 Andrew Folick
Andrew Folick Suneil K. Koliwad
Suneil K. Koliwad Martin Valdearcos
Martin Valdearcos- 1Diabetes Center, University of California, San Francisco, San Francisco, CA, United States
- 2Department of Medicine, University of California, San Francisco, San Francisco, CA, United States
In mammals, myeloid cells help maintain the homeostasis of peripheral metabolic tissues, and their immunologic dysregulation contributes to the progression of obesity and associated metabolic disease. There is accumulating evidence that innate immune cells also serve as functional regulators within the mediobasal hypothalamus (MBH), a critical brain region controlling both energy and glucose homeostasis. Specifically, microglia, the resident parenchymal myeloid cells of the CNS, play important roles in brain physiology and pathology. Recent studies have revealed an expanding array of microglial functions beyond their established roles as immune sentinels, including roles in brain development, circuit refinement, and synaptic organization. We showed that microglia modulate MBH function by transmitting information resulting from excess nutrient consumption. For instance, microglia can sense the excessive consumption of saturated fats and instruct neurons within the MBH accordingly, leading to responsive alterations in energy balance. Interestingly, the recent emergence of high-resolution single-cell techniques has enabled specific microglial populations and phenotypes to be profiled in unprecedented detail. Such techniques have highlighted specific subsets of microglia notable for their capacity to regulate the expression of lipid metabolic genes, including lipoprotein lipase (LPL), apolipoprotein E (APOE) and Triggering Receptor Expressed on Myeloid Cells 2 (TREM2). The discovery of this transcriptional signature highlights microglial lipid metabolism as a determinant of brain health and disease pathogenesis, with intriguing implications for the treatment of brain disorders and potentially metabolic disease. Here we review our current understanding of how changes in microglial lipid metabolism could influence the hypothalamic control of systemic metabolism.
Introduction
The brain contains the second highest lipid concentration in the body, behind adipose tissue, and lipids constitute 50% of the brain’s dry weight (1). Beyond serving as energy substrates, brain lipids play a wide range of roles in cellular physiology, including membrane organization, protein modification, cell-cell interactions, membrane trafficking, energy storage and signal transduction. Lipid metabolism within the brain is therefore highly regulated, and disruption of central nervous system (CNS) lipid homeostasis can produce devastating neurological consequences. For instance, impaired cholesterol or fatty acid metabolism leads to severe neurodevelopmental defects, intellectual disabilities, and motor dysfunction (2, 3). Neurons themselves engage in relatively low levels of lipid synthesis, in contrast to recent studies which suggest that glial cells are critical for both the synthesis and metabolism of lipids in the brain (4). For example, an “astrocyte-neuron lactate shuttle” has been postulated, in which astrocytes metabolize lipids in order to provide energy substrates for neurons (5) and regulate neurite outgrowth and synaptogenesis (6). Oligodendrocytes are also highly active in lipid metabolism, and have been shown to synthesize the cholesterol necessary for myelin sheath formation (7). Importantly, microglia can both synthesize and accumulate lipids, and both microglial lipid composition and lipid metabolic capability are increasingly implicated in determining their ability to regulate neuronal functions as well as their contributions to brain pathology (8–10).
Integration of lipidomic and genomic datasets can elucidate gene-environment (e.g. diet) interactions regulating lipid metabolism as a means to reveal biomarkers predictive of metabolic disease (11). Recent studies utilizing lipidomics and single-cell RNA sequencing (scRNA-seq) have revealed intriguing heterogeneity among microglia, and the importance of lipid and lipoprotein metabolism in microglial physiology (12, 13). This work had been done primarily in the context of specific neurodegenerative diseases, while our lab and others have investigated the role of dietary lipids in the immunological activation of microglia in the context of obesity and metabolism (14–16). This review summarizes our current knowledge of lipid metabolism in microglia, with a focus on its potential contribution to hypothalamic physiology and dysfunction in the context of metabolic disease.
Lipid Metabolism in the Brain
The brain has a high energy demand, and historical consensus has been that its energy requirements are almost entirely satisfied by glucose metabolism. However, this dogma has been recently challenged, as it was shown that approximately 20% of the brain’s total energy requirement is met though the oxidation of fatty acids (FAs) (17). Additionally, FA oxidation by cultured mouse brain slices is increased by withdrawing extracellular glucose (18). Astrocytes and microglia likely contribute significantly to brain utilization of FAs as energy substrates (18, 19). Astrocytes express higher levels of key FA oxidation enzymes, however detailed cell-type specific experiments comparing the capacity to oxidize fatty acids in vivo have not been reported (18). Neurons may have also the capacity to utilize FAs as an energy source, as a recent study using rat brain demonstrated that isolated neuronal mitochondria utilize FAs as an energy source even in the presence of other mitochondrial substrates (20). By contrast, the capacity of neurons to oxidize FAs for energy is known to be quite limited (21). One reason for this limited capacity may be that neurons are highly susceptible to reactive oxygen stress (ROS) generated by FA oxidation, and it is widely accepted that mitochondrial oxidative stress and dysfunction contribute to neurologic disorders (22). Thus, the selective pressure to avoid oxidative stress may underlie the neuronal preference to oxidize glucose as their primary fuel source (21).
Neuron-Glia Interactions in Brain Lipid Metabolism
Given the importance of lipids to overall brain physiology, the limited lipid metabolic capacity of neurons themselves has prompted exploration into the essential roles of glial cells in lipid metabolism, storage and synthesis. This effort has revealed the importance of coordinated lipid metabolism and trafficking between neurons and glia, as exemplified by work done to establish a genetic link between Parkinson’s disease (PD) and genes controlling lipid metabolism (23, 24). Indeed, both PD patients and experimental animal models of PD exhibit abnormal lipid accumulation in dopaminergic neurons and their surrounding microglia, but have a reduced lipid load in adjacent astrocytes. One recent study found that a Western diet impairs recovery from demyelinating injuries, by inhibiting microglial phagocytosis and clearance of lipid debris (25). Another study found that in the setting of demyelination, microglia synthesize desmosterol, the immediate cholesterol precursor and liver X receptor (LXR) agonist, and that microglial sterol synthesis is essential for efficient remyelination (26). As oligodendrocytes were thought to be the primary synthesizers of sterols in the brain, and require sterols for myelination, this indicates a new role for intercellular trafficking of sterols. Together, these findings indicate that a disturbance in the multicellular handling and trafficking of lipids plays may play a key role in PD pathogenesis (27).
However a broader, more systematic understanding of the regulation of lipid metabolism and flux between brain cell types in different physiological and pathological states is limited, with few detailed lipidomic profiles of CNS cell types having been published to date. However, recent studies have revealed that certain lipid species are enriched in distinct cell types and brain regions. For instance, microglia are enriched in sphingolipids and characterized by high levels of sphingomyelin species, which are almost absent in neurons and oligodendrocytes (13). Microglia in particular are essential for the clearance and recycling of lipid debris, and recent work has shown that aging-related defects in microglial lipid handling contribute both to their inflammatory activation and to the impairment of their response to demyelination (28). Further insights into microglial lipid metabolism will be essential to understanding how lipids impact brain function.
Effects of Diet on Brain Lipid Composition and Metabolism
In addition to having a relatively high absolute lipid content, the composition of brain lipids is also distinct from that of other tissues in the body. Indeed, 75% of lipids in mammals are present exclusively in neural tissues, underscoring that brain function has unique lipid requirements (29). The brain is the most cholesterol-rich organ in the body, and brain cholesterol is primarily supplied by local de novo synthesis (30). Cholesterol is essential for neuronal physiology, and defects in cholesterol metabolism leads to neurological diseases (31). Despite the primary de novo synthesis, diet may also affect sterol metabolism in the brain. The cholesterol metabolite 27-hydroxycholesterol (27-OHC) can pass through the BBB, and 27-OHC is significantly increased in plasma and adipose tissue of animals on HFD (32). Excess 27-OHC impairs brain glucose uptake (33). Additionally, peripheral cholesterol contained in circulating HDL, undergoes selective uptake mediated by the scavenger receptor class B type 1 for entry into the brain (34). In humans, low HDL levels are associated with increased risk for PD (35). Interestingly, genetic HDL deficiency caused increased astrogliosis, but not microgliosis, in the hypothalamus (36).
On the other hand, some FAs must be transported into the brain from the systemic circulation in a dynamic process (37). For instance, the brain is rich in long-chain polyunsaturated fatty acids (LC-PUFAs), particularly arachidonic acid (AA), eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA) but the brain has limited capacity to synthesize LC-PUFAs (38, 39). These have to therefore be provided through the diet, either as precursors, n-6 linoleic acid (LA) and n-3 α−linolenic acid (ALA), or as preformed AA and DHA (40, 41). Indeed, several radiolabeled studies have shown incorporation of circulating FAs into neurons (37). FAs could passively diffuse across the blood brain barrier (BBB), as shown for palmitate (PA), AA and DHA (42–44), however FA transporters such as FAT/CD36 may also play a key role in promoting the dissociation of FAs albumin in order to facilitate their diffusion across the BBB (45). In summary, brain lipid composition is highly regulated, and while distinct from peripheral lipid composition, is importantly influenced by circulating lipids including those from dietary sources.
The obesogenic high-fat diets (HFD) commonly used in mice, including the so-called “Western” diet with increased cholesterol levels, are characterized by a markedly high saturated fatty acid (SFA) content and a relatively low n-3 polyunsaturated fatty acids (PUFA) content, resulting in a high n-6/n-3 ratio (46). Given this, it is notable that studies suggest that not every type of fat is equally obesogenic when consumed in an isocaloric manner. Indeed, the profile of consumed fats, rather than strictly the energy they contain, may be critical for the development of obesity (47). Circulating lipid levels are affected by dietary fat composition; for example, one lipidomic analysis of postprandial plasma showed significant changes in the levels of 316 different lipids species after an individual switched from eating a breakfast based on dairy foods to one that was soy oil-based (48). Recent studies have also investigated the effect of dietary fat on the brain lipidome. Mice consuming a HFD have reduced EPA content in cerebral phospholipids and sphingolipids, in association with increased inflammation and consequently impaired brain function (49). By contrast, diets enriched in the n-3 fatty acids EPA and DHA induce a different set of alterations in the FA composition of brain phospholipids, including increasing the number of double bonds in several phospholipid species (13). Indeed, supplementing a standard saturated fat-rich HFD with a daily gavage of fish oil rich in EPA and DHA is sufficient to increase brain PUFAs and reduce brain gliosis in obese mice (50). In probing this further, it is notable that both EPA and DHA are precursors of pro-resolving lipid mediators with anti-inflammatory properties (51). In contrast, n-3 deficient neonatal mice exhibit increased microglial phagocytosis of synaptic elements resulting in altered neuronal morphology and function (10). Thus, dietary changes in EPA and DHA, as detected by monitoring dietary n-6/n-3 ratios, may directly modulate microglial polarization states in a manner relevant to CNS diseases associated with microglial dysfunction.
Integration of Lipid Signals by the Mediobasal Hypothalamus (MBH)
The MBH, defined here as the hypothalamic region containing the arcuate nucleus (ARC) and median eminence (ME), is strategically located to directly sense and coordinate a response to nutritional signals. The structure and function of the BBB within the ME and ventromedial ARC, as a circumventricular organs, is unique, being supplied by fenestrated capillaries (52–55). Thus, substances that do not cross into the brain parenchyma in other regions of the brain may pass into the ME and ARC with relative ease (56, 57). For example, very low density lipoproteins (VLDL) are not thought to cross the BBB (34), however we demonstrated rapid accumulation of VLDL within the MBH after intravenous administration, and this was predominantly localized to microglia in the ME and ARC (14). Indeed, a recent study showed that triglyceride (TG)-rich lipoproteins are sensed in the hypothalamus by an LPL-dependent mechanism (58), and the uptake of dietary PA, a common SFA, into the hypothalamus is remarkably higher than for other brain regions (59). Thus, specialized fuel-sensing neurons that form critical hypothalamic circuits are uniquely positioned to sense circulating glucose and lipid species, including FAs (60).
Whereas lipid sensing in the MBH may create responsiveness to nutritional lipids, there may be roles for lipid sensing in other brain regions as well. For instance, recent work has shown that both nutritional and parenteral TG exposure modulated activity of neurons in the mesocorticolimbic system (MCL) and affects behavioral and reward responses (61, 62). These effects were dependent on neuronal lipoprotein lipase, suggesting a direct response to TGs (61, 62). Radiolabeled triolein was able to be detected in whole brain after peripheral injection, suggesting that some intact TG may pass through the BBB, however the location of triolein accumulation within the brain was not determined (63). This sensing capacity may play a role in the context of autophagy, lipids housed within locally-generated lipoproteins (e.g. APOE), or perhaps by context-specific selective permeability of the vasculature in certain brain regions to circulating lipids. Further research is needed to tease apart these possibilities.
