
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 04 November 2021
Sec. Reproduction
Volume 12 - 2021 | https://doi.org/10.3389/fendo.2021.664645
This article is part of the Research Topic Female infertility: Genetics of Reproductive Ageing, Menopause and Primary Ovarian Insufficiency View all 14 articles
 Raffaella Rossetti1*†
Raffaella Rossetti1*† Silvia Moleri1†
Silvia Moleri1† Fabiana Guizzardi1,2
Fabiana Guizzardi1,2 Davide Gentilini3
Davide Gentilini3 Laura Libera1
Laura Libera1 Anna Marozzi4
Anna Marozzi4 Costanzo Moretti5
Costanzo Moretti5 Francesco Brancati6,7
Francesco Brancati6,7 Marco Bonomi1,4
Marco Bonomi1,4 Luca Persani1,4*
Luca Persani1,4*Primary ovarian insufficiency (POI) is one of the major causes of female infertility associated with the premature loss of ovarian function in about 3.7% of women before the age of 40. This disorder is highly heterogeneous and can manifest with a wide range of clinical phenotypes, ranging from ovarian dysgenesis and primary amenorrhea to post-pubertal secondary amenorrhea, with elevated serum gonadotropins and hypoestrogenism. The ovarian defect still remains idiopathic in some cases; however, a strong genetic component has been demonstrated by the next-generation sequencing (NGS) approach of familiar and sporadic POI cases. As recent evidence suggested an oligogenic architecture for POI, we developed a target NGS panel with 295 genes including known candidates and novel genetic determinants potentially involved in POI pathogenesis. Sixty-four patients with early onset POI (range: 10–25 years) of our cohort have been screened with 90% of target coverage at 50×. Here, we report 48 analyzed patients with at least one genetic variant (75%) in the selected candidate genes. In particular, we found the following: 11/64 patients (17%) with two variants, 9/64 (14%) with three variants, 9/64 (14%) with four variants, 3/64 (5%) with five variants, and 2/64 (3%) with six variants. The most severe phenotypes were associated with either the major number of variations or a worse prediction in pathogenicity of variants. Bioinformatic gene ontology analysis identified the following major pathways likely affected by gene variants: 1) cell cycle, meiosis, and DNA repair; 2) extracellular matrix remodeling; 3) reproduction; 4) cell metabolism; 5) cell proliferation; 6) calcium homeostasis; 7) NOTCH signaling; 8) signal transduction; 9) WNT signaling; 10) cell death; and 11) ubiquitin modifications. Consistently, the identified pathways have been described in other studies dissecting the mechanisms of folliculogenesis in animal models of altered fertility. In conclusion, our results contribute to define POI as an oligogenic disease and suggest novel candidates to be investigated in patients with POI.
Female factors account for one-third of all causes of infertility. Besides tubal disease and endometrial pathology, the dysregulation of any essential step involved in the ovulation of a competent oocyte may cause primary ovarian insufficiency (POI), a clinical syndrome defined by the premature loss of ovarian function. A recent meta-analysis of 31 epidemiological studies on the prevalence of POI in different countries between 1987 and 2018 reports an overall occurrence up to 3.7% in women younger than 40 years (1). At present, this disease is currently diagnosed when fertility is irreversibly affected. POI can manifest with a wide variety of clinical phenotypes, ranging from ovarian dysgenesis (OD) and primary amenorrhea (PA) to post-pubertal secondary amenorrhea (SA) for more than 4 months with raised gonadotrophins and low estradiol. The consequent long-standing estrogen deficiency exposes these women to an increased risk of complications such as cardiovascular diseases, reduced bone mineral density, and cognitive impairment (2). This disorder is highly heterogeneous in its etiology and several causes have been reported, mainly genetic, associated with chromosomal abnormalities (especially including X chromosome, such as in Turner syndrome), but also autoimmune, infectious, or iatrogenic. However, most causes of POI are still unknown, and the identification of novel causative genes is challenging. More recently, the advent of next-generation sequencing (NGS) technique and, especially, the whole exome screening (WES) of large POI families expanded the list of candidate genes to be screened in patients and consequently empowered the possibilities of a genetic diagnosis in idiopathic cases (3). Some WES studies demonstrated that pathogenic variants in meiotic chromosome pairing and synaptonemal complex (4–6) or alterations of other proteins of DNA recombination and repair (7, 8) could be responsible for POI onset by usually impairing meiotic progression and triggering oocyte death, as further evidenced by murine models (9). Other studies identified variants in the folliculogenesis players of all stages of ovarian follicle maturation, which involves the precise interaction of hundreds of genes: from the primordial follicle stock establishment of ovarian reserve (10) to the primordial to primary follicle activation (11, 12), throughout the follicular development in the gonadotropin-independent (13, 14) and gonadotropin-dependent stages (15, 16). Furthermore, alterations in genes contributing to extracellular matrix (ECM) remodeling by proteolytic activity on specific substrates within the ovarian context have been linked to the ECM turnover of abnormal somatic cells, thus leading to defects in follicular development (17). Since biological processes related to metabolism and immune system activation resulted to be enhanced in gene expression dynamics along with ovary development, including pathways associated with cell cycle, proliferation, apoptosis, ovulation, angiogenesis, and steroidogenesis (18), the disturbance of any of these pathways has been associated with reproductive diseases like POI (19–22). Moreover, the remarkable point that emerged from recent NGS studies is the occurrence of oligogenic defects (6, 23). From this perspective, various interacting genes might affect several mechanisms and pathways, and the synergistic and/or cumulative effect of several variants may contribute to POI phenotype. The NGS results in 64 patients with PA or early onset SA based on our panel of 295 POI candidate genes presented here aims to contribute to expand the list of potentially causative candidate genes and to support a genetically heterogeneous architecture of POI, resulting from defects in multiple complementary pathways.
Genomic DNA (gDNA) was extracted from peripheral blood of enrolled patients. The Ampliseq Custom DNA panel (Illumina) was designed ad hoc to include the coding exons and flanking splice sites of 295 genes. The NGS panel, called OVO-Array (Table S1), has been obtained by joining data from: a) literature search of genes known to be previously associated with female infertility, with a role in ovary and in menopause onset (n = 159) (23); b) transcriptomic analysis of human granulosa cells treated with the oocyte-derived growth factor BMP15 (n of genes: 19; focus of another study with manuscript under review); and c) WES on 10 Italian women selected with the most severe phenotypes of PA and OD, six of them with familial inheritance (n = 117). The latter were identified among a total of 18,570 rare variants (missense, 86%; nonsense, 3.5%; indels, 5.6%). Briefly, pathway analysis (by Reactome v.74) of 1,916 genetic identifiers, found mutated at least once in WES, revealed an enrichment in the following biological processes with a key role in ovarian functionality (e.g., chromatin organization, cell cycle and meiosis, extracellular matrix organization, and cell–cell communication). Within these pathways, we further selected 117 genes potentially correlated with POI onset. The total coverage of the target genes by the designed amplicons was 100%. Library was prepared using enzymatic DNA fragmentation, with 50 ng of total gDNA and quantified with Quant-iT PicoGreen (Thermo Fisher Scientific). Nextera Rapid Capture Enrichment protocol (Illumina) was followed to tagment gDNA, amplify tagmented gDNA, hybridize probes, capture hybridized probes and for library capture and amplification. The library was then loaded onto the reagent cartridge (Illumina) and sequencing was performed on a NextSeq 500 (Illumina). Reads were then examined to identify single nucleotide alterations or small insertions/deletions, and a first in silico analysis (including base calling and demultiplexing) has been performed using MiSeq provided software (Real Time Analysis RTA v.1.18.54 and Casava v.1.8.2, Illumina). FastQ files for each sample, containing mate paired-end reads after demultiplexing and adapter removal, have been used as input for MiSeq pipeline. Briefly, FastQ files have been processed with MiSeq Reporter v2.0.26 using the Custom Amplicon workflow. This analytical method required FastQ files, a “Manifest file” containing information about the sequences of primer pairs, the expected sequence of the amplicons, and the coordinates of the reference genome (Homo sapiens, hg19, build 37.2) as input. Each read pair has been aligned using the MEM algorithm of the BWA software. Local Indel realignment and base recalibration step were performed using the software GATK. The realigned and recalibrated BAM file was used as input to GATK Unified Genotyper thus generating a VCFv4.2 file for each sample. Quality control of sequencing data was performed directly on FastQ files using the FastQC software. Reads were also filtered based on quality mapping and removed if their quality mapping was <20. Genetic variants showing a PHredScore lower than 20 were also filtered out.
We collected a cohort of patients to be analyzed through the OVO-Array panel, following IRCCS Istituto Auxologico Italiano Ethics Committee approval (BIOEFFECT, code 05M101_2014), composed of a total of 64 women with either primary (PA, n = 21) or early onset secondary amenorrhea (SA, n = 43), with onset before 25 years, all characterized by FSH values >40 IU/L and low estradiol. We excluded karyotype abnormalities, FMR1 premutations, and ovarian autoimmunity in all of them. Moreover, we selected additional 43 patients (PA, n = 18 and SA, n = 25) with the same inclusion criteria and early onset POI before 25 years of age. These patients have been analyzed between the years 2013 and 2020 for variants on a selected subset of only nine causative genes for POI, mainly involved in folliculogenesis and meiosis (BMP15, FIGLA, FOXL2, FSHR, GDF9, NOBOX, NR5A1, SYCE1, STAG3) for diagnostic purposes at IRCCS Istituto Auxologico Italiano. The results obtained with the different screening approaches (OVO-Array vs. diagnostic routine) have been then compared. Informed consent was obtained from all patients prior to blood sample collection and molecular studies.
