 Omar Al Rifai1,2
Omar Al Rifai1,2 Delia Susan-Resiga3Rachid Essalmani3John W. M. Creemers4Nabil G. Seidah3
Delia Susan-Resiga3Rachid Essalmani3John W. M. Creemers4Nabil G. Seidah3 Mathieu Ferron1,2,5,6*
Mathieu Ferron1,2,5,6*- 1Unité de recherche en physiologie moléculaire, Institut de Recherches Cliniques de Montréal, Montréal, QC, Canada
- 2Programme de biologie moléculaire, Université de Montréal, Montréal, QC, Canada
- 3Unité de recherche en biochimie neuroendocrinienne, Institut de Recherches Cliniques de Montréal, Montréal, QC, Canada
- 4Department of Human Genetics, Katholieke Universiteit (KU) Leuven, Leuven, Belgium
- 5Département de Médecine, Université de Montréal, Montréal, QC, Canada
- 6Division of Experimental Medicine, McGill University, Montréal, QC, Canada
Fibroblast growth factor 23 (FGF23) is a hormone secreted from fully differentiated osteoblasts and osteocytes that inhibits phosphate reabsorption by kidney proximal tubules. The full-length (i.e., intact) protein mediates FGF23 endocrine functions, while endoproteolytic cleavage at a consensus cleavage sequence for the proprotein convertases (PCs) inactivates FGF23. Two PCs, furin and PC5, were shown to cleave FGF23 in vitro at RHTR179↓, but whether they are fulfilling this function in vivo is currently unknown. To address this question, we used here mice lacking either or both furin and PC5 in cell-specific manners and mice lacking the paired basic amino acid-cleaving enzyme 4 (PACE4) in all cells. Our analysis shows that furin inactivation in osteoblasts and osteocytes results in a 25% increase in circulating intact FGF23, without any significant impact on serum phosphate levels, whether mice are maintained on a normal or a low phosphate diet. Under conditions of iron deficiency, FGF23 is normally processed in control mice, but its processing is impaired in mice lacking furin in osteoblasts and osteocytes. In contrast, FGF23 is normally cleaved following erythropoietin or IL-1β injections in mice lacking furin or both furin and PC5, and in PACE4-deficient mice. Altogether, these studies suggest that furin is only partially responsible for FGF23 cleavage under certain conditions in vivo. The processing of FGF23 may therefore involve the redundant action of multiple PCs or of other peptidases in osteoblasts, osteocytes and hematopoietic cells.
Introduction
Fibroblast growth factor 23 (FGF23) is a bone-derived hormone (i.e., osteokine) secreted by fully differentiated osteoblasts and osteocytes. FGF23 plays a fundamental role in phosphate and vitamin D metabolism through a direct action on the kidney (1, 2). In this tissue, its signaling is mediated through interaction with the FGF receptor 1 (FGFR1) and its co-receptor α-klotho (3). FGF23 signaling through FGFR1/α-klotho in the kidney proximal tubule suppresses the expression of Slc34a1 and Slc34a3, the genes encoding for the sodium phosphate co-transporters NaPi2A and NaPi2C, thereby increasing urinary phosphate excretion and decreasing serum phosphate levels. In addition, FGF23 reduces the 1,25-dihydroxyvitamin D3 (calcitriol) level by decreasing expression of Cyp27b1 which encodes the 25-hydroxyvitamin D-1 alpha hydroxylase and increasing expression of Cyp24a1 which encodes the 1,25-dihydroxyvitamin D3 24-hydroxylase (4–7). These endocrine functions are mediated through full-length or intact FGF23 (amino acids [aa] 25-251). Endoproteolytic cleavage of FGF23 at the basic motif R176HTR179↓SA, a consensus cleavage site for proprotein convertases (PCs), releases N-terminal (aa 25-179) and C-terminal (aa 180-251) fragments and inactivates FGF23.
The importance of endoproteolysis in the regulation of FGF23 activity is exemplified by rare human diseases. Mutations in the FGF23 cleavage site cause autosomal dominant hypophosphatemic rickets (ADHR) which is characterized by hypophosphatemia and osteomalacia due to the increased level of active intact FGF23 (8, 9). Conversely, other mutations in FGF23 which result in an increase in FGF23 processing and subsequent decrease in circulating intact FGF23 levels lead to familial tumoral calcinosis (FTC), a human disease characterized by hyperphosphatemia and heterotopic calcification (10–12).
Iron deficiency, erythropoietin treatment or interleukin-1 beta (IL-1β) stimulate FGF23 production by osteocytes and bone marrow hematopoietic cells (13–18). However, this increase in FGF23 production only results in the accumulation of intact FGF23 levels and hypophosphatemia in ADHR patients or in mice with an ADHR-like mutation in Fgf23. In wildtype animals or healthy subjects, the same stimuli result in increased circulating levels of cleaved FGF23, revealing the importance of endoproteolysis by PC for the regulation of this hormone.
The PCs of the subtilisin/kexin type include proprotein convertase 1 (PC1), proprotein convertase 2 (PC2), furin, proprotein convertase 4 (PC4), proprotein convertase 5A and B (PC5A/B), PACE4 and proprotein convertase 7 (PC7) (19, 20). Previous studies suggest that only furin, PACE4, PC5A and PC7 are expressed in the osteoblastic lineage (21, 22). In addition, furin and PC5 can cleave FGF23 in vitro (21), and FURIN inactivation in the human osteosarcomas U2-OS cell line impaired the cleavage of ectopically expressed human FGF23 (23). Nevertheless, the identity of the enzyme(s) responsible for FGF23 endoproteolysis in vivo remains undetermined. In the present study we assessed whether furin and PC5 cleave FGF23 in vivo and affect phosphate metabolism. We showed that unlike what is the case in vitro, furin inactivation in the osteoblastic lineage results only in a modest increase in intact FGF23 under normal or low phosphate diet, but impairs FGF23 cleavage in conditions of iron deficiency. We also show that furin, PC5 and PACE4, another PC expressed in osteoblasts, which all can cleave FGF23 in vitro, failed to do so in vivo following treatment with erythropoietin or IL-1β. Together, our data suggest that furin, PC5 and PACE4 may not be the main PCs cleaving FGF23 in vivo.
Materials and Methods
Mice and Diets
Furinflox/flox and Pcsk5flox/flox mice were generated by introducing loxP site flanking exon 2 of furin gene and exon 1 of Pcsk5 gene as described previously (24, 25). To generate conditional knockout of furin (Furinosb-/-) or PC5 (Pcsk5osb-/-) in osteoblasts and osteocytes, Furinflox/flox and Pcsk5flox/flox were crossed with hOCN-Cre transgenic mice (22), which express the Cre-recombinase under the control of human osteocalcin promoter (26). Furin;Pcsk5osb-/- mice are deficient in both Furin and Pcsk5 in osteoblasts/osteocytes and were generated by breeding Furinosb-/- and Pcsk5flox/flox mice. FurinBM-/- and Furinosb;BM-/- mice were generated by breeding Furinosb-/- mice with Furinflox/+;Vav-iCre mice. The Vav-iCre expresses the Cre recombinase under the control of the Vav promoter, allowing the efficient deletion of furin in fetal and adult hematopoietic stem cells (HSC) (27). Pcsk6-/- mice were provided by Dr. Robert Day (Université de Sherbrooke), these mice were generated as described previously (28). All mice were backcrossed at least 10 time with the C57BL/6J genetic background, they were maintained at the IRCM pathogen free animal facility on 12h light/dark cycle and fed a normal chow diet. In all experiments, unless otherwise indicated, female mice were used.
In some indicated experiments mice were fed synthetic diet to induce iron or phosphate deficiency. To induce iron deficiency, after weaning at 21 days old, mice were fed either with normal chow diet or an iron deficient diet (Research diet, AIN-76A modified rodent diet, D09070102i) for 14 weeks in combination with bleeding equivalent to one blood capillary (70 µl) every two weeks. To induce phosphate deficiency, mice were fed for 5 days a normal phosphate diet (Envigo custom diet TD. 98243, 0.6% phosphorus), then divided into two groups. The first group was maintained for one more week on 0.6% PI diet while the second group was fed a phosphate deficient diet (Envigo custom diet, TD. 86128, 0.02% phosphorus). In other experiments, mice fed a normal chow diet (Envigo 2018 Teklad global 18% protein rodent diets, containing ~0.7% inorganic phosphate) were injected with vehicle (saline or PBS), 300U/kg or 3000U/kg of recombinant human erythropoietin (EPREX, Janssen-Cilag) or 50ng/g IL-1β (STEMCELL Technologies). At the end of experiments, mice were anesthetized by injecting i.p. Ketamine (10 mg/kg)/Xylazine (1mg/kg), and serum was collected by heart puncture. All animal use complied with the guidelines of the Canadian Committee for Animal Protection and was approved by IRCM Animal Care Committee.
Serum Biochemistry
Intact and C-terminal FGF23 measurements were performed on EDTA plasma collected from mice under fed conditions using ELISA assays specific for mouse intact or C-terminal FGF23 (Quidel, cat#: 60-6800 and cat#: 60-6300). Since C-terminal FGF23 ELISA detects both intact and processed FGF23, it reflects the total concentration of FGF23 in the circulation. We also calculated the level of cleaved FGF23 by subtracting intact from C-terminal values. Serum and urine phosphate level was determined using a phosphorus assay kit (Sekisui Diagnostic, cat#:117-30). For urine phosphate measurements, urine was collected in the morning for 3 consecutive days, early in the morning. Mice were handled gently on the cage grid and the tail was gently pull up to allow urine excretion. Urine was collected on a clean piece of parafilm, transferred in a tube and immediately frozen at -80°C. Urine phosphate level was normalized to creatinine measured using a creatinine assay (Quidel, cat#: 8009).
RNA Extraction
Liver, kidney and bone marrow RNA was extracted using the standard protocol previously described by Chomczynski et al. (29), while bone RNA was extracted using Trizol reagent (Thermo Fisher). cDNA was generated by RNA reverse transcription using M-MLV Reverse Transcriptase (Thermo Fisher). Quantitative real-time PCR (qPCR) was performed using specific primers (Appendix Table) and SYBR Green qPCR Master Mix (BiMake). Expression levels were normalized to Actb expression levels.