Lipid Sensing by Hypothalamic Neurons
Circulating FA levels are increased after consumption of a HFD (64, 65) and the rate of entry of FAs into the brain is proportional to their plasma concentration (59). Indeed, hypothalamic levels of free FAs are increased by HFD feeding, suggesting an important role for these FAs in hypothalamic lipid-sensing pathways (66). Supporting this, it has been shown that FAs modify neuronal firing rates in the ARC (67). Moreover, intracerebroventricular (ICV) infusion of the monounsaturated FA, oleic acid (OA), suppresses food intake and hepatic glucose production (68) indicating that FAs can signal nutrient availability to the brain. Furthermore, increased LCFA-CoA levels in hypothalamic neurons suppress endogenous glucose production suggesting that hypothalamic lipid sensing regulates glucose homeostasis through a mechanism involving the esterification of LCFAs to LCFA-CoAs (69). Also, ICV infusions of OA or DHA, but not PA, reduce food intake and body weight indicating a selective hypothalamic response to specific unsaturated fatty acids (UFAs). However, ICV and direct infusions of FAs into the brain are not physiological. Short-term (3 days) of HFD can cause rewiring of anorexigenic proopiomelanocortin (POMC) neurons in the ARC, suggesting a physiological role for lipid sensing in the hypothalamus (70). In further support of this concept, a recent elegant study found that intragastric administration of lipids inhibited the activity of hunger-promoting Agouti-related protein (AgRP) neurons in the MBH (71). Furthermore, the ability of lipid infusion to inhibit the activity of AgRP neurons was blunted in HFD-fed animals, suggesting a reduction in the lipid sensitivity of AgRP neurons in this context (72). In summary, there is clear evidence that a HFD, in particular dietary FAs, is sensed by hypothalamic neuronal pathways to regulate energy homeostasis. The precise mechanisms of lipid sensing by hypothalamic neurons have been well studied, and have been recently reviewed (73–75).
Dysregulated lipid metabolism in the hypothalamus may affect neuronal FA sensing and therefore contribute to the development of metabolic diseases. In particular, excessive lipid accumulation and resultant activation of cellular stress pathways can lead to disruption of hypothalamic function. During both acute and chronic HFD feeding, multiple inflammatory and stress response pathways are activated in the hypothalamus, leading the dysfunction of hypothalamic circuits regulating energy and glucose homeostasis, resulting in leptin and insulin resistance (76). In evaluating the specific changes in hypothalamic lipid composition induced by overconsumption, specific attention has been paid to how excess lipid accumulation drives ER stress in the hypothalamus (77). Rodent studies have shown that HFD feeding induces ER stress in multiple metabolic tissues including the hypothalamus (78). This response is not uniform across all hypothalamic nuclei and seems to be specific to the ARC but not other regions such as the paraventricular nucleus (PVN) (78). Induction of hypothalamic ER stress leads to increased food intake, reduced energy expenditure and resultant obesity, and this is mediated at least in part by defective α-MSH production among POMC neurons (79) and development of leptin resistance (80). Fat composition is important in this regard, because saturated fats (e.g., PA) are more deleterious than unsaturated fats to hypothalamic neurons (81), and ER stress sensors are specifically activated by increasing ER membrane lipid saturation (82). Interestingly, PA-induced ER stress in hypothalamic neurons decreases protein abundance and function of the melanocortin 4 receptor (MC4R) (83), and central inhibition of lipid oxidation and ER stress is sufficient to restore hypothalamic lipid sensing and energy homeostasis in mice (84).
ER stress and inflammatory pathways are functionally coupled, and induction of CNS ER stress in lean mice is sufficient to activate NF-κB signaling (85). Furthermore, there is convincing evidence that ER stress activates the NLRP3 inflammasome in myeloid cells through different pathways in a context-dependent manner. We demonstrated that IRE1α, a critical ER sensor of both unfolded protein and saturated lipid stress, mediates SFA-induced IL-1β secretion in macrophages upon sensing increasing saturation of cellular phospholipids (86). However, most metabolic studies in the hypothalamus have focused on neuronal ER stress, and the potential contribution of ER stress in glial cells to hypothalamic dysfunction has not been explored yet. However, a recent study did show that disrupting proteasome activity in microglia triggers the induction of a type I interferon (IFN) response in an IRE1-dependent manner (87), suggesting that microglial ER stress is worth studying in the context of hypothalamic regulation.
Lipid Sensing by Non-Neuronal Cells
Hypothalamic neurons are critical to the regulation of energy and glucose homeostasis, and our understanding of neuronal circuits controlling metabolism has advanced greatly over the past decade. However, recent studies implicate non-neuronal cells, including microglia, as physiologic regulators of hypothalamic function as well. For instance, astrocytes are the most abundant glial cells in the CNS and are involved in multiple fundamental processes, including metabolic homeostasis, neurovascular coupling, and BBB maintenance (88). Recent studies show that disrupting astrocyte lipid homeostasis may contribute to neurological disorders (89, 90). In addition, astrocytes participate in immune responses, and HFD consumption induces morphological changes in hypothalamic astrocytes (91). Astrocytes can influence hypothalamic circuits involved in the control of feeding and energy metabolism, at least in part by regulating extracellular levels of adenosine (92, 93). Furthermore, a recent study suggested that astrocytic insulin signaling regulates hypothalamic glucose sensing and systemic glucose metabolism (94). Also, astrocytes in the MBH can respond to acute changes in nutritional exposure, with morphological changes after overnight fasting (91), or as soon as 1-hr post-prandially (95). Interestingly, post-prandial retraction of astrocytes surrounding POMC neurons was only seen with standard chow diet, but not HFD (95). Tanycytes are radial glia-like cells that line the wall of the third ventricle in the brain, a privileged position to integrate multiple peripheral inputs (96, 97). Tanycytes can sense nutrients such as FAs in the cerebrospinal fluid (CSF), facilitate the transport of metabolic hormones across the BBB, and integrate signals to regulate appetite and energy balance (98, 99).
Microglia are increasingly being recognized as highly dynamic cells that continuously monitor for alterations to their environment, and assume different states of activation according to the unique CNS microenvironment in which they reside. Within the hypothalamus, microglia are emerging as key physiological mediators, both in the context of normal hypothalamic function and regulating the metabolic response to HFD. As such, key details of microglial lipid sensing and metabolic regulation have gained considerable interest, and are therefore reviewed below.
Microglia as Novel Regulators of Hypothalamic Function
Recent findings reveal an expanding array of functions for microglia, beyond their established roles as immune sentinels and phagocytic removers of cellular debris. These include roles in synaptic organization (100), neuronal excitability (101) and trophic support for brain repair (102) (Figure 1). Given the vital role of microglia in maintaining CNS homeostasis, it is not surprising that several brain disorders are associated with microglial dysfunction (103). Disrupting the interactions between neurons and microglia has devastating effects on memory, anxiety and other behavioral domains, demonstrating the importance of myeloid cells in brain physiology (104). Furthermore, interactions between microglia and astrocytes have been implicated in brain health and disease (105), and their cross-talk may play an important role in HFD-induced hypothalamic dysfunction. Activated microglia can induce reactive astrocytes by secreting proinflammatory molecules, such as IL-1α, TNF and C1q as previously demonstrated in a lipopolysaccharide (LPS)-induced murine neuroinflammation model (106). In addition, recent work suggests that microglial activity is directly regulated by metabolites of dietary tryptophan metabolism produced by commensal flora, and that this response controls a downstream inflammatory response among astrocytes (107). Recently, we showed that the inflammatory signaling of microglia dictates susceptibility to diet-induced hypothalamic dysfunction and obesity (15).
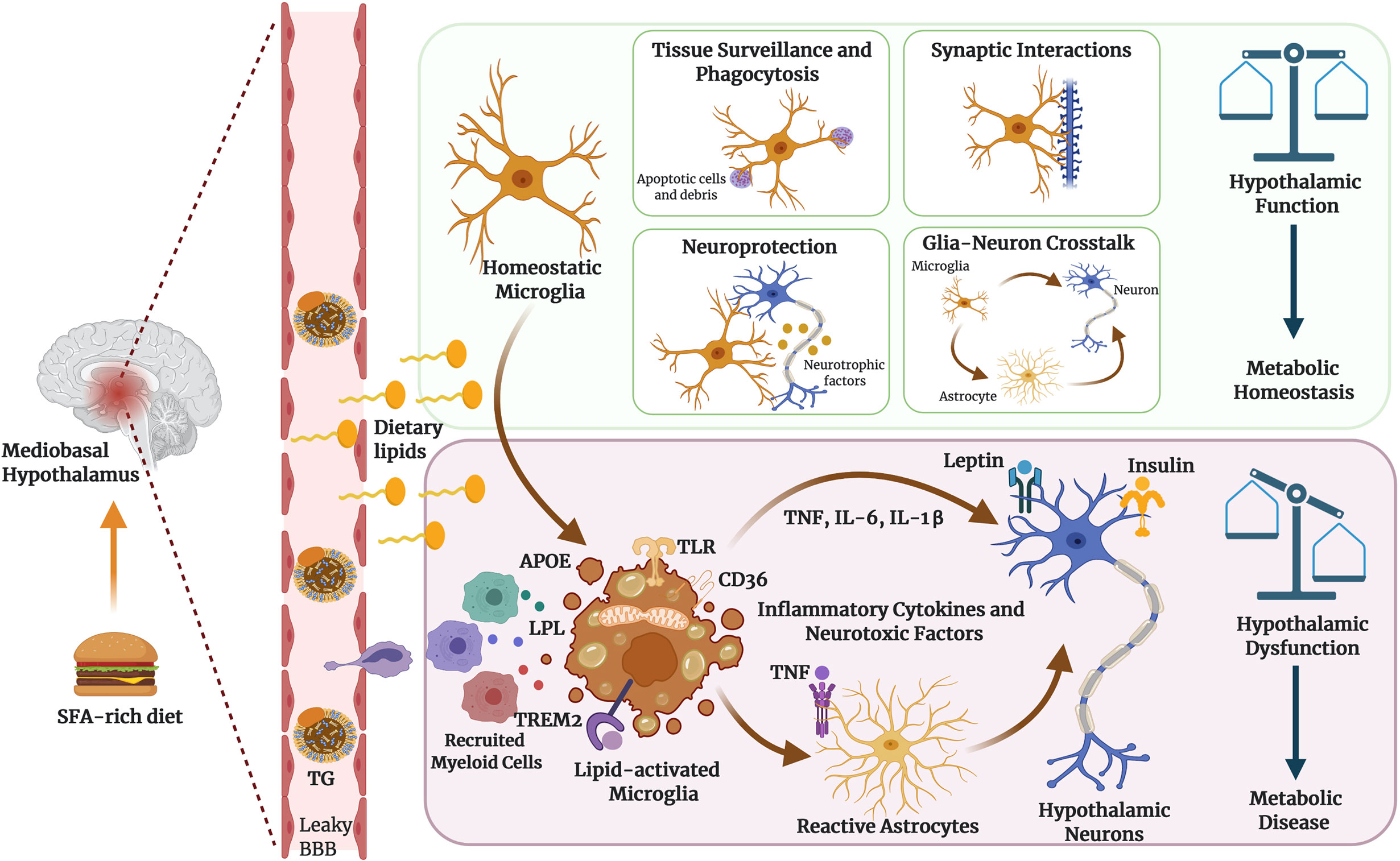
Figure 1 Microglia as dynamic cellular mediators of hypothalamic function. Microglia can perform diverse functions to maintain brain homeostasis, actively screening the surroundings, intercellular communication and remodeling the brain circuits through synaptic pruning and neuronal plasticity. Hypothalamic microglia integrate systemic metabolic signals such as dietary lipids to establish functional states that influence neuronal control of energy homeostasis. TG: refers to triglyceride-rich lipoproteins, including chylomicrons and VLDL particles. Figure created with BioRender.com.
Microglial Inflammatory Signaling Regulates Hypothalamic Immune Response to Dietary Excess
Chronic low-grade inflammation is considered one of the hallmarks of metabolic disease, and activation of inflammatory pathways have been described in several metabolic tissues. Animal and human studies have identified white adipose tissue (WAT) as the primary site where inflammation is initiated and exacerbated in response to weight gain (108). Obesity promotes drastic changes in the resident immune cell profile and function in WAT. Adipose tissue macrophages adopt a metabolically activated (MMe) phenotype distinct from that associated with classical “M1” activation, upregulating proteins involved in lipid processing including ABCA1, PLIN2 and CD36 to maintain adipose tissue homeostasis (109). Moreover, a novel and conserved macrophage population called lipid-associated macrophages (LAMs) is involved in controlling WAT lipid homeostasis has been recently described in multiple obesity-related mouse models (110).
