
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol. , 14 April 2021
Sec. Cancer Endocrinology
Volume 12 - 2021 | https://doi.org/10.3389/fendo.2021.665631
This article is part of the Research Topic NIKE: Neuroendocrine Tumors, Innovation in Knowledge and Education View all 11 articles
 Giovanni Vitale1,2*
Giovanni Vitale1,2* Alessia Cozzolino3
Alessia Cozzolino3 Pasqualino Malandrino4
Pasqualino Malandrino4 Roberto Minotta5
Roberto Minotta5 Giulia Puliani3,6Davide Saronni2
Giulia Puliani3,6Davide Saronni2 Antongiulio Faggiano7
Antongiulio Faggiano7 Annamaria Colao5 on behalf of NIKE
Annamaria Colao5 on behalf of NIKENeuroendocrine neoplasms (NENs) are a heterogeneous group of tumors originating from neuroendocrine cells dispersed in different organs. Receptor tyrosine kinases are a subclass of tyrosine kinases with a relevant role in several cellular processes including proliferation, differentiation, motility and metabolism. Dysregulation of these receptors is involved in neoplastic development and progression for several tumors, including NENs. In this review, we provide an overview concerning the role of the fibroblast growth factor (FGF)/fibroblast growth factor receptor (FGFR) system in the development and progression of NENs, the occurrence of fibrotic complications and the onset of drug-resistance. Although no specific FGFR kinase inhibitors have been evaluated in NENs, several clinical trials on multitarget tyrosine kinase inhibitors, acting also on FGF system, showed promising anti-tumor activity with an acceptable and manageable safety profile in patients with advanced NENs. Future studies will need to confirm these issues, particularly with the development of new tyrosine kinase inhibitors highly selective for FGFR.
Neuroendocrine neoplasms (NENs) are a heterogeneous group of tumors originating from neuroendocrine cells dispersed in different organs (1–5).
Receptor tyrosine kinases are a subclass of tyrosine kinases with a relevant role in several cellular processes including proliferation, differentiation, motility and metabolism. Dysregulation of these receptors plays a relevant role in neoplastic development and progression for several tumors, including NENs (6, 7).
In this review, we provide an overview concerning the role of the fibroblast growth factor (FGF)/fibroblast growth factor receptor (FGFR) system in NENs.
FGFs and related receptors are members of a large family with a wide range of effects. This system is involved in organogenesis (during development), homeostasis and repair of adult tissues. Moreover, FGF family promotes angiogenesis, growth, differentiation and migration of cells mainly through the activation of RAS-MAPK, PI3K-AKT and PLCγ pathways, with a relevant role in the development and progression of several tumors (8). These effects are mediated by the interaction of FGFs with four tyrosine kinase receptors: FGFR1, FGFR2, FGFR3 and FGFR4, which are composed by an extracellular domain, a transmembrane domain and an intracellular domain. The binding of ligands induces conformational changes that lead to a dimerization of these receptors. This event activates the intracellular tyrosine kinase domain, which in turn triggers the signalling cascade (8). FGFs, based on their biochemical functions, sequence similarity and evolutionary relationships, are classified into different subfamilies: FGF1, FGF4, FGF7, FGF8, FGF9, FGF15/19 and FGF11.
FGF1 and FGF2 are members of the FGF1 subfamily. FGF1 is the only FGF that can activate all FGFRs splice variants. It is involved in cell cycle regulation, cell differentiation, survival and apoptosis. FGF1 plays a central role in neuroprotection and axon regeneration and appears to improve functional recovery after spinal cord injury (9). FGF2 has known angiogenic properties (10, 11). The FGF4 subfamily (FGF4, 5,6) can activate FGFR1-3(IIIc) and FGFR4. These molecules are fundamental in embryonic development and muscle regeneration (8, 9). FGF7 subfamily (FGF3, 7, 10, 22) preferentially activates FGFR2(IIIb), although FGF3 and FGF10 can also interact with FGFR1(IIIb). FGF3 is involved in the neural development, while FGF7 is required for lung, kidney and neuronal synapses development. The development of epithelial components, such as limb and lungs, and mammary gland requires epithelial-mesenchymal interactions granted by FGF10. Finally, FGF22 regulates the circuit remodeling in the injured spinal cord (12–15). FGF8 subfamily members (FGF8, 17 and 18) activate FGFR4 and FGFR1-3(IIIc). They are involved in the skeletal and brain development and in odontogenesis (8, 14, 16–18). The FGF9 subfamily (FGF9, 16, 20) interacts with FGFR1-3(IIIc), FGFR3(IIIb) and FGFR4. These proteins are involved in a proper heart, kidney and skeletal development (8, 14, 19, 20). The FGF15/19 subfamily comprises FGF15/19, 21 and 23. FGF15/19 bind FGFR1-3(IIIc) and FGFR4. FGF21 can activate FGFR1(IIIc) and 3(IIIc), as well as FGF23, which can also interact with FGFR4. This subfamily acts as hormones and regulates hepatocyte and adipocyte metabolism (8, 14). FGF11 subfamily members (FGF11, 12, 13, 14) are known as intracellular FGFs. These peptides are not secreted and interact with the cytosolic carboxy terminal tail of voltage gated sodium channels. They cover an important role in the development of the nervous system (8, 21).
A deregulation of the FGF/FGFR system can be involved in cancer development and progression through modulation of cell proliferation, migration and angiogenesis (22).
Besides its role in physiological angiogenesis, FGF2 is implied in tumor-induced angiogenesis and metastatic process and appears to direct tumor-associated macrophages toward a pro-tumorigenic state (23–25). FGF4 promotes cancer cell proliferation, invasion and migration by causing a switch of the receptor FGFR2-IIIb, a splice variant expressed in epithelial cells, into FGFR2-IIIc, expressed in mesenchymal cells and able to induce epithelial-mesenchymal transition (26). FGF5 can promote osteosarcoma proliferation by activating the MAPK signaling pathway (27) and the FGF5/FGFR1 axis contributes to melanoma progression (28). FGF6 can stimulate proliferation of prostate cancer cells through the activation of FGFR4 (29). Among the FGF7 subfamily, FGF3 and FGF7 have been reported to be highly expressed in breast cancer (30, 31) and gastric adenocarcinoma (32), respectively. In addition, the FGF10/FGFR-IIb signaling appears to have a role in breast and pancreatic tumors (15, 33). Although the mechanism is unclear, Jarosz et al. observed a potential role of FGF22 in skin tumorigenesis (34). In a recent study, FGF22 and its receptor FGFR-IIb appear to be associated with the development of lung adenocarcinoma through the MAPK and Rap I signaling pathways (35). A deregulation of FGF18, caused by an altered expression of its negative regulator miR-590-5p, is able to stimulate proliferation and epithelial-mesenchymal transition, with enhanced invasion abilities, in gastric cancer cells (36). In HER+ breast cancer cell lines, overexpression of FGF18 stimulates the expression of genes involved in migration and cancer metastasis through Akt/GSK3β pathway (37). By the interaction with FGFR2 and FGFR3 and the activation of the ERK/Akt pathway, FGF18 is able to induce proliferation and invasion in endometrial carcinoma (38).
