 Ryan A. Lafferty
Ryan A. Lafferty Finbarr P. M. O’Harte
Finbarr P. M. O’Harte Nigel Irwin
Nigel Irwin Victor A. Gault
Victor A. Gault Peter R. Flatt
Peter R. Flatt
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 18 May 2021
Sec. Gut Endocrinology
Volume 12 - 2021 | https://doi.org/10.3389/fendo.2021.689678
This article is part of the Research TopicProglucagon-Derived PeptidesView all 25 articles
Initially discovered as an impurity in insulin preparations, our understanding of the hyperglycaemic hormone glucagon has evolved markedly over subsequent decades. With description of the precursor proglucagon, we now appreciate that glucagon was just the first proglucagon-derived peptide (PGDP) to be characterised. Other bioactive members of the PGDP family include glucagon-like peptides -1 and -2 (GLP-1 and GLP-2), oxyntomodulin (OXM), glicentin and glicentin-related pancreatic peptide (GRPP), with these being produced via tissue-specific processing of proglucagon by the prohormone convertase (PC) enzymes, PC1/3 and PC2. PGDP peptides exert unique physiological effects that influence metabolism and energy regulation, which has witnessed several of them exploited in the form of long-acting, enzymatically resistant analogues for treatment of various pathologies. As such, intramuscular glucagon is well established in rescue of hypoglycaemia, while GLP-2 analogues are indicated in the management of short bowel syndrome. Furthermore, since approval of the first GLP-1 mimetic for the management of Type 2 diabetes mellitus (T2DM) in 2005, GLP-1 therapeutics have become a mainstay of T2DM management due to multifaceted and sustainable improvements in glycaemia, appetite control and weight loss. More recently, longer-acting PGDP therapeutics have been developed, while newfound benefits on cardioprotection, bone health, renal and liver function and cognition have been uncovered. In the present article, we discuss the physiology of PGDP peptides and their therapeutic applications, with a focus on successful design of analogues including dual and triple PGDP receptor agonists currently in clinical development.
While the gut hormones secretin and gastrin were discovered almost two decades earlier (1, 2), it was the extraction, isolation and purification of insulin from canine pancreatic extracts in Toronto in 1921, that truly signifies the advent of peptide-based therapeutics (3). Indeed, the first clinical use of animal-derived insulin began the following year. Continued innovation has led to the production of longer-acting formulations (4), as well as biosynthetic, recombinant DNA human insulins in the 1980’s (5). In this respect, it is incredible to think that a century later, insulin remains a vital mainstay in the management of Type 1 diabetes mellitus (T1DM).
Although insulin therapy is often indicated in poorly controlled Type 2 diabetes mellitus (T2DM), this condition is more often managed with diet plus an array of medications that augment remaining endogenous insulin production and function. Indeed, peptide-based therapeutics have become important tools in the management of T2DM, emulating the success of insulin in T1DM. In particular, enzymatically stable analogues, based on the endogenous incretin hormone glucagon-like peptide 1 (GLP-1), are now widely prescribed second- and third-line agents for T2DM (6). Furthermore, orally-available inhibitors of the enzyme dipeptidyl peptidase-4 (DPP-4), which degrades incretins including GLP-1, have been increasingly prescribed since their approval in 2007 (7).
As its name suggests, GLP-1 is related to the glucose-elevating hormone, glucagon. Indeed, a family of glucagon-related peptides exists, all of which are derived from differential processing of a common prohormone, proglucagon (8). Whilst glucagon and its hyperglycaemic actions were discovered in 1922 (9), its amino acid sequence was not elucidated until 1957 (10). Furthermore, proglucagon went undiscovered until the early 1980’s, when its cDNA was initially identified in anglerfish (11, 12), with discovery of a proglucagon equivalent in rat (13, 14), hamster (15), cow and human several years later (16). These discoveries were made possible with the advent of lab-scale cDNA cloning techniques, which made it feasible to accurately predict amino acid sequences of proteins by decoding the nucleotide sequences of cloned recombinant cDNA copies of mRNAs. Such experiments highlighted that glucagon and several peptides with a high degree of sequence homology were encoded by this prohormone (11, 12).
Interestingly, anglerfish islets were demonstrated to express two separate proglucagon peptides, meaning a hybrid approach was taken to identify cDNA encoding the 29 amino acid (aa), anglerfish glucagon (11, 12). From there, cDNA encoding for previously sequenced proteins, glicentin and oxyntomodulin was uncovered (17, 18), with glucagon located within the middle portion of this sequence (11). However, the proposed proglucagon sequence exhibited unexpected C-terminal elongation, containing an additional 34-residue glucagon-related carboxyl-terminal peptide, which exhibited structural similarity with another previously sequenced hormone, glucose-dependent insulinotropic polypeptide (GIP) (11, 19). Further study of anglerfish proglucagon led to the characterisation of a second proglucagon cDNA, derived from a different mRNA and gene which encoded glucagon. This shared significant homology with mammalian glucagons, but also a second C-terminal glucagon-related peptide, again comprised of 34 residues with significant sequence homology to glucagon (12).
Whilst work in anglerfish provided an excellent starting point, particularly in highlighting the presence of these carboxy glucagon-related peptides (11, 12), it was the elucidation of the structure of mammalian proglucagon which truly sparked interest in proglucagon-derived peptides (PGDP’s). While sequence homology with anglerfish proglucagon was high, isolation of the first mammalian proglucagon from hamster unveiled organisational differences, with the 158 amino-acid mammalian precursor containing three PGDP arranged in tandem, namely glucagon and what the authors termed, glucagon-like peptides 1 and 2 (GLP-1 and GLP-2) (15). The biological importance of these carboxy-peptides was initially unclear. Through a combined approach employing immunoassays, immunohistochemistry and chromatography of tissue extracts, it was established that GLP-1 and GLP-2 coexisted with glucagon in pancreatic islet cells and with oxyntomodulin in intestinal L-cells, where they are present at vastly greater concentrations than islets (20).
We now understand that proglucagon is expressed in both alpha-cells of the pancreatic islets (21, 22), as well as neuroendocrine L-cells (23), primarily located in the distal ileum and colon ( (24); Figure 1). However, the PGDP profile is not identical in the pancreas and gut, due to differential post-translational processing of proglucagon by tissue-specific enzymes termed prohormone convertases (PC) ( (25); Figure 1). Broadly speaking, it is accepted that pancreatic alpha-cells mainly possess PC2, which cleaves dibasic Lys-Arg sites within proglucagon to generate glicentin-related pancreatic peptide (GRPP), glucagon, intervening peptide-1 (IP-1) and major proglucagon fragment (MPGF) ( (26, 27); Figure 1). In contrast, in the L-cell, proglucagon is cleaved by PC1/3 at Arg-Arg sites to yield glicentin, GRPP, oxyntomodulin (OXM), GLP-1, intervening peptide-2 (IP-2) and GLP-2 ( (23, 26); Figure 1). It is important to note that these distinctions are not totally sacrosanct, with a degree of crossover existing. As such, recent evidence has highlighted that the gut is a possible extrapancreatic source of glucagon ( (28); Figure 2), while local intra-islet GLP-1 production has also been established in alpha cells, particularly in times of beta-cell stress (29). Moreover, it is now understood that proglucagon-containing neurons are located in the solitary nucleus of the medulla oblongata (30), which utilises PC1/3 in a similar fashion to the gut to generate PGDP’s in the central nervous system (CNS) ( (31); Figure 1). These PGDP’s and their therapeutic exploitation will be discussed in due course.
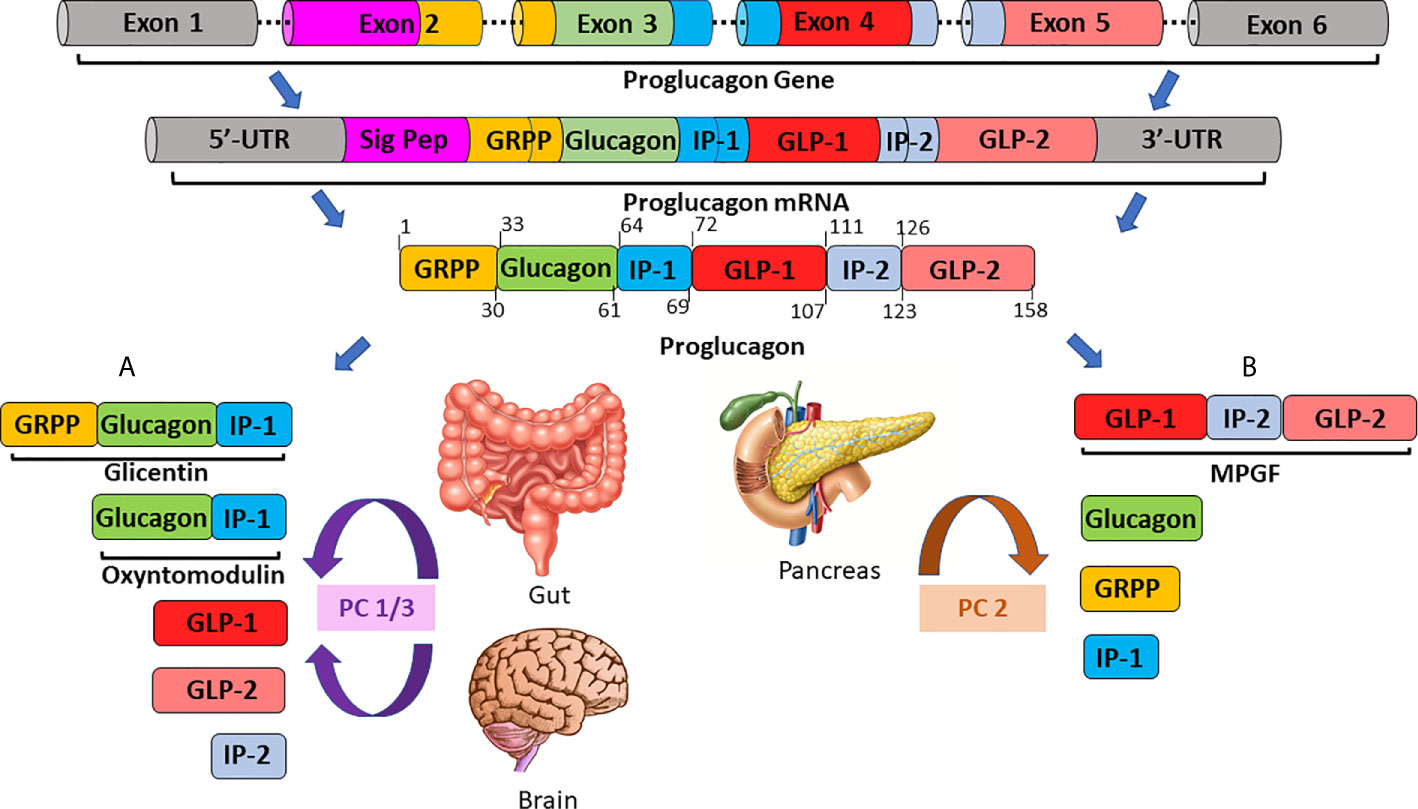
Figure 1 A schematic overview of tissue-specific proglucagon processing in the gut/brain (A) and in the pancreas (B). The proglucagon gene, located on chromosome 2 and comprised of 6 exons, is transcribed to generate proglucagon messenger RNA (mRNA). Proglucagon mRNA is subsequently translated to yield the 158 residue, precursor protein, proglucagon. In enteroendocrine L-cells of the ileum and colon (A) proglucagon is processed by prohormone convertase 1/3 (PC1/3) to generate glicentin, oxyntomodulin, glucagon-like peptides-1 and -2 (GLP-1, GLP-2) and intervening peptide-2 (IP-2). Conversely, in pancreatic alpha-cells (B), post-translational modification by prohormone convertase 2 (PC2) is responsible for the generation of the major proglucagon fragment (MPGF), glucagon, glicentin-related pancreatic polypeptide (GRPP) and intervening peptide-1 (IP-1).
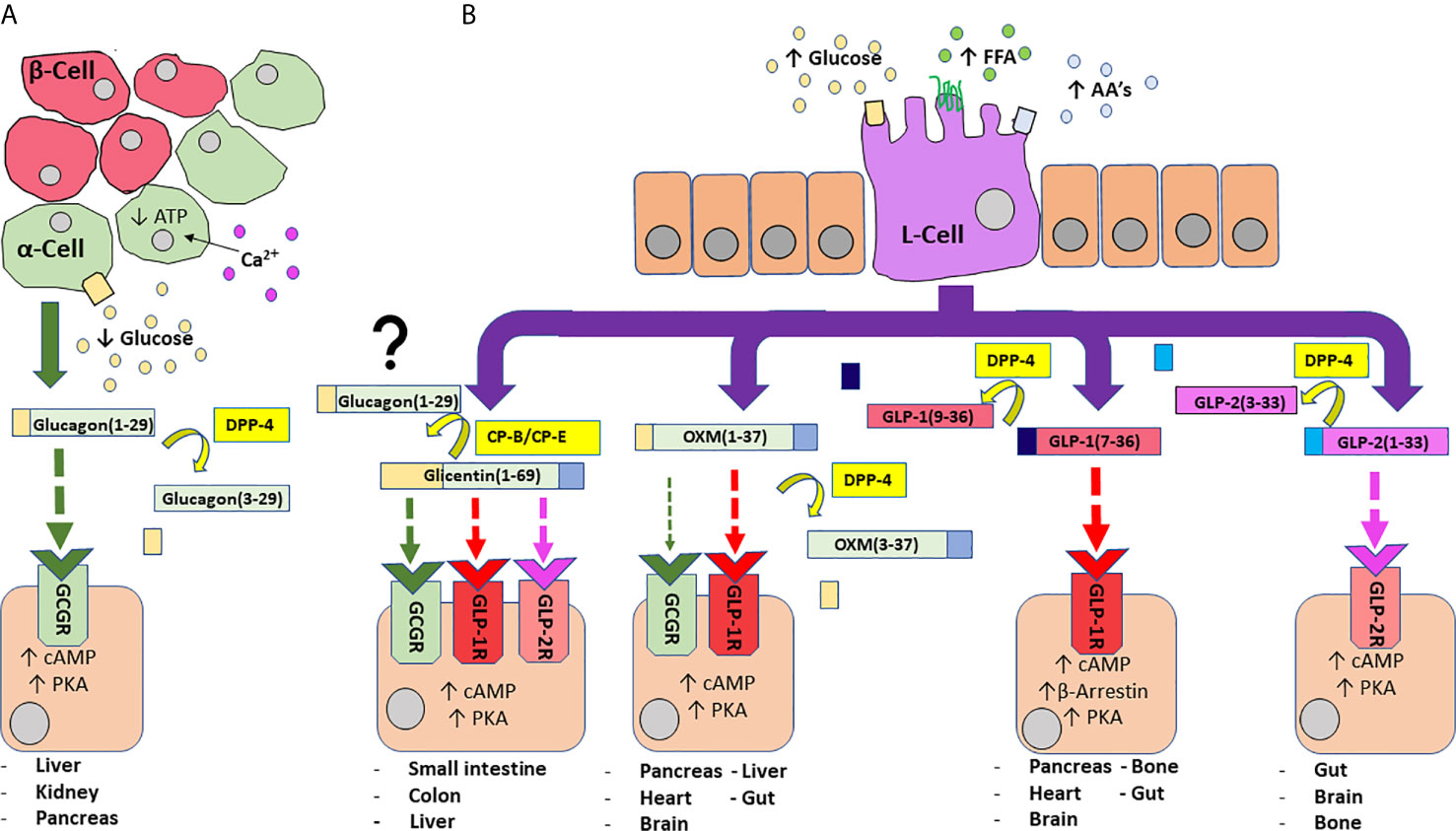
Figure 2 An overview of PGDP actions and secretion from pancreatic alpha-cells (A) and enteroendocrine L-cells (B)). A fall in circulating glucose concentration sees a reduction in intracellular adenosine triphosphate (ATP) levels and resultant closure of ATP-sensitive K+ channels to depolarise the plasma membrane and trigger the influx of Ca2+ ions, the primary stimulus for glucagon release (A). Glucagon is subject to N-terminal dipeptide removal by dipeptidyl-peptidase 4 (DPP-4). Glucagon(1-29) agonises glucagon receptors (GCGR) to evoke protein kinase A (PKA) activation and subsequent mobilisation of cyclic adenosine monophosphate (cAMP). Enteroendocrine L-cells of the distal gut are an open-type cell, rich in chemoreceptors and respond to digestion products of dietary carbohydrate, free fatty acids (FFA) and amino acids (AA’s) to release a number of PGDP’s into circulation (B). Glicentin(1-69) is an agonist for GCGR, GLP-1R and GLP-2R, although with less affinity than their primary hormonal ligands. Additionally, glicentin may serve as a precursor to glucagon in the gut, facilitated enzymatic degradation by enzymes such as carboxypeptidases-B and -E (CP-B, CP-E). Oxyntomodulin (OXM) is a dual agonist for GCGR and GLP-1R, but shows bias towards GLP-1R. It is cleaved by DPP-4 to yield inactive OXM(3-37). Bioactive glucagon-like peptide 1 (GLP-1(7-36)) agonises target GLP-1R to evoke PKA-mediated rises in cAMP, while activation of β-arrestin is also implicated in insulin secretion. DPP-4 cleaved GLP-1(9-36) is inactive. Glucagon-like peptide 2 (GLP-2) agonises target GLP-2R to evoke rises in PKA/cAMP. It is inactivated by DPP-4 to generate GLP-2(3-23). Enzymes are indicated by yellow boxes/arrows. Receptor interactions are indicated by dashed lines, with affinity indicated by increasing thickness of the arrow. Major tissues expressing receptors are also provided.
The 29 aa polypeptide hormone glucagon (Table 1) is the most widely recognised PGDP (9, 10), produced by PC2-mediated cleavage of proglucagon in pancreatic alpha cells ( (26, 27); Figure 1).

Table 1 Glucagon and related analogues in the management of hypoglycaemia in T1DM.
Discovered shortly after insulin (9), glucagon and insulin are intrinsically linked, with the major metabolic actions of glucagon counteracting those of insulin (35). As such, insulin secretion from pancreatic beta-cells is stimulated largely by elevated glucose concentrations, reducing circulating glucose levels via inhibition of glycogenolysis and gluconeogenesis, accompanied by stimulation of glycogen synthesis in the liver (36). Furthermore, insulin stimulates glucose uptake via GLUT-4 translocation in adipose and muscle (37), which in turn promotes efficient metabolism of protein, lipids and carbohydrate (favouring glycolysis) (38). Conversely, hypoglycaemia following fasting, or exercise is the most potent stimulus for glucagon secretion [(39, 40); Figure 2].
The hyperglycaemic action of glucagon is well-established, being demonstrated as early as its discovery, with the hormone’s name reflecting this; glucagon – “the glucose agonist” (9). Hyperglycaemic actions of glucagon are mediated through promotion of glycogenolysis and gluconeogenesis in liver, whilst also inhibiting glycolysis and glycogenesis (41). Furthermore, in times of limited carbohydrate availability, glucagon promotes non-carbohydrate energy formation in the generation of lipids and ketone bodies or through the breakdown of fatty acids to acetyl-coenzyme A (42). Further research into the actions of glucagon has demonstrated a role in satiety, with acute administration in humans diminishing hunger and reducing food intake (43), whilst also stimulating energy expenditure and cardiac contractility (44, 45).
There is some debate over the receptor interactions at play in some of these biological actions, for example: given that circulating glucagon concentrations rise following a period of fasting, its involvement in food reduction seems counter-intuitive, suggesting cross-reactivity with the GLP-1 receptor (GLP-1R) (42). In the context of this article, we will consider glucagon actions mediated through agonism of its own specific G protein-coupled receptor (GPCR) the glucagon receptor (GCGR). This receptor is widely expressed, particularly in the liver, but is also found in the adrenal glands, heart, adipose tissue, GIT, and pancreas (46, 47). Binding with the receptor activates adenylyl cyclase that leads to intracellular production of cyclic adenosine monophosphate (cAMP) and subsequent activation of protein kinase A (PKA). PKA stimulates the synthesis of transcription factors including cAMP response element-binding protein (CREB) in the nucleus, a promoter of gene expression. Simultaneously, GCGR activation of phospholipase C (PLC) and subsequent increase in inositol 1,4,5-triphosphate (IP3), facilitates release of calcium ions from the endoplasmic reticulum to influence CREB-regulated transcription co-activator (CRTC2), which enhances CREB-dependent gene expression (42). Importantly, glucagon is rapidly inactivated in the circulation by enzymes, including DPP-4, to generate inactive glucagon (3–29) (48); Figure 2).
While considered for many years as solely a consequence of insulin deficiency, in the 1970’s the “bihormonal hypothesis”, proposed by Roger Unger, highlighted the role of an imbalance in the complex interplay between glucagon and insulin in instigating diabetic hyperglycaemia (35). Indeed, the rationale behind this longstanding hypothesis inspired research into the development of dual pump systems, sometimes termed “dual-hormone artificial pancreas”. Such pumps are regulated by a glucose sensor to deliver insulin or glucagon, as necessary, from independent pumps and are thought to be possibly more efficacious than insulin-only pumps (49), although none have successfully reached the clinic to date. We now understand that T2DM is characterised by elevated fasting glucagon levels (50), while glucose suppression following a glucose challenge is stunted (51). Furthermore, it has been suggested that postprandial hyperglucagonaemia and impaired glucagon response to hypoglycaemia are features of T1DM (52).
Given glucagon’s ability to rapidly mobilise glucose from tissue stores, GCGR agonism has found valuable application in countering severe hypoglycaemia in T1DM patients, an adverse consequence of insulin therapy (53). Mild-to-moderate hypoglycaemia is defined as an event that can be self-treated, irrespective of symptom severity, or an asymptomatic blood glucose measurement of ≤3.9 mmol/L (54). It is usually managed via ingestion of rapidly absorbed carbohydrates, such as drinks or foods high in glucose, whereas severe hypoglycaemia requires immediate, emergency intervention (32). While intravenous (i.v.) infusion of dextrose is an option, it is now more common for patients or carers to possess an injectable glucagon preparation, which can be administered subcutaneously (s.c.) or intramuscularly (i.m.) (55). Such intervention is reliable and faster than the dextrose method, greatly reducing the risk of hypoglycaemic-induced coma and death. Rather than requiring a potentially lengthy wait for arrival of a qualified healthcare professional to perform an i.v. infusion, glucagon emergency kits simply involve reconstitution of glucagon powder, which can be injected into the patient’s leg or abdomen (32, 55). Moreover, a ready-to use autoinjector preparation termed “Zegalogue®” has recently gained FDA approval for management of hypoglycaemia (33), further improving ease of use. I.v. dextrose may then be required to prevent rebound hypoglycaemia (34), a potential consequence of the rapid in vivo inactivation of administered native glucagon (48).
Longer-acting, DPP-4 resistant analogues are in development that may address the issue of rebound hypoglycaemia. Two such analogues are the fatty-acid incorporating, NNC9204-0043 currently listed at Novo Nordisk ((34); Table 1), and dasiglucagon, which employs several amino acid substitutions to infer improved stability [(56); Table 1]. The former has only shown promise in in vitro settings (34), whereas dasiglucagon has very recently gained FDA approval in T1DM (56). Indeed, dasiglucagon is the active component of Zegalogue, and beyond application in prefilled injector pens, is currently in phase 3 trials as a subcutaneous infusion for treating congenital hyperinsulinaemia, and in phase 2 trials as part of a bihormonal artificial pancreas pump system alongside insulin (57). Glucagon emergency kits have been further improved with the development of intranasal (i.n.) glucagon. While not entirely novel, having been in development since the 1990’s (34), the first such product was only approved in 2019 (58). Termed Baqsimi®, the ready-to-use i.n. formulation has been proposed to lead to resolution of hypoglycaemia up to four times faster than injectable glucagon kits (59). The single-use preparation simply requires the user to administer one spray into either nostril, which is reported to deliver a 3 mg dose of glucagon (57).
The next PC1/3-mediated (Figure 1), L-cell-derived PGDP to be discussed has become a mainstay of T2DM management, representing one of the principal modern success stories of peptide therapeutic development. GLP-1 is a 29-residue (Table 2), gut-derived incretin hormone (77). GLP-1 is released post-prandially from L-cells [(77–79); Figure 2], with release influenced by the composition of each meal ingested; in particular, meals that are rich in fat and carbohydrate are known to be the primary physiological stimulus for GLP-1 secretion (78–81). Additionally, GLP-1 secretion can be triggered, not only by mixed nutrient load, but also via individual nutrients and bile acids. For example, oral administration of glucose alone has been shown to stimulate GLP-1 secretion in humans (82), as well as amino acids such as glutamine (83). Sodium-glucose transporter 1 (SGLT1) plays a glucose-sensing role on the L-cell surface, and although a contributor, is thought to play a lesser role than glucose transporters (GLUT) in relation to GLP-1 release (84). GLP-1 secretion is biphasic, with an early phase occurring 10-15 min after ingestion of nutrients and a second, more prolonged phase occurring 30-60 min after ingestion (81). Given the distal location of L-cells in the gut, it is unlikely that direct nutrient contact with these cells can be the sole mechanism initiating GLP-1 secretion. Thus, the autonomic nervous system, in particular the vagus nerve (which innervates a significant portion of the gut), is thought to play a role in this early phase of release, with nutrient content being more important for the second phase (85).