Interestingly, recent studies have provided evidence that HFD consumption also increases the expression of genes governing inflammatory signaling in the hypothalamus (111–113). This phenomenon has also been described in human obesity, and obese individuals without a systemic disease showed markedly increased levels of inflammatory markers in the hypothalamus compared to healthy non-obese individuals (114, 115). With this in mind, it is notable that HFD consumption in mice rapidly increases the accumulation and activation of microglial populations secreting inflammatory cytokines specifically in the MBH (14). Moreover, the activation of hypothalamic inflammatory pathways in response to HFD consumption is much more rapid than it is in peripheral tissues such as WAT, even preceding any significant diet-induced weight gain, suggesting that the inflammatory response of the MBH to dietary excess is a cause, rather than a consequence, of obesity (113). Indeed, a single high-fat meal is sufficient to induce morphological changes and increased Iba1 expression in hypothalamic microglia (16).
We have shown that either pharmacologically depleting resident microglia, or genetically restraining their inflammatory capacity via NF-κB signaling, protects mice from diet-induced hyperphagia and weight gain, whereas specifically forcing NF-κB-dependent microglial inflammatory activation reduces energy expenditure and increases both food intake and weight gain even in absence of a dietary challenge (15). Microglial inflammatory signaling may induce obesity by causing hypothalamic neuronal dysfunction, including the induction of neuronal insulin and leptin resistance (76). Moreover, prolonged microglial activation may also induce apoptosis of anorexigenic/catabolic POMC neurons (116).
Metabolic Plasticity of Microglia
Microglia have the ability to adapt their metabolic pathways to use the energy substrates available in their local environment, and to acquire diverse and complex phenotypes during inflammatory activation in response to an insult or injury (18)(Figure 2). A comparative transcriptional profiling of genes related to energy metabolism in different brain cell types revealed that microglia express specific sets of genes required for both glycolytic and oxidative energy metabolism (117). For instance, microglia express the long-chain fatty acyl-CoA synthetase, which catalyzes the formation of fatty acyl-CoAs that are, in turn, β-oxidized into acetyl-CoA units and can be further metabolized in the TCA cycle. Additionally, a recent study showed that microglia are able to maintain oxidative phosphorylation and homeostatic function during periods of hypoglycemia by shifting fuel utilization to glutamine (18). Homeostatic microglia, which are tasked with regulating day-to-day aspects of tissue homeostasis throughout the CNS, rely mainly on oxidative phosphorylation for ATP production, while microglia activated in the context of pro-inflammatory circumstances favor glycolysis (118, 119). When specifically activated, microglia are able to release several metabolites into the extracellular milieu (e.g.: succinate, itaconate, lactate) that modulate neuronal functionality and survival. For instance, a recent study showed that succinate produced by CNS myeloid cells is sensed by neural stem cells during the chronic phase of a mouse model of experimental autoimmune encephalitis (EAE) to ameliorate neuroinflammation via succinate-dependent mechanisms (120). Experiments on cultured microglia consistently show that they respond to proinflammatory stimuli by increasing glycolytic flux (121, 122). The metabolic alterations of isolated cells in vitro may differ from those in vivo. However, a novel approach to image NADH fluorescence has been recently employed to detect an enhanced glycolytic response of microglia to LPS treatment in mouse brain slices (18). Glycolysis is less efficient than oxidative phosphorylation (OXPHOS), however this glycolytic shift may redirect metabolites to provide the cell with precursor molecules for the production of inflammatory factors. Indeed, it has been shown that glycolysis is indispensable to stimulate secretion of pro-inflammatory cytokines by macrophages, the peripheral tissue analogs of microglia (123). Conversely, fatty acid β-oxidation and mitochondrial function are necessary for microglia to manifest relatively anti-inflammatory polarization states (124).
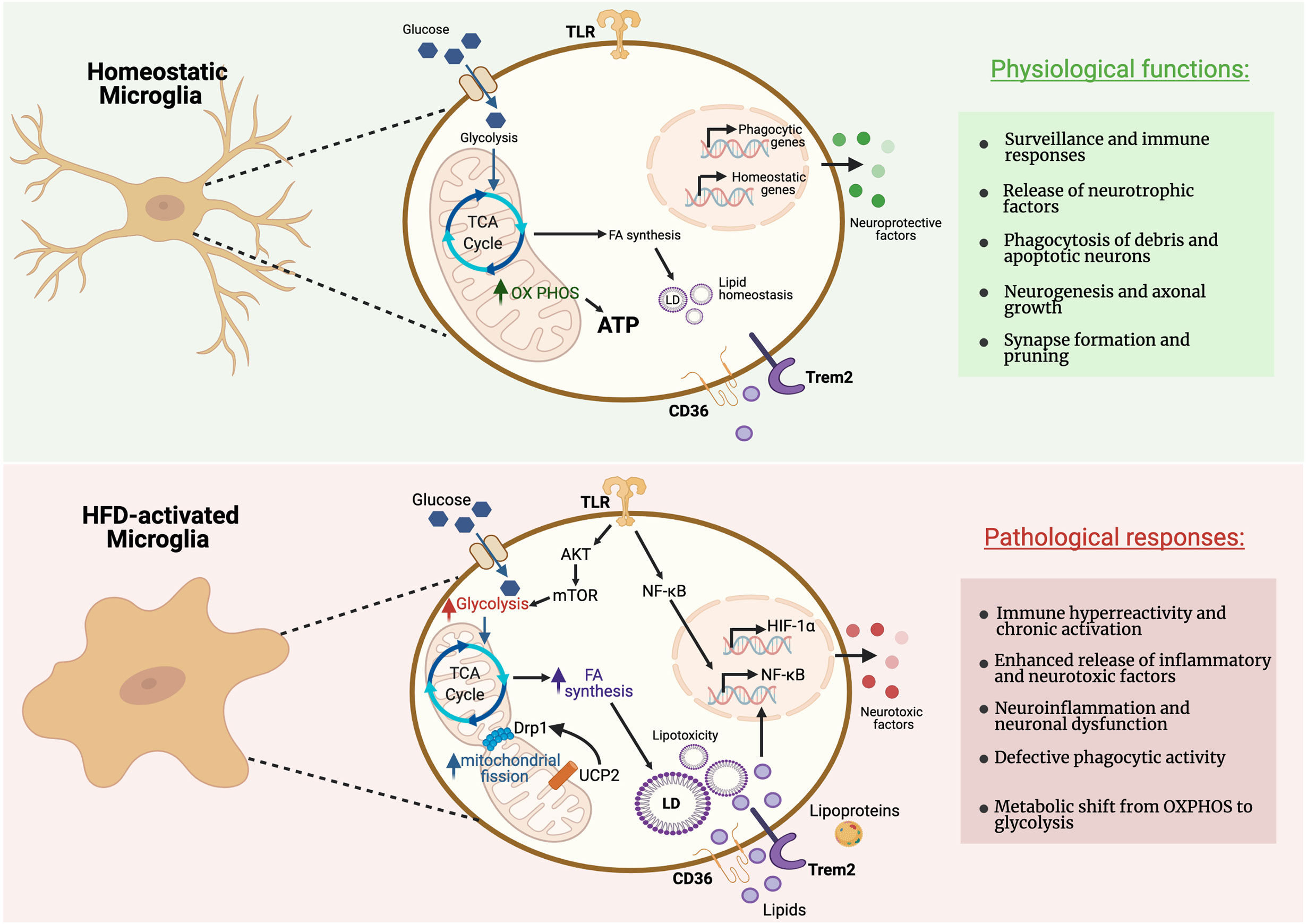
Figure 2 Metabolic pathways regulating microglial activity during homeostasis and pathological responses. Microglia can rapidly adapt their energy metabolism to nutrient availability and transcriptomic analyses revealed that microglia express genes necessary for both glycolysis and oxidative metabolism. Microglia in their homeostatic status show reliance on oxidative metabolism to maintain their neuroprotective properties. However, microglia in proinflammatory states preferentially use glycolysis for energy production. This metabolic switch towards glycolysis allows microglia to produce ATP rapidly, despite being comparatively less efficient, for the secretion of inflammatory cytokines. High-fat diet (HFD) triggers a microglial inflammatory response leading to neuronal dysfunction in the MBH. Figure created with BioRender.com.
In considering what might control broad shifts in fuel metabolism among microglia, it is notable that epigenetic changes, including histone modifications and DNA methylation, are important modifiers of gene expression and are known to mediate the metabolic reprogramming of myeloid cells. For instance, feeding mice a HFD for 4 weeks is sufficient to induce lasting epigenetic modifications in myeloid progenitor cells in the bone marrow, leading to increased immune responses to LPS challenge even after the mice were returned to a regular low-fat chow diet (125). Despite the fact that metabolic alterations have been implicated in several disease models (126), more knowledge is needed to understand which specific metabolic pathways can be targeted to restore the homeostatic microglia phenotype in chronic inflammatory diseases, including obesity.
Recent studies have shed light on how changes in mitochondrial morphology and function may impact microglia polarization and function. Microglia stimulated with LPS, demonstrate increased mitochondrial fragmentation, which was dependent on ROS-mediated activation of adenosine monosphosphate-activated protein kinase (AMPK) (127). Mitochondrial fragmentation in reactive microglia requires dynamin-related protein 1 (DRP1), an essential component of mitochondrial fission (127). Short-term HFD (3 days) caused decreased size and increased number of mitochondria in microglia in the MBH, associated with increased levels of activation of DRP1 (128) (Figure 2).
Mitochondrial uncoupling protein 2 (UCP2) plays a key role in reactive microglia. Knockdown of UCP2 modulates microglia response to both LPS and IL-4 (129). Deletion of microglial UCP2 prevented HFD-induced increases in mitochondrial fission in MBH microglia, and reduced microglial activation in the MBH and HFD-induced obesity (128). UCP2 has been shown to effect both ROS production (130, 131) and fuel utilization (132). Microglia in culture showed increased mitochondrial respiration in the presence of high glucose and palmitate, dependent on the presence of UCP2 (128). Further mechanistic studies are necessary to explore the impact of mitochondrial function and fuel utilization in the regulation of MBH microglia.
The Impact of Sex on Microglial Phenotypes in Metabolic Regulation
The study of sex differences in physiology has gained attention, and sexual dimorphism in obesity and metabolic disease has been described (133, 134). HFD induces activation of microglia in the MBH of rodents, in a sexually dimorphic manner, affecting male differently than females (135). However, the mechanism underlying these differences are not well understood. Interestingly, recent studies suggest that male and female mice differentially metabolize lipids acquired from the diet. For instance, HFD feeding increases PA and sphingolipids levels in the hypothalamic tissue of male mice but not in the females (135). Alterations in sphingolipid-mediated signaling pathways might provide an additional mechanism by which SFAs induce hypothalamic dysfunction in the MBH (136). On the other hand, microglia in the adult mouse brain have sex-specific features and that could explain sex differences in neurological disease susceptibility (137). Moreover, it has been recently shown that microbiota influences adult microglia in a sex-specific manner (138). For instance, short-chain fatty acids (SCFAs) are the main metabolites produced by bacterial fermentation of dietary fiber in the gastrointestinal tract, and these SCFAs influence gut-brain communication and brain function directly or indirectly through immune, endocrine, and vagal pathways (139). Although the SCFAs have been shown to protect against diet-induced obesity in mice (140) and overweight humans (141), the underlying mechanisms are not well understood. SCFAs are important regulators of innate immune responses and recently have been involved in the regulation of microglial function (142). Thus, regulating CNS myeloid cell functions by manipulating the gut microbiota may represent a promising therapeutic approach to mitigate metabolic diseases.
Dietary Lipids Regulate Microglial Polarization and Responses in the MBH
Bioactive dietary FAs are potent modulators of microglial inflammatory responses. Lipid accumulation in myeloid cell types more broadly, is well demonstrated to be associated with the activation of inflammatory signaling cascades (143). Moreover, microglia express a wide range of lipid metabolism-related genes such as those encoding fatty acid oxidation enzymes (144), lipoprotein lipases, lipid transporters, and lipid-sensitive receptors (e.g. receptors for endocannabinoids, prostaglandins or phospholipids), suggesting that lipids are important regulators of microglial physiology. Microglia can store FAs within lipid droplets, which are known to control their inflammatory responsiveness and phagocytic activity (145). Some reports suggest that dietary lipids in the context of the whole mammal, can also influence microglial function through indirect mechanisms including microbially-derived metabolites, hormonal control, and gut and systemically-derived inflammatory signals. For instance, treating microglia with insulin in vitro decreases LPS-induced TNF production and phagocytic activity in a dose-dependent manner (146). Moreover, ghrelin, an orexigenic hormone produced by the stomach and duodenum, directly exerts anti-inflammatory and anti-oxidative effects on LPS-activated microglia when introduced to them in culture (147).
