 Juan Ignacio Burgos1
Juan Ignacio Burgos1 Ludovic Vallier
Ludovic Vallier Santiago A. Rodríguez-Seguí
Santiago A. Rodríguez-Seguí
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 06 July 2021
Sec. Diabetes: Molecular Mechanisms
Volume 12 - 2021 | https://doi.org/10.3389/fendo.2021.692596
This article is part of the Research Topic Advances in Stem Cell Technology to Model and Treat Diabetes View all 13 articles
The occurrence of diabetes mellitus is characterized by pancreatic β cell loss and chronic hyperglycemia. While Type 1 and Type 2 diabetes are the most common types, rarer forms involve mutations affecting a single gene. This characteristic has made monogenic diabetes an interesting disease group to model in vitro using human pluripotent stem cells (hPSCs). By altering the genotype of the original hPSCs or by deriving human induced pluripotent stem cells (hiPSCs) from patients with monogenic diabetes, changes in the outcome of the in vitro differentiation protocol can be analyzed in detail to infer the regulatory mechanisms affected by the disease-associated genes. This approach has been so far applied to a diversity of genes/diseases and uncovered new mechanisms. The focus of the present review is to discuss the latest findings obtained by modeling monogenic diabetes using hPSC-derived pancreatic cells generated in vitro. We will specifically focus on the interpretation of these studies, the advantages and limitations of the models used, and the future perspectives for improvement.
Diabetes mellitus (DM) is characterized by pancreatic β cell loss and chronic hyperglycemia. Type 1 diabetes (T1D) is caused by the autoimmune reaction against β cells (1), and Type 2 diabetes originates from insulin resistance and β cell overload (2–4). In addition, rarer monogenic forms of diabetes account for approximately 1–5% of diabetes cases, depending on the population studied (5, 6). Over 30 subtypes of monogenic diabetes have been identified to date, each having a characteristic phenotype and a specific pattern of inheritance (6, 7). The identification of genes implicated in the pathogenesis of monogenic diabetes, including components of the insulin secretory pathway and transcription factors, has provided important insights into human pancreas and β cell development and function.
Monogenic diabetes is caused by either splice-site, non-sense, missense, or frame-shift mutations, and more rarely partial or full deletions, affecting a single gene (8–14). The disease phenotype and associated extra-pancreatic features vary depending on the affected gene (15). These characteristics have made monogenic diabetes an interesting disease subtype to model using human pluripotent stem cells (hPSCs). Indeed, hPSCs can be differentiated into pancreatic cells following key steps of differentiation induced by well-established combinations of growth factors and small molecules, thereby respecting a natural path of development [recently reviewed in (16, 17)]. Thus, differentiation of human induced pluripotent stem cells (hiPSCs) either derived from patients with monogenic diabetes or genetically edited to carry the mutation of interest can be used to study the potential regulatory mechanisms affected by each of the disease-associated genes (Figure 1). Applying this approach to a diversity of genes has led to the discovery of new mechanisms associated with specific regulators of pancreatic development. The focus of this review is to discuss the latest findings obtained by modeling monogenic diabetes using hPSC-derived pancreatic cells generated in vitro. We will focus on the interpretation of these studies, the advantages and limitations of the models used, and the future perspectives for improvement. Of note, the reader is referred to recent reviews concerning: 1) the state-of-the-art knowledge in pancreatic β cell development in mice and humans (18–21); 2) the tools for hPSC genome editing (22, 23); 3) a comparison of the in vitro pancreatic differentiation protocols including the latest advances to achieve functional β cells from hPSCs (16, 17, 24); 4) analyzing the intrinsic variation in the protocol outcomes from different sources of hPSCs (25, 26); and 5) the use of in vitro pancreatic differentiation from hPSCs to discover new mechanisms underlying human pancreas development (24, 27), or to model other types of diabetes (23, 28–31). The later also summarize findings on monogenic diabetes modeling. Our review adds up on top of these by exclusively focusing on the modeling of monogenic diabetes, discussing in more detail the different approaches taken and including extremely recent works which provide insightful information for the interpretation of the results published so far.
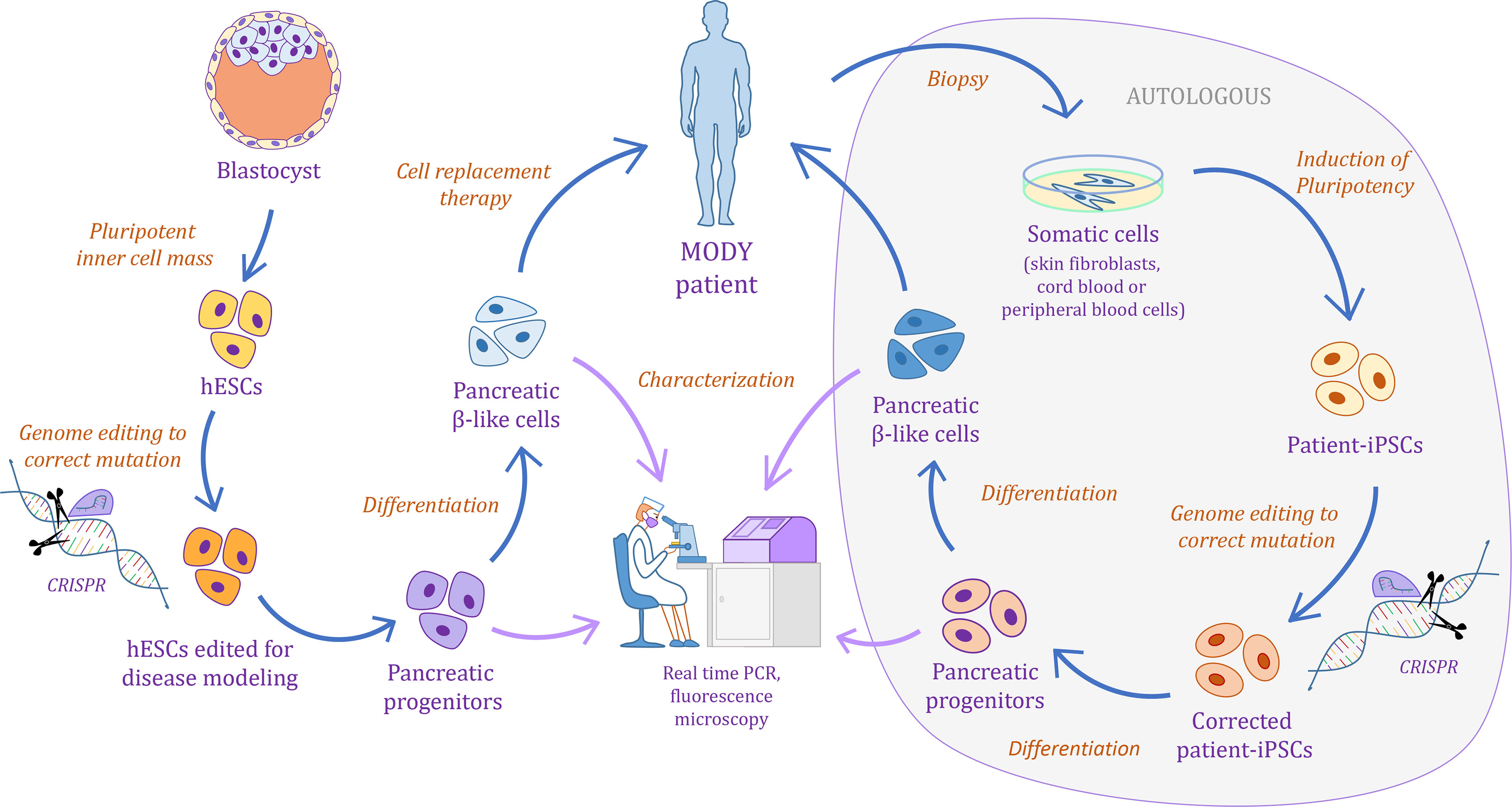
Figure 1 Schematic illustrating the pipeline for monogenic diabetes disease modeling using human pluripotent stem cells.
Pancreas development begins with the establishment of the pancreatic bud containing multipotent pancreatic progenitor cells (MPCs) at ~E8.5 in the mouse (19, 20) or ~29 days post conception in humans (20, 21, 32) and progresses until E18.5 in the mouse (19, 20) or 24 weeks post conception (wpc) estimated in humans (20, 21, 33–35). By this time, most of the pancreatic progenitor cells are terminally fate-committed. The MPCs are capable of differentiating along the three main lineages of the adult pancreas, namely the ductal, the exocrine (comprising acinar cells that secrete digestive enzymes), and the endocrine (including the β cells that produce insulin, but also the α, δ, γ, and PP cells) (19, 36, 37). As the pancreas develops, MPCs differentiate into acinar or endocrine-ductal bipotent progenitor (BP) cells, and eventually to endocrine-committed progenitors (EPs) that will give rise to β cells. Importantly, recent single cell RNA-seq (scRNA-seq) studies have described the transcriptional profiles that characterize these pancreatic progenitor cell stages in the mouse and identified additional progenitor cell sub stages (38–42). Thus, we can now rely on a precise transcriptomic fingerprint for several progenitors that arise during mouse pancreas development.
Cumulative knowledge has revealed the role and stage-specific functions of signaling pathways in the pancreatic developmental program, including Wnt, TGF-β, Notch, FGF and, more recently, the Hippo pathway (19, 36, 37, 43–46). The pancreatic mesenchyme has also been shown to play important roles during development, by fine-tuning the crosstalk with the pancreatic epithelium through these pathways (47–50). This knowledge has been exploited to develop protocols to differentiate human embryonic stem cells (hESCs) into the pancreatic lineage (51–55), opening the possibility to produce large quantities of β cells for cell-based therapies and also providing a new avenue for research in human pancreas development.
Differentiation protocols currently available to produce β cells from hPSCs take advantage of the cell signaling events that occur during fetal development (18–21). Despite this knowledge-based approach, the generation of fully functional β cells in vitro has remained elusive. We thus refer to the cells produced in vitro so far as β-like cells. This limitation could be in part explained by the fact that the function of these pathways during pancreas development has been mainly studied in rodents, and it has recently been reported that human pancreas development could differ in several aspects (20, 21, 32, 56, 57). We are still lacking systematic studies comparing islet development between human and mouse, although recent reports are moving forward to address this gap (57–60). It is expected that a deeper understanding of these inter-species differences in islet development will probably be critical for the production of fully functional and mature β cells from hPSCs. As well, this will allow a more precise dissection of the molecular mechanisms driving diabetes predisposition by genetic mutations and/or external stimuli. Noteworthy, recent protocols allow the derivation of monohormonal insulin-producing cells expressing key β cell transcription factors, including PDX1, NKX6.1, and MAFA (61–65). Fine tuning of such protocols could further improve the glucose-response of in vitro derived β-like cells (66–69) without the need to involve a step of cell transplantation in mice to ensure proper maturation.
The use of hPSCs as in vitro model system to study human pancreatic development has gained momentum and several important discoveries have been made using this approach [recently reviewed in (27)].
By comparing the transcriptomes and key epigenomic features of MPCs derived in vitro from hESCs with human fetal primary pancreatic tissue of six wpc embryos, we were able to show that in vitro derived cells closely recapitulated the main expression profile and regulatory landscape of their in vivo counterparts (45). Furthermore, this epigenomic characterization was extended to include ChIP-seq profiling of several transcription factors by taking advantage of the in vitro system. A combined analysis of these data led to the discovery that TEAD1 was an integral component of the enhancer network in human embryonic pancreatic progenitors (45, 46). The relevance of TEAD protein binding for the activation of MPC enhancers was mechanistically validated using the platform provided by hPSC-derived pancreatic cells in vitro. We concluded that, while highly tissue specific enhancers were defined by co-binding of pancreas-specific transcription factors, TEAD proteins conferred these regions the ability to be regulated by YAP (an effector of the Hippo pathway) during human pancreas development. These results were consistent with reports for a role of the Hippo pathway in mouse pancreas development (43, 44). More recent reports support these findings by showing that YAP and other components of the Hippo pathway are active and highly enriched within the SOX9+/PTF1A+ progenitor cells of the human fetal pancreas (58).