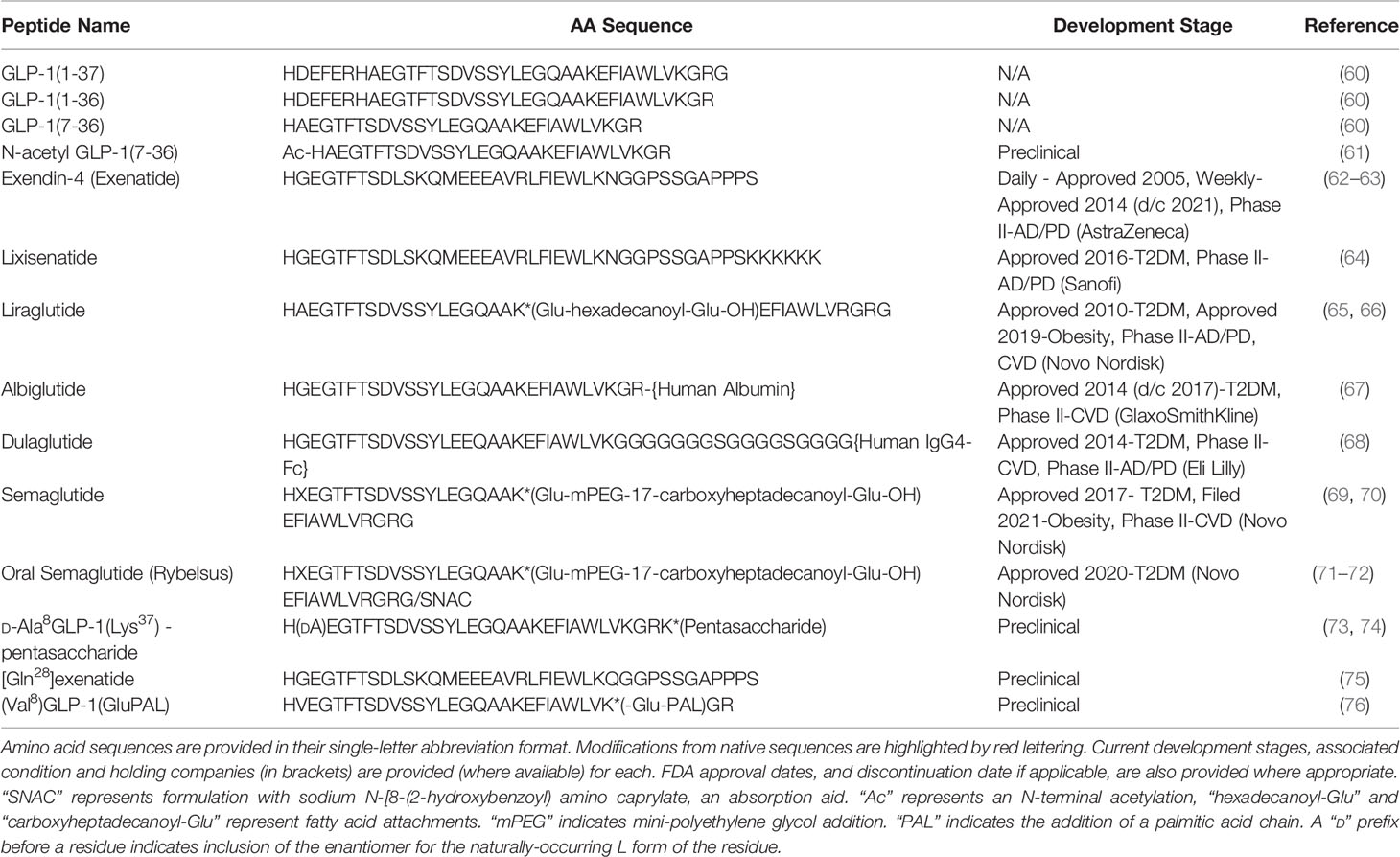
Table 2 GLP-1-based therapeutic peptides.
The biologically active forms of GLP-1 are GLP-1 (7–36)-amide and GLP-1 (7–37) which are equipotent in terms of their incretin effects [(60); Table 2 and Figure 2]. However, they do not circulate equally, with GLP-1 (7–36)-amide accounting for ~80% (20, 82). Both forms of circulating GLP-1 are subject to rapid N-terminal degradation by DPP-4 (86, 87), cleaving after Ala2 to generate GLP-1(9-36) or (9-37) metabolites (86, 87). While GLP-1(9-36) is considered a weak antagonist of beta-cell GLP-1R (88), there is evidence suggesting that this metabolite may reduce inflammation in cardiac tissue following myocardial infarction (89). GLP-1(9-36) has also been demonstrated to promote cardiac glucose uptake similar to GLP-1(7-36)-amide (90), so the descriptor “inactive” may not be entirely accurate. Additionally, a recent study suggests that GLP-1(9-36)-amide may indirectly influence glycaemia through antagonism of GCGR on alpha-cells to influence the glucagonostatic effects of GLP-1 (91). However, the implications of any GLP-1(9-36) effects on glycaemia are thought to be relatively inconsequential in comparison to GLP-1(7-36)-amide (92).
The GLP-1R is a family B, or secretin-like G-protein coupled receptor (GPCR) (93). A structurally identical GLP-1R has been identified in various tissues, for example: pancreatic tissue (alpha-, beta-, delta-cells), stomach, and intestine, as well as CNS regions including the hypothalamus and brainstem ( (81, 93); Figure 2). Binding of GLP-1 to its target-receptor on the beta-cell surface leads to activation of several intracellular transduction pathways (Figure 2). The hormone augments insulin secretion, mainly via stimulation of intracellular cAMP-mediated events and promotes glucose-induced biosynthesis of insulin, resulting in replenishment of insulin stores within beta-cells and reducing cell exhaustion (81, 94–96). Conversely, GLP-1 is known to suppress glucagon secretion from alpha-cells ( (97); Figure 3). The mechanisms behind this have been hotly debated, with it claimed to be an indirect effect mediated through increased insulin or somatostatin secretion (98, 99), while some have indicated the effect is more direct (100), especially given the presence, albeit at low expression (~10%), of GLP-1R on alpha-cells (101). Beyond this, activation of pancreas duodenum homeobox 1 (Pdx-1), a transcription factor essential for pancreatic development and beta-cell function (activated downstream from GLP-1R via cAMP activation), is thought to be a shared influence in these three processes (102). Prevention of beta-cell exhaustion may indirectly prevent cell death, but GLP-1 also directly influences proliferation by a number of proposed pathways including phosphatidylinositol 3-kinase (PI3-K) mediated rises in extracellular signal-related kinase (ERK) 1/2 and p38 mitogen-activated protein kinase (MAPK), as well as Pdx-1 (103). In keeping with this, exendin-4 has been shown to have no effect on proliferation or inhibition of apoptosis in beta-cell specific, Pdx-1 knockout (KO) mice (104).
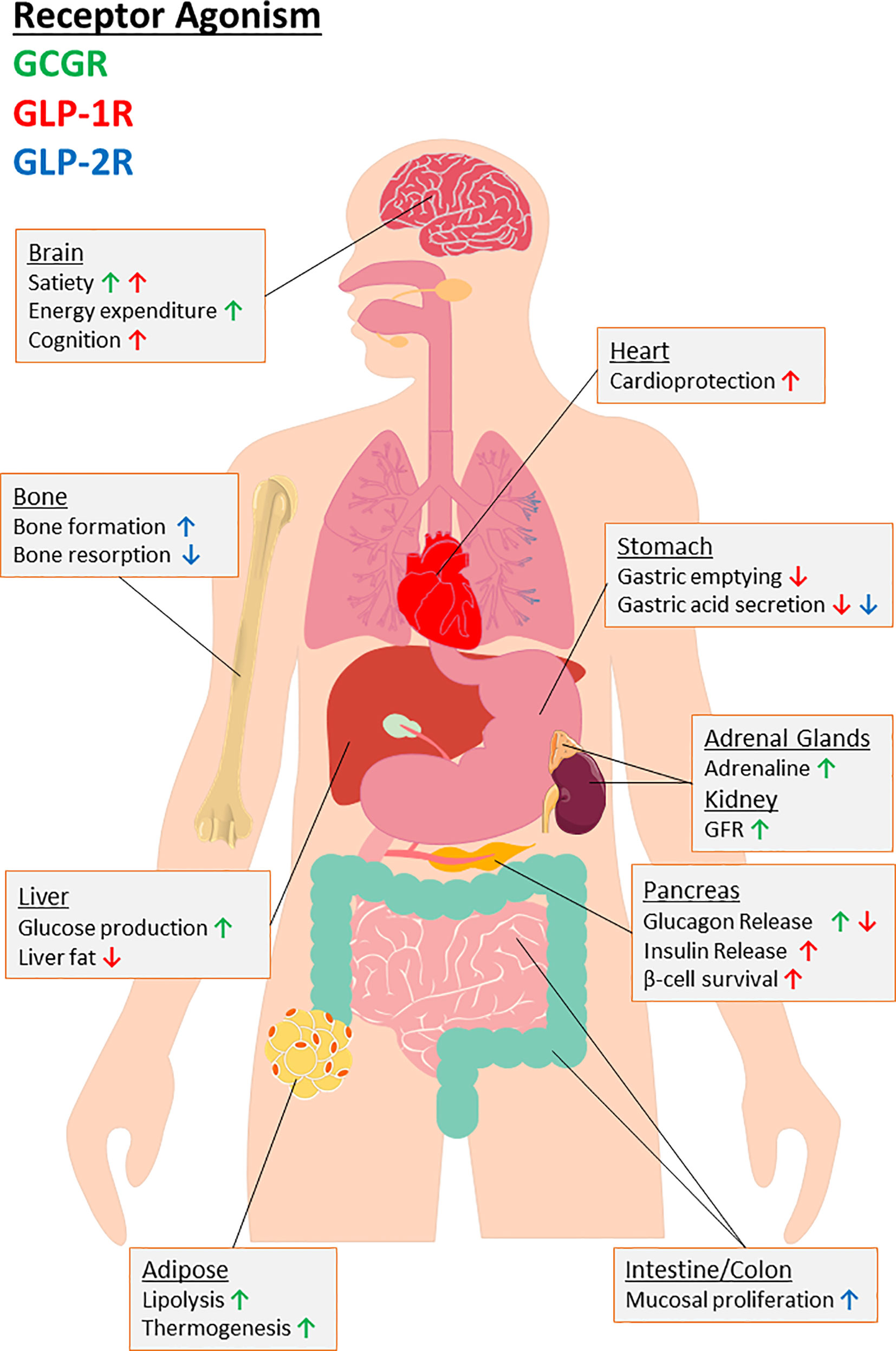
Figure 3 An overview of the biological consequences for agonism of target receptors of major PGDP’s, namely glucagon receptor (GCGR) and glucagon-like peptide-1 and -2 receptors (GLP-1R, GLP-2R). Organ-specific actions are provided with arrows indicating up or downregulation of specific effects to highlight the therapeutic potential for multiagonism in relation to PGDP’s. As indicated by the key, the colour of arrow indicates the receptor interactions responsible. “GFR” indicates glomerular filtration rate.
Since entry into the clinic, research on GLP-1 has continued, unveiling new mechanisms behind the various benefits of GLP-1R agonists, as well as possible new applications in other conditions. With regards to diabetes, it is now well established that chronic administration of GLP-1R mimetics not only enhances insulin secretion but also positively influences overall islet function, restoring normal morphology in even severe models of diabetes (105). Additionally, the ability of exogenous GLP-1R mimetics to maintain and promote beta-cell mass through reductions in apoptosis and increases in proliferation are well established (105–108). Culture of DPP-4 resistant, N-acetyl-GLP-1 (Table 2) with pancreatic ductal-cells has also been shown to induce expression of genes indicative of a transition to a beta-cell like phenotype (61, 109), but translation to humans requires further study. Advances in cell-lineage tracing technology have seen the development of transgenic animal models that employ fluorescently tagged alpha- or beta-cells to identify such islet cell transitioning events in the in vivo setting (110, 111). Recent studies have shown that administration of liraglutide to such mice with diabetes can prevent beta- to alpha-cell transdifferentiation (112), whilst also actively driving alpha- to beta-cell conversion to help restore beta-cell mass (113–115).
GLP-1 also inhibits glucagon secretion and exerts additional extra-pancreatic actions of therapeutic value including inhibition of gastric acid secretion and gastric emptying (Figure 3), which help reduce post-prandial spiking of blood glucose by slowing transit of nutrients from the stomach to the small intestine (81). In addition to locally produced GLP-1 (116), GLP-1 crosses the blood-brain barrier to agonise GLP-1R within hypothalamic CNS centres, where ingestive behaviour and satiety is dictated [(117); Figure 3]. Increased satiety reduces food intake, with resultant weight loss being an important benefit in overweight or obese-T2DM patients. Moreover, the widespread tissue presence of GLP-1R has witnessed new physiological roles for GLP-1 beyond glycaemia and satiety such as cardioprotection ( (118, 119); Figure 3), enhancing bone mass and strength in preclinical models of T2DM (120), and is thought to play an important role in enhancing cognition ( (121); Figure 3). Additionally, a possible role for GLP-1 in resolution of hepatic steatosis (122–124) through reduction in fatty acid accumulation by activation of both macroautophagy and chaperone-mediated autophagy (125), has attracted much interest.
GLP-1 was not the first incretin hormone to be discovered, with GIP being identified almost two decades previously in 1969 (126). However, with a proposed role for GIP in development of obesity coupled with a loss of insulinotropic effect in T2DM (127), therapeutic application did not follow such a straightforward path. Thus, when a preservation of the insulinotropic effects of GLP-1 in obesity was established (128), excitement surrounding the possible therapeutic application of this newly discovered incretin hormone began to grow. Furthermore, direct comparisons of analogues of these two incretins often resulted in more favourable outcomes for GLP-1 compared to GIP (129). Nonetheless, current evidence regarding GIP-based therapy looks more promising in T2DM once glycaemic control has been re-established (130). This is perhaps evident with new compounds being developed that operate through combined activation of GLP-1R and GIPR (130), as discussed in more detail below.
Initial therapeutic investigations into GLP-1 were promising, highlighting that delivery of exogenous, native peptide had the ability to improve overall glycaemia, insulin sensitivity, beta-cell function and reduce both appetite and food intake when administered by continuous s.c. infusion over a 6 week period in patients with diabetes (131). Moreover, tachyphylaxis was not reported and the side-effect profile was favourable (131). However, due to rapid inactivation by DPP-4 (132), continuous infusion was required, making it unsuitable for regular use in a “real world” setting.
With the discovery of exendin-4, an unexpected GLP-1R mimetic isolated from the saliva of the Gila monster lizard (Heloderma suspectum) (62), the tide began to turn. The first 30 residues of this 39 aa peptide demonstrated 53% sequence identity with human GLP-1 (Table 2), but despite such variance, the peptide was proven to be a potent agonist for mammalian GLP-1R (62), effectively bringing about GLP-1R-mediated benefits on glycaemia, body weight and appetite (133). Importantly, the substitution of Ala2 with Gly2 in exendin-4 conferred resistance to DPP-4, while further sequence variations rendered the peptide less susceptible to ectopeptidases like neprilysin (NEP) (134). Studies in anaesthetised pigs has shown that GLP-1 clearance involves multiple organs including hepatic, peripheral and renal extraction, whereas exendin-4 is subject solely to glomerular filtration, which also appears to be up to two-fold slower than native GLP-1 (134). This results in an in vivo action of ~5 hours (63), allowing for twice daily administration as opposed to continuous infusion. Synthetic exendin-4 reached full approval for therapeutic use in humans in 2005 (Byetta™), being prescribed under the generic trade name “exenatide” and has become a highly influential and widely prescribed second- and third-line agent in T2DM, generally following failure of metformin or metformin/sulphonylurea combination (135). Indeed, oral DPP-4 inhibitors, such as sitagliptin, were approved as second-line agents in 2007 (7), while a plethora of additional GLP-1 mimetics have since gained regulatory approval for diabetes in addition to exenatide, namely the longer-acting mimetics: liraglutide, semaglutide, albiglutide and dulaglutide (Table 2). In contrast, attempts to discover suitable bioactive small molecule agonists of GLP-1R have failed, despite considerable efforts, due to poor potency and allosteric alteration of receptor conformation (136, 137).
Beyond glucose homeostasis, exciting research has highlighted extra-pancreatic benefits and new applications for established GLP-1 mimetics, many of which are exciting prospects. For example, despite the enormous upsurge in the incidence of obesity and associated complications including T2DM (138), existing drug therapies for obesity are grossly insufficient, with bariatric surgery being far more effective (139). Against this background, in 2019 liraglutide became the first GLP-1 analogue approved by the FDA, EMA and MHRA as a treatment option for obesity (65). Importantly, while glycaemic improvements undoubtedly influence weight loss, pharmacokinetic investigation in human participants suggested the effects of liraglutide on weight loss are primarily mediated through increased energy expenditure (66). Prior to regulatory approval, the “SCALE”, phase III trials demonstrated a sustained 2-year weight loss with liraglutide treatment as an adjunct to diet and exercise in non-diabetic participants (140, 141), strengthening the argument that effects are largely independent of glycaemic modulation. Additionally, 3-year follow-up demonstrated that liraglutide delayed diabetes development in patients with pre-diabetes, taking almost 3 times longer in patients receiving liraglutide (142).
Given the successful application of liraglutide in this regard and the scale of the obesity problem, other GLP-1R mimetics are beginning to be touted as treatment options for obesity. Indeed, a phase III clinical programme assessing efficacy and safety of once-weekly semaglutide (SUSTAIN) in T2DM was completed recently for s.c. semaglutide, manifesting a substantial average weight loss of 14.9% (-15.3 kg) following 68 weeks treatment (69). Additionally, a direct comparison between liraglutide and semaglutide indicated superior weight loss was attained with the latter (143). FDA approval has now been sought for semaglutide use in obesity, meaning we may be on the verge of witnessing a new treatment option available for obesity that rivals bariatric surgery.
There is also increasing interest in the therapeutic potential of combining currently available GLP-1R mimetics (Table 2) with other currently prescribed antidiabetic drugs. The combination of exenatide with the sodium–glucose co-transporter 2 (SGLT2) inhibitor, dapagliflozin, was investigated in the DURATION-8, phase III clinical trial which demonstrated a degree of synergy between the two agents, with improvements in short- and long-term glycaemia and weight loss exceeding either agent alone (144). Moreover, a 2-year follow-up demonstrated long-term efficacy of this combination (145). An additional phase II trial, ENERGIZE, has sought to identify the mechanism behind the apparent synergy (146), the findings of which may influence whether such a combination is advanced further.
The growing strength of the cardiovascular and renal benefits of established GLP-1 mimetics add another string to their bow in the management of T2DM, with cardiovascular disease (CVD) being the number one cause of death in patients with T2DM (147). As demonstrated by long-term prospective cardiovascular outcomes trials (CVOTs), which have reported over the last four years, liraglutide (LEADER), semaglutide (SUSTAIN-6), albiglutide (HARMONY OUTCOMES) and dulaglutide (REWIND) have all shown significant reductions in composite cardiovascular outcomes [(64, 119, 148); Table 2], indicating they may be the agents of choice when macrovascular complication risk is high in T2DM patients. These longer-acting GLP-1R mimetics elicit more favourable cardiovascular outcomes than shorter-acting agents like exenatide or its analogue lixisenatide (EXSCEL and ELIXA), which demonstrated non-inferiority, but no obvious cardiovascular benefit [(64, 119, 148); Table 2]. Additionally, proposed renal benefits of SGLT2 inhibitors have seen trials such as “DECLARE-TIMI 58” report reduced rates of hospitalisation due to heart failure in dapagliflozin-treated groups of T2DM patients (148). Thus, given the exploration of exenatide and dapagliflozin in the DURATION-8 and ENERGIZE trials (144–147), it may stand to reason that such a combination may be studied in relation to CVD, perhaps with a more favourable GLP-1R mimetic than exenatide. Indeed, the phase III FLOW trial is currently recruiting patients to assess the renoprotective actions of semaglutide. Thus, we await the results of this trial to determine whether semaglutide may be the GLP-1R mimetic of choice in this regard (149).
Vascular deterioration in T2DM can also be linked to cognitive impairment, with growing evidence highlighting cross-sectional and prospective associations between T2DM and cognitive impairment and diminished memory and executive function (150). Clinical studies have concluded that T2DM is a significant risk factor that can double the likelihood of developing dementia (151). It appears that a loss of insulin sensitivity in the brain (152), coupled with impaired insulin function (153), results in impaired growth factor secondary messenger cascades that are vital for cell growth, repair and synaptic function (154). GLP-1 receptor mimetics such as exendin-4 or liraglutide can reverse insulin desensitisation in the brain (155, 156). Key biomarkers for cognitive impairment such as phosphorylation of protein kinase B (AKT) and glycogen synthase kinase-3beta (GSK-3B), were reduced by liraglutide administration in diabetic rats in a time-dependent manner (153). In more practical terms, exendin-4 administration in a diet-induced obese (DIO) model reversed impaired memory formation in mice (157) and liraglutide normalised object recognition memory impairment in a similar model (158). Similar findings have been observed with DPP-4 inhibitors (159), although it is important to note that other gut hormones, particularly GIP (157), are also implicated here. Additionally, similar to CVD (145, 146), it appears that the combination of GLP-1 mimetic with SGLT-2 inhibitor may too be beneficial with regards to cognition, with DIO/STZ-mice receiving liraglutide/SGLT-2 combination therapy presenting with improved recognition and hippocampal morphology (160).
Importantly, evidence suggests that the beneficial effects of GLP-1 in relation to cognition may be independent from glycaemic improvement, with a study comparing metformin and the GLP-1 analogue (Val8)GLP-1(GluPAL) demonstrating that only the latter reversed memory impairment in DIO mice (76). This hypothesis is supported by the finding that GLP-1R agonists have also shown neuroprotective effects in non-diabetic patients with Alzheimer’s (AD) or Parkinson’s disease (PD) (161, 162). Long-term potentiation (LTP) of synaptic activity, the cellular correlate of memory (163), is impaired in diabetes. Liraglutide administration reversed diabetes-related LTP blockade and actively promoted LTP formation in DIO mice (157, 158), while rescuing hippocampal LTP loss in an ob/ob murine model of obesity-diabetes (164).
While the close relation between GLP-1 and insulin signalling is undoubtedly important in cognition, it is crucial to highlight that beyond this mechanism, GLP-1R mimetics upregulated several neuroprotective growth factors such as: insulin-like growth factor 1 (IGF-1) (165), brain-derived neurotrophic factor (BDNF) (166), glia-derived neurotrophic factor (GDNF) (164), as well as vascular endothelial growth factor (VEGF) (157, 158).
Indeed, preclinical work in rodents has illuminated both the associations between cognitive decline in AD/PD and T2DM, whilst implicating the potential of GLP-1R activation in curbing such decline (167). As such, exenatide was employed in small-scale, proof of concept, human trials in PD patients, with these trials of <100 participants indicating exenatide treatment elicited improved scores in tests of cognitive function over the course of 12 months treatment (168, 169). Moreover, a further 12 months after study conclusion, those patients receiving exenatide still achieved significantly improved cognition scores than those receiving placebo (170). With such promising results, it is unsurprising that larger scale trials were conducted, such as the phase II, ELAD trial (171), which employed liraglutide in patients with moderate AD and associated dementia. Outcomes were disappointing, with it announced in late 2020 that no difference in cerebral glucose metabolic rate or improvement in daily activity was apparent between treatment or placebo (171), although some scores of cognitive function were improved by liraglutide. Despite such disappointment, interest in GLP-1R mimetics in relation to cognitive function has not been perturbed, with a number of phase II trials recruiting in 2020 to study currently available GLP-1R mimetics in AD and PD (172). Notably, a common theme of these trials is an adjustment of treatment demographic towards patients with relatively recently diagnosed AD/PD (172).
Increased bone fragility is a further complication associated with diabetes, with the aetiology suspected to be due to an increase in porosity of bone, impacting on bone quality (173). Bone fragility also appears to be a feature in both T1DM and T2DM (174–176). Like cardiovascular complications, effects on bone have the potential to limit physical activity in T2DM patients. Furthermore, a role for endogenous GLP-1 in the development of diabetes-associated bone fragility has been identified, with GLP-1R KO mice presenting with reduced bone mass through increased osteoclast activity (177, 178). Given the implication of GLP-1R involvement in the aetiology of bone fragility in diabetes, research has explored the possibility of GLP-1R agonist or DPP-4 inhibitor use in the management of the condition with favourable outcomes (175, 179). Exenatide has been shown to enhance bone strength by increasing trabecular bone mass, bone formation and trabecular microarchitecture, whilst also improving collagen maturity in rodent models of diabetes (180, 181). Similarly, liraglutide significantly prevented deterioration of the quality of the bone matrix in a streptozotocin-induced, rodent model of T1DM (175). Importantly, GLP-1 is not the only incretin involved in the pathogenesis of bone fragility in diabetes, with single GIP receptor (GIPR) KO and dual GLP-1R/GIPR KO mice presenting with enhanced bone fragility (182, 183). Indeed, the unimolecular GIPR/GLP-1R/GCGR agonist, [D-Ala2]GIP–Oxm (Table 4), significantly improved bone strength and mass at both organ and tissue levels in leptin receptor-deficient, ob/ob obese diabetic mice (184). Possible translation of these findings from animals to humans is still required.
There is increasing evidence in support of incretin-analogue use in polycystic ovary syndrome (PCOS) (185), an endocrine disorder which greatly impacts fertility in women, with over 10% of women of reproductive age affected by the condition (186). PCOS is a metabolic disorder that has overlap with T2DM, with patients often being overweight, and presenting with symptoms such as severe insulin resistance, hyperinsulinaemia and dyslipidaemia (187). The interrelation between PCOS and T2DM is further highlighted by the ability of bariatric surgery, specifically Roux-en-Y bariatric surgery (RYGB), to totally ameliorate both T2DM and PCOS (188, 189). Moreover, incretin function has been shown to be impaired in PCOS (187), thus application of GLP-1 mimetics in this condition is a hypothesis built on firm physiological reasoning. Although in relative infancy compared to application in T2DM, the study of application of GLP-1 mimetics in PCOS has been overwhelmingly positive (190). Liraglutide was shown to normalise irregular menstrual bleeding in PCOS patients (191), whilst improving conception rates when used at low dosage in combination with metformin (192). Indeed, it has been suggested that in obese PCOS patients with concurrent insulin resistance, GLP-1 analogues may be a better treatment option than metformin (193). Possible application of PGDPs in female fertility is worthy of further exploration.