The FGF/FGFR pathway has also a key role in the onset of drug-resistance (39). FGF/FGFR pathway is the first compensatory mechanism in tumors resistant to drugs targeting the vascular endothelial growth factor (VEGF) system (40–42). Indeed, VEGF-dependent vessels are suppressed during prolonged anti-VEGF therapy, while the expression of FGF2 is increased, leading to a novel angiogenesis dependent on FGF2 signaling pathway. This condition drives the tumor toward drug-resistance (42). Boichuk et al. (43) showed that FGF signaling is activated in gastrointestinal stromal tumors after the acquisition of imatinib resistance. Interestingly, the use of a potent FGF inhibitor markedly reduced cell growth in resistant cells compared to imatinib-sensitive cells. This effect increased when the two molecules were combined in resistant cells, showing also that the FGF-inhibitor can restore sensitivity to imatinib.
The role of the FGF/FGFR system has been analyzed also in NENs and several lines of evidence support its function in the modulation of tumor fibrosis, proliferation, angiogenesis and drug resistance, through a dynamic cross talk between NEN cells, fibroblasts, endothelial cells and inflammatory cells (44).
Bordi et al. identified FGF2 by immunohistochemistry in endocrine cells of the gastric oxyntic mucosa and mRNA of FGF2 in enterochromaffin-like carcinoid tumors (45). Immunohistochemical studies demonstrated the staining for FGF-2 in 100% of NEN cells from the midgut and the pancreas, while FGF2 receptors were observed only in the stromal component (46). La Rosa et al. found cytoplasmic immunoreactivity for FGF1 in 26 (43%) out of 60 GEP-NENs and FGFR1-4 were found in 68-88% of tumors with tumor microenvironment components also expressing FGFRs (47). The authors observed also that normal endocrine cells of the gut rarely expressed FGFRs thus hypothesizing that in normal mucosa the FGF/FGFR system has not an autocrine role on modulating endocrine cells functions. Therefore, de novo expression of FGFRs by NEN cells may play a role in the autocrine/paracrine signaling responsible of tumorigenesis, stromal fibrosis and tumor-induced angiogenesis.
NEN are often characterized by the development of fibrosis, local or distant. The best-known fibrotic complications are carcinoid heart disease, which develops in about 20% of patients with carcinoid syndrome (48), and mesenteric fibrosis, which affects up to 40-50% of small bowel NENs (49, 50). Less known complications are represented by retroperitoneal fibrosis (50), scleroderma (51), infiltration of the pleura (52) or alveoli (53) and fibrosis of the bladder (54). Although the pathogenesis of fibrotic complications is unclear, serotonin, with a relevant mitogenic power on fibroblasts, mesangial cells, smooth muscle cells, endothelial cells and NEN cells, may have a role in these events (55). The FGF system appears to be also implicated in the mechanism of gastrointestinal NEN fibrosis (56). In fact, Bordi (45) showed that among the 10 patients suffering from type 3 gastric NEN and with positive immunohistochemistry for FGF2, some had diffuse stromal fibrosis. Another study (57), which analyzed a pool of 41 gastrointestinal NENs, showed a positive correlation between FGF1 and the amount of fibrous stroma in tumors. The FGF is responsible of cell proliferation and stroma formation and its action is potentiated by serotonin (58). Moreover, FGF may activate also the expression of the connective tissue growth factor genes that regulate myofibroblast differentiation, collagen synthesis and fibroblast proliferation (59).
The mRNA expression of FGF receptor was found more frequently in functioning NENs (including gastrinomas and insulinomas) than in functionally inactive NENs (53.6% vs. 22.2%) (60). Although this difference was not statistically significant (p=0.10), speculating on the association between FGFR expression and hormone production may be not totally irrational, but further evidence is required to corroborate these findings.
The FGFR4-G388R single-nucleotide polymorphism was investigated in 71 patients with pancreatic NEN (61). The authors observed that FGFR4-R388 allele was independently associated with liver metastases. To further analyze the impact of the FGFR4 SNP, the same authors transfected BON1 cells with either FGFR4-G388 or FGFR4-R388 and injected them in SCID mice. They found that xenografts expressing FGFR4-R388 displayed a more aggressive biological behavior and were resistant to everolimus treatment. This latter aspect was investigated also among 17 patients previously treated with everolimus in a clinical trial. Patients harboring FGFR4-R388 allele achieved a worse tumor response (9% vs. 25%) and a reduced median PFS (4.8 vs. 16.6 months) and OS (9.3vs 40 months) compared to patients homozygous for FGFR4-G388. Although decreased drug response was related to persistently high mTOR and STAT3 phosphorylation despite of everolimus treatment, these data were not confirmed by Cros et al., who reported no modification of the mTOR pathway in patients with pancreatic or ileal NENs harboring FGFR4-R388 allele (62). This apparent inconsistency corroborates the need for further studies validating the identification of molecular parameters useful to predict drug efficacy and resistance (63).
The FGF/FGFR system collaborates with the VEGF signaling pathway in the initiation and maintenance of tumor angiogenesis. These mechanisms have been demonstrated in allograft transplantation experiments and in mouse model of pancreatic NEN (the Rip1Tag2 transgenic mice), where interfering with the FGF function by a soluble form of the FGFR2 IIIb significantly inhibited tumor-induced angiogenesis and tumor growth (64). The FGF system acts as a second proangiogenic circuit, indeed VEGF is the main regulator of angiogenesis but, as reported by Casanovas et al., experiments in the Rip1Tag2 model of pancreatic islet carcinoma documented that initial inhibition of the angiogenesis achieved by VEGF signaling blockade was restored by the upregulation of the FGF system (65). Therefore, blocking both VEGF and FGF signaling pathways may reveal synergic antiangiogenic effects and inhibit tumor progression secondary to compensatory feedback loops driving tumor revascularization. For instance, Allen et al. investigated the effect of brivanib, a selective inhibitor targeting both VEGF and FGF receptors, in a mouse model of pancreatic NEN. Brivanib was effective not only as first-line therapy, but also as second-line treatment after failure of two agents inhibiting VEGF receptors (DC101 and sorafenib) (66).
In the last few years, the therapeutic approach for NENs has changed following the approval of several innovative targeted treatments such as tyrosine kinase inhibitors (TKIs). Although no specific FGFR kinase inhibitors have been evaluated in NENs, several clinical trials on multitarget TKIs, acting also on FGF, are ongoing and few published studies have demonstrated their efficacy in NENs (44). The interest in FGF pathway inhibitors relies also in the possibility to overcome resistance to VEGF inhibition that may arise after long term use of these drugs or could be intrinsic in tumor expressing FGF2 (67–69). The results of clinical trials in NENs evaluating multitarget TKI, acting also on FGF, are described below (Table 1).
