP)-Binding Folds of the Cystic Fibrosis Gene. Proc Natl Acad Sci USA (1990) 87(21):8447–51. doi: 10.1073/pnas.87.21.8447
135. Zielenski J, Tsui LC. Cystic Fibrosis: Genotypic and Phenotypic Variations. Annu Rev Genet (1995) 29:777–807. doi: 10.1146/annurev.ge.29.120195.004021
136. Saint-Criq V, Gray MA. Role of CFTR in Epithelial Physiology. Cell Mol Life Sci (2017) 74(1):93–115. doi: 10.1007/s00018-016-2391-y
137. Tarran R, Loewen ME, Paradiso AM, Olsen JC, Gray MA, Argent BE, et al. Regulation of Murine Airway Surface Liquid Volume by CFTR and Ca2+-Activated Cl- Conductances. J Gen Physiol (2002) 120(3):407–18. doi: 10.1085/jgp.20028599
138. Knowles MR, Boucher RC. Mucus Clearance as a Primary Innate Defense Mechanism for Mammalian Airways. J Clin Invest (2002) 109(5):571–7. doi: 10.1172/JCI15217
139. Johnson DC. Airway Mucus Function and Dysfunction. N Engl J Med (2011) 364(10):978. doi: 10.1056/NEJMc1014719
140. Knapp EA, Fink AK, Goss CH, Sewall A, Ostrenga J, Dowd C, et al. The Cystic Fibrosis Foundation Patient Registry. Design and Methods of a National Observational Disease Registry. Ann Am Thorac Soc (2016) 13(7):1173–9. doi: 10.1513/AnnalsATS.201511-781OC
141. Kelly A, Moran A. Update on Cystic Fibrosis-Related Diabetes. J Cyst Fibros (2013) 12(4):318–31. doi: 10.1016/j.jcf.2013.02.008
142. Quinton PM. Cystic Fibrosis: Impaired Bicarbonate Secretion and Mucoviscidosis. Lancet (2008) 372(9636):415–7. doi: 10.1016/S0140-6736(08)61162-9
143. Keogh RH, Szczesniak R, Taylor-Robinson D, Bilton D. Up-To-Date and Projected Estimates of Survival for People With Cystic Fibrosis Using Baseline Characteristics: A Longitudinal Study Using UK Patient Registry Data. J Cyst Fibros (2018) 17(2):218–27. doi: 10.1016/j.jcf.2017.11.019
144. Harness-Brumley CL, Elliott AC, Rosenbluth DB, Raghavan D, Jain R. Gender Differences in Outcomes of Patients With Cystic Fibrosis. J Womens Health (Larchmt) (2014) 23(12):1012–20. doi: 10.1089/jwh.2014.4985
145. Chotirmall SH, Smith SG, Gunaratnam C, Cosgrove S, Dimitrov BD, O'Neill SJ, et al. Effect of Estrogen on Pseudomonas Mucoidy and Exacerbations in Cystic Fibrosis. N Engl J Med (2012) 366(21):1978–86. doi: 10.1056/NEJMoa1106126
146. Moran A, Dunitz J, Nathan B, Saeed A, Holme B, Thomas W. Cystic Fibrosis-Related Diabetes: Current Trends in Prevalence, Incidence, and Mortality. Diabetes Care (2009) 32(9):1626–31. doi: 10.2337/dc09-0586
147. Lewis C, Blackman SM, Nelson A, Oberdorfer E, Wells D, Dunitz J, et al. Diabetes-Related Mortality in Adults With Cystic Fibrosis. Role of Genotype and Sex. Am J Respir Crit Care Med (2015) 191(2):194–200. doi: 10.1164/rccm.201403-0576OC
148. Ahmed N, Corey M, Forstner G, Zielenski J, Tsui LC, Ellis L, et al. Molecular Consequences of Cystic Fibrosis Transmembrane Regulator (CFTR) Gene Mutations in the Exocrine Pancreas. Gut (2003) 52(8):1159–64. doi: 10.1136/gut.52.8.1159
149. Moran A, Hardin D, Rodman D, Allen HF, Beall RJ, Borowitz D, et al. Diagnosis, Screening and Management of Cystic Fibrosis Related Diabetes Mellitus: A Consensus Conference Report. Diabetes Res Clin Pract (1999) 45(1):61–73. doi: 10.1016/s0168-8227(99)00058-3
150. Milla CE, Billings J, Moran A. Diabetes is Associated With Dramatically Decreased Survival in Female But Not Male Subjects With Cystic Fibrosis. Diabetes Care (2005) 28(9):2141–4. doi: 10.2337/diacare.28.9.2141
151. Pezzulo AA, Gutierrez J, Duschner KS, McConnell KS, Taft PJ, Ernst SE, et al. Glucose Depletion in the Airway Surface Liquid is Essential for Sterility of the Airways. PloS One (2011) 6(1):e16166. doi: 10.1371/journal.pone.0016166
152. Edlund A, Esguerra JL, Wendt A, Flodstrom-Tullberg M, Eliasson L. CFTR and Anoctamin 1 (ANO1) Contribute to cAMP Amplified Exocytosis and Insulin Secretion in Human and Murine Pancreatic Beta-Cells. BMC Med (2014) 12:87. doi: 10.1186/1741-7015-12-87
153. Guo JH, Chen H, Ruan YC, Zhang XL, Zhang XH, Fok KL, et al. Glucose-Induced Electrical Activities and Insulin Secretion in Pancreatic Islet Beta-Cells Are Modulated by CFTR. Nat Commun (2014) 5:4420. doi: 10.1038/ncomms5420