Since the approval of exendin-4 for T2DM, increasingly longer acting formulations of GLP-1 analogues have been developed. The first, liraglutide, a mammalian GLP-1 analogue employing conjugation to a palmitic acid chain via a linker coupled to the Lys26 residue was approved in 2010 [(194); Table 2]. This modification increased half-life to ~12 h, through promoting non-covalent binding to albumin and reduced renal clearance, permitting once daily administration (195). Indeed, further longer-acting analogues were developed employing several strategies. The conjugation of the native GLP-1 analogue, D-Ala8GLP-1(Lys37), to an antithrombin III (ATIII)-binding pentasaccharide, known as CarboCarrier®, produced a peptide with potential for once-weekly dosing [(73, 74); Table 2], while a once-weekly exenatide preparation (Bydureon™) which employs microspheres to form a slowly released, peptide-depot gained regulatory approval in 2014 [(196); Table 2]. Additionally, the once weekly preparations albiglutide and dulaglutide employ covalent interactions to attach the peptide to human albumin or a tail fragment of an IgG 4 antibody respectively, which impedes clearance (67, 68), while semaglutide achieves the same pharmacokinetic profile with non-covalent interaction with albumin (70). Such advancement has continued with a once-monthly, hydrogel preparation utilising the analogue [Gln28]exenatide currently undergoing development (75), while a novel osmotic minipump, termed Itca 650, is currently in phase III clinical trials (FREEDOM-1) (197). This pump administers a constant infusion of exenatide following subcutaneous implantation, reported to last for up to 12 months before requiring replacement (197).
In addition to this novel delivery method, there is growing interest in development of oral GLP-1 therapies, with preclinical data now describing bioactivity of orally delivered exendin-4 (198, 199), albeit requiring a considerably larger dose than intraperitoneal injection in mice. Most notable is a novel formulation of semaglutide that makes use of an absorption enhancer, sodium N-(8-[2-hydroxylbenzoyl] amino) caprylate (SNAC), designed to protect peptides from proteolytic degradation and promote absorption across the gastric mucosa [(71); Table 2]. Phase II trials comparing oral to s.c. semaglutide in diabetes management revealed comparable improvements in glycaemia when compared to placebo, but notably oral treatment attained slightly greater weight loss over the 26 week study (-6.9 kg/-7.6%, compared to -6.4 kg/-7.2%) (71). This therapeutic has recently gained FDA approval following successful phase III trials (PIONEER-7) in T2DM patients and provides significantly better improvements in glycated haemoglobin (HbA1c) than sitagliptin in T2DM (200). Like previously available oral antidiabetics (7), oral semaglutide is taken once-daily as a tablet formulation, being prescribed under the brand name Rybelsus® (201). Moreover, as part of the PIONEER trial program, oral semaglutide was studied in patients with renal impairment and demonstrated favourable outcomes (202), possibly indicating that like s.c. semaglutide there was cardiovascular benefit (118). However, when outcomes were assessed upon completion of PIONEER-6 non-inferiority compared to placebo was evident (72), but there was no obvious cardiovascular benefit. These new findings are highly relevant and should lead to greater patient acceptability and compliance in treatment of T2DM and other disorders, as compared to traditional injection route for peptide therapies.
The discovery of GLP-1 and GLP-2 occurred simultaneously following the cloning of cDNAs and genes encoding mammalian proglucagon in the early 1980s, with experiments unveiling the sequences of two novel glucagon-like peptides (15, 16). At that time, the biological functions had not been described for either hormone, with the insulinotropic actions of GLP-1 reported in 1987 (96). This delay was due to the lack of bioactivity of GLP-1 (1–37) (203), which hampered progress until the truncated peptide GLP-1 (7–36)-amide was uncovered (204). Perhaps, as a result of subsequent research focusing on the exciting prospect of exploiting GLP-1 as a potential antidiabetic agent, GLP-2 based research may be considered somewhat less intense, with the biological action as a growth promoter in gut not being uncovered until almost a decade after actions of GLP-1 (205).
The development of the first GLP-1/GLP-2 secreting GLUTag cell-line represents a starting point in the elucidation of the biological function of GLP-2. This cell line was produced via the creation of a transgenic mouse model with GLP-1/2 secreting tumours in the colon, from which L-cells could be extracted and immortalised (206). An observation was made that these animals all exhibited marked enlargement of the small bowel following tumour-induction, inspiring the hypothesis that a PGDP secreted by these tumours must have been responsible for the intestinotrophic activity (205). Interestingly, Bloom had reported the first enteroglucagonoma patient with small intestinal villous hypertrophy, malabsorption, as well as colonic and jejunal stasis some 20 years earlier (207). However, the question remained as to which hormone, or hormones, were responsible. Initially, the intermediary peptide, glicentin, was identified to elicit intestinotrophic action (208). However, subsequent administration of synthetic GLP-2 into mice indicated that GLP-2-mediated increases in small bowel weight surpassed those seen with glicentin (209), making it the more likely instigator.
As their name suggests, both GLP hormones are closely related with both being synthesised by the action of PC1/3 and secreted from intestinal L-cells of the distal gut (Figure 1) (25, 210). Following liberation from proglucagon, the 33 residue GLP-2 is secreted post-prandially in a biphasic fashion from nutrient-sensing L-cells (Figure 2), particularly in response to carbohydrates and lipids contained within luminal contents [(211); Figure 2]. Notably, the distal location of these cells indicates a neural pathway must be involved, given plasma GLP-2 levels (along with other L-cell-derived hormones) are shown to rise rapidly following ingestion (212).
GLP-2 exerts its actions through agonism of its own target receptor, a GPCR termed the GLP-2 receptor (GLP-2R) [(25); Figure 2]. The receptor is widely expressed throughout the entirety of the gut and is highly specific for GLP-2, with other PGDPs demonstrating relatively low affinity (213). Similar to GLP-1, agonism of the GLP-2R evokes a rise in intracellular cAMP and subsequent PKA activation, however, intracellular calcium remains unchanged [(214); Figure 2]. Activation of the receptor directly reduces enterocyte apoptosis and increases crypt cell proliferation, which operates in tandem to increase microvilli height [(215); Figure 3]. The hormone has also been demonstrated to improve intestinal blood flow, decrease gut motility and inhibit gastric acid secretion [(216); Figure 3]. There is some evidence that GLP-2 is produced in small functional amounts within pancreatic islets, but the alternative processing of proglucagon by PC1/3 in alpha-cells to give GLP-1 under conditions of cellular stress is likely much more significant (217).
The intestinotrophic properties of GLP-2 were an attractive prospect in development of therapeutics for conditions such as short-bowel syndrome (SBS), usually a consequence of surgical removal of a section of the bowel in Crohn’s disease (218). This condition is characterised by malabsorption as a result of chronic diarrhoea with further dehydration and weight loss, and depending on severity, the overall quality of life can be greatly impaired. The condition can be managed by parenteral nutrition (PN) and hydration, however, in the long-term this increases the likelihood of infection and potentially sepsis (219). Additionally, patients have a strict reliance on PN which can impede mobility, further impacting on quality of life. Hence, a medication with the ability to manage the condition and reduce the need for PN was highly sought after.
In support of GLP-2 use in SBS, endogenous levels have been shown to rise following excision of bowel (220), while preclinical data showed promising improvements in bowel mass in rats receiving GLP-2 infusion following 75% removal of the mid jejuno-ileum (221). Moreover, infusion of GLP-2 in patients in whom the terminal ileum and colon had been resected, improved intestinal absorption and nutritional status (222). Thus, GLP-2R has clear application in treatment of the condition. As is the case with GLP-1(7–36), GLP-2 is rendered inactive by enzymatic N-terminal dipeptide (His1-Ala2) removal by DPP-4, producing the major fragment GLP-2 (3–33) (205). Thus, in order to be therapeutically viable, the native hormone must be modified to facilitate exogenous administration.
Substitution of the penultimate Ala2 for Gly2 (as found in exendin-4) enabled the development of [Gly2]GLP-2 (Table 3), a DPP-4 resistant, long-acting GLP-2 mimetic (214). The peptide employed single amino acid substitution and presented a more specific approach than blanket DPP-4 inhibition (222). The analogue was later named “teduglutide” and demonstrated early promise in a dose-range pilot study in human SBS patients (227). Subsequent phase III clinical trials confirmed beneficial effects in several cohorts of SBS patients, manifesting in improved intestinal morphology, renal function as well as a favourable side-effect profile (223, 228). Furthermore, treatment reduced reliance on PN in many patients (223), while a portion of previously dependent patients was able to completely discontinue PN (229). Teduglutide was subsequently approved by the FDA in 2012 and is prescribed under the trade names Gattex® in the USA and Revestive® in Europe (Table 3).

Table 3 GLP-2-based therapeutic peptides.
Following the success of teduglutide, further GLP-2 analogues are currently in development, with research aimed to improve the ~5 h circulating half-life of teduglutide (230). Apraglutide ([Gly2, Nle10, D-Phe11, Leu16]-GLP-2) employed further substitutions (Table 3), identified through structure-activity relationship studies of lipophilic amino acid substitutions in positions 11 and 16 of teduglutide, and has been shown to prolong in vivo bioactivity through reduced renal clearance in rodents (224). Similar findings were observed in monkey and mini-pig (225), whilst exhibiting excellent specificity and potency for the GLP-2R. The peptide was more efficacious than both teduglutide and another GLP-2 analogue in development, glepaglutide [(226); Table 3], and has started recruiting for phase III clinical trials in SBS patients (231). That said, glepaglutide has a reported half-life of 50 h and has also entered phase III clinical trials (226). It employs nine amino acid substitutions and a C-terminal tail of six Lys residues (Table 3). The analogue forms a subcutaneous depot at the injection site, from which glepaglutide and its active metabolites are gradually released into the circulation. Phase II trials indicated the analogue was well absorbed, effective and tolerated (226). Thus, apraglutide and glepaglutide may represent an exciting new step in development of GLP-2 analogues, emulating the success of long-acting GLP-1 analogues, which can be administered at less frequent intervals than currently available once-daily preparation, teduglutide.
An additional similarity to GLP-1 research is the pursuit of new therapeutic applications. With the widespread expression of GLP-2R (213), it was postulated that GLP-2 may have application in the management of osteoporosis. Osteoporosis is a condition characterised by bone mass reduction and microarchitecture impairment caused by an imbalance in bone formation and resorption, increasing the risk of fractures (232). Moreover, the prevalence of osteoporosis continues to surge in accordance with an increasingly ageing population (233). A number of the widely-prescribed, anti-resorptive drugs, particularly bisphosphonates, are believed to possess unfavourable side-effect profiles (234), thus alternative treatment options are being sought. Indeed, the involvement of gut hormones in bone mass and formation has been widely researched, with the roles of GLP-1, as well as GIP (175), discussed above.
However, unlike these related gut hormones, the role and indeed application of GLP-2 with respect to bone mass is more divisive. In initial studies of GLP-2 in SBS, an additional observation was made that, following 5 weeks treatment, patients presented with significantly increased spinal areal bone mineral density (222). Subsequently, it was demonstrated that s.c. GLP-2 administration reduced bone resorption in post-menopausal women while not affecting bone formation (235). However, the findings in SBS patients were refuted, with a later study reporting that an intact bowel is required for exogenous GLP-2 administration to have such an effect (236). Additionally, unlike GIPR, an equivalent GLP-2R has not been identified on human osteoclasts (237), indicating that its actions are indirect, with inhibition of parathyroid hormone (PTH), mediated by activation of GLP-2R on PTH gland, suggested to be the mediator of its effects on bone resorption (236). Moreover, a small-scale trial in healthy males employed GIPR antagonists to confirm the antiresorptive effects of GLP-2 are independent of this receptor (238). The mechanisms behind the bone actions of GLP-2 require further investigation to firmly establish a link.
Despite this, several studies support the involvement and potential use of GLP-2 in bone formation in some capacity. In a study of postmenopausal women with concurrent T2DM, it was revealed that ingestion of a mixed nutrient meal saw a reduction in biomarkers for bone fragility, coupled with a rise in GLP-2 levels (239), indicating the importance of the gut. However, this study did not ascertain the involvement of other gut hormones. These findings are supported by more recent work in ovariectomised rats, an animal model replicating postmenopausal osteoporosis. It was established that 4 weeks s.c. administration of GLP-2 resulted in improvements of bone architecture and mass through both promotion of bone formation and a reduction in resorption (240). Interestingly, studies of GLP-2 effects on bone have all employed human GLP-2, as opposed to longer-acting analogues. Furthermore, i.v. administration of a high dose of GLP-2 was outperformed by lower doses of s.c. GLP-2 in terms of reducing bone resorption (241). Thus, given longer acting, s.c. teduglutide is currently available, as well as other enzyme resistant analogues in development, their potential use for therapy of osteoporosis is exciting. Moreover, given the involvement of several gut hormones in this gut-bone axis (242), coupled with the success of unimolecular multiagonists with relation to bone improvements (184), it stands to reason that incorporation of a GLP-2R agonising component may improve the efficacy of such agents in promoting bone density.
Oxyntomodulin (OXM) was discovered as a fragment of glicentin (243, 244), sharing substantial sequence homology and essentially the entire 29 amino acid glucagon molecule with an additional C-terminal octapeptide, IP-1, resulting in 37 residue OXM ( (245, 246); Figure 1 and Table 4).

Table 4 Oxyntomodulin-based therapeutic peptides.
Like other gut-based PGDPs, OXM is released post-prandially from L-cells (254). OXM increases energy expenditure and physical activity, promotes weight loss and improves glycaemia in humans (254, 255). No specific OXM receptor is known to exist; rather, the peptide acts as a dual agonist for GCGR and GLP-1R (Figure 2), although it binds to both with lower affinity than either of their primary ligands (256, 257). In the current thinking, OXM-mediated weight loss is believed to be elicited through activation of the GCGR, bringing about anorectic actions and increased energy expenditure [(258); Figure 3]. In contrast, GLP-1R agonism accounts for improved glucose homeostasis through augmented insulin secretion, overcoming the hyperglycaemic actions of GCGR activation [(259); Figure 3]. Mechanistic studies reveal that OXM behaves as a differential agonist depending on the receptor, acting as a full agonist in recruiting β-arrestin 2 to the GCGR, but partial agonist in recruiting β-arrestin 1 and 2 and GPCR kinase 2 to the GLP-1R (260). Furthermore, some data suggests that OXM is a GLP-1R-biased agonist relative to GCGR (260).
As alluded to above, the ability of OXM to effectively activate both GCGR and GLP-1R, thereby improving blood glucose and body weight, is attractive for the development of peptide therapeutics for obesity/T2DM, provided an appropriate receptor balance is struck. Like all other PGDPs, OXM is subject to rapid inactivation by DPP-4 which targets cleavage after the N-terminal Ser2 residue (261). This rapid inactivation precludes use of the unmodified hormone as a therapeutic. Thus, while initial studies demonstrated that native OXM decreases food intake and enhances energy expenditure in both healthy and obese human volunteers, these employed undesirably frequent dosing of three- or four-times daily (262, 263).
As with GLP-1, DPP-4 resistant forms of OXM are required therapeutically and given the sequence similarities between the two peptides, successful approaches taken with GLP-1 can be applied to OXM (261, 262). One example, (D-Ser2)Oxm[mPEG-PAL] (Table 4), employed substitution of the naturally occurring L-Ser2 with the enantiomer D-Ser2 to promote DPP-4 resistance, while further utilising C-16 palmitic acid conjugation via a mini-PEG linker at the C-terminus to reduce renal clearance and improve circulating half-life (247). The resulting peptide was fully resistant to DPP-4, whilst clearly retaining bioactivity: increasing cAMP in both GLP-1R and GCGR transfected cell lines, as well as enhancing insulin release from clonal pancreatic beta-cells (247). Additionally, daily administration of (D-Ser2)Oxm[mPEG-PAL] to ob/ob mice decreased food intake and body weight, whilst increasing plasma and pancreatic insulin and improving glucose tolerance (247). Several biomarkers of obesity were also improved, including increased adiponectin with reductions in both visfatin and triglyceride concentrations (247). The OXM analogue also exerted beneficial effects on blood glucose control in STZ-diabetic mice, including elevations in total islet and beta-cell areas associated with an increase in the number of smaller-sized islets and enhanced islet proliferation (264). A follow-up study with (D-Ser2)Oxm[mPEG-PAL] in transgenic mice with fluorescently tagged alpha cells also demonstrated highly favourable effects on islet cell transdifferentiation (265). Interestingly, another study employed dogfish and ratfish oxyntomodulin peptides (Table 4), which despite numerous sequence variations from human OXM, remained effective at mammalian GCGR and GLP-1R (248). This suggests a possible early advantage of such dual receptor actions in evolutionary terms.
The therapeutic applicability of enzymatically stable OXM analogues is clear and a number of analogues are in various stages of development for potential use in T2DM therapy (Table 4). However, it has been demonstrated that a balance in GCGR/GLP-1R agonism must be reached when designing OXM analogues, with a number of examples demonstrated to induce hyperphagia (266). OXM analogues with Glu3 substitution favour GLP-1R activation and do not exhibit an orexigenic effect (266), hence, it is assumed that such an effect must be mediated via GCGR agonism (266, 267). However, with the development of OX-SR, a sustained-release oxyntomodulin analogue which employs 5 central, depot-forming, amino acid substitutions between residues 16-27 of the human peptide (exact sequence not disclosed by authors), an OXM analogue capable of bringing about GCGR-mediated increases in energy expenditure was developed, and despite having an orexigenic effect actually elicited 2% weight loss following 3 days administration in rats (249). In this respect, while OX-SR was proven to agonise both receptors in vitro, the analogue showed greater affinity for GCGR than GLP-1R (249). More prolonged studies, including those in models of diabetes are required to investigate the long-term consequences of such prolonged exposure to GCGR and GLP-1R activation by OX-SR, but the peptide does represent a potential once-weekly OXM formulation (249). Excitingly, regulatory approval of the first OXM analogue may be on the horizon, with the long acting, fatty-acid derivatised analogue LY3305677 (sometimes termed IBI362) currently in separate phase II clinical trials investigating management of T2DM and obesity (250, 251).
Glicentin is a product of PC1/3 proglucagon processing, while GRPP glicentin-related pancreatic peptide (GRPP) is a product of PC2 processing in the pancreas [(22, 25); Figure 1]. Radioimmunoassay of gut extracts revealed substances with glucagon-like immunoreactivity that cross-reacted with antibodies directed towards the N-terminus of glucagon (268), with further investigation identifying two related proteins, one appearing to be a fragment of the other. Firstly, the 69 residue, N-terminal proglucagon fragment glicentin (243), which contained the entire glucagon sequence attached to an N-terminal portion later identified as GRPP (269, 270). The smaller fragment was essentially glucagon attached to a C-terminal octapeptide called intervening peptide-1 (IP-1) (271), later this C-terminally extended glucagon was denominated as oxyntomodulin.
We now know glicentin is released post-prandially from L-cells of the distal ileum and colon, particularly in response to glucose, lipids and amino acids, especially arginine, entering the duodenum [(272–274); Figure 2]. The hormone elicits a number of physiological effects such as a paracrine role in promoting intestinal growth and regulating motility (275), as well as playing a role in glucose homeostasis through augmenting insulin secretion and inhibiting glucagon secretion (276). However, no glicentin receptor has yet been identified, but the hormone has been shown to agonise and elicit cAMP production following binding to glucagon, GLP-1 and GLP-2 receptors [(277, 278); Figure 2]. Additionally, earlier work with glicentin suggested that its actions were largely dependent upon the degradation of the hormone into smaller molecular fragments (279), possibly including carboxylase-mediated generation of glucagon (Figure 2). This may, in part, explain why there has been relatively little research exploring development of glicentin-based therapeutics. Furthermore, it is likely that a lack of commercialised detection methods for glicentin have hindered its overall investigation and therapeutic application (280). However, with increasing availability and affordability of capable assays and given the increasing interest in peptide therapeutics, we may see renewed interest in this PGDP (281). Moreover, it has recently been put forward that post-surgery rises in glicentin, along with OXM, are the best predictors of decreased in intake of energy-dense foods and weight loss following RYGB, more so than even GLP-1 (282). Whether this translates to functional involvement remains unclear, and instead it is postulated that increased glicentin levels are a useful indicator of improved overall L-cell function (282).
As discussed above, GRPP is one of the products of PC2 processing of proglucagon in pancreatic alpha-cells (25, 26). A 30 residue, N-terminal fragment of proglucagon (Figure 1), GRPP was discovered after glicentin using glicentin-specific antibodies in pancreatic extracts (269). Structural elucidation highlighted that the peptide was identical to the N-terminus of gut-derived glicentin, hence the name glicentin-related pancreatic peptide (269). Despite its discovery almost four decades ago, research on this PGDP is sparse, but earlier experiments in dogs suggest the peptide may influence glucose homeostasis through increasing plasma insulin and decreasing plasma glucagon (283). A more recent study utilised isolated-perfused pancreas and liver from rats to pursue a detailed investigation of the physiology of this peptide (284). In contradiction of initial findings, this study demonstrated that while glucose output from the liver remained unaffected, GRPP brought about potent inhibition of glucose‐stimulated insulin secretion in perfused pancreas, with cAMP assay indicating that these actions were not mediated through either GLP-1R or GCGR, meaning an unidentified receptor may be at play (284). Given the lack of physiological data surrounding GRPP, it is unsurprising that no therapeutic exploration has been made on this PGDP.
Unimolecular multiagonists represent an exciting future in the therapeutic application of PGDPs, with increasingly complex and experimental molecules being developed. As briefly mentioned above, RYGB surgery induces rapid remission of T2DM in 70-80% patients (285). Importantly, secretion and action of a number of gut hormones, including the PGDPs GLP-1, GLP-2, OXM and glicentin, together with PYY, GIP, cholecystokinin (CCK), neurotensin (NT) and secretin, are positively modulated in concert following RYGB (286). These are thought to be major determinants in the improvements of appetite, body weight, glucose tolerance and insulin sensitivity demonstrated post-surgery (286). Thus, given high costs, limited availability and potential risks associated with surgical procedures, there is a current focus on designing multiagonist molecules with the ability to emulate the post-surgical, hormonal mechanisms of RYGB, which have the potential to be more widely available to patients than surgery. Additionally, they have the potential to evoke an array of positive actions within various organs (Figure 3), and such molecules could surpass advantages observed with individual peptides.
Earlier research employing combinations of single gut hormones or analogues provided a sound basis for the application of multi-agonism in T2DM (287). Indeed, with the combination of liraglutide plus an acylated GIP analogue (288), synergy was demonstrated leading to improved glucose-lowering and insulinotropic actions in obese-diabetic mice compared to either of the individual incretin analogues alone. Furthermore, recent combination studies have further strengthened the idea that combined exogenous peptide administration can effectively emulate the benefits of RYGB. As such, infusion of a multi-peptide preparation of GLP-1, OXM and PYY (3–31, 35–39) termed “GOP”, can replicate the postprandial levels of these hormones observed after RYGB, and can safely bring about 32% reduction in food intake in a standardised meal test (289). Moreover, continuous GOP infusion, delivered by pump over a 4-week period in obese patients with prediabetes or diabetes, resulted in improvements in glucose tolerance which surpassed those of RYGB (290).
To date, a number of unimolecular double- and triple-agonists have been developed with several being actively pursued for clinical application (Table 5). The majority of these typically employing a GLP-1R agonist component combined with another gut hormone, often an incretin or other PGDP.
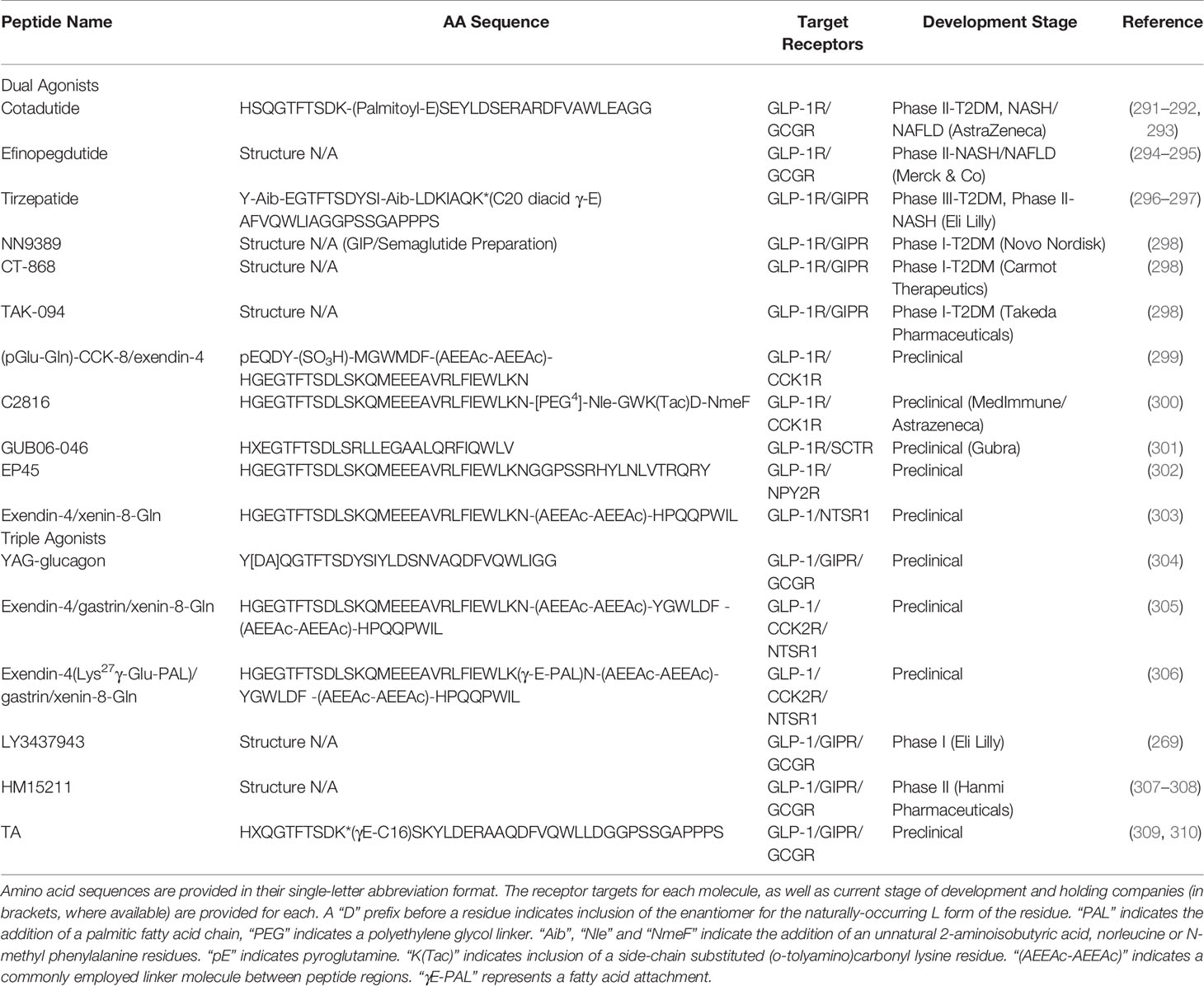
Table 5 Multiagonists based on proglucagon-derived peptides in development.
