 Qian Li
Qian Li Dan Tian2
Dan Tian2- 1Department of Endocrinology, Key Laboratory of Endocrinology, NHC, State Key Laboratory of Complex Severe and Rare Diseases, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences, Beijing, China
- 2Department of Nuclear Medicine, The First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, China
- 3Department of Medical Cell Biology, Uppsala University, Uppsala, Sweden
Objective: Mutations in AQP2 (aquaporin-2) lead to rare congenital nephrogenic diabetes insipidus (NDI), which has been limitedly studied in Chinese population.
Methods: Twenty-five subjects from seven families with NDI in a department (Beijing, PUMCH) were screened for AQP2 mutations. Clinical characteristics were described and genotype-phenotype correlation analysis was performed.
Results: We identified 9 AQP2 mutations in 13 patients with NDI, including 3 novel AQP2 mutations (p.G165D, p.Q255RfsTer72 and IVS3-3delC). Missense mutations were the most common mutation type, followed by splicing mutations, and frameshift mutations caused by small deletion or insertion. The onset-age in our patients was younger than 1 year old. Common manifestations included polydipsia, polyuria (7/7) and intermittent fever (6/7). Less common presentations included short stature (3/7) and mental impairment (1/7). High osmotic hypernatremia and low osmotic urine were the main biochemical features. Dilation of the urinary tract was a common complication of NDI (3/6). Level of serum sodium in NDI patients with compound het AQP2 mutations was higher than non-compound het mutations.
Conclusion: In the first and largest case series of NDI caused by AQP2 mutation in Chinese population, we identified 9 AQP2 mutations, including 3 novel mutations. Phenotype was found to correlate with genotypes, revealed by higher level of serum sodium in patients with compound het AQP2 mutations than non-compound het mutations. This knowledge broadens genotypic and phenotypic spectrum for rare congenital NDI and provided basis for studying molecular biology of AQP2.
Introduction
Congenital Nephrogenic Diabetes Insipidus (NDI) is characterized by impaired water resorption in the collecting duct due to insensitivity to arginine vasopressin (AVP) of principal cells, leading to polyuria and polydipsia (1). Mutations in the two key genes-AQP2 (aquaporin-2) and AVPR2 (vasopressin V2 receptor) were identified in most patients with a clear phenotype of NDI (2). Approximately 10% of patients with inherited NDI have a mutation in AQP2 with an autosomal recessive or dominant pattern (OMIM 125800, 107777)) (3). The manifestations generally emerge within the first weeks of life. Infants with congenital NDI may suffer from hypernatremic dehydration, intermittent fever, irritaility, poor feeding and constipation (4). Repeated episodes of hypernatraemic dehydration and rehydration may cause brain tissue damage, seizure and even impair mental development (5, 6). Dilation of the urinary tract was present in 67% of the reported cases, which should be treated timely to avoid end-stage renal disease (7). Current therapeutic methods for NDI focus on relieving symptoms, which are limited and only partially beneficial (8).
AQP2 is a key factor for maintaining normal body water homeostasis. When the plasma osmolality increases, antidiuretic hormone (AVP) is released from the pituitary gland and binds to AVPR2 in the principal cells of the kidney collecting duct, resulting in accumulation of AQP2 in the apical plasma membrane, which is responsible for water reabsorption (9, 10). AQP2 forms a homotetramer and each monomer is composed of 271 amino acids, containing six transmembrane spanning regions with intracellular COOH terminus, which is essential for correct routing of AQP2. The structure of AQP2 has been clarified at 4.9-Å resolution, except for the structure of intracellular domains (11). The trafficking and degradation of AQP2 are regulated by multiple post-translational modifications, including phosphorylation, ubiquitination and glycosylation (12, 13). For example, AVP increases phosphorylation of AQP2 at ser256 and ser269, which is important for the accumulation of AQP2 (14–16). More than 90% of mutations are inherited in autosomal recessive mode, which can be categorized into three types according to the structural analysis: (i) the pore features (e.g. A70D), (ii) the tetramer assembly (e.g. T126M), and (iii) the monomer folding (e.g. A47V). The remaining 10% of mutations are dominant, mainly involving the C-terminal tail for AQP2 routing (e.g. R254L) (17–21).
In this study, we performed gene mutation analysis in 7 Chinese families and identified 9 AQP2 mutations, including 3 novel AQP2 mutations (p.G165D, p.Q255RfsTer72 and IVS3-3delC). Further, we characterized the clinical, biochemical and imaging features in these patients, and performed genotype-phenotype correlation analysis.
Materials and Methods
Subjects
All patients included in this study were referred to our department in Peking Union Medical College Hospital (PUMCH) for diagnosis and treatment of NDI. Diagnosis of NDI was made according to family histories, clinical symptoms (polyuria and polydipsia, fever due to dehydration), laboratory data (urine and serum osmolality, serum sodium), and responses to water deprivation and/or minirin administration. Patients were enrolled after approval from the Institutional Review Board and Ethics Committee of PUMCH and written informed consents were achieved.