154. Ntimbane T, Mailhot G, Spahis S, Rabasa-Lhoret R, Kleme ML, Melloul D, et al. CFTR Silencing in Pancreatic Beta-Cells Reveals a Functional Impact on Glucose-Stimulated Insulin Secretion and Oxidative Stress Response. Am J Physiol Endocrinol Metab (2016) 310(3):E200–12. doi: 10.1152/ajpendo.00333.2015
155. Edlund A, Barghouth M, Huhn M, Abels M, Esguerra J, Mollet I, et al. Defective Exocytosis and Processing of Insulin in a Cystic Fibrosis Mouse Model. J Endocrinol (2019) 241(1):45–57. doi: 10.1530/JOE-18-0570
156. Khan D, Kelsey R, Maheshwari RR, Stone VM, Hasib A, Manderson Koivula FN, et al. Short-Term CFTR Inhibition Reduces Islet Area in C57BL/6 Mice. Sci Rep (2019) 9(1):11244. doi: 10.1038/s41598-019-47745-w
157. Li A, Vigers T, Pyle L, Zemanick E, Nadeau K, Sagel SD, et al. Continuous Glucose Monitoring in Youth With Cystic Fibrosis Treated With Lumacaftor-Ivacaftor. J Cyst Fibros (2019) 18(1):144–9. doi: 10.1016/j.jcf.2018.07.010
158. Thomassen JC, Mueller MI, Alejandre Alcazar MA, Rietschel E, van Koningsbruggen-Rietschel S. Effect of Lumacaftor/Ivacaftor on Glucose Metabolism and Insulin Secretion in Phe508del Homozygous Cystic Fibrosis Patients. J Cyst Fibros (2018) 17(2):271–5. doi: 10.1016/j.jcf.2017.11.016
159. Boom A, Lybaert P, Pollet JF, Jacobs P, Jijakli H, Golstein PE, et al. Expression and Localization of Cystic Fibrosis Transmembrane Conductance Regulator in the Rat Endocrine Pancreas. Endocrine (2007) 32(2):197–205. doi: 10.1007/s12020-007-9026-x
160. Rotti PG, Xie W, Poudel A, Yi Y, Sun X, Tyler SR, et al. Pancreatic and Islet Remodeling in Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Knockout Ferrets. Am J Pathol (2018) 188(4):876–90. doi: 10.1016/j.ajpath.2017.12.015
161. White MG, Maheshwari RR, Anderson SJ, Berlinguer-Palmini R, Jones C, Richardson SJ, et al. In Situ Analysis Reveals That CFTR Is Expressed in Only a Small Minority of Beta-Cells in Normal Adult Human Pancreas. J Clin Endocrinol Metab (2020) 105(5):1366–74. doi: 10.1210/clinem/dgz209
162. Hart NJ, Aramandla R, Poffenberger G, Fayolle C, Thames AH, Bautista A, et al. Cystic Fibrosis-Related Diabetes Is Caused by Islet Loss and Inflammation. JCI Insight (2018) 3(8):e98240. doi: 10.1172/jci.insight.98240
163. Sun X, Yi Y, Xie W, Liang B, Winter MC, He N, et al. CFTR Influences Beta Cell Function and Insulin Secretion Through Non-Cell Autonomous Exocrine-Derived Factors. Endocrinology (2017) 158(10):3325–38. doi: 10.1210/en.2017-00187
164. Colomba J, Boudreau V, Lehoux-Dubois C, Desjardins K, Coriati A, Tremblay F, et al. The Main Mechanism Associated With Progression of Glucose Intolerance in Older Patients With Cystic Fibrosis Is Insulin Resistance and Not Reduced Insulin Secretion Capacity. J Cyst Fibros (2019) 18(4):551–6. doi: 10.1016/j.jcf.2019.01.009
165. Edlund A, Pedersen MG, Lindqvist A, Wierup N, Flodstrom-Tullberg M, Eliasson L. CFTR Is Involved in the Regulation of Glucagon Secretion in Human and Rodent Alpha Cells. Sci Rep (2017) 7(1):90. doi: 10.1038/s41598-017-00098-8
166. Huang WQ, Guo JH, Yuan C, Cui YG, Diao FY, Yu MK, et al. Abnormal CFTR Affects Glucagon Production by Islet Alpha Cells in Cystic Fibrosis and Polycystic Ovarian Syndrome. Front Physiol (2017) 8:835. doi: 10.3389/fphys.2017.00835
Keywords: diabetes of the exocrine pancreas (DEP), acute pancreatitis (AP), chronic pancreatitis (CP), cystic fibrosis (CF), interaction
Citation: Gál E, Dolenšek J, Stožer A, Czakó L, Ébert A and Venglovecz V (2021) Mechanisms of Post-Pancreatitis Diabetes Mellitus and Cystic Fibrosis-Related Diabetes: A Review of Preclinical Studies. Front. Endocrinol. 12:715043. doi: 10.3389/fendo.2021.715043
Received: 26 May 2021; Accepted: 19 August 2021;
Published: 10 September 2021.
Edited by:
Ondřej Šeda, Charles University, CzechiaReviewed by:
Max Petrov, The University of Auckland, New ZealandErling Tjora, Haukeland University Hospital, Norway
Copyright © 2021 Gál, Dolenšek, Stožer, Czakó, Ébert and Venglovecz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Viktória Venglovecz, dmVuZ2xvdmVjei52aWt0b3JpYUBtZWQudS1zemVnZWQuaHU=