In particular, long-chain SFAs have emerged as a potential nutritional triggers of microglial activation in the MBH, exerting effects in the brain analogous to those documented for peripheral tissues. HFD intake increases brain SFA levels, and more specifically those of lipids containing PA (14). Indeed, PA levels are increased in the CSF of overweight and obese humans (148). We showed that microglia in the MBH can sense rising levels of saturated fats, when consumed in excess, and transduce this to instruct local neurons. Moreover, enteric isocaloric gavage of SFAs, but not UFAs, for only 3 days is sufficient to induce microglial activation in the MBH, reproducing the response seen in the MBH of mice fed a HFD (14). These findings support the idea that SFAs trigger this response. However, the HFD commonly used for animal studies also contains high amounts of sugars, and another study suggested that dietary sugars, instead of fat, drive hypothalamic inflammation (149). One caveat of this study was that the authors did not control for calories and the sources of fat vs. carbohydrates across diets. Intriguingly, a recent comprehensive study of 29 different diets with different macronutrient compositions showed that only dietary fat, but not protein or carbohydrates, regulates hypothalamic control of energy intake and promotes adiposity (150). Besides macronutrient distribution, the specific source of dietary FA is can modulate microglial inflammatory responses. For instance, the substitution of dietary lard for flaxseed oil or olive oil reduced food intake and inflammatory markers in the MBH, highlighting a specific pro-inflammatory impact of SFAs (151).
SFAs were initially thought to induce inflammation as direct agonists of the toll-like receptor 4(TLR4), a member of the interleukin-1 receptor superfamily with a prominent role in innate immune responses. In support of this hypothesis, pharmacological and genetic approaches to inhibit hypothalamic TLR4 signaling suppressed SFA-induced microglial activation and inflammatory cytokines expression in rodent models fed a HFD (112, 152). However, a recent study showed that TLR4 is not the receptor for SFAs. Rather, TLR4-dependent priming alters cellular metabolism, lipid metabolic pathways and membrane lipid composition, changes that are required for engagement of SFA-induced inflammatory pathways (153). The fatty acid translocase CD36 is another potential mediator of microglial lipid-sensing. Indeed, CD36 has been shown to be essential for microglia-mediated uptake of myelin debris (154), and microglia response to beta-amyloid (155). However, while CD36 is known to be involved in long-chain fatty acid uptake and sensing in other tissues, its role in lipid-sensing in microglia has not been reported. Unlike SFAs, PUFAs have beneficial effects on the brain and reduce neuroinflammation. PUFAs, when incorporated into cell membranes, increasing membrane fluidity in a manner that was shown to help microglia engage in phagocytosis (156). Microglial movement was remarkably impaired in mice fed a diet deficient in n-3 PUFAs (157). Also, PUFAs are endogenous ligands of the G-couple receptor GPR120, which may explain, at least in part, how they activate anti-inflammatory signaling pathways (158). Moreover, GPR120 is primarily expressed by microglia in the hypothalamus and is suggested to be involved in regulating microglial inflammatory responses that influence energy homeostasis (159).
Lipoprotein Metabolism and Lipid Mediators Regulating Microglial Phenotypes
The emergence of new technology such as scRNA-seq has enabled the identification and characterization of the diversity of microglial populations. These studies have revealed that the heterogeneity of microglia in both normal and disease states exists beyond the simplistic M1/M2 paradigm, with a spectrum of cellular states existing from homeostatic microglia to pathology-associated microglia (160, 161). In addition, scRNA-seq of myeloid cells has revealed extensive regional heterogeneity in both microglia and non-parenchymal brain myeloid cells including so-called “border-associated” macrophages found proximal to, and within, meningeal lining tissue (162). Recently, several comprehensive ex vivo scRNA-seq analyses of microglia have defined specific transcriptional clusters with common metabolic characteristics. For instance, a novel microglial population called disease-associated microglia (DAM) was recently identify in a mouse models of AD and amyotrophic lateral sclerosis (ALS) expressing a distinct set of genes associated with lipid and lipoprotein metabolism (163). This transcriptional signature represents a preference for lipids as fuel substrates, ostensibly to meet the increased bioenergetic demands of this form of activated microglia (163). A similar signature is also observed in microglia in the context of demyelination, suggesting engagement of a transcriptional microglial phenotype that enables the ability to phagocytose and clear lipid debris (164). Moreover, human microglia from white matter adjacent to chronic multiple sclerosis (MS) lesions showed upregulation of scavenger receptor and lipid metabolism genes including LPL and PPARG (165). Additionally, analyses of non-diseased human brain revealed clusters of microglia enriched for expression of metabolism-encoding genes, including APOE and LPL, in white- vs. grey matter, and increased with aging (166). While the transcriptional signature of activated microglial populations in these studies have shown variability in the response to different stimuli and experimental conditions, there is a clear consistent implication of alterations in lipid metabolism in analyses of microglia activated by stimuli other than those associated with acute infection.
Lipoprotein Lipase (LPL)
Lipoprotein lipase (LPL), an enzyme needed for the hydrolytic cleavage and release of FAs from TGs, and a number of recent reports have highlighted LPL as a key feature of reparative microglia, which are recruited to restore tissue homeostasis in the context of injury, for example. scRNA-seq of DAM, in a murine model of Alzheimer’s disease (AD), revealed that LPL levels are markedly increased in a unique microglial subset associated with phagocytosis and protection in AD (163). Furthermore, LPL gene transcription is elevated in a cuprizone model of demyelination (167), and a recent study suggested that LPL is a novel feature of a the supportive microglial phenotype that emerges during remyelination and repair via clearance of lipid debris (9).
LPL is expressed in the brain, spinal cord, and peripheral nerves but is predominantly expressed by macrophages and microglia in the human and murine brain (117, 168). Although the function of LPL in the microglial response to neurodegenerative disease is not well understood, LPL polymorphisms are been implicated in disease risk, such as an association with AD risk. For instance, loss-of-function LPL polymorphisms with reduced enzymatic activity are associated with increased AD risk as well as with increased VLDL-TG levels (169). Conversely, patients with LPL polymorphisms leading to increased LPL activity have reduced hippocampal amyloid plaque formation (170). Microglia-specific knockdown of Lpl exhibited decreased cell number and soma size of microglia in the ARC of mice fed a hypercaloric diet (168), supporting the hypothesis that lipoprotein metabolism is important in the regulation of MBH microglial function. In these mice, POMC neuronal loss was accelerated and they gained more weight than control mice. Microglia lacking Lpl demonstrated a shift in fuel utilization towards glutamine and decreased phagocytic capacity, suggestive of an immunometabolic shift (168). Taken together, these data suggest that LPL regulates lipid and lipoprotein uptake, which may provide the lipids needed to maintain homeostatic microglial functions in the MBH.
Apolipoprotein E (APOE)
Apolipoprotein E (APOE) is the major carrier for lipids in the brain, and APOE genotype is the most profound genetic risk factor for AD, predominantly by modulating microglial activation (171). In the brain, APOE is expressed predominantly by astrocytes and microglia and a major role for APOE in the brain is to maintain a consistent supply of essential lipids to neurons (172). Extensive studies have established the role of APOE in mediating inter-cellular cholesterol transport from glia to neuronal cells (173). The human APOE gene exists as three different alleles, ε2, ε3 and ε4 and these isoforms change the lipid and receptor binding ability of APOE.
Microglial APOE production is strongly induced during injury and disease, including in AD (174). APOE is a key component of transcriptional signature of activated microglia, as demonstrated in post-mortem human brain studies, AD mouse models and studies of cultured microglia (163, 171). APOE induces an anti-inflammatory phenotype in macrophages and similarly an APOE peptide inhibits inflammatory processes in isolated microglia through the APOE receptor, LRP1 (175). In APOE-deficient mouse models, peptides based on the APOE receptor-binding domain prevent LPS-induced inflammation (176). Interestingly, blocking inflammatory signaling increases APOE expression in microglia (177), suggesting a negative feedback loop between APOE levels and inflammation.
The mechanistic role of APOE expression in hypothalamic microglia has not been explored in models of diet-induced obesity, but data from studies in neurodegenerative disease lend clues towards the potential function.
Triggering Receptor Expressed on Myeloid Cells 2 (TREM2)
The Triggering Receptor Expressed on Myeloid Cells 2 (TREM2) is a type 1 transmembrane receptor protein expressed on myeloid cells. This receptor binds a wide array of ligands including extracellular lipids and lipoproteins, and loss of function variants in TREM2 are also associated with increased risk of AD. TREM2 modulates inflammatory signaling in myeloid cells, and in the brain is primarily expressed by microglia. TREM2 is crucial for induction of the transcriptomic and functional program of DAMs, by activation of phagocytosis and lipid metabolism-related pathways. TREM2-deficient microglia have strong metabolic defects, characterized by impaired lipid metabolism, accumulation of cholesterol esters, aberrant autophagy, altered mTOR signaling, and reduced ATP production (8, 178). Also, TREM2-deficient microglia have reduced mitochondrial mass and increased phosphorylation of AMPK (178), a key regulator of energy metabolism that is activated in response to low glucose and inhibits a shift in the cellular metabolism from oxidative phosphorylation to glycolysis (179). A recent study revealed that TREM2 activation by APOE, drives a neurodegenerative phenotype in microglia, characterized by suppression of transcription factors regulating homeostatic microglia (171). Thus, targeting of the TREM2-APOE pathway may represent a novel therapeutic approach to restore homeostatic microglia in neurological disease.
TREM2 signaling in peripheral macrophages has recently been linked to metabolic disease. TREM2 KO mice exhibit increased obesity, insulin resistance and altered adipose tissue remodeling in response to HFD feeding (180). TREM2 is required for induction of monocyte-derived LAMs, in which LPL and APOE are induced by a TREM2-dependent mechanism as a consequence of HFD-induced obesity in mice (110). Similar transcriptional signatures were also identified in aortic macrophages during atherosclerosis (181) and fatty livers of mice fed a HFD (110). TREM2 activation via DAP12 antagonizes TLR signaling and inflammatory cytokine production in cultured macrophages and, conversely, TREM2 expression is abrogated by pro-inflammatory signaling (182, 183). However, the role of TREM2 in lipid-induced microglial activation in the MBH has not been investigated.
Concluding Remarks and Future Perspectives
Both microglia and lipid metabolism are now known to play keys role in the onset and progression of the pathology of a wide variety of neurological diseases. The traditional view of the brain as an immune privileged organ has undergone a paradigm shift. In recent years, it has become increasingly clear that immune cells actively contribute to homeostatic processes in the CNS. Furthermore, dysfunctional microglial subsets characterized by excessive droplet- and membrane-associated lipid accumulation and attenuated lipid efflux have recently been the subject of considerable investigation (28, 184). Based on exciting data from other fields, it is increasingly becoming likely that a better understanding of how lipid mediators regulate the interaction between the immune and nervous systems may help uncover novel therapeutic targets to prevent and treat metabolic diseases as well. Indeed, many of the advances in determining of the role of lipid and lipoprotein metabolism that have occurred in the context of neurodegenerative disease (12) have the capacity to provide direct insight into the mechanisms by which microglia are activated in the MBH by nutritional signals.
The CNS hosts a heterogeneous population of myeloid cells, including parenchymal homeostatic microglia, and perivascular and meningeal border-associated macrophages. These myeloid cells share the expression of numerous markers, and a major obstacle has been the lack of tools to discriminate between specific microglial as well as other brain myeloid populations. However, new approaches for single-cell profiling have revealed a remarkable functional complexity in the CNS myeloid compartment in both homeostatic and disease contexts. Microglia are highly dependent on environmental signals to maintain their polarization. Given that such signals may vary across brain regions, it is notable that immune profiling of human brain microglia by single-cell proteomics revealed remarkable regional heterogeneity (185). Myeloid cells strategically located in close proximity to fenestrated blood vessels in the MBH may be able to sense metabolic factors including circulating lipids. To this end, we showed that HFD feeding induces the accumulation of a unique mix of myeloid cells in the MBH (15). This immunological response also includes the accumulation of perivascular macrophages involved in alterations systemic glucose metabolism (186). However, methods using marker-based analyses have technical limitations, and unbiased approaches are needed to resolve the heterogeneity and complexity of myeloid cell types within different CNS regions. Understanding the contribution of individual diet-responsive myeloid cell types will be critical for the development of novel therapeutics for obesity and T2D.
In summary, the emergence of a new field focused on microglial function, heterogeneity, and cell-cell crosstalk is providing us with an unprecedented understanding of how dietary lipids modulate microglial functions and their engagement with other cell types within the brain, including the MBH. This information has tremendous potential to help us identify new therapeutic targets to prevent overnutrition-induced hypothalamic dysfunction and metabolic disease.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was funded by the NIDDK (Diabetes, Endocrinology & Metabolism Training Grant T32DK007418 to AF, R01DK103175 to SK, K01DK113064, and R03DK125627 to MV).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank members of their laboratories for helpful discussions on the topic and the Nutrition Obesity Research Centers at UCSF (DK098722).