The human in vitro differentiation system has provided additional mechanistic evidence for the relevance of the Hippo pathway in pancreas differentiation by showing that YAP links extracellular matrix-mediated mechano-signals to regulate gene expression. Integration of such signaling plays a key role in the fate choice of bipotent pancreatic progenitors, whereby YAP downregulation favors endocrine cell commitment (70). In agreement, sustained in vitro YAP activation impairs β cell differentiation while inhibition of YAP enhances differentiation of functional β cells derived from hPSCs (71). Combined together, these results illustrate how in vitro pancreatic differentiation can help in the discovery of new regulatory mechanisms that are relevant for human β cell development.
Mapping the dynamic changes in histone modifications and chromatin accessibility across different stages of the in vitro pancreatic differentiation protocol has provided important insights into the suitability and current limitations of this model (72–75). It has to be noted that, despite successful applications, the in vitro pancreatic differentiation system does not exactly replicate all the epigenomic features of their in vivo counterparts. Sander and colleagues have profiled selected chromatin modifications and the transcriptome of these cells, at different stages of the pancreatic endocrine differentiation protocol using hESCs (72). They showed that removal of Polycomb group (PcG)-mediated repression on stage-specific genes was a key mechanism for the induction of developmental regulators in the in vitro system, consistent with the in vivo relevance of this mechanism in mouse endocrine pancreas development (76–78). However, they also reported that elimination of PcG-mediated repression on endocrine-specific genes was not fully recapitulated by the in vitro derived endocrine cells. This was particularly evident at genes involved in organ morphogenesis, underscoring a current limitation (i.e. the lack of tissue-specific contextual cell signaling) of the in vitro protocols for studying some aspects of in vivo development. Noteworthy, these experiments were performed using 2D in vitro differentiation protocols which have been shown to be less efficient to produce functional β-like cells than three-dimensional suspension culture systems (61–63, 66–68). This “second generation” protocols could be more efficient in allowing the proper deposition of epigenetic marks.
Another example of the complexity of the in vitro endocrine differentiation process was provided by Petersen et al. who profiled single-cell transcripts by qRT-PCR at selected stages of the protocol used to produce β-like cells from hPSCs (79). This analysis identified two distinct progenitor cell populations with the potential to differentiate into monohormonal β-like cells. NKX6.1 expression prior or after the onset of NEUROG3 (the gene coding for the EP transcription factor marker NGN3) was the main difference between these progenitors. Building up on these results, Ramond et al. performed a combined analysis of single-cell qRT-PCR datasets obtained from pancreatic progenitor and endocrine cells from in vitro and in vivo samples (59, 60). Their observations suggest that these distinct progenitor cell populations identified in vitro could indeed exist during in vivo development. This finding contrasts with knowledge gained from mouse studies where Nkx6 factors systematically specify endocrine cell fate upstream of Ngn3 in the MPC stage (80, 81). Still, the function and characteristics of these two populations of progenitors in the developing human pancreas remain to be fully elucidated.
Taking advantage of novel single-cell technologies can help to match some of the transcriptional signatures from in vivo pancreatic progenitor cell stages identified in the mouse (38–41), with pancreatic progenitors derived from hPSCs (38, 66, 82). However, such exercise remains challenging due to inter-species differences and also the impact of the in vitro culture. New tools are being quickly developed to address this limitation, making the bioinformatic analyses of these data an exciting area of research (83, 84). More recently, the first scRNA-seq experiments using human embryonic pancreas from 15.2 and 17.1 wpc have been reported (58, 85). Integration of these scRNA-seq datasets with those derived from the mouse embryonic pancreas will help to identify differences and similarities in the transcriptional fingerprints of the distinct pancreatic progenitor cell types. This will ultimately contribute to validate the identity of pancreatic progenitors produced from hPSCs.
The most frequently affected maturity-onset diabetes of the young (MODY) genes include the enzyme glucokinase (GCK, MODY2) (86, 87) and the transcription factor genes hepatic nuclear factor 1 alpha (HNF1A, MODY3) (88), hepatic nuclear factor 4 alpha (HNF4A, MODY1) (89), and hepatic nuclear factor 1 beta (HNF1B, MODY5) (90). Other MODY genes include PDX1 (MODY4), NEUROD1 (MODY6), KLF11 (MODY7), CEL (MODY8), PAX4 (MODY9), INS (MODY10), BLK (MODY11), ABCC8 (MODY12), KCNJ11 (MODY13), APPL1 (MODY14) (91). On the other hand, homozygous mutations at several lineage determining transcription factors, such as PTF1A, PDX1, NEUROG3, RFX6, NEUROD1, MNX1, NKX2.2 and GLIS3 result in permanent neonatal diabetes mellitus (PNDM) in humans (13, 92–100). Interestingly, heterozygous mutations in these genes rarely result in diabetes in mice, thereby suggesting an important divergence in the activity or function for these factors between human and mouse (19, 21, 101). The importance of haploinsufficiency and the mechanisms by which a decrease in transcription factor activity causes a disease in humans is poorly understood, mostly due to the lack of an appropriate model system. As an example, MODY5 diabetes (HNF1B-associated) can be induced by a diversity of mutations including several splice-site, non-sense, missense, and frame-shift mutations or whole gene deletions, all of which result in a diabetes (102). The heterozygous mutation in mouse has no effect on pancreatic development, while homozygous mutation blocks foregut specification thereby masking its downstream function in the differentiation of MPCs. Ultimately, haploinsufficiency may reflect the functional effects of different gene anomalies, stochastic variation in temporal gene expression during early development or additional genetic and/or environmental modifiers that may influence the disease phenotype (102–104).
As mentioned above, mouse models often do not recapitulate the disease phenotype associated with heterozygous mutations of HNF1A, HNF4A, or HNF1B in humans. The genetic discrepancy between the mouse and monogenic diabetes gene haploinsufficient patients and the difficulty in accessing patient samples have reinforced the interest in using hPSCs (Figure 1). Genome-editing tools combined with directed differentiation of hPSCs offer a unique platform for generating patient-specific disease models to elucidate novel genes and molecular pathways that underlie monogenic diseases with complex traits, such as diabetes, and ultimately lead to the development of novel therapeutic strategies [recently reviewed in (22, 25)]. Several studies in the last decade have used genetically engineered hPSC culture systems for differentiation into pancreatic cells to further expand our understanding of the roles of various genes associated with monogenic diabetes. Their findings are summarized in Tables 1, 2, and these will be discussed in more detail next.

Table 1 Summary of reports modeling maturity-onset diabetes of the young (MODY) mutation effects.

Table 2 Summary of reports modeling monogenic mutations associated with permanent neonatal diabetes mellitus (PNDM) or pancreatic agenesis.
Egli and colleagues provided the first example for the use of hiPSCs to create insulin-producing cells from patients with Wolfram Syndrome (WS) (119). hiPSCs were generated from individuals with diabetes caused by mutations in the WFS1 gene and healthy-donor controls. Differentiation of these cells towards β-like cells revealed increased levels of ER stress molecules and decreased insulin content in WFS1-deficient β-like cells. Overall, insulin processing and secretion in response to various secretagogues was comparable to healthy controls, but the former displayed increased activity of unfolded protein response (UPR) pathways.
More recently, Maxwell et al. used CRISPR/Cas9 to correct a diabetes-causing pathogenic variant in WFS1 hiPSCs (120). Noteworthy, β-like cells differentiated from WFS1-corrected hiPSCs showed robust and dynamic insulin secretion in response to glucose, and reversed streptozocin-induced diabetes when transplanted into mice. Single-cell RNA-seq transcriptome profiling showed that indeed these cells displayed increased insulin levels and decreased expression of genes associated with endoplasmic reticulum stress. Taken together, these studies illustrate the potential of in vitro pancreatic differentiation from hPSCs to study how mechanisms related to cellular stress can affect diabetes onset.
Homozygous null mutations in PDX1 result in pancreatic agenesis both in mice and humans (13, 121–123). Human patients with PDX1 heterozygous inactivating mutations exhibit MODY4 diabetes caused by defects in β cell function and/or the maintenance of β cell mass in adults (36). In rodents, it has been reported that Pdx1+/− mice can develop a functional pancreas (121, 122) but become diabetic in adulthood due to β cell apoptosis (124).
Another pioneer study to model monogenic diabetes was reported by Huangfu and colleagues, who used TALEN and CRISPR-Cas-mediated gene editing combined with hPSC-directed differentiation. These researchers provide a systematic analysis of the role for PDX1 and seven additional pancreatic transcription factors (RFX6, PTF1A, GLIS3, MNX1, NGN3, HES1 and ARX) in pancreatic cell commitment (111). Noteworthy, they created mono- or biallelic frameshift mutations in all these genes and used untargeted isogenic cell lines as controls. This analysis not only defined the specific developmental steps affected by these mutations in a model of human pancreas differentiation, but also revealed new mechanisms. Tables 1, 2 show a summary of their results for the genes previously associated with MODY and/or PNDM. An interesting finding of this work was that monoallelic frameshift translation mutations disrupting the PDX1 protein sequence cause a reduction (up to 65%) in the number of insulin+ cells derived in vitro. These findings suggest a haploinsufficient requirement for PDX1 in pancreatic endocrine development. Importantly, this phenotype correlates with the observation that patients with heterozygous mutations in PDX1 present with diabetes from an early age (125). These results further validate that decreased amounts of PDX1 could lead to β cell dysfunction, a decrease in β cell mass during fetal development and/or the maintenance of β cell mass in adults (124, 126, 127).
More recently, Lickert and colleagues generated hiPSCs from two patients with heterozygous missense mutations in the PDX1 coding region (PDX1P33T/+ and PDX1C18R/+) leading to single amino acid exchanges in its transactivation domain (112). By comparing with a control hiPSC line derived from a healthy donor, the authors showed that MPC differentiation was not affected in patient-derived hiPSCs. However, the PDX1 heterozygous point mutations impaired the differentiation of β-like cells and affected their response to glucose. A more severe effect was observed when artificially introducing the same point mutations in homozygosis (i.e. PDX1P33T/P33T and PDX1C18R/C18R) in isogenic cell lines derived from the original control cell. Interestingly, this resulted in impaired NKX6.1 induction in MPCs just in one of the cell lines (PDX1P33T/P33T). Nevertheless, when differentiated towards insulin producing cells, both homozygous cell lines yielded a decreased number of β-like cells with impaired glucose response. The authors also generated additional isogenic lines carrying different heterozygous mutations in the PDX1 transactivation domain, to generate a frame-shift mutation (PDX1+/−). This created a more severe phenotype to the one observed in the patient-derived hiPSCs, leading to similar outcomes as obtained from the homozygous isogenic PDX1P33T/P33T point mutated cells. Further transcriptomic analyses of MPCs differentiated from these cell lines ascribed the observed effects to downregulation of key PDX1-bound genes including MEG3 and NNA, which are involved in pancreas development and insulin secretion.
Taken together, these results illustrate how predisposition to develop diabetes can be provoked at the stage of pancreatic endocrine lineage development by genetic mutations on a gene that plays a key role at this timepoint. These anomalies could impair the glucose-responsive function of β-like cells through misregulation of genes involved in β cell development, maturation, and function. These results also emphasize that the choice between patient-derived hiPSCs or healthy donor hiPSCs with mutations artificially introduced, as well as the choice of the control cell line used, can affect experimental outcomes and their interpretations. In this context, patient-derived hiPSCs could carry additional mutations in non-coding regulatory regions and/or other genes which might further impair the in vitro differentiation outcomes. This effects have been elegantly exposed in a recent work by Gadue and colleagues (117), which will be discussed in more detail below, in the GATA6 section of this review. In contrast, the use of healthy donor hiPSCs with mutations artificially introduced has the advantage of enabling the use of isogenic cell lines (i.e. non-mutated hiPSCs) to exclude additional effects of the genetic background.
Lack of Rfx6 in mice blocks differentiation of all islet cell types, with the exception of pancreatic-polypeptide-producing cells, while RFX6 mutations in humans result in PNDM (93, 100, 128). Modeling of the RFX6 requirement for human endocrine pancreas development has been addressed by Zhu et al. Their findings, in agreement with current knowledge, show a reduction of endocrine cell commitment from pancreatic progenitor cells derived from RFX6−/− mutant hPSCs (111).