As previously discussed, the notion of GCGR agonism in pursuit of a therapeutic for T2DM, or its related conditions, seems counterintuitive. However, given the surprising beneficial effects of OXM agonists in T2DM, the benefits of targeting these two receptors in tandem was clearly demonstrated (Figure 3). Additionally, the structural similarity between the two PGDPs was clearly demonstrated (Table 4). As such, this combination pioneered unimolecular PGDP-based research, with the cholesterol-conjugated OXM analogue DualAG and the glucagon, GLP-1 chimeric peptide “Aib2 C24 chimera 2 lactam 40K” both showing preclinical promise in murine, DIO models of obesity-diabetes [(252, 311); Table 4].
While a number of GLP-1/glucagon based peptides have been generated (Table 5), many have witnessed therapeutic pursuit abandoned. Currently, a molecule of particular promise is cotadutide (formerly MEDI0382). Cotadutide is a linear, chimeric peptide employing important residues from both glucagon and GLP-1 into its sequence (Table 5), with a palmitoyl FA attachment on Lys10 to prolong circulating half-life (253). The peptide is reported to be a balanced dual-agonist for GLP-1 and GCGR, which brought about significant weight loss through improved glycaemia in DIO mice and non-human primates, being more effective than liraglutide alone (253). The concept of balance in respect to such molecules is crucial, as it is important to maximise weight loss whilst minimising the potential to cause hyperglycaemia, with as little as 10% relative GLP-1 sequence contribution minimising hyperglycaemia whilst retaining weight loss (312). The effects on glycaemia were supported by acute administration studies in humans, however a slower dose titration was deemed necessary to avoid adverse effects on gastric emptying (291).
When assessed in phase II trials in T2DM patients, slower titration of cotadutide was employed to reflect such findings (313). This study revealed that daily administration in patients with controlled T2DM improved overall glycaemic control, as measured by HbA1c, which was associated with sustained weight loss following 41 days daily administration (313). Subsequently, it was revealed that these positive effects on glycaemia were likely the result of improved gastric emptying and postprandial insulin response (292). Additionally, patients presented with significant improvements in liver fat, with levels falling by 39% (313), which was notable given an equivalent fall in levels with liraglutide takes around 6 months (314). These findings on liver fat have seen a refocus of research toward application in non-alcoholic fatty liver disease (NAFLD) and steatohepatitis (NASH) (293), both common consequences of uncontrolled T2DM (315). The study revealed that cotadutide’s actions on the liver to reduce lipid content, drive glycogen flux and improve mitochondrial turnover and function are directly mediated through modulation of hepatic GCGRs, while metabolic improvements mediated via agonism of extrahepatic GLP-1Rs further enhanced improvement (293). A similar story is unfolding for the GLP-1/GCGR agonist efinopegdutide (formerly HM12525A), a longer acting agonist which employs modified exendin-4 conjugated to human IgG, facilitating once-weekly administration ( (294); Table 5). The peptide appealed as a treatment for T2DM due to promising preclinical results which demonstrated lipolytic and insulinotropic effects in diabetic mice (316). However, potent lowering effects on cholesterol and liver fat have seen this analogue also repurposed as a potential NAFLD/NASH medication (295).
With synergy demonstrated by administration of liraglutide plus an acylated, enzyme resistant GIP analogue (288), the value of developing molecules targeting these two incretin receptors was evident. As such, a number of unimolecular GLP-1/GIP agonists have been developed and are at various stages of clinical testing (Table 5).
One particular success story involves a molecule termed tirzepatide (formerly LY3298176) (296). The peptide is a linear, 39 aa peptide containing two unnatural residues and a C20 diacid fatty acid attached via a linker to Lys20 (Table 5), all of which contribute to a circulating half-life of ~5 days, which permits once weekly dosing (296). Tirzepatide may be considered a GIP-based analogue, sharing greater sequence homology with GIP than GLP-1 (particularly at the N-terminus) (Table 5), with GLP-1R agonism induced via aa substitution (296, 317). The peptide was shown to effectively lower blood glucose via insulinotropic actions at both receptors in preclinical studies in mice, while phase I trials revealed effective weight loss in T2DM patients and good tolerability (296). Interestingly, in vitro mechanistic studies suggest the peptide is biased towards the GIPR, activating with equipotency to native GIP whilst having 5-fold weaker affinity than native GLP-1 at GLP-1R, with a preference to initiate cAMP mobilisation to enhance insulin secretion (317), which may be of particular benefit in obesity-diabetes. These results were supported in phase II trials in T2DM patients with HbA1c reductions of 2%, highly impressive body weight reductions of 5-10% (max 11.3 kg) and significant reductions in waist-circumference demonstrated following 12 weeks treatment (297). Moreover, comparison to the established GLP-1R mimetic, dulaglutide, proved tirzepatide to elicit more significant reductions in body weight (-4.52 kg/6.4% compared to -1.3 kg/1.8% for dulaglutide after 4 weeks), with the authors concluding inclusion of GIPR agonism builds upon sole GLP-1R activation to enhance weight loss via modulating appetite and gastric emptying, with the antiemetic effect of GIPR also improving tolerability (296). It is likely further mechanistic investigation will be pursued to fully elucidate the biological processes at play, especially as no single effect could be entirely attributed to GIPR/GLP-1R agonism (318). Thus, while important synergy is likely to be occurring, confirmation is required.
Tirzepatide has now progressed to phase III clinical trials in T2DM, and we await data from these studies with great anticipation. In similar fashion to the GLP-1/glucagon analogues discussed, tirzepatide has also found application in the treatment of NASH, with a follow-on study in T2DM patients revealing that several biomarkers of liver inflammation were reduced in patients receiving higher doses of the analogue (319). Indeed, a number of other analogues such as NN9389 (GIP and semaglutide combination), CT-868 and TAK-094 are all currently in phase I clinical trials as potential T2DM treatments (298), but any detailed literature on these analogues remains elusive at the time of writing.
A literature search for dual agonists also reveals some slightly left-field combinations with GLP-1, although importantly these involve other gut hormones shown to be upregulated by bariatric surgery (286). The combinations explored so far in preclinical studies all have the potential to elicit a range of additional effects on various systems in the body (Figure 3). For example, a long-acting GLP-1/CCK hybrid peptide has been developed which employs the key regions of (pGlu-Gln)-CCK-8, a stabilised form of CCK (320), and exendin-4 attached to one another via a linker molecule (Table 5). Through simultaneous activation of both GLP-1 and CCK-2 receptors, this co-agonist outperformed exendin-4 in terms of satiety and body weight reductions in obese-diabetic mice (299). A similar molecule, essentially reversing the configuration of GLP-1 and CCK components [(300); Table 5], also highlights the potential of dual receptor activation in this regard, outperforming (pGlu-Gln)-CCK-8 in terms of body weight reduction following 10 weeks treatment in DIO mice (300).
A GLP-1/secretin chimeric peptide, based on the sequence of secretin with GLP-1R activity induced via substitution of important GLP-1 residues (Table 5), has been developed. This peptide decreased food intake and body weight more effectively than liraglutide alone (301). Moreover, this analogue improved short-term glycaemic control (39% fall in fasting blood glucose), HbA1c (-1.6%) and promoted a 78% rise in beta cell mass following twice daily s.c. administration over an 8 week period in diabetic, db/db mice (301).
Another successful, but seemingly counterintuitive pairing is the combination of GLP-1 and PYY. PYY is insulinostatic but holds therapeutic potential due to induction of beta cell rest, promotion of beta-cell mass, satiety and weight loss (321). Moreover, a synergistic effect between PYY and GLP-1 has been established (322), supporting their incorporation in a co-agonist. One such peptide, termed EP45, has been developed as a chimeric peptide employing PYY (25–36) incorporated with exendin-4(1–33); Table 5). Indeed, the peptide was demonstrated to effectively activate both GLP-1R and NPY2R in transfected cell lines (302), but in vivo application is yet to be published.
Finally, an enzyme resistant GLP-1/xenin dual-agonist, Exendin-4/xenin-8-Gln (303), has been developed with xenin, which is a regulatory peptide co-secreted postprandially with GIP from intestinal K-cells [(323); Table 5]. Xenin is known to potentiate the actions of GIP (324), and in addition to positive glycaemic outcomes, through reduced appetite and augmented insulin secretion, the peptide also restored GIP sensitivity (303) that is dampened in obesity (325). Consistent with these actions, Exendin-4/xenin-8-Gln induced substantial benefits in DIO diabetic mice (303).
Beyond the incorporation of GLP-1 with other gut hormones, there is also growing interest concerning the conjugation of GLP-1 with nuclear hormones like oestrogen, thyroid hormone (T3) and dexamethasone (326). In particular, the conjugation of GLP-1-estrogen allows selective targeting of oestrogen receptors (ER) in GLP-1R expressing cells. This reduces obesity and improves dyslipidaemia and hyperglycaemia more so than sole activation of either GLP-1R or ER (327). In relation to the metabolic effects of these conjugates, preclinical studies in rodents have demonstrated that these conjugates act on reward centres within the supramammillary nucleus to induce an anorectic effect (328), which positively influences glycaemia. Moreover, such conjugates were demonstrated to improve beta-cell function and survival (329), which in a study employing a combination of GLP-1-estrogen and insulin in a DIO model of diabetes, allowed for a 60% reduction in the insulin dose compared to a control group of animals receiving insulin monotherapy (330). While conjugation to GLP-1 proves an effective method to prevent the oncogenic and gynaecological actions of oestrogen (331), distinct differences in the hormonal aetiologies of obesity in males and females have demonstrated that administration of such agents in different sexes of mice elicit subtle differences in obesity-related inflammation pathways (332, 333). Thus, the impact of gender in relation to the applicability of these agents needs to be further explored.
Given the successful development of GLP-1/GIP and GLP-1/glucagon dual-agonists (293, 296), the next obvious step was to develop triple-agonists based on these three gut hormones [(318); Table 5]. One such molecule, termed YAG-glucagon (Table 5), is an analogue based on human glucagon with a number of amino acid substitutions to impart GIPR and GLP-1R agonism (304). The DPP-4 resistant analogue was demonstrated to be an effective tri-agonist in vitro, while twice-daily administration in DIO mice manifested in improved blood glucose, circulating insulin and enhanced insulin sensitivity (304). While this molecule has not surpassed preclinical stage, a couple of examples appear to be progressing well at present. LY3437943, a reported tri-agonist is currently undergoing phase I trials in management of obesity-diabetes (307). More data is available for HM15211, a tri-agonist employing a GLP-1/GIP/glucagon peptide (sequence not available) attached to a human aglycosylate Fc fragment which prolongs half-life to permit once weekly administration [(334); Table 5]. HM15211was more effective than daily administration of liraglutide in increasing energy expenditure, with improvements in weight loss and hepatic inflammation markers in rodent models (334), and is currently recruiting for phase II trials as a treatment for NASH (308).
Similar to single GLP-1 agonists, a tri-agonist termed TA is also finding application with regards to neuroprotection [(309); Table 5]. This hybridised GLP-1/GIP/glucagon activator was initially developed for management of obesity-diabetes and showed promising preclinical results in rodent models of diabetes-obesity (310). However, the more recent repurposing of this molecule towards management of AD is particularly exciting, with daily administration of the analogue in a murine model of AD over a 2 month period reversing memory deficit, reducing pro-mitochondrial apoptosis markers and upregulating growth factors involved in synaptic function (309). The preclinical study has not been followed-up to date, but represents a potentially fruitful new avenue for the application of PGDP-based multiagonists.
Interestingly, the aforementioned GLP-1/xenin combination has been exploited further with the development of the triple-agonist exendin‐4/gastrin/xenin‐8‐Gln (335), a direct descendent of the previously discussed dual-agonist (303). This incorporates the hexapeptide gastrin into its sequence (Table 5), evoking the ability to agonise GLP-1R, CCK2R and NTSR1 in tandem (335). Preclinical studies with this peptide were promising, eliciting improved glycaemic control when administered twice daily in DIO diabetic mice over 21 days, through elevation in circulating insulin levels, improved insulin and GIP sensitivity, with encouraging reductions in fat mass, triglycerides and cholesterol levels (335). Moreover, this analogue has been further modified via the covalent attachment of a hexadecanoyl fatty acid to improve circulating half-life and duration of effect (Table 5), with twice daily administration in obese-diabetic ob/ob mice recapitulating the metabolic benefits attained with the non-acylated form (305).
The recognised role of hyperglucagonaemia in the pathophysiology of diabetes, and the effectiveness of concomitant activation of GCGR, alongside GLP-1R by oxyntomodulin, raises an apparently conflicting question: can glucagon antagonists or glucagon agonists be utilised as a diabetes or obesity therapy? A similar question exists for therapeutic GIP analogues (306). In fact, both aspects are being explored although neither is, as yet, fully understood, or nearing final stages of development.
The therapeutic potential for glucagon suppression is clear, especially given that a synthetic analogue of the glucagon suppressing hormone amylin, termed “Pramlinitide”, is currently prescribed in the USA as an adjunct to insulin therapy (336). However, pramlintide is not a specific inhibitor of glucagon secretion, as it also is well known to slow the rate of gastric emptying and induce satiety. Thus, direct glucagon receptor antagonism may represent a more specific alternative in this regard. While it is true that many small-molecule glucagon antagonists exist (337–339), these have been discounted due to undesirable pharmacokinetic properties which led to rapid renal clearance and diminished effects (337, 340). Moreover, off-target safety concerns were present, including activation of peroxisome proliferator-activated receptor-delta (PPAR-δ) (337), a transcription factor which plays roles in inflammation and certain cancers (341). That said, a few small molecules such as Eli Lilly and Co’s GRA LY2409021 have made it as far as phase II trials (342). These demonstrated promising reductions in HbA1c but were ultimately let down by undesirable side-effect profiles, often eliciting potentially dangerous elevations in liver enzymes (343). Hence, a view was taken that development of glucagon peptide-based antagonists could herald better tolerated compounds with improved pharmacokinetic and safety profiles.
Logical design of such compounds took the sequence of native glucagon (Table 6), modifying the structure, paying particular attention to previously identified important residues for GCGR agonism, namely N-terminal His1, Gly4 and Asp9 residues (349–353), whilst also ensuring that they are resistant to the actions of DPP-4 [(349); Figure 2]. Two such analogues termed desHis1Pro4Glu9(Lys12PAL)-glucagon and desHis1Pro4Glu9(Lys30PAL)-glucagon [(346); Table 6], employed simple amino acid substitutions at residues 4 and 9, while His1 was deleted to produce compounds with the potential to effectively block the receptor and palmitic acid (PAL) was attached via linker molecules to substituted Lys residues at differing positions (346), a means of prolonging circulating half-life (351). Indeed, both molecules, as well as their non-acylated counterparts, were shown to be resistant to DPP-4 (346, 353). The compounds possessed strong antagonist properties, dose-dependently reducing glucagon-mediated cAMP production and insulin secretion together with counteracting glucagon-mediated hyperglycaemia in vivo (346).

Table 6 Glucagon antagonist peptides for T2DM.
Further related analogue development resulted in synthesis and characterisation of desHis1Glu9(Lys30PAL)-glucagon and desHis1Glu9-glucagon-[mPEG] [(299, 354); Table 6]. These peptides were resistant to DPP-4 degradation (344, 348, 355) and lacked adverse metabolic or islet morphological effects when administered twice daily to lean mice (347). Preclinical testing of desHis1Glu9-glucagon and desHis1Glu9(Lys30PAL)-glucagon in HFF obese mice reversed obesity-driven hyperinsulinaemia and insulin resistance together with improvements in lipid profile, glucose tolerance and increased pancreatic insulin stores (345).
These studies are typical of others that have led to development of peptidergic glucagon receptor antagonists for T2DM, in particular the first reported antagonistic, glucagon analogue [l-N alphatrinitrophenylhistidine, 12-homoarginine]-glucagon, which elicited decreases in circulating glucose of up to 65% with continuous infusion in anaesthetised rats (356). However, despite this preclinical promise peptide-based agents were largely abandoned at this point possibly due to short half-life in pursuit of small-molecule antagonists (357).
To date, no glucagon antagonist has reached regulatory approval, with previous safety concerns raised over hypoglycaemia (358), unfavourable alterations in serum lipid levels and liver enzymes (359, 360), as well as the potential for malignant hyperplasia of alpha-cells (361). Intriguingly, a similar tale is true for GLP-1R agonists, with a number of small molecule examples dotted through the literature (362), however none have managed to recapitulate the success of peptidergic agents. Preclinical data with peptide-based antagonists indicate more favourable side-effect profiles than small-molecules (345, 347). Thus, while work continues on small-molecule antagonists, such as RVT-1502, which has recently progressed through phase II trials, demonstrating reductions in HbA1c of up to 1% over 12 weeks treatment, concerns over liver function still remain, and the compound has not ascended to phase III trials (363). Such concerns may lead to an upsurge in interest for peptide-based glucagon antagonists.
Given the use of glucagon to rescue severe insulin-induced hypoglycaemia T1DM (53) and its ascribed role in the hyperglycaemia of diabetes (35, 51), the concept of using glucagon agonists therapeutically initially seems illogical. However, the surprising effectiveness of dual or triple agonism indicates that weight loss and increased energy expenditure associated with GCGR agonism can be exploited when the hyperglycaemic actions of the hormone are counteracted by the incretins GLP-1 and/or GIP (357, 364).
Several approaches have been explored to generate such, potentially useful, enzyme-resistant GCGR agonists including (D-Ser2)glucagon, where a D-amino acid substitution has been employed to impart DPP-4 resistance more effectively than N-acetyl-glucagon [(365); Table 7]. Insulin-releasing activity was maintained, but when further modified to generate (D-Ser2)glucagon-exe, an analogue with the nine C-terminal amino acid residues of exendin(1–39) (Table 7), clear antidiabetic benefits were induced (365). Importantly, inclusion of the C-terminal nonapeptide from exendin(1–39) in this molecule imparts the ability to agonise GLP-1R as well as GCGR, as demonstrated by reduced effectiveness in GLP-1R KO mice (365). Thus, twice daily administration of (D-Ser2)glucagon-exe in HFF mice improved glucose tolerance, insulin sensitivity and islet morphology, while improvements in energy expenditure, O2 consumption and physical activity together with reduced food-intake led to decreased body weight and influenced glycaemic improvement (365).

Table 7 Glucagon and related peptide analogues at preclinical stage for T2DM.
A number of naturally occurring, piscine-derived, glucagon peptides such as dogfish glucagon (and its analogues) and paddlefish glucagon have also been shown to possess potent antidiabetic/anti-obesity potential in cellular and animal models of diabetes [(248, 366, 367); Table 7]. Furthermore, studies using GLP1-R KO mice and cell lines indicated that benefits on glucose tolerance, beta-cell function, insulin sensitivity and circulating triglycerides were mediated via dual GCGR and GLP-1R agonism (248, 366). Thus, in relation to management of T2DM, inclusion of GCGR agonist in multiagonist molecules appears to be where the future novelty lies for such agents.
The application of proglucagon-derived peptides (PGDPs) in the management of conditions such as T2DM represents the pinnacle of a remarkable story in peptide discovery and rational drug design. It was shear perseverance which led to the elucidation of proglucagon almost six decades after that of glucagon (9, 11–16). Rapid discoveries followed of GLP-1 and GLP-2 (15, 16), both of which have been successfully exploited by peptide chemistry approaches to generate fully approved medications. While innovation has witnessed the production of increasingly long-acting agents, multi-action unimolecular agonists and novel delivery methods (67, 68, 70, 73–75, 195–197), there is still a growing need for ever more effective agents to counter obesity, diabetes and a host of other degenerative diseases. Better understanding of the physiology of PGDPs and their various roles in the likes of cognition, bone turnover, cardiovascular function, fertility and liver function (157, 174, 185, 334, 368, 369), may herald important future uses for proglucagon-derived therapeutics.
All authors contributed to the article and approved the submitted version.
PF, VG, NI,and FO’H are named on patents filed by Ulster University for the exploitation of incretin-based drugs and other peptide therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Research in the authors’ laboratories on gut peptide therapeutics has been generously supported over many years by Diabetes UK, European Foundation for the Study of Diabetes, Diabetes Research and Wellness Foundation, Invest Northern Ireland, Northern Ireland Department for Education, and Ulster University Strategic Funding.
1. Edkins JS. The Chemical Mechanism of Gastric Secretion. J Physiol (1905) 34:133–44. doi: 10.1113/jphysiol.1906.sp001146
2. Bayliss WM, Starling EH. The Mechanism of Pancreatic Secretion. J Physiol (1902) 28:325–53. doi: 10.1113/jphysiol.1902.sp000920
3. Banting FG, Best CH, Collip JB, Campbell WR, Fletcher AA. Pancreatic Extracts in the Treatment of Diabetes Mellitus. Can Med Assoc J (1922) 12:141–6.
4. Lawrence RD, Archer N. Zinc Protamine Insulin a Clinical Trial of the New Preparation. Br Med J (1937) 1:487–91. doi: 10.1136/bmj.1.3974.487
5. Bell GI, Pictet RL, Rutter WJ, Cordell B, Tischer E, Goodman HM. Sequence of the Human Insulin Gene. Nature (1980) 284:26–32. doi: 10.1038/284026a0
6. Davies MJ, D’Alessio DA, Fradkin J, Kernan WN, Mathieu C, Mingrone G, et al. Management of Hyperglycemia in Type 2 Diabetes, 2018. a Consensus Report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care (2018) 41:2669–701. doi: 10.2337/dci18-0033
7. Wilkinson S, Douglas I, Stirnadel-Farrant H, Fogarty D, Pokrajac A, Smeeth L, et al. Changing Use of Antidiabetic Drugs in the UK: Trends in Prescribing 2000-2017. BMJ Open (2018) 8:e022768. doi: 10.1136/bmjopen-2018-022768
8. Lund PK, Goodman RH, Habener JF. Pancreatic Pre-Proglucagons are Encoded by Two Separate Mrnas. J Biol Chem (1981) 256:6515–8. doi: 10.1016/S0021-9258(19)69015-0
9. Murlin JR, Clough HD, Gibbs CBF, Stokes AM. Aqueous Extracts of the Pancreas. Influence on the Carbohydrate Metabolism of Depancreatized Animals. J Biol Chem (1923) 56:253–96. doi: 10.1016/S0021-9258(18)85619-8
10. Bromer WW, Sinn LG, Staub A, Behrens OK. The Amino Acid Sequence of Glucagon. Diabetes (1957) 6:234–8. doi: 10.2337/diab.6.3.234
11. Lund PK, Goodman RH, Dee PC, Habener JF. Pancreatic Preproglucagon cDNA Contains Two Glucagon-Related Coding Sequences Arranged in Tandem. Proc Natl Acad Sci USA (1982) 79:345–9. doi: 10.1073/pnas.79.2.345
12. Lund PK, Goodman RH, Montminy MR, Dee PC, Habener JF. Anglerfish Islet Pre-Proglucagon II: Nucleotide and Corresponding Amino Acid Sequence of the Cdna. J Biol Chem (1983) 258:3280–4. doi: 10.1016/S0021-9258(18)32858-8
13. Heinrich G, Gros P, Habener JF. Glucagon Gene Sequence. Four of Six Exons Separate Functional Domains of Rat Pre-Proglucagon. J Biol Chem (1984) 259:14082–7. doi: 10.1016/S0021-9258(18)89859-3
14. Heinrich G, Gros P, Kay Lund P, Bentley RC, Habener JF. Pre-Proglucagon Messenger Ribonucleic Acid: Nucleotide and Encoded Amino Acid Sequences of the Rat Pancreatic Complementary Deoxyribonucleic Acid. Endocrinology (1984) 115:2176–81. doi: 10.1210/endo-115-6-2176
15. Bell GI, Santerre RF, Mullenbach GT. Hamster Preproglucagon Contains the Sequence of Glucagon and Two Related Peptides. Nature (1983) 302:716–8. doi: 10.1038/302716a0
16. Bell GI, Sanchez-Pescador R, Laybourn PJ, Najarian RC. Exon Duplication and Divergence in the Human Preproglucagon Gene. Nature (1983) 304:368–71. doi: 10.1038/304368a0
17. Sundby F, Jacobsen H, Moody AJ. Purification and Characterization of a Protein From Porcine Gut With Glucagon Like Immunoreactivity. Horm Metab Res (1976) 8:366–71. doi: 10.1055/s-0028-1093615
18. Bataille D, Gespach C, Coudray AM, Rosselin G. “Enteroglucagon”: A Specific Effect on Gastric Glands Isolated From the Rat Fundus. Evidence for an “Oxyntomodulin” Action. Biosci Rep (1981) 1:151–5. doi: 10.1007/BF01117012
19. Brown JC, Dryburgh JR. A Gastric Inhibitory Polypeptide. II. The Complete Amino Acid Sequence. Can J Biochem (1971) 49:867–72. doi: 10.1139/o71-122
20. Ørskov C, Holst JJ, Poulsen SS, Kirkegaard P. Pancreatic and Intestinal Processing of Proglucagon in Man. Diabetologia (1987) 30:874–81. doi: 10.1007/BF00274797
21. Rouillé Y, Westermarki G, Martin SK, Steiner DF. Proglucagon is Processed to Glucagon by Prohormone Convertase PC2 in αtc1-6 Cells. Proc Natl Acad Sci USA (1994) 91:3242–6. doi: 10.1073/pnas.91.8.3242
22. Da Silva Xavier G. The Cells of the Islets of Langerhans. J Clin Med (2018) 7:54. doi: 10.3390/jcm7030054
23. Damholt AB, Buchan AMJ, Holst JJ, Kofod H. Proglucagon Processing Profile in Canine L Cells Expressing Endogenous Prohormone Convertase 1/3 and Prohormone Convertase 2. Endocrinology (1999) 140:4800–8. doi: 10.1210/endo.140.10.7068
24. Spreckley E, Murphy KG. The L-cell in Nutritional Sensing and the Regulation of Appetite. Front Nutr (2015) 2:23. doi: 10.3389/fnut.2015.00023
25. Steiner DF, Smeekens SP, Ohagi S. Shu Jin Chan. The New Enzymology of Precursor Processing Endoproteases. J Biol Chem (1992) 267:23435–8. doi: 10.1016/S0021-9258(18)35852-6
26. Holst JJ. Enteroglucagon. Annu Rev Physiol (1997) 59:257–71. doi: 10.1146/annurev.physiol.59.1.257
27. Rouillé Y, Bianchi M, Irminger JC, Halban PA. Role of the Prohormone Convertase PC2 in the Processing of Proglucagon to Glucagon. FEBS Lett (1997) 413:119–23. doi: 10.1016/S0014-5793(97)00892-2
28. Lund A, Bagger JI, Albrechtsen NJW, Christensen M, Grøndahl M, Hartmann B, et al. Evidence of Extrapancreatic Glucagon Secretion in Man. Diabetes (2016) 65:585–97. doi: 10.2337/db15-1541
29. de Souza AH, Tang J, Yadev AK, Saghafi ST, Kibbe CR, Linnemann AK, et al. Intra-Islet GLP-1, But Not CCK, is Necessary for β-Cell Function in Mouse and Human Islets. Sci Rep (2020) 10:2823. doi: 10.1038/s41598-020-59799-2
30. Trapp S, Richards JE. The Gut Hormone Glucagon-Like Peptide-1 Produced in Brain: Is This Physiologically Relevant? Curr Opin Pharmacol (2013) 13:964–9. doi: 10.1016/j.coph.2013.09.006
31. Vrang N, Larsen PJ. Preproglucagon Derived Peptides GLP-1, GLP-2 and Oxyntomodulin in the CNS: Role of Peripherally Secreted and Centrally Produced Peptides. Prog Neurobiol (2010) 92:442–62. doi: 10.1016/j.pneurobio.2010.07.003
32. Kedia. Treatment of Severe Diabetic Hypoglycemia With Glucagon: An Underutilized Therapeutic Approach. Diabetes Metab Syndr Obes Targets Ther (2011) 4:337–46. doi: 10.2147/dmso.s20633
33. Hövelmann U, Olsen MB, Mouritzen U, Lamers D, Kronshage B, Heise T. Low Doses of Dasiglucagon Consistently Increase Plasma Glucose Levels From Hypoglycaemia and Euglycaemia in People With Type 1 Diabetes Mellitus. Diabetes Obes Metab (2019) 21:601–10. doi: 10.1111/dom.13562
34. Number CAS, Aldrich S. Compound Details (2013). Available at: https://www.novonordisk.com/partnering-and-open-innovation/compound-sharing/compound-details.31606777675869.html (Accessed March 12, 2021). 1935439.