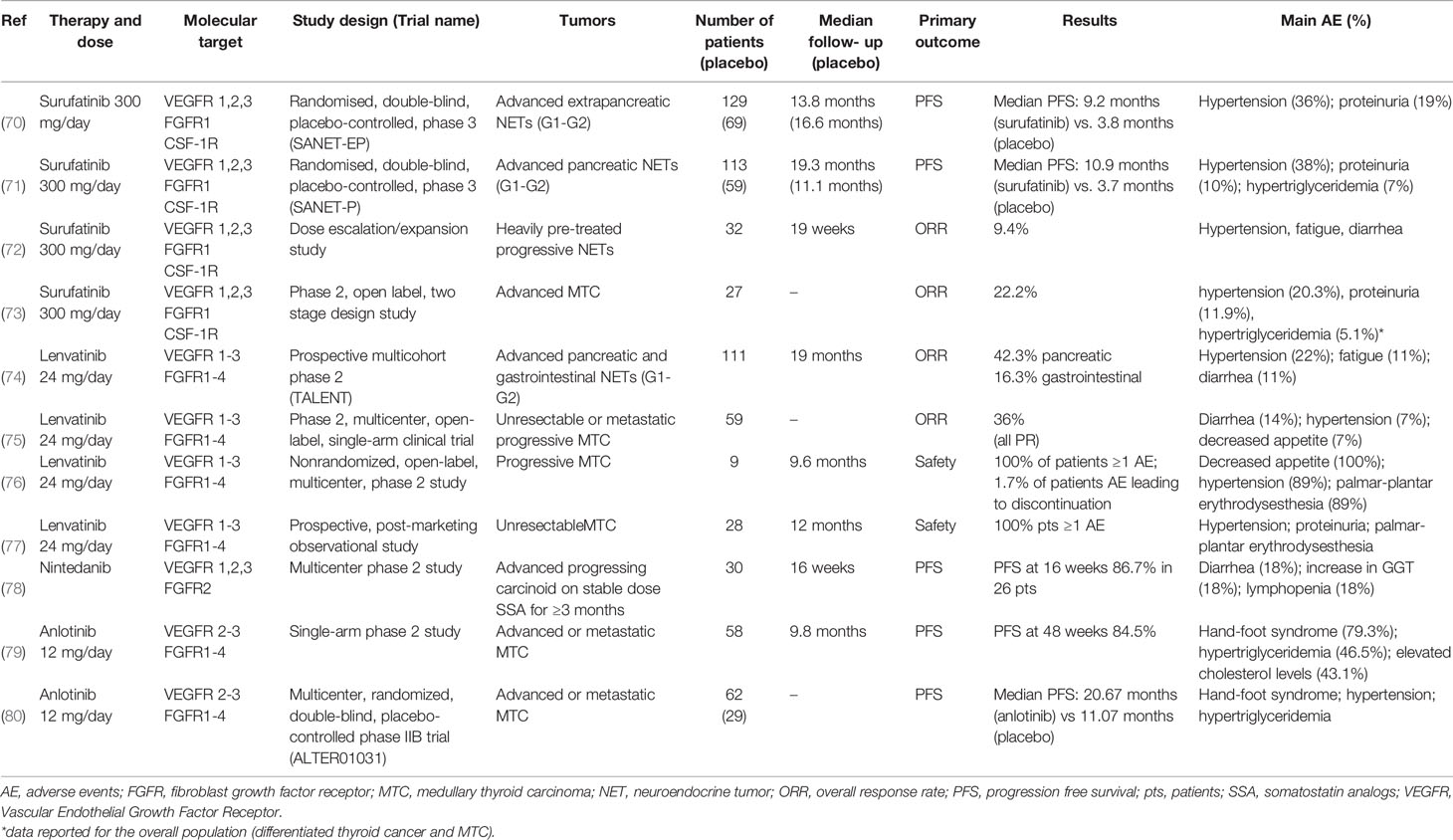
Table 1 Clinical trials evaluating the effects of multitarget tyrosine kinase inhibitors, acting also on FGFR, in patients with NENs.
Surufatinib is a potent TKI targeting VEGF receptors (VEGFR) 1, 2, and 3, FGFR1, and CSF-1R. In preliminary phase I and Ib/II studies surufatinib showed encouraging anti-tumor activity in advanced NENs (81, 82).
Two randomized phase III placebo controlled trials evaluated safety and efficacy of surufatinib in patients with well differentiated NENs of extra-pancreatic (SANET-ep) and pancreatic (SANET-p) origin (70, 71).
In SANET-ep study (70) 198 patients were randomly assigned to surufatinib 300 mg/day (n=129) or placebo (n=69). Median progression-free survival (PFS) was 9.2 months in the surufatinib group versus 3.8 months in the placebo group. The overall response rate (ORR) was 10% in the surufatinib group versus zero in the placebo group. The most common treatment-related adverse events (AE) of grade ≥ 3 were hypertension (36% surufatinib vs 13% placebo) and proteinuria (19% vs. 0%). In SANET-p study (71) 113 patients were randomly assigned to surufatinib (300 mg/day) and 59 to placebo. The median PFS was 10.9 months for surufatinib versus 3.7 months for placebo; ORR was 19% in the surufatinib group and 2% in the placebo group. The most common AE of grade ≥ 3 were hypertension (38% surufatinib vs. 7% placebo), proteinuria (10% vs. 2%) and hypertriglyceridemia (7% vs. none).
Another study evaluated the effect of surufatinib dose escalation/expansion in 32 patients with heavily pre-treated progressive NENs, 16 patients with pancreatic NENs and 16 with extra-pancreatic NENs. Nineteen patients remained on active treatment (13 extra-pancreatic and 6 pancreatic), 9 patients discontinued due to disease progression, 2 withdrew consent and 2 discontinued due to AE. An ORR of 9.4% was observed (72).
An open label phase II study evaluated efficacy and tolerability of surufatinib (300 mg/day) in 27 patients with progressive medullary thyroid cancer (MTC). Objective response was observed in 22.2% of patients with MTC, and the majority (88.9%) achieved disease control. The therapy was well tolerated (73).
Therefore, surufatinib demonstrated promising anti-tumor activity with an acceptable and manageable safety profile in advanced NENs.
Lenvatinib is a potent VEGFR1-3 and FGFR1-4 inhibitor. The TALENT trial, a prospective phase II study, evaluated efficacy, safety and tolerability of lenvatinib (24 mg once daily) in G1/G2 advanced pancreatic (n=55) and gastrointestinal (n=56) NENs resistant to previous targeted agents. The ORR was 29% (42.3% for pancreatic NENs and 16.3% for gastrointestinal NENs). PFS and overall survival (OS) for pancreatic NENs were 15.5 months and 29.2 months, while for gastrointestinal NENs were 15.4 months and not reached, respectively. The most frequent grade 3/4 AE were hypertension (22%), fatigue (11%) and diarrhea (11%) (74). Thus, lenvatinib showed a promising PFS and OS in a pretreated population.
A phase II, multicenter, open-label, single-arm clinical trial evaluated efficacy and tolerability of lenvatinib (24-mg daily, 28-day cycles) in 59 patients with MTC. ORR was 36%, all PR. Disease control rate (DCR) was 80%, 44% had SD. Median time to response was 3.5 months. Median PFS was 9.0 months. Grade 3/4 AE included diarrhea (14%), hypertension (7%), decreased appetite (7%), fatigue, dysphagia and increased alanine aminotransferase levels (5% each) (75).