Von Kossa/Van Gieson Staining
Mouse skeletons were collected and fixed in 10% formalin for 24 hours and in 70% ethanol for at least an additional 24 hours. Lumbar vertebrae were collected and dehydrated by gradual increase of ethanol percentage. Vertebrae were then embedded in methyl methacrylate resin as described previously (30). Vertebrae were sectioned at 7-µm sections and stained with Von kossa/Van Gieson and images were analyzed using the Osteomeasure analysis system (Osteometrics, Atlanta, GA).
FGF23 Processing in Cell Culture
The cDNAs coding for human furin, PC7, and PACE4, and mouse PC5A and PC5B were cloned into the pIRES2-EGFP vector (31), while the cDNA of hFGF23 was cloned in pIRES2-EGFP-V5. CHO-K1 and furin-deficient CHO-FD11 cells were cultured at 37°C and 5% CO2 in Ham’s F-12 medium supplemented with 10% (v/v) FBS (32) and transfected with 3 µg of plasmid following the standard protocol for Lipofectamine 2000 (Thermo Fisher). For the PCs inhibition, CHO-K1 cells were transfected with cDNAs encoding for hFGF23-V5 and PCs in the ratio of 5:1 and 24 hours later pretreated for 5 hours with Dec-RVKR-CMK (50 μM; Tocris) or D6R (20 μM; Calbiochem) followed by 22 hours of treatment in serum free media. Cells supernatant was then collected in the presence of 1x complete protease inhibitor cocktail (Roche) and FGF23 processing was analyzed by SDS-PAGE (15% Tris-glycine) followed by western blot using V5 antibody (Thermo Fisher).
Statistics
Statistical analyses were performed using GraphPad Prism software version 7.03. Results are shown as the mean ± SEM. Unpaired, 2-tailed Student’s t test was used for single measurements or comparison between two groups, while 1-way or 2-way ANOVA followed by Bonferroni’s post-test were used for comparison of more than 2 groups. A P value of less than 0.05 was considered statistically significant. All experiments were performed at least on 3 independent animals. Before starting each in vivo experiment, the mice were randomized into experimental groups with similar average body weight.
Results
In Vivo Inactivation of Furin in Osteoblasts and Osteocytes Partially Impairs FGF23 Cleavage, but Decreases Phosphate Excretion
To investigate the role of furin in the regulation of FGF23 processing in vivo, we generated mice in which the Furin gene was specifically inactivated in osteoblasts and osteocytes (i.e., Furinosb-/-) by breeding Furinfl/fl mice with hOCN-Cre transgenic animals which express the Cre recombinase under the control of human osteocalcin promoter (26). We previously reported that the Furin gene was efficiently and specifically inactivated in osteoblasts and bones in these mice (22). Although the precursor of osteocalcin, another bone-derived hormone involved in the control of glucose homeostasis (33), is not processed in Furinosb-/- mice, the same animals do not develop glucose intolerance before 9 months of age (22). Thus, to avoid a potential confounding effect of hyperglycemia on FGF23 and phosphate metabolism, all the following experiments were performed on mice aged between 3 and 6 months. When fed a standard chow diet containing ~0.7% inorganic phosphate (Pi), 4-month-old Furinosb-/- female mice displayed a significant 25% rise in circulating intact FGF23 (Figure 1A). This rise was however more moderate than expected in view of in vitro-based studies (23) and did not significantly alter the expression of Slc34a1, Slc34a3, Cyp27b1 or Cyp24a1 (Figure 1B). Accordingly, Furinosb-/- mice maintained normal serum phosphate levels and bone histology did not reveal presence of osteomalacia (Figures 1C, D).
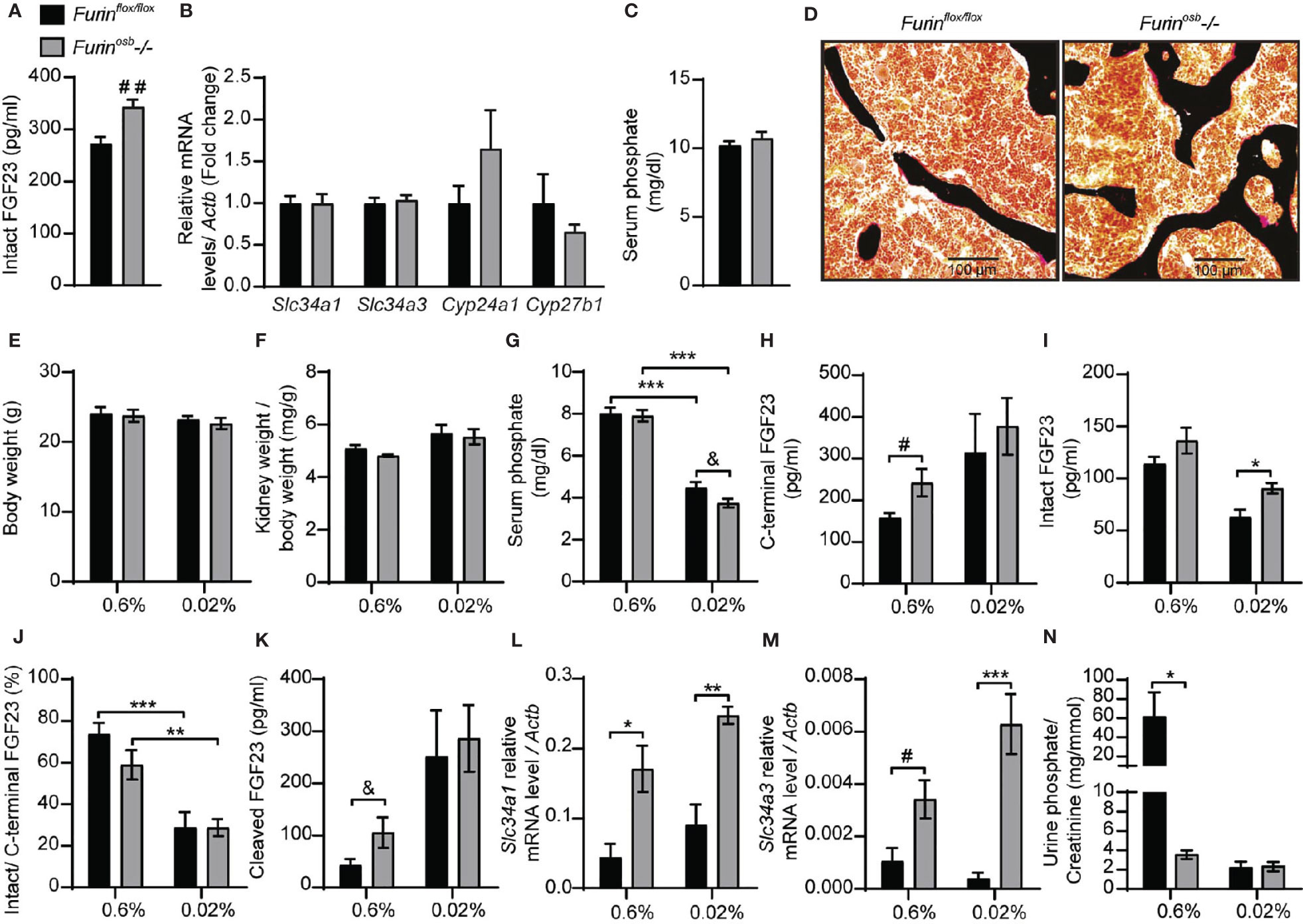
Figure 1 Increased FGF23 level and reduced phosphate excretion in Furinosb-/- mice. FGF23 and phosphate levels in in 4-month-old Furinflox/flox (n=6) and Furinosb-/- (n=6) mice fed on standard chow diet (see Material and Methods) (A–D). Intact FGF23 levels in plasma (A). FGF23 target genes expression in kidney (B). Serum phosphate level (C). Von Kossa/van Gieson staining on vertebrae sections (D). FGF23 and phosphate parameters in 6-months-old Furinflox/flox (n=6) and Furinosb-/- (n=6-7) mice fed a normal phosphate diet (0.6%; see Material and Methods) or a low phosphate diet (0.02%; see Material and Methods) for one week (E–N). Body weight (E). Kidney weight normalize to body weight (F). Serum phosphate levels (G). Plasma level of C-terminal FGF23 (H), intact FGF23 (I), percentage (%) of intact over C-terminal FGF23 (J) and cleaved FGF23 levels (K) calculated as (C-terminal FGF23) - (intact FGF23). Expression level in kidney of the sodium phosphate cotransporter Slc34a1 (L) and Slc34a3 (M). (N) Urine phosphate measurements normalized to creatinine. Results represent the mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001, by 2-way ANOVA with Bonferroni’s multiple comparisons test or by Student’s t-test (A–C, G, H, K, M) #: P<0.05; ##: P<0.01; &: 0.05<P<0.1.
Serum phosphate level is a potent regulator of FGF23 and in mice fed a low-phosphate synthetic diet the circulating intact FGF23 levels are reduced, while those of the processed fraction are increased, presumably due to the action of one or several PCs (34–36). We therefore next assessed if severe hypophosphatemia would be induced in Furinosb-/- mice by reducing their phosphate intake. We found that feeding control and Furinosb-/- mice a low-phosphate diet (0.02% Pi) for one week did not affect body weight or kidney weight (Figures 1E, F), but significantly decreased serum phosphate level (Figure 1G). Under the same conditions, Furinosb-/- mice tend to have a lower serum phosphate level compared to control mice, although this did not reach statistical significance. Furinosb-/- mice on normal diet had >25% increase in intact and C-terminal (total) FGF23, while maintaining normal intact/C-terminal FGF23 ratio compared to control littermates (Figures 1H–J). Under low phosphate diet circulating intact FGF23 was reduced in control mice but remains significantly higher in Furinosb-/- mice (Figure 1I). However, the ratio of intact over C-terminal FGF23 was decreased in both control and Furinosb-/- mice under low phosphate diet (Figure 1J). Moreover, the amount of circulating cleaved FGF23 (i.e., C-terminal FGF23 minus intact FGF23) was increased on low phosphate diet regardless of the genotype, suggesting efficient cleavage of FGF23 even in absence of furin (Figure 1K). These results suggest that in conditions of normal or low phosphate intake, processing of FGF23 still occurs in absence of furin in osteoblasts and osteocytes.