References
1. Hamilton JA, Hillard CJ, Spector AA, Watkins PA. Brain Uptake and Utilization of Fatty Acids, Lipids and Lipoproteins: Application to Neurological Disorders. J Mol Neurosci (2007) 33:2–11. doi: 10.1007/s12031-007-0060-1
2. Karasinska JM, Hayden MR. Cholesterol Metabolism in Huntington Disease. Nat Rev Neurol (2011) 7:561–72. doi: 10.1038/nrneurol.2011.132
3. Van Veldhoven PP. Biochemistry and Genetics of Inherited Disorders of Peroxisomal Fatty Acid Metabolism. J Lipid Res (2010) 51:2863–95. doi: 10.1194/jlr.R005959
4. Barber CN, Raben DM. Lipid Metabolism Crosstalk in the Brain: Glia and Neurons. Front Cell Neurosci (2019) 13:212. doi: 10.3389/fncel.2019.00212
5. Pellerin L, Bouzier-Sore A-K, Aubert A, Serres S, Merle M, Costalat R, et al. Activity-Dependent Regulation of Energy Metabolism by Astrocytes: An Update. Glia (2007) 55:1251–62. doi: 10.1002/glia.20528
6. Bazan NG. Synaptic Lipid Signaling: Significance of Polyunsaturated Fatty Acids and Platelet-Activating Factor. J Lipid Res (2003) 44:2221–33. doi: 10.1194/jlr.R300013-JLR200
7. Saher G, Brügger B, Lappe-Siefke C, Möbius W, Tozawa R, Wehr MC, et al. High Cholesterol Level Is Essential for Myelin Membrane Growth. Nat Neurosci (2005) 8:468–75. doi: 10.1038/nn1426
8. Nugent AA, Lin K, van Lengerich B, Lianoglou S, Przybyla L, Davis SS, et al. Trem2 Regulates Microglial Cholesterol Metabolism Upon Chronic Phagocytic Challenge. Neuron (2020) 105:837–54.e9. doi: 10.1016/j.neuron.2019.12.007
9. Bruce KD, Gorkhali S, Given K, Coates AM, Boyle KE, Macklin WB, et al. Lipoprotein Lipase Is a Feature of Alternatively-Activated Microglia and May Facilitate Lipid Uptake in the CNS During Demyelination. Front Mol Neurosci (2018) 11:57. doi: 10.3389/fnmol.2018.00057
10. Madore C, Leyrolle Q, Morel L, Rossitto M, Greenhalgh AD, Delpech JC, et al. Essential Omega-3 Fatty Acids Tune Microglial Phagocytosis of Synaptic Elements in the Mouse Developing Brain. Nat Commun (2020) 11:6133. doi: 10.1038/s41467-020-19861-z
11. Jha P, McDevitt MT, Halilbasic E, Williams EG, Quiros PM, Gariani K, et al. Genetic Regulation of Plasma Lipid Species and Their Association With Metabolic Phenotypes. Cell Syst (2018) 6:709–21.e6. doi: 10.1016/j.cels.2018.05.009
12. Loving BA, Bruce KD. Lipid and Lipoprotein Metabolism in Microglia. Front Physiol (2020) 11:393. doi: 10.3389/fphys.2020.00393
13. Fitzner D, Bader JM, Penkert H, Bergner CG, Su M, Weil M-T, et al. Cell-Type- and Brain-Region-Resolved Mouse Brain Lipidome. Cell Rep (2020) 32:108132. doi: 10.1016/j.celrep.2020.108132
14. Valdearcos M, Robblee MM, Benjamin DI, Nomura DK, Xu AW, Koliwad SK. Microglia Dictate the Impact of Saturated Fat Consumption on Hypothalamic Inflammation and Neuronal Function. Cell Rep (2014) 9:2124–38. doi: 10.1016/j.celrep.2014.11.018
15. Valdearcos M, Douglass JD, Robblee MM, Dorfman MD, Stifler DR, Bennett ML, et al. Microglial Inflammatory Signaling Orchestrates the Hypothalamic Immune Response to Dietary Excess and Mediates Obesity Susceptibility. Cell Metab (2017) 26:185–97.e3. doi: 10.1016/j.cmet.2017.05.015
16. Cansell C, Stobbe K, Sanchez C, Le Thuc O, Mosser C-A, Ben-Fradj S, et al. Dietary Fat Exacerbates Postprandial Hypothalamic Inflammation Involving Glial Fibrillary Acidic Protein-Positive Cells and Microglia in Male Mice. Glia (2020) 69(1):42–60. doi: 10.1002/glia.23882
17. Ebert D, Haller RG, Walton ME. Energy Contribution of Octanoate to Intact Rat Brain Metabolism Measured by 13C Nuclear Magnetic Resonance Spectroscopy. J Neurosci (2003) 23:5928–35. doi: 10.1523/JNEUROSCI.23-13-05928.2003
18. Bernier L-P, York EM, Kamyabi A, Choi HB, Weilinger NL, MacVicar BA. Microglial Metabolic Flexibility Supports Immune Surveillance of the Brain Parenchyma. Nat Commun (2020) 11:1559. doi: 10.1038/s41467-020-15267-z
19. Bouyakdan K, Taïb B, Budry L, Zhao S, Rodaros D, Neess D, et al. A Novel Role for Central ACBP/DBI as a Regulator of Long-Chain Fatty Acid Metabolism in Astrocytes. J Neurochem (2015) 133:253–65. doi: 10.1111/jnc.13035
20. Panov A, Orynbayeva Z, Vavilin V, Lyakhovich V. Fatty Acids in Energy Metabolism of the Central Nervous System. BioMed Res Int (2014) 2014:472459. doi: 10.1155/2014/472459
21. Schönfeld P, Reiser G. Why Does Brain Metabolism Not Favor Burning of Fatty Acids to Provide Energy? Reflections on Disadvantages of the Use of Free Fatty Acids as Fuel for Brain. J Cereb Blood Flow Metab (2013) 33:1493–9. doi: 10.1038/jcbfm.2013.128
22. Waldbaum S, Patel M. Mitochondria, Oxidative Stress, and Temporal Lobe Epilepsy. Epilepsy Res (2010) 88:23–45. doi: 10.1016/j.eplepsyres.2009.09.020
23. Alecu I, Bennett SAL. Dysregulated Lipid Metabolism and Its Role in α-Synucleinopathy in Parkinson’s Disease. Front Neurosci (2019) 13:328. doi: 10.3389/fnins.2019.00328
24. Billingsley KJ, Bandres-Ciga S, Saez-Atienzar S, Singleton AB. Genetic Risk Factors in Parkinson’s Disease. Cell Tissue Res (2018) 373:9–20. doi: 10.1007/s00441-018-2817-y
25. Bosch-Queralt M, Cantuti-Castelvetri L, Damkou A, Schifferer M, Schlepckow K, Alexopoulos I, et al. Diet-Dependent Regulation of Tgfβ Impairs Reparative Innate Immune Responses After Demyelination. Nat Metab (2021) 3:211–27. doi: 10.1038/s42255-021-00341-7
26. Berghoff SA, Spieth L, Sun T, Hosang L, Schlaphoff L, Depp C, et al. Microglia Facilitate Repair of Demyelinated Lesions Via Post-Squalene Sterol Synthesis. Nat Neurosci (2021) 24:47–60. doi: 10.1038/s41593-020-00757-6
27. Brekk OR, Honey JR, Lee S, Hallett PJ, Isacson O. Cell Type-Specific Lipid Storage Changes in Parkinson’s Disease Patient Brains Are Recapitulated by Experimental Glycolipid Disturbance. Proc Natl Acad Sci USA (2020) 117:27646–54. doi: 10.1073/pnas.2003021117
28. Cantuti-Castelvetri L, Fitzner D, Bosch-Queralt M, Weil M-T, Su M, Sen P, et al. Defective Cholesterol Clearance Limits Remyelination in the Aged Central Nervous System. Science (2018) 359:684–8. doi: 10.1126/science.aan4183
29. Bozek K, Wei Y, Yan Z, Liu X, Xiong J, Sugimoto M, et al. Organization and Evolution of Brain Lipidome Revealed by Large-Scale Analysis of Human, Chimpanzee, Macaque, and Mouse Tissues. Neuron (2015) 85:695–702. doi: 10.1016/j.neuron.2015.01.003
30. Zhang J, Liu Q. Cholesterol Metabolism and Homeostasis in the Brain. Protein Cell (2015) 6:254–64. doi: 10.1007/s13238-014-0131-3
31. Petrov AM, Kasimov MR, Zefirov AL. Brain Cholesterol Metabolism and Its Defects: Linkage to Neurodegenerative Diseases and Synaptic Dysfunction. Acta Naturae (2016) 8:58–73. doi: 10.32607/20758251-2016-8-1-58-73
32. Wooten JS, Wu H, Raya J, Perrard XD, Gaubatz J, Hoogeveen RC. The Influence of an Obesogenic Diet on Oxysterol Metabolism in C57BL/6J Mice. Cholesterol (2014) 2014:843468. doi: 10.1155/2014/843468
33. Ismail M-A-M, Mateos L, Maioli S, Merino-Serrais P, Ali Z, Lodeiro M, et al. 27-Hydroxycholesterol Impairs Neuronal Glucose Uptake Through an IRAP/GLUT4 System Dysregulation. J Exp Med (2017) 214:699–717. doi: 10.1084/jem.20160534
34. Wang H, Eckel RH. What Are Lipoproteins Doing in the Brain? Trends Endocrinol Metab (2014) 25:8–14. doi: 10.1016/j.tem.2013.10.003
35. Park J-H, Lee C-W, Nam MJ, Kim H, Kwon D-Y, Yoo JW, et al. Association of High-Density Lipoprotein Cholesterol Variability and the Risk of Developing Parkinson Disease. Neurology (2021) 96:e1391–401. doi: 10.1212/WNL.0000000000011553
36. Götz A, Lehti M, Donelan E, Striese C, Cucuruz S, Sachs S, et al. Circulating HDL Levels Control Hypothalamic Astrogliosis Via Apoa-I. J Lipid Res (2018) 59:1649–59. doi: 10.1194/jlr.M085456
37. Rapoport SI, Chang MC, Spector AA. Delivery and Turnover of Plasma-Derived Essential PUFAs in Mammalian Brain. J Lipid Res (2001) 42:678–85. doi: 10.1016/S0022-2275(20)31629-1
38. Plourde M, Cunnane SC. Extremely Limited Synthesis of Long Chain Polyunsaturates in Adults: Implications for Their Dietary Essentiality and Use as Supplements. Appl Physiol Nutr Metab (2007) 32:619–34. doi: 10.1139/H07-034
39. DeMar JC, Lee H-J, Ma K, Chang L, Bell JM, Rapoport SI, et al. Brain Elongation of Linoleic Acid Is a Negligible Source of the Arachidonate in Brain Phospholipids of Adult Rats. Biochim Biophys Acta (2006) 1761:1050–9. doi: 10.1016/j.bbalip.2006.06.006
40. Alashmali SM, Hopperton KE, Bazinet RP. Lowering Dietary N-6 Polyunsaturated Fatty Acids: Interaction With Brain Arachidonic and Docosahexaenoic Acids. Curr Opin Lipidol (2016) 27:54–66. doi: 10.1097/MOL.0000000000000255
41. Domenichiello AF, Kitson AP, Chen CT, Trépanier M-O, Stavro PM, Bazinet RP. The Effect of Linoleic Acid on the Whole Body Synthesis Rates of Polyunsaturated Fatty Acids From α-Linolenic Acid and Linoleic Acid in Free-Living Rats. J Nutr Biochem (2016) 30:167–76. doi: 10.1016/j.jnutbio.2015.11.016
42. Ouellet M, Emond V, Chen CT, Julien C, Bourasset F, Oddo S, et al. Diffusion of Docosahexaenoic and Eicosapentaenoic Acids Through the Blood-Brain Barrier: An in Situ Cerebral Perfusion Study. Neurochem Int (2009) 55:476–82. doi: 10.1016/j.neuint.2009.04.018
43. Williams WM, Chang MC, Hayakawa T, Grange E, Rapoport SI. In Vivo Incorporation From Plasma of Radiolabeled Palmitate and Arachidonate Into Rat Brain Microvessels. Microvasc Res (1997) 53:163–6. doi: 10.1006/mvre.1996.1984
44. Spector R. Fatty Acid Transport Through the Blood-Brain Barrier. J Neurochem (1988) 50:639–43. doi: 10.1111/j.1471-4159.1988.tb02958.x
45. Le Foll C, Dunn-Meynell A, Musatov S, Magnan C, Levin BE. FAT/CD36: A Major Regulator of Neuronal Fatty Acid Sensing and Energy Homeostasis in Rats and Mice. Diabetes (2013) 62:2709–16. doi: 10.2337/db12-1689
46. Simopoulos AP. The Importance of the omega-6/omega-3 Fatty Acid Ratio in Cardiovascular Disease and Other Chronic Diseases. Exp Biol Med (2008) 233:674–88. doi: 10.3181/0711-MR-311
47. Hariri N, Thibault L. High-Fat Diet-Induced Obesity in Animal Models. Nutr Res Rev (2010) 23:270–99. doi: 10.1017/S0954422410000168
48. Sun T, Wang X, Cong P, Xu J, Xue C. Mass Spectrometry-Based Lipidomics in Food Science and Nutritional Health: A Comprehensive Review. Comp Rev Food Sci Food Saf (2020) 19:2530–58. doi: 10.1111/1541-4337.12603
49. Pakiet A, Jakubiak A, Czumaj A, Sledzinski T, Mika A. The Effect of Western Diet on Mice Brain Lipid Composition. Nutr Metab (Lond) (2019) 16:81. doi: 10.1186/s12986-019-0401-4
50. Demers G, Roy J, Machuca-Parra AI, Dashtehei Pour Z, Bairamian D, Daneault C, et al. Fish Oil Supplementation Alleviates Metabolic and Anxiodepressive Effects of Diet-Induced Obesity and Associated Changes in Brain Lipid Composition in Mice. Int J Obes (2020) 44:1936–45. doi: 10.1038/s41366-020-0623-6
51. Serhan CN, Chiang N, Van Dyke TE. Resolving Inflammation: Dual Anti-Inflammatory and Pro-Resolution Lipid Mediators. Nat Rev Immunol (2008) 8:349–61. doi: 10.1038/nri2294
52. Sánchez-Lasheras C, Könner AC, Brüning JC. Integrative Neurobiology of Energy Homeostasis-Neurocircuits, Signals and Mediators. Front Neuroendocrinol (2010) 31:4–15. doi: 10.1016/j.yfrne.2009.08.002
53. Lechan RM, Toni R. Functional Anatomy of the Hypothalamus and Pituitary. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, et al, editors. Endotext. South Dartmouth, MA: MDText.com, Inc (2016).