Another phase II study evaluated lenvatinib treatment in 9 patients with MTC. The most frequently reported AE were decreased appetite (100%), hypertension (89%), palmar-plantar erythrodysesthesia (89%), diarrhea (89%), fatigue (78%) and proteinuria (67%). Median PFS was 9.2 months. Median OS was 12.1 months. ORR was 22% and DCR was 100% (76).
Recently, a prospective, post-marketing observational study evaluated, in daily clinical practice, the safety and effectiveness of lenvatinib in 28 patients with MTC. Hypertension, proteinuria and palmar-plantar erythrodysesthesia syndrome were the most frequently reported AE. The 12-months OS rate was 83%. ORR was 45% (77).
Nintedanib is a dual inhibitor of VEGFR1, -2, and -3 as well as FGFR2 and showed both antiangiogenic and antitumor activity in the RIP1-Tag2 transgenic mouse model of tumorigenesis for pancreatic NEN (44). A multicenter phase II study evaluated efficacy, safety and tolerability of nintedanib in 30 patients with unresectable/metastatic carcinoids on stable dose of SSA for ≥3 months. PFS at 16 weeks was 86.7% in 26 patients. PR was observed in 4%, SD in 83%, disease progression in 8% of patients. Quality of life was maintained or improved in at least 50% of subjects. The most common grade 3 AE were hypertension and decreased appetite (78).
A prospective randomized double-blind phase II study evaluated the efficacy and tolerability of nintedanib in progressing MTC after prior TKI treatment. The study was stopped due to slow accrual with 32/67 patients enrolled, without reaching the targeted statistical power. The most common AE were diarrhea (18%), nausea (9%), GGT increase (18%) and lymphopenia (18%) (83).
Anlotinib is a novel TKI targeting VEGFR2-3 and FGFR1-4 with high affinity. Anlotinib has previously shown promising antitumor activity on MTC in preclinical models and a phase I study (84). A phase II clinical trial showed a relevant antitumor activity of anlotinib (12 mg once daily, two weeks on/one week off) in 58 patients with advanced MTC. PFS rates at 24, 36, and 48 weeks were 92.2%, 87.8% and 84.5%, respectively. Significant decreases in serum calcitonin (≥50%) occurred in 57.5% of patients. The most common AE included hand-foot syndrome (79.3%), hypertriglyceridemia (46.5%), hypercholesterolemia (43.1%), fatigue (41.4%), proteinuria (39.7%), hypertension (39.7%), sore throat (37.9%), diarrhea (34.5%) and anorexia (34.5%) (79).
These data have been confirmed in a phase IIb study (ALTER01031), enrolling a larger cohort of patients (80). Ninety-one patients with advanced MTC were randomized: 62 to anlotinib arm and 29 to placebo arm (12 mg/die from day 1 to 14 of a 21-day cycle). Median PFS was 20.7 months in anlotinib arm vs. 11.1 months in placebo arm. The most common AE after anlotinib arm were hand-foot syndrome, hypertension, hypertriglyceridemia and diarrhea (80).
Several clinical trials on the use of multi target TKI, with an action also on FGFR, in patients with NENs are currently ongoing. Table 2 reports the main characteristics of trials registered on clinicaltrials.gov.
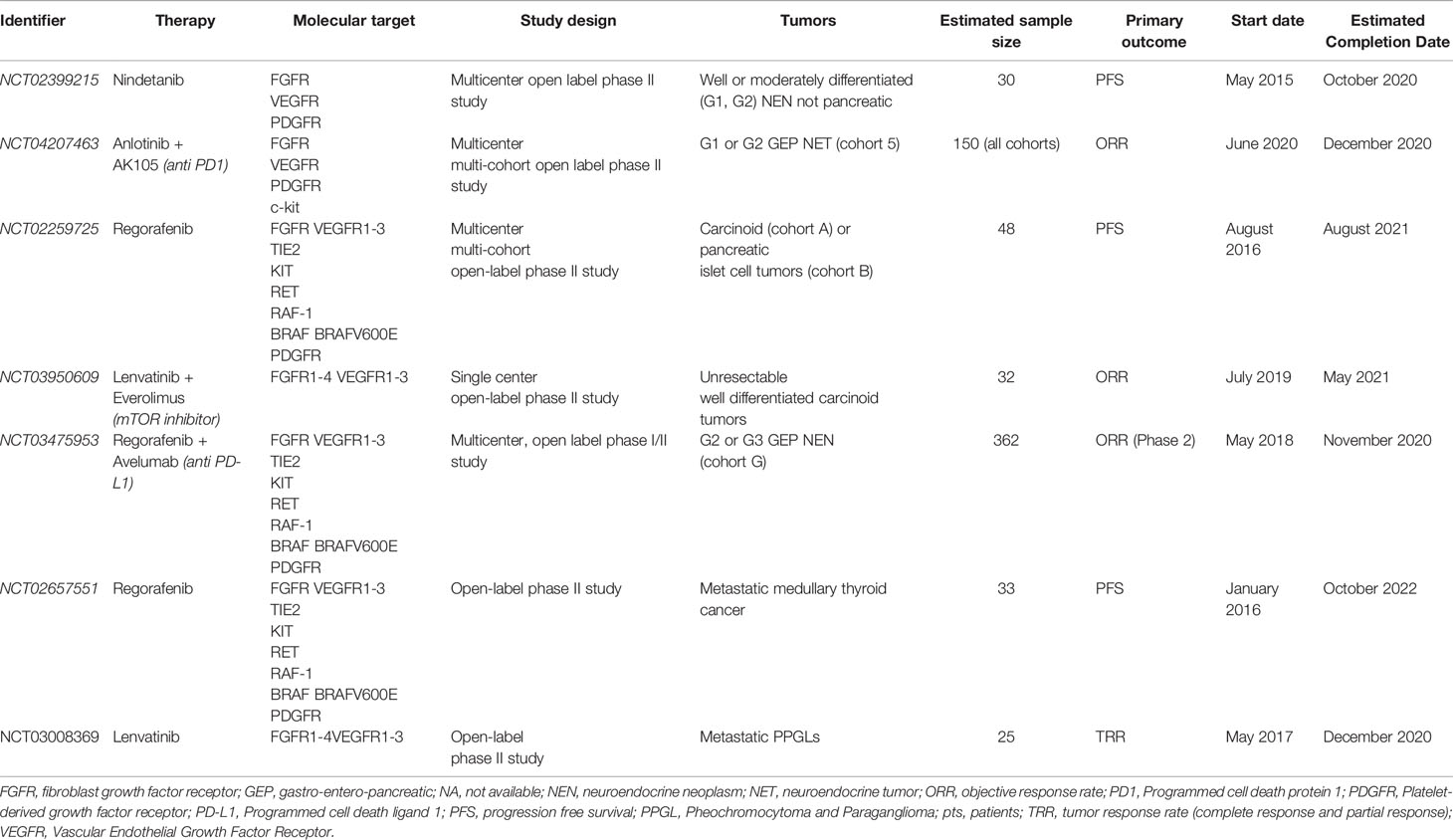
Table 2 Ongoing clinical trials evaluating the effects of multitarget tyrosine kinase inhibitors, acting also on FGFR, in patients with NENs.