Surprisingly, when fed synthetic normal or low phosphate diets, Furinosb-/- mice displayed an increased expression in the kidney of the sodium phosphate transporter genes Slc34a1 and Slc34a3 compared to control mice, regardless of the level of phosphate in the diet (Figures 1L, M). In addition, Furinosb-/- mice fed the normal phosphate diet showed a decrease in urinary phosphate compared to control littermate (Figure 1N). Challenging the mice for one week on the low phosphate diet reduced urinary phosphate in control mice as expected, but not in Furinosb-/- mice, as these mice maintained the low urinary phosphate observed on normal phosphate diet (Figure 1N). Together these results show that unlike what was observed in vitro, in both physiological conditions or during decreased phosphate intake, inactivation of Furin in the osteoblast lineage has only a modest impact on FGF23 processing. They also suggest that furin may influence phosphate excretion through FGF23-independent mechanisms.
Furin Contributes to FGF23 Processing During Iron Deficiency
It was previously reported that mice carrying an ADHR mutation (R176Q, i.e., loss of P4 Arg in the motif R176HTR179↓SA) in the Fgf23 gene did not develop hypophosphatemia unless they were fed a low-iron diet (14). In view of these observations, we induced iron-deficiency in control and Furinosb-/- mice by feeding them a low-iron diet in combination with repeated tail bleedings (i.e., ̴70 µl every other week) for 14 weeks, before assessing the impact on FGF23 and phosphate metabolism. Control and Furinosb-/- mice fed this diet grew normally (Figure 2A), but both developed iron deficiency as reflected by the decrease in hepcidin (Hamp) expression and the increase in transferrin receptor (Tfrc) expression in liver (Figures 2B, C). As previously shown (14), iron deficient control and Furinosb-/- mice displayed an increase in Fgf23 gene expression in bone (Figure 2D). However, iron-deficiency resulted in increased in circulating C-terminal FGF23 only in control mice, although both genotypes showed an increase in circulating intact FGF23 (Figures 2E, F). These results indicate that FGF23 processing appears to be severely impaired in Furinosb-/- mice under conditions of iron deficiency as these mice maintained in the circulation a 1:1 ratio (i.e., 100%) of intact over C-terminal FGF23 and very low level of cleaved FGF23 (Figures 2G, H). Despite this near complete inhibition in FGF23 processing, iron deficient Furinosb-/- mice displayed a paradoxical increase in serum phosphate level compared to the control littermates fed the same diet (Figure 2I). Altogether these results suggest that furin may be the main PC responsible of FGF23 processing under iron deficiency.
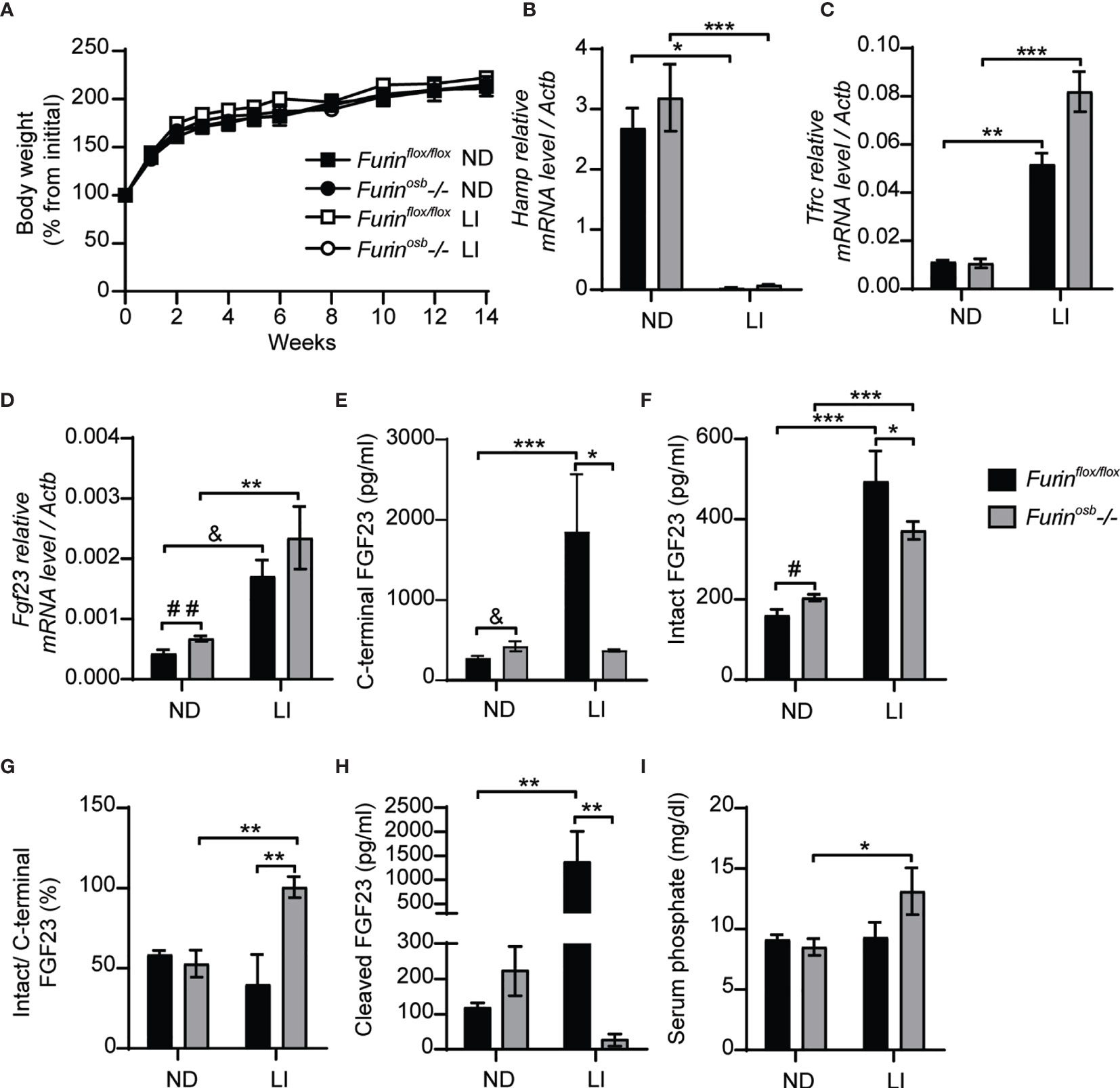
Figure 2 Impaired FGF23 processing in Furinosb-/- mice following iron restriction. Furinflox/flox (n=3-5) and Furinosb-/- (n=7-8) 21-day-old mice were fed normal chow diet (ND) or a low iron diet (LI) for 14 weeks. (A) Body weight growth curve presented as percentage of initial body weight. (B, C) Expression of iron-regulated genes in liver. Hepcidin (Hamp) expression (B). Transferrin receptor (Tfrc) expression (C). (D) Fgf23 expression in long bone. (E–H) Plasma level of C-terminal FGF23 (E), intact FGF23 (F), percentage (%) of intact over C-terminal FGF23 (G) and cleaved FGF23 levels (H), calculated as (C-terminal FGF23) - (intact FGF23). (I) Serum phosphate levels. Results represent the mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001, by 2-way ANOVA with Bonferroni’s multiple comparisons test or by Student’s t-test (D–F) #: P<0.05; ##: P<0.01; &: 0.05<P<0.1.
Furin Does Not Process FGF23 in Osteoblasts and Osteocytes Following rhEPO and IL-1β Injections
Erythropoietin and interleukin 1 beta (IL-1β), which are increased in condition of anemia and inflammation respectively, are two potent inducers of Fgf23 at the transcriptional level (15–18, 37, 38). In wild type mice, the majority of circulating FGF23 (i.e., >80%) is cleaved following injection of either recombinant human erythropoietin (rhEPO) or IL-1β (15, 16, 18), suggesting the action of one or several PCs. Hence, we tested if furin contributes to FGF23 processing after a single injection of rhEPO or IL-1β. We first confirmed that two relatively low doses of rhEPO (i.e., 300U/kg and 3000U/kg) were sufficient to induce in bones and bone marrow of wild type C57BL/6J mice the expression of two known EPO target genes, erythropoietin receptor (Epor) and erythroferrone (Erfe) (Figures 3A–D). These two doses also robustly induced Fgf23 expression in both tissues (Figures 3E, F). We next assessed the effect of the same dose of rhEPO in control and Furinosb-/- mice on the various circulating forms of FGF23. Circulating levels of total (i.e., C-terminal) FGF23 were increased dose-dependently following rhEPO injections, while circulating intact FGF23 levels were only modestly changed in both control and Furinosb-/- mice (Figures 3G, H). The ratio of intact over C-terminal FGF23 level was robustly reduced by the same treatment (Figure 3I) and cleaved FGF23 was increased more than 10-fold by rhEPO in both control and Furinosb-/- mice (Figure 3J), suggesting that following rhEPO, FGF23 is normally processed even in absence of furin in osteoblasts and osteocytes.