35. Unger RH, Orci L. The Essential Role of Glucagon in the Pathogenesis of Diabetes Mellitus. Lancet (1975) 305:14–6. doi: 10.1016/S0140-6736(75)92375-2
36. Fu Z R, Gilbert E, Liu D. Regulation of Insulin Synthesis and Secretion and Pancreatic Beta-Cell Dysfunction in Diabetes. Curr Diabetes Rev (2012) 9:25–53. doi: 10.2174/15733998130104
37. Huang S, Czech MP. The GLUT4 Glucose Transporter. Cell Metab (2007) 5:237–52. doi: 10.1016/j.cmet.2007.03.006
38. Dimitriadis G, Mitron P, Lambadiari V, Maratou E, Raptis SA. Insulin Effects in Muscle and Adipose Tissue. Diabetes Res Clin Pract (2011) 93:S52–9. doi: 10.1016/S0168-8227(11)70014-6
39. Gerich JE, Lorenzi M, Hane S, Gustafson G, Guillemin R, Forsham PH. Evidence for a Physiologic Role of Pancreatic Glucagon in Human Glucose Homeostasis: Studies With Somatostatin. Metabolism (1975) 24:175–82. doi: 10.1016/0026-0495(75)90018-9
40. Quesada I, Todorova MG, Soria B. Different Metabolic Responses in α-, β-, and δ-Cells of the Islet of Langerhans Monitored by Redox Confocal Microscopy. Biophys J (2006) 90:2641–50. doi: 10.1529/biophysj.105.069906
41. Unger RH. Glucagon Physiology and Pathophysiology in the Light of New Advances. Diabetologia (1985) 28:574–8. doi: 10.1007/BF00281991
42. Müller TD, Finan B, Clemmensen C, Di Marchi RD, Tschöp MH. The New Biology and Pharmacology of Glucagon. Physiol Rev (2017) 97:721–66. doi: 10.1152/physrev.00025.2016
43. Bagger JI, Holst JJ, Hartmann B, Andersen B, Knop FK, Vilsbøll T. Effect of Oxyntomodulin, Glucagon, GLP-1, and Combined Glucagon +GLP-1 Infusion on Food Intake, Appetite, and Resting Energy Expenditure. J Clin Endocrinol Metab (2015) 100:4541–52. doi: 10.1210/jc.2015-2335
44. Nair KS. Hyperglucagonemia Increases Resting Metabolic Rate in Man During Insulin Deficiency. J Clin Endocrinol Metab (1987) 64:896–901. doi: 10.1210/jcem-64-5-896
45. Ceriello A, Genovese S, Mannucci E, Gronda E. Glucagon and Heart in Type 2 Diabetes: New Perspectives. Cardiovasc Diabetol (2016) 15:123. doi: 10.1186/s12933-016-0440-3
46. Svoboda M, Tastenoy M, Vertongen P, Robberecht P. Relative Quantitative Analysis of Glucagon Receptor mRNA in Rat Tissues. Mol Cell Endocrinol (1994) 105:131–7. doi: 10.1016/0303-7207(94)90162-7
47. Salem V, Izzi-Engbeaya C, Coello C, Thomas DB, Chambers ES, Comninos AN, et al. Glucagon Increases Energy Expenditure Independently of Brown Adipose Tissue Activation in Humans. Diabetes Obes Metab (2016) 18:72–81. doi: 10.1111/dom.12585
48. Hinke SA, Pospisilik JA, Demuth HU, Mannhart S, Kühn-Wache K, Hoffmann T, et al. Dipeptidyl Peptidase IV (DPIV/CD26) Degradation of Glucagon. Characterization of Glucagon Degradation Products and DPIV-resistant Analogs. J Biol Chem (2000) 275:3827–34. doi: 10.1074/jbc.275.6.3827
49. Haidar A. Insulin-and-Glucagon Artificial Pancreas Versus Insulin-Alone Artificial Pancreas: A Short Review. Diabetes Spectr (2019) 32:215–21. doi: 10.2337/ds18-0097
50. Reaven GM, Chen YDI, Golay A, Swislocki ALM, Jaspan JB. Documentation of Hyperglucagonemia Throughout the Day in Nonobese and Obese Patients With Noninsulin-Dependent Diabetes Mellitus. J Clin Endocrinol Metab (1987) 64:106–10. doi: 10.1210/jcem-64-1-106
51. Knop FK, Vilsbøll T, Madsbad S, Holst JJ, Krarup T. Inappropriate Suppression of Glucagon During OGTT But Not During Isoglycaemic I.V. Glucose Infusion Contributes to the Reduced Incretin Effect in Type 2 Diabetes Mellitus. Diabetologia (2007) 50:797–805. doi: 10.1007/s00125-006-0566-z
52. Cryer PE. Minireview: Glucagon in the Pathogenesis of Hypoglycemia and Hyperglycemia in Diabetes. Endocrinology (2012) 153:1039–48. doi: 10.1210/en.2011-1499
53. Muhlhauser I, Koch J, Berger M. Pharmacokinetics and Bioavailability of Injected Glucagon: Differences Between Intramuscular, Subcutaneous, and Intravenous Administration. Diabetes Care (1985) 8:39–42. doi: 10.2337/diacare.8.1.39
54. Clarke W, Jones T, Rewers A, Dunger D, Klingensmith GJ. Assessment and Management of Hypoglycemia in Children and Adolescents With Diabetes. Pediatr Diabetes (2009) 10:134–45. doi: 10.1111/j.1399-5448.2009.00583.x
55. Pearson T. Glucagon as a Treatment of Severe Hypoglycemia: Safe and Efficacious But Underutilized. Diabetes Educ (2008) 34:128–34. doi: 10.1177/0145721707312400
56. Hövelmann U, Bysted BV, Mouritzen U, Macchi F, Lamers D, Kronshage B, et al. Pharmacokinetic and Pharmacodynamic Characteristics of Dasiglucagon, a Novel Soluble and Stable Glucagon Analog. Diabetes Care (2018) 41:531–7. doi: 10.2337/dc17-1402
57. Beato-Víbora PI, Arroyo-Díez FJ. New Uses and Formulations of Glucagon for Hypoglycaemia. Drugs Context (2019) 8:212599. doi: 10.7573/dic.212599
58. Lowe RN, Trujillo JM. Intranasal Glucagon: A New Way to Treat Hypoglycemic Emergencies. Ann Pharmacother (2020) 54:780–7. doi: 10.1177/1060028020905846
59. Rosenfalck AM, Bendtson I, Jørgensen S, Binder C. Nasal Glucagon in the Treatment of Hypoglycaemia in Type 1 (Insulin-Dependent) Diabetic Patients. Diabetes Res Clin Pract (1992) 17:43–50. doi: 10.1016/0168-8227(92)90042-P
60. Orskov C, Wettergren A, Holst JJ. Biological Effects and Metabolic Rates of Glucagonlike Peptide-1 7-36 Amide and Glucagonlike Peptide-1 7-37 in Healthy Subjects are Indistinguishable. Diabetes (1993) 42:658–61. doi: 10.2337/diab.42.5.658
61. Liu HK, Green BD, Gault VA, McCluskey JT, McClenaghan NH, O’Harte FPM, et al. N-Acetyl-GLP-1: A Dpp IV-resistant Analogue of Glucagon-Like Peptide-1 (GLP-1) With Improved Effects on Pancreatic β-Cell-Associated Gene Expression. Cell Biol Int (2004) 28:69–73. doi: 10.1016/j.cellbi.2003.10.004
62. Eng J, Kleinman WA, Singh L, Singh G, Raufman JP. Isolation and Characterization of exendin-4, an Exendin-3 Analogue, From Heloderma Suspectum Venom. Further Evidence for an Exendin Receptor on Dispersed Acini From Guinea Pig Pancreas. J Biol Chem (1992) 267:7402–5. doi: 10.1016/s0021-9258(18)42531-8
63. Kolterman OG, Kim DD, Shen L, Ruggles JA, Nielsen LL, Fineman MS, et al. Pharmacokinetics, Pharmacodynamics, and Safety of Exenatide in Patients With Type 2 Diabetes Mellitus. Am J Heal Pharm (2005) 62:173–81. doi: 10.1093/ajhp/62.2.173
64. Brown E, Cuthbertson DJ, Wilding JP. Newer GLP-1 Receptor Agonists and Obesity-Diabetes. Peptides (2018) 100:61–7. doi: 10.1016/j.peptides.2017.12.009
65. Kelly AS, Auerbach P, Barrientos-Perez M, Gies I, Hale PM, Marcus C, et al. A Randomized, Controlled Trial of Liraglutide for Adolescents With Obesity. N Engl J Med (2020) 382:2117–28. doi: 10.1056/NEJMoa1916038
66. Van Can J, Sloth B, Jensen CB, Flint A, Blaak EE, Saris WHM. Effects of the Once-Daily GLP-1 Analog Liraglutide on Gastric Emptying, Glycemic Parameters, Appetite and Energy Metabolism in Obese, non-Diabetic Adults. Int J Obes (2014) 38:784–93. doi: 10.1038/ijo.2013.162
67. Trujillo JM, Nuffer W. Albiglutide: A New GLP-1 Receptor Agonist for the Treatment of Type 2 Diabetes. Ann Pharmacother (2014) 48:1494–501. doi: 10.1177/1060028014545807
68. Jendle J, Grunberger G, Blevins T, Giorgino F, Hietpas RT, Botros FT. Efficacy and Safety of Dulaglutide in the Treatment of Type 2 Diabetes: A Comprehensive Review of the Dulaglutide Clinical Data Focusing on the AWARD Phase 3 Clinical Trial Program. Diabetes Metab Res Rev (2016) 32:776–90. doi: 10.1002/dmrr.2810
69. Wilding JPH, Batterham RL, Calanna S, Davies M, Van Gaal LF, Lingvay I, et al. Once-Weekly Semaglutide in Adults With Overweight or Obesity. N Engl J Med (2021) 384:989. doi: 10.1056/NEJMoa2032183
70. Lingvay I, Catarig AM, Frias JP, Kumar H, Lausvig NL, le Roux CW, et al. Efficacy and Safety of Once-Weekly Semaglutide Versus Daily Canagliflozin as Add-on to Metformin in Patients With Type 2 Diabetes (SUSTAIN 8): A Double-Blind, Phase 3b, Randomised Controlled Trial. Lancet Diabetes Endocrinol (2019) 7:834–44. doi: 10.1016/S2213-8587(19)30311-0
71. Davies M, Pieber TR, Hartoft-Nielsen ML, Hansen OKH, Jabbour S, Rosenstock J. Effect of Oral Semaglutide Compared With Placebo and Subcutaneous Semaglutide on Glycemic Control in Patients With Type 2 Diabetes a Randomized Clinical Trial. JAMA J Am Med Assoc (2017) 318:1460–70. doi: 10.1001/jama.2017.14752
72. Husain M, Birkenfeld AL, Donsmark M, Dungan K, Eliaschewitz FG, Franco DR, et al. Oral Semaglutide and Cardiovascular Outcomes in Patients With Type 2 Diabetes. N Engl J Med (2019) 381:841–51. doi: 10.1056/NEJMoa1901118
73. Irwin N, Patterson S, De Kort M, Moffett RC, Wisse JAJ, Dokter WHA, et al. Synthesis and Evaluation of a Series of Long-Acting Glucagon-Like Peptide-1 (GLP-1) Pentasaccharide Conjugates for the Treatment of Type 2 Diabetes. Chem Med Chem (2015) 10:1424–34. doi: 10.1002/cmdc.201500140
74. Patterson S, de Kort M, Irwin N, Moffett RC, Dokter WHA, Bos ES, et al. Pharmacological Characterization and Antidiabetic Activity of a Long-Acting Glucagon-Like Peptide-1 Analogue Conjugated to an Antithrombin III-binding Pentasaccharide. Diabetes Obes Metab (2015) 17:760–70. doi: 10.1111/dom.12483
75. Schneider EL, Hearn BR, Pfaff SJ, Reid R, Parkes DG, Vrang N, et al. A Hydrogel-Microsphere Drug Delivery System That Supports Once-Monthly Administration of a GLP-1 Receptor Agonist. ACS Chem Biol (2017) 12:2107–16. doi: 10.1021/acschembio.7b00218
76. Lennox R, Porter DW, Flatt PR, Holscher C, Irwin N, Gault VA. Comparison of the Independent and Combined Effects of Sub-Chronic Therapy With Metformin and a Stable GLP-1 Receptor Agonist on Cognitive Function, Hippocampal Synaptic Plasticity and Metabolic Control in High-Fat Fed Mice. Neuropharmacology (2014) 86:22–30. doi: 10.1016/j.neuropharm.2014.06.026
77. Buhls T, Thimq L, Kofodll H, Brskovs C, Harlingll H, Holsts JJ. Naturally Occurring Products of Proglucagon 11 1-160 in the Porcine and Human Small Intestine. J Biol Chem (1988) 263:8621–4. doi: 10.1016/S0021-9258(18)68350-4
78. Reimann F, Gribble FM. Glucose-Sensing in Glucagon-Like peptide-1-secreting Cells. Diabetes (2002) 51:2757–63. doi: 10.2337/diabetes.51.9.2757
79. Reimann F, Habib AM, Tolhurst G, Parker HE, Rogers GJ, Gribble FM. Glucose Sensing in L Cells: A Primary Cell Study. Cell Metab (2008) 8:532–9. doi: 10.1016/j.cmet.2008.11.002
80. Tolhurst G, Reimann F, Gribble FM. Nutritional Regulation of Glucagon-Like Peptide-1 Secretion. J Physiol (2009) 587:27–32. doi: 10.1113/jphysiol.2008.164012
81. Baggio LL, Drucker DJ. Biology of Incretins: GLP-1 and GIP. Gastroenterology (2007) 132:2131–57. doi: 10.1053/j.gastro.2007.03.054
82. Unger RH, Ohneda A, Valverde I, Eisentraut AM, Exton J. Characterization of the Responses of Circulating Glucagon-Like Immunoreactivity to Intraduodenal and Intravenous Administration of Glucose. J Clin Invest (1968) 47:48–65. doi: 10.1172/JCI105714
83. Greenfield JR, Farooqi IS, Keogh JM, Henning E, Habib AM, Blackwood A, et al. Oral Glutamine Increases Circulating Glucagon-Like Peptide 1, Glucagon, and Insulin Concentrations in Lean, Obese, and Type 2 Diabetic Subjects. Am J Clin Nutr (2009) 89:106–13. doi: 10.3945/ajcn.2008.26362
84. Parker HE, Adriaenssens A, Rogers G, Richards P, Koepsell H, Reimann F, et al. Predominant Role of Active Versus Facilitative Glucose Transport for Glucagon-Like Peptide-1 Secretion. Diabetologia (2012) 55:2445–55. doi: 10.1007/s00125-012-2585-2
85. Krieger JP, Langhans W, Lee SJ. Vagal Mediation of GLP-1’s Effects on Food Intake and Glycemia. Physiol Behav (2015) 152:372–80. doi: 10.1016/j.physbeh.2015.06.001
86. Hansen L, Deacon CF, Ørskov C, Holst JJ. Glucagon-Like peptide-1-(7-36)amide is Transformed to Glucagon-Like peptide-1-(9-36)amide by Dipeptidyl Peptidase IV in the Capillaries Supplying the L Cells of the Porcine Intestine. Endocrinology (1999) 140:5356–63. doi: 10.1210/endo.140.11.7143
87. Holst JJ. On the Physiology of GIP and GLP-1. Horm Metab Res (2004) 36:747–54. doi: 10.1055/s-2004-826158
88. Knudsen LB, Pridal L. Glucagon-Like peptide-1-(9-36) Amide is a Major Metabolite of Glucagon-Like peptide-1-(7-36) Amide After In Vivo Administration to Dogs, and it Acts as an Antagonist on the Pancreatic Receptor. Eur J Pharmacol (1996) 318:429–35. doi: 10.1016/S0014-2999(96)00795-9
89. Robinson E, Tate M, Lockhart S, McPeake C, O’Neill KM, Edgar KS, et al. Metabolically-Inactive Glucagon-Like peptide-1(9-36)amide Confers Selective Protective Actions Against Post-Myocardial Infarction Remodelling. Cardiovasc Diabetol (2016) 15:65. doi: 10.1186/s12933-016-0386-5
90. Nikolaidis LA, Elahi D, Shen YT, Shannon RP. Active Metabolite of GLP-1 Mediates Myocardial Glucose Uptake and Improves Left Ventricular Performance in Conscious Dogs With Dilated Cardiomyopathy. Am J Physiol Hear Circ Physiol (2005) 289:58–66. doi: 10.1152/ajpheart.00347.2005
91. Guida C, Miranda C, Asterholm IW, Basco D, Benrick A, Chanclon B, et al. Promiscuous Receptor Activation Mediates Glucagonostatic Effects of GLP-1(9-36) and GLP-1(7-36). bioRxiv (2019) 785667. doi: 10.1101/785667. PPR94312.