In a more recent study, Trott et al. used hiPSCs derived from individuals with Mitchell–Riley syndrome (MRS) to specifically associate the role of RFX6 mutations and the lack of pancreatic endocrine cells in a human model of pancreas development (118). X-ray microtomography of one of these patients confirmed the spectrum of congenital defects typical of MRS (loss of the pancreas body and tail), and exome sequencing identified a homozygous non-sense mutation in RFX6. hiPSCs derived from this patient and differentiated along the pancreatic cell lineage revealed that these cells efficiently differentiate into posterior foregut cells but exhibited a reduction in the pancreatic endoderm differentiation, which was accompanied by expression of genes associated with mesoderm differentiation. These findings indicate that RFX6 is crucial for maintaining the transcriptional program that specifies early pancreatic endoderm in humans.
While loss of Ngn3 function has been associated with complete lack of pancreatic endocrine cells in mice (129), the phenotype in humans is variable [recently reviewed in (21)]. In this sense, while some patients with homozygous or compound heterozygous NEUROG3 mutations show glycemic control into adulthood, indicating a functional endocrine pancreas, others present neonatal diabetes (96, 130, 131). A recent study suggests that each mutation could have unique effects on the structure and function of NGN3 (132). To further understand this divergence, the requirement of NGN3 for the generation of insulin-producing cells during human development has been addressed using hPSC differentiation. Zhu et al. reported that in vitro endocrine pancreatic differentiation of hPSCs with biallelic mutations in NEUROG3 formed some insulin-producing cells (111), whereas another study reported a total lack of endocrine cells differentiated from NEUROG3−/− hPSCs (133). The latter work described that as little as 10% NEUROG3 expression is sufficient for the formation of pancreatic endocrine cells, supporting that NGN3 is essential for endocrine pancreas development in humans. The divergence between differentiation protocols used in each laboratory and the influence of genetic background could explain the varied phenotypes observed between these two studies. Interestingly, a new adult mouse islet resident pancreatic endocrine progenitor cell population has been recently reported (134). These cells express the surface marker Procr, are Neurog3 negative and, when isolated and co-cultured with endothelial cells, are able to give rise to islet-like clusters containing all endocrine cell types. Apparently, differentiation of this adult progenitor cell population into endocrine cells does not involve Neurog3 expression, raising the intriguing question of whether such a population exists in humans and, if so, whether in vitro pancreatic differentiation from hPSCs is able to follow this “alternative” path for endocrine cell production. Such possibility could explain the divergence between different reports concerning the requirement of NGN3 in endocrine cell production. Taken together, these studies illustrate the complexity, as well as the potential, associated with hPSC differentiation for modeling the impact of genetic mutations on human development.
It has been reported that global Glis3−/− mice die of severe neonatal diabetes shortly after birth (135). Minor differences in gene dosage of Glis3 produce substantive changes in the expression levels of Neurog3 and Ins1, leading to a variable phenotype among the multiple Glis3-KO mouse lines (136). In agreement with these phenotypes, human biallelic mutations in GLIS3 underlie a rare clinical syndrome, characterized by neonatal diabetes and congenital hypothyroidism (92).
The first report of the in vitro modeling for the requirement of GLIS3 in human pancreas development was provided by Zhu et al. These researchers did not find defects in pancreatic endocrine differentiation using GLIS3-/- mutant hESCs, when using a first generation in vitro pancreatic differentiation protocol that allows producing poly-hormonal cells (111).
More recently, Amin et al. developed an improved differentiation protocol that allowed the production of monohormonal β-like cells with enhanced functionality (64). Noteworthy, this protocol allowed the generation of robust GLIS3 expression at the PDX1+/NKX6.1+ pancreatic progenitor cell stage, in contrast with previously reported protocols (52, 111). Using this improved protocol, they were able to demonstrate that differentiation of GLIS3−/− mutant hESCs presented impaired expression of pancreatic endocrine-associated genes, including PDX1, NEUROD1, NKX6.1, and MAFA. These cells also showed increased β-like cell death. These findings contrast with those reported by Zhu et al. (111). The difference could be explained by the improvements in the differentiation protocol, which allow a closer recapitulation of the differentiation steps to produce β-like cells. Furthermore, providing an illustrative example of the utility of the in vitro β cell differentiation protocols, these researchers performed a chemical screen that allowed the identification of a novel drug candidate that rescued mutant GLIS3-associated β-cell death both in vitro and in vivo (64).
Hnf1a has been shown to regulate the expression pattern of islet-specific genes involved in key functions of this tissue (137). In the mouse, while homozygous knockout (Hnf1a−/−) results in insulin secretory defects and higher blood glucose concentrations, heterozygous knockout (Hnf1a+/−) do not display this phenotype (138). This is in sharp contrast with the MODY3 pathology in humans, in which heterozygous mutations result in diabetes (139). In an attempt to elucidate the mechanisms by which dysfunctional HNF1A affects pancreatic development and/or β cell function, Gadue and colleagues have modeled MODY3 using CRISPR-Cas9 genome-edited hESCs and EndoC-BH human cell lines (109). Loss of HNF1A function was accomplished by deletion and premature termination in one or both HNF1A alleles, resulting in heterozygous and homozygous KO mutations. Their results suggest that HNF1A plays an essential role in endocrine cell development, as its loss leads to abnormal expression of genes related to β cell function and diabetes. Noteworthy, complete loss of HNF1A did not impair the production of pancreatic progenitors, but this factor was necessary for proper endocrine cell development as revealed by decreased expression of PAX4, and impaired insulin expression and secretion. Interestingly, HNF1A loss of function (deletion in one or both alleles of HNF1A) led to increased expression of α cell markers, including glucagon. The authors suggest that the increase found in α cells derived from this model system appears to be human-specific, since Hnf1a knockout mice do not display this phenotype.
Another key finding of this work was the identification of a previously unannotated human-specific long intergenic non-coding RNA (lncRNA). The LINC01139, designated LINKA, was shown to act as a downstream target of HNF1A. In vitro endocrine pancreatic differentiation of LINKA-deficient hESCs showed no effect on the production of pancreatic progenitors, but revealed a limited bias towards the production of α cells. Furthermore, β-like cells produced from LINKA-deficient hESCs showed a decrease in maximal respiration capacity to a similar extent as seen in the HNF1A heterozygous cells. Taken together, their findings point to a role for LINKA in the regulation of a subset of HNF1A target genes with implications in cellular respiration. The in vivo relevance of LINKA for diabetes onset remains to be explored. Of note, a significant variability was observed in the expression changes among the hESC lines used in this study. These could be partially explained by the impact of the genetic background, which could lead to differences in the efficiency of differentiation protocol when applied to each cell line.
A more recent report was provided by Teo and colleagues (110). These researchers used MODY3 patient-derived hiPSCs to study the impact of a recently reported patient-specific heterozygous HNF1A+/H126D mutation (140). The authors used hiPSCs reprogrammed from a healthy donor and H9 hESCs as two independent wild type controls. Molecular dynamics simulations predicted that the H126D mutation could compromise DNA binding and gene target transcription. Indeed, RNA-seq and ChIP-seq analyses performed on MODY3 hiPSC-derived endocrine progenitors revealed that the expression of several HNF1A gene targets was affected by the mutation. An in-depth analysis of the effects on the β-like cells derived from HNF1A+/H126D hiPSCs demonstrated that the HNF1A mutation causes a GLUT2 deficiency, that is associated with reduced glucose uptake and ATP production. Their findings reveal the importance of HNF1A in regulating GLUT2 and several genes involved in the MODY3 pathology that may partly account for the lack of insulin secretion clinically observed in these patients. This report extends the findings reported by Cardenas-Diaz et al. (109) by revealing additional mechanisms triggered by the HNF1A mutations on the rest of the stimulus-secretion coupling pathway and on HNF1A transcriptional targets in human β-like cells. Noteworthy, Teo and colleagues performed RNA-seq and ChIP-seq at the endocrine progenitor cell stage. They did not found a differential regulation of the LINC01139 (LINKA) at this stage, and unfortunately the expression of this lncRNA in β-like cells derived from HNF1A+/H126D hiPSCs is not reported. It remains to be elucidated whether LINC01139 is also downregulated in the latter model. Potential discrepancies on the regulation of this lncRNA could be accounted by the different approaches followed in each work to evaluate the effects of HNF1A haploinsufficiency. On one hand Cardenas-Diaz et al. artificially introduced KO mutations by generating a genomic deletion leading to premature termination in one or both HNF1A alleles, and non-mutated isogenic cell lines were used as controls. This approach has the advantage of using an isogenic control cell line, which neutralizes contributions from the genomic background. However, the mutations introduced generate a strong HNF1A loss of function that might not appropriately recapitulate the mechanisms that take place in MODY3 patients. On the other hand, Teo and colleagues used MODY3 patient-derived hiPSCs carrying a mutation that causes an amino acid substitution (HNF1A+/H126D) and used hiPSCs derived from a healthy donor and H9 hESCs as wild type controls. This approach has the advantage of using hiPSCs derived from patient cells, accounting for a closer model to the MODY3 disease. However, the use of non-isogenic hPSCs as controls does not allow accounting for potential effects derived from the different genomic backgrounds. As presented in more detail in the next section, these might introduce an additional bias in the differentiation outcome. In summary, further studies are required to elucidate whether deregulation of LINC01139 plays a relevant role in MODY3 diabetes.
Mono allelic mutations in GATA6 have been linked with pancreas agenesis in humans (141) while the knockout of the same gene has little effect on pancreatic development in the mouse. Indeed, only knockout of both Gata4 and Gata6 results in pancreatic agenesis (142, 143). Thus, GATA6 seems to have a different or at least a more extensive function in human development. To confirm this observation, Shi et al. used CRISPR/Cas9 to create hPSCs carrying frameshift mutations in GATA6, alone or in combination with GATA4 mutations (115). Their results show that GATA6+/− haploinsufficiency alters pancreatic progenitor cell differentiation leading to a reduced number of glucose-responsive β-like cells. Given that heterozygous inactivating mutations in GATA6 have been linked with pancreas agenesis, these findings suggest that the severity of the phenotype could vary according to additional genetic, epigenetic, and/or environmental factors that were not accounted by the differentiation process. Interestingly, the authors also describe dosage-sensitive requirements for GATA6 and GATA4 in the formation of both definitive endoderm and pancreatic progenitor cells, confirming the complex interplays between these factors observed in genetic studies in the mouse.
In another study, Tiyaboonchai et al. used hiPSCs derived from a patient with pancreatic agenesis associated with a heterozygous GATA6 frameshift mutation, which leads to production of a truncated protein. These researchers also used CRISPR/Cas9 genome editing to introduce this mutation on both alleles of the same hiPSC line (114). Noteworthy, hiPSC lines with homozygous mutations failed to differentiate into endoderm. Re-expression of GATA6 or other GATA family members restored this defect. The use of endodermal progenitor cell lines established from the hiPSC allelic series, which expressed GATA6 at lower levels but GATA4 and GATA3 at higher levels, allowed bypassing the endoderm defect and focusing on pancreatic β cell differentiation. The authors found that all mutant lines were able to differentiate into pancreatic β-like cells, but the response to glucose in these cells was functionally defective. Also, they showed that the clear decrease in pancreas specification and β-like cell generation was associated with limited endogenous retinoic acid signaling during in vitro pancreas induction using the GATA6 mutant cell lines.
Additional information was provided by Chia et al. who combined both gene-edited and patient-derived hPSCs to study the function of GATA6 (116). These authors found that GATA6 heterozygous hPSCs show a limited reduction in endoderm formation, while GATA6-null hPSCs can only form mesoderm-like cells. Thus, GATA6 seems to be upstream of the endoderm program in humans. Consistent with this hypothesis, genome-wide studies showed that GATA6 binds and cooperates with EOMES/SMAD2/3 to regulate the expression of master endoderm genes. In addition, the early deficit of GATA6+/− in definitive endoderm was accompanied by a significant reduction in PDX1+ pancreatic progenitors and C-peptide+ β-like cells. These findings show that, in humans, the formation of definitive endoderm and acquisition of pancreatic fate are exquisitely sensitive to GATA6 gene dosage.