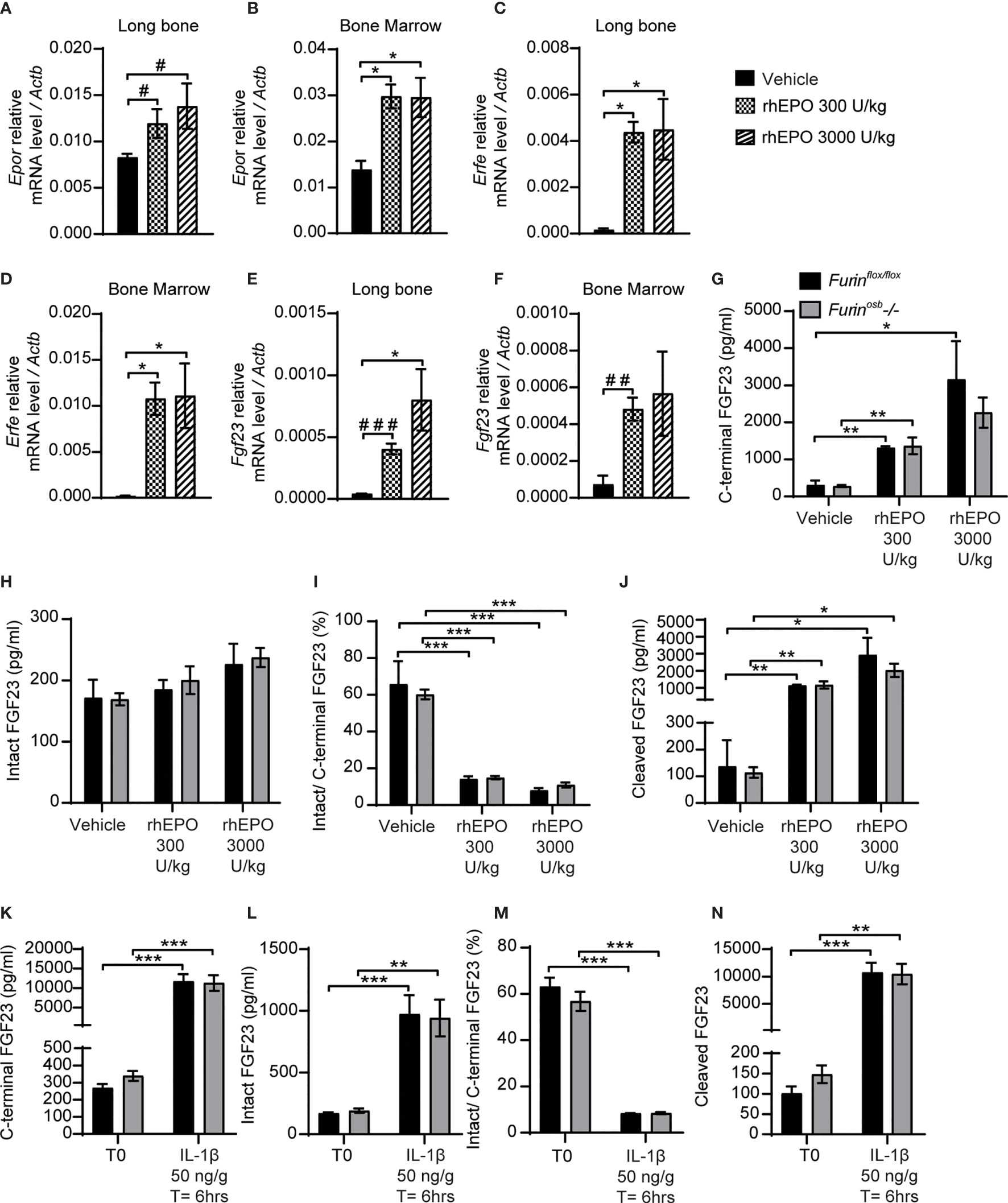
Figure 3 FGF23 is normally processed following erythropoietin and IL-1β injection in Furinosb-/- mice. Genes expression in long bone (A, B, E) and bone marrow (C, D, F) of 2-month-old C57BL/6J mice 6 hours following the injection of vehicle (n=4), 300 U/kg (n=4) or 3000 U/kg (n=4) of recombinant human erythropoietin (rhEPO). Erythropoietin receptor (Epor) (A, C), erythroferrone (Erfe) (B, D) and FGF23 (Fgf23) (C, F). (G–J) Plasma FGF23 measurements in 4-month-old Furinflox/flox (n=3) and Furinosb-/- (n=3) mice 6 hours following the injection of vehicle, 300 U/kg or 3000 U/kg of rhEPO. C-terminal FGF23 (G), intact FGF23 (H), percentage (%) of intact over C-terminal FGF23 (I) and cleaved FGF23 levels (J), calculated as (C-terminal FGF23) - (intact FGF23). (K–N) Plasma FGF23 measurements in 4-month-old Furinflox/flox (n=4) and Furinosb-/- (n=4) mice before (T0) and 6 hours following the injection of 50 ng/g IL-1β (T=6hrs). C-terminal FGF23 (K), intact FGF23 (L), percentage (%) of intact over C-terminal FGF23 (M) and cleaved FGF23 levels (N). Results represent the mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001, by 1-way ANOVA (A–F) or 2-way ANOVA (G–N) with Bonferroni’s multiple comparisons test, or by Student’s t-test (A, E, F) #: P<0.05; ##: P<0.01, ###: P<0.001.
In separate experiments, control and Furinosb-/- mice were administered a single dose of IL-1β (50ng/g), that induces Fgf23 expression in bone and raises circulating total and cleaved FGF23 levels (15). Six hours after the injection of IL-1β, the circulating level of total (C-terminal) FGF23 increased over 30-fold, while intact FGF23 increased around 10-fold, in both control and Furinosb-/- mice (Figures 3K, L). Following the IL-1β treatment the ratio of intact over C-terminal FGF23 level decreased from 60% to less than 10% in both control and mutant mice (Figure 3M) and the plasma concentration of cleaved FGF23 increased more than 10 times (Figure 3N). Together, these data show that FGF23 processing occurs normally following rhEPO or IL-1β injections in the absence of furin in osteoblasts and osteocytes.
Normal FGF23 Processing in Mice Lacking Furin in Osteoblasts, Osteocytes, and Hematopoietic Cells Following rhEPO Injections
The data presented above suggest that FGF23 was normally processed following rhEPO or IL-1β injection in Furinosb-/- mice. Osteoblasts and osteocytes are the main source of circulating FGF23 in normal conditions (38, 39), but bone marrow hematopoietic cells contribute to approximately 40% of circulating FGF23 following rhEPO treatment (38). Therefore, in Furinosb-/- mice, bone marrow hematopoietic cells could still be a significant source of cleaved FGF23 following rhEPO injections. To test this possibility, we generated mice lacking furin in all bone marrow hematopoietic cells (FurinBM-/-) by breeding Furinflox/flox mice with Vav1-Cre. Vav1-Cre transgenic animals expresses the Cre recombinase under the control of the Vav1 promoter, allowing the efficient deletion of furin in fetal and adult hematopoietic stem cells (HSC) (27) and hence in all hematopoietic cells. To address any potential redundancy between osteocytes and hematopoietic cells in the cleavage of FGF23, we also generated mice lacking Furin in both cell types, i.e., Furinflox/flox;hOCN-Cre;Vav1-Cre mice (Furinosb;BM-/-). Furin deficiency in T cells impairs the function of regulatory and effector T cells, causing at around 6 month of age severe inflammatory bowel disease, weight loss, ruffled hair and hunched appearance (40). Confirming efficient inactivation of Furin in hematopoietic cells, these defects were also observed in our FurinBM-/- mice between 4 and 6 months of age (data not shown). Hence, in the following experiments all mice were 8 weeks of age, an age when FurinBM-/- mice are still healthy.
A single injection of rhEPO (3000U/kg) increased circulating total (C-terminal) FGF23 by 20-30 fold and intact FGF23 by about 2 fold in Furinflox/flox, Furinosb-/-, FurinBM-/- and Furinosb;BM-/- mice (Figures 4A, B). The same treatment also reduced the ratio of intact over C-terminal FGF23 from 40% to <10%, while plasma cleaved FGF23 level increased to the same extent (>10 fold) in mice of all genotypes (Figures 4C, D). Together these results indicate that in vivo, FGF23 is still processed in absence of furin in osteoblasts/osteocytes and bone marrow hematopoietic cells in physiological condition and following rhEPO injections.
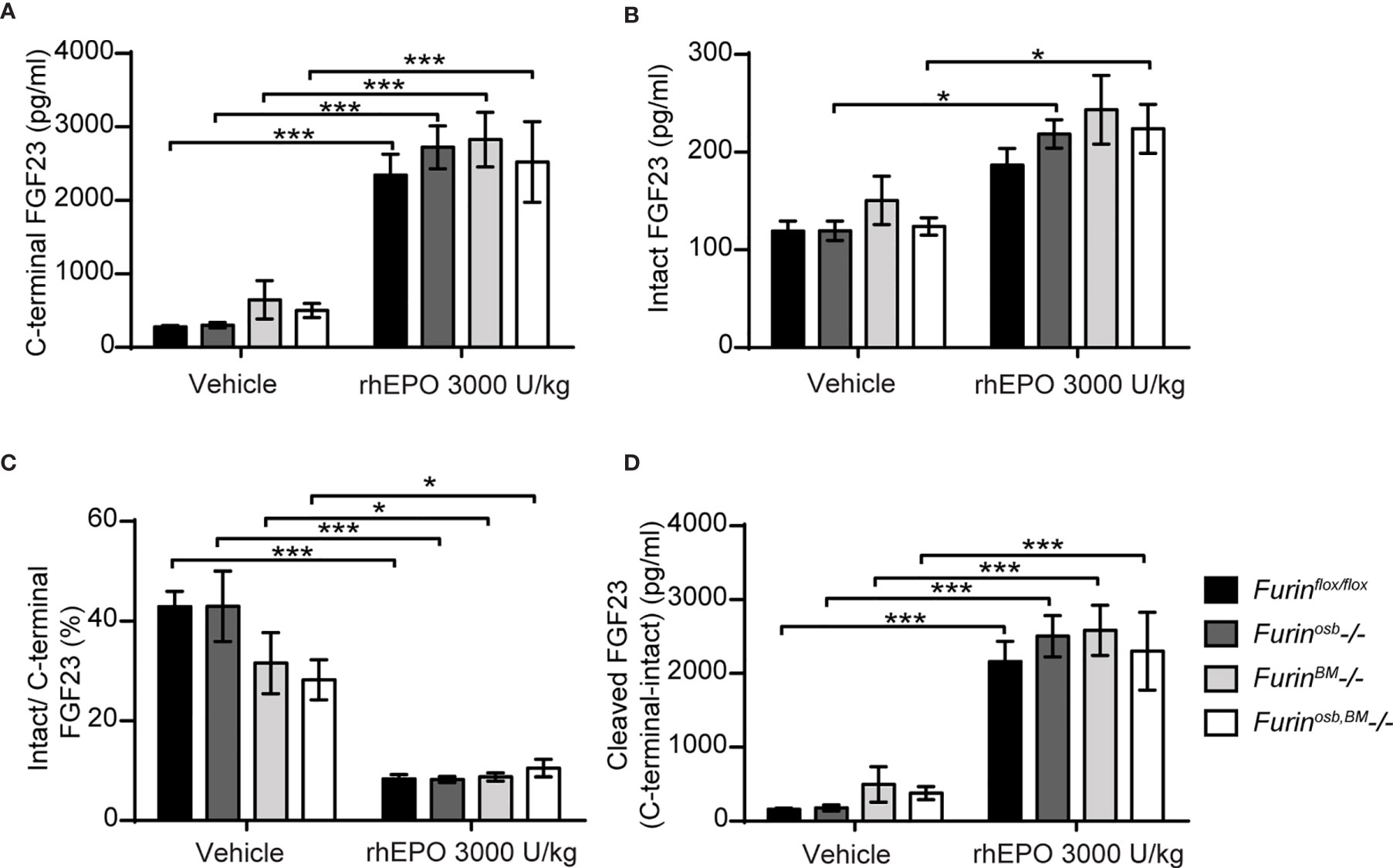
Figure 4 Furin inactivation in osteoblasts, osteocytes and hematopoietic cells did not impair FGF23 processing following erythropoietin injection. Plasma FGF23 measurements in 8-week-old Furinflox/flox (n=7), Furinosb-/- (n=5), Furinosb-/- (n=5) and Furinosb;BM-/- (n=6) male and female mice 6 hours following injection of vehicle or 3000 U/kg of recombinant human erythropoietin (rhEPO): C-terminal FGF23 (A), intact FGF23 (B), percentage (%) of intact over C-terminal FGF23 (C) and cleaved FGF23 levels (D), calculated as (C-terminal FGF23) - (intact FGF23). Results represent the mean ± SEM. *P < 0.05 and ***P < 0.001, by 2-way ANOVA with Bonferroni’s multiple comparisons test.