The genetic variants resulting from the OVO-Array experiment and those derived from the diagnostic routine were annotated using Annovar software (24) and then classified as rare if resulting unknown or with a minor frequency allele (MAF) <0.01 in 1000 genomes, dbSNP, or EXAC databases. By checking for pathogenicity prediction the VarSome database until July 2021 (25), only those rare variants classified as likely pathogenic (LP), pathogenic (P), or variant of unknown significance (VUS), according to the American College of Medical Genetics (ACMG) classification guidelines (26), were considered for further analysis and confirmed by using Sanger sequencing. Gene ontology analysis of all the significatively altered genes was performed against DAVID Bioinformatics v6.8 (27) and Reactome Pathway Browser version 74 (28) on October 26, 2020. Both databases cross-reference other resources (e.g., NCBI, Ensembl, UniProt, KEGG, ChEBI, PubMed, and GO).
We recruited 64 patients presenting PA or SA with early onset (from 10 to 25 years) and performed NGS analysis on 295 selected genes, including known candidates and novel potentially causative genes. The mean coverage depth of the target regions was greater than 90% at 50× for all patients. Variants with MAF >0.01 were filtered out. We considered the identified variants relevant only if they were rare (MAF <0.01) or never described; predicted as P, LP, or VUS; and already known to be associated with POI. We also considered nine variants predicted as likely benign, found in recurring genes with a higher frequency in our patients than in the general female population or previously associated to POI and functionally characterized.
We report a total of 114 rare variants in 78 different genes (Figure 1A, Table S2) in 64 patients. In 75% of patients analyzed by means of OVO-Array, we could find at least one alteration (48/64) possibly related to POI. Noteworthy, 2 or more variants have been identified in 34 of them; in particular, we found 11 patients (11/64, 17%) with two variants, 9 patients (9/64, 14%) with three variants, 9 patients (9/64, 14%) with four variants, 3 patients (3/64, 5%) with five variants, and 2 patients (2/64, 3%) with six variants (Figure 2A, Table S3). No significative alterations in the selected 295 genes were found in only 16 patients of the cohort.
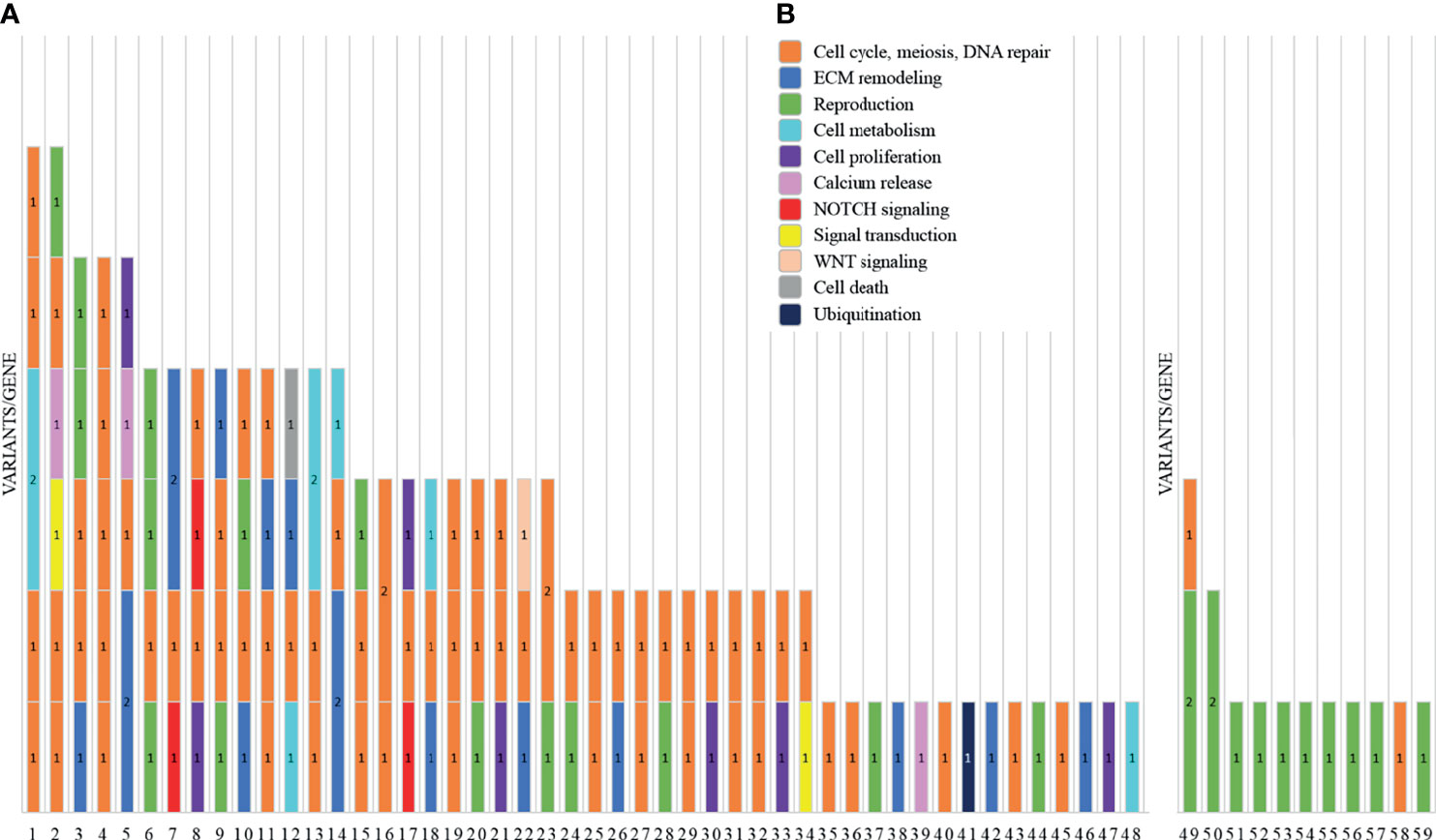
Figure 1 Graph representations of the number of variants identified by NGS analysis in two subgroups of primary ovarian insufficiency (POI) patients. For each patient, indicated are the ID number (x-axis) and the number of variants/genes found altered (1 = 1 variant/gene; 2 = 2 variants/gene). Each color represents a different pathway: cell cycle, meiosis, and DNA repair (orange); ECM remodeling (blue); reproduction (green); cell metabolism (cyan); cell proliferation (purple); calcium release (pink); NOTCH signaling (red); signal transduction (yellow); WNT signaling (light orange); cell death (gray); and ubiquitination (dark blue). (A) The histogram reports the variants found by screening 295 candidate genes included in the OVO-Array panel. (B) NGS results of the analysis for diagnostic purposes of nine known POI genes.
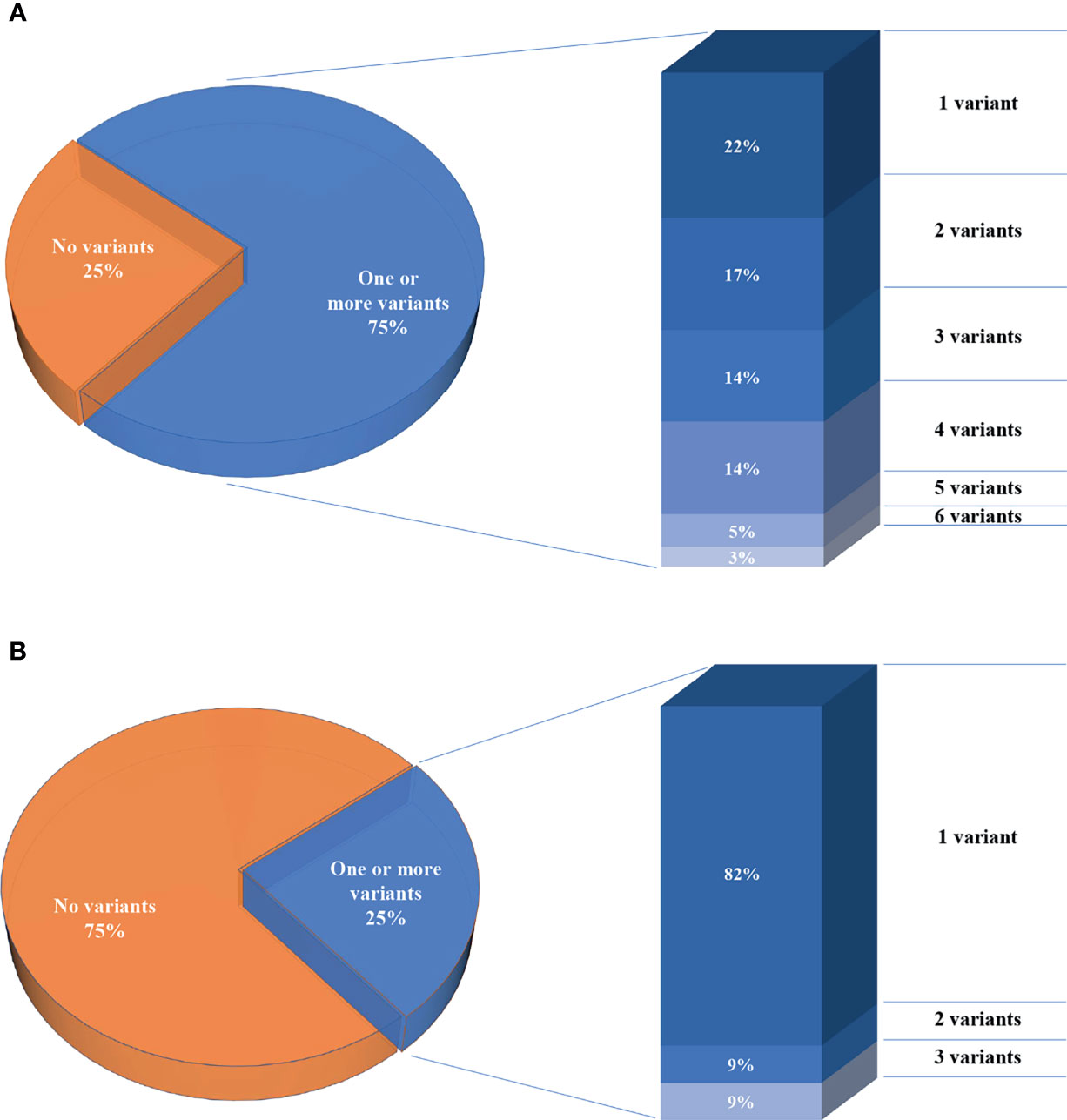
Figure 2 Percentages of the number of patients per number of variants. (A) Seventy-five percent of patients analyzed through the OVO-Array panel harbor one or more variants, left. Histogram representing the percentages of patients carrying variants, right. (B) Twenty-five percent of patients analyzed in only nine known POI genes.