Clinical, Biochemical and Imaging Analyses
Medical records were reviewed to extract information, consisting of demographic data, manifestations, biochemical and imaging results. Short stature was defined as a condition in which the height of an individual is within the 3rd percentile of the height for Chinese population of the same age and sex (22). Mental impairment was assessed by qualified doctors. Estimated urine volume (L) in 24 hours was recorded according to detailed medical histories. Routine laboratory tests, including serum sodium (Na), creatinine (Cr(E)), uric acid (UA), serum and urine osmolality, and urine specific gravity (SG), were measured by auto-analyzers according to standard methods at the central laboratory of PUMCH. Reference ranges were provided by the central laboratory of PUMCH. Urological ultrasonography was performed in 6 of 7 patients by qualified doctors.
Genomic DNA Extraction, Amplification, and Sequencing
Whole blood samples of patients and their family members were collected. Genomic DNA was purified using DNA extraction kit (QIAamp DNA Micro; Qiagen, Germany) according to the manufacturer’s protocol. Primers were designed targeting exons and flanking intron sequences of AQP2 and AVPR2. The primer sets and annealing temperature are given in detail in Supplementary Table 1. All polymerase chain reactions (PCR) were performed using 100 ng of genomic DNA template in a total volume of 50 μl containing 10 pmol of each primer, and 25 μl of 2x Easytaq mix (Tiangen Biotech, Beijing, China), under the following conditions: initial denaturation at 94°C for 5 min, followed by 30 cycles at 94°C for 30 s, 60–63°C for 30 s and 72°C for 60 s, finally elongation at 72°C for 7 min. Direct DNA sequence analysis was performed by automated DNA sequencing in an ABI DNA sequencer (Model 377, Applied Biosystems, Foster City, CA), and data were compared with published sequences (AQP2: NC_000012, NM_000486, NP_000477; AVPR2: NC_000023, NM_000054, NP_000045).Gene SNPs (single nucleotide polymorphism) were excluded. In silico prediction of missense variants was performed by Polyphen-2 and SIFT (23, 24), splicing mutations by Spliceman (25). The variants were classified according to the latest American College of Medical Genetics guidelines (26).
Statistical Analysis
Data was presented as mean ± SD or median (range) as appropriate. All statistical analyses were performed using Statistical Product and Service Solutions (SPSS) for window version 19.0 (SPSS Inc., Chicago, IL). Differences between two groups were compared with Student’s t test and the Mann-Whitney U test for normally distributed variables and non-normally distributed variables, respectively. Categorical data were presented as frequencies and percentages (%) and analyzed using the chi-square test and Fisher’s exact test. p<0.05 was considered as significant.
Results
AQP2 Mutations in 7 Chinese NDI Families
Sanger sequencing was applied to screen AQP2 in 25 individuals (7 probands, 13 patients and their relatives). A total of nine different types of AQP2 mutations were identified in 7 families (Details shown in Table 1). Missense mutations were the most common mutation type, followed by splicing mutations, frameshift mutations caused by small deletion or insertion. Of these AQP2 mutations, 3 were novel mutations (p.G165D, p.Q255RfsTer72 and IVS3-3delC) (Table 1), which were predicted to be pathogenic according to ACMG guidelines. The pedigrees of these NDI families were shown in Figure 1. One novel splicing mutation caused by small deletion in intron3 (IVS3-3delC) was shared by 2 independent families (Family5 and Family6), and inherited in an autosomal dominant pattern. One novel frameshift mutation (p.Q255RfsTer72) involving C-terminal of AQP2 was also dominant. The other mutations were recessive and NDI patients carried compound heterozygous mutations (e.g., Case3: p.G165D and IVS3-1 G>A) or homozygous mutations (e.g., Case2: p.G215S). Most mutations were inherited from parents, while Case3 had a de novo mutation (IVS3-1G>A). The distribution of nine AQP2 mutations was shown in Figure 2. Three out of the nine mutations were located in exon2, two in exon1, two in exon4, and two in intron3 (Figure 2A). The locations of amino acids were shown in the schematic diagram of AQP2, three in TMD5 (transmembrane domain-5), one in TMD-2, one in TMD-3, one in TMD-6 and one in ICT (intracellular C terminal) (Figure 2B).

Table 1 Mutation analysis of AQP2 in patients with NDI.