Several PCs Can Process FGF23 in Cell Culture
Our data suggests that in some conditions (e.g., low phosphate diet, rhEPO injection or IL-1β injection), more than one PC may cleave FGF23 in vivo. In order to identify additional PC(s) that might process FGF23 in vivo, we tested FGF23 processing in cell lines expressing or not furin. In Chinese hamster ovary (CHO-K1) cells, which endogenously express furin (41), ectopically transfected human FGF23 is secreted and partially processed (Figure 5A). In the same cells, cleavage of FGF23 was inhibited by decanoyl-RVKR-CMK (RVKR) a pan-PC cell-permeable inhibitor, but not by hexa-D-arginine (D6R), a cell-surface PC inhibitor (Figure 5A). These observations suggest that cleavage of FGF23 occurs before it is secreted, in an intracellular secretory compartment, likely the trans-Golgi network (41) where furin, but also PC5A, PACE4 and PC7 are present. In addition, FGF23 was not cleaved in CHO-FD11 cells, which do not endogenously express furin (41), and co-transfection with a furin expressing vector was sufficient to restore efficient cleavage (Figure 5B, first two lanes). We next tested the capacity of other PCs, i.e., PC5A, PC5B, PACE4 and PC7 (19), to process FGF23 in absence of furin in the same cells. Among all these PCs, PC5A was the most efficient in processing FGF23 although PC5B, PACE4 and PC7 were also able to process FGF23 albeit less efficiently than PC5A (Figure 5B). Altogether, these results indicate that in addition to furin, several other PCs have the capacity to process FGF23 in vitro.
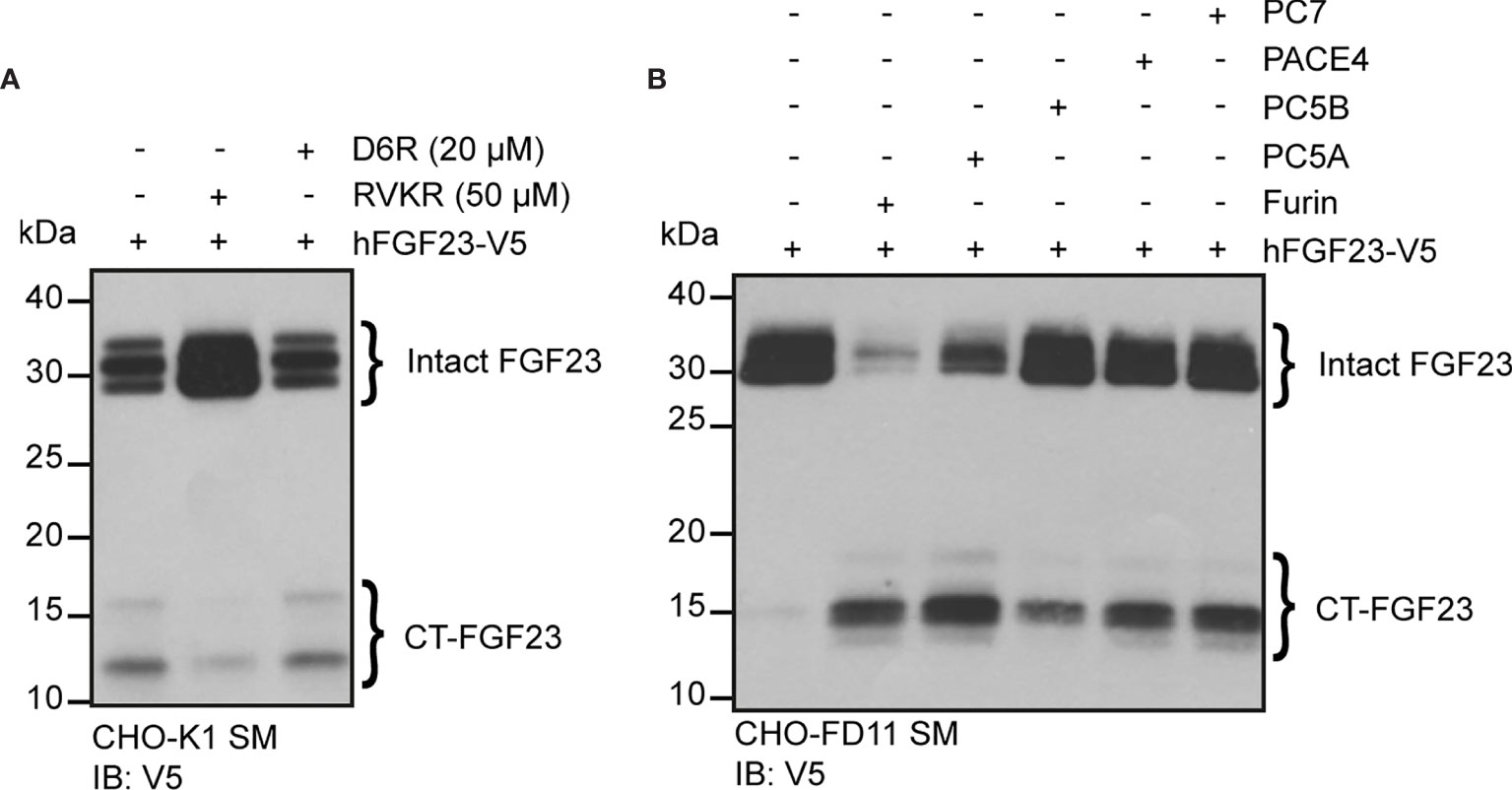
Figure 5 Multiple proprotein convertases can process FGF23 in mammalian cells. (A) Western blot analysis of the secretion media (SM) from CHO-K1 cells transfected with V5 tagged human FGF23 (hFGF23-V5) and cultured in the absence (–) or presence (+) of PC inhibitors, decanoyl-RVKR-CMK (RVKR) (50 μM) or hexa-D-arginine (D6R) (20 μM). (B) Western blot analysis of the secretion media (SM) from CHO furin-deficient FD11 cells co-transfected with V5 tagged human FGF23 (hFGF23-V5) and proprotein convertase Furin, PC5A, PC5B, PACE4 or PC7. Intact and processed C-terminal (CT) FGF23 forms are detected with V5 antibody.
Genetic Ablation of PC5 and Furin in Osteoblasts and Osteocytes Does Not Impair FGF23 Processing
Since PC5A and PC5B, which are both encoded by Pcsk5 (19), can cleave FGF23 in cell culture, and PC5A is expressed in differentiated osteoblasts and in osteocytes (21, 22), we generated mice in which Pcsk5 was inactivated specifically in osteoblasts and osteocytes by breeding Pcsk5fl/fl mice (24) with hOCN-Cre transgenic animals. When fed a standard chow diet, control (Pcsk5fl/fl) and Pcsk5osb-/- mice displayed similar circulating level of intact FGF23 and serum phosphate (Figures 6A, B). Von Kossa/van Gieson staining on non-decalcified bone sections did not reveal any sign of osteomalacia in these mice (Figure 6C). These analyses suggest that PC5 in the osteoblast lineage is dispensable for the regulation of FGF23 and phosphate levels in vivo.