92. Meier JJ, Gethmann A, Nauck MA, Götze O, Schmitz F, Deacon CF, et al. The Glucagon-Like Peptide-1 Metabolite GLP-1-(9–36) Amide Reduces Postprandial Glycemia Independently of Gastric Emptying and Insulin Secretion in Humans. Am J Physiol Metab (2006) 290:E1118–23. doi: 10.1152/ajpendo.00576.2005
93. Cordomí A, Fourmy D, Tikhonova IG. Gut Hormone GPCRs: Structure, Function, Drug Discovery. Curr Opin Pharmacol (2016) 31:63–7. doi: 10.1016/j.coph.2016.09.001
94. Doyle ME, Egan JM. Mechanisms of Action of Glucagon-Like Peptide 1 in the Pancreas. Pharmacol Ther (2007) 113:546–93. doi: 10.1016/j.pharmthera.2006.11.007
95. Irwin N, Flatt PR. Enteroendocrine Hormone Mimetics for the Treatment of Obesity and Diabetes. Curr Opin Pharmacol (2013) 13:989–95. doi: 10.1016/j.coph.2013.09.009
96. Kreymann B, Ghatei MA, Williams G, Bloom SR. Glucagon-Like Peptide-1 7-36: A Physiological Incretin in Man. Lancet (1987) 330:1300–4. doi: 10.1016/S0140-6736(87)91194-9
97. Schirra J, Nicolaus M, Roggel R, Katschinski M, Storr M, Woerle HJ, et al. Endogenous Glucagon-Like Peptide 1 Controls Endocrine Pancreatic Secretion and Antro-Pyloro-Duodenal Motility in Humans. Gut (2006) 55:243–51. doi: 10.1136/gut.2004.059741
98. Unger RH, Orci L. Paracrinology of Islets and the Paracrinopathy of Diabetes. Proc Natl Acad Sci USA (2010) 107:16009–12. doi: 10.1073/pnas.1006639107
99. Ørgaard A, Holst JJ. The Role of Somatostatin in GLP-1-induced Inhibition of Glucagon Secretion in Mice. Diabetologia (2017) 60:1731–9. doi: 10.1007/s00125-017-4315-2
100. De Marinis YZ, Salehi A, Ward CE, Zhang Q, Abdulkader F, Bengtsson M, et al. GLP-1 Inhibits and Adrenaline Stimulates Glucagon Release by Differential Modulation of N- and L-type Ca2+ Channel-Dependent Exocytosis. Cell Metab (2010) 11:543–53. doi: 10.1016/j.cmet.2010.04.007
101. Richards P, Parker HE, Adriaenssens AE, Hodgson JM, Cork SC, Trapp S, et al. Identification and Characterization of GLP-1 Receptor-Expressing Cells Using a New Transgenic Mouse Model. Diabetes (2014) 63:1224–33. doi: 10.2337/db13-1440
102. Kornelius E, Li HH, Peng CH, Yang YS, Chen WJ, Chang YZ, et al. Liraglutide Protects Against Glucolipotoxicity-Induced RIN-m5F β-Cell Apoptosis Through Restoration of PDX1 Expression. J Cell Mol Med (2019) 23:619–29. doi: 10.1111/jcmm.13967
103. MacDonald PE, El-kholy W, Riedel MJ, Salapatek AMF, Light PE, Wheeler MB. The Multiple Actions of GLP-1 on the Process of Glucose-Stimulated Insulin Secretion. Diabetes (2002) 51:S434–42. doi: 10.2337/diabetes.51.2007.s434
104. Li Y, Cao X, Li LX, Brubaker PL, Edlund H, Drucker DJ. β-Cell Pdx1 Expression is Essential for the Glucoregulatory, Proliferative, and Cytoprotective Actions of Glucagon-Like Peptide-1. Diabetes (2005) 54:482–91. doi: 10.2337/diabetes.54.2.482
105. Moffett RC, Patterson S, Irwin N, Flatt PR. Positive Effects of GLP-1 Receptor Activation With Liraglutide on Pancreatic Islet Morphology and Metabolic Control in C57BL/KsJ Db/Db Mice With Degenerative Diabetes. Diabetes Metab Res Rev (2015) 31:248–55. doi: 10.1002/dmrr.2608
106. Vasu S, Moffett RC, Thorens B, Flatt PR. Role of Endogenous GLP-1 and GIP in Beta Cell Compensatory Responses to Insulin Resistance and Cellular Stress. PloS One (2014) 9:e101005. doi: 10.1371/journal.pone.0101005
107. Li Y, Hansotia T, Yusta B, Ris F, Halban PA, Drueker DJ. Glucagon-Like Peptide-1 Receptor Signaling Modulates β Cell Apoptosis. J Biol Chem (2003) 278:471–8. doi: 10.1074/jbc.M209423200
108. Li Y, Cao X, Li LX, Brubaker PL, Edlund H, Drucker DJ. β-Cell Pdx1 Expression is Essential for the Glucoregulatory, Proliferative, and Cytoprotective Actions of Glucagon-Like Peptide-1. Diabetes (2005) 54:482–91. doi: 10.2337/diabetes.54.2.482
109. Hui H, Wright C, Perfetti R. Glucagon-Like Peptide 1 Induces Differentiation of Islet Duodenal homeobox-1-positive Pancreatic Ductal Cells Into Insulin-Secreting Cells. Diabetes (2001) 50:785–96. doi: 10.2337/diabetes.50.4.785
110. Thorens B, Tarussio D, Maestro MA, Rovira M, Heikkilä E, Ferrer J. Ins1 Cre Knock-in Mice for Beta Cell-Specific Gene Recombination. Diabetologia (2015) 58:558–65. doi: 10.1007/s00125-014-3468-5
111. Campbell JR, Martchenko A, Sweeney ME, Maalouf MF, Psichas A, Gribble FM, et al. Essential Role of Syntaxin-Binding Protein-1 in the Regulation of Glucagon-Like Peptide-1 Secretion. Endocrinology (2020) 161:1–13. doi: 10.1210/endocr/bqaa039
112. Tanday N, Flatt PR, Irwin N, Charlotte Moffett R. Liraglutide and Sitagliptin Counter Beta- to Alpha-Cell Transdifferentiation in Diabetes. J Endocrinol (2020) 245:53–64. doi: 10.1530/JOE-19-0451
113. Thorel F, Népote V, Avril I, Kohno K, Desgraz R, Chera S, et al. Conversion of Adult Pancreatic α-Cells to β-Cells After Extreme β-Cell Loss. Nature (2010) 464:1149–54. doi: 10.1038/nature08894
114. Lee YS, Lee C, Choung JS, Jung HS, Jun HS. Glucagon-Like Peptide 1 Increases β-Cell Regeneration by Promoting α- to β-Cell Transdifferentiation. Diabetes (2018) 67:2601–14. doi: 10.2337/db18-0155
115. Sarnobat D, Moffett CR, Tanday N, Reimann F, Gribble FM, Flatt PR, et al. Antidiabetic Drug Therapy Alleviates Type 1 Diabetes in Mice by Promoting Pancreatic α-Cell Transdifferentiation. Biochem Pharmacol (2020) 182:114216. doi: 10.1016/j.bcp.2020.114216
116. Trapp S, Richards JE. The Gut Hormone Glucagon-Like Peptide-1 Produced in Brain: Is This Physiologically Relevant? Curr Opin Pharmacol (2013) 13:964–9. doi: 10.1016/j.coph.2013.09.006
117. Dailey MJ, Moran TH. Glucagon-Like Peptide 1 and Appetite. Trends Endocrinol Metab (2013) 24:85–91. doi: 10.1016/j.tem.2012.11.008
118. Sheahan KH, Wahlberg EA, Gilbert MP. An Overview of GLP-1 Agonists and Recent Cardiovascular Outcomes Trials. Postgrad Med J (2020) 96:156–61. doi: 10.1136/postgradmedj-2019-137186
119. Gulsin GS, Graham-Brown MPM, Davies MJ, McCann GP. Emerging Glucose-Lowering Therapies: A Guide for Cardiologists. Heart (2020) 106:18–23. doi: 10.1136/heartjnl-2019-315758
120. Mabilleau G, Pereira M, Chenu C. Novel Skeletal Effects of Glucagon-Like Peptide-1 (GLP-1) Receptor Agonists. J Endocrinol (2018) 236:R29–42. doi: 10.1530/JOE-17-0278
121. Gault VA, Hölscher C. GLP-1 Receptor Agonists Show Neuroprotective Effects in Animal Models of Diabetes. Peptides (2018) 100:101–7. doi: 10.1016/j.peptides.2017.11.017
122. Grupta NA, Mells J, Dunham RM, Grakoui A, Handy J, Saxena NK, et al. Glucagon-Like Peptide-1 Receptor is Present on Human Hepatocytes and has a Direct Role in Decreasing Hepatic Steatosis In Vitro by Modulating Elements of the Insulin Signaling Pathway. Hepatology (2010) 51:1584–92. doi: 10.1002/hep.23569
123. Armstrong MJ, Gaunt P, Aithal GP, Barton D, Hull D, Parker R, et al. Liraglutide Safety and Efficacy in Patients With non-Alcoholic Steatohepatitis (LEAN): A Multicentre, Double-Blind, Randomised, Placebo-Controlled Phase 2 Study. Lancet (2016) 387:679–90. doi: 10.1016/S0140-6736(15)00803-X
124. Newsome PN, Buchholtz K, Cusi K, Linder M, Okanoue T, Ratziu V, et al. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N Engl J Med (2021) 384:1113–24. doi: 10.1056/nejmoa2028395
125. Wang XC, Gusdon AM, Liu H, Qu S. Effects of Glucagon-Like Peptide-1 Receptor Agonists on non-Alcoholic Fatty Liver Disease and Inflammation. World J Gastroenterol (2014) 20:14821–30. doi: 10.3748/wjg.v20.i40.14821
126. Brown JC, Pederson RA, Jorpes E, Mutt V. Preparation of Highly Active Enterogastrone. Can J Physiol Pharmacol (1969) 47:113–4. doi: 10.1139/y69-020
127. McClean PL, Irwin N, Cassidy RS, Holst JJ, Gault VA, Flatt PR. GIP Receptor Antagonism Reverses Obesity, Insulin Resistance, and Associated Metabolic Disturbances Induced in Mice by Prolonged Consumption of High-Fat Diet. Am J Physiol Endocrinol Metab (2007) 293:1746–55. doi: 10.1152/ajpendo.00460.2007
128. Egan JM, Meneilly GS, Habener JF, Elahi D. Glucagon-Like Peptide-1 Augments Insulin-Mediated Glucose Uptake in the Obese State. J Clin Endocrinol Metab (2002) 87:3768–73. doi: 10.1210/jcem.87.8.8743
129. Green BD, Gault VA, Flatt PR, Harriott P, Greer B, O’Harte FPM. Comparative Effects of GLP-1 and GIP on cAMP Production, Insulin Secretion, and In Vivo Antidiabetic Actions Following Substitution of Ala 8/Ala2 With 2-Aminobutyric Acid. Arch Biochem Biophys (2004) 428:136–43. doi: 10.1016/j.abb.2004.05.005
130. Irwin N, Flatt PR. New Perspectives on Exploitation of Incretin Peptides for the Treatment of Diabetes and Related Disorders. World J Diabetes (2015) 6:1285–95. doi: 10.4239/wjd.v6.i15.1285
131. Zander M, Madsbad S, Madsen JL, Holst JJ. Effect of 6-Week Course of Glucagon-Like Peptide 1 on Glycaemic Control, Insulin Sensitivity, and β-Cell Function in Type 2 Diabetes: A Parallel-Group Study. Lancet (2002) 359:824–30. doi: 10.1016/S0140-6736(02)07952-7
132. Deacon CF, Johnsen AH, Holst JJ. Degradation of Glucagon-Like Peptide-1 by Human Plasma In Vitro Yields an N-terminally Truncated Peptide That is a Major Endogenous Metabolite In Vivo. J Clin Endocrinol Metab (1995) 80:952–7. doi: 10.1210/jcem.80.3.7883856
133. Edwards CMB, Stanley SA, Davis R, Brynes AE, Frost GS, Seal LJ, et al. Exendin-4 Reduces Fasting and Postprandial Glucose and Decreases Energy Intake in Healthy Volunteers. Am J Physiol Endocrinol Metab (2001) 281:155–61. doi: 10.1152/ajpendo.2001.281.1.e155
134. Simonsen L, Holst JJ, Deacon CF. Exendin-4, But Not Glucagon-Like peptide-1, is Cleared Exclusively by Glomerular Filtration in Anaesthetised Pigs. Diabetologia (2006) 49:706–12. doi: 10.1007/s00125-005-0128-9
135. Cvetković RS, Plosker GL. Exenatide: A Review of its Use in Patients With Type 2 Diabetes Mellitus (as an Adjunct to Metformin and/or a Sulfonylurea). Drugs (2007) 67:935–54. doi: 10.2165/00003495-200767060-00008
136. Knudsen LB, Kiel D, Teng M, Behrens C, Bhumralkar D, Kodra JT, et al. Small-Molecule Agonists for the Glucagon-Like Peptide 1 Receptor. Proc Natl Acad Sci USA (2007) 104:937–42. doi: 10.1073/pnas.0605701104
137. Thompson A, Stephens JW, Bain SC, Kanamarlapudi V. Molecular Characterisation of Small Molecule Agonists Effect on the Human Glucagon Like Peptide-1 Receptor Internalisation. PloS One (2016) 11:e0154229. doi: 10.1371/journal.pone.0154229
138. Wang YC, McPherson K, Marsh T, Gortmaker SL, Brown M. Health and Economic Burden of the Projected Obesity Trends in the USA and the UK. Lancet (2011) 378:815–25. doi: 10.1016/S0140-6736(11)60814-3
139. Maciejewski ML, Arterburn DE, Van Scoyoc L, Smith VA, Yancy WS, Weidenbacher HJ, et al. Bariatric Surgery and Long-Term Durability of Weight Loss. JAMA Surg (2016) 151:1046–55. doi: 10.1001/jamasurg.2016.2317
140. Davies MJ, Bergenstal R, Bode B, Kushner RF, Lewin A, Skjøth TV, et al. Efficacy of Liraglutide for Weight Loss Among Patients With Type 2 Diabetes: The SCALE Diabetes Randomized Clinical Trial. JAMA J Am Med Assoc (2015) 314:687–99. doi: 10.1001/jama.2015.9676
141. Pi-Sunyer X, Astrup A, Fujioka K, Greenway F, Halpern A, Krempf M, et al. A Randomized, Controlled Trial of 3.0 Mg of Liraglutide in Weight Management. N Engl J Med (2015) 373:11–22. doi: 10.1056/NEJMoa1411892
142. Le Roux CW, Astrup AV, Fujioka K, Greenway F, Lau DCW, Van Gaal L, et al. 3 Years of Liraglutide Versus Placebo for Type 2 Diabetes Risk Reduction and Weight Management in Individuals With Prediabetes: A Randomised, Double-Blind Trial. Lancet (2017) 389:1399–409. doi: 10.1016/S0140-6736(17)30069-7
143. Nauck MA, Petrie JR, Sesti G, Mannucci E, Courrèges JP, Lindegaard ML, et al. A Phase 2, Randomized, Dose-Finding Study of the Novel Once-Weekly Human GLP-1 Analog, Semaglutide, Compared With Placebo and Open-Label Liraglutide in Patients With Type 2 Diabetes. Diabetes Care (2016) 39:231–41. doi: 10.2337/dc15-0165
144. Frías JP, Guja C, Hardy E, Ahmed A, Dong F, Öhman P, et al. Exenatide Once Weekly Plus Dapagliflozin Once Daily Versus Exenatide or Dapagliflozin Alone in Patients With Type 2 Diabetes Inadequately Controlled With Metformin Monotherapy (DURATION-8): A 28 Week, Multicentre, Double-Blind, Phase 3, Randomised Controlled Trial. Lancet Diabetes Endocrinol (2016) 4:1004–16. doi: 10.1016/S2213-8587(16)30267-4
145. Jabbour SA, Frías JP, Ahmed A, Hardy E, Choi J, Sjöström CD, et al. Efficacy and Safety Over 2 Years of Exenatide Plus Dapagliflozin in the DURATION-8 Study: A Multicenter, Double-Blind, Phase 3, Randomized Controlled Trial. Diabetes Care (2020) 43:2528–36. doi: 10.2337/dc19-1350
146. Rajeev SP, Sprung VS, Roberts C, Harrold JA, Halford JCG, Stancak A, et al. Compensatory Changes in Energy Balance During Dapagliflozin Treatment in Type 2 Diabetes Mellitus: A Randomised Double-Blind, Placebo-Controlled, Cross-Over Trial (ENERGIZE) - Study Protocol. BMJ Open (2017) 7:13539. doi: 10.1136/bmjopen-2016-013539
147. Matheus ASDM, Tannus LRM, Cobas RA, Palma CCS, Negrato CA, Gomes MDB. Impact of Diabetes on Cardiovascular Disease: An Update. Int J Hypertens (2013) 2013:653789. doi: 10.1155/2013/653789
148. Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N Engl J Med (2019) 380:347–57. doi: 10.1056/NEJMoa1812389
149. Williams DM, Evans M. Semaglutide: Charting New Horizons in GLP-1 Analogue Outcome Studies. Diabetes Ther (2020) 11:2221–35. doi: 10.1007/s13300-020-00917-8
150. Stewart R, Liolitsa D. Type 2 Diabetes Mellitus, Cognitive Impairment and Dementia. Diabetes Med (1999) 16:93–112. doi: 10.1046/j.1464-5491.1999.00027.x
151. Janson J, Laedtke T, Parisi JE, O’Brien P, Petersen RC, Butler PC. Increased Risk of Type 2 Diabetes in Alzheimer Disease. Diabetes (2004) 53:474–81. doi: 10.2337/diabetes.53.2.474
152. Gispen WH, Biessels GJ. Cognition and Synaptic Plasticity in Diabetes Mellitus. Trends Neurosci (2000) 23:542–9. doi: 10.1016/S0166-2236(00)01656-8
153. Yang Y, Zhang J, Ma D, Zhang M, Hu S, Shao S, et al. Subcutaneous Administration of Liraglutide Ameliorates Alzheimer-Associated Tau Hyperphosphorylation in Rats With Type 2 Diabetes. J Alzheimer’s Dis (2013) 37:637–48. doi: 10.3233/JAD-130491
154. Hölscher C. Insulin, Incretins and Other Growth Factors as Potential Novel Treatments for Alzheimer’s and Parkinson’s Diseases. Biochem Soc Trans (2014) 42:593–9. doi: 10.1042/BST20140016
155. Long-Smith CM, Manning S, McClean PL, Coakley MF, O’Halloran DJ, Holscher C, et al. The Diabetes Drug Liraglutide Ameliorates Aberrant Insulin Receptor Localisation and Signalling in Parallel With Decreasing Both Amyloid-β Plaque and Glial Pathology in a Mouse Model of Alzheimer’s Disease. Neuro Mol Med (2013) 15:102–14. doi: 10.1007/s12017-012-8199-5
156. Bomfim TR, Forny-Germano L, Sathler LB, Brito-Moreira J, Houzel JC, Decker H, et al. An Anti-Diabetes Agent Protects the Mouse Brain From Defective Insulin Signaling Caused by Alzheimer’s Disease-Associated Aβ Oligomers. J Clin Invest (2012) 122:1339–53. doi: 10.1172/JCI57256
157. Gault VA, Porter WD, Flatt PR, Hölscher C. Actions of Exendin-4 Therapy on Cognitive Function and Hippocampal Synaptic Plasticity in Mice Fed a High-Fat Diet. Int J Obes (2010) 34:1341–4. doi: 10.1038/ijo.2010.59
158. Porter DW, Kerr BD, Flatt PR, Holscher C, Gault VA. Four Weeks Administration of Liraglutide Improves Memory and Learning as Well as Glycaemic Control in Mice With High Fat Dietary-Induced Obesity and Insulin Resistance. Diabetes Obes Metab (2010) 12:891–9. doi: 10.1111/j.1463-1326.2010.01259.x
159. Gault VA, Lennox R, Flatt PR. Sitagliptin, a Dipeptidyl Peptidase-4 Inhibitor, Improves Recognition Memory, Oxidative Stress and Hippocampal Neurogenesis and Upregulates Key Genes Involved in Cognitive Decline. Diabetes Obes Metab (2015) 17:403–13. doi: 10.1111/dom.12432
160. Millar P, Pathak N, Parthsarathy V, Bjourson AJ, O’Kane M, Pathak V, et al. Metabolic and Neuroprotective Effects of Dapagliflozin and Liraglutide in Diabetic Mice. J Endocrinol (2017) 234:255–67. doi: 10.1530/JOE-17-0263
161. Gejl M, Gjedde A, Egefjord L, Møller A, Hansen SB, Vang K, et al. In Alzheimer’s Disease, 6-Month Treatment With GLP-1 Analog Prevents Decline of Brain Glucose Metabolism: Randomized, Placebo-Controlled, Double-Blind Clinical Trial. Front Aging Neurosci (2016) 8:108. doi: 10.3389/fnagi.2016.00108
162. Athauda D, Maclagan K, Skene SS, Bajwa-Joseph M, Letchford D, Chowdhury K, et al. Exenatide Once Weekly Versus Placebo in Parkinson’s Disease: A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet (2017) 390:1664–75. doi: 10.1016/S0140-6736(17)31585-4
163. Hölscher C. Synaptic Plasticity and Learning and Memory: LTP and Beyond. J Neurosci Res (1999) 58:62–75. doi: 10.1002/(SICI)1097-4547(19991001)58:1<62::AID-JNR7>3.0.CO;2-G177
164. Porter WD, Flatt PR, Hölscher C, Gault VA. Liraglutide Improves Hippocampal Synaptic Plasticity Associated With Increased Expression of Mash1 in Ob/Ob Mice. Int J Obes (2013) 37:678–84. doi: 10.1038/ijo.2012.91
165. Moloney AM, Griffin RJ, Timmons S, O’Connor R, Ravid R, O’Neill C. Defects in IGF-1 Receptor, Insulin Receptor and IRS-1/2 in Alzheimer’s Disease Indicate Possible Resistance to IGF-1 and Insulin Signalling. Neurobiol Aging (2010) 31:224–43. doi: 10.1016/j.neurobiolaging.2008.04.002
166. Park HR, Park M, Choi J, Park KY, Chung HY, Lee J. A High-Fat Diet Impairs Neurogenesis: Involvement of Lipid Peroxidation and Brain-Derived Neurotrophic Factor. Neurosci Lett (2010) 482:235–9. doi: 10.1016/j.neulet.2010.07.046
167. Athauda D, Foltynie T. Protective Effects of the GLP-1 Mimetic Exendin-4 in Parkinson’s Disease. Neuropharmacology (2018) 136:260–70. doi: 10.1016/j.neuropharm.2017.09.023
168. Aviles-Olmos I, Dickson J, Kefalopoulou Z, Djamshidian A, Ell P, Soderlund T, et al. Exenatide and the Treatment of Patients With Parkinson’s Disease. J Clin Invest (2013) 123:2730–6. doi: 10.1172/JCI68295
169. Athauda D, Maclagan K, Skene SS, Bajwa-Joseph M, Letchford D, Chowdhury K, et al. Exenatide Once Weekly Versus Placebo in Parkinson’s Disease: A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet (2017) 390:1664–75. doi: 10.1016/S0140-6736(17)31585-4
170. Aviles-Olmos I, Dickson J, Kefalopoulou Z, Djamshidian A, Kahan J, Ell P, et al. Motor and Cognitive Advantages Persist 12 Months After Exenatide Exposure in Parkinson’s Disease. J Parkinsons Dis (2015) 4:337–44. doi: 10.3233/JPD-140364
171. Femminella GD, Frangou E, Love SB, Busza G, Holmes C, Ritchie C, et al. Evaluating the Effects of the Novel GLP-1 Analogue Liraglutide in Alzheimer’s Disease: Study Protocol for a Randomised Controlled Trial (ELAD Study). Trials (2019) 20:191. doi: 10.1186/s13063-019-3259-x
172. Mulvaney CA, Duarte GS, Handley J, Evans DJW, Menon S, Wyse R, et al. GLP-1 Receptor Agonists for Parkinson’s Disease. Cochrane Database Syst Rev (2020) CD012990. doi: 10.1002/14651858.CD012990.pub2
173. Vestergaard P, Rejnmark L, Mosekilde L. Relative Fracture Risk in Patients With Diabetes Mellitus, and the Impact of Insulin and Oral Antidiabetic Medication on Relative Fracture Risk. Diabetologia (2005) 48:1292–9. doi: 10.1007/s00125-005-1786-3
174. Mieczkowska A, Mansur A, Irwin N, Flatt PR, Chappard D, Mabilleau G. Alteration of the Bone Tissue Material Properties in Type 1 Diabetes Mellitus: A Fourier Transform Infrared Microspectroscopy Study. Bone (2015) 76:31–9. doi: 10.1016/j.bone.2015.03.010
175. Mansur SA, Mieczkowska A, Bouvard B, Flatt PR, Chappard D, Irwin N, et al. Stable Incretin Mimetics Counter Rapid Deterioration of Bone Quality in Type 1 Diabetes Mellitus. J Cell Physiol (2015) 230:3009–18. doi: 10.1002/jcp.25033
176. Mabilleau G, Perrot R, Flatt PR, Irwin N, Chappard D. High Fat-Fed Diabetic Mice Present With Profound Alterations of the Osteocyte Network. Bone (2016) 90:99–106. doi: 10.1016/j.bone.2016.06.008
177. Yamada C, Yamada Y, Tsukiyama K, Yamada K, Udagawa N, Takahashi N, et al. The Murine Glucagon-Like Peptide-1 Receptor is Essential for Control of Bone Resorption. Endocrinology (2008) 149:574–9. doi: 10.1210/en.2007-1292
178. Mabilleau G, Mieczkowska A, Irwin N, Flatt PR, Chappard D. Optimal Bone Mechanical and Material Properties Require a Functional Glucagon-Like Peptide-1 Receptor. J Endocrinol (2013) 219:59–68. doi: 10.1530/JOE-13-0146
179. Mansur SA, Mieczkowska A, Flatt PR, Chappard D, Irwin N, Mabilleau G. Sitagliptin Alters Bone Composition in High-Fat-Fed Mice. Calcif Tissue Int (2019) 104:437–48. doi: 10.1007/s00223-018-0507-0
180. Pereira M, Gohin S, Roux JP, Fisher A, Cleasby ME, Mabilleau G, et al. Exenatide Improves Bone Quality in a Murine Model of Genetically Inherited Type 2 Diabetes Mellitus. Front Endocrinol (2017) 8:327. doi: 10.3389/fendo.2017.00327
181. Mansur SA, Mieczkowska A, Flatt PR, Chappard D, Irwin N, Mabilleau G. The GLP-1 Receptor Agonist Exenatide Ameliorates Bone Composition and Tissue Material Properties in High Fat Fed Diabetic Mice. Front Endocrinol (2019) 10:51. doi: 10.3389/fendo.2019.00051
182. Mieczkowska A, Irwin N, Flatt PR, Chappard D, Mabilleau G. Glucose-Dependent Insulinotropic Polypeptide (GIP) Receptor Deletion Leads to Reduced Bone Strength and Quality. Bone (2013) 56:337–42. doi: 10.1016/j.bone.2013.07.003
183. Mieczkowska A, Mansur S, Bouvard B, Flatt PR, Thorens B, Irwin N, et al. Double Incretin Receptor Knock-Out (DIRKO) Mice Present With Alterations of Trabecular and Cortical Micromorphology and Bone Strength. Osteoporos Int (2014) 26:209–18. doi: 10.1007/s00198-014-2845-8
184. Mansur SA, Mieczkowska A, Flatt PR, Bouvard B, Chappard D, Irwin N, et al. A New Stable GIP-Oxyntomodulin Hybrid Peptide Improved Bone Strength Both at the Organ and Tissue Levels in Genetically-Inherited Type 2 Diabetes Mellitus. Bone (2016) 87:102–13. doi: 10.1016/j.bone.2016.04.001
185. Moffett RC, Naughton V. Emerging Role of GIP and Related Gut Hormones in Fertility and PCOS. Peptides (2020) 125:170233. doi: 10.1016/j.peptides.2019.170233
186. March WA, Moore VM, Willson KJ, Phillips DIW, Norman RJ, Davies MJ. The Prevalence of Polycystic Ovary Syndrome in a Community Sample Assessed Under Contrasting Diagnostic Criteria. Hum Reprod (2010) 25:544–51. doi: 10.1093/humrep/dep399
187. Rojas J, Chávez M, Olivar L, Rojas M, Morillo J, Mejías J, et al. Polycystic Ovary Syndrome, Insulin Resistance, and Obesity: Navigating the Pathophysiologic Labyrinth. Int J Reprod Med (2014) 2014:1–17. doi: 10.1155/2014/719050
188. Schauer PR, Burguera B, Ikramuddin S, Cottam D, Gourash W, Hamad G, et al. Effect of Laparoscopic Roux-En Y Gastric Bypass on Type 2 Diabetes Mellitus. Ann Surg (2003) 4:467–85. doi: 10.1097/01.sla.0000089851.41115.1b
189. Jamal M, Gunay Y, Capper A, Eid A, Heitshusen D, Samuel I. Roux-En-Y Gastric Bypass Ameliorates Polycystic Ovary Syndrome and Dramatically Improves Conception Rates: A 9-Year Analysis. Surg Obes Relat Dis (2012) 8:440–4. doi: 10.1016/j.soard.2011.09.022
190. Abdalla MA, Deshmukh H, Atkin S, Sathyapalan T. The Potential Role of Incretin-Based Therapies for Polycystic Ovary Syndrome: A Narrative Review of the Current Evidence. Ther Adv Endocrinol Metab (2021) 12:204201882198923. doi: 10.1177/2042018821989238
191. Nylander M, Frøssing S, Clausen HV, Kistorp C, Faber J, Skouby SO. Effects of Liraglutide on Ovarian Dysfunction in Polycystic Ovary Syndrome: A Randomized Clinical Trial. Reprod BioMed Online (2017) 35:121–7. doi: 10.1016/j.rbmo.2017.03.023
192. Salamun V, Jensterle M, Janez A, Bokal EV. Liraglutide Increases IVF Pregnancy Rates in Obese PCOS Women With Poor Response to First-Line Reproductive Treatments: A Pilot Randomized Study. Eur J Endocrinol (2018) 179:1–11. doi: 10.1530/EJE-18-0175
193. Han Y, Li Y, He B. GLP-1 Receptor Agonists Versus Metformin in PCOS: A Systematic Review and Meta-Analysis. Reprod BioMed Online (2019) 39:332–42. doi: 10.1016/j.rbmo.2019.04.017