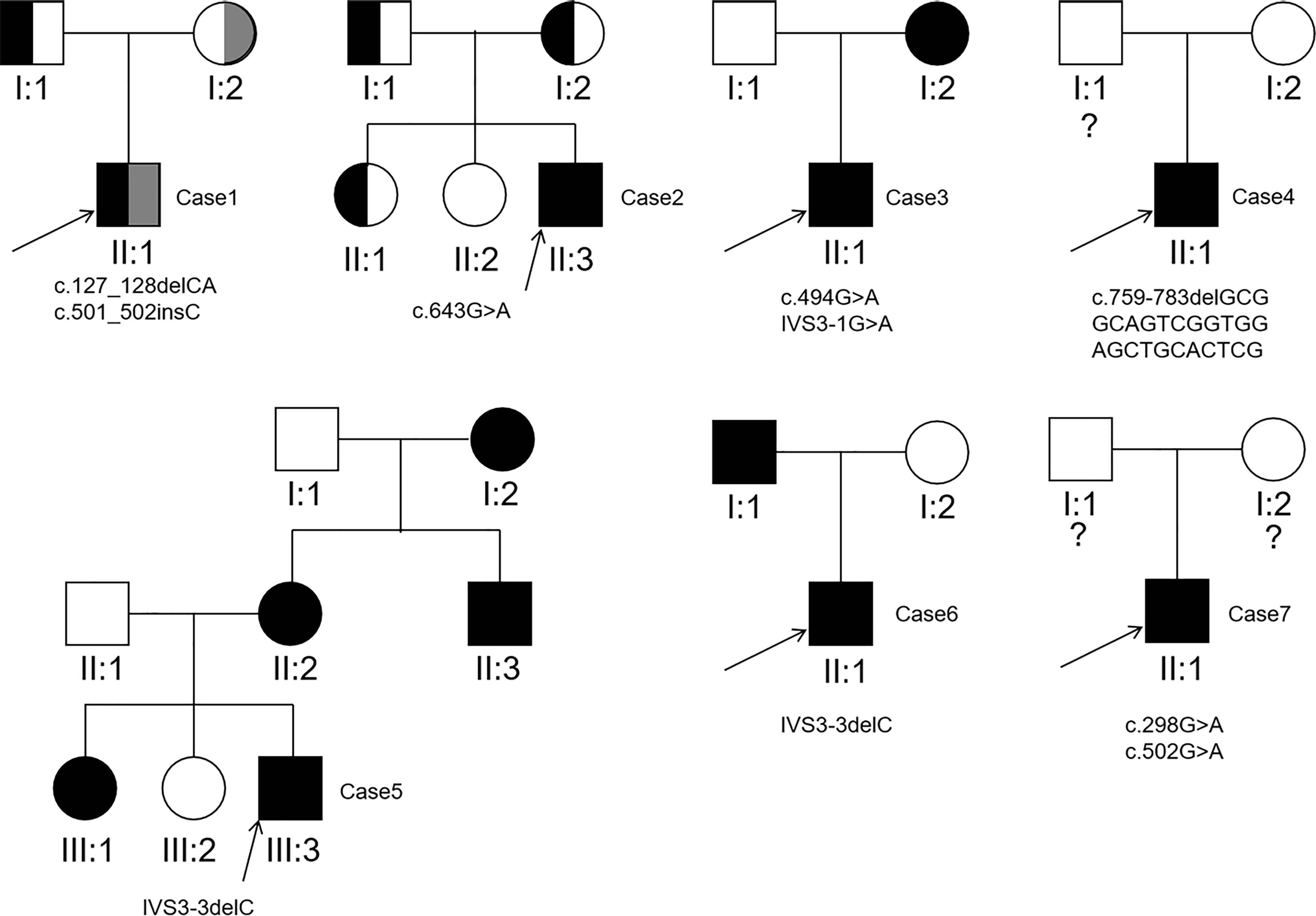
Figure 1 Pedigrees of the 7 families detected to carry pathogenic AQP2 mutations. The question mark indicates AQP2 mutation analysis of the individual was not performed.
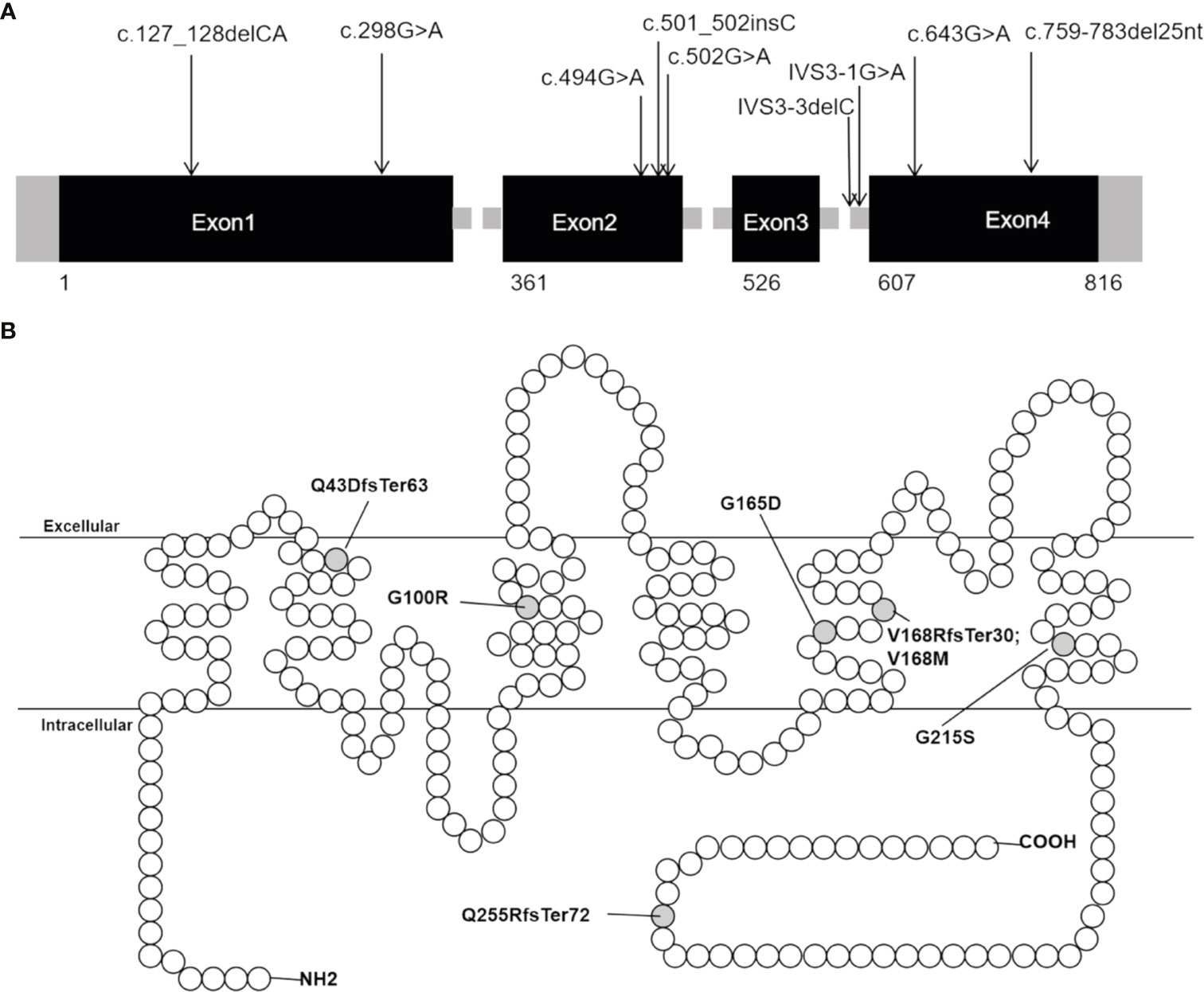
Figure 2 Distribution of AQP2 mutations detected in our patients with NDI. (A) Distribution of the nine mutations in AQP2 gene. (B) Schematic diagram of the primary structure of AQP2, showing the location of changed amino acid. The black rectangles indicate DNA coding sequence; the empty circles indicate amino acid of AQP2; the grey half-filled circles indicate amino acid alterations found in our study.
Clinical Presentations in NDI Patients
Detailed demographic data, manifestations, biochemical characteristics and imaging results of the 7 probands with congenital NDI were reviewed and described. The demographic features and manifestations were shown in Table 2. All probands included were male, and 4 out of 13 patients were female in all families. The mean age of patients was 14.1 years (14.1 ± 13.8, range 2-42 years). The onset age was younger than 1 year old in all patients. The most common clinical presentations were polydipsia and polyuria (7/7) and intermittent fever (6/7). Among 7 patients, 3 had short stature and 1 had mental impairment.

Table 2 Clinical characteristics in patients with NDI.
Biochemical and Imaging Characteristics in NDI Patients
Biochemical and imaging characteristics were shown in Table 3. The urine output was increased in all patients, ranging from 4.3 ml/kg/h to 6.9ml/kg/h in children and adolescents, and 12.8 ml/kg/h to 13.9 ml/kg/h in adults. Urine SG and osmolality were decreased in all patients (6/6 and 7/7, respectively). However, serum osmolality and sodium were maintained at a relatively high level (307±23 mOsm/kgH2O; 145±6 mmol/L). Level of UA was elevated in 2 patients, and level of creatinine was normal in all patients (7/7). The urological ultrasonography was assessed in 6 patients except for the youngest child. Mild hydronephrosis of the left kidney and mild dilation of the right ureter were found in Case4. Hydronephrosis and dilation of the kidney and ureter as well as irregularly thickened bladder wall were found in Case6 and Case7.