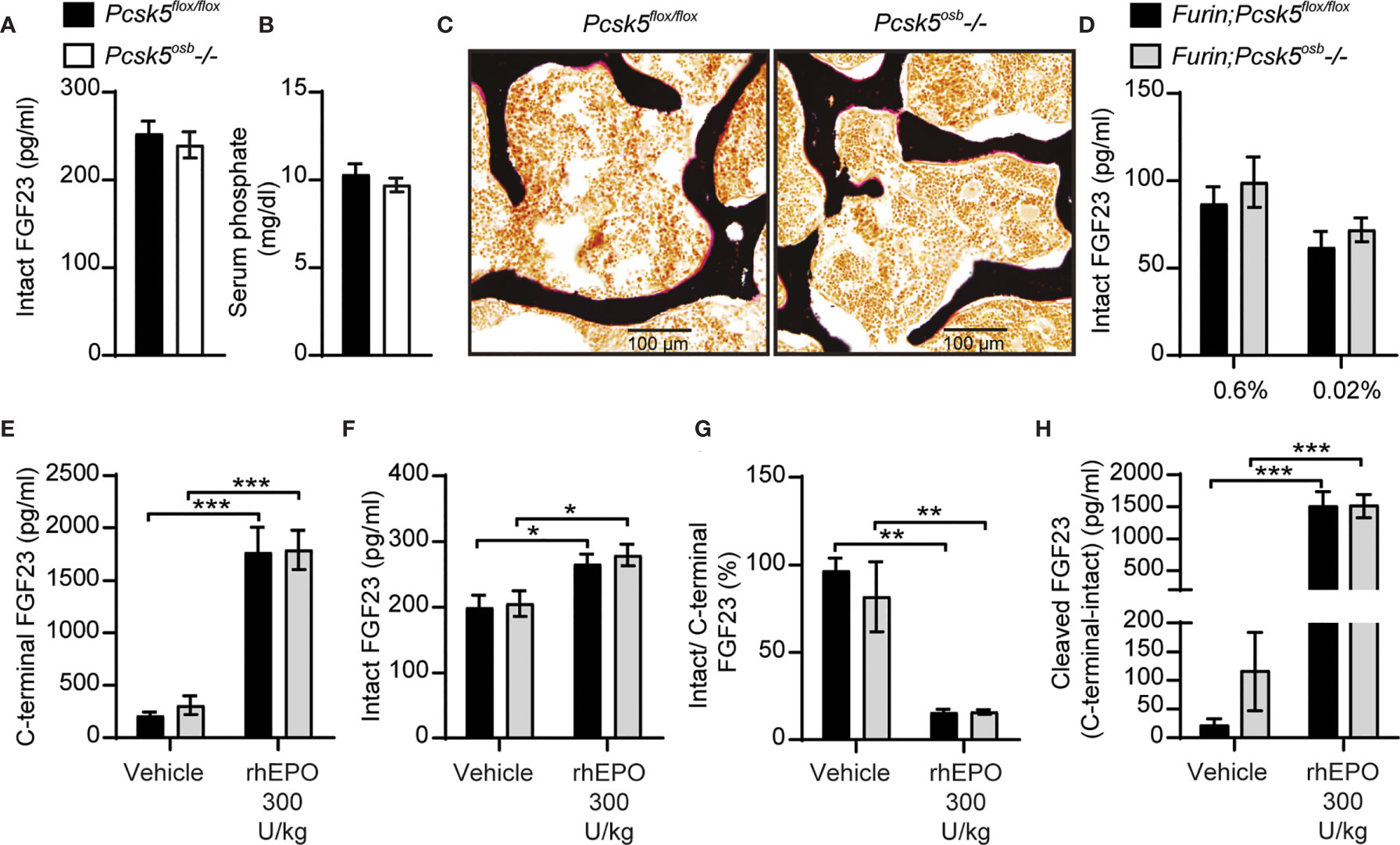
Figure 6 Genetic ablation of PC5 alone or in combination with Furin does not impair FGF23 processing in vivo. Plasma intact FGF23 (A) and serum phosphate levels (B) in 4-month-old Pcsk5flox/flox (n=6) and Pcsk5osb-/- (n=7) mice fed a normal chow diet. (C) Von Kossa/van Gieson staining of vertebrae sections of Pcsk5flox/flox and Pcsk5osb-/-. (D) Plasma intact FGF23 in 6-month-old Furin;Pcsk5flox/flox (n=7-8) and Furin;Pcsk5osb-/- (n=8) fed a normal phosphate diet (0.6%) or a low phosphate diet (0.02%) for one week. (E, H) Plasma FGF23 measurements in 4-month-old Furin;Pcsk5flox/flox (n=4) and Furin;Pcsk5osb-/- (n=4) 6 hours following an injection of vehicle and 300 U/kg of recombinant human erythropoietin (rhEPO): C-terminal FGF23 (E), intact FGF23 (F), percentage (%) of intact over C-terminal FGF23 (G) and cleaved FGF23 levels (H), calculated as (C-terminal FGF23) - (intact FGF23). Results represent the mean ± SEM. *P < 0.01, **P < 0.01 and ***P < 0.001, by 2-way ANOVA with Bonferroni’s multiple comparisons test, or by Student’s t-test (A, B).
To test if PC5 and furin redundantly cleave FGF23 in vivo, we next generated mice lacking both furin and PC5 specifically in osteoblasts and osteocytes (i.e., Furinflox/flox; Pcsk5flox/flox; hOCN-Cre or Furin;Pcsk5osb-/- mice). Similar to the single inactivation of furin in the same cells, osteoblast and osteocytes-specific inactivation of furin and PC5 resulted in a modest increase in circulating intact FGF23 levels whether mice were fed normal or low phosphate diet (Figure 6D). Likewise, injection of rhEPO (300U/kg) increased circulating total (C-terminal) FGF23 by almost 10-fold and intact FGF23 by 25% in both control and Furin;Pcsk5osb-/- mice (Figures 6E, F). In these conditions, plasma intact over C-terminal ratio decreased by approximately 80%, while plasma cleaved FGF23 increased more than 10-fold, regardless of the mice genotype (Figures 6G, H). Altogether, these results show that FGF23 is still efficiently processed in vivo even when both furin and PC5 were inactivated in osteoblasts and osteocytes.
Whole Body PACE4 Deletion Does Not Impair FGF23 Processing Under Physiological Conditions or Following IL-1β Injections
PACE4 is strongly expressed in mouse osteoblasts (22) and can cleave FGF23 in vitro (Figure 5B). We therefore next asked whether PACE4 may be involved in processing FGF23 in vivo. When fed a standard chow diet, Pcsk6-/- mice showed circulating levels of total (C-terminal) and intact FGF23 that were comparable to those of wild type littermates. Following a single IL-1β (50ng/g) injection, Pcsk6-/- mice displayed a significant decrease in circulating total (C-terminal) FGF23 while intact FGF23 was not significantly changed (Figures 7A, B). The ratio of intact over C-terminal FGF23 was equally decreased after IL-1β treatment in wild type and Pcsk6-/- mice plasma (Figure 7C). Circulating processed FGF23 was robustly increased by the same treatment regardless of the genotype, although PACE4 deficient mice did displayed a significant 30% reduction compared to wild type mice (Figure 7D). Altogether, these results show that PACE4 may partially contribute to FGF23 expression or secretion in vivo in response to IL-1β. Yet, FGF23 is still processed in absence of PACE4, suggesting the redundant action of other PCs.
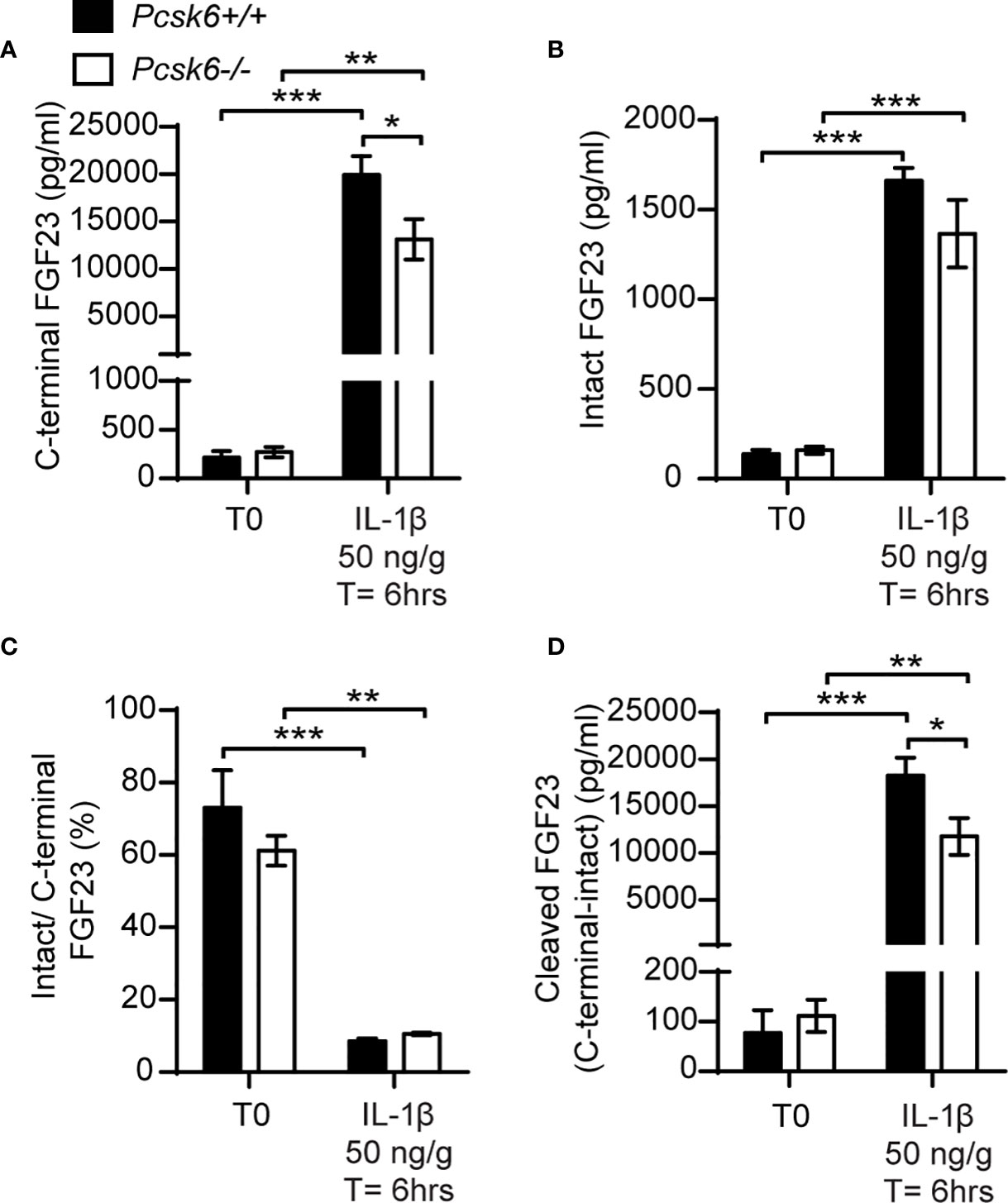
Figure 7 PACE4 inactivation in mice does not impair FGF23 processing. Plasma FGF23 measurements in 4-month-old Pcsk6 +/+ (n=4) and Pcsk6 -/- (n=4) male mice before (T0) and 6 hours following the injection of 50 ng/g of IL-1β: C-terminal FGF23 (A), intact FGF23 (B), percentage (%) of intact over C-terminal FGF23 (C), and cleaved FGF23 levels (D), calculated as (C-terminal FGF23) - (intact FGF23). Results represent the mean ± SEM. *P < 0.01, **P < 0.01 and ***P < 0.001, by 2-way ANOVA with Bonferroni’s multiple comparisons test.