194. Knudsen LB, Nielsen PF, Huusfeldt PO, Johansen NL, Madsen K, Pedersen FZ, et al. Potent Derivatives of Glucagon-Like Peptide-1 With Pharmacokinetic Properties Suitable for Once Daily Administration. J Med Chem (2000) 43:1664–9. doi: 10.1021/jm9909645
195. Vilsbøll T, Zdravkovic M, Le-Thi T, Krarup T, Schmitz O, Courrèges JP, et al. Liraglutide, a Long-Acting Human Glucagon-Like Peptide-1 Analog, Given as Monotherapy Significantly Improves Glycemic Control and Lowers Body Weight Without Risk of Hypoglycemia in Patients With Type 2 Diabetes. Diabetes Care (2007) 30:1608–10. doi: 10.2337/dc06-2593
196. Cai Y, Wei L, Ma L, Huang X, Tao A, Liu Z, et al. Long-Acting Preparations of Exenatide. Drug Des Devel Ther (2013) 7:963–70. doi: 10.2147/DDDT.S46970
197. Rosenstock J, Buse JB, Azeem R, Prabhakar P, Kjems L, Huang H, et al. Efficacy and Safety of ITCA 650, a Novel Drug-Device GLP-1 Receptor Agonist, in Type 2 Diabetes Uncontrolled With Oral Antidiabetes Drugs: The FREEDOM-1 Trial. Diabetes Care (2018) 41:333–40. doi: 10.2337/dc17-1306
198. Drucker DJ. Advances in Oral Peptide Therapeutics. Nat Rev Drug Discovery (2020) 19:277–89. doi: 10.1038/s41573-019-0053-0
199. Graham GV, McLaughlin CM, Flatt PR. Role of Exendin-4 in the Gila Monster: Further Lessons Regarding Human Oral Glucagon-Like Peptide-1 Therapy? Diabetes Obes Metab (2020) 22:2509–11. doi: 10.1111/dom.14171
200. Pieber TR, Bode B, Mertens A, Cho YM, Christiansen E, Hertz CL, et al. Efficacy and Safety of Oral Semaglutide With Flexible Dose Adjustment Versus Sitagliptin in Type 2 Diabetes (PIONEER 7): A Multicentre, Open-Label, Randomised, Phase 3a Trial. Lancet Diabetes Endocrinol (2019) 7:528–39. doi: 10.1016/S2213-8587(19)30194-9
201. Anderson SL, Beutel TR, Trujillo JM. Oral Semaglutide in Type 2 Diabetes. J Diabetes Complications (2020) 34:107520. doi: 10.1016/j.jdiacomp.2019.107520
202. Rodbard HW, Dougherty T, Taddei-Allen P. Efficacy of Oral Semaglutide: Overview of the Pioneer Clinical Trial Program and Implications for Managed Care. Am J Manag Care (2020) 26:S335–43. doi: 10.37765/AJMC.2020.88554
203. Bailey CJ, Flatt PR. Glucagon-Like Peptide-1 and the Entero-Insular Axis in Obese Hyperglycaemic (Ob/Ob)Mice. Life Sci (1987) 40:521–25. doi: 10.1016/0024-3205(87)90364-X
204. Holst JJ, Ørskov C, Vagn Nielsen O, Schwartz TW. Truncated Glucagon-Like Peptide I, an Insulin-Releasing Hormone From the Distal Gut. FEBS Lett (1987) 211:169–74. doi: 10.1016/0014-5793(87)81430-8
205. Drucker DJ, Lee YC, Asa SL, Brubaker PL. Inhibition of Pancreatic Glucagon Gene Expression in Mice Bearing a Subcutaneous Glucagon-Producing Glutag Transplantable Tumor. Mol Endocrinol (1992) 6:2175–84. doi: 10.1210/mend.6.12.1491697
206. Lee YC, Asa SL, Drucker DJ. Glucagon Gene 5’-Flanking Sequences Direct Expression of Simian Virus 40 Large T Antigen to the Intestine, Producing Carcinoma of the Large Bowel in Transgenic Mice. J Biol Chem (1992) 267:10705–8. doi: 10.1016/s0021-9258(19)50075-8
208. Myojo S, Tsujikawa T, Sasaki M, Fujiyama Y, Bamba T. Trophic Effects of Glicentin on Rat Small-Intestinal Mucosa. Vivo Vitro J Gastroenterol (1997) 32:300–5. doi: 10.1007/BF02934484
209. Drucker DJ, Ehrlich P, Asa SL, Brubaker PL. Induction of Intestinal Epithelial Proliferation by Glucagon-Like Peptide 2. Proc Natl Acad Sci USA (1996) 93:7911–6. doi: 10.1073/pnas.93.15.7911
210. Lim GE, Brubaker PL. Glucagon-Like Peptide 1 Secretion by the L-cell: The View From Within. Diabetes (2006) 55:S70–7. doi: 10.2337/db06-S020
211. Burrin DG, Petersen Y, Stoll B, Sangild P. Glucagon-Like Peptide 2: A Nutrient-Responsive Gut Growth Factor. J Nutr (2001) 131:709–12. doi: 10.1093/jn/131.3.709
212. Rocca AS, Brubaker PL. Role of the Vagus Nerve in Mediating Proximal Nutrient-Induced Glucagon- Like Peptide-1 Secretion. Endocrinology (1999) 140:1687–94. doi: 10.1210/endo.140.4.6643
213. Munroe DG, Gupta AK, Kooshesh F, Vyas TB, Rizkalla G, Wang H, et al. Prototypic G Protein-Coupled Receptor for the Intestinotrophic Factor Glucagon-Like Peptide 2. Proc Natl Acad Sci USA (1999) 96:1569–73. doi: 10.1073/pnas.96.4.1569
214. Yusta B, Somwar R, Wang F, Munroe D, Grinstein S, Klip A, et al. Identification of Glucagon-Like Peptide-2 (GLP-2)-activated Signaling Pathways in Baby Hamster Kidney Fibroblasts Expressing the Rat GLP-2 Receptor. J Biol Chem (1999) 274:30459–67. doi: 10.1074/jbc.274.43.30459
215. Benjamin MA, McKay DM, Yang PC, Cameron H, Perdue MH. Glucagon-Like Peptide-2 Enhances Intestinal Epithelial Barrier Function of Both Transcellular and Paracellular Pathways in the Mouse. Gut (2000) 47:112–9. doi: 10.1136/gut.47.1.112
216. Lovshin J, Drucker DJ. New Frontiers in the Biology of GLP-2. Regul Pept (2000) 90:27–32. doi: 10.1016/S0167-0115(00)00117-8
217. Khan D, Vasu S, Moffett RC, Irwin N, Flatt PR. Differential Expression of Glucagon-Like Peptide-2 (GLP-2) is Involved in Pancreatic Islet Cell Adaptations to Stress and Beta-Cell Survival. Peptides (2017) 95:68–75. doi: 10.1016/j.peptides.2017.07.011
218. Thompson JS, Iyer KR, Dibaise JK, Young RL, Brown CR, Langnas AN. Short Bowel Syndrome and Crohn’s Disease. J Gastrointest Surg (2003) 7:1069–72. doi: 10.1016/j.gassur.2003.08.007
219. Seetharam P, Rodrigues G. Short Bowel Syndrome: A Review of Management Options. Saudi J Gastroenterol (2011) 17:229–35. doi: 10.4103/1319-3767.82573
220. Dahly EM, Gillingham MB, Guo Z, Murali SG, Nelson DW, Holst JJ, et al. Role of Luminal Nutrients and Endogenous GLP-2 in Intestinal Adaptation to Mid-Small Bowel Resection. Am J Physiol Liver Physiol (2003) 284:G670–82. doi: 10.1152/ajpgi.00293.2002
221. Scott RB, Kirk D, Macnaughton WK, Meddings JB. GLP-2 Augments the Adaptive Response to Massive Intestinal Resection in Rat. Am J Physiol Gastrointest Liver Physiol (1998) 275:38–45. doi: 10.1152/ajpgi.1998.275.5.g911
222. Jeppesen PB, Hartmann B, Thulesen J, Graff J, Lohmann J, Hansen BS, et al. Glucagon-Like Peptide 2 Improves Nutrient Absorption and Nutritional Status in Short-Bowel Patients With No Colon. Gastroenterology (2001) 120:806–15. doi: 10.1053/gast.2001.22555
223. Jeppesen PB, Gilroy R, Pertkiewicz M, Allard JP, Messing B, O’Keefe SJ. Randomised Placebo-Controlled Trial of Teduglutide in Reducing Parenteral Nutrition and/or Intravenous Fluid Requirements in Patients With Short Bowel Syndrome. Gut (2011) 60:902–14. doi: 10.1136/gut.2010.218271
224. Wiśniewski K, Sueiras-Diaz J, Jiang G, Galyean R, Lu M, Thompson D, et al. Synthesis and Pharmacological Characterization of Novel Glucagon-Like Peptide-2 (GLP-2) Analogues With Low Systemic Clearance. J Med Chem (2016) 59:3129–39. doi: 10.1021/acs.jmedchem.5b01909
225. Hargrove DM, Alagarsamy S, Croston G, Laporte R, Qi S, Srinivasan K, et al. Pharmacological Characterization of Apraglutide, a Novel Long-Acting Peptidic Glucagon-Like Peptide-2 Agonist, for the Treatment of Short Bowel Syndrome. J Pharmacol Exp Ther (2020) 373:193–203. doi: 10.1124/jpet.119.262238
226. Naimi RM, Hvistendahl M, Enevoldsen LH, Madsen JL, Fuglsang S, Poulsen SS, et al. Glepaglutide, a Novel Long-Acting Glucagon-Like Peptide-2 Analogue, for Patients With Short Bowel Syndrome: A Randomised Phase 2 Trial. Lancet Gastroenterol Hepatol (2019) 4:354–63. doi: 10.1016/S2468-1253(19)30077-9
227. Jeppesen PB, Sanguinetti EL, Buchman A, Howard L, Scolapio JS, Ziegler TR, et al. Teduglutide (ALX-0600), a Dipeptidyl Peptidase IV Resistant Qlucagon-Like Peptide 2 Analogue, Improves Intestinal Function in Short Bowel Syndrome Patients. Gut (2005) 54:1224–31. doi: 10.1136/gut.2004.061440
228. Jeppesen PB, Lund P, Gottschalck IB, Nielsen HB, Holst JJ, Mortensen J, et al. Short Bowel Patients Treated for Two Years With Glucagon-Like Peptide 2: Effects on Intestinal Morphology and Absorption, Renal Function, Bone and Body Composition, and Muscle Function. Gastroenterol Res Pract (2009) 2009:616054. doi: 10.1155/2009/616054
229. Iyer KR, Kunecki M, Boullata JI, Fujioka K, Joly F, Gabe S, et al. Independence From Parenteral Nutrition and Intravenous Fluid Support During Treatment With Teduglutide Among Patients With Intestinal Failure Associated With Short Bowel Syndrome. J Parenter Enter Nutr (2017) 41:946–51. doi: 10.1177/0148607116680791
230. Marier JF, Beliveau M, Mouksassi MS, Shaw P, Cyran J, Kesavan J, et al. Pharmacokinetics, Safety, and Tolerability of Teduglutide, a Glucagon-Like Peptide-2 (GLP-2) Analog, Following Multiple Ascending Subcutaneous Administrations in Healthy Subjects. J Clin Pharmacol (2008) 48:1289–99. doi: 10.1177/0091270008320605
231. Trial to Evaluate Efficacy and Safety of Apraglutide in SBS-IF. Clinicaltrials.Gov . Available at: https://clinicaltrials.gov/ct2/show/NCT04627025 (Accessed March 15, 2021).
232. Kanis JA, Cooper C, Rizzoli R, Reginster JY. European Guidance for the Diagnosis and Management of Osteoporosis in Postmenopausal Women. Osteoporos Int (2019) 30:3–44. doi: 10.1007/s00198-018-4704-5
233. Demontiero O, Vidal C, Duque G. Aging and Bone Loss: New Insights for the Clinician. Ther Adv Musculoskelet Dis (2012) 4:61–76. doi: 10.1177/1759720X11430858
234. Khosla S, Hofbauer LC. Osteoporosis Treatment: Recent Developments and Ongoing Challenges. Lancet Diabetes Endocrinol (2017) 5:898–907. doi: 10.1016/S2213-8587(17)30188-2
235. Henriksen DB, Alexandersen P, Bjarnason NH, Vilsbøll T, Hartmann B, Henriksen EE, et al. Role of Gastrointestinal Hormones in Postprandial Reduction of Bone Resorption. J Bone Miner Res (2003) 18:2180–9. doi: 10.1359/jbmr.2003.18.12.2180
236. Gottschalck IB, Jeppesen PB, Holst JJ, Henriksen DB. Reduction in Bone Resorption by Exogenous Glucagon-Like Peptide-2 Administration Requires an Intact Gastrointestinal Tract. Scand J Gastroenterol (2008) 43:929–37. doi: 10.1080/00365520801965381
237. Pacheco-Pantoja EL, Ranganath LR, Gallagher JA, Wilson PJ, Fraser WD. Receptors and Effects of Gut Hormones in Three Osteoblastic Cell Lines. BMC Physiol (2011) 11:12. doi: 10.1186/1472-6793-11-12
238. Skov-Jeppesen K, Svane MS, Martinussen C, Gabe MBN, Gasbjerg LS, Veedfald S, et al. GLP-2 and GIP Exert Separate Effects on Bone Turnover: A Randomized, Placebo-Controlled, Crossover Study in Healthy Young Men. Bone (2019) 125:178–5. doi: 10.1016/j.bone.2019.05.014
239. Lopes LSG, Schwartz RP, Ferraz-De-Souza B, Da Silva MER, Corrêa PHS, Nery M. The Role of Enteric Hormone GLP-2 in the Response of Bone Markers to a Mixed Meal in Postmenopausal Women With Type 2 Diabetes Mellitus. Diabetol Metab Syndr (2015) 7:13. doi: 10.1186/s13098-015-0006-7
240. Xu B, He Y, Lu Y, Ren W, Shen J, Wu K, et al. Glucagon Like Peptide 2 has a Positive Impact on Osteoporosis in Ovariectomized Rats. Life Sci (2019) 226:47–56. doi: 10.1016/j.lfs.2019.04.013
241. Askov-Hansen C, Jeppesen PB, Lund P, Hartmann B, Holst JJ, Henriksen DB. Effect of Glucagon-Like Peptide-2 Exposure on Bone Resorption: Effectiveness of High Concentration Versus Prolonged Exposure. Regul Pept (2013) 181:4–8. doi: 10.1016/j.regpep.2012.11.002
242. Schiellerup SP, Skov-Jeppesen K, Windeløv JA, Svane MS, Holst JJ, Hartmann B, et al. Gut Hormones and Their Effect on Bone Metabolism. Potential Drug Therapies in Future Osteoporosis Treatment. Front Endocrinol (2019) 10:75. doi: 10.3389/fendo.2019.00075
243. Holst JJ. Evidence That Glicentin Contains the Entire Sequence of Glucagon. Biochem J (1980) 187:337–43. doi: 10.1042/bj1870337
244. Baldissera FGA, Holst JJ, Knuhtsen S, Hilsted L, Nielsen OV. Oxyntomodulin (Glicentin-(33-69)): Pharmacokinetics, Binding to Liver Cell Membranes, Effects on Isolated Perfused Pig Pancreas, and Secretion From Isolated Perfused Lower Small Intestine of Pigs. Regul Pept (1988) 21:151–66. doi: 10.1016/0167-0115(88)90099-7
245. Bataille D, Gespach C, Coudray AM, Rosselin G. “Enteroglucagon”: A Specific Effect on Gastric Glands Isolated From the Rat Fundus. Evidence for an “Oxyntomodulin” Action. Biosci Rep (1981) 1:151–5. doi: 10.1007/BF01117012
246. Bataille D, Gespach C, Tatemoto K, Marie JC, Coudray AM, Rosselin G, et al. Bioactive Enteroglucagon (Oxyntomodulin): Present Knowledge on its Chemical Structure and its Biological Activities. Peptides (1981) 2:41–4. doi: 10.1016/0196-9781(81)90008-
247. Kerr BD, Flatt PR, Gault VA. (D-Ser2)Oxm[Mpeg-PAL]: A Novel Chemically Modified Analogue of Oxyntomodulin With Antihyperglycaemic, Insulinotropic and Anorexigenic Actions. Biochem Pharmacol (2010) 80:1727–35. doi: 10.1016/j.bcp.2010.08.010
248. Graham GV, Conlon JM, Abdel-Wahab YH, Flatt PR. Glucagon-Related Peptides From Phylogenetically Ancient Fish Reveal New Approaches to the Development of Dual GCGR and GLP1R Agonists for Type 2 Diabetes Therapy. Peptides (2018) 110:19–29. doi: 10.1016/j.peptides.2018.10.013
249. Scott R, Minnion J, Tan T, Bloom SR. Oxyntomodulin Analogue Increases Energy Expenditure. Via Glucagon Receptor Peptides (2018) 104:70–7. doi: 10.1016/j.peptides.2018.04.008
250. A Study of LY3305677 in Participants With Type 2 Diabetes. Clinicaltrials.Gov. Available at: https://clinicaltrials.gov/ct2/show/NCT03928379 (Accessed March 15, 2021).
251. Evaluate the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of IBI362 in Overweight or Obesity Subjects. Clinicaltrials.Gov . Available at: https://clinicaltrials.gov/ct2/show/NCT04440345 (Accessed March 15, 2021).
252. Day JW, Ottaway N, Patterson JT, Gelfanov V, Smiley D, Gidda J, et al. A New Glucagon and GLP-1 Co-Agonist Eliminates Obesity in Rodents. Nat Chem Biol (2009) 5:749–57. doi: 10.1038/nchembio.209
253. Henderson SJ, Konkar A, Hornigold DC, Trevaskis JL, Jackson R, Fritsch Fredin M, et al. Robust Anti-Obesity and Metabolic Effects of a Dual GLP-1/glucagon Receptor Peptide Agonist in Rodents and non-Human Primates. Diabetes Obes Metab (2016) 18:1176–90. doi: 10.1111/dom.12735
254. Wynne K, Field BC, Bloom SR. The Mechanism of Action for Oxyntomodulin in the Regulation of Obesity. Curr Opin Investig Drugs (2010) 11:1151–7.
255. Pocai A. Action and Therapeutic Potential of Oxyntomodulin. Mol Metab (2014) 3:241–51. doi: 10.1016/j.molmet.2013.12.001
256. Baldissera FGA, Holst JJ, Knuhtsen S, Hilsted L, Nielsen OV. Oxyntomodulin (Glicentin-(33-69)): Pharmacokinetics, Binding to Liver Cell Membranes, Effects on Isolated Perfused Pig Pancreas, and Secretion From Isolated Perfused Lower Small Intestine of Pigs. Regul Pept (1988) 21:151–66. doi: 10.1016/0167-0115(88)90099-7
257. Gros L, Thorens B, Bataille D, Kervran A. Glucagon-Like peptide-1-(7-36) Amide, Oxyntomodulin, and Glucagon Interact With a Common Receptor in a Somatostatin-Secreting Cell Line. Endocrinology (1993) 133:631–8. doi: 10.1210/endo.133.2.8102095
258. Baggio LL, Huang Q, Brown TJ, Drucker DJ. Oxyntomodulin and Glucagon-Like Peptide-1 Differentially Regulate Murine Food Intake and Energy Expenditure. Gastroenterology (2004) 127:546–58. doi: 10.1053/j.gastro.2004.04.063
259. Kosinski JR, Hubert J, Carrington PE, Chicchi GG, Mu J, Miller C, et al. The Glucagon Receptor is Involved in Mediating the Body Weight-Lowering Effects of Oxyntomodulin. Obesity (2012) 20:1566–71. doi: 10.1038/oby.2012.67
260. Jorgensen R, Kubale V, Vrecl M, Schwartz TW, Elling CE. Oxyntomodulin Differentially Affects Glucagon-Like Peptide-1 Receptor β-Arrestin Recruitment and Signalling Through Gαs. J Pharmacol Exp Ther (2007) 322:148–54. doi: 10.1124/jpet.107.120006
261. Deacon CF. Physiology and Pharmacology of DPP-4 in Glucose Homeostasis and the Treatment of Type 2 Diabetes. Front Endocrinol (2019) 10:80. doi: 10.3389/fendo.2019.00080
262. Cohen MA, Ellis SM, Le Roux CW, Batterham RL, Park A, Patterson M, et al. Oxyntomodulin Suppresses Appetite and Reduces Food Intake in Humans. J Clin Endocrinol Metab (2003) 88:4696–701. doi: 10.1210/jc.2003-030421
263. Wynne K, Park AJ, Small CJ, Meeran K, Ghatei MA, Frost GS, et al. Oxyntomodulin Increases Energy Expenditure in Addition to Decreasing Energy Intake in Overweight and Obese Humans: A Randomised Controlled Trial. Int J Obes (2006) 30:1729–36. doi: 10.1038/sj.ijo.0803344
264. Irwin N, Pathak V, Pathak NM, Gault VA, Flatt PR. Sustained Treatment With a Stable Long-Acting Oxyntomodulin Analogue Improves Metabolic Control and Islet Morphology in an Experimental Model of Type 1 Diabetes. Diabetes Obes Metab (2015) 17:887–95. doi: 10.1111/dom.12508
265. Sarnobat D, Moffett RC, Gault VA, Tanday N, Reimann F, Gribble FM, et al. Effects of Long-Acting GIP, Xenin and Oxyntomodulin Peptide Analogues on Alpha-Cell Transdifferentiation in Insulin-Deficient Diabetic GluCreERT2;ROSA26-eYFP Mice. Peptides (2020) 125:170205. doi: 10.1016/j.peptides.2019.170205
266. Price SL, Minnion JS, Bloom SR. Increased Food Intake With Oxyntomodulin Analogues. Peptides (2015) 73:95–100. doi: 10.1016/j.peptides.2015.09.006
267. Druce MR, Minnion JS, Field BCT, Patel SR, Shillito JC, Tilby M, et al. Investigation of Structure-Activity Relationships of Oxyntomodulin (Oxm) Using Oxm Analogs. Endocrinology (2009) 150:1712–21. doi: 10.1210/en.2008-0828
268. Heding LG. Radioimmunological Determination of Pancreatic and Gut Glucagon in Plasma. Diabetologia (1971) 7:10–9. doi: 10.1007/bf02346248
269. Thim L, Moody AJ. Purification and Chemical Characterization of a Glicentin-Related Pancreatic Peptide (Proglucagon Fragment) From Porcine Pancreas. Biochim Biophys Acta (BBA)/Protein Struct Mol (1982) 703:134–41. doi: 10.1016/0167-4838(82)90041-3
270. Thim L, Moody AJ. The Primary Structure of Porcine Glicentin (Proglucagon). Regul Pept (1981) 2:139–50. doi: 10.1016/0167-0115(81)90007-0
271. Baldissera FGA, Holst JJ, Knuhtsen S, Hilsted L, Nielsen OV. Oxyntomodulin (Glicentin-(33-69)): Pharmacokinetics, Binding to Liver Cell Membranes, Effects on Isolated Perfused Pig Pancreas, and Secretion From Isolated Perfused Lower Small Intestine of Pigs. Regul Pept (1988) 21:151–66. doi: 10.1016/0167-0115(88)90099-7
272. Ohneda A. Response of Plasma Glicentin to Intraduodenal Administration of Glucose in Piglets. Diabetes Res Clin Pract (1987) 3:97–102. doi: 10.1016/S0168-8227(87)80013-X
273. Ohneda A, Takahashi H, Maruyama Y. Response of Plasma Glicentin to Fat Ingestion in Piglets. Diabetes Res Clin Pract (1987) 3:103–9. doi: 10.1016/S0168-8227(87)80014-1
274. Ohneda A, Kobayashi T, Nihei J, Takahashi H. Effect of Intraluminal Administration of Amino Acids Upon Plasma Glicentin. Diabetes Res Clin Pract (1988) 5:265–70. doi: 10.1016/S0168-8227(88)80061-5
275. Shibata C, Naito H, Jin XL, Ueno T, Funayama Y, Fukushima K, et al. Effect of Glucagon, Glicentin, Glucagon-Like Peptide-1 and -2 on Interdigestive Gastroduodenal Motility in Dogs With a Vagally Denervated Gastric Pouch. Scand J Gastroenterol (2001) 36:1049–55. doi: 10.1080/003655201750422648
276. Ohneda A, Ohneda K, Nagsaki T, Sasaki K. Insulinotropic Action of Human Glicentin in Dogs. Metabolism (1995) 44:47–51. doi: 10.1016/0026-0495(95)90288-0
277. Geneviève R, Magous R, Mochizuki T, Le ND, Martinez J, Bali JP, et al. Glicentin and Oxyntomodulin Modulate Both the Phosphoinositide and Cyclic Adenosine Monophosphate Signalling Pathways in Gastric Myocytes. Endocrinology (1999) 140:22–8. doi: 10.1210/endo.140.1.6424
278. Ayachi SE, Borie F, Magous R, Sasaki K, Le Nguyen D, Bali JP, et al. Contraction Induced by Glicentin on Smooth Muscle Cells From the Human Colon is Abolished by Exendin (9-39). Neurogastroenterol Motil (2005) 17:302–9. doi: 10.1111/j.1365-2982.2004.00628.x
279. Thieden HID, Holst JJ, Dich J, Moody A, Sundby F. Effect of Highly Purified Porcine Gut Glucagon-Like Immunoreactivity (Glicentin) on Glucose Release From Isolated Rat Hepatocytes. BBA Gen Subj (1981) 675:163–70. doi: 10.1016/0304-4165(81)90222-1
280. Raffort J, Lareyre F, Massalou D, Fénichel P, Panaïa-Ferrari P, Chinetti G. Insights on Glicentin, a Promising Peptide of the Proglucagon Family. Biochem Med (2017) 27:308–24. doi: 10.11613/BM.2017.034
281. Hasib A. Multiagonist Unimolecular Peptides for Obesity and Type 2 Diabetes: Current Advances and Future Directions. Clin Med Insights Endocrinol Diabetes (2020) 13:1179551420905844. doi: 10.1177/1179551420905844
282. Nielsen MS, Ritz C, Wewer Albrechtsen NJ, Holst JJ, le Roux CW, Sjödin A. Oxyntomodulin and Glicentin may Predict the Effect of Bariatric Surgery on Food Preferences and Weight Loss. J Clin Endocrinol Metab (2020) 105:e1064–74. doi: 10.1210/clinem/dgaa061
283. Ohneda A, Ohneda M. Effect of Glicentin-Related Peptides Upon the Secretion of Insulin and Glucagon in the Canine Pancreas. Tohoku J Exp Med (1988) 155:197–204. doi: 10.1620/tjem.155.197
284. Whiting L, Stewart KW, Hay DL, Harris PW, Choong YS, Phillips ARJ, et al. Glicentin-Related Pancreatic Polypeptide Inhibits Glucose-Stimulated Insulin Secretion From the Isolated Pancreas of Adult Male Rats. Physiol Rep (2015) 3:12638. doi: 10.14814/phy2.12638
285. Mingrone G, Castagneto-Gissey L. Mechanisms of Early Improvement/Resolution of Type 2 Diabetes After Bariatric Surgery. Diabetes Metab (2009) 35:518–23. doi: 10.1016/S1262-3636(09)73459-7
286. Moffett RC, Docherty NG, le Roux CW. The Altered Enteroendocrine Repertoire Following roux-en-Y-gastric Bypass as an Effector of Weight Loss and Improved Glycaemic Control. Appetite (2021) 156:104807. doi: 10.1016/j.appet.2020.104807
287. Irwin N, McClean PL, Hunter K, Flatt RP. Metabolic Effects of Sustained Activation of the GLP-1 Receptor Alone and in Combination With Background GIP Receptor Antagonism in High Fat-Fed Mice. Diabetes Obes Metab (2009) 11:603–10. doi: 10.1111/j.1463-1326.2009.01036.x
288. Gault VA, Kerr BD, Harriott P, Flatt PR. Administration of an Acylated GLP-1 and GIP Preparation Provides Added Beneficial Glucose-Lowering and Insulinotropic Actions Over Single Incretins in Mice With Type 2 Diabetes and Obesity. Clin Sci (2011) 121:107–17. doi: 10.1042/CS20110006
289. Tan T, Behary P, Tharakan G, Minnion J, Al-Najim W, Wewer Albrechtsen NJ, et al. The Effect of a Subcutaneous Infusion of GLP-1, OXM, and PYY on Energy Intake and Expenditure in Obese Volunteers. J Clin Endocrinol Metab (2017) 102:2364–72. doi: 10.1210/jc.2017-00469
290. Behary P, Tharakan G, Alexiadou K, Johnson N, Wewer Albrechtsen NJ, Kenkre J, et al. Combined GLP-1, Oxyntomodulin, and Peptide YY Improves Body Weight and Glycemia in Obesity and Prediabetes/Type 2 Diabetes: A Randomized, Single-Blinded, Placebo-Controlled Study. Diabetes Care (2019) 42:1446–53. doi: 10.2337/dc19-0449
291. Ambery PD, Klammt S, Posch MG, Petrone M, Pu W, Rondinone C, et al. MEDI0382, a GLP-1/glucagon Receptor Dual Agonist, Meets Safety and Tolerability Endpoints in a Single-Dose, Healthy-Subject, Randomized, Phase 1 Study. Br J Clin Pharmacol (2018) 84:2325–35. doi: 10.1111/bcp.13688
292. Parker VER, Robertson D, Wang T, Hornigold DC, Petrone M, Cooper AT, et al. Efficacy, Safety, and Mechanistic Insights of Cotadutide, a Dual Receptor Glucagon-Like Peptide-1 and Glucagon Agonist. J Clin Endocrinol Metab (2020) 105:dgz047. doi: 10.1210/clinem/dgz047
293. Boland ML, Laker RC, Mather K, Nawrocki A, Oldham S, Boland BB, et al. Resolution of NASH and Hepatic Fibrosis by the GLP-1R and GCGR Dual-Agonist Cotadutide Via Modulating Mitochondrial Function and Lipogenesis. Nat Metab (2020) 2:413–31. doi: 10.1038/s42255-020-0209-6
294. Kang JH, Kim JH, Yi J, Han O, Kim Y, Baek E, et al. The Ultra-Long Acting LAPSGLP/GCGdual Agonist, HM12525A, Demonstrated Safety and Prolonged Pharmacokinetics in Healthy Volunteers: A Phase 1 First-in-Human Study. Diabetologia (2015) 58:S380–1. doi: 10.2337/db151385
295. Jung SY, Lee JS, Kim JY, Lee YM, Kim YH, Kang JH, et al. Potent Weight Loss Mechanism of the Novel Long-Acting GLP-1/glucagon Dual Receptor Agonist (HM12525A). Diabetes (2015) 64:A287.