Moreover, among the 43 patients screened for diagnostic purposes, we could identify at least one genetic variant in known POI genes in 11 of them, thus providing a genetic diagnosis in about 25% of POI patients through NGS. Only one patient harbored two different alterations and another one harbored three variants (Figures 1B, 2B, Tables S4, S5). All the identified variants were confirmed by Sanger sequencing.
We summarized the prioritized variants harbored by each patient analyzed by OVO-Array NGS panel in Supplementary Tables S2 and S3. The phenotype of each patient is also reported. Nomenclature validation was performed using Mutalyzer 2.0.33 (https://www.mutalyzer.nl/) according to HGVS nomenclature version 2.0.
Tables 1–6 display all the genetic variations identified by OVO-Array by the number of variants per patient. Briefly, each table prioritizes patients depending on their phenotype severity and shows a detailed characterization (including origin, karyotype, age of menarche and POI onset, and familiarity). The frequency of each variant is then compared with those of the female general population available in the gnomAD ver.2.1.1 public dataset. The number of variants per patient is thus correlated with the VarSome predicted pathogenicity and phenotype of the patients. For each variation, VarSome criteria and its relative updated link are shown.
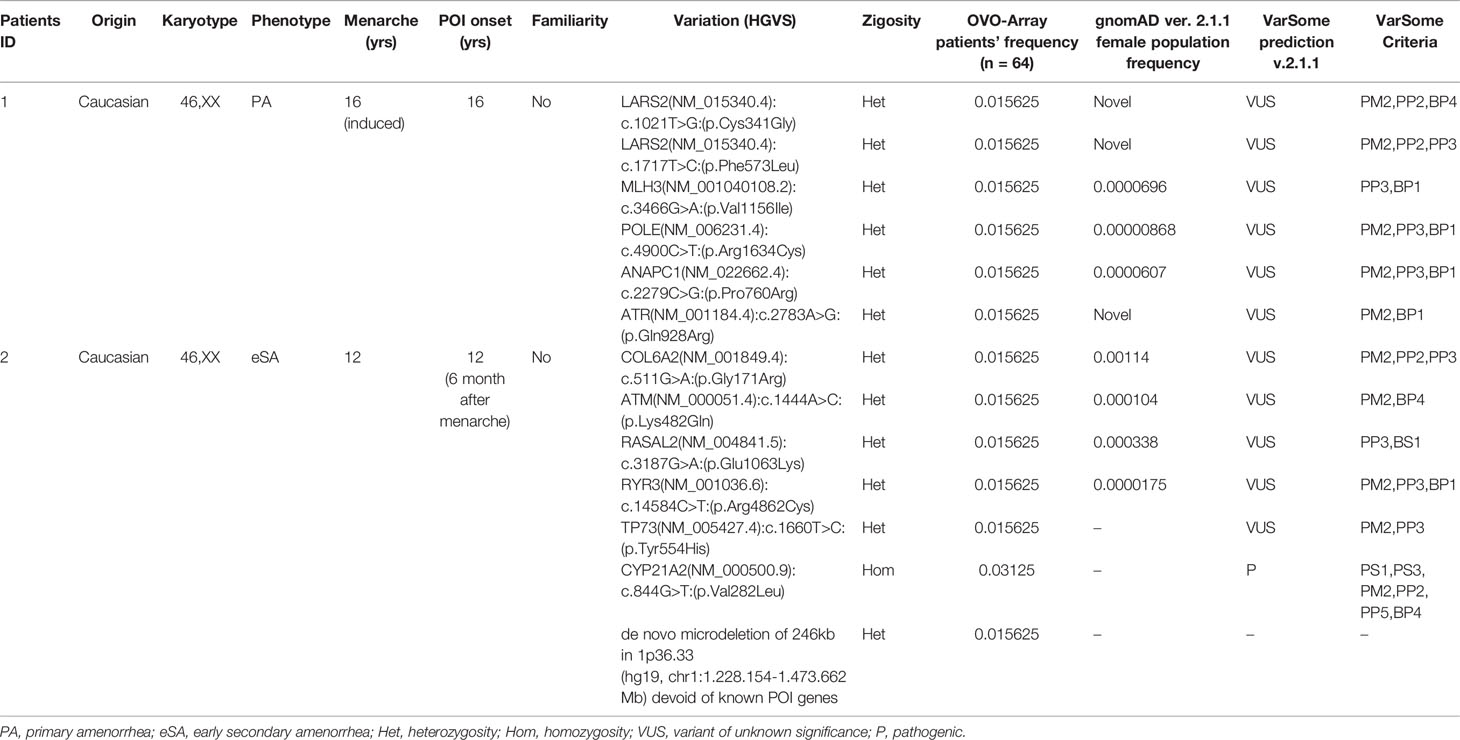
Table 1 Correlation among severity, phenotype and pathogenicity predictions in patients with 6 variants in potentially POI genes.
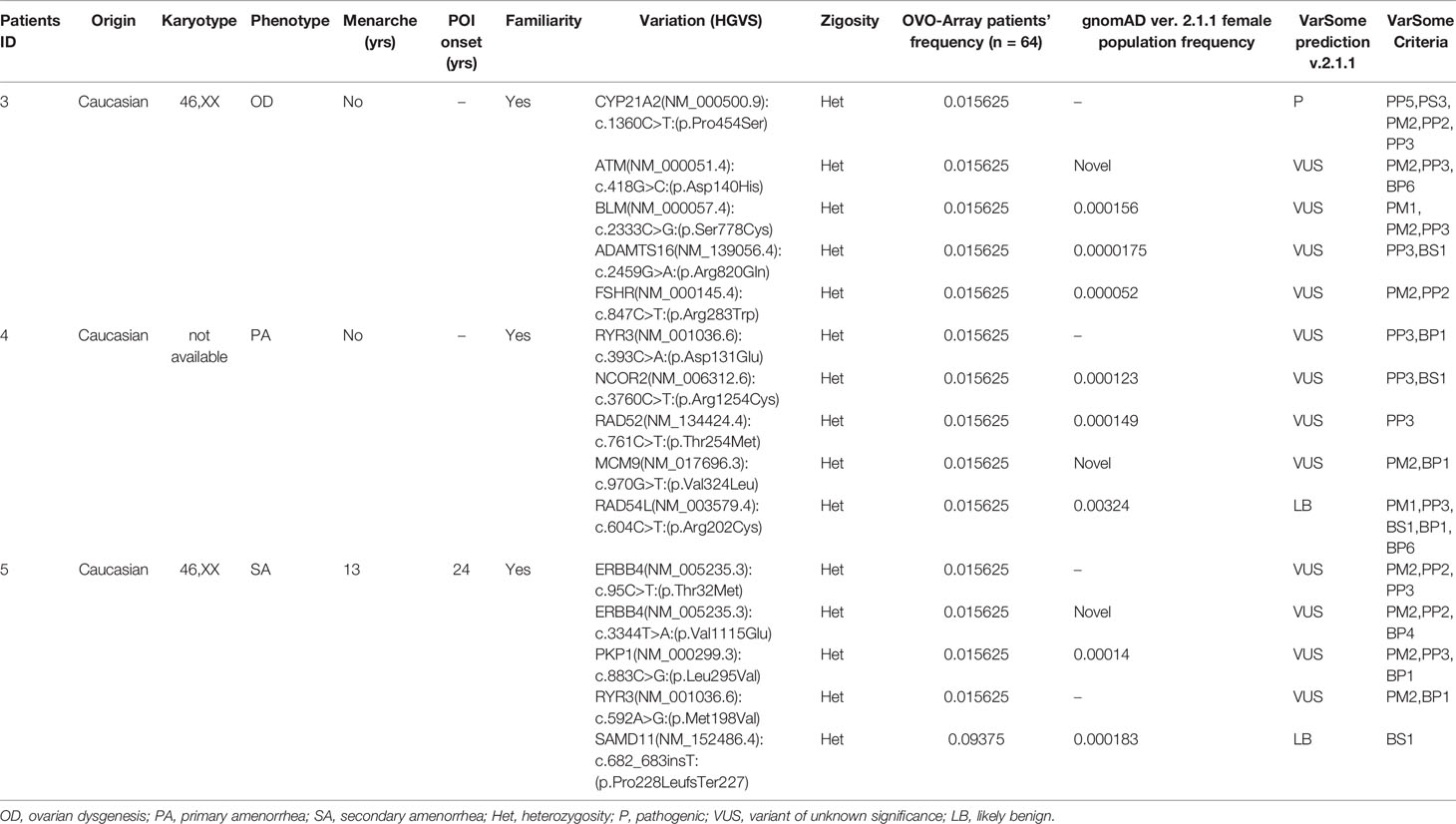
Table 2 Correlation among severity, phenotype and pathogenicity predictions in patients with 5 variants in potentially POI genes.