Taken together, the above-mentioned reports revealed different levels of requirement of GATA6 for pancreatic differentiation between protocols, labs and cell lines (Table 2). In this context, a very recent report by Gadue and colleagues provides an illustrative example which might help to understand this apparent divergence. These researchers generated a hiPSC line derived from a pancreatic agenesis patient, harboring a heterozygous 4 bp duplication in exon 2 of GATA6 leading to a premature STOP codon, a genetically matched control line, and an identically artificially-mutated ESC line. Using these cell lines the authors identified a minor allele frequency of a SNP located downstream of GATA6 which was associated with the level of expression of this gene (117). In their in vitro model, the expression of the GATA6 protein remained depressed in pancreatic progenitor cells even after correction of the coding mutation. Screening the regulatory regions of the GATA6 gene in the patient cells and an additional pancreas agenesis hiPSC line revealed the above-mentioned SNP. Noteworthy, introducing this non-coding disease modifier SNP by CRISPR/Cas9 in control hESCs confirmed that it depressed GATA6 expression in pancreas precursors. Thus, the phenotypic diversity found in GATA6 heterozygous patients and the outcome of in vitro studies could be explained in part by this genetic variant.
The findings reported by Gadue and colleagues suggest that caution has to be taken when interpreting the results of monogenic diabetes modeling using patient-derived hiPSCs. Additional genomic variants might contribute to the in vitro differentiation outcomes, making it difficult to compare the results obtained by different groups. Nevertheless, some of the studies mentioned above did use the same hPSC line, including the original H9 line derived by JA Thomson and colleagues (144). In such cases, it is worth to underline that each group used different protocols of differentiation. Specific additives could compensate for the decrease in GATA6 expression. For example, retinoic acid seems to support GATA6 function in pancreatic specification. Addition/increase of this morphogen could modulate the effect of GATA6 haploinsufficiency. Taken together, these results illustrate the challenges and, at the same time, highlight the unique interest of investigating the function of key transcription factors in pancreatic development using hPSCs.
In mouse, it has been shown that full inactivation Hnf4a is embryonically lethal, while heterozygote knockout mice are normoglycemic and do not present diabetes features (145–147). In contrast, MODY1 patients carrying heterozygous mutations in HNF4A present diabetes due to impaired β cell function (148). Patient-derived hiPSCs have been recently used to address the potential mechanisms involved in this phenotype. Ræder and colleagues reported the use of hiPSCs derived from patients carrying a non-sense HNF4A mutation associated with MODY1 to study its effect on pancreas and β cell differentiation (105). Noteworthy, the mutation studied in this work (p. Ile271fs) generates a truncated HNF4A product from one of the alleles. The authors show that insulin-positive cells could be generated in vitro from these cells, suggesting that this human HNF4A mutation neither blocked the expression of the insulin gene nor the production of insulin-producing cells in vitro. However, they acknowledge that the insulin-producing cells derived are immature as a result of the β cell differentiation protocol per se, leaving open the possibility that HNF4A could have more subtle effects on the functionality of fully mature β cells.
In another study, Braverman-Gross et al. generated hiPSCs from MODY1 patients harboring a different non-sense mutation in the HNF4A gene and evaluated its differentiation along the pancreatic lineage (106). In this case, the mutation studied affects all HNF4A transcripts and impairs the protein dimerization and transactivation domains. Pancreatic progenitors differentiated from these cells exhibited an upregulation of other key pancreatic transcription factors, including PAX6, NEUROD1, and NEUROG3. The authors suggest that such gene expression increase could be a compensatory mechanism utilized by MODY1 cells to overcome the reduction in HNF4A expression. Interestingly, they also note that the differential expression of HNF4A target genes in posterior foregut progenitors derived from mutant cells is affected by the number of HNF4A DNA binding sites, its transcription start site distance, and the number of other transcription factor binding sites. Unfortunately, the authors of this work did not extend the differentiation protocol to evaluate proportion and functionality of β-like cells derived from these hiPSC samples.
MODY1 disease modeling was also more recently accomplished by Teo and colleagues using hiPSCs derived from patients with frameshift mutations that introduce a premature stop codon in HNF4A, leading to an unstable mRNA and overall lowered HNF4A levels (107). This mutation is the same one (p. Ile271fs) studied by Ræder and colleagues. Control hiPSC lines were derived from a non-diabetic patient family member. The resulting cell lines were differentiated into liver and pancreatic endocrine cells. Phenotypic analyses showed that HNF4A haploinsufficiency affects foregut endoderm gene expression signatures, contributing to long-term consequences on hepatic and pancreatic cell fates. While key developmental genes were perturbed by HNF4A haploinsufficiency at the pancreatic progenitor stage (including HNF1B, PDX1, GATA4, and RFX6), these mutant hiPSCs were still able to procure β-like cells expressing specific markers, including insulin and C-peptide. However, the β-like cells derived with the assayed in vitro protocol were not fully mature. More critical effects of HNF4A mutations taking place during the β cell maturation process or on already mature β cells could not be properly evaluated with the protocol described in this work.
Taken together, the results reported so far from hiPSC models used to study the effects of different HNF4A mutations suggest that the effects of such mutations might be more relevant at the functional level of the β cells produced. The generation of fully functional β-like cells from in vitro differentiation protocols still remains a challenge. Thus, evaluating the functionality of the β-like cells produced from control or patient-derived hiPSC cannot be appropriately assessed with the current differentiation protocols. On the other hand, it should be noted that while two of these studies evaluated the effects of the same mutation, the results described by Braverman-Gross et al. analyzed a different HNF4A mutation. These mutations lead to HNF4A loss-of-function through different mechanisms, thus potentially explaining the different outcomes obtained in each of the reports. Last, but not least, it should be kept in mind when using patient-derived hiPSCs that additional mutations in other genes or in HNF4A regulatory regions could also modulate the outcome of the in vitro differentiation experiments, as illustrated above for GATA6. To conclude, additional studies are necessary to address how HNF4A mutations cause MODY in humans, especially using the next generation of pancreatic differentiation protocols that improve the production of fully mature β-like cells.
Teo and colleagues established a well-controlled patient-derived hiPSC pancreatic differentiation model to elucidate the molecular mechanisms underlying MODY5 pancreatic hypoplasia (113). Differentiation of MODY5-hiPSCs into pancreatic progenitors showed that the HNF1BS148L/+ mutation causes the up-regulation of several key endocrine pancreas-enriched transcription factors including PDX1. Pancreatic differentiation using these cells did not block PDX1, PTF1A, GATA4, and GATA6 expression, suggesting that MODY5-mediated pancreatic hypoplasia in this case is mechanistically independent from the effect associated with these transcription factors. On the other hand, the point mutation in HNF1B caused an indirect reduction in the expression of the insulin gene activator PAX6, suggesting that loss of one copy of HNF1B in humans impairs β cell development and function. Although these findings are consistent with the potential occurrence of maturity-onset diabetes, they fail to uncover the mechanism by which HNF1B haploinsufficiency results in pancreatic hypoplasia.
To further address this question, we recently used an alternative hiPSC pancreatic differentiation model to elucidate the molecular mechanisms underlying HNF1B-associated diabetes (Khairi et al, manuscript submitted). To evaluate the transcriptional differences in the HNF1B haploinsufficient cells, we used bulk RNA-seq at several stages of the pancreatic differentiation protocol (from DE to β-like cells), immunofluorescence staining, and scRNA-seq at the MPC stage. Our analyses show that absence of HNF1B blocks the specification of the pancreatic fate from the foregut progenitor stage. In contrast, HNF1B haploinsufficiency allows differentiation of MPCs and the generation of functional β-like cells although at a lower frequency than the control isogenic cell line. We further report that HNF1B haploinsufficiency impairs cell proliferation in foregut progenitors and MPCs. Our results show that HNF1B plays a key role in the production and expansion of pancreatic progenitors and suggest that this factor could regulate the expression of several Hippo pathway components in MPCs. Thus, the level of HNF1B, combined with environmental stimuli, could define the number of pancreatic progenitor cells generated during development and therefore contribute to the susceptibility to diabetes during childhood/adulthood.
It has been described that homozygous inactivating mutations in PTF1A cause pancreatic and cerebellar agenesis (98). In agreement, Ptf1a null mice present a complete absence of exocrine pancreatic tissue, but all islet endocrine cell types are present until the late stages of embryogenesis (149). Zhu et al. reported the in vitro modeling of the PTF1A requirement for human pancreas development. Using PTF1A−/− hESCs and a first generation in vitro pancreatic differentiation protocol, these researchers did not find defects in pancreatic endocrine differentiation (111). This finding is in agreement with previous reports showing that Ptf1a is not required for the specification of Ngn3+ endocrine progenitors or the differentiation of mature β cells in mice (150).
The study of PTF1A regulation provides another example of how human in vitro pancreatic differentiation can guide the discovery of a developmental regulatory mechanism, in this case consisting in the identification of recessive mutations in a distal non-coding region (151). Identification of genetic mutations resulting in pancreatic agenesis can be challenging as these can be located in regulatory regions far away from known regulators. Accordingly, genome sequencing of a cohort of patients presenting pancreatic agenesis revealed several mutations in a distal non-coding region located >1 Mb upstream the PTF1A gene. Enhancer profiling in MPCs, derived in vitro from hPSCs, confirmed the functional importance of this regulatory sequence in humans (151). The mutation sites coincided with a FOXA2 binding site profiled by ChIP-seq in in vitro MPCs. Further mechanistic experiments performed in vitro confirmed that the targeted region acts as an enhancer in human MPCs, and that patient mutations affect PDX1 and FOXA2 binding. These findings allowed us to propose that the mutated enhancer region is in charge of triggering the early PTF1A expression in the gut region where the pancreas is specified. This study illustrates how human genetic and in vitro differentiation of hPSCs can be combined to define mechanisms driving developmental diseases.
Balboa et al. generated a model based on hiPSCs from patients carrying INS mutations and engineered isogenic CRISPR-Cas9 mutation-corrected lines. These cells were differentiated to β-like cells (152). Using this model, the authors show that the INS mutations lead to accumulation of proinsulin misfolding, increased signs of ER-stress, and reduced proliferation in INS-mutant β-like cells compared with corrected controls. Following transplantation into mice, INS-mutant grafts presented reduced insulin secretion and further increased ER-stress, associated with decreased PDX1 expression and β cell size, as well as mitochondrial alterations. The authors conclude that neonatal diabetes-associated INS-mutations lead to defective β cell mass expansion, contributing to neonatal diabetes development.
In another recent study, Egli and colleagues generated hiPSCs from fibroblasts of a patient with PNDM and undetectable insulin at birth due to a homozygous mutation in the translation start site of the insulin gene (153). Their results show that the differentiation of INS mutant cells resulted in hormone-negative hiPSCs, and the correction of this mutation by CRISPR-Cas9 restored insulin production and secretion to levels comparable to those of wild type endocrine cells. The authors also demonstrate that the insulin-producing cells of corrected patient hiPSCs protect mice from diabetes, providing a proof-of-principle study for the use of replacement therapy as a treatment for monogenic diabetes.
Saarimäki-Vire et al. used hiPSCs derived from a patient with PNDM and pancreatic hypoplasia to investigate the effects of an activating STAT3 mutation on pancreatic development (154). Noteworthy, the mutation studied has been identified as the cause of PNDM in association with early onset autoimmunity. These authors demonstrate that the mutation in STAT3 leads to premature endocrine differentiation through binding and direct induction of NEUROG3 by the increased nuclear shuttling of the mutated protein. They also showed that correction of the STAT3 mutation using CRISPR/Cas9 completely reversed the disease phenotype. These results demonstrate that, in addition to the early onset autoimmunity, the same mutation leads to a primary developmental defect in pancreatic organogenesis.
The field of hPSCs has allowed important advances in our understanding of the molecular mechanisms underlying the different forms of monogenic diabetes. Indeed, the establishment of hPSC-based in vitro platforms offers a unique opportunity to study pancreas development and to investigate the pathophysiology underlying monogenic diabetes. This basic knowledge paves the way to the development of new treatments, not only for diabetes induced by genetic mutations, but also more broadly for personalized medicine therapies in the context of type I and type II diabetes. Nonetheless, several challenges require attention. Current in vitro β-like cell differentiation protocols have been markedly improved and may be sufficient to recapitulate several of the MODY phenotypes in the hPSC-based model. However, one of their greatest limitations remains the lack of metabolic maturation of the β-like cells derived. A solution to turn the differentiated cells into fully mature and functional β cells has been their transplantation in mouse to allow for the latest steps of cell differentiation to take place in vivo. Alternative methods involve culture in 3D and cell self-aggregation into islet-like clusters to produce β-like cells with improved functionality. The emergence of scRNA-seq is expected to lead to the identification of new markers involved in pancreatic β cell maturation, thus allowing improved benchmarking of the in vitro differentiation protocol outcomes. Also, scRNA-seq applied to human embryonic pancreatic tissue might provide additional insights into the developmental cues that differ among mice and humans. This will provide additional input to improve the in vitro differentiation protocols by modulating yet unknown signaling cues.