Discussion
Over 20 years ago, mutations in a putative proprotein convertase (PC) target sequence within human FGF23 were found to cause ADHR in humans. Yet, despite the critical importance of this question for our understanding of phosphate metabolism, the identity of the specific PC(s) involved in FGF23 cleavage in vivo has remained elusive. We provide evidence here suggesting that furin, but not PC5, partially regulates FGF23 processing in vivo under normal conditions although this processing is by no means complete. The requirement of furin for FGF23 processing appears to be context or cell-type dependent, since FGF23 is still properly cleaved following rhEPO or IL-1β injection when furin is inactivated alone or in combination with PC5. Our results therefore suggest that additional yet to be characterized peptidases cleave FGF23 in vivo.
In mice fed standard chow diet, the modest increase in circulating intact FGF23 observed in Furinosb-/- mice did not cause hypophosphatemia and osteomalacia. This result is consistent with the observation that ADHR patients show variable age of onset and penetrance of the disease (42). Since iron deficiency positively correlates with intact FGF23 circulating levels in ADHR patient and results in hypophosphatemia and osteomalacia in mice carrying an AHDR mutation (13, 14), we induced iron deficiency in control and furin deficient mice. Even under these conditions, furin deficient mice did not develop hypophosphatemia and in fact displayed higher serum phosphate level despite a near complete impairment of FGF23 cleavage. These observations suggest that although furin may be important for the cleavage of FGF23 in osteocytes in the context of iron deficiency, it may also be involved in the regulation of phosphate metabolism through FGF23-independent mechanism(s).
The results obtained in the context of iron deficiency raised the question as to whether, under normal conditions, FGF23 may also be cleaved by another PC that would be downregulated in osteocytes following iron deficiency. To clarify whether FGF23 cleavage by furin is context specific, we injected controls and furin-deficient mice with rhEPO or IL-1β. These two circulating factors are produced in response to anemia and inflammation respectively (15, 43), and induce cleaved FGF23 secretion in bone and bone marrow cells in rodents (15–18, 37, 38). Herein, despite a strong increase in circulating total (C-terminal) FGF23 level following rhEPO or IL-1β injection, Furinosb-/- mice did not show a concurrent increase in intact FGF23 suggesting that either cleavage of FGF23 occurs by one or several other PC(s) in osteocytes, or that cleaved FGF23 originates mainly from bone marrow hematopoietic cells in this context. To address this second possibility, we generated mice deficient in furin in osteoblast-derived cells and/or in all hematopoietic cells. Our data shows that in these two cellular compartments, furin is dispensable for FGF23 cleavage following the injection of rhEPO further supporting the notion that furin is not the only peptidase involved in FGF23 processing in vivo.
PC5 (Pcsk5), PACE4 (Pcsk6) and PC7 (Pcsk7) are expressed in differentiated osteoblasts (22) and these PCs also target the minimal consensus cleavage site (i.e., RXXR) recognized by furin (19), suggesting that these enzymes may also cleave FGF23. Despite that, inactivation of Pcsk5 specifically in osteoblasts and osteocytes did not impact intact FGF23 level and serum phosphate concentrations. In addition, mice deficient in both furin and PC5 in the osteoblast lineage are comparable to control mice in terms of FGF23 circulating level under physiological conditions and following rhEPO injection, suggesting that PC5 does not compensate for furin absence in vivo. Finally, even though PACE4 is highly expressed in differentiated osteoblasts (22) and cleaves FGF23 in mammalian cells, Pcsk6 inactivation in mice only reduced FGF23 production following IL-1β injection and did not impact its cleavage.
Interestingly, in human hepatocyte and myeloid cell lines iron depletion increases furin expression and concomitantly decreases PC7 expression (44). If a similar regulation of furin and PC7 by iron also exists in osteocytes and/or hematopoietic cells, it could potentially explain why FGF23 cleavage is totally dependent on furin in the context of iron deficiency, but not in normal physiological conditions or following injections of IL-1β or rhEPO. Since no conditional allele of Pcsk7 is currently available in mice, a potential redundant action of furin and PC7 in FGF23 cleavage in bone cells in vivo was not investigated in the current study. Another limitation of our study is that we did not measured the impact of the inactivation of furin, PC5 or PACE4 on circulating levels of PTH, calcium and calcitriol, which are also regulated by FGF23.
Altogether, these findings provide genetic evidence that furin in osteoblasts and osteocytes is only partially responsible for FGF23 processing in vivo. Our data also showed that FGF23 cleavage is differentially regulated depending on the physiological context. Under conditions of iron deficiency furin is responsible for this process, while following single injection of rhEPO or IL-1β, additional proteases may be involved in FGF23 processing. Alternately, the increase in cleaved FGF23 following rhEPO and IL-1β injection could originate from non-osteoblastic cells such as bone marrow hematopoietic cells and involve multiple proteases including or not furin. It is also possible that cleavage of FGF23 occurs mostly extracellularly or in the circulation. Our study also excluded PC5 and PACE4 as major FGF23 processing enzymes in vivo. In conclusion, additional studies are required to determine precisely which enzyme(s) are involved in FGF23 cleavage in vivo and how furin regulates phosphate metabolism independently of FGF23.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by IRCM Animal Care Committee.
Author Contributions
MF, DS-R, and OAR conceived and planned the experiments. OAR and DS-R carried out the experiments. OAR and MF analyzed the data. RE, JC, and NS provided critical mouse lines. OAR and MF wrote the manuscript with input from all authors. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by funding from the Canada Research Chair program (MF) and the Natural Sciences and Engineering Research Council of Canada (RGPIN-2016-05213, to MF) and a CIHR Foundation grant (# 148363 to NS). OAR received scholarships from IRCM and FRQS.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. R. Day for providing Pcsk6-/- mice, Dr. T. Clemens for sharing the hOC-Cre mice and Dr. T. Möröy for the iVav-Cre strain.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2021.690681/full#supplementary-material
Supplementary Table | Primers used.
References
1. Meyer RA Jr., Meyer MH, Gray RW. Parabiosis Suggests a Humoral Factor Is Involved in X-Linked Hypophosphatemia in Mice. J Bone Miner Res (1989) 4(4):493–500. doi: 10.1002/jbmr.5650040407
2. Miyauchi A, Fukase M, Tsutsumi M, Fujita T. Hemangiopericytoma-Induced Osteomalacia: Tumor Transplantation in Nude Mice Causes Hypophosphatemia and Tumor Extracts Inhibit Renal 25-Hydroxyvitamin D 1-Hydroxylase Activity. J Clin Endocrinol Metab (1988) 67(1):46–53. doi: 10.1210/jcem-67-1-46
3. Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, et al. Klotho Converts Canonical FGF Receptor Into a Specific Receptor for FGF23. Nature (2006) 444(7120):770–4. doi: 10.1038/nature05315
4. Larsson T, Marsell R, Schipani E, Ohlsson C, Ljunggren O, Tenenhouse HS, et al. Transgenic Mice Expressing Fibroblast Growth Factor 23 Under the Control of the alpha1(I) Collagen Promoter Exhibit Growth Retardation, Osteomalacia, and Disturbed Phosphate Homeostasis. Endocrinology (2004) 145(7):3087–94. doi: 10.1210/en.2003-1768
5. Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, et al. Fgf-23 Is a Potent Regulator of Vitamin D Metabolism and Phosphate Homeostasis. J Bone Miner Res (2004) 19(3):429–35. doi: 10.1359/JBMR.0301264
6. Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, et al. Targeted Ablation of Fgf23 Demonstrates an Essential Physiological Role of FGF23 in Phosphate and Vitamin D Metabolism. J Clin Invest (2004) 113(4):561–8. doi: 10.1172/JCI200419081
7. Shimada T, Urakawa I, Yamazaki Y, Hasegawa H, Hino R, Yoneya T, et al. Fgf-23 Transgenic Mice Demonstrate Hypophosphatemic Rickets With Reduced Expression of Sodium Phosphate Cotransporter Type IIa. Biochem Biophys Res Commun (2004) 314(2):409–14. doi: 10.1016/j.bbrc.2003.12.102
8. Consortium A. Autosomal Dominant Hypophosphataemic Rickets Is Associated With Mutations in FGF23. Nat Genet (2000) 26(3):345–8. doi: 10.1038/81664