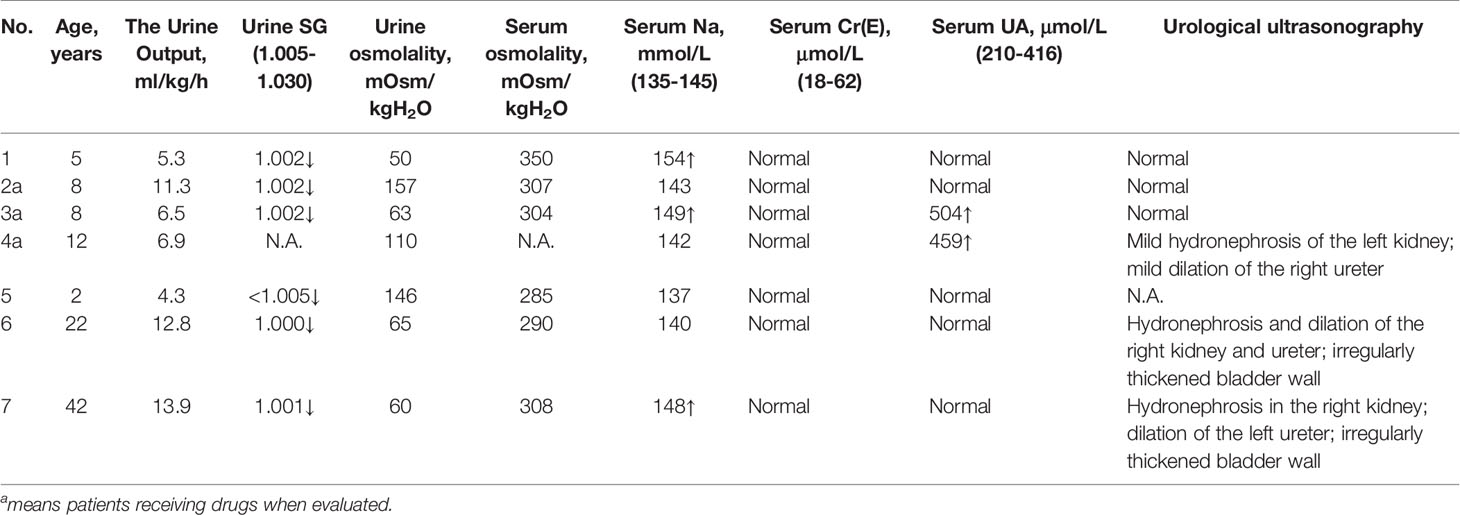
Table 3 Biochemical and radiological characteristics in patients with NDI.
Drug Treatment
According to available data, for Case1, low-salt diet and good habits of drinking and voiding were recommended. Oral administration of hydrochlorothiazide (12.5 mg) and amiloride (1.25 mg) were given four times per day. The urine output and serum sodium were decreased when he visited us again after 3 months (5 to 3 L/24hours; 154 to 150 mmol/L). For Case6, hydrochlorothiazide (25 mg) and amiloride (2.5 mg) were given four times per day. The urine volume in 24h was decreased (20 to 9 L), urine osmolarity was increased (65 to 134 mOsm/kg H2O), and serum sodium was decreased (140 to 138 mmol/L) at the second visit.
Genotype-Phenotype Correlation Analysis
Level of serum sodium in NDI patients with compound het mutations was higher than non-compound het mutations (150.3 ± 3.2 vs 140.5 ± 2.6 mmol/L, p=0.007, shown in Supplementary Table 2). There was no significant difference in onset-age, urine osmolality or other manifestations between two groups (Supplementary Table 2).
Discussion
To the best of our knowledge, this is the first and largest case series of congenital NDI caused by AQP2 mutation in Chinese population. So far, there were 71 putative disease-causing AQP2 mutations reported to cause NDI (February 25th, 2021; The Human Gene Mutation Database at the Institute of Medical Genetics in Cardiff; http://www.hgmd.cf.ac.uk), including 55 missense/nonsense mutations, 4 splicing mutations, 9 small deletions, 2 small insertions, and 1 gross mutation. Here, we established genetic profile of AQP2 in our patients, including 4 missense mutations, 2 splicing mutations and 3 frameshift mutations, showing a relatively even distribution in multiple domains. Further, we identified 3 novel AQP2 mutations (p.G165D, p.Q255RfsTer72 and IVS3-3delC), which updated AQP2 mutation spectrum and provided basis for functional studies and drug development.
Among these mutations, seven out of nine AQP2 mutations caused recessive NDI and two caused dominant NDI. The novel missense mutation (G165D, Case3) exhibiting a recessive mode was located in transmembrane domain-5 of AQP2 and predicted to be pathogenic by software, which may lead to retention in the endoplasmic reticulum (ER), the main molecular pathogenesis by recessive mutations reported previously (13). The novel frameshift mutation (p.Q255RfsTer72, Case4) located in the C-terminal and the novel splicing mutation (IVS-3delC) identified in two independent families with NDI (Case6 and Case7) exhibited dominant pattern, which may cause misrouting of AQP2 heterotetramer formed by wild-type and mutant monomer according to previous studies (17, 20). The heterozygous (p.Q43DfsTer63 and p.V168RfsTer30, Case1) and homozygous mutations (G215S, Case2) were reported by us before (27). The splicing mutation (IVS3-1G>A) was identified in a sporadic patient by other groups (28). Missense mutations (G100R and V168M) were predicted to impair monomer folding or pore features by previous studies (18, 29, 30). All mutations identified in our patients explained the NDI phenotype and provided persuasive evidence for studying molecular biology of AQP2. There were also limitations in this study, including the lack of next-generation sequencing and molecular characterization of mutations. Clinical characteristics of NDI patients were described and genotype-phenotype correlation analysis was performed. We firstly found level of serum sodium was higher in patients with compound het AQP2 mutations than non-compound het mutations, suggesting more severe phenotype due to more damage to AQP2 tetramer functions, which may be explained by functional analysis in a larger sample population (20). In our patients, the onset-age was younger than 1 year old. Common presentations were polydipsia, polyuria, and intermittent fever, and biochemical features were characterized by high osmotic hypernatremia and low osmotic urine. Results also indicated elevated level of serum uric acid was found in 2 cases, possibly due to inhibition of uric acid excretion by hydrochlorothiazide. Dilation of the urinary tract was found in 3 of 7 patients and the youngest was only 12 years old, which was a severe complicaton of NDI, especially common in patients with poor voiding habits. Anatomic obstructions should be considered in patients with hydronephrosis (1, 31). Compared to previous studies (32–35), we characterized phenotypes from more cases in a different population and found similar spectrum.