296. Coskun T, Sloop KW, Loghin C, Alsina-Fernandez J, Urva S, Bokvist KB, et al. LY3298176, a Novel Dual GIP and GLP-1 Receptor Agonist for the Treatment of Type 2 Diabetes Mellitus: From Discovery to Clinical Proof of Concept. Mol Metab (2018) 18:3–14. doi: 10.1016/j.molmet.2018.09.009
297. Frias JP, Nauck MA, Van J, Benson C, Bray R, Cui X, et al. Efficacy and Tolerability of Tirzepatide, a Dual Glucose-Dependent Insulinotropic Peptide and Glucagon-Like Peptide-1 Receptor Agonist in Patients With Type 2 Diabetes: A 12-Week, Randomized, Double-Blind, Placebo-Controlled Study to Evaluate Different Dose-Escalation Regimens. Diabetes Obes Metab (2020) 22:938–46. doi: 10.1111/dom.13979
298. Sloop KW, Briere DA, Emmerson PJ, Willard FS. Beyond Glucagon-Like Peptide-1: Is G-Protein Coupled Receptor Polypharmacology the Path Forward to Treating Metabolic Diseases? ACS Pharmacol Transl Sci (2018) 1:3–11. doi: 10.1021/acsptsci.8b00009
299. Irwin N, Pathak V, Flatt PR. A Novel CCK-8/GLP-1 Hybrid Peptide Exhibiting Prominent Insulinotropic, Glucose-Lowering, and Satiety Actions With Significant Therapeutic Potential in High-Fat-Fed Mice. Diabetes (2015) 64:2996–3009. doi: 10.2337/db15-0220
300. Hornigold DC, Roth E, Howard V, Will S, Oldham S, Coghlan MP, et al. A GLP-1:CCK Fusion Peptide Harnesses the Synergistic Effects on Metabolism of CCK-1 and GLP-1 Receptor Agonism in Mice. Appetite (2018) 127:334–40. doi: 10.1016/j.appet.2018.05.131
301. van Witteloostuijn SB, Dalbøge LS, Hansen G, Midtgaard SR, Jensen GV, Jensen KJ, et al. Gub06-046, a Novel Secretin/Glucagon-Like Peptide 1 Co-Agonist, Decreases Food Intake, Improves Glycemic Control, and Preserves Beta Cell Mass in Diabetic Mice. J Pept Sci (2017) 23:845–54. doi: 10.1002/psc.3048
302. Chepurny OG, Bonaccorso RL, Leech CA, Wöllert T, Langford GM, Schwede F, et al. Chimeric Peptide EP45 as a Dual Agonist at GLP-1 and NPY2R Receptors. Sci Rep (2018) 8:6192. doi: 10.1038/s41598-018-24359-2
303. Hasib A, Ng MT, Khan D, Gault VA, Flatt PR, Irwin N. A Novel GLP-1/xenin Hybrid Peptide Improves Glucose Homeostasis, Circulating Lipids and Restores GIP Sensitivity in High Fat Fed Mice. Peptides (2018) 100:202–11. doi: 10.1016/j.peptides.2017.10.015
304. Bhat VK, Kerr BD, Vasu S, Flatt PR, Gault VA. A DPP-IV-resistant Triple-Acting Agonist of GIP, GLP-1 and Glucagon Receptors With Potent Glucose-Lowering and Insulinotropic Actions in High-Fat-Fed Mice. Diabetologia (2013) 56:1417–24. doi: 10.1007/s00125-013-2892-2
305. Hasib A, Ng MT, Tanday N, Craig SL, Gault VA, Flatt PR, et al. Exendin-4(Lys27PAL)/Gastrin/Xenin-8-Gln: A Novel Acylated GLP-1/gastrin/xenin Hybrid Peptide That Improves Metabolic Status in Obese-Diabetic (Ob/Ob) Mice. Diabetes Metab Res Rev (2019) 35:e3106. doi: 10.1002/dmrr.3106
306. Irwin N, Flatt PR. Evidence for Beneficial Effects of Compromised Gastric Inhibitory Polypeptide Action in Obesity-Related Diabetes and Possible Therapeutic Implications. Diabetologia (2009) 52:1724–31. doi: 10.1007/s00125-009-1422-8
307. A Study of LY3437943 in Participants With Type 2 Diabetes Mellitus (T2DM). Case Med Res (2019). doi: 10.31525/ct1-nct04143802
308. Study to Evaluate Efficacy, Safety and Tolerability of HM15211 in Subjects . Available at: https://clinicaltrials.gov/ct2/show/NCT04505436 (Accessed February 12, 2021).
309. Tai J, Liu W, Li Y, Li L, Hölscher C. Neuroprotective Effects of a Triple GLP-1/GIP/glucagon Receptor Agonist in the APP/PS1 Transgenic Mouse Model of Alzheimer’s Disease. Brain Res (2018) 1678:64–74. doi: 10.1016/j.brainres.2017.10.012
310. Finan B, Yang B, Ottaway N, Smiley DL, Ma T, Clemmensen C, et al. A Rationally Designed Monomeric Peptide Triagonist Corrects Obesity and Diabetes in Rodents. Nat Med (2015) 21:27–36. doi: 10.1038/nm.3761
311. Pocai A, Carrington PE, Adams JR, Wright M, Eiermann G, Zhu L, et al. Glucagon-Like Peptide 1/Glucagon Receptor Dual Agonism Reverses Obesity in Mice. Diabetes (2009) 58:2258–66. doi: 10.2337/db09-0278
312. Day JW, Gelfanov V, Smiley D, Carrington PE, Eiermann G, Chicchi G, et al. Optimization of Co-Agonism at GLP-1 and Glucagon Receptors to Safely Maximize Weight Reduction in DIO-Rodents. Biopolymers (2012) 98:443–50. doi: 10.1002/bip.22072
313. Ambery P, Parker VE, Stumvoll M, Posch MG, Heise T, Plum-Moerschel L, et al. MEDI0382, a GLP-1 and Glucagon Receptor Dual Agonist, in Obese or Overweight Patients With Type 2 Diabetes: A Randomised, Controlled, Double-Blind, Ascending Dose and Phase 2a Study. Lancet (2018) 391:2607–18. doi: 10.1016/S0140-6736(18)30726-8
314. Petit JM, Cercueil JP, Loffroy R, Denimal D, Bouillet B, Fourmont C, et al. Effect of Liraglutide Therapy on Liver Fat Content in Patients With Inadequately Controlled Type 2 Diabetes: The Lira-NAFLD Study. J Clin Endocrinol Metab (2017) 102:407–15. doi: 10.1210/jc.2016-2775
315. Amarapurkar DN, Amarapurkar AD, Patel ND, Agal S, Baigal R, Gupte P, et al. Nonalcoholic Steatohepatitis (NASH) With Diabetes: Predictors of Liver Fibrosis. Ann Hepatol (2006) 5:30–3. doi: 10.1016/s1665-2681(19)32036-8
316. Park YJ, Jung SY, Kim JK, Lee JS, Lee YM, Kim YH, et al. Lipolytic and Insulinotropic Effects of HM12525A, a Novel Long-Acting GLP-1/glucagon Dual Agonist. Diabetologia (2014) 57:S352.
317. Willard FS, Douros JD, Gabe MBN, Showalter AD, Wainscott DB, Suter TM, et al. Tirzepatide is an Imbalanced and Biased Dual GIP and GLP-1 Receptor Agonist. JCI Insight (2020) 5:e140532. doi: 10.1172/jci.insight.140532
318. Knerr PJ, Mowery SA, Finan B, Perez-Tilve D, Tschöp MH, DiMarchi RD, et al. Selection and Progression of Unimolecular Agonists atGlp-1, and Glucagon Receptors as Drug Candidates. Peptides (2020) 125:170225. doi: 10.1016/j.peptides.2019.170225
319. Hartman ML, Sanyal AJ, Loomba R, Wilson JM, Nikooienejad A, Bray R, et al. Effects of Novel Dual GIP and GLP-1 Receptor Agonist Tirzepatide on Biomarkers of Nonalcoholic Steatohepatitis in Patients With Type 2 Diabetes. Diabetes Care (2020) 43:1352–5. doi: 10.2337/dc19-1892
320. Irwin N, Frizelle P, O’Harte FPM, Flatt PR. (Pglu-Gln)-CCK-8[Mpeg]: A Novel, Long-Acting, mini-PEGylated Cholecystokinin (CCK) Agonist That Improves Metabolic Status in Dietary-Induced Diabetes. Biochim Biophys Acta Gen Subj (2013) 1830:4009–16. doi: 10.1016/j.bbagen.2013.04.004
321. Lafferty RA, Flatt PR, Irwin N. Established and Emerging Roles Peptide YY (PYY) and Exploitation in Obesity-Diabetes. Curr Opin Endocrinol Diabetes Obes (2020) 28:253–61. doi: 10.1097/MED.0000000000000612
322. Rangwala SM, D’Aquino K, Zhang YM, Bader L, Edwards W, Zheng S, et al. A Long-Acting Pyy 3–36 Analog Mediates Robust Anorectic Efficacy With Minimal Emesis in Nonhuman Primates. Cell Metab (2019) 29:837–43. doi: 10.1016/j.cmet.2019.01.017
323. Taylor AI, Irwin N, McKillop AM, Patterson S, Flatt PR, Gault VA. Evaluation of the Degradation and Metabolic Effects of the Gut Peptide Xenin on Insulin Secretion, Glycaemic Control and Satiety. J Endocrinol (2010) 207:87–93. doi: 10.1677/JOE-10-0085
324. Martin CMA, Gault VA, McClean S, Flatt PR, Irwin N. Degradation, Insulin Secretion, Glucose-Lowering and GIP Additive Actions of a Palmitate-Derivatised Analogue of Xenin-25. Biochem Pharmacol (2012) 84:312–19. doi: 10.1016/j.bcp.2012.04.015
325. McClean PL, Irwin N, Cassidy RS, Holst JJ, Gault VA, Flatt PR. GIP Receptor Antagonism Reverses Obesity, Insulin Resistance, and Associated Metabolic Disturbances Induced in Mice by Prolonged Consumption of High-Fat Diet. Am J Physiol Endocrinol Metab (2007) 293:1746–55. doi: 10.1152/ajpendo.00460.2007
326. Brandt SJ, Müller TD, DiMarchi RD, Tschöp MH, Stemmer K. Peptide-Based Multi-Agonists: A New Paradigm in Metabolic Pharmacology. J Intern Med (2018) 284:581–602. doi: 10.1111/joim.12837
327. Finan B, Yang B, Ottaway N, Stemmer K, Müller TD, Yi CX, et al. Targeted Estrogen Delivery Reverses the Metabolic Syndrome. Nat Med (2012) 18:1847–56. doi: 10.1038/nm.3009
328. Vogel H, Wolf S, Rabasa C, Rodriguez-Pacheco F, Babaei CS, Stöber F, et al. GLP-1 and Estrogen Conjugate Acts in the Supramammillary Nucleus to Reduce Food-Reward and Body Weight. Neuropharmacology (2016) 110:396–406. doi: 10.1016/j.neuropharm.2016.07.039
329. Schwenk RW, Baumeier C, Finan B, Kluth O, Brauer C, Joost H-G, et al. Glp-1–oestrogen Attenuates Hyperphagia and Protects From Beta Cell Failure in Diabetes-Prone New Zealand Obese (NZO) Mice. Diabetologia (2015) 58:604–14. doi: 10.1007/s00125-014-3478-3
330. Sachs S, Bastidas-Ponce A, Tritschler S, Bakhti M, Böttcher A, Sánchez-Garrido MA, et al. Targeted Pharmacological Therapy Restores β-Cell Function for Diabetes Remission. Nat Metab (2020) 2:192–209. doi: 10.1038/s42255-020-0171-3
331. Brandt SJ, Götz A, Tschöp MH, Müller TD. Gut Hormone Polyagonists for the Treatment of Type 2 Diabetes. Peptides (2018) 100:190–201. doi: 10.1016/j.peptides.2017.12.021
332. Fuente-Martin E, Garcia-Caceres C, Morselli E, Clegg DJ, Chowen JA, Finan B, et al. Estrogen, Astrocytes and the Neuroendocrine Control of Metabolism. Rev Endocr Metab Disord (2013) 14:331–8. doi: 10.1007/s11154-013-9263-7
333. Morselli E, Fuente-Martin E, Finan B, Kim M, Frank A, Garcia-Caceres C, et al. Hypothalamic PGC-1α Protects Against High-Fat Diet Exposure by Regulating Erα. Cell Rep (2014) 9:633–45. doi: 10.1016/j.celrep.2014.09.025
334. Choi J, Kim JK, Lee SM, Kwon H, Lee J, Bae S, et al. Therapeutic Effect of a Novel Long-Acting GLP-1/GIP/Glucagon Triple Agonist (HM15211) in CDHFD-induced NASH and Fibrosis Mice. Diabetes (2020) 69:1830–P. doi: 10.2337/db20-1830-p
335. Hasib A, Ng MT, Khan D, Gault VA, Flatt PR, Irwin N. Characterisation and Antidiabetic Utility of a Novel Hybrid Peptide, Exendin-4/Gastrin/Xenin-8-Gln. Eur J Pharmacol (2018) 834:126–35. doi: 10.1016/j.ejphar.2018.07.027
336. Edelman S, Maier H, Wilhelm K. Pramlintide in the Treatment of Diabetes Mellitus. BioDrugs (2008) 22:375–86. doi: 10.2165/0063030-200822060-00004
337. Guzman-Perez A, Pfefferkorn JA, Lee ECY, Stevens BD, Aspnes GE, Bian J, et al. The Design and Synthesis of a Potent Glucagon Receptor Antagonist With Favorable Physicochemical and Pharmacokinetic Properties as a Candidate for the Treatment of Type 2 Diabetes Mellitus. Bioorg Med Chem Lett (2013) 23:3051–8. doi: 10.1016/j.bmcl.2013.03.014
338. Qureshi SA, Candelore MR, Xie D, Yang X, Tota LM, Ding VDH, et al. A Novel Glucagon Receptor Antagonist Inhibits Glucagon-Mediated Biological Effects. Diabetes (2004) 53:3267–73. doi: 10.2337/diabetes.53.12.3267
339. Mu J, Qureshi SA, Brady EJ, Muise ES, Candelore MR, Jiang G, et al. Anti-Diabetic Efficacy and Impact on Amino Acid Metabolism of GRA1, a Novel Small-Molecule Glucagon Receptor Antagonist. PloS One (2012) 7:e49572. doi: 10.1371/journal.pone.0049572
340. Filipski KJ, Bian J, Ebner DC, Lee ECY, Li JC, Sammons MF, et al. A Novel Series of Glucagon Receptor Antagonists With Reduced Molecular Weight and Lipophilicity. Bioorg Med Chem Lett (2012) 22:415–20. doi: 10.1016/j.bmcl.2011.10.113
341. Liu Y, Colby JK, Zuo X, Jaoude J, Wei D, Shureiqi I. The Role of Ppar-δ in Metabolism, Inflammation, and Cancer: Many Characters of a Critical Transcription Factor. Int J Mol Sci (2018) 19:3339. doi: 10.3390/ijms19113339
342. Guzman CB, Zhang XM, Liu R, Regev A, Shankar S, Garhyan P, et al. Treatment With LY2409021, a Glucagon Receptor Antagonist, Increases Liver Fat in Patients With Type 2 Diabetes. Diabetes Obes Metab (2017) 19:1521–8. doi: 10.1111/dom.12958
343. Pearson MJ, Unger RH, Holland WL. Clinical Trials, Triumphs, and Tribulations of Glucagon Receptor Antagonists. Diabetes Care (2016) 39:1075–7. doi: 10.2337/dci15-0033
344. Unson CG, Gurzenda EM, Merrifield RB. Biological Activities of des-His1[Glu9]glucagon Amide, a Glucagon Antagonist. Peptides (1989) 10:1171–7. doi: 10.1016/0196-9781(89)90010-7
345. McShane LM, Franklin ZJ, O’Harte FPM, Irwin N. Ablation of Glucagon Receptor Signalling by Peptide-Based Glucagon Antagonists Improves Glucose Tolerance in High Fat Fed Mice. Peptides (2014) 60:95–101. doi: 10.1016/j.peptides.2014.08.002
346. O’Harte FPM, Franklin ZJ, Rafferty EP, Irwin N. Characterisation of Structurally Modified Analogues of Glucagon as Potential Glucagon Receptor Antagonists. Mol Cell Endocrinol (2013) 381:26–34. doi: 10.1016/j.mce.2013.07.014
347. Franklin ZJ, O’Harte FPM, Irwin N. Effects of Short-Term Chemical Ablation of Glucagon Signalling by Peptide-Based Glucagon Receptor Antagonists on Insulin Secretion and Glucose Homeostasis in Mice. Biol Chem (2014) 395:433–42. doi: 10.1515/hsz-2013-0224
348. Irwin N, Franklin ZJ, O’Harte FPM. DesHis1Glu9-glucagon-[mPEG] and desHis 1Glu9(Lys30PAL)-Glucagon: Long-acting Peptide-Based PEGylated and Acylated Glucagon Receptor Antagonists With Potential Antidiabetic Activity. Eur J Pharmacol (2013) 709:43–51. doi: 10.1016/j.ejphar.2013.03.041
349. Hruby VJ. Structure-Conformation-Activity Studies of Glucagon and Semi-Synthetic Glucagon Analogs. Mol Cell Biochem (1982) 44:49–64. doi: 10.1007/BF00573846
350. Unson CG, Macdonald D, Merrifield RB. The Role of Histidine-1 in Glucagon Action. Arch Biochem Biophys (1993) 300:747–50. doi: 10.1006/abbi.1993.1103
351. Ahn JM, Medeiros M, Trivedi D, Hruby VJ. Development of Potent Glucagon Antagonists: Structure–Activity Relationship Study of Glycine at Position 4. J Pept Res (2001) 58:151–8. doi: 10.1034/j.1399-3011.2001.00880.x
352. Unson CG, Macdonald D, Ray K, Durrah TL, Merrifield RB. Position 9 Replacement Analogs of Glucagon Uncouple Biological Activity and Receptor Binding. J Biol Chem (1991) 266:2763–6. doi: 10.1016/S0021-9258(18)49911-5
353. Hinke SA, Pospisilik JA, Demuth HU, Mannhart S, Kühn-Wache K, Hoffmann T, et al. Dipeptidyl Peptidase IV (DPIV/CD26) Degradation of Glucagon. Characterization of Glucagon Degradation Products and DPIV-resistant Analogs. J Biol Chem (2000) 275:3827–34. doi: 10.1074/jbc.275.6.3827
354. Lee J, Lee C, Kim I, Moon HR, Kim TH, Oh KT, et al. Preparation and Evaluation of Palmitic Acid-Conjugated Exendin-4 With Delayed Absorption and Prolonged Circulation for Longer Hypoglycemia. Int J Pharm (2012) 424:50–7. doi: 10.1016/j.ijpharm.2011.12.050
355. Yang Z, Wang J, Lu Q, Xu J, Kobayashi Y, Takakura T, et al. Pegylation Confers Greatly Extended Half-Life and Attenuated Immunogenicity to Recombinant Methioninase in Primates. Cancer Res (2004) 64:6673–8. doi: 10.1158/0008-5472.CAN-04-1822
356. Johnson DG, Goebel CU, Hruby VJ, Bregman MD, Trivedi D. Hyperglycemia of Diabetic Rats Decreased by a Glucagon Receptor Antagonist. Science (1982) 215:1115–6. doi: 10.1126/science.6278587
357. Wewer Albrechtsen NJ. Glucagon Receptor Signalling in Metabolic Diseases. Peptides (2018) 100:42–7. doi: 10.1016/j.peptides.2017.11.016
358. Kelly RP, Garhyan P, Raddad E, Fu H, Lim CN, Prince MJ, et al. Short-Term Administration of the Glucagon Receptor Antagonist LY2409021 Lowers Blood Glucose in Healthy People and in Those With Type 2 Diabetes. Diabetes Obes Metab (2015) 17:414–22. doi: 10.1111/dom.12446
359. Guan HP, Yang X, Lu K, Wang SP, Castro-Perez JM, Previs S, et al. Glucagon Receptor Antagonism Induces Increased Cholesterol Absorption. J Lipid Res (2015) 56:2183–95. doi: 10.1194/jlr.M060897
360. Kazda CM, Ding Y, Kelly RP, Garhyan P, Shi C, Lim CN, et al. Evaluation of Efficacy and Safety of the Glucagon Receptor Antagonist LY2409021 in Patients With Type 2 Diabetes: 12-and 24-Week Phase 2 Studies. Diabetes Care (2016) 39:1241–9. doi: 10.2337/dc15-1643
361. Larger E, Wewer Albrechtsen NJ, Hansen LH, Gelling RW, Capeau J, Deacon CF, et al. Pancreatic α-Cell Hyperplasia and Hyperglucagonemia Due to a Glucagon Receptor Splice Mutation. Endocrinol Diabetes Metab Case Rep (2016) 2016:16–0081. doi: 10.1530/EDM-16-0081
362. Sloop KW, Willard FS, Brenner MB, Ficorilli J, Valasek K, Showalter AD, et al. Novel Small Molecule Glucagon-Like Peptide-1 Receptor Agonist Stimulates Insulin Secretion in Rodents and From Human Islets. Diabetes (2010) 59:3099–107. doi: 10.2337/db10-0689
363. Pettus JH, D’Alessio D, Frias JP, Vajda EG, Pipkin JD, Rosenstock J, et al. Efficacy and Safety of the Glucagon Receptor Antagonist RVT-1502 in Type 2 Diabetes Uncontrolled on Metformin Monotherapy: A 12-Week Dose-Ranging Study. Diabetes Care (2020) 43:161–8. doi: 10.2337/dc19-1328
364. Scott RV, Bloom SR. Problem or Solution: The Strange Story of Glucagon. Peptides (2018) 100:36–41. doi: 10.1016/j.peptides.2017.11.013
365. Lynch AM, Pathak N, Pathak V, O’Harte FPM, Flatt PR, Irwin N, et al. A Novel DPP IV-Resistant C-terminally Extended Glucagon Analogue Exhibits Weight-Lowering and Diabetes-Protective Effects in High-Fat-Fed Mice Mediated Through Glucagon and GLP-1 Receptor Activation. Diabetologia (2014) 57:1927–36. doi: 10.1007/s00125-014-3296-7
366. O’Harte FPM, Ng MT, Lynch AM, Conlon JM, Flatt PR. Novel Dual Agonist Peptide Analogues Derived From Dogfish Glucagon Show Promising In Vitro Insulin Releasing Actions and Antihyperglycaemic Activity in Mice. Mol Cell Endocrinol (2016) 431:133–44. doi: 10.1016/j.mce.2016.05.012
367. O’Harte FPM, Ng MT, Lynch AM, Conlon JM, Flatt PR. Dogfish Glucagon Analogues Counter Hyperglycaemia and Enhance Both Insulin Secretion and Action in Diet-Induced Obese Diabetic Mice. Diabetes Obes Metab (2016) 18:1013–24. doi: 10.1111/dom.12713
368. Allen SJ, Watson JJ, Shoemark DK, Barua NU, Patel NK. GDNF. NGF and BDNF as Therapeutic Options for Neurodegeneration. Pharmacol Ther (2013) 138:155–75. doi: 10.1016/j.pharmthera.2013.01.004
Keywords: proglucagon, glucagon, GLP-1, GLP-2, oxyntomodulin, diabetes, obesity, multi-agonist
Citation: Lafferty RA, O’Harte FPM, Irwin N, Gault VA and Flatt PR (2021) Proglucagon-Derived Peptides as Therapeutics. Front. Endocrinol. 12:689678. doi: 10.3389/fendo.2021.689678
Received: 01 April 2021; Accepted: 05 May 2021;
Published: 18 May 2021.
Edited by:
Aylin Carla Hanyaloglu, Imperial College London, United KingdomReviewed by:
Sanjay Kalra, Independent researcher, Karnal, IndiaCopyright © 2021 Lafferty, O’Harte, Irwin, Gault and Flatt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Victor A. Gault, dmEuZ2F1bHRAdWxzdGVyLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.