54. Cheunsuang O, Stewart AL, Morris R. Differential Uptake of Molecules From the Circulation and CSF Reveals Regional and Cellular Specialisation in CNS Detection of Homeostatic Signals. Cell Tissue Res (2006) 325:397–402. doi: 10.1007/s00441-006-0162-z
55. Campbell JN, Macosko EZ, Fenselau H, Pers TH, Lyubetskaya A, Tenen D, et al. A Molecular Census of Arcuate Hypothalamus and Median Eminence Cell Types. Nat Neurosci (2017) 20:484–96. doi: 10.1038/nn.4495
56. Ciofi P, Garret M, Lapirot O, Lafon P, Loyens A, Prévot V, et al. Brain-Endocrine Interactions: A Microvascular Route in the Mediobasal Hypothalamus. Endocrinology (2009) 150:5509–19. doi: 10.1210/en.2009-0584
57. Ciofi P. The Arcuate Nucleus as a Circumventricular Organ in the Mouse. Neurosci Lett (2011) 487:187–90. doi: 10.1016/j.neulet.2010.10.019
58. Wang H, Astarita G, Taussig MD, Bharadwaj KG, DiPatrizio NV, Nave K-A, et al. Deficiency of Lipoprotein Lipase in Neurons Modifies the Regulation of Energy Balance and Leads to Obesity. Cell Metab (2011) 13:105–13. doi: 10.1016/j.cmet.2010.12.006
59. Rapoport SI. In Vivo Labeling of Brain Phospholipids by Long-Chain Fatty Acids: Relation to Turnover and Function. Lipids (1996) 31 Suppl:S97–101. doi: 10.1007/BF02637059
60. Jordan SD, Könner AC, Brüning JC. Sensing the Fuels: Glucose and Lipid Signaling in the CNS Controlling Energy Homeostasis. Cell Mol Life Sci (2010) 67:3255–73. doi: 10.1007/s00018-010-0414-7
61. Cansell C, Castel J, Denis RGP, Rouch C, Delbes AS, Martinez S, et al. Dietary Triglycerides Act on Mesolimbic Structures to Regulate the Rewarding and Motivational Aspects of Feeding. Mol Psychiatry (2014) 19:1095–105. doi: 10.1038/mp.2014.31
62. Berland C, Montalban E, Perrin E, Di Miceli M, Nakamura Y, Martinat M, et al. Circulating Triglycerides Gate Dopamine-Associated Behaviors Through DRD2-Expressing Neurons. Cell Metab (2020) 31:773–90. doi: 10.1016/j.cmet.2020.02.010
63. Banks WA, Farr SA, Salameh TS, Niehoff ML, Rhea EM, Morley JE, et al. Triglycerides Cross the Blood-Brain Barrier and Induce Central Leptin and Insulin Receptor Resistance. Int J Obes (2018) 42:391–7. doi: 10.1038/ijo.2017.231
64. Weigert C, Brodbeck K, Staiger H, Kausch C, Machicao F, Häring HU, et al. Palmitate, But Not Unsaturated Fatty Acids, Induces the Expression of Interleukin-6 in Human Myotubes Through Proteasome-Dependent Activation of Nuclear Factor-Kappab. J Biol Chem (2004) 279:23942–52. doi: 10.1074/jbc.M312692200
65. Roberts R, Bickerton AS, Fielding BA, Blaak EE, Wagenmakers AJ, Chong MF-F, et al. Reduced Oxidation of Dietary Fat After a Short Term High-Carbohydrate Diet. Am J Clin Nutr (2008) 87:824–31. doi: 10.1093/ajcn/87.4.824
66. Yue JTY, Lam TKT. Lipid Sensing and Insulin Resistance in the Brain. Cell Metab (2012) 15:646–55. doi: 10.1016/j.cmet.2012.01.013
67. Wang R, Cruciani-Guglielmacci C, Migrenne S, Magnan C, Cotero VE, Routh VH. Effects of Oleic Acid on Distinct Populations of Neurons in the Hypothalamic Arcuate Nucleus Are Dependent on Extracellular Glucose Levels. J Neurophysiol (2006) 95:1491–8. doi: 10.1152/jn.00697.2005
68. Obici S, Feng Z, Morgan K, Stein D, Karkanias G, Rossetti L. Central Administration of Oleic Acid Inhibits Glucose Production and Food Intake. Diabetes (2002) 51:271–5. doi: 10.2337/diabetes.51.2.271
69. Obici S, Feng Z, Arduini A, Conti R, Rossetti L. Inhibition of Hypothalamic Carnitine Palmitoyltransferase-1 Decreases Food Intake and Glucose Production. Nat Med (2003) 9:756–61. doi: 10.1038/nm873
70. Benani A, Hryhorczuk C, Gouazé A, Fioramonti X, Brenachot X, Guissard C, et al. Food Intake Adaptation to Dietary Fat Involves PSA-Dependent Rewiring of the Arcuate Melanocortin System in Mice. J Neurosci (2012) 32:11970–9. doi: 10.1523/JNEUROSCI.0624-12.2012
71. Beutler LR, Chen Y, Ahn JS, Lin Y-C, Essner RA, Knight ZA. Dynamics of Gut-Brain Communication Underlying Hunger. Neuron (2017) 96:461–75. doi: 10.1016/j.neuron.2017.09.043
72. Beutler LR, Corpuz TV, Ahn JS, Kosar S, Song W, Chen Y, et al. Obesity Causes Selective and Long-Lasting Desensitization of AgRP Neurons to Dietary Fat. Elife (2020) 9. doi: 10.7554/eLife.55909
73. Dragano NR, Monfort-Pires M, Velloso LA. Mechanisms Mediating the Actions of Fatty Acids in the Hypothalamus. Neuroscience (2020) 447:15–27. doi: 10.1016/j.neuroscience.2019.10.012
74. Magnan C, Levin BE, Luquet S. Brain Lipid Sensing and the Neural Control of Energy Balance. Mol Cell Endocrinol (2015) 418(Pt 1):3–8. doi: 10.1016/j.mce.2015.09.019
75. Bruce KD, Zsombok A, Eckel RH. Lipid Processing in the Brain: A Key Regulator of Systemic Metabolism. Front Endocrinol (Lausanne) (2017) 8:60. doi: 10.3389/fendo.2017.00060
76. Jais A, Brüning JC. Hypothalamic Inflammation in Obesity and Metabolic Disease. J Clin Invest (2017) 127(1):24–32. doi: 10.1172/JCI88878
77. Volmer R, Ron D. Lipid-Dependent Regulation of the Unfolded Protein Response. Curr Opin Cell Biol (2015) 33:67–73. doi: 10.1016/j.ceb.2014.12.002
78. Cakir I, Cyr NE, Perello M, Litvinov BP, Romero A, Stuart RC, et al. Obesity Induces Hypothalamic Endoplasmic Reticulum Stress and Impairs Proopiomelanocortin (POMC) Post-Translational Processing. J Biol Chem (2013) 288:17675–88. doi: 10.1074/jbc.M113.475343
79. Schneeberger M, Dietrich MO, Sebastián D, Imbernón M, Castaño C, Garcia A, et al. Mitofusin 2 in POMC Neurons Connects ER Stress With Leptin Resistance and Energy Imbalance. Cell (2013) 155:172–87. doi: 10.1016/j.cell.2013.09.003
80. Hosoi T, Sasaki M, Miyahara T, Hashimoto C, Matsuo S, Yoshii M, et al. Endoplasmic Reticulum Stress Induces Leptin Resistance. Mol Pharmacol (2008) 74:1610–9. doi: 10.1124/mol.108.050070
81. Tse EK. Belsham DD. Palmitate Induces Neuroinflammation, ER Stress, and Pomc mRNA Expression in Hypothalamic mHypoA-POMC/GFP Neurons Through Novel Mechanisms That Are Prevented by Oleate. Mol Cell Endocrinol (2018) 472:40–9. doi: 10.1016/j.mce.2017.11.017
82. Volmer R, van der Ploeg K, Ron D. Membrane Lipid Saturation Activates Endoplasmic Reticulum Unfolded Protein Response Transducers Through Their Transmembrane Domains. Proc Natl Acad Sci USA (2013) 110:4628–33. doi: 10.1073/pnas.1217611110
83. Cragle FK, Baldini G. Mild Lipid Stress Induces Profound Loss of MC4R Protein Abundance and Function. Mol Endocrinol (2014) 28:357–67. doi: 10.1210/me.2013-1357
84. Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, et al. Endoplasmic Reticulum Stress Plays a Central Role in Development of Leptin Resistance. Cell Metab (2009) 9:35–51. doi: 10.1016/j.cmet.2008.12.004
85. Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF-kappaB and ER Stress Link Overnutrition to Energy Imbalance and Obesity. Cell (2008) 135:61–73. doi: 10.1016/j.cell.2008.07.043
86. Robblee MM, Kim CC, Porter Abate J, Valdearcos M, Sandlund KLM, Shenoy MK, et al. Saturated Fatty Acids Engage an IRE1α-Dependent Pathway to Activate the NLRP3 Inflammasome in Myeloid Cells. Cell Rep (2016) 14:2611–23. doi: 10.1016/j.celrep.2016.02.053
87. Studencka-Turski M, Çetin G, Junker H, Ebstein F, Krüger E. Molecular Insight Into the IRE1α-Mediated Type I Interferon Response Induced by Proteasome Impairment in Myeloid Cells of the Brain. Front Immunol (2019) 10:2900. doi: 10.3389/fimmu.2019.02900
88. Sofroniew MV, Vinters HV. Astrocytes: Biology and Pathology. Acta Neuropathol (2010) 119:7–35. doi: 10.1007/s00401-009-0619-8
89. Liu L, Zhang K, Sandoval H, Yamamoto S, Jaiswal M, Sanz E, et al. Glial Lipid Droplets and ROS Induced by Mitochondrial Defects Promote Neurodegeneration. Cell (2015) 160:177–90. doi: 10.1016/j.cell.2014.12.019
90. Ioannou MS, Jackson J, Sheu S-H, Chang C-L, Weigel AV, Liu H, et al. Neuron-Astrocyte Metabolic Coupling Protects Against Activity-Induced Fatty Acid Toxicity. Cell (2019) 177:1522–35.e14. doi: 10.1016/j.cell.2019.04.001
91. Zhang Y, Reichel JM, Han C, Zuniga-Hertz JP, Cai D. Astrocytic Process Plasticity and Ikkβ/Nf-κb in Central Control of Blood Glucose, Blood Pressure, and Body Weight. Cell Metab (2017) 25:1091–102.e4. doi: 10.1016/j.cmet.2017.04.002
92. Kim JG, Suyama S, Koch M, Jin S, Argente-Arizon P, Argente J, et al. Leptin Signaling in Astrocytes Regulates Hypothalamic Neuronal Circuits and Feeding. Nat Neurosci (2014) 17:908–10. doi: 10.1038/nn.3725
93. Yang L, Qi Y, Yang Y. Astrocytes Control Food Intake by Inhibiting AGRP Neuron Activity Via Adenosine A1 Receptors. Cell Rep (2015) 11:798–807. doi: 10.1016/j.celrep.2015.04.002
94. García-Cáceres C, Quarta C, Varela L, Gao Y, Gruber T, Legutko B, et al. Astrocytic Insulin Signaling Couples Brain Glucose Uptake With Nutrient Availability. Cell (2016) 166:867–80. doi: 10.1016/j.cell.2016.07.028
95. Nuzzaci D, Cansell C, Liénard F, Nédélec E, Ben Fradj S, Castel J, et al. Postprandial Hyperglycemia Stimulates Neuroglial Plasticity in Hypothalamic Pomc Neurons After a Balanced Meal. Cell Rep (2020) 30:3067–78.e5. doi: 10.1016/j.celrep.2020.02.029
96. Bolborea M, Dale N. Hypothalamic Tanycytes: Potential Roles in the Control of Feeding and Energy Balance. Trends Neurosci (2013) 36:91–100. doi: 10.1016/j.tins.2012.12.008
97. Rodríguez E, Guerra M, Peruzzo B, Blázquez JL. Tanycytes: A Rich Morphological History to Underpin Future Molecular and Physiological Investigations. J Neuroendocrinol (2019) 31:e12690. doi: 10.1111/jne.12690
98. Bolborea M, Pollatzek E, Benford H, Sotelo-Hitschfeld T, Dale N. Hypothalamic Tanycytes Generate Acute Hyperphagia Through Activation of the Arcuate Neuronal Network. Proc Natl Acad Sci USA (2020) 117:14473–81. doi: 10.1073/pnas.1919887117
99. Gao Y, Tschöp MH, Luquet S. Hypothalamic Tanycytes: Gatekeepers to Metabolic Control. Cell Metab (2014) 19:173–5. doi: 10.1016/j.cmet.2014.01.008
100. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science (2011) 333:1456–8. doi: 10.1126/science.1202529
101. Coull JAM, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, et al. BDNF From Microglia Causes the Shift in Neuronal Anion Gradient Underlying Neuropathic Pain. Nature (2005) 438:1017–21. doi: 10.1038/nature04223
102. Lalancette-Hébert M, Gowing G, Simard A, Weng YC, Kriz J. Selective Ablation of Proliferating Microglial Cells Exacerbates Ischemic Injury in the Brain. J Neurosci (2007) 27:2596–605. doi: 10.1523/JNEUROSCI.5360-06.2007
103. Bachiller S, Jiménez-Ferrer I, Paulus A, Yang Y, Swanberg M, Deierborg T, et al. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front Cell Neurosci (2018) 12:488. doi: 10.3389/fncel.2018.00488
104. Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, et al. Deficient Neuron-Microglia Signaling Results in Impaired Functional Brain Connectivity and Social Behavior. Nat Neurosci (2014) 17:400–6. doi: 10.1038/nn.3641
105. Jha MK, Jo M, Kim J-H, Suk K. Microglia-Astrocyte Crosstalk: An Intimate Molecular Conversation. Neuroscientist (2019) 25:227–40. doi: 10.1177/1073858418783959
106. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic Reactive Astrocytes Are Induced by Activated Microglia. Nature (2017) 541:481–7. doi: 10.1038/nature21029
107. Rothhammer V, Borucki DM, Tjon EC, Takenaka MC, Chao C-C, Ardura-Fabregat A, et al. Microglial Control of Astrocytes in Response to Microbial Metabolites. Nature (2018) 557:724–8. doi: 10.1038/s41586-018-0119-x
108. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity Is Associated With Macrophage Accumulation in Adipose Tissue. J Clin Invest (2003) 112:1796–808. doi: 10.1172/JCI19246
109. Kratz M, Coats BR, Hisert KB, Hagman D, Mutskov V, Peris E, et al. Metabolic Dysfunction Drives a Mechanistically Distinct Proinflammatory Phenotype in Adipose Tissue Macrophages. Cell Metab (2014) 20:614–25. doi: 10.1016/j.cmet.2014.08.010
110. Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, et al. Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2-Dependent Manner. Cell (2019) 178:686–98. doi: 10.1016/j.cell.2019.05.054
111. De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, Boschero AC, et al. Consumption of a Fat-Rich Diet Activates a Proinflammatory Response and Induces Insulin Resistance in the Hypothalamus. Endocrinology (2005) 146:4192–9. doi: 10.1210/en.2004-1520
112. Milanski M, Degasperi G, Coope A, Morari J, Denis R, Cintra DE, et al. Saturated Fatty Acids Produce an Inflammatory Response Predominantly Through the Activation of TLR4 Signaling in Hypothalamus: Implications for the Pathogenesis of Obesity. J Neurosci (2009) 29:359–70. doi: 10.1523/JNEUROSCI.2760-08.2009
113. Thaler JP, Yi C-X, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO, et al. Obesity Is Associated With Hypothalamic Injury in Rodents and Humans. J Clin Invest (2012) 122:153–62. doi: 10.1172/JCI59660
114. Puig J, Blasco G, Daunis-I-Estadella J, Molina X, Xifra G, Ricart W, et al. Hypothalamic Damage Is Associated With Inflammatory Markers and Worse Cognitive Performance in Obese Subjects. J Clin Endocrinol Metab (2015) 100:E276–81. doi: 10.1210/jc.2014-2682
115. Schur EA, Melhorn SJ, Oh S-K, Lacy JM, Berkseth KE, Guyenet SJ, et al. Radiologic Evidence That Hypothalamic Gliosis Is Associated With Obesity and Insulin Resistance in Humans. Obes (Silver Spring) (2015) 23:2142–8. doi: 10.1002/oby.21248
116. Moraes JC, Coope A, Morari J, Cintra DE, Roman EA, Pauli JR, et al. High-Fat Diet Induces Apoptosis of Hypothalamic Neurons. PloS One (2009) 4:e5045. doi: 10.1371/journal.pone.0005045
117. Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, et al. An RNA-sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J Neurosci (2014) 34:11929–47. doi: 10.1523/JNEUROSCI.1860-14.2014
118. Gimeno-Bayón J, López-López A, Rodríguez MJ, Mahy N. Glucose Pathways Adaptation Supports Acquisition of Activated Microglia Phenotype. J Neurosci Res (2014) 92:723–31. doi: 10.1002/jnr.23356
119. Voloboueva LA, Emery JF, Sun X, Giffard RG. Inflammatory Response of Microglial BV-2 Cells Includes a Glycolytic Shift and Is Modulated by Mitochondrial Glucose-Regulated Protein 75/Mortalin. FEBS Lett (2013) 587:756–62. doi: 10.1016/j.febslet.2013.01.067
120. Peruzzotti-Jametti L, Bernstock JD, Vicario N, Costa ASH, Kwok CK, Leonardi T, et al. Macrophage-Derived Extracellular Succinate Licenses Neural Stem Cells to Suppress Chronic Neuroinflammation. Cell Stem Cell (2018) 22:355–68.e13. doi: 10.1016/j.stem.2018.01.020
121. Nair S, Sobotka KS, Joshi P, Gressens P, Fleiss B, Thornton C, et al. Lipopolysaccharide-Induced Alteration of Mitochondrial Morphology Induces a Metabolic Shift in Microglia Modulating the Inflammatory Response In Vitro and In Vivo. Glia (2019) 67:1047–61. doi: 10.1002/glia.23587
122. Hu Y, Mai W, Chen L, Cao K, Zhang B, Zhang Z, et al. mTOR-mediated Metabolic Reprogramming Shapes Distinct Microglia Functions in Response to Lipopolysaccharide and ATP. Glia (2020) 68:1031–45. doi: 10.1002/glia.23760
123. Wang F, Zhang S, Jeon R, Vuckovic I, Jiang X, Lerman A, et al. Interferon Gamma Induces Reversible Metabolic Reprogramming of M1 Macrophages to Sustain Cell Viability and Pro-Inflammatory Activity. EBioMedicine (2018) 30:303–16. doi: 10.1016/j.ebiom.2018.02.009
124. Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative Metabolism and PGC-1beta Attenuate Macrophage-Mediated Inflammation. Cell Metab (2006) 4:13–24. doi: 10.1016/j.cmet.2006.05.011
125. Christ A, Günther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell (2018) 172:162–75.e14. doi: 10.1016/j.cell.2017.12.013
126. Na YR, Je S, Seok SH. Metabolic Features of Macrophages in Inflammatory Diseases and Cancer. Cancer Lett (2018) 413:46–58. doi: 10.1016/j.canlet.2017.10.044
127. Katoh M, Wu B, Nguyen HB, Thai TQ, Yamasaki R, Lu H, et al. Polymorphic Regulation of Mitochondrial Fission and Fusion Modifies Phenotypes of Microglia in Neuroinflammation. Sci Rep (2017) 7:4942. doi: 10.1038/s41598-017-05232-0
128. Kim JD, Yoon NA, Jin S, Diano S. Microglial UCP2 Mediates Inflammation and Obesity Induced by High-Fat Feeding. Cell Metab (2019) 30:952–62.e5. doi: 10.1016/j.cmet.2019.08.010
129. De Simone R, Ajmone-Cat MA, Pandolfi M, Bernardo A, De Nuccio C, Minghetti L, et al. The Mitochondrial Uncoupling Protein-2 Is a Master Regulator of Both M1 and M2 Microglial Responses. J Neurochem (2015) 135:147–56. doi: 10.1111/jnc.13244
130. Nègre-Salvayre A, Hirtz C, Carrera G, Cazenave R, Troly M, Salvayre R, et al. A Role for Uncoupling Protein-2 as a Regulator of Mitochondrial Hydrogen Peroxide Generation. FASEB J (1997) 11:809–15. doi: 10.1096/fasebj.11.10.9271366
131. Arsenijevic D, Onuma H, Pecqueur C, Raimbault S, Manning BS, Miroux B, et al. Disruption of the Uncoupling Protein-2 Gene in Mice Reveals a Role in Immunity and Reactive Oxygen Species Production. Nat Genet (2000) 26:435–9. doi: 10.1038/82565
132. Andrews ZB, Liu Z-W, Walllingford N, Erion DM, Borok E, Friedman JM, et al. UCP2 Mediates Ghrelin’s Action on NPY/AgRP Neurons by Lowering Free Radicals. Nature (2008) 454:846–51. doi: 10.1038/nature07181
133. Palmer BF, Clegg DJ. The Sexual Dimorphism of Obesity. Mol Cell Endocrinol (2015) 402:113–9. doi: 10.1016/j.mce.2014.11.029
134. Griffin C, Lanzetta N, Eter L, Singer K. Sexually Dimorphic Myeloid Inflammatory and Metabolic Responses to Diet-Induced Obesity. Am J Physiol Regul Integr Comp Physiol (2016) 311:R211–6. doi: 10.1152/ajpregu.00136.2016
135. Morselli E, Fuente-Martin E, Finan B, Kim M, Frank A, Garcia-Caceres C, et al. Hypothalamic PGC-1α Protects Against High-Fat Diet Exposure by Regulating Erα. Cell Rep (2014) 9:633–45. doi: 10.1016/j.celrep.2014.09.025
136. Kang S-C, Kim B-R, Lee S-Y, Park T-S. Sphingolipid Metabolism and Obesity-Induced Inflammation. Front Endocrinol (Lausanne) (2013) 4:67. doi: 10.3389/fendo.2013.00067
137. Villa A, Gelosa P, Castiglioni L, Cimino M, Rizzi N, Pepe G, et al. Sex-Specific Features of Microglia From Adult Mice. Cell Rep (2018) 23:3501–11. doi: 10.1016/j.celrep.2018.05.048
138. Thion MS, Low D, Silvin A, Chen J, Grisel P, Schulte-Schrepping J, et al. Microbiome Influences Prenatal and Adult Microglia in a Sex-Specific Manner. Cell (2018) 172:500–16.e16. doi: 10.1016/j.cell.2017.11.042
139. Dalile B, Van Oudenhove L, Vervliet B, Verbeke K. The Role of Short-Chain Fatty Acids in Microbiota-Gut-Brain Communication. Nat Rev Gastroenterol Hepatol (2019) 16:461–78. doi: 10.1038/s41575-019-0157-3
140. den Besten G, Bleeker A, Gerding A, van Eunen K, Havinga R, van Dijk TH, et al. Short-Chain Fatty Acids Protect Against High-Fat Diet-Induced Obesity Via a Pparγ-Dependent Switch From Lipogenesis to Fat Oxidation. Diabetes (2015) 64:2398–408. doi: 10.2337/db14-1213
141. Chambers ES, Viardot A, Psichas A, Morrison DJ, Murphy KG, Zac-Varghese SEK, et al. Effects of Targeted Delivery of Propionate to the Human Colon on Appetite Regulation, Body Weight Maintenance and Adiposity in Overweight Adults. Gut (2015) 64:1744–54. doi: 10.1136/gutjnl-2014-307913
142. Erny D, Hrabě de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, et al. Host Microbiota Constantly Control Maturation and Function of Microglia in the CNS. Nat Neurosci (2015) 18:965–77. doi: 10.1038/nn.4030
143. den Brok MH, Raaijmakers TK, Collado-Camps E, Adema GJ. Lipid Droplets as Immune Modulators in Myeloid Cells. Trends Immunol (2018) 39:380–92. doi: 10.1016/j.it.2018.01.012
144. Mauerer R, Walczak Y, Langmann T. Comprehensive mRNA Profiling of Lipid-Related Genes in Microglia and Macrophages Using Taqman Arrays. Methods Mol Biol (2009) 580:187–201. doi: 10.1007/978-1-60761-325-1_10
145. Melo RCN, Weller PF. Lipid Droplets in Leukocytes: Organelles Linked to Inflammatory Responses. Exp Cell Res (2016) 340:193–7. doi: 10.1016/j.yexcr.2015.10.028