For treatment in NDI, low-salt diet, good habits of drinking and voiding were recommended. Adequate hydration should be noted for infants. Hypotonic fluids are usually appropriate for intravenous infusion, but hyponatremia should be avoided when other salt losses exist, such as diarrhea. Common drugs included hydrochlorothiazide and amiloride (7, 17, 36). Thiazides blocked sodium-chloride cotransporter in the distal convoluted tubule and activated renin-angiotensin-aldosterone system, leading to increased sodium and water resorption in proximal tubule and decreased urine excretion (37). Amiloride was a combination drug with thiazides to prevent hypokalemia. Here, urine output was decreased by nearly 50% after treatment with hydrochlorothiazide in two cases, and level of serum sodium was decreased. However, lack of long-term follow-up data and the limited sample size were also limitations. Novel treatments including molecular chaperones and gene therapy may cure congenital NDI, but few clinical data are currently available (7, 17, 36).
In conclusion, we performed gene mutation analysis in 7 Chinese families and identified 9 AQP2 mutations, including 3 novel mutations (p.G165D, p.Q255RfsTer72 and IVS3-3delC). Then, we characterized clinical, biochemical and imaging features in these patients, and found similar phenotype spectrum compared to previous study. Further, we found level of serum sodium was higher in patients with compound het AQP2 mutations than non-compound het mutations. This knowledge broadens genotypic and phenotypic spectrum for rare congenital NDI and provided basis for studying molecular biology of AQP2.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by the Institutional Review Board and Ethics Committee of PUMCH. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
Study design: LD and WX. Study conduct: QL, DT, and JC. Data collection: QL, DT, and JC. Data analysis: QL, DT, JC, LD, and WX. Data interpretation: QL, DT, JC, LD, and WX. Drafting manuscript: QL. Revising manuscript content: QL, DT, JC, LD, and WX. Approving final version of manuscript: QL, LD, and WX. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Natural Science Fund (No. 81670814), National Key R&D Program of China (2018YFA 0800801) and National Natural Science Fund (No. 81970757).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We appreciate our patients and their families for their participating in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2021.686818/full#supplementary-material
Supplementary Table 1 | The PCR primer sets and conditions for AQP2 and AVPR2 gene.
Supplementary Table 2 | Comparison of phenotypes in patients with compound het AQP2 mutations and non-compound het mutations. Data presents as Mean ± SD or Median (minimum, maximum), p<0.05 was considered as significant.
References
1. Bockenhauer D, Bichet DG. Pathophysiology, Diagnosis and Management of Nephrogenic Diabetes Insipidus. Nat Rev Nephrol (2015) 11(10):576–88. doi: 10.1038/nrneph.2015.89
2. Fushimi K US, Hara Y, Hirata Y, Marumo F, Sasaki S. Cloning and Expression of Apical Membrane Water Channel of Rat Kidney Collecting Tubule. Nature (1993) 361:549–52. doi: 10.1038/361549a0
3. Sasaki. S CM, Kikuchi E, Rai T, Uchida S. Hereditary Nephrogenic Diabetes Insipidus in Japanese Patients: Analysis of 78 Families and Report of 22 New Mutations in AVPR2 and AQP2. Clin Exp Nephrol (2013) 17:338–44. doi: 10.1007/s10157-012-0726-z
4. Wesche D DP, Knoers NV. Congenital Nephrogenic Diabetes Insipidus: The Current State of Affairs. Pediatr Nephrol (2012) 27:2183–204. doi: 10.1007/s00467-012-2118-8
5. Hillman DA, Neyzi O, Porter P, Cushman A, Talbot NB. Renal (Vasopressin-Resistant) Diabetes Insipidus; Definition of the Effects of a Homeostatic Limitation in Capacity to Conserve Water on the Physical, Intellectual and Emotional Development of a Child. Pediatrics (1958) 21:430–5. doi: 10.1016/S0022-3476(58)80276-0