The other growing challenge is the divergence of results between different groups studying the same mutation/genes but using either different hPSC lines and/or different protocols. Indeed, genetic background and culture conditions can have a strong effect on phenotype, thus leading to different experimental outcomes. New hiPSC lines derived from monogenic diabetic patients continue to be reported. A very recent study described the generation of hiPSCs derived from MODY2 patients, but in this case its differentiation into the pancreatic lineage was not evaluated so far (108). Thus, there is a need to develop standard hiPSC lines which could be shared between laboratories. More importantly, the use of “universal” culture conditions to grow and to differentiate hPSC lines would be incredibly useful to allow the comparison of data generated and to precisely establish the importance of genetic background on the phenotype observed in vitro. Such standardization implies that culture conditions are fully described and shared between laboratories. The use of isogenic control hPSC lines is also essential and is helping to overcome the limitations related to the variability between lines, especially when compared with the use of family controls, which is inherent to differences in genetic background. Numerous studies have successfully used CRISPR/Cas9 tools to generate isogenic hPSC lines by introducing patient-specific mutations, editing genes in control-hPSC lines to investigate the implication of a single genetic variant on β cell differentiation and function. Here, we revisited the latest advances in the application of in vitro pancreatic cell differentiation from hPSCs to model several types of monogenic diabetes. Much work remains to be done to improve the modeling of monogenic diabetes but, as it stems from this review, in vitro pancreatic differentiation from hPSCs is definitely gaining momentum.
All authors reviewed the literature, wrote, and edited the paper. JIB prepared the table. LV and SAR-S conceptualized the review topic and contents, and approved the paper. All authors contributed to the article and approved the submitted version.
This work was funded by Office of the Royal Society and the Consejo Nacional de Investigaciones Científicas y Técnicas of Argentina (IEC\R2\181023 to SAR-S and LV). Work in the Rodríguez-Seguí laboratory is supported by grants from the Agencia Nacional de Promoción Científica y Tecnológica of Argentina (ANPCyT: PICT-2015 3605, PICT-2017 2071) and the University of Buenos Aires (UBACyT 20020170200156BA). Work in the Vallier laboratory is supported by grants from European Research Council Grant New-Chol, the Cambridge Hospitals National Institute for Health Research Biomedical Research Center and core support grant from the Wellcome and Medical Research Council to the Wellcome-Medical Research Council Cambridge Stem Cell Institute.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We apologize to our colleagues whose references were omitted owing to space constraints. SAR-S is a career investigator from the Consejo Nacional de Investigaciones Científicas y Técnicas of Argentina (CONICET). JIB is supported by a postdoctoral fellowship from the ANPCyT.
1. Atkinson MA, Eisenbarth GS, Michels AW. Type 1 Diabetes. Lancet (2014) 383:69–82. doi: 10.1016/S0140-6736(13)60591-7
2. Matveyenko AV, Butler PC. Beta-Cell Deficit Due to Increased Apoptosis in the Human Islet Amyloid Polypeptide Transgenic (HIP) Rat Recapitulates the Metabolic Defects Present in Type 2 Diabetes. Diabetes (2006) 55:2106–14. doi: 10.2337/db05-1672
3. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-Cell Deficit and Increased Beta-Cell Apoptosis in Humans With Type 2 Diabetes. Diabetes (2003) 52:102–10. doi: 10.2337/diabetes.52.1.102
4. Weyer C, Bogardus C, Mott DM, Pratley RE. The Natural History of Insulin Secretory Dysfunction and Insulin Resistance in the Pathogenesis of Type 2 Diabetes Mellitus. J Clin Invest (1999) 104:787–94. doi: 10.1172/JCI7231
5. Malecki MT. The Search for Undiagnosed MODY Patients: What Is the Next Step? Diabetologia (2010) 53:2465–7. doi: 10.1007/s00125-010-1908-4
6. Murphy R, Ellard S, Hattersley AT. Clinical Implications of a Molecular Genetic Classification of Monogenic Beta-Cell Diabetes. Nat Clin Pract Endocrinol Metab (2008) 4:200–13. doi: 10.1038/ncpendmet0778
7. Rubio-Cabezas O, Hattersley AT, Njolstad PR, Mlynarski W, Ellard S, White N, et al. ISPAD Clinical Practice Consensus Guidelines 2014. The Diagnosis and Management of Monogenic Diabetes in Children and Adolescents. Pediatr Diabetes (2014) 15 Suppl 20:47–64. doi: 10.1111/pedi.12192
8. Bansal V, Gassenhuber J, Phillips T, Oliveira G, Harbaugh R, Villarasa N, et al. Spectrum of Mutations in Monogenic Diabetes Genes Identified From High-Throughput DNA Sequencing of 6888 Individuals. BMC Med (2017) 15:213–3. doi: 10.1186/s12916-017-0977-3
9. Fujita Y, Tanaka D, Tatsuoka H, Matsubara M, Hyo T, Hamamoto Y, et al. A Novel Splice-Site Mutation of the HNF1B Gene in a Family With Maturity Onset Diabetes of the Young Type 5 (MODY5). Endocrinol Diabetes Metab Case Rep (2020) 2020:20-0092. doi: 10.1530/edm-20-0092
10. Gragnoli C, Stanojevic V, Gorini A, Preussenthal G, Thomas M, Habener J. IPF-1/MODY4 Gene Missense Mutation in an Italian Family With Type 2 and Gestational Diabetes. Metabolism: Clin Exp (2005) 54:983–8. doi: 10.1016/j.metabol.2005.01.037
11. Patel KA, Kettunen J, Laakso M, Stančáková A, Laver TW, Colclough K, et al. Heterozygous RFX6 Protein Truncating Variants Are Associated With MODY With Reduced Penetrance. Nat Commun (2017) 8:888. doi: 10.1038/s41467-017-00895-9
12. Xiao X, Liu L, Xiao Y, Xie Z, Li L, Zhou H, et al. A Novel Frameshift Mutation in the Insulin Gene in a Family With Maturity Onset Diabetes of the Young. J Diabetes (2019) 11:83–6. doi: 10.1111/1753-0407.12849
13. Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF. Pancreatic Agenesis Attributable to a Single Nucleotide Deletion in the Human IPF1 Gene Coding Sequence. Nat Genet (1997) 15:106–10. doi: 10.1038/ng0197-106
14. Horikawa Y, Iwasaki N, Hara M, Furuta H, Hinokio Y, Cockburn BN, et al. Mutation in Hepatocyte Nuclear Factor 1β Gene (TCF2) Associated With MODY. Nat Genet (1997) 17:384–5. doi: 10.1038/ng1297-384
15. Hattersley AT, Patel KA. Precision Diabetes: Learning From Monogenic Diabetes. Diabetologia (2017) 60:769–77. doi: 10.1007/s00125-017-4226-2
16. Loo LSW, Lau HH, Jasmen JB, Lim CS, Teo AKK. An Arduous Journey From Human Pluripotent Stem Cells to Functional Pancreatic β Cells. Diabetes Obes Metab (2017) 20:3–13. doi: 10.1111/dom.12996
17. Santosa MM, Low BSJ, Pek NMQ, Teo AKK. Knowledge Gaps in Rodent Pancreas Biology: Taking Human Pluripotent Stem Cell-Derived Pancreatic Beta Cells Into Our Own Hands. Front Endocrinol (2016) 6:194. doi: 10.3389/fendo.2015.00194
18. Bakhti M, Böttcher A, Lickert H. Modelling the Endocrine Pancreas in Health and Disease. Nat Rev Endocrinol (2019) 15:155–71. doi: 10.1038/s41574-018-0132-z
19. Pan FC, Wright C. Pancreas Organogenesis: From Bud to Plexus to Gland. Dev Dyn (2011) 240:530–65. doi: 10.1002/dvdy.22584
20. Nair G, Hebrok M. Islet Formation in Mice and Men: Lessons for the Generation of Functional Insulin-Producing Beta-Cells From Human Pluripotent Stem Cells. Curr Opin Genet Dev (2015) 32:171–80. doi: 10.1016/j.gde.2015.03.004
21. Jennings RE, Scharfmann R, Staels W. Transcription Factors That Shape the Mammalian Pancreas. Diabetologia (2020) 63:1974–80. doi: 10.1007/s00125-020-05161-0
22. Millette K, Georgia S. Gene Editing and Human Pluripotent Stem Cells: Tools for Advancing Diabetes Disease Modeling and Beta-Cell Development. Curr Diabetes Rep (2017) 17:116. doi: 10.1007/s11892-017-0947-3
23. Hu M, Cherkaoui I, Misra S, Rutter GA. Functional Genomics in Pancreatic Beta Cells: Recent Advances in Gene Deletion and Genome Editing Technologies for Diabetes Research. Front Endocrinol (2020) 11:576632. doi: 10.3389/fendo.2020.576632
24. Petersen MBK, Gonçalves CAC, Kim YH, Grapin-Botton A. Chapter Six - Recapitulating and Deciphering Human Pancreas Development From Human Pluripotent Stem Cells in a Dish. Curr Topics Dev Biol Acad Press (2018) 129:143–90. pp. doi: 10.1016/bs.ctdb.2018.02.009
25. Yiangou L, Ross ADB, Goh KJ, Vallier L. Human Pluripotent Stem Cell-Derived Endoderm for Modeling Development and Clinical Applications. Cell Stem Cell (2018) 22:485–99. doi: 10.1016/j.stem.2018.03.016
26. Efrat S. Epigenetic Memory: Lessons From Ips Cells Derived From Human β Cells. Front Endocrinol (2020) 11:614234. doi: 10.3389/fendo.2020.614234
27. Gaertner B, Carrano AC, Sander M. Human Stem Cell Models: Lessons for Pancreatic Development and Disease. Genes Dev (2019) 33:1475–90. doi: 10.1101/gad.331397.119
28. Balboa D, Saarimäki-Vire J, Otonkoski T. Concise Review: Human Pluripotent Stem Cells for the Modeling of Pancreatic β-Cell Pathology. Stem Cells (2019) 37:33–41. doi: 10.1002/stem.2913
29. Abdelalim EM. Modeling Different Types of Diabetes Using Human Pluripotent Stem Cells. Cell Mol Life Sci (2020) 78:2459–83. doi: 10.1007/s00018-020-03710-9
30. Amirruddin NS, Low BSJ, Lee KO, Tai ES, Teo AKK. New Insights Into Human Beta Cell Biology Using Human Pluripotent Stem Cells. Semin Cell Dev Biol (2019) 103:31–40. doi: 10.1016/j.semcdb.2019.11.004
31. Balboa D, Iworima DG, Kieffer TJ. Human Pluripotent Stem Cells to Model Islet Defects in Diabetes. Front Endocrinol (2021) 12:642152. doi: 10.3389/fendo.2021.642152