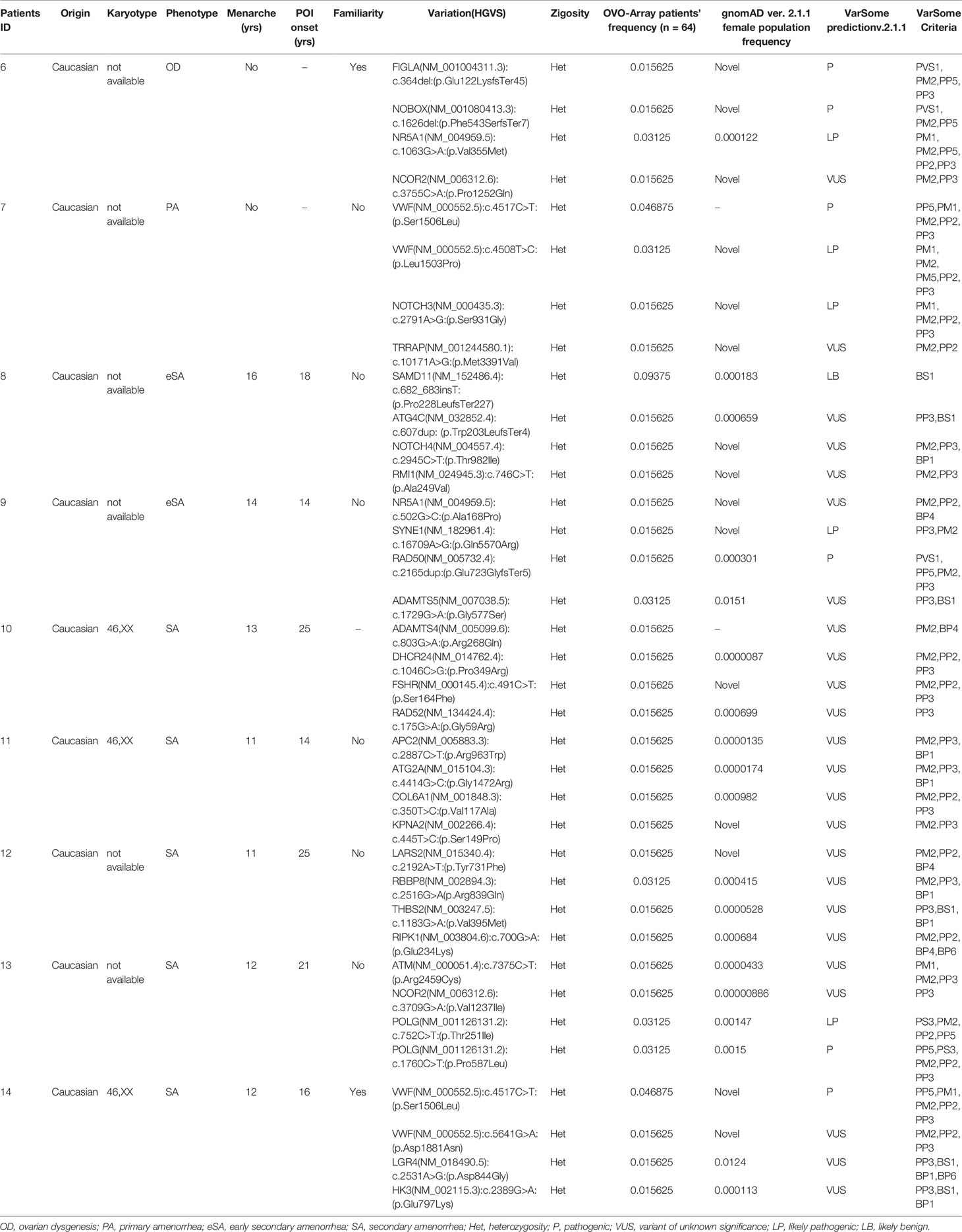
Table 3 Correlation among severity, phenotype and pathogenicity predictions in patients with 4 variants in potentially POI genes.
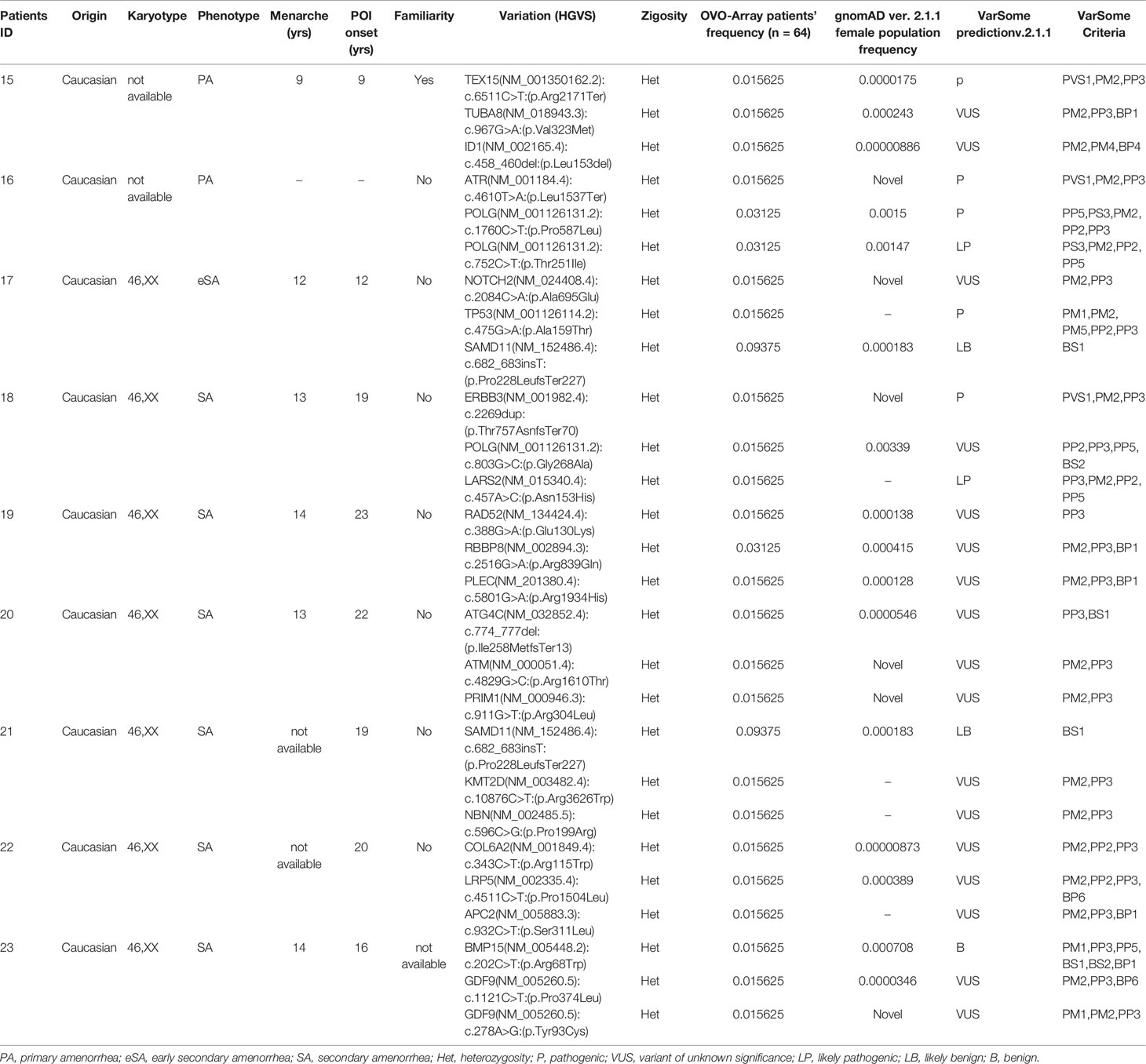
Table 4 Correlation among severity, phenotype and pathogenicity predictions in patients with 3 variants in potentially POI genes.