In the last years there is mounting evidence supporting the role of FGF/FGFR system in the development and progression of NENs and probably in the occurrence of fibrotic complications (mesenteric and/or retroperitoneal fibrosis). In addition, the FGF/FGFR pathway could also have a key role in the onset of drug-resistance. Indeed, FGF/FGFR pathway is a main compensatory mechanism in anti-VEGF-therapy-resistant tumors.
Currently no specific FGFR kinase inhibitors have been evaluated in patients affected by advanced NENs. Although recent clinical trials have reported a significant antitumor activity and manageable safety profile of several multitarget TKIs, which are able to block many molecular pathways including FGFR, it is not possible to isolate the efficacy of FGFR inhibition alone. Future studies should better confirm these issues and clarify the role of FGF/FGFR pathway in promoting drug-resistance in NENs. The development of new TKIs, highly selective for FGFR and with less toxicity, may open an innovative therapeutic strategy to be integrated into a personalized approach for this heterogeneous class of tumors. In addition, recent preclinical studies showed a potent inhibition in tumor growth both in hepatocellular carcinoma (85) and in ovarian cancer (86), through the simultaneous blockade of mTOR and FGFR pathways. Considering the pivotal role of deregulated mTOR signaling activation in the proliferation of NENs, particularly in pancreatic tumors, combining mTOR inhibitors and TKIs targeting FGFRs could represent a future therapeutic approach in NENs.
GV, AlC, PM, RM, GP, and DS conceptualized and wrote the manuscript. AF and AnC contributed to draft the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Italian Ministry of Education, University and Research (MIUR): PRIN 2017Z3N3YC.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This review is part of the ‘Neuroendocrine Tumors Innovation Knowledge and Education’ project led by Prof. Annamaria Colao and Prof. Antongiulio Faggiano, which aims at increasing the knowledge on NET. We would like to acknowledge all the Collaborators of the “NIKE” project: Manuela Albertelli - Genova; Barbara Altieri – Wurzburg; Luigi Barrea – Napoli; Filomena Bottiglieri – Napoli; Severo Campione – Napoli; Federica Campolo – Roma, Roberta Centello – Roma; Alessia Cozzolino – Roma; Federica De Cicco – Napoli; Sergio Di Molfetta - Bari; Valentina Di Vito – Roma; Alessandra Dicitore – Milano; Andrea Dotto – Genova; Giuseppe Fanciulli – Sassari; Tiziana Feola - Roma; Diego Ferone - Genova; Francesco Ferraù - Messina; Marco Gallo – Alessandria; Elisa Giannetta - Roma; Federica Grillo - Genova; Erika Maria Grossrubatscher – Milano; Elia Guadagno - Napoli; Valentina Guarnotta - Palermo; Andrea M. Isidori - Roma; Andrea Lania - Milano; Andrea Lenzi - Roma; Fabio Lo Calzo – Avellino; Pasqualino Malandrino - Catania; Erika Messina – Messina; Roberto Minotta – Napoli; Roberta Modica - Napoli; Giovanna Muscogiuri – Napoli; Riccardo Pofi - Roma; Giulia Puliani - Roma; Alberto Ragni – Torino; Paola Razzore – Torino; Laura Rizza - Roma; Manila Rubino - Milano; Rosaria Maria Ruggeri – Messina; Davide Saronni – Milano; Emilia Sbardella – Roma; Concetta Sciammarella – Verona; Francesca Sciarra – Roma; Franz Sesti – Roma; Maria Grazia Tarsitano – Roma; Mary Anna Venneri - Roma; Giovanni Vitale – Milano; Isabella Zanata - Ferrara.
1. La Rosa S, Uccella S. Classification of neuroendocrine neoplasms: lights and shadows. Rev Endocr Metab Disord (2020). doi: 10.1007/s11154-020-09612-2
2. Barriuso J, Lamarca A. Clinical and Translational Research Challenges in Neuroendocrine Tumours. Curr Med Chem (2020) 27:4823–39. doi: 10.2174/0929867327666200207120725
3. Alexandraki KI, Tsoli M, Kyriakopoulos G, Angelousi A, Nikolopoulos G, Kolomodi D, et al. Current concepts in the diagnosis and management of neuroendocrine neoplasms of unknown primary origin. Minerva Endocrinol (2019) 44:378–86. doi: 10.23736/S0391-1977.19.03012-8
4. Fuentes-Fayos AC, García-Martínez A, Herrera-Martínez AD, Jiménez-Vacas JM, Vázquez-Borrego MC, Castaño JP, et al. Molecular determinants of the response to medical treatment of growth hormone secreting pituitary neuroendocrine tumors. Minerva Endocrinol (2019) 44:109–28. doi: 10.23736/S0391-1977.19.02970-5
5. Tamagno G, Bennett A, Ivanovski I. Lights and darks of neuroendocrine tumors of the appendix. Minerva Endocrinol (2020) 45:381–92. doi: 10.23736/S0391-1977.20.03206-X
6. Kiesewetter B, Raderer M. How I treat neuroendocrine tumours. ESMO Open (2020) 5:e000811. doi: 10.1136/esmoopen-2020-000811
7. Walenkamp A, Crespo G, Fierro Maya F, Fossmark R, Igaz P, Rinke A, et al. Hallmarks of gastrointestinal neuroendocrine tumours: implications for treatment. Endocr Relat Cancer (2014) 21:R445–60. doi: 10.1530/ERC-14-0106
8. Ornitz DM, Itoh N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip Rev Dev Biol (2015) 4(3):215–66. doi: 10.1002/wdev.176
9. Li J, Wang Q, Cai H, He Z, Wang H, Chen J, et al. FGF1 improves functional recovery through inducing PRDX1 to regulate autophagy and anti-ROS after spinal cord injury. J Cell Mol Med (2018) 22(5):2727–38. doi: 10.1111/jcmm.13566
10. Presta M, Andrés G, Leali D, Dell’Era P, Ronca R. Inflammatory cells and chemokines sustain FGF2-induced angiogenesis. Eur Cytokine Netw (2009) 20(2):39–50. doi: 10.1684/ecn.2009.0155
11. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature (2011) 473(7347):298–307. doi: 10.1038/nature10144
12. Jacobi A, Loy K, Schmalz AM, Hellsten M, Umemori H, Kerschensteiner M, et al. FGF22 signaling regulates synapse formation during post-injury remodeling of the spinal cord. EMBO J (2015) 34(9):1231–43. doi: 10.15252/embj.201490578