32. Jennings RE, Berry AA, Kirkwood-Wilson R, Roberts NA, Hearn T, Salisbury RJ, et al. Development of the Human Pancreas From Foregut to Endocrine Commitment. Diabetes (2013) 62:3514–22. doi: 10.2337/db12-1479
33. Jeon J, Correa-Medina M, Ricordi C, Edlund H, Diez JA. Endocrine Cell Clustering During Human Pancreas Development. J Histochem Cytochem (2009) 57:811–24. doi: 10.1369/jhc.2009.953307
34. Salisbury RJ, Blaylock J, Berry AA, Jennings RE, De Krijger R, Piper Hanley K, et al. The Window Period of NEUROGENIN3 During Human Gestation. Islets (2014) 6:e954436. doi: 10.4161/19382014.2014.954436
35. Sarkar SA, Kobberup S, Wong R, Lopez AD, Quayum N, Still T, et al. Global Gene Expression Profiling and Histochemical Analysis of the Developing Human Fetal Pancreas. Diabetologia (2008) 51:285–97. doi: 10.1007/s00125-007-0880-0
36. Oliver-Krasinski JM, Stoffers DA. On the Origin of the Beta Cell. Genes Dev (2008) 22:1998–2021. doi: 10.1101/gad.1670808
37. Zaret KS. Genetic Programming of Liver and Pancreas Progenitors: Lessons for Stem-Cell Differentiation. Nat Rev Genet (2008) 9:329–40. doi: 10.1038/nrg2318
38. Byrnes LE, Wong DM, Subramaniam M, Meyer NP, Gilchrist CL, Knox SM, et al. Lineage Dynamics of Murine Pancreatic Development at Single-Cell Resolution. Nat Commun (2018) 9:3922. doi: 10.1038/s41467-018-06176-3
39. Scavuzzo MA, Hill MC, Chmielowiec J, Yang D, Teaw J, Sheng K, et al. Endocrine Lineage Biases Arise in Temporally Distinct Endocrine Progenitors During Pancreatic Morphogenesis. Nat Commun (2018) 9:3356. doi: 10.1038/s41467-018-05740-1
40. Yu XX, Qiu WL, Yang L, Zhang Y, He MY, Li LC, et al. Defining Multistep Cell Fate Decision Pathways During Pancreatic Development at Single-Cell Resolution. EMBO J (2019) 38:e100164. doi: 10.15252/embj.2018100164
41. Bastidas-Ponce A, Tritschler S, Dony L, Scheibner K, Tarquis-Medina M, Salinno C, et al. Comprehensive Single Cell Mrna Profiling Reveals a Detailed Roadmap for Pancreatic Endocrinogenesis. Development (2019) 146:dev173849. doi: 10.1242/dev.173849
42. van Gurp L, Muraro MJ, Dielen T, Seneby L, Dharmadhikari G, Gradwohl G, et al. A Transcriptomic Roadmap to Alpha- and Beta-Cell Differentiation in the Embryonic Pancreas. Development (2019) 146:dev173716. doi: 10.1242/dev.173716
43. George NM, Day CE, Boerner BP, Johnson RL, Sarvetnick NE. Hippo Signaling Regulates Pancreas Development Through Inactivation of Yap. Mol Cell Biol (2012) 32:5116–28. doi: 10.1128/MCB.01034-12
44. Gao T, Zhou D, Yang C, Singh T, Penzo-Mendez A, Maddipati R, et al. Hippo Signaling Regulates Differentiation and Maintenance in the Exocrine Pancreas. Gastroenterology (2013) 144:1543–53.e1. doi: 10.1053/j.gastro.2013.02.037
45. Cebola I, Rodriguez-Segui SA, Cho CH, Bessa J, Rovira M, Luengo M, et al. TEAD and YAP Regulate the Enhancer Network of Human Embryonic Pancreatic Progenitors. Nat Cell Biol (2015) 17:615–26. doi: 10.1038/ncb3160
46. Rodriguez-Segui SA, Bessa J. Gatekeepers of Pancreas: TEAD and YAP. Oncotarget (2015) 6:15736–7. doi: 10.18632/oncotarget.4607
47. Guo T, Landsman L, Li N, Hebrok M. Factors Expressed by Murine Embryonic Pancreatic Mesenchyme Enhance Generation of Insulin-Producing Cells From Hescs. Diabetes (2013) 62:1581–92. doi: 10.2337/db12-0167
48. Landsman L, Nijagal A, Whitchurch TJ, Vanderlaan RL, Zimmer WE, Mackenzie TC, et al. Pancreatic Mesenchyme Regulates Epithelial Organogenesis Throughout Development. PloS Biol (2011) 9:e1001143. doi: 10.1371/journal.pbio.1001143
49. Sneddon JB, Borowiak M, Melton DA. Self-Renewal of Embryonic-Stem-Cell-Derived Progenitors by Organ-Matched Mesenchyme. Nature (2012) 491:765–8. doi: 10.1038/nature11463
50. Cozzitorto C, Mueller L, Ruzittu S, Mah N, Willnow D, Darrigrand J-F, et al. A Specialized Niche in the Pancreatic Microenvironment Promotes Endocrine Differentiation. Dev Cell (2020) 55:150–162.e6. doi: 10.1016/j.devcel.2020.08.003
51. D’Amour KA, Agulnick AD, Eliazer S, Kelly OG, Kroon E, Baetge EE. Efficient Differentiation of Human Embryonic Stem Cells to Definitive Endoderm. Nat Biotechnol (2005) 23:1534–41. doi: 10.1038/nbt1163
52. D’Amour KA, Bang AG, Eliazer S, Kelly OG, Agulnick AD, Smart NG, et al. Production of Pancreatic Hormone Expressing Endocrine Cells From Human Embryonic Stem Cells. Nat Biotechnol (2006) 24:1392–401. doi: 10.1038/nbt1259
53. Kroon E, Martinson LA, Kadoya K, Bang AG, Kelly OG, Eliazer S, et al. Pancreatic Endoderm Derived From Human Embryonic Stem Cells Generates Glucose-Responsive Insulin-Secreting Cells In Vivo. Nat Biotechnol (2008) 26:443–52. doi: 10.1038/nbt1393
54. Nostro MC, Sarangi F, Ogawa S, Holtzinger A, Corneo B, Li X, et al. Stage-Specific Signaling Through Tgfβ Family Members and WNT Regulates Patterning and Pancreatic Specification of Human Pluripotent Stem Cells. Development (2011) 138:861–71. doi: 10.1242/dev.055236
55. Cho CH, Hannan NR, Docherty FM, Docherty HM, Joao Lima M, Trotter MW, et al. Inhibition of Activin/Nodal Signalling Is Necessary for Pancreatic Differentiation of Human Pluripotent Stem Cells. Diabetologia (2012) 55:3284–95. doi: 10.1007/s00125-012-2687-x
56. Jennings RE, Berry AA, Strutt JP, Gerrard DT, Hanley NA. Human Pancreas Development. Development (2015) 142:3126–37. doi: 10.1242/dev.120063
57. Jennings RE, Berry AA, Gerrard DT, Wearne SJ, Strutt J, Withey S, et al. Laser Capture and Deep Sequencing Reveals the Transcriptomic Programmes Regulating the Onset of Pancreas and Liver Differentiation in Human Embryos. Stem Cell Rep (2017) 9:1387–94. doi: 10.1016/j.stemcr.2017.09.018
58. Villani V, Thornton ME, Zook HN, Crook CJ, Grubbs BH, Orlando G, et al. SOX9+/PTF1A+ Cells Define the Tip Progenitor Cells of the Human Fetal Pancreas of the Second Trimester. Stem Cells Trans Med (2019) 8:1249–64. doi: 10.1002/sctm.19-0231
59. Ramond C, Beydag-Tasoz BS, Azad A, van de Bunt M, Petersen MBK, Beer NL, et al. Understanding Human Fetal Pancreas Development Using Subpopulation Sorting, RNA Sequencing and Single-Cell Profiling. Development (2018) 145:dev165480. doi: 10.1242/dev.165480
60. Ramond C, Glaser N, Berthault C, Ameri J, Kirkegaard JS, Hansson M, et al. Reconstructing Human Pancreatic Differentiation by Mapping Specific Cell Populations During Development. Elife (2017) 6:e27564. doi: 10.7554/eLife.27564
61. Pagliuca FW, Millman JR, Gurtler M, Segel M, Van Dervort A, Ryu JH, et al. Generation of Functional Human Pancreatic Beta Cells In Vitro. Cell (2014) 159:428–39. doi: 10.1016/j.cell.2014.09.040
62. Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, et al. Reversal of Diabetes With Insulin-Producing Cells Derived In Vitro From Human Pluripotent Stem Cells. Nat Biotechnol (2014) 32:1121–33. doi: 10.1038/nbt.3033
63. Russ HA, Parent AV, Ringler JJ, Hennings TG, Nair GG, Shveygert M, et al. Controlled Induction of Human Pancreatic Progenitors Produces Functional Beta-Like Cells. Vitro EMBO J (2015) 34:1759–72. doi: 10.15252/embj.201591058
64. Amin S, Cook B, Zhou T, Ghazizadeh Z, Lis R, Zhang T.A.-O.X., et al. Discovery of a Drug Candidate for GLIS3-Associated Diabetes. Nat Commun (2018) 9:2681. doi: 10.1038/s41467-018-04918-x
65. Sui L, Danzl N, Campbell SR, Viola R, Williams D, Xing Y, et al. β-Cell Replacement in Mice Using Human Type 1 Diabetes Nuclear Transfer Embryonic Stem Cells. Diabetes (2017) 67:26–35. doi: 10.2337/db17-0120
66. Veres A, Faust AL, Bushnell HL, Engquist EN, Kenty JH, Harb G, et al. Charting Cellular Identity During Human In Vitro Beta-Cell Differentiation. Nature (2019) 569:368–73. doi: 10.1038/s41586-019-1168-5
67. Nair GG, Liu JS, Russ HA, Tran S, Saxton MS, Chen R, et al. Recapitulating Endocrine Cell Clustering in Culture Promotes Maturation of Human Stem-Cell-Derived β Cells. Nat Cell Biol (2019) 21:263–74. doi: 10.1038/s41556-018-0271-4
68. Velazco-Cruz L, Song J, Maxwell KG, Goedegebuure MM, Augsornworawat P, Hogrebe NJ, et al. Acquisition of Dynamic Function in Human Stem Cell-Derived β Cells. Stem Cell Rep (2019) 12:351–65. doi: 10.1016/j.stemcr.2018.12.012
69. Hogrebe NA-O, Augsornworawat PA-O, Maxwell KA-O, Velazco-Cruz L, Millman JA-O. Targeting the Cytoskeleton to Direct Pancreatic Differentiation of Human Pluripotent Stem Cells. Nat Biotechnol (2020) 38:460–70. doi: 10.1038/s41587-020-0430-6
70. Mamidi A, Prawiro C, Seymour PA, de Lichtenberg KH, Jackson A, Serup P, et al. Mechanosignalling via Integrins Directs Fate Decisions of Pancreatic Progenitors. Nature (2018) 564:114–8. doi: 10.1038/s41586-018-0762-2
71. Rosado-Olivieri EA, Anderson K, Kenty JH, Melton DA. YAP Inhibition Enhances the Differentiation of Functional Stem Cell-Derived Insulin-Producing β Cells. Nat Commun (2019) 10:1464. doi: 10.1038/s41467-019-09404-6
72. Xie R, Everett LJ, Lim HW, Patel NA, Schug J, Kroon E, et al. Dynamic Chromatin Remodeling Mediated by Polycomb Proteins Orchestrates Pancreatic Differentiation of Human Embryonic Stem Cells. Cell Stem Cell (2013) 12:224–37. doi: 10.1016/j.stem.2012.11.023
73. Loh KM, Ang LT, Zhang J, Kumar V, Ang J, Auyeong JQ, et al. Efficient Endoderm Induction From Human Pluripotent Stem Cells by Logically Directing Signals Controlling Lineage Bifurcations. Cell Stem Cell (2014) 14:237–52. doi: 10.1016/j.stem.2013.12.007
74. Wang A, Yue F, Li Y, Xie R, Harper T, Patel NA, et al. Epigenetic Priming of Enhancers Predicts Developmental Competence of Hesc-Derived Endodermal Lineage Intermediates. Cell Stem Cell (2015) 16:386–99. doi: 10.1016/j.stem.2015.02.013
75. Li J, Wu X, Zhou Y, Lee M, Guo L, Han W, et al. Decoding the Dynamic DNA Methylation and Hydroxymethylation Landscapes in Endodermal Lineage Intermediates During Pancreatic Differentiation of Hesc. Nucleic Acids Res (2018) 46:2883–900. doi: 10.1093/nar/gky063