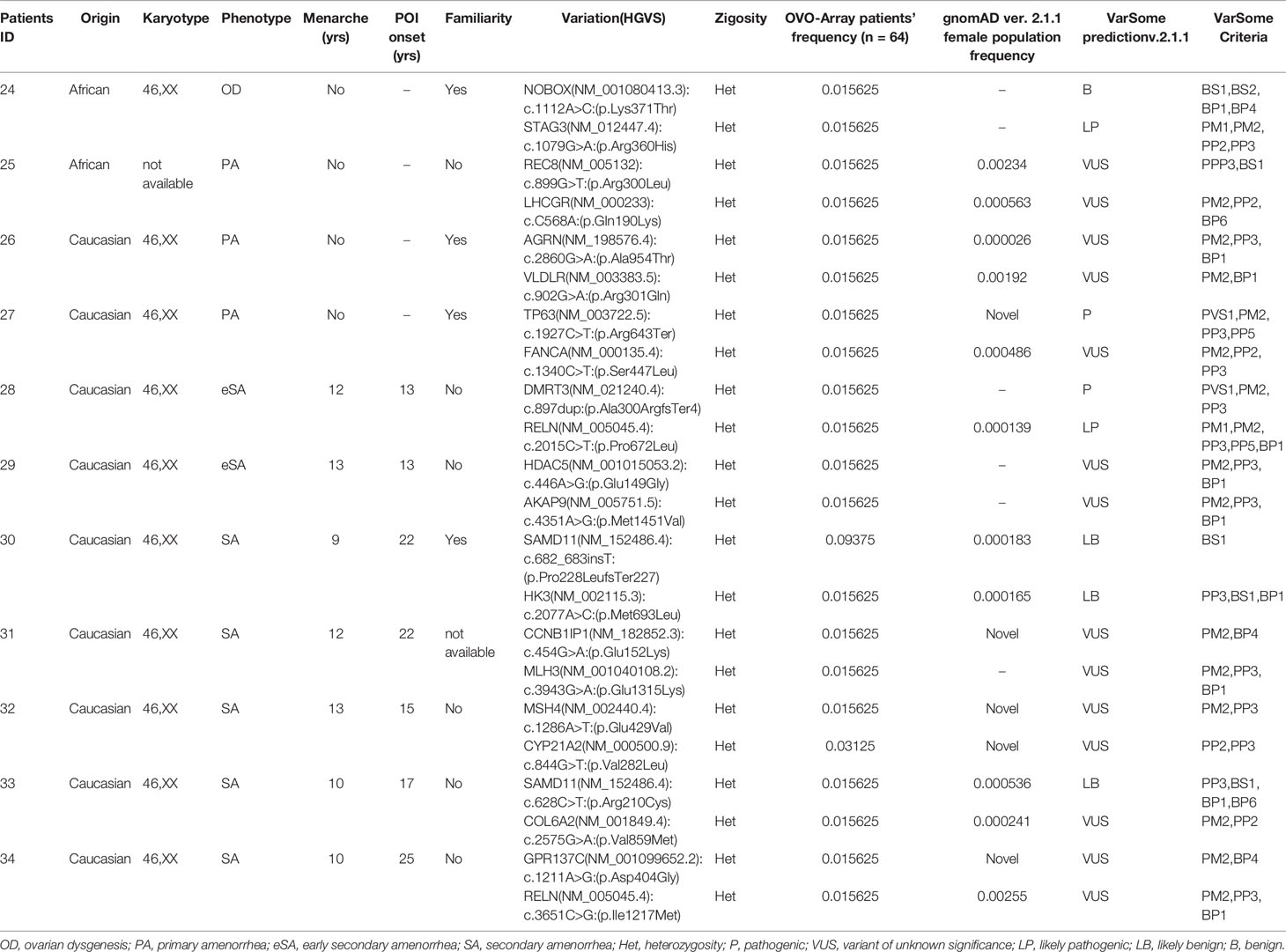
Table 5 Correlation among severity, phenotype and pathogenicity predictions in patients with 2 variants in potentially POI genes.
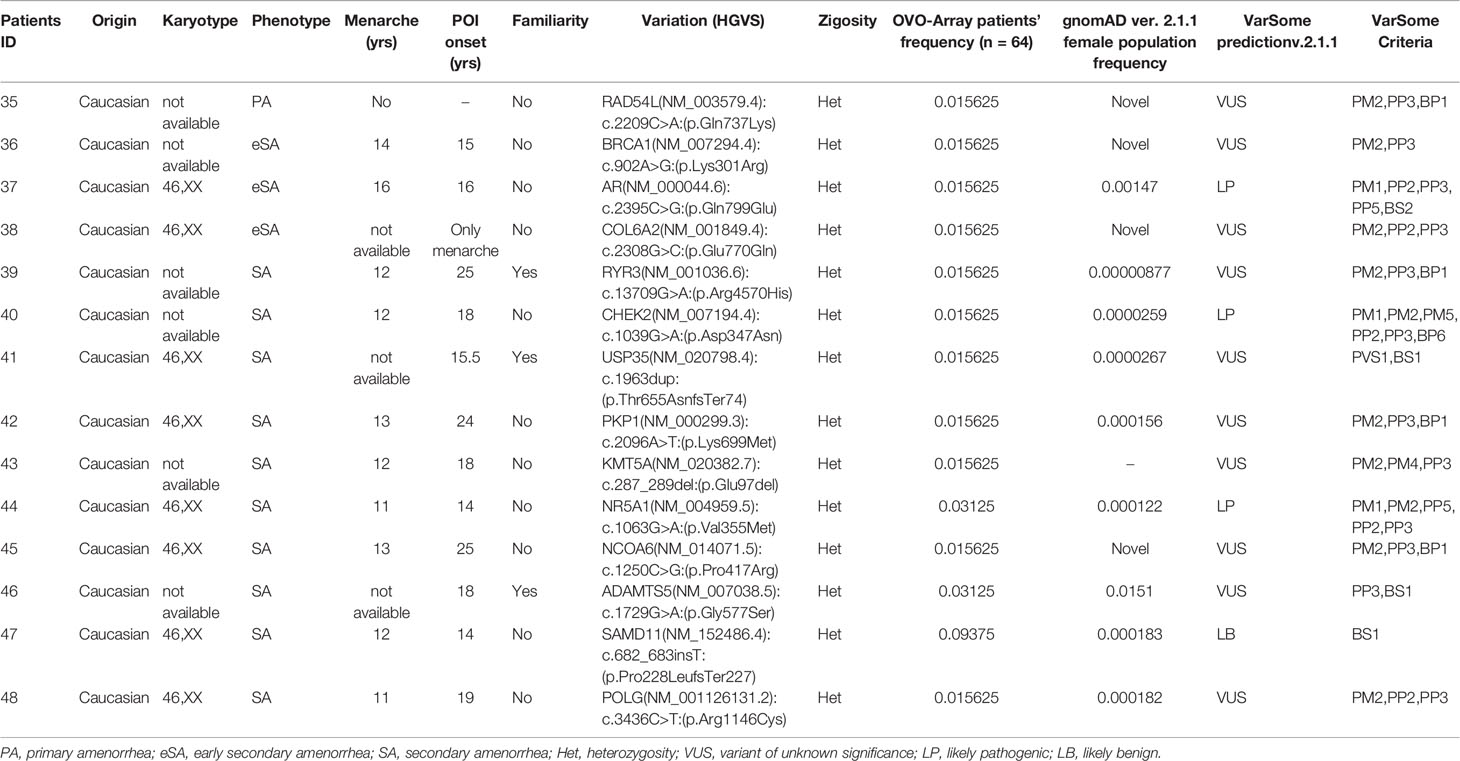
Table 6 Correlation among severity, phenotype, and pathogenicity predictions in patients with only 1 variant in potentially POI genes.
Taken together, our results uncovered 12 already known variants associated to POI: p.Arg300Leu in REC8; p.Arg68Trp in BMP15; p.Glu122Lysfs*45 in FIGLA; p.Pro103Ser, p.Thr121Ile, and p.Pro374Leu in GDF9; p.Phe543Serfs*7, p.Gly111Arg, and p.Lys371Thr in NOBOX; p.Val355Met in NR5A1; p.Arg643* in TP63; and p.Asn153His in LARS2 (green colored genetic variants in Tables S2, S4). Moreover, we could identify 41 novel rare variants in genes already associated to POI etiology (red colored in Tables S2, S4) and 74 novel rare variants in additional genes participating in key steps of ovarian follicle development, which may be considered putative candidates involved in POI pathogenesis (Table S2). Interestingly, 74 variants have been identified in 27 recurring genes among our two POI populations, and some of these genes/variants have already been associated to POI: 9 genetic alterations and 10 genes (green and red colored in Table 7, respectively). Furthermore, we found 12 alterations in recurrent genes which affected more than one patient and resulted at higher frequency in our POI populations, with respect to the general female population reported in the gnomAD (ver. 2.1.1) database (Table 7): c.1729G>A (p.Gly577Ser) in ADAMTS5, c.202C>T (p.Arg68Trp) in BMP15, c.844G>T (p.Val282Leu) in CYP21A2, c.926G>C (p.Gly309Ala) in FSHR, c.1063G>A (p.Val355Met) in NR5A1, c.752C>T (p.Thr251Ile) and c.1760C>T (p.Pro587Leu) in POLG, c.2516G>A (p.Arg839Gln) in RBBP8, c.682_683insT (p.Pro228Leufs*227) in SAMD11, and c.4508T>C (p.Leu1503Pro) and c.4517C>T (p.Ser1506Leu) in VWF.
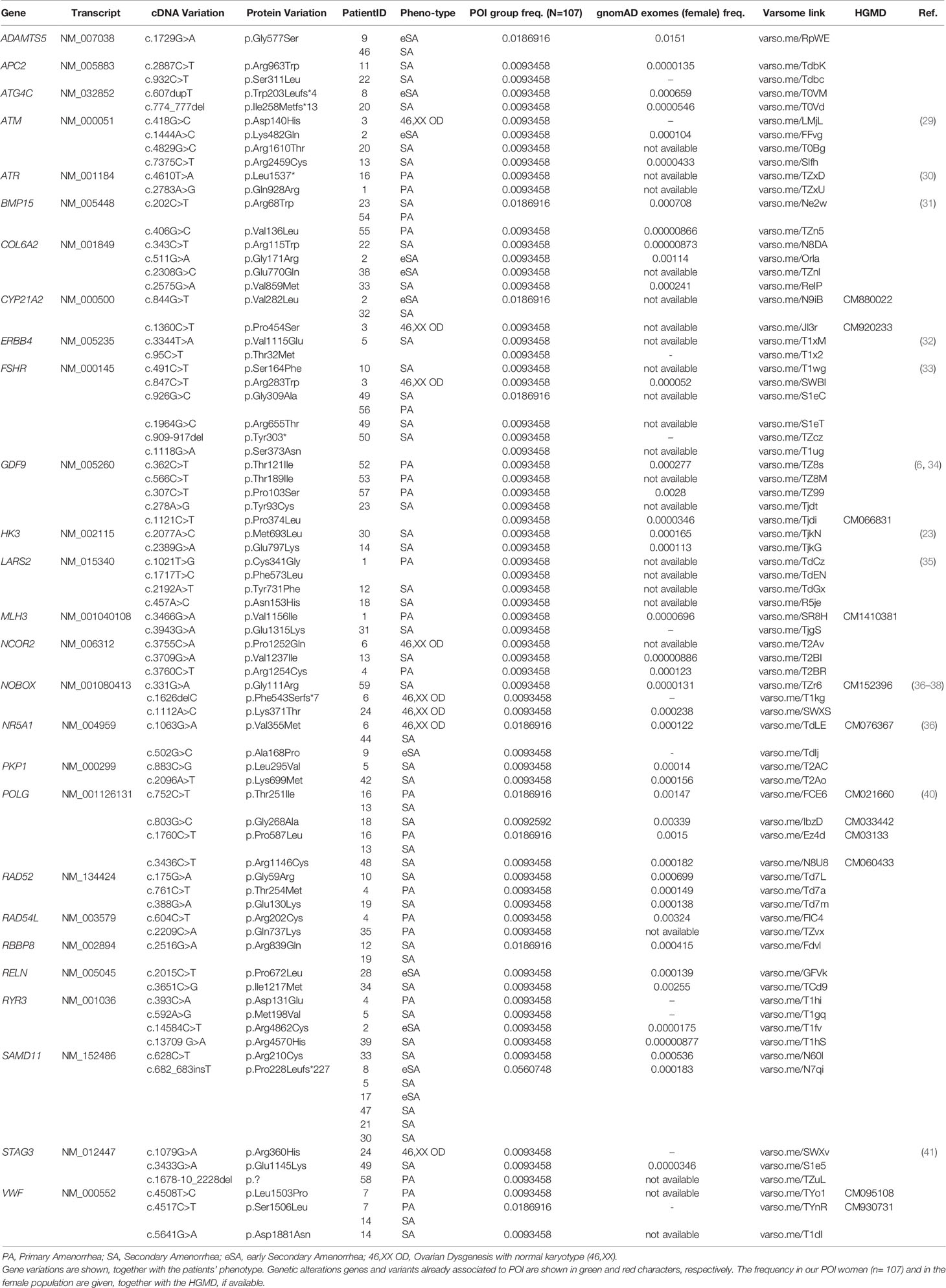
Table 7 Summary of the recurring variants in our POI cohorts.