13. Anderson MJ, Schimmang T, Lewandoski M. An FGF3-BMP Signaling Axis Regulates Caudal Neural Tube Closure, Neural Crest Specification and Anterior-Posterior Axis Extension. PloS Genet (2016) 12(5):e1006018. doi: 10.1371/journal.pgen.1006018
14. Xie Y, Su N, Yang J, Tan Q, Huang S, Jin M, et al. FGF/FGFR signaling in health and disease. Signal Transduct Target Ther (2020) 5(1):181. doi: 10.1038/s41392-020-00222-7
15. Rivetti S, Chen C, Chen C, Bellusci S. Fgf10/Fgfr2b Signaling in Mammary Gland Development, Homeostasis, and Cancer. Front Cell Dev Biol (2020) 8:415. doi: 10.3389/fcell.2020.00415
16. Chan WK, Price DJ, Pratt T. FGF8 morphogen gradients are differentially regulated by heparan sulphotransferases Hs2st and Hs6st1 in the developing brain. Biol Open (2017) 6(12):1933–42. doi: 10.1242/bio.028605
17. Hao Y, Tang S, Yuan Y, Liu R, Chen Q. Roles of FGF8 subfamily in embryogenesis and oral−maxillofacial diseases (Review). Int J Oncol (2019) 54(3):797–806. doi: 10.3892/ijo.2019.4677
18. Boylan M, Anderson MJ, Ornitz DM, Lewandoski M. The Fgf8 subfamily (Fgf8, Fgf17 and Fgf18) is required for closure of the embryonic ventral body wall. Development (2020) 147(21):dev189506. doi: 10.1242/dev.189506
19. Charoenlarp P, Rajendran AK, Iseki S. Role of fibroblast growth factors in bone regeneration. Inflammation Regen (2017) 37:10. doi: 10.1186/s41232-017-0043-8
20. Wang S, Li Y, Jiang C, Tian H. Fibroblast growth factor 9 subfamily and the heart. Appl Microbiol Biotechnol (2018) 102(2):605–13. doi: 10.1007/s00253-017-8652-3
21. Zhang X, Bao L, Yang L, Wu Q, Li S. Roles of intracellular fibroblast growth factors in neural development and functions. Sci China Life Sci (2012) 55(12):1038–44. doi: 10.1007/s11427-012-4412-x
22. Tanner Y, Grose RP. Dysregulated FGF signalling in neoplastic disorders. Semin Cell Dev Biol (2016) 53:126–35. doi: 10.1016/j.semcdb.2015.10.012
23. Cao Y, Cao R. Hedlund EM. R Regulation of tumor angiogenesis and metastasis by FGF and PDGF signaling pathways. J Mol Med (Berl) (2008) 86(7):785–9. doi: 10.1007/s00109-008-0337-z
24. Hosaka K, Yang Y, Seki T, Du Q, Jing X, He X, et al. Therapeutic paradigm of dual targeting VEGF and PDGF for effectively treating FGF-2 off-target tumors. Nat Commun (2020) 11(1):3704. doi: 10.1038/s41467-020-17525-6
25. Im JH, Buzzelli JN, Jones K, Franchini F, Gordon-Weeks A, Markelc B, et al. FGF2 alters macrophage polarization, tumour immunity and growth and can be targeted during radiotherapy. Nat Commun (2020) 11(1):4064. doi: 10.1038/s41467-020-17914-x
26. Qi L, Song W, Li L, Cao L, Yu Y, Song C, et al. FGF4 induces epithelial-mesenchymal transition by inducing store-operated calcium entry in lung adenocarcinoma. Oncotarget (2016) 7(45):74015–30. doi: 10.18632/oncotarget.12187
27. Han D, Wang M, Yu Z, Yin L, Liu C, Wang J, et al. FGF5 promotes osteosarcoma cells proliferation via activating MAPK signaling pathway. Cancer Manag Res (2019) 11:6457–66. doi: 10.2147/CMAR.S200234
28. Ghassemi S, Vejdovszky K, Sahin E, Ratzinger L, Schelch K, Mohr T, et al. FGF5 is expressed in melanoma and enhances malignancy in vitro and in vivo. Oncotarget (2017) 8(50):87750–62. doi: 10.18632/oncotarget.21184
29. Ropiquet F, Giri D, Kwabi-Addo B, Mansukhani A, Ittmann M. Increased expression of fibroblast growth factor 6 in human prostatic intraepithelial neoplasia and prostate cancer. Cancer Res (2000) 60(15):4245–50.
30. Roy D, Calaf GM. Allelic loss at chromosome 11q13 alters FGF3 gene expression in a human breast cancer progression model. Oncol Rep (2014) 32(6):2445–52. doi: 10.3892/or.2014.3502
31. Parish A, Schwaederle M, Daniels G, Piccioni D, Fanta P. Schwab Ret al. Fibroblast growth factor family aberrations in cancers: clinical and molecular characteristics. Cell Cycle (2015) 14(13):2121–8. doi: 10.1080/15384101.2015.1041691
32. Huang T, Wang L, Liu D, Li P, Xiong H, Zhuang L, et al. FGF7/FGFR2 signal promotes invasion and migration in human gastric cancer through upregulation of thrombospondin-1. Int J Oncol (2017) 50(5):1501–12. doi: 10.3892/ijo.2017.3927
33. Ndlovu R, Deng LC, Wu J, Li XK, Zhang JS. Fibroblast Growth Factor 10 in Pancreas Development and Pancreatic Cancer. Front Genet (2018) 9:482. doi: 10.3389/fgene.2018.00482
34. Jarosz M, Robbez-Masson L, Chioni AM, Cross B, Rosewell I, Grose R. Fibroblast growth factor 22 is not essential for skin development and repair but plays a role in tumorigenesis. PloS One (2012) 7(6):e39436. doi: 10.1371/journal.pone.0039436
35. Liu HY, Zhao H, Li WX. Integrated Analysis of Transcriptome and Prognosis Data Identifies FGF22 as a Prognostic Marker of Lung Adenocarcinoma. Technol Cancer Res Treat (2019) 18:1533033819827317. doi: 10.1177/1533033819827317
36. Zhang J, Zhou Y, Huang T, Wu F, Pan Y, Dong Y, et al. FGF18, a prominent player in FGF signaling, promotes gastric tumorigenesis through autocrine manner and is negatively regulated by miR-590-5p. Oncogene (2019) 38(1):33–46. doi: 10.1038/s41388-018-0430-x
37. Song N, Zhong J, Hu Q, Gu T, Yang B, Zhang J, et al. FGF18 Enhances Migration and the Epithelial-Mesenchymal Transition in Breast Cancer by Regulating Akt/GSK3β/B-Catenin Signaling. Cell Physiol Biochem (2018) 49(3):1019–32. doi: 10.1159/000493286
38. Wu J, Tao X, Zhang H, Yi XH, Yu YH. Estrogen-Induced Stromal FGF18 Promotes Proliferation and Invasion of Endometrial Carcinoma Cells Through ERK and Akt Signaling. Cancer Manag Res (2020) 12:6767–77. doi: 10.2147/CMAR.S254242
39. Zhou Y, Wu C, Lu G, Hu Z, Chen Q, Du X. FGF/FGFR signaling pathway involved resistance in various cancer types. J Cancer (2020) 11(8):2000–7. doi: 10.7150/jca.40531