146. Maldonado-Ruiz R, Fuentes-Mera L, Camacho A. Central Modulation of Neuroinflammation by Neuropeptides and Energy-Sensing Hormones During Obesity. BioMed Res Int (2017) 2017:7949582. doi: 10.1155/2017/7949582
147. Lee JY, Yune TY. Ghrelin Inhibits Oligodendrocyte Cell Death by Attenuating Microglial Activation. Endocrinol Metab (Seoul) (2014) 29:371–8. doi: 10.3803/EnM.2014.29.3.371
148. Melo HM, Seixas da Silva G da S, Sant’Ana MR, Teixeira CVL, Clarke JR, Miya Coreixas VS, et al. Palmitate Is Increased in the Cerebrospinal Fluid of Humans With Obesity and Induces Memory Impairment in Mice Via Pro-inflammatory Tnf-α. Cell Rep (2020) 30:2180–94.e8. doi: 10.1016/j.celrep.2020.01.072
149. Gao Y, Bielohuby M, Fleming T, Grabner GF, Foppen E, Bernhard W, et al. Dietary Sugars, Not Lipids, Drive Hypothalamic Inflammation. Mol Metab (2017) 6:897–908. doi: 10.1016/j.molmet.2017.06.008
150. Hu S, Wang L, Yang D, Li L, Togo J, Wu Y, et al. Dietary Fat, But Not Protein or Carbohydrate, Regulates Energy Intake and Causes Adiposity in Mice. Cell Metab (2018) 28:415–31.e4. doi: 10.1016/j.cmet.2018.06.010
151. Cintra DE, Ropelle ER, Moraes JC, Pauli JR, Morari J, de SCT, et al. Unsaturated Fatty Acids Revert Diet-Induced Hypothalamic Inflammation in Obesity. PloS One (2012) 7:e30571. doi: 10.1371/journal.pone.0030571
152. Zhao Y, Li G, Li Y, Wang Y, Liu Z. Knockdown of Tlr4 in the Arcuate Nucleus Improves Obesity Related Metabolic Disorders. Sci Rep (2017) 7:7441. doi: 10.1038/s41598-017-07858-6
153. Lancaster GI, Langley KG, Berglund NA, Kammoun HL, Reibe S, Estevez E, et al. Evidence That TLR4 Is Not a Receptor for Saturated Fatty Acids But Mediates Lipid-Induced Inflammation by Reprogramming Macrophage Metabolism. Cell Metab (2018) 27:1096–110.e5. doi: 10.1016/j.cmet.2018.03.014
154. Grajchen E, Wouters E, van de Haterd B, Haidar M, Hardonnière K, Dierckx T, et al. CD36-Mediated Uptake of Myelin Debris by Macrophages and Microglia Reduces Neuroinflammation. J Neuroinflamm (2020) 17:224. doi: 10.1186/s12974-020-01899-x
155. Moore KJ, El Khoury J, Medeiros LA, Terada K, Geula C, Luster AD, et al. A CD36-initiated Signaling Cascade Mediates Inflammatory Effects of Beta-Amyloid. J Biol Chem (2002) 277:47373–9. doi: 10.1074/jbc.M208788200
156. Bazinet RP, Layé S. Polyunsaturated Fatty Acids and Their Metabolites in Brain Function and Disease. Nat Rev Neurosci (2014) 15:771–85. doi: 10.1038/nrn3820
157. Madore C, Nadjar A, Delpech J-C, Sere A, Aubert A, Portal C, et al. Nutritional N-3 PUFAs Deficiency During Perinatal Periods Alters Brain Innate Immune System and Neuronal Plasticity-Associated Genes. Brain Behav Immun (2014) 41:22–31. doi: 10.1016/j.bbi.2014.03.021
158. Milligan G, Alvarez-Curto E, Hudson BD, Prihandoko R, Tobin AB. FFA4/GPR120: Pharmacology and Therapeutic Opportunities. Trends Pharmacol Sci (2017) 38:809–21. doi: 10.1016/j.tips.2017.06.006
159. Dragano NRV, Solon C, Ramalho AF, de Moura RF, Razolli DS, Christiansen E, et al. Polyunsaturated Fatty Acid Receptors, GPR40 and GPR120, Are Expressed in the Hypothalamus and Control Energy Homeostasis and Inflammation. J Neuroinflamm (2017) 14:91. doi: 10.1186/s12974-017-0869-7
160. von Maydell D, Jorfi M. The Interplay Between Microglial States and Major Risk Factors in Alzheimer’s Disease Through the Eyes of Single-Cell RNA-Sequencing: Beyond Black and White. J Neurophysiol (2019) 122:1291–6. doi: 10.1152/jn.00395.2019
161. Masuda T, Sankowski R, Staszewski O, Prinz M. Microglia Heterogeneity in the Single-Cell Era. Cell Rep (2020) 30:1271–81. doi: 10.1016/j.celrep.2020.01.010
162. Van Hove H, Martens L, Scheyltjens I, De Vlaminck K, Pombo Antunes AR, De Prijck S, et al. A Single-Cell Atlas of Mouse Brain Macrophages Reveals Unique Transcriptional Identities Shaped by Ontogeny and Tissue Environment. Nat Neurosci (2019) 22:1021–35. doi: 10.1038/s41593-019-0393-4
163. Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A Unique Microglia Type Associated With Restricting Development of Alzheimer’s Disease. Cell (2017) 169:1276–90. doi: 10.1016/j.cell.2017.05.018
164. Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, et al. Single-Cell RNA Sequencing of Microglia Throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity (2019) 50:253–71. doi: 10.1016/j.immuni.2018.11.004
165. van der Poel M, Ulas T, Mizee MR, Hsiao C-C, Miedema SSM, Adelia, et al. Transcriptional Profiling of Human Microglia Reveals Grey-White Matter Heterogeneity and Multiple Sclerosis-Associated Changes. Nat Commun (2019) 10:1139. doi: 10.1038/s41467-019-08976-7
166. Sankowski R, Böttcher C, Masuda T, Geirsdottir L, Sagar, Sindram E, et al. Mapping Microglia States in the Human Brain Through the Integration of High-Dimensional Techniques. Nat Neurosci (2019) 22:2098–110. doi: 10.1038/s41593-019-0532-y
167. Olah M, Amor S, Brouwer N, Vinet J, Eggen B, Biber K, et al. Identification of a Microglia Phenotype Supportive of Remyelination. Glia (2012) 60:306–21. doi: 10.1002/glia.21266
168. Gao Y, Vidal-Itriago A, Kalsbeek MJ, Layritz C, García-Cáceres C, Tom RZ, et al. Lipoprotein Lipase Maintains Microglial Innate Immunity in Obesity. Cell Rep (2017) 20:3034–42. doi: 10.1016/j.celrep.2017.09.008
169. Ren L, Ren X. Meta-Analyses of Four Polymorphisms of Lipoprotein Lipase Associated With the Risk of Alzheimer’s Disease. Neurosci Lett (2016) 619:73–8. doi: 10.1016/j.neulet.2016.03.021
170. Baum L, Wiebusch H, Pang CP. Roles for Lipoprotein Lipase in Alzheimer’s Disease: An Association Study. Microsc Res Tech (2000) 50:291–6. doi: 10.1002/1097-0029(20000815)50:4<291::AID-JEMT8>3.0.CO;2-L
171. Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity (2017) 47:566–581.e9. doi: 10.1016/j.immuni.2017.08.008
172. Holtzman DM, Herz J, Bu G. Apolipoprotein E and Apolipoprotein E Receptors: Normal Biology and Roles in Alzheimer Disease. Cold Spring Harb Perspect Med (2012) 2:a006312. doi: 10.1101/cshperspect.a006312
173. Mauch DH, Nägler K, Schumacher S, Göritz C, Müller EC, Otto A, et al. CNS Synaptogenesis Promoted by Glia-Derived Cholesterol. Science (2001) 294:1354–7. doi: 10.1126/science.294.5545.1354
174. Rangaraju S, Dammer EB, Raza SA, Gao T, Xiao H, Betarbet R, et al. Quantitative Proteomics of Acutely-Isolated Mouse Microglia Identifies Novel Immune Alzheimer’s Disease-Related Proteins. Mol Neurodegener (2018) 13:34. doi: 10.1186/s13024-018-0266-4
175. Pocivavsek A, Mikhailenko I, Strickland DK, Rebeck GW. Microglial Low-Density Lipoprotein Receptor-Related Protein 1 Modulates C-Jun N-terminal Kinase Activation. J Neuroimmunol (2009) 214:25–32. doi: 10.1016/j.jneuroim.2009.06.010
176. Lynch JR, Tang W, Wang H, Vitek MP, Bennett ER, Sullivan PM, et al. APOE Genotype and an ApoE-mimetic Peptide Modify the Systemic and Central Nervous System Inflammatory Response. J Biol Chem (2003) 278:48529–33. doi: 10.1074/jbc.M306923200
177. Pocivavsek A, Rebeck GW. Inhibition of C-Jun N-terminal Kinase Increases Apoe Expression In Vitro and In Vivo. Biochem Biophys Res Commun (2009) 387:516–20. doi: 10.1016/j.bbrc.2009.07.048
178. Ulland TK, Song WM, Huang SC-C, Ulrich JD, Sergushichev A, Beatty WL, et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell (2017) 170:649–663.e13. doi: 10.1016/j.cell.2017.07.023
179. Hardie DG. AMP-Activated Protein Kinase: An Energy Sensor That Regulates All Aspects of Cell Function. Genes Dev (2011) 25:1895–908. doi: 10.1101/gad.17420111
180. Liu C, Li P, Li H, Wang S, Ding L, Wang H, et al. TREM2 Regulates Obesity-Induced Insulin Resistance Via Adipose Tissue Remodeling in Mice of High-Fat Feeding. J Transl Med (2019) 17:300. doi: 10.1186/s12967-019-2050-9
181. Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, et al. Single-Cell RNA-Seq Reveals the Transcriptional Landscape and Heterogeneity of Aortic Macrophages in Murine Atherosclerosis. Circ Res (2018) 122:1661–74. doi: 10.1161/CIRCRESAHA.117.312509
182. Hamerman JA, Jarjoura JR, Humphrey MB, Nakamura MC, Seaman WE, Lanier LL. Cutting Edge: Inhibition of TLR and FcR Responses in Macrophages by Triggering Receptor Expressed on Myeloid Cells (TREM)-2 and DAP12. J Immunol (2006) 177:2051–5. doi: 10.4049/jimmunol.177.4.2051
183. Gao X, Dong Y, Liu Z, Niu B. Silencing of Triggering Receptor Expressed on Myeloid Cells-2 Enhances the Inflammatory Responses of Alveolar Macrophages to Lipopolysaccharide. Mol Med Rep (2013) 7:921–6. doi: 10.3892/mmr.2013.1268
184. Marschallinger J, Iram T, Zardeneta M, Lee SE, Lehallier B, Haney MS, et al. Lipid-Droplet-Accumulating Microglia Represent a Dysfunctional and Proinflammatory State in the Aging Brain. Nat Neurosci (2020) 23:194–208. doi: 10.1038/s41593-019-0566-1
185. Miedema A, Wijering MHC, Eggen BJL, Kooistra SM. High-Resolution Transcriptomic and Proteomic Profiling of Heterogeneity of Brain-Derived Microglia in Multiple Sclerosis. Front Mol Neurosci (2020) 13:583811. doi: 10.3389/fnmol.2020.583811
Keywords: microglia, hypothalamus, lipids, obesity, diabetes
Citation: Folick A, Koliwad SK and Valdearcos M (2021) Microglial Lipid Biology in the Hypothalamic Regulation of Metabolic Homeostasis. Front. Endocrinol. 12:668396. doi: 10.3389/fendo.2021.668396
Received: 16 February 2021; Accepted: 05 May 2021;
Published: 27 May 2021.
Edited by:
Rachel Nicole Lippert, German Institute of Human Nutrition Potsdam-Rehbruecke (DIfE), GermanyReviewed by:
Alexandre Benani, Centre National de la Recherche Scientifique (CNRS), FranceXavier Fioramonti, INRA UMR1286 Laboratoire NutriNeuro, France
Copyright © 2021 Folick, Koliwad and Valdearcos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martin Valdearcos, bWFydGluLnZhbGRlYXJjb3Njb250cmVyYXNAdWNzZi5lZHU=