The gene ontology analysis was carried out by interrogating the Reactome pathway database and DAVID gene functional annotation tool (refer to Tables S6, S7 for the statistical evaluation of the enrichment analysis). Considering both bioinformatic tools, we identified five main pathways likely affected by multiple genes found altered in our OVO-Array cohort and with functions in ovarian development and physiological maturation of oocytes (Figure 1A). Most altered genes, 34 in total, participated in meiosis, cell cycle, and DNA repair processes, whereas 15 genes were involved in reproductive pathways (folliculogenesis, oocyte maturation, and follicular development). Both groups resulted to be affected in all manifestations of POI, from 46,XX OD to SA, but especially in the most severe phenotypes. Twelve genes with variations were identified in the ECM remodeling pathway, which influences relevant processes during follicle development, such as cell morphology, communication, proliferation, survival, and steroidogenesis, and appeared to be mostly affected with later POI onset (SA <25 years), but also in few exceptional 46,XX OD or PA cases. Fourteen altered genes were involved in the control of specific aspects of cell metabolism with roles in ovarian function, in both PA and SA patients. A rare frameshift alteration of SAMD11, which was demonstrated as a promoter of cell proliferation, has been found in several patients mostly affected with SA. Four different variants of RYR3, coding for a calcium channel with a role in the homeostasis of calcium, have been identified among all phenotypes. Furthermore, another identified pathway, fundamental in granulosa cell differentiation and proliferation, is the NOTCH signaling, represented by NOTCH2, NOTCH3, and NOTCH4 genes found to be altered in both PA and early SA cases. Finally, we also identified alterations in genes belonging to WNT signaling, cell death, and post-translational modifications mainly affecting SA patients (Table S2, Figure 3).
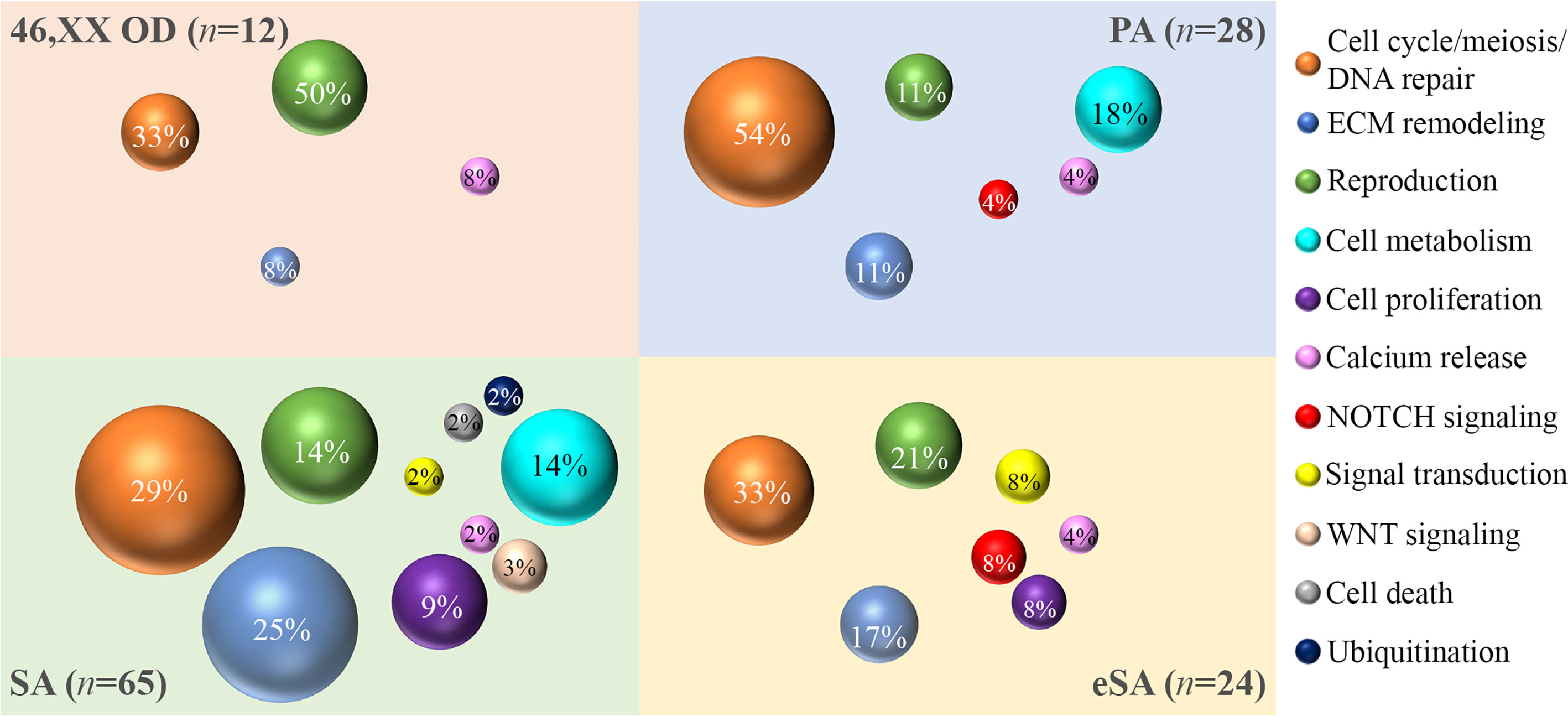
Figure 3 Bubble chart of the pathways identified by the gene ontology analysis. The sample numerosity (n) is reported for each phenotype (46,XX OD, light red; PA, light blue; eSA, light yellow; and SA <25 years, light green). Pathways are represented by colored bubbles (refer to the legend on the right). The size of the bubbles depends on the number of variants identified in each pathway for each phenotype. The percentages within the bubbles indicate the ratio between the number of variants per pathway associated to a phenotype and the total of variants associated to that phenotype. 46,XX OD = ovarian dysgenesis with normal karyotype (46,XX); PA = primary amenorrhea with functional ovarian defect, eSA = secondary amenorrhea onset few months after menarche; SA = secondary amenorrhea <25 years of age.
Most patients showed at least two pathways affected by variable genetic variations and a few showed several variations in genes from only one pathway (e.g., patient 27, presenting with PA and one variant in TP63 and FANCA genes, both related to meiosis, cell cycle, and DNA repair processes). Among SA, we also found three patients with the involvement of several variations in a singular pathway: patient 19 with three variants and patient 31 with two variants affecting genes related to meiosis, cell cycle, and DNA repair processes and patient 23 with a variant in BMP15 and a compound heterozygosity in GDF9, both key actors of folliculogenesis (Table S3).
In this study, we used the OVO-Array panel, a targeted NGS approach, to investigate the genetic cause of POI in a cohort of 64 patients with early onset of the disorder. The sequencing was focused on 295 candidate genes selected from literature and our previous data. Through this approach, we could identify 9 already known variants (6, 31, 34, 36–38, 42, 43) and 32 novel rare variants in genes already associated to POI etiology. Moreover, 34 novel rare variants have been found in additional genes participating in pathways important for ovarian physiology.
Our data are consistent with several lines of evidence recently emerging pointing to an oligogenic architecture for POI, according to which the presence of multiple genetic variants may partially explain the heterogenicity of phenotypes observed in POI patients. Indeed, we observed at least two variants in 35 patients, 5% of the cohort screened with our OVO-Array panel. Noteworthy, two of these patients harbored six variants. On the contrary, the screening of nine POI genes for diagnostic routine permitted to identify only 2 out of the 11 patients with alterations (18%) carrying either two or three variants.