6. Vest M, Talbot NB, Crawford JD. Hypocaloric Dwarfism and Hydronephrosis in Diabetes Insipidus. Am J Dis Child (1963) 105:175–81. doi: 10.1001/archpedi.1963.02080040177008
7. Uribarri J KM. Hereditary Nephrogenic Diabetes Insipidus and Bilateral Nonobstructive Hydronephrosis. Nephron (1993) 653(3):346–89. doi: 10.1159/000187510
8. Milano S, Carmosino M, Gerbino A, Svelto M, Procino G. Hereditary Nephrogenic Diabetes Insipidus: Pathophysiology and Possible Treatment. An Update. Int J Mol Sci (2017) 18(11):2385. doi: 10.3390/ijms18112385
9. Jung HJ KT. Molecular Mechanisms Regulating Aquaporin-2 in Kidney Collecting Duct. Am J Physiol Renal Physiol (2016) 311(6):F1318–F28. doi: 10.1152/ajprenal.00485.2016
10. Bourque CW. Central Mechanisms of Osmosensation and Systemic Osmoregulation. Nat Rev Neurosci (2008) 9(7):519–31. doi: 10.1038/nrn2400
11. Schenk AD WP, Scheuring S, de Groot BL, Muller SA, Stahlberg H, Philippsen A, et al. The 4.5 A Structure of Human AQP2. J Mol Biol (2005) 350:278–89. doi: 10.1016/j.jmb.2005.04.030
12. Moeller HB OE, Fenton RA. Regulation of the Water Channel Aquaporin-2 by Posttranslational Modification. Am J Physiol Renal Physiol (2011) 300:F1062–F73. doi: 10.1152/ajprenal.00721.2010
13. Chao Gao PJH, Wenzheng Z. AQP2: Mutations Associated With Congenital Nephrogenic Diabetes Insipidus and Regulation by Post-Translational Modifications and Protein-Protein Interactions. Cells (2020) 2172:1–18. doi: 10.3390/cells9102172
14. Fushimi K SS, Marumo F. Phosphorylation of Serine 256 is Required for cAMP-Dependent Regulatory Exocytosis of the Aquaporin-2 Water Channel. J Biol Chem (1997) 272(23):14800–4. doi: 10.1074/jbc.272.23.14800
15. Trimpert C, van den Berg DT, Fenton RA, Klussmann E, Deen PM. Vasopressin Increases S261 Phosphorylation in AQP2-P262L, a Mutant in Recessive Nephrogenic Diabetes Insipidus. Nephrol Dial Transplant (2012) 27(12):4389–97. doi: 10.1093/ndt/gfs292
16. Hoffert JD PT, Wang G, Shen RF, Knepper MA. Quantitative Phosphoproteomics of Vasopressin-Sensitive Renal Cells: Regulation of Aquaporin-2 Phosphorylation at Two Sites. Proc Natl Acad Sci USA (2006) 103(18):7159–64. doi: 10.1073/pnas.0600895103
17. Moeller HB, Rittig S, Fenton RA. Nephrogenic Diabetes Insipidus: Essential Insights Into the Molecular Background and Potential Therapies for Treatment. Endocr Rev (2013) 34(2):278–301. doi: 10.1210/er.2012-1044
18. Calvanese L, D’Auria G, Vangone A, Falcigno L, Oliva R. Structural Basis for Mutations of Human Aquaporins Associated to Genetic Diseases. Int J Mol Sci (2018) 19(6):1577. doi: 10.3390/ijms19061577
19. Frick A, Eriksson UK, de Mattia F, Oberg F, Hedfalk K, Neutze R, et al. X-Ray Structure of Human Aquaporin 2 and its Implications for Nephrogenic Diabetes Insipidus and Trafficking. Proc Natl Acad Sci USA (2014) 111(17):6305–10. doi: 10.1073/pnas.1321406111
20. El Tarazi A, Lussier Y, Da Cal S, Bissonnette P, Bichet DG. Functional Recovery of AQP2 Recessive Mutations Through Hetero-Oligomerization With Wild-Type Counterpart. Sci Rep (2016) 6:33298. doi: 10.1038/srep33298
21. De Mattia F, Savelkoul PJ, Kamsteeg EJ, Konings IB, van der Sluijs P, Mallmann R, et al. Lack of Arginine Vasopressin-Induced Phosphorylation of Aquaporin-2 Mutant AQP2-R254L Explains Dominant Nephrogenic Diabetes Insipidus. J Am Soc Nephrol (2005) 16(10):2872–80. doi: 10.1681/ASN.2005010104
22. Hui Li C-YJ, Zong X-N, Zhang Y-Q. Height and Weight Standardized Growth Charts for Chinese Children and Adolescents Aged 0 to 18 Years. Zhonghua Er Ke Za Zhi (2009) 47(7):487–92. doi: 10.3760/cma.j.issn.0578-1310.2009.07.003
23. Adzhubei IA SS, Peshkin L, Ramensky VE, Gerasimova A, Bork P. A Method and Server for Predicting Damaging Missense Mutations. Nat Methods (2010) 7:248–9. doi: 10.1038/nmeth0410-248