76. van Arensbergen J, Garcia-Hurtado J, Moran I, Maestro MA, Xu XB, Van de Casteele M, et al. Derepression of Polycomb Targets During Pancreatic Organogenesis Allows Insulin-Producing Beta-Cells to Adopt a Neural Gene Activity Program. Genome Res (2010) 20:722–32. doi: 10.1101/gr.101709.109
77. van Arensbergen J, Garcia-Hurtado J, Maestro MA, Correa-Tapia M, Rutter GA, Vidal M, et al. Ring1b Bookmarks Genes in Pancreatic Embryonic Progenitors for Repression in Adult Beta Cells. Genes Dev (2013) 27:52–63. doi: 10.1101/gad.206094.112
78. Xu CR, Li LC, Donahue G, Ying L, Zhang YW, Gadue P, et al. Dynamics of Genomic H3k27me3 Domains and Role of EZH2 During Pancreatic Endocrine Specification. EMBO J (2014) 33:2157–70. doi: 10.15252/embj.201488671
79. Petersen MBK, Azad A, Ingvorsen C, Hess K, Hansson M, Grapin-Botton A, et al. Single-Cell Gene Expression Analysis of a Human ESC Model of Pancreatic Endocrine Development Reveals Different Paths to Beta-Cell Differentiation. Stem Cell Rep (2017) 9:1246–61. doi: 10.1016/j.stemcr.2017.08.009
80. Schaffer AE, Freude KK, Nelson SB, Sander M. Nkx6 Transcription Factors and Ptf1a Function as Antagonistic Lineage Determinants in Multipotent Pancreatic Progenitors. Dev Cell (2010) 18:1022–9. doi: 10.1016/j.devcel.2010.05.015
81. Aigha I, Memon B, Elsayed A, Abdelalim E. Differentiation of Human Pluripotent Stem Cells Into Two Distinct NKX6.1 Populations of Pancreatic Progenitors. Stem Cell Res Ther (2018) 9:83. doi: 10.1186/s13287-018-0834-0
82. Krentz NAJ, Lee MYY, Xu EE, Sproul SLJ, Maslova A, Sasaki S, et al. Single-Cell Transcriptome Profiling of Mouse and Hesc-Derived Pancreatic Progenitors. Stem Cell Rep (2018) 11:1551–64. doi: 10.1016/j.stemcr.2018.11.008
83. Barkas N, Petukhov V, Nikolaeva D, Lozinsky Y, Demharter S, Khodosevich K, et al. Joint Analysis of Heterogeneous Single-Cell RNA-Seq Dataset Collections. Nat Methods (2019) 16:695–8. doi: 10.1038/s41592-019-0466-z
84. Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, et al. Loh, P.-R., and Raychaudhuri, s., Fast, Sensitive and Accurate Integration of Single-Cell Data With Harmony. Nat Methods (2019) 16:1289–96. doi: 10.1038/s41592-019-0619-0
85. Helman A, Cangelosi AL, Davis JC, Pham Q, Rothman A, Faust AL, et al. A Nutrient-Sensing Transition at Birth Triggers Glucose-Responsive Insulin Secretion. Cell Metab (2020) 31:1004–1016.e5. doi: 10.1016/j.cmet.2020.04.004
86. Hattersley AT, Turner RC, Patel P, O’Rahilly S, Wainscoat JS, Permutt MA, et al. Linkage of Type 2 Diabetes to the Glucokinase Gene. Lancet (1992) 339:1307–10. doi: 10.1016/0140-6736(92)91958-B
87. Froguel P, Vaxillaire M, Sun F, Velho G, Zouali H, Butel MO, et al. Close Linkage of Glucokinase Locus on Chromosome 7p to Early-Onset non-Insulin-Dependent Diabetes Mellitus. Nature (1992) 356:162–4. doi: 10.1038/356162a0
88. Yamagata K, Oda N, Kaisaki PJ, Menzel S, Furuta H, Vaxillaire M, et al. Mutations in the Hepatocyte Nuclear Factor-1 Alpha Gene in Maturity-Onset Diabetes of the Young (MODY3). Nature (1996) 384:455–8. doi: 10.1038/384455a0
89. Yamagata K, Furuta H, Oda N, Kaisaki PJ, Menzel S, Cox NJ, et al. Mutations in the Hepatocyte Nuclear Factor-4 Alpha Gene in Maturity-Onset Diabetes of the Young (MODY1). Nature (1996) 384:458–60. doi: 10.1038/384458a0
90. Horikawa Y, Iwasaki N, Hara M, Furuta H, Hinokio Y, Cockburn BN, et al. Mutation in Hepatocyte Nuclear Factor-1 Beta Gene (TCF2) Associated With MODY. Nat Genet (1997) 17:384–5. doi: 10.1038/ng1297-384
91. Firdous P, Nissar K, Ali S, Ganai BA, Shabir U, Hassan T, et al. Genetic Testing of Maturity-Onset Diabetes of the Young Current Status and Future Perspectives. Front Endocrinol (2018) 9:253. doi: 10.3389/fendo.2018.00253
92. Senee V, Chelala C, Duchatelet S, Feng D, Blanc H, Cossec JC, et al. Mutations in GLIS3 Are Responsible for a Rare Syndrome With Neonatal Diabetes Mellitus and Congenital Hypothyroidism. Nat Genet (2006) 38:682–7. doi: 10.1038/ng1802
93. Smith SB, Qu HQ, Taleb N, Kishimoto NY, Scheel DW, Lu Y, et al. Rfx6 Directs Islet Formation and Insulin Production in Mice and Humans. Nature (2010) 463:775–80. doi: 10.1038/nature08748
94. Flanagan SE, De Franco E, Lango Allen H, Zerah M, Abdul-Rasoul MM, Edge JA, et al. Analysis of Transcription Factors Key for Mouse Pancreatic Development Establishes NKX2-2 and MNX1 Mutations as Causes of Neonatal Diabetes in Man. Cell Metab (2014) 19:146–54. doi: 10.1016/j.cmet.2013.11.021
95. Bonnefond A, Vaillant E, Philippe J, Skrobek B, Lobbens S, Yengo L, et al. Transcription Factor Gene MNX1 Is a Novel Cause of Permanent Neonatal Diabetes in a Consanguineous Family. Diabetes Metab (2013) 39:276–80. doi: 10.1016/j.diabet.2013.02.007
96. Rubio-Cabezas O, Jensen JN, Hodgson MI, Codner E, Ellard S, Serup P, et al. Permanent Neonatal Diabetes and Enteric Anendocrinosis Associated With Biallelic Mutations in NEUROG3. Diabetes (2011) 60:1349. doi: 10.2337/db10-1008
97. Rubio-Cabezas O, Minton JAL, Kantor I, Williams D, Ellard S, Hattersley AT. Homozygous Mutations in NEUROD1 Are Responsible for a Novel Syndrome of Permanent Neonatal Diabetes and Neurological Abnormalities. Diabetes (2010) 59:2326. doi: 10.2337/db10-0011
98. Sellick GS, Barker KT, Stolte-Dijkstra I, Fleischmann C, J Coleman R, Garrett C, et al. Mutations in PTF1A Cause Pancreatic and Cerebellar Agenesis. Nat Genet (2004) 36:1301–5. doi: 10.1038/ng1475
99. Dimitri P, Warner JT, Minton JA, Patch AM, Ellard S, Hattersley AT, et al. Novel GLIS3 Mutations Demonstrate an Extended Multisystem Phenotype. Eur J Endocrinol (2011) 164:437–43. doi: 10.1530/EJE-10-0893
100. Concepcion JP, Reh CS, Daniels M, Liu X, Paz VP, Ye H, et al. Neonatal Diabetes, Gallbladder Agenesis, Duodenal Atresia, and Intestinal Malrotation Caused by a Novel Homozygous Mutation in RFX6. Pediatr Diabetes (2014) 15:67–72. doi: 10.1111/pedi.12063
101. Rieck S, Bankaitis ED, Wright CV. Lineage Determinants in Early Endocrine Development. Semin Cell Dev Biol (2012) 23:673–84. doi: 10.1016/j.semcdb.2012.06.005
102. Clissold RL, Hamilton AJ, Hattersley AT, Ellard S, Bingham C. HNF1B-Associated Renal and Extra-Renal Disease-an Expanding Clinical Spectrum. Nat Rev Nephrol (2014) 11:102–12. doi: 10.1038/nrneph.2014.232
103. Edghill EL, Bingham C, Slingerland AS, Minton JA, Noordam C, Ellard S, et al. Hepatocyte Nuclear Factor-1 Beta Mutations Cause Neonatal Diabetes and Intrauterine Growth Retardation: Support for a Critical Role of HNF-1beta in Human Pancreatic Development. Diabetes Med (2006) 23:1301–6. doi: 10.1111/j.1464-5491.2006.01999.x
104. Bellanne-Chantelot C, Clauin S, Chauveau D, Collin P, Daumont M, Douillard C, et al. Large Genomic Rearrangements in the Hepatocyte Nuclear Factor-1beta (TCF2) Gene Are the Most Frequent Cause of Maturity-Onset Diabetes of the Young Type 5. Diabetes (2005) 54:3126–32. doi: 10.2337/diabetes.54.11.3126
105. Vethe H, Bjørlykke Y, Ghila LM, Paulo JA, Scholz H, Gygi SP, et al. Probing the Missing Mature β-Cell Proteomic Landscape in Differentiating Patient Ipsc-Derived Cells. Sci Rep (2017) 7:4780. doi: 10.1038/s41598-017-04979-w
106. Braverman-Gross C, Nudel N, Ronen D, Beer NL, McCarthy MI, Benvenisty N. Derivation and Molecular Characterization of Pancreatic Differentiated MODY1-Ipscs. Stem Cell Res (2018) 31:16–26. doi: 10.1016/j.scr.2018.06.013
107. Ng NHJ, Jasmen JB, Lim CS, Lau HH, Krishnan VG, Kadiwala J, et al. HNF4A Haploinsufficiency in MODY1 Abrogates Liver and Pancreas Differentiation From Patient-Derived Induced Pluripotent Stem Cells. iScience (2019) 16:192–205. doi: 10.1016/j.isci.2019.05.032
108. Aqel YWA, Ali G, Elsayed AK, Al-Khawaga S, Hussain K, Abdelalim EM. Generation of Two Human Ipsc Lines From Patients With Maturity-Onset Diabetes of the Young Type 2 (MODY2) and Permanent Neonatal Diabetes Due to Mutations in the GCK Gene. Stem Cell Res (2020) 48:101991. doi: 10.1016/j.scr.2020.101991
109. Cardenas-Diaz FL, Osorio-Quintero C, Diaz-Miranda MA, Kishore S, Leavens K, Jobaliya C, et al. Modeling Monogenic Diabetes Using Human Escs Reveals Developmental and Metabolic Deficiencies Caused by Mutations in HNF1A. Cell Stem Cell (2019) 25:273–289.e5. doi: 10.1016/j.stem.2019.07.007
110. Low BSJ, Lim CS, Ding SSL, Tan YS, Ng NHJ, Krishnan VG, et al. Decreased GLUT2 and Glucose Uptake Contribute to Insulin Secretion Defects in MODY3/HNF1A Hipsc-Derived Mutant β Cells. Nat Commun (2021) 12:3133. doi: 10.1038/s41467-021-22843-4
111. Zhu Z, Li QV, Lee K, Rosen BP, González F, Soh C-L, et al. Genome Editing of Lineage Determinants in Human Pluripotent Stem Cells Reveals Mechanisms of Pancreatic Development and Diabetes. Cell Stem Cell (2016) 18:755–68. doi: 10.1016/j.stem.2016.03.015
112. Wang X, Sterr M, Ansarullah, Burtscher I, Böttcher A, Beckenbauer J, et al. Point Mutations in the PDX1 Transactivation Domain Impair Human β-Cell Development and Function. Mol Metab (2019) 24:80–97. doi: 10.1016/j.molmet.2019.03.006
113. Teo AK, Lau HH, Valdez IA, Dirice E, Tjora E, Raeder H, et al. Early Developmental Perturbations in a Human Stem Cell Model of MODY5/HNF1B Pancreatic Hypoplasia. Stem Cell Rep (2016) 6:357–67. doi: 10.1016/j.stemcr.2016.01.007
114. Tiyaboonchai A, Cardenas-Diaz FL, Ying L, Maguire JA, Sim X, Jobaliya C, et al. GATA6 Plays an Important Role in the Induction of Human Definitive Endoderm, Development of the Pancreas, and Functionality of Pancreatic β Cells. Stem Cell Rep (2017) 8:589–604. doi: 10.1016/j.stemcr.2016.12.026