We observed that almost all patients screened by the OVO-Array, who carried the major number of variants, presented with a severe phenotype (PA or early SA onset just after menarche), whereas the majority of women carrying only one or two alterations (15 out of 25) were affected by SA. However, we also found that some patients harboring fewer pathogenic or likely pathogenic variants displayed a severe phenotype. These findings suggest that POI could emerge either by the disruption of a single fundamental genetic function or as a result of multilocus variations in genes interacting within different pathways, as proposed by other authors for oligogenic diseases (44). On this line, patient 6 of this cohort was previously reported for the potential synergic detrimental effect of a complex pattern of multiple inherited genetic variants in FIGLA, NOBOX, and NR5A1 (36). In the present study, we included this patient into the OVO-Array analysis, and by sequencing additional genes, we could identify another alteration (c.3755C>A, p.Pro1252Gln) in NCOR2. This gene encodes for a nuclear receptor corepressor that, together with its paralog NCOR1, mediates the retinoic acid-dependent repression of Fgf8 in mouse during organogenesis and, if this complex is mutated, exhibits increased Fgf8 expression, similar to retinoic acid deficiency (45). On one hand, retinoic acid is critical for the entrance in meiosis of ovarian germ cells (46); on the other hand, FGF8 cooperates with BMP15 to promote glycolysis in cumulus cells (47). Therefore, we could hypothesize that an alteration affecting these fundamental pathways might combine with the other three already described variants and play a synergic effect leading to the severe phenotype of the patient. This evidence supports multilocus analysis as a fundamental tool to explain the full phenotypic spectrum of women with POI.
Since NGS has demonstrated to be a powerful tool for the identification of new molecular players and pathways in POI onset, the information derived from the analysis of large NGS panels, such as the OVO-Array, might increase the diagnostic power up to 75% of POI cases, in contrast to the current 25% of positive diagnosis obtained by screening few POI genes. The actual limit of this approach is the necessity of validating new putative candidates by further functional testing and sequencing in larger cohorts. Nonetheless, in this study, we could observe both the presence of the same variant in more than one patient at an increased frequency in women with POI with respect to the general female population and different alterations affecting the same genes in several patients. Although the fertility history of the controls reported in public datasets is unknown, thus introducing a potential bias in the analysis, our findings support the relevance of these candidates in POI pathogenesis and deserve further investigations.
A first hint pointing at the possible involvement of the newly identified candidates derives from gene ontology, which highlighted pathways fundamental for the reproductive success in mammals.
Meiotic recombination is a complex process, which requires the combination of a large plethora of factors (48). Furthermore, during the arrest at meiotic prophase I, primordial follicle oocytes are more vulnerable to DNA double stand breaks emanating from endogenous and exogenous sources (49); thus, pathogenic variations in one or more of their encoding genes might explain the onset of POI, as previously proposed (50). Noteworthy, we found 34 altered genes participating in meiosis, cell cycle, and DNA repair as possible candidates for POI. This pathway resulted to be mainly affected in patients with the most severe phenotypes, and in several cases, we found variants in at least two genes encoding DNA repair and meiotic factors. Moreover, this was the only pathway affected by genetic variants in some patients. Our data indicate that, in line with its oligogenic nature, POI might also result from alterations in more loci belonging to the same pathway.
The next major pathway in which we have found several altered genes was folliculogenesis, encompassing all stages of ovarian follicle development. In one case of SA (patient 23), we found variations in two major factors of folliculogenesis: a variant of BMP15 (p.Arg68Trp) with deleterious functional effects (31), together with a compound heterozygosity in GDF9 [p.Tyr93Cys and the previously reported p.Pro374Leu (34)], supporting the possibility that the disruption of a unique pathway at variable levels can also cause the disorder. Besides the known actors (i.e., FIGLA, NOBOX, BMP15, GDF9, FSHR, LHCGR, NR5A1, AR), we found variants in two genes involved in autophagosome assembly (ATG2A and ATG4C). Their role may deserve further attention because autophagy is an important mechanism in mammalian ovarian development, regulating follicular atresia (51). We also identified genes causing disorders of sex development or with roles in spermatogenesis. Indeed, CYP21A2 alterations are responsible for congenital adrenal hyperplasia, caused by diminished aldosterone and cortisol production that result in ambiguous genitalia in female affected (52). DMRT3 encodes for a transcription factor with evolutionary conserved roles in sex development, whose haploinsufficiency leads to 46,XY male to female sex reversal (53). TUBA8 encodes an isoform of α-tubulin highly expressed in ovarian follicle (www.proteinatlas.org). This gene has an ortholog in mouse which is expressed in the brain and testis, with a role in spermatid development (54), and can cause asthenozoospermia in men (55). ID1 has been described as potential AMH downstream target genes (56, 57), and since it is involved in the regulation of follicular growth, further characterization of its molecular function in this pathway would be needed. KMT2D alterations are the main cause of Kabuki syndrome, a congenital intellectual disability with genitourinary anomalies among other additional features. Interestingly, a reduced number of dominant families was reported in a Kabuki cohort (58), thus supporting a possible role of KMT2D variations in female fertility.
We also identified alterations in genes involved in ECM remodeling in the OVO-Array POI cohort, such as ADAMTS4, ADAMTS5, ADAMTS16, AGRN, COL6A1, COL6A2, ERBB3, ERBB4, PKP1, RELN, THBS2, and VWF. Given the ECM contribution to granulosa cell survival and proliferation, the ECM is required for maintaining the follicular cell morphology in all phases of ovarian follicle function (59). This ECM role is supposed to be mediated, at least in part, by the distinct ADAMTS subtypes and collagens. The ECM regulates cell aggregation and intracellular communication between the oocyte, granulosa, and theca cells within the follicle. The communication of small metabolites, ions, and second messengers between each cell type is made possible by a network of gap junctions, such as desmosomes containing among others the accessory plaque protein plakophilin 1. The ECM proteins are also necessary to support granulosa cell survival and steroidogenesis; for example, reelin contributes to follicular stability. Factors within the ECM are important in the control of follicular development and atresia. Included in this group of ECM proteins are thrombospondins, which are thought to contribute to the regulation of angiogenesis, and von Willebrand factor (59).
Several elements of Notch signaling appear to be involved in the POI pathogenesis in the OVO-Array cohort (NOTCH2, NOTCH3, NOTCH4). The Notch pathway is a contact-dependent signaling system active in the mammalian developing ovary which has multiple functions in follicle assembly, maturation, development, and meiotic entry (60). Notch proteins (NOTCH1, NOTCH2, NOTCH3, and NOTCH4) function as transmembrane receptors for one of the membrane-bound ligands (JAG1, JAG2, DLL1, DLL3, and DLL4) and their binding causes a conformational change of the Notch protein starting a series of sequential proteolytic cleavages at the receptor juxtamembrane region allowing the release of the Notch intracellular domain that is eventually free to translocate to the nucleus. Within the nucleus, it activates the transcription of Notch target genes. Receptors, ligands, modulators, and activated genes belonging to this system are expressed and finely regulated during folliculogenesis. Although Notch1 and Notch4 expression is limited to the ovarian vasculature, Notch2 and Notch3 are expressed in the granulosa cells of developing follicles and mediate follicle assembly and growth in mammals. Consistent with its role within the mammalian ovarian follicle, genetic alterations in NOTCH2 were previously identified via whole-exome sequencing and functional evidence demonstrated the correlation of NOTCH2 missense variations in POI (23, 61).
Several rare variants have been identified in genes regulating different aspects of follicular cell metabolism. Hexokinase 3 (HK3) converts glucose to glucose-6-phosphate in the first step of glucose metabolism, exerting a protective effect against oxidative stress (62), and it was associated with age at natural menopause (63). A recent transcriptomic study in primates demonstrated that genes most regulated in aged oocytes and granulosa cells are related to antioxidant defenses (64), further supporting the importance of cellular damage in infertility. POLG and LARS2 play roles in mitochondrial DNA replication, gene expression, and protein synthesis and degradation (65). Mutations in POLG can cause among others the neurological conditions Alpers syndrome and progressive external ophthalmoplegia (PEO), and the latter can associate with POI (35). Four POLG variants emerged by our analysis: a PA (patient 23, see 3.1.1 section) and a SA patient with c. c.752C>T (p.Thr251Ile) and c.1760C>T (p.Pro587Leu) in compound heterozygosity; c.803G>C (p.Gly268Ala) and c.3436C>T (p.Arg1146Cys) in two patients with SA. Unfortunately, no further neurological information was available for these patients. Mutations in LARS2, encoding mitochondrial leucyl-tRNA synthetase, lead to POI and hearing loss in Perrault syndrome (66). We identified the c.457A>C (p.Asn153His) variant (ClinVar VCV000191173.2) of LARS2 in a patient with SA, who was not previously associated with Perrault syndrome, and other three missense variants in two SA patients. Unfortunately, we have no further clinical information also in these patients. The cholesterol synthetase DHCR24 belongs to the pathway of steroid biosynthesis which regulates many physiological processes (i.e., stress response, ovarian cycle, and endocrine system) (67), and this is the first report of a variant in DHCR24 in a patient with POI. The VLDLR gene in humans is relevant for steroidogenesis as its expression in granulosa cells of pre-ovulatory follicles keeps a relevant role in lipoprotein endocytosis during follicular growth (19). Evidence in hens correlates the VLDLR function with fertility (68) and its haploinsufficiency in women has been associated to impaired folliculogenesis (69). Here, we confirm previous findings by describing a novel missense variant in a patient with PA.
WNT signaling has essential functions in ovary differentiation, follicle development, and hormone synthesis (70). In this study, we identified missense variants in LGR4, one of the receptors for R-spondins, which augment the WNT signaling pathway (71), and LRP5, a co-receptor for the canonical Wnt–β-catenin signaling with a known role in the regulation of bone mineral density (72), but we could not find any further evidence in the ovary.