40. Ellis LM, Hicklin DJ. Pathways mediating resistance to vascular endothelial growth factor-targeted therapy. Clin Cancer Res (2008) 14(20):6371–5. doi: 10.1158/1078-0432.CCR-07-5287
41. Zhao Y, Adjei AA. Targeting Angiogenesis in Cancer Therapy: Moving Beyond Vascular Endothelial Growth Factor. Oncologist (2015) 20(6):660–73. doi: 10.1634/theoncologist.2014-0465
42. Ichikawa K, Watanabe Miyano S, Minoshima Y, Matsui J, Funahashi Y. Activated FGF2 signaling pathway in tumor vasculature is essential for acquired resistance to anti-VEGF therapy. Sci Rep (2020) 10(1):2939. doi: 10.1038/s41598-020-59853-z
43. Boichuk S, Galembikova A, Dunaev P, Micheeva E, Valeeva E, Novikova M, et al. Targeting of FGF-Signaling Re-Sensitizes Gastrointestinal Stromal Tumors (GIST) to Imatinib In Vitro and In Vivo. Molecules (2018) 23(10):2643. doi: 10.3390/molecules23102643
44. Cives M, Pelle’ E, Quaresmini D, Rizzo FM, Tucci M, Silvestris F. The Tumor Microenvironment in Neuroendocrine Tumors: Biology and Therapeutic Implications. Neuroedocrinology (2019) 109:83–99. doi: 10.1159/000497355
45. Bordi C, Falchetti A, Buffa R, Azzoni C, D’Adda T, Caruana P, et al. Production of basic fibroblast growth factor by gastric carcinoid tumors and their putative cells of origin. Hum Pathol (1994) 25:175–80. doi: 10.1016/0046-8177(94)90275-5
46. Chaudhry A, Funa K, Oberg K. Expression of growth factor peptides and their receptors in neuroendocrine tumors of the digestive system. Acta Oncol (1993) 32:107–14. doi: 10.3109/02841869309083898
47. La Rosa S, Uccella S, Erba S, Capella C, Sessa F. Immunohistochemical Detection of Fibroblast Growth Factor Receptors in Normal Endocrine Cells and Related Tumors of the Digestive System. Appl Immunohistochem Mol Morphol (2001) 9:319–28. doi: 10.1097/00129039-200112000-00006
48. Bhattacharyya S, Toumpanakis C, Chilkunda D, Caplin ME, Davar J. Risk factors for the development and progression of carcinoid heart disease. Am J Cardiol (2011) 107(8):1221–6. doi: 10.1016/j.amjcard.2010.12.025
49. Koumarianou A, Alexandraki KI, Wallin G, Kaltsas G. Pathogenesis and Clinical Management of Mesenteric Fibrosis in Small Intestinal Neuroendocine Neoplasms: A Systematic Review. J Clin Med (2020) 9(6):1777. doi: 10.3390/jcm9061777
50. Daskalakis K, Karakatsanis A, Stålberg P, Norlén O, Hellman P. Clinical signs of fibrosis in small intestinal neuroendocrine tumours. Br J Surg (2017) 104:69–75. doi: 10.1002/bjs.10333
51. Ratnavel RC, Burrows NP, Pye RJ. Scleroderma and the carcinoid syndrome. Clin Exp Dermatol (1994) 19:83–5. doi: 10.1111/j.1365-2230.1994.tb01126.x
52. Moss SF, Lehner PJ, Gilbey SG, Kennedy A, Hughes JM, Bloom SR, et al. Pleural involvement in the carcinoid syndrome. Q J Med (1993) 86:49–53. doi: 10.1093/oxfordjournals.qjmed.a068737
53. Seo JW, Im JG, Kim YW, Kim JH, Sheppard MN. Synchronous double primary lung cancers of squamous and neuroendocrine type associated with cryptogenic fibrosing alveolitis. Thorax (1991) 46:857–8. doi: 10.1136/thx.46.11.857
54. Hallen A. FIBROSIS IN THE CARCINOID SYNDROME. Lancet (1964) 1:746–7. doi: 10.1016/s0140-6736(64)92853-3
55. Laskaratos F-M, Rombouts K, Caplin M, Toumpanakis C, Thirlwell C, Mandair D. Neuroendocrine tumors and fibrosis: An unsolved mystery? Cancer (2017) 123:4770–90. doi: 10.1002/cncr.31079
56. Zuetenhorst JM, Bonfrer JMGM, Korse CM, Bakker R, van Tinteren H, Taal BG. Carcinoid heart disease: the role of urinary 5-hydroxyindoleacetic acid excretion and plasma levels of atrial natriuretic peptide, transforming growth factor-beta and fibroblast growth factor. Cancer (2003) 97:1609–15. doi: 10.1002/cncr.11226
57. La Rosa S, Chiaravalli AM, Capella C, Uccella S, Sessa F. Immunohistochemical localization of acidic fibroblast growth factor in normal human enterochromaffin cells and related gastrointestinal tumours. Virchows Archiv (1997) 430:117–24. doi: 10.1007/BF01008032
58. Svejda B, Kidd M, Giovinazzo F, Eltawil K, Gustafsson BI, Pfragner R, et al. The 5-HT(2B) receptor plays a key regulatory role in both neuroendocrine tumor cell proliferation and the modulation of the fibroblast component of the neoplastic microenvironment. Cancer (2010) 116:2902–12. doi: 10.1002/cncr.25049
59. Koumarianou A, Alexandraki KI, Wallin G, Kaltsas G, Daskalakis K. Pathogenesis and Clinical Management of Mesenteric Fibrosis in Small Intestinal Neuroendocine Neoplasms: A Systematic Review. J Clin Med (2020) 9. doi: 10.3390/jcm9061777
60. Wulbrand U, Wied M, Zöfel P, Göke B, Arnold R, Fehmann H-C. Growth factor receptor expression in human gastroenteropancreatic neuroendocrine tumours. Eur J Clin Invest (1998) 28:1038–49. doi: 10.1046/j.1365-2362.1998.00397.x
61. Serra S, Zheng L, Hassan M, Phan AT, Woodhouse LJ, Yao JC, et al. The FGFR4-G388R single-nucleotide polymorphism alters pancreatic neuroendocrine tumor progression and response to mTOR inhibition therapy. Cancer Res (2012) 72:5683–91. doi: 10.1158/0008-5472.CAN-12-2102
62. Cros J, Moati E, Raffenne J, Hentic O, Svrcek M, de Mestier L, et al. Gly388Arg FGFR4 Polymorphism Is Not Predictive of Everolimus Efficacy in Well-Differentiated Digestive Neuroendocrine Tumors. Neuroendocrinology (2016) 103:495–9. doi: 10.1159/000440724
63. Zatelli MC, Fanciulli G, Malandrino P, Ramundo V, Faggiano A, Colao A, et al. Predictive factors of response to mTOR inhibitors in neuroendocrine tumours. Endocr Relat Cancer (2016) 23:R173–83. doi: 10.1530/ERC-15-0413
64. Compagni A, Wilgenbus P, Impagnatiello MA, Cotten M, Christofori G. Fibroblast growth factors are required for efficient tumor angiogenesis. Cancer Res (2000) 60:7163–9.
65. Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell (2005) 8:299–309. doi: 10.1016/j.ccr.2005.09.005
66. Allen MD, Neumann S, Gershengorn MC. Occupancy of both sites on the thyrotropin (TSH) receptor dimer is necessary for phosphoinositide signaling. FASEB J (2011) 25:3687–94. doi: 10.1096/fj.11-188961
67. Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer (2010) 10(2):116–29. doi: 10.1038/nrc2780
68. Arai H, Battaglin F, Wang J, Lo JH, Soni S, Zhang W, et al. Molecular insight of regorafenib treatment for colorectal cancer. Cancer Treat Rev (2019) 81:101912. doi: 10.1016/j.ctrv.2019.101912
69. Kopetz S, Hoff PM, Morris JS, Wolff RA, Eng C, Glover KY, et al. Phase II trial of infusional fluorouracil, irinotecan, and bevacizumab for metastatic colorectal cancer: efficacy and circulating angiogenic biomarkers associated with therapeutic resistance. J Clin Oncol (2010) 28(3):453–9. doi: 10.1200/JCO.2009.24.8252
70. Xu J, Shen L, Zhou Z, Li J, Bai C, Chi Y, et al. Surufatinib in advanced extrapancreatic neuroendocrine tumours (SANET-ep): a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol (2020) 21(11):1500–12. doi: 10.1016/S1470-2045(20)30496-4
71. Xu J, Shen L, Bai C, Wang W, Li J, Yu X, et al. Surufatinib in advanced pancreatic neuroendocrine tumours (SANET-p): a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol (2020) 21(11):1489–99. doi: 10.1016/S1470-2045(20)30493-9
72. Dasari A, Li D, Sung M, Tucci C, Kauh J, Kania M, et al. Efficacy and safety of surufatinib in United States (US) patients (pts) with neuroendocrine tumors (NETs). J Clin Oncol (2020) 38(15):4610. doi: 10.1200/JCO.2020.38.15_suppl.4610
73. Chen J, Ji Q, Bai C, Lin Y, Zheng X, Zhang Y, et al. Surufatinib in Chinese Patients with Locally Advanced or Metastatic Differentiated Thyroid Cancer and Medullary Thyroid Cancer: A Multicenter, Open-Label, Phase II Trial. Thyroid (2020) 30(9):1245–53. doi: 10.1089/thy.2019.0453
74. Capdevila J, Fazio N, Lopez Lopez C, Teule A, Valle J, Tafuto S, et al. Final results of the TALENT trial (GETNE1509): a prospective multicohort phase II study of lenvatinib in patients (pts) with G1/G2 advanced pancreatic (panNETs) and gastrointestinal (giNETs) neuroendocrine tumors (NETs). J Clin Oncol (2019) 37(15):4106. doi: 10.1200/JCO.2019.37.15_suppl.4106
75. Schlumberger M, Jarzab B, Cabanillas ME, Robinson B, Pacini F, Ball DW, et al. A Phase II Trial of the Multitargeted Tyrosine Kinase Inhibitor Lenvatinib (E7080) in Advanced Medullary Thyroid Cancer. Clin Cancer Res (2016) 22(1):44–53. doi: 10.1158/1078-0432.CCR-15-1127
76. Takahashi S, Kiyota N, Yamazaki T, Chayahara N, Nakano K, Inagaki L, et al. A Phase II study of the safety and efficacy of lenvatinib in patients with advanced thyroid cancer. Future Oncol (2019) 15(7):717–26. doi: 10.2217/fon-2018-0557
77. Takahashi S, Tahara M, Ito K, Tori M, Kiyota N, Yoshida K, et al. Safety and Effectiveness of Lenvatinib in 594 Patients with Unresectable Thyroid Cancer in an All-Case Post-Marketing Observational Study in Japan. Adv Ther (2020) 37(9):3850–62. doi: 10.1007/s12325-020-01433-8
78. Iyer R, Konda B, Hall Owen D, Attwood K, Sarker S, Suffren S, et al. Multicenter phase 2 study of nintedanib in patients (pts) with advanced progressing carcinoid tumors. J Clin Oncol (2018) 36(15):4105. doi: 10.1200/JCO.2018.36.15_suppl.4105
79. Sun Y, Du F, Gao M, Ji Q, Li Z, Zhang Y, et al. Anlotinib for the Treatment of Patients with Locally Advanced or Metastatic Medullary Thyroid Cancer. Thyroid (2018) 28(11):1455–61. doi: 10.1089/thy.2018.0022
80. Li D, Tang P, Chen X, Ge M, Zhang Y, Guo Z, et al. Anlotinib treatment in locally advanced or metastatic medullary thyroid carcinoma: A multicenter, randomized, double-blind, placebo-controlled phase IIB trial. J Clin Oncol (2019) 37(15):6019. doi: 10.1200/JCO.2019.37.15_suppl.6019
81. Xu JM, Wang Y, Chen YL, Jia R, Li J, Gong JF, et al. Sulfatinib, a novel kinase inhibitor, in patients with advanced solid tumors: results from a phase I study. Oncotarget (2017) 8(26):42076–86. doi: 10.18632/oncotarget.14942
82. Xu J, Li J, Bai C, Xu N, Zhou Z, Li Z, et al. Surufatinib in Advanced Well-Differentiated Neuroendocrine Tumors: A Multicenter, Single-Arm, Open-Label, Phase Ib/II Trial. Clin Cancer Res (2019) 25(12):3486–94. doi: 10.1158/1078-0432.CCR-18-2994
83. Newbold K, Schoeffski P, Hasan B, Locati L, Godbert Y, De la Fouchardiere C, et al. Nintedanib (BIBF1120) after first line therapy in progressive medullary thyroid cancer: A multicenter EORTC prospective randomized double-blind phase II study (NCT01788982). Ann Oncol (2020) 31(4):S1087–S8. doi: 10.1016/j.annonc.2020.08.1407
84. Sun Y, Niu W, Du F, Du C, Li S, Wang J, et al. Safety, pharmacokinetics, and antitumor properties of anlotinib, an oral multi-target tyrosine kinase inhibitor, in patients with advanced refractory solid tumors. J Hematol Oncol (2016) 9(1):105. doi: 10.1186/s13045-016-0332-8
85. Scheller T, Hellerbrand C, Moser C, Schmidt K, Kroemer A, Brunner SM, et al. mTOR inhibition improves fibroblast growth factor receptor targeting in hepatocellular carcinoma. Br J Cancer (2015) 112(5):841–50. doi: 10.1038/bjc.2014.638
Keywords: neuroendocrine neoplasms, FGFR (fibroblast growth factor receptor), FGF (fibroblast growth factor), VEGF - vascular endothelial growth factor, VEGFR - vascular endothelial growth factor receptor
Citation: Vitale G, Cozzolino A, Malandrino P, Minotta R, Puliani G, Saronni D, Faggiano A and Colao A (2021) Role of FGF System in Neuroendocrine Neoplasms: Potential Therapeutic Applications. Front. Endocrinol. 12:665631. doi: 10.3389/fendo.2021.665631
Received: 08 February 2021; Accepted: 22 March 2021;
Published: 14 April 2021.
Edited by:
Michele Caraglia, University of Campania Luigi Vanvitelli, ItalyReviewed by:
Davide Gentilini, University of Pavia, ItalyCopyright © 2021 Vitale, Cozzolino, Malandrino, Minotta, Puliani, Saronni, Faggiano and Colao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giovanni Vitale, Z2lvdmFubmkudml0YWxlQHVuaW1pLml0; Zy52aXRhbGVAYXV4b2xvZ2ljby5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.