115. Shi Z-D, Lee K, Yang D, Amin S, Verma N, Li QV, et al. Genome Editing in Hpscs Reveals GATA6 Haploinsufficiency and a Genetic Interaction With GATA4 in Human Pancreatic Development. Cell Stem Cell (2017) 20:675–688.e6. doi: 10.1016/j.stem.2017.01.001
116. Chia CY, Madrigal P, Denil SLIJ, Martinez I, Garcia-Bernardo J, El-Khairi R, et al. GATA6 Cooperates With EOMES/SMAD2/3 to Deploy the Gene Regulatory Network Governing Human Definitive Endoderm and Pancreas Formation. Stem Cell Rep (2019) 12:57–70. doi: 10.1016/j.stemcr.2018.12.003
117. Kishore S, De Franco E, Cardenas-Diaz FL, Letourneau-Freiberg LR, Sanyoura M, Osorio-Quintero C, et al. A non-Coding Disease Modifier of Pancreatic Agenesis Identified by Genetic Correction in a Patient-Derived Ipsc Line. Cell Stem Cell (2020) 27:137–146.e6. doi: 10.1016/j.stem.2020.05.001
118. Trott J, Alpagu Y, Tan EK, Shboul M, Dawood Y, Elsy M, et al. Mitchell-Riley Syndrome Ipscs Exhibit Reduced Pancreatic Endoderm Differentiation Due to a Mutation in RFX6. Development (2020) 147:dev194878. doi: 10.1242/dev.194878
119. Shang L, Hua H, Foo K, Martinez H, Watanabe K, Zimmer M, et al. β-Cell Dysfunction Due to Increased ER Stress in a Stem Cell Model of Wolfram Syndrome. Diabetes (2014) 63:923. doi: 10.2337/db13-0717
120. Maxwell KG, Augsornworawat P, Velazco-Cruz L, Kim MH, Asada R, Hogrebe NJ, et al. Gene-Edited Human Stem Cell-Derived β Cells From a Patient With Monogenic Diabetes Reverse Preexisting Diabetes in Mice. Sci Trans Med (2020) 12:eaax9106. doi: 10.1126/scitranslmed.aax9106
121. Offield MF, Jetton TL, Labosky PA, Ray M, Stein RW, Magnuson MA, et al. PDX-1 Is Required for Pancreatic Outgrowth and Differentiation of the Rostral Duodenum. Development (1996) 122:983–95. doi: 10.1242/dev.122.3.983
122. Jonsson J, Carlsson L, Edlund T, Edlund H. Insulin-Promoter-Factor 1 Is Required for Pancreas Development in Mice. Nature (1994) 371:606–9. doi: 10.1038/371606a0
123. Ahlgren U, Jonsson J, Edlund H. The Morphogenesis of the Pancreatic Mesenchyme Is Uncoupled From That of the Pancreatic Epithelium in IPF1/PDX1-Deficient Mice. Development (1996) 122:1409–16. doi: 10.1242/dev.122.5.1409
124. Johnson JD, Ahmed NT, Luciani DS, Han Z, Tran H, Fujita J, et al. Increased Islet Apoptosis in Pdx1+/- Mice. J Clin Invest (2003) 111:1147–60. doi: 10.1172/JCI16537
125. Stoffers DA, Ferrer J, Clarke WL, Habener JF. Early-Onset Type-II Diabetes Mellitus (MODY4) Linked to IPF1. Nat Genet (1997) 17:138–9. doi: 10.1038/ng1097-138
126. Brissova M, Blaha M, Spear C, Nicholson W, Radhika A, Shiota M, et al. Reduced PDX-1 Expression Impairs Islet Response to Insulin Resistance and Worsens Glucose Homeostasis. Am J Physiol-Endocrinol Metab (2005) 288:E707–14. doi: 10.1152/ajpendo.00252.2004
127. Brissova M, Shiota M, Nicholson WE, Gannon M, Knobel SM, Piston DW, et al. Reduction in Pancreatic Transcription Factor PDX-1 Impairs Glucose-Stimulated Insulin Secretion. J Biol Chem (2002) 277:11225–32. doi: 10.1074/jbc.M111272200
128. Zegre Amorim M, Houghton JAL, Carmo S, Salva IS, Pita A, Pereira-da-Silva L. Mitchell-Riley Syndrome: A Novel Mutation in RFX6 Gene. Case Rep Genet (2015) 2015:937201. doi: 10.1155/2015/937201
129. Gradwohl G, Dierich A, LeMeur M, Guillemot F. Neurogenin3 Is Required for the Development of the Four Endocrine Cell Lineages of the Pancreas. Proc Natl Acad Sci (2000) 97:1607. doi: 10.1073/pnas.97.4.1607
130. Rubio-Cabezas O, Codner E, Flanagan SE, Gómez JL, Ellard S, Hattersley AT. Neurogenin 3 Is Important But Not Essential for Pancreatic Islet Development in Humans. Diabetologia (2014) 57:2421–4. doi: 10.1007/s00125-014-3349-y
131. Solorzano-Vargas RS, Bjerknes M, Wang J, Wu SV, Garcia-Careaga MG, Pitukcheewanont P, et al. Null Mutations of NEUROG3 Are Associated With Delayed-Onset Diabetes Mellitus. JCI Insight (2020) 5:e127657. doi: 10.1172/jci.insight.127657
132. Zhang X, McGrath PS, Salomone J, Rahal M, McCauley HA, Schweitzer J, et al. A Comprehensive Structure-Function Study of Neurogenin3 Disease-Causing Alleles During Human Pancreas and Intestinal Organoid Development. Dev Cell (2019) 50:367–80.e7. doi: 10.1016/j.devcel.2019.05.017
133. McGrath PS, Watson CL, Ingram C, Helmrath MA, Wells JM. The Basic Helix-Loop-Helix Transcription Factor NEUROG3 Is Required for Development of the Human Endocrine Pancreas. Diabetes (2015) 64:2497. doi: 10.2337/db14-1412
134. Wang D, Wang J, Bai L, Pan H, Feng H, Clevers H, et al. Long-Term Expansion of Pancreatic Islet Organoids From Resident Procr(+) Progenitors. Cell (2020) 180:1198–1211 e19. doi: 10.1016/j.cell.2020.02.048
135. Watanabe N, Hiramatsu K, Miyamoto R, Yasuda K, Suzuki N, Oshima N, et al. A Murine Model of Neonatal Diabetes Mellitus in Glis3-Deficient Mice. FEBS Lett (2009) 583:2108–13. doi: 10.1016/j.febslet.2009.05.039
136. Yang Y, Bush SP, Wen X, Cao W, Chan L. Differential Gene Dosage Effects of Diabetes-Associated Gene GLIS3 in Pancreatic β Cell Differentiation and Function. Endocrinology (2017) 158:9–20. doi: 10.1210/en.2016-1541
137. Servitja JM, Pignatelli M, Maestro MA, Cardalda C, Boj SF, Lozano J, et al. Hnf1alpha (MODY3) Controls Tissue-Specific Transcriptional Programs and Exerts Opposed Effects on Cell Growth in Pancreatic Islets and Liver. Mol Cell Biol (2009) 29:2945–59. doi: 10.1128/MCB.01389-08
138. Pontoglio M, Sreenan S, Roe M, Pugh W, Ostrega D, Doyen A, et al. Defective Insulin Secretion in Hepatocyte Nuclear Factor 1alpha-Deficient Mice. J Clin Invest (1998) 101:2215–22. doi: 10.1172/JCI2548
139. Ellard S, Colclough K. Mutations in the Genes Encoding the Transcription Factors Hepatocyte Nuclear Factor 1 Alpha (HNF1A) and 4 Alpha (HNF4A) in Maturity-Onset Diabetes of the Young. Hum Mutat (2006) 27:854–69. doi: 10.1002/humu.20357
140. Haliyur R, Tong X, Sanyoura M, Shrestha S, Lindner J, Saunders DC, et al. Human Islets Expressing HNF1A Variant Have Defective β Cell Transcriptional Regulatory Networks. J Clin Invest (2019) 129:246–51. doi: 10.1172/jci121994
141. Allen HL, Flanagan SE, Shaw-Smith C, De Franco E, Akerman I, Caswell R, et al. GATA6 Haploinsufficiency Causes Pancreatic Agenesis in Humans. Nat Genet (2012) 44:20–2. doi: 10.1038/ng.1035
142. Xuan S, Borok MJ, Decker KJ, Battle MA, Duncan SA, Hale MA, et al. Pancreas-Specific Deletion of Mouse Gata4 and Gata6 Causes Pancreatic Agenesis. J Clin Invest (2012) 122:3516–28. doi: 10.1172/JCI63352
143. Carrasco M, Delgado I, Soria B, Martin F, Rojas A. GATA4 and GATA6 Control Mouse Pancreas Organogenesis. J Clin Invest (2012) 122:3504–15. doi: 10.1172/JCI63240
144. Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, et al. Embryonic Stem Cell Lines Derived From Human Blastocysts. Science (1998) 282:1145. doi: 10.1126/science.282.5391.1145
145. Chen WS, Manova K, Weinstein DC, Duncan SA, Plump AS, Prezioso VR, et al. Disruption of the HNF-4 Gene, Expressed in Visceral Endoderm, Leads to Cell Death in Embryonic Ectoderm and Impaired Gastrulation of Mouse Embryos. Genes Dev (1994) 8:2466–77. doi: 10.1101/gad.8.20.2466[doi
146. Stoffel M, Duncan SA. The Maturity-Onset Diabetes of the Young (MODY1) Transcription Factor HNF4α Regulates Expression of Genes Required for Glucose Transport and Metabolism. Proc Natl Acad Sci (1997) 94:13209. doi: 10.1073/pnas.94.24.13209
147. Shih DQ, Heimesaat M, Kuwajima S, Stein R, Wright CVE, Stoffel M. Profound Defects in Pancreatic Beta-Cell Function in Mice With Combined Heterozygous Mutations in Pdx-1, Hnf-1alpha, and Hnf-3beta. Proc Natl Acad Sci (2002) 99:3818. doi: 10.1073/pnas.062605899
148. Fajans SS, Bell GI, Polonsky KS. Molecular Mechanisms and Clinical Pathophysiology of Maturity-Onset Diabetes of the Young. New Engl J Med (2001) 345:971–80. doi: 10.1056/NEJMra002168
149. Krapp A, Knöfler M, Ledermann B, Bürki K, Berney C, Zoerkler N, et al. The Bhlh Protein PTF1-P48 Is Essential for the Formation of the Exocrine and the Correct Spatial Organization of the Endocrine Pancreas. Genes Dev (1998) 12:3752–63. doi: 10.1101/gad.12.23.3752
150. Burlison JS, Long Q, Fujitani Y, Wright CV, Magnuson MA. Pdx-1 and Ptf1a Concurrently Determine Fate Specification of Pancreatic Multipotent Progenitor Cells. Dev Biol (2008) 316:74–86. doi: 10.1016/j.ydbio.2008.01.011
151. Weedon MN, Cebola I, Patch AM, Flanagan SE, De Franco E, Caswell R, et al. Recessive Mutations in a Distal PTF1A Enhancer Cause Isolated Pancreatic Agenesis. Nat Genet (2014) 46:61–4. doi: 10.1038/ng.2826
152. Balboa D, Saarimäki-Vire J, Borshagovski D, Survila M, Pulkkila P, Galli E, et al. Insulin Mutations Impair Beta-Cell Development in a Patient-Derived Ipsc Model of Neonatal Diabetes. Elife (2018) 7:e38519. doi: 10.7554/eLife.38519
153. Ma S, Viola R, Sui L, Cherubini V, Barbetti F, Egli D. β Cell Replacement After Gene Editing of a Neonatal Diabetes-Causing Mutation at the Insulin Locus. Stem Cell Rep (2018) 11:1407–15. doi: 10.1016/j.stemcr.2018.11.006
Keywords: pancreas, beta cell, human, pluripotent stem cell, monogenic, modeling, diabetes, in vitro differentiation
Citation: Burgos JI, Vallier L and Rodríguez-Seguí SA (2021) Monogenic Diabetes Modeling: In Vitro Pancreatic Differentiation From Human Pluripotent Stem Cells Gains Momentum. Front. Endocrinol. 12:692596. doi: 10.3389/fendo.2021.692596
Received: 08 April 2021; Accepted: 15 June 2021;
Published: 06 July 2021.
Edited by:
Timo Otonkoski, University of Helsinki, FinlandReviewed by:
Marcia Hiriart, Universidad Nacional Autonoma de Mexico, MexicoCopyright © 2021 Burgos, Vallier and Rodríguez-Seguí. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ludovic Vallier, bHYyMjVAY2FtLmFjLnVr; Santiago A. Rodríguez-Seguí, c3JvZHJpZ3VlekBmYm1jLmZjZW4udWJhLmFy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.