9. Nesbitt T, Coffman TM, Griffiths R, Drezner MK. Crosstransplantation of Kidneys in Normal and Hyp Mice. Evidence That the Hyp Mouse Phenotype Is Unrelated to an Intrinsic Renal Defect. J Clin Invest (1992) 89(5):1453–9. doi: 10.1172/JCI115735
10. Benet-Pages A, Orlik P, Strom TM, Lorenz-Depiereux B. An FGF23 Missense Mutation Causes Familial Tumoral Calcinosis With Hyperphosphatemia. Hum Mol Genet (2005) 14(3):385–90. doi: 10.1093/hmg/ddi034
11. Topaz O, Shurman DL, Bergman R, Indelman M, Ratajczak P, Mizrachi M, et al. Mutations in GALNT3, Encoding a Protein Involved in O-Linked Glycosylation, Cause Familial Tumoral Calcinosis. Nat Genet (2004) 36(6):579–81. doi: 10.1038/ng1358
12. Ichikawa S, Gray AK, Padgett LR, Allen MR, Clinkenbeard EL, Sarpa NM, et al. Genetic Rescue of Glycosylation-Deficient Fgf23 in the Galnt3 Knockout Mouse. Endocrinology (2014) 155(10):3891–8. doi: 10.1210/en.2014-1199
13. Imel EA, Peacock M, Gray AK, Padgett LR, Hui SL, Econs MJ. Iron Modifies Plasma FGF23 Differently in Autosomal Dominant Hypophosphatemic Rickets and Healthy Humans. J Clin Endocrinol Metab (2011) 96(11):3541–9. doi: 10.1210/jc.2011-1239
14. Farrow EG, Yu X, Summers LJ, Davis SI, Fleet JC, Allen MR, et al. Iron Deficiency Drives an Autosomal Dominant Hypophosphatemic Rickets (ADHR) Phenotype in Fibroblast Growth factor-23 (Fgf23) Knock-in Mice. Proc Natl Acad Sci USA (2011) 108(46):E1146–55. doi: 10.1073/pnas.1110905108
15. David V, Martin A, Isakova T, Spaulding C, Qi L, Ramirez V, et al. Inflammation and Functional Iron Deficiency Regulate Fibroblast Growth Factor 23 Production. Kidney Int (2016) 89(1):135–46. doi: 10.1038/ki.2015.290
16. Hanudel MR, Eisenga MF, Rappaport M, Chua K, Qiao B, Jung G, et al. Effects of Erythropoietin on Fibroblast Growth Factor 23 in Mice and Humans. Nephrol Dial Transplant (2018) 34(12):2057–65. doi: 10.1093/ndt/gfy189
17. Flamme I, Ellinghaus P, Urrego D, Kruger T. FGF23 Expression in Rodents Is Directly Induced Via Erythropoietin After Inhibition of Hypoxia Inducible Factor Proline Hydroxylase. PloS One (2017) 12(10):e0186979. doi: 10.1371/journal.pone.0186979
18. Daryadel A, Bettoni C, Haider T, Imenez Silva PH, Schnitzbauer U, Pastor-Arroyo EM, et al. Erythropoietin Stimulates Fibroblast Growth Factor 23 (FGF23) in Mice and Men. Pflugers Arch (2018) 470(10):1569–82. doi: 10.1007/s00424-018-2171-7
19. Seidah NG, Prat A. The Biology and Therapeutic Targeting of the Proprotein Convertases. Nat Rev Drug Discov (2012) 11(5):367–83. doi: 10.1038/nrd3699
20. Stijnen P, Ramos-Molina B, O’Rahilly S, Creemers JW. Pcsk1 Mutations and Human Endocrinopathies: From Obesity to Gastrointestinal Disorders. Endocr Rev (2016) 37(4):347–71. doi: 10.1210/er.2015-1117
21. Yamamoto H, Ramos-Molina B, Lick AN, Prideaux M, Albornoz V, Bonewald L, et al. Posttranslational Processing of FGF23 in Osteocytes During the Osteoblast to Osteocyte Transition. Bone (2016) 84:120–30. doi: 10.1016/j.bone.2015.12.055
22. Al Rifai O, Chow J, Lacombe J, Julien C, Faubert D, Susan-Resiga D, et al. Proprotein Convertase Furin Regulates Osteocalcin and Bone Endocrine Function. J Clin Invest (2017) 127(11):4104–17. doi: 10.1172/JCI93437
23. Tagliabracci VS, Engel JL, Wiley SE, Xiao J, Gonzalez DJ, Nidumanda Appaiah H, et al. Dynamic Regulation of FGF23 by Fam20C Phosphorylation, GalNAc-T3 Glycosylation, and Furin Proteolysis. Proc Natl Acad Sci USA (2014) 111(15):5520–5. doi: 10.1073/pnas.1402218111
24. Essalmani R, Zaid A, Marcinkiewicz J, Chamberland A, Pasquato A, Seidah NG, et al. In Vivo Functions of the Proprotein Convertase PC5/6 During Mouse Development: Gdf11 Is a Likely Substrate. Proc Natl Acad Sci USA (2008) 105(15):5750–5. doi: 10.1073/pnas.0709428105
25. Roebroek AJ, Taylor NA, Louagie E, Pauli I, Smeijers L, Snellinx A, et al. Limited Redundancy of the Proprotein Convertase Furin in Mouse Liver. J Biol Chem (2004) 279(51):53442–50. doi: 10.1074/jbc.M407152200
26. Zhang M, Xuan S, Bouxsein ML, von Stechow D, Akeno N, Faugere MC, et al. Osteoblast-Specific Knockout of the Insulin-Like Growth Factor (IGF) Receptor Gene Reveals an Essential Role of IGF Signaling in Bone Matrix Mineralization. J Biol Chem (2002) 277(46):44005–12. doi: 10.1074/jbc.M208265200
27. de Boer J, Williams A, Skavdis G, Harker N, Coles M, Tolaini M, et al. Transgenic Mice With Hematopoietic and Lymphoid Specific Expression of Cre. Eur J Immunol (2003) 33(2):314–25. doi: 10.1002/immu.200310005
28. Constam DB, Robertson EJ. SPC4/PACE4 Regulates a TGFbeta Signaling Network During Axis Formation. Genes Dev (2000) 14(9):1146–55. doi: 10.1101/gad.14.9.1146
29. Chomczynski P, Sacchi N. The Single-Step Method of RNA Isolation by Acid Guanidinium Thiocyanate-Phenol-Chloroform Extraction: Twenty-Something Years on. Nat Protoc (2006) 1(2):581–5. doi: 10.1038/nprot.2006.83
30. Chappard D, Palle S, Alexandre C, Vico L, Riffat G. Bone Embedding in Pure Methyl Methacrylate At Low Temperature Preserves Enzyme Activities. Acta Histochem (1987) 81(2):183–90. doi: 10.1016/S0065-1281(87)80012-0
31. Bergeron E, Vincent MJ, Wickham L, Hamelin J, Basak A, Nichol ST, et al. Implication of Proprotein Convertases in the Processing and Spread of Severe Acute Respiratory Syndrome Coronavirus. Biochem Biophys Res Commun (2005) 326(3):554–63. doi: 10.1016/j.bbrc.2004.11.063
32. Gordon VM, Rehemtulla A, Leppla SH. A Role for PACE4 in the Proteolytic Activation of Anthrax Toxin Protective Antigen. Infect Immun (1997) 65(8):3370–5. doi: 10.1128/IAI.65.8.3370-3375.1997
33. Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, Confavreux C, et al. Endocrine Regulation of Energy Metabolism by the Skeleton. Cell (2007) 130(3):456–69. doi: 10.1016/j.cell.2007.05.047
34. Bon N, Frangi G, Sourice S, Guicheux J, Beck-Cormier S, Beck L. Phosphate-Dependent FGF23 Secretion is Modulated by Pit2/Slc20a2. Mol Metab (2018) 11:197–204. doi: 10.1016/j.molmet.2018.02.007
35. Yu X, Sabbagh Y, Davis SI, Demay MB, White KE. Genetic Dissection of Phosphate- and Vitamin D-mediated Regulation of Circulating Fgf23 Concentrations. Bone (2005) 36(6):971–7. doi: 10.1016/j.bone.2005.03.002
36. Perwad F, Azam N, Zhang MY, Yamashita T, Tenenhouse HS, Portale AA. Dietary and Serum Phosphorus Regulate Fibroblast Growth Factor 23 Expression and 1,25-Dihydroxyvitamin D Metabolism in Mice. Endocrinology (2005) 146(12):5358–64. doi: 10.1210/en.2005-0777
37. Toro L, Barrientos V, Leon P, Rojas M, Gonzalez M, Gonzalez-Ibanez A, et al. Erythropoietin Induces Bone Marrow and Plasma Fibroblast Growth Factor 23 During Acute Kidney Injury. Kidney Int (2018) 93(5):1131–41. doi: 10.1016/j.kint.2017.11.018
38. Clinkenbeard EL, Hanudel MR, Stayrook KR, Appaiah HN, Farrow EG, Cass TA, et al. Erythropoietin Stimulates Murine and Human Fibroblast Growth Factor-23, Revealing Novel Roles for Bone and Bone Marrow. Haematologica (2017) 102(11):e427–30. doi: 10.3324/haematol.2017.167882
39. Clinkenbeard EL, Cass TA, Ni P, Hum JM, Bellido T, Allen MR, et al. Conditional Deletion of Murine Fgf23: Interruption of the Normal Skeletal Responses to Phosphate Challenge and Rescue of Genetic Hypophosphatemia. J Bone Miner Res (2016) 31(6):1247–57. doi: 10.1002/jbmr.2792
40. Pesu M, Watford WT, Wei L, Xu L, Fuss I, Strober W, et al. T-Cell-Expressed Proprotein Convertase Furin Is Essential for Maintenance of Peripheral Immune Tolerance. Nature (2008) 455(7210):246–50. doi: 10.1038/nature07210
41. Susan-Resiga D, Essalmani R, Hamelin J, Asselin MC, Benjannet S, Chamberland A, et al. Furin Is the Major Processing Enzyme of the Cardiac-Specific Growth Factor Bone Morphogenetic Protein 10. J Biol Chem (2011) 286(26):22785–94. doi: 10.1074/jbc.M111.233577
42. Imel EA, Hui SL, Econs MJ. FGF23 Concentrations Vary With Disease Status in Autosomal Dominant Hypophosphatemic Rickets. J Bone Miner Res (2007) 22(4):520–6. doi: 10.1359/jbmr.070107
43. Jelkmann W. Regulation of Erythropoietin Production. J Physiol (2011) 589(Pt 6):1251–8. doi: 10.1113/jphysiol.2010.195057
Keywords: FGF23 = fibroblast growth factor 23, phosphate, endoproteolysis, proprotein convertases, subtilisin–kexin family, furin, PC5, PACE4
Citation: Al Rifai O, Susan-Resiga D, Essalmani R, Creemers JWM, Seidah NG and Ferron M (2021) In Vivo Analysis of the Contribution of Proprotein Convertases to the Processing of FGF23. Front. Endocrinol. 12:690681. doi: 10.3389/fendo.2021.690681
Received: 03 April 2021; Accepted: 13 May 2021;
Published: 04 June 2021.
Edited by:
Giovanni Lombardi, Galeazzi Orthopedic Institute (IRCCS), ItalyReviewed by:
Nadia Rucci, University of L’Aquila, ItalyGiulia Battafarano, Bambino Gesù Children Hospital (IRCCS), Italy
Copyright © 2021 Al Rifai, Susan-Resiga, Essalmani, Creemers, Seidah and Ferron. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mathieu Ferron, bWF0aGlldS5mZXJyb25AaXJjbS5xYy5jYQ==