24. Kumar P HS, Ng PC. Predicting the Effects of Coding non-Synonymous Variants on Protein Function Using the SIFT Algorithm. Nat Protoc (2009) 4:1073–81. doi: 10.1038/nprot.2009.86
25. Kian Huat Lim WGF. Spliceman—a Computational Web Server That Predicts Sequence Variations in pre-mRNA Splicing. Bioinformatics (2012) 28(7):1031–2. doi: 10.1093/bioinformatics/bts074
26. Richards S AN, Bale S, Bick D, Das S, Gastier-Foster J. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med (2015) 17:(405–424). doi: 10.1038/gim.2015.30
27. Jing Cen MN, Duan L, Gu F. Novel Autosomal Recessive Gene Mutations in Aquaporin-2 in Two Chinese Congenital Nephrogenic Diabetes Insipidus Pedigrees. Int J Clin Exp Med (2015) 8(3):3629–39. doi: 10.1507/endocrj.50.473
28. Toshihiro Tajima KO, Satoh K, Nakae J, Fujieda K. Two Novel Aquaporin-2 Mutations in a Sporadic Japanese Patient With Autosomal Recessive Nephrogenic Diabetes Insipidus. Endocr J (2003) 50(4):473–6. doi: 10.1507/endocrj.50.473
29. Carroll P A-MH, Al-Abbad A, Al-Hassoun I, Al-Hamed M, Al-Amr R, Butt AI, et al. Novel Mutations Underlying Nephrogenic Diabetes Insipidus in Arab Families. Genet Med (2006) 87(7):443–7. doi: 10.1097/01.gim.0000223554.46981.7a
30. Cristina Boccalandro FDM, Guo D-C, Xue L, Orlander P, King TM, Gupta P, et al. Characterization of an Aquaporin-2 Water Channel Gene Mutation Causing Partial Nephrogenic Diabetes Insipidus in a Mexican Family: Evidence of Increased Frequency of the Mutation in the Town of Origin. J Am Soc Nephrol (2004) 15(5):1223–31. doi: 10.1097/01.ASN.0000125248.85135.43
31. Yoo TH RD, Song YS, Lee SC, Kim HJ, Kim JS, Choi HY, et al. Congenital Nephrogenic Diabetes Insipidus Presented With Bilateral Hydronephrosis: Genetic Analysis of V2R Gene Mutations. Yonsei Med (2006) 47:126–30. doi: 10.3349/ymj.2006.47.1.126
32. García Castaño A, Pérez de Nanclares G, Madariaga L, Aguirre M, Chocron S, Ariceta G, et al. Novel Mutations Associated With Nephrogenic Diabetes Insipidus. A Clinical-genetic study. Eur J Pediatr (2015) 174(10):1373–85. doi: 10.1007/s00431-015-2534-4
33. Ramón Peces JN. Severe Congenital Nephrogenic Diabetes Insipidus in a Compound Heterozygote With a New Large Deletion of the AQP2 Gene. A case report. Mol Genet Genomic Med (2019) 7(4):e00568. doi: 10.1002/mgg3.568
34. Musone D, Nicosia V, Treglia A, Amoroso F, de Gemmis P, Hladnik U, et al. Compound Heterozygosis With Novel AQP2 Gene Mutation in Sisters Affected by Autosomal Congenital Nephrogenic Diabetes Insipidus. G Ital Nefrol (2020) 37(2). doi: 10.1515/jpem-2013-0097
35. Long BC, Weber ZJ, Oberin JM, Sutter DE, Berg JM, et al. Nephrogenic Diabetes Insipidus in a 15-Year-Old Hispanic Female With a Novel AQP2 Mutation. J Pediatr Endocrinol Metab (2019) 32(9):1031–4. doi: 10.1515/jpem-2019-0099
36. Babey M KP, Robertson GL. Familial Forms of Diabetes Insipidus: Clinical and Molecular Characteristics. Nat Rev Endocrinol (2011) 7:701–14. doi: 10.1038/nrendo.2011.100
Keywords: Nephrogenic diabetes insipidus, aquaporin-2, mutation, water resorption, vasopressin V2 receptor
Citation: Li Q, Tian D, Cen J, Duan L and Xia W (2021) Novel AQP2 Mutations and Clinical Characteristics in Seven Chinese Families With Congenital Nephrogenic Diabetes Insipidus. Front. Endocrinol. 12:686818. doi: 10.3389/fendo.2021.686818
Received: 28 March 2021; Accepted: 24 May 2021;
Published: 10 June 2021.
Edited by:
Valentino Cherubini, Azienda Ospedaliero Universitaria Ospedali Riuniti, ItalyReviewed by:
Bellamkonda K. Kishore, University of Utah Health Care, United StatesGeorge Paltoglou, National and Kapodistrian University of Athens, Greece
Andras Balla, Semmelweis University, Hungary
Copyright © 2021 Li, Tian, Cen, Duan and Xia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weibo Xia, eGlhd2VpYm84MzAxQDE2My5jb20=; Lian Duan, ZHVhbmxpYW5wdW1jQDE2My5jb20=