Cell proliferation and death pathways were represented by the identification in our cohort of variants in SAMD11 and RIPK1, respectively. SAMD11 has never been associated with ovarian phenotypes before. This gene encodes for a transcriptional modulator relatively uncharacterized but phylogenetically conserved from zebrafish to humans, widely expressed in various tissues (73), where it might promote cell proliferation. Conversely, RIPK1 can mediate apoptosis during embryonic development (74). This is the first report of variants in these genes associated with an ovarian phenotype, and their role in the ovary should be investigated in further studies. Another gene involved in mitosis control that we found altered in POI was USP35, encoding a deubiquitinating enzyme which regulates the stability and function of Aurora B kinase and whose depletion results in inhibited metaphase chromosome alignment (75).
In mammalian oocytes, calcium is one of the major signal molecules involved in meiotic cell cycle resumption, arrest, and apoptosis. RYR3 encodes a calcium channel that mediates the intracellular release of Ca2+, and the ryanodine receptors (RyR) family has been identified in mammalian oocytes (76). Here, we reported several RYR3 variants with predicted pathogenic effect, which might dysregulate the homeostasis of calcium within oocytes, thus supporting a role for this gene in the disorder.
We propose novel candidate gene-disease variants likely causative or conferring susceptibility to POI onset. Our findings, by supporting the oligogenic nature of POI, suggest that in the presence of the most severe forms of ovarian insufficiency, it is necessary to screen multiple candidates or to perform WES analysis, since more alterations in different genes may synergize for the determination of the phenotype. Moreover, we show that multilocus analysis could increase the diagnostic power and the accuracy of POI diagnosis, thus ameliorating genetic counseling in patients. The systematic application of the OVO-Array multilocus analysis shall improve the management of POI including a personalized approach to the fertility defect and to the associated extra-ovarian abnormalities that can frequently anticipate or follow the POI onset.
The original contributions presented in the study are included in the article/Supplementary Material and online at https://doi.org/10.5281/zenodo.4543275 (Digital Object Identifier 10.5281/zenodo.4543275). Further inquiries can be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by IRCCS Istituto Auxologico Italiano Ethics Committee. Written informed consent to participate in this study was provided by the patients or, if minors, by the legal guardian/next of kin of the participants.
RR was responsible for the conception and design, OVO-Array experiment, and interpretation of data and drafted the manuscript. SM was responsible for the sequencing analysis, carried out the in silico predictions, and helped in drafting the manuscript. FG was responsible for NGS for diagnostic screening. DG was responsible for the bioinformatic NGS analysis. LL contributed to the OVO-Array experiment. AM, CM, FB, and MB were responsible for the enrollment of patients and critical revision of the manuscript. LP designed and supervised the research and revised the manuscript critically for important intellectual content. All authors contributed to the article and approved the submitted version.
This research was funded by the Italian Ministry of Health with the grant “BIOEFFECT” (GR‐2011‐02351636 Giovani Ricercatori to RR). The funders had no role in the design of the study, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2021.664645/full#supplementary-material
1. Golezar S, Ramezani Tehrani F, Khazaei S, Ebadi A, Keshavarz Z. The Global Prevalence of Primary Ovarian Insufficiency and Early Menopause: A Meta-Analysis. Climacteric (2019) 22:403–11. doi: 10.1080/13697137.2019.1574738
2. Tsiligiannis S, Panay N, Stevenson JC. Premature Ovarian Insufficiency and Long-Term Health Consequences. Curr Vasc Pharmacol (2019) 17:604–9. doi: 10.2174/1570161117666190122101611
3. Huhtaniemi I, Hovatta O, La Marca A, Livera G, Monniaux D, Persani L, et al. Advances in the Molecular Pathophysiology, Genetics, and Treatment of Primary Ovarian Insufficiency. Trends Endocrinol Metab (2018) 29:400–19. doi: 10.1016/j.tem.2018.03.010
4. Caburet S, Arboleda VA, Llano E, Overbeek PA, Barbero JL, Oka K, et al. Mutant Cohesin in Premature Ovarian Failure. N Engl J Med (2014) 370:943–9. doi: 10.1056/NEJMoa1309635
5. de Vries L, Behar DM, Smirin-Yosef P, Lagovsky I, Tzur S, Basel-Vanagaite L. Exome Sequencing Reveals SYCE1 Mutation Associated With Autosomal Recessive Primary Ovarian Insufficiency. J Clin Endocrinol Metab (2014) 99:E2129–32. doi: 10.1210/jc.2014-1268
6. Bouilly J, Beau I, Barraud S, Bernard V, Azibi K, Fagart J, et al. Identification of Multiple Gene Mutations Accounts for a New Genetic Architecture of Primary Ovarian Insufficiency. J Clin Endocrinol Metab (2016) 101:4541–50. doi: 10.1210/jc.2016-2152
7. Bouali N, Francou B, Bouligand J, Imanci D, Dimassi S, Tosca L, et al. New MCM8 Mutation Associated With Premature Ovarian Insufficiency and Chromosomal Instability in a Highly Consanguineous Tunisian Family. Fertil Steril (2017) 108:694–702. doi: 10.1016/j.fertnstert.2017.07.015
8. Fauchereau F, Shalev S, Chervinsky E, Beck-Fruchter R, Legois B, Fellous M, et al. A non-Sense MCM9 Mutation in a Familial Case of Primary Ovarian Insufficiency. Clin Genet (2016) 89:603–7. doi: 10.1111/cge.12736
9. Yuan L, Liu JG, Hoja MR, Wilbertz J, Nordqvist K, Höög C. Female Germ Cell Aneuploidy and Embryo Death in Mice Lacking the Meiosis-Specific Protein SCP3. Science (2002) 296:1115–8. doi: 10.1126/science.1070594
10. Chen B, Li L, Wang J, Li T, Pan H, Liu B, et al. Consanguineous Familial Study Revealed Biallelic FIGLA Mutation Associated With Premature Ovarian Insufficiency. J Ovarian Res (2018) 11:48. doi: 10.1186/s13048-018-0413-0
11. Li L, Wang B, Zhang W, Chen B, Luo M, Wang J, et al. A Homozygous NOBOX Truncating Variant Causes Defective Transcriptional Activation and Leads to Primary Ovarian Insufficiency. Hum Reprod (2017) 32:248–55. doi: 10.1093/humrep/dew271
12. França MM, Funari MFA, Lerario AM, Nishi MY, Pita CC, Fontenele EGP, et al. A Novel Homozygous 1-Bp Deletion in the NOBOX Gene in Two Brazilian Sisters With Primary Ovarian Failure. Endocrine (2017) 58:442–7. doi: 10.1007/s12020-017-1459-2
13. Zhang W, Wang J, Wang X, Li L, Pan H, Chen B, et al. A Novel Homozygous Mutation of Bone Morphogenetic Protein 15 Identified in a Consanguineous Marriage Family With Primary Ovarian Insufficiency. Reprod BioMed Online (2018) 36:206–9. doi: 10.1016/j.rbmo.2017.10.104
14. França MM, Funari MFA, Nishi MY, Narcizo A, Domenice S, Costa E, et al. Identification of the First Homozygous 1-Bp Deletion in GDF9 Gene Leading to Primary Ovarian Insufficiency by Using Targeted Massively Parallel Sequencing. Clin Genet (2018) 93:408–11. doi: 10.1111/cge.13156
15. Bramble MS, Goldstein EH, Lipson A, Ngun T, Eskin A, Gosschalk JE, et al. A Novel Follicle-Stimulating Hormone Receptor Mutation Causing Primary Ovarian Failure: A Fertility Application of Whole Exome Sequencing. Hum Reprod (2016) 31:905–14. doi: 10.1093/humrep/dew025
16. Yariz KO, Walsh T, Uzak A, Spiliopoulos M, Duman D, Onalan G, et al. Inherited Mutation of the Luteinizing Hormone/Choriogonadotropin Receptor (LHCGR) in Empty Follicle Syndrome. Fertil Steril (2011) 96:e125–30. doi: 10.1016/j.fertnstert.2011.05.057
17. Fonseca DJ, Patiño LC, Suárez YC, Rodríguez ADJ, Mateus HE, Jiménez KM, et al. Next Generation Sequencing in Women Affected by Nonsyndromic Premature Ovarian Failure Displays New Potential Causative Genes and Mutations. Fertil Steril (2015) 104:154–62.e2. doi: 10.1016/j.fertnstert.2015.04.016
18. Bian X, Xie Q, Zhou Y, Wu H, Cui J, Jia L, et al. Transcriptional Changes of Mouse Ovary During Follicle Initial or Cyclic Recruitment Mediated by Extra Hormone Treatment. Life Sci (2021) 264:118654. doi: 10.1016/j.lfs.2020.118654
19. Murata M, Tamura A, Kodama H, Hirano H, Takahashi O, Tanaka T. Possible Involvement of Very Low Density Lipoproteins in Steroidogenesis in the Human Ovary. Mol Hum Reprod (1998) 4:797–801. doi: 10.1093/molehr/4.8.797
20. Jaillard S, Bell K, Akloul L, Walton K, Mcelreavy K, Stocker WA, et al. New Insights Into the Genetic Basis of Premature Ovarian Insufficiency: Novel Causative Variants and Candidate Genes Revealed by Genomic Sequencing. Maturitas (2020) 141:9–19. doi: 10.1016/j.maturitas.2020.06.004
21. Cai X, Fu H, Wang Y, Liu Q, Wang X. Depletion of GPSM1 Enhances Ovarian Granulosa Cell Apoptosis via cAMP-PKA-CREB Pathway In Vitro. J Ovarian Res (2020) 13:136. doi: 10.1186/s13048-020-00740-6
22. Cho J, Kim TH, Seok J, Jun JH, HPark H, Kweon M, et al. Vascular Remodeling by Placenta-Derived Mesenchymal Stem Cells Restores Ovarian Function in Ovariectomized Rat Model via the VEGF Pathway. Lab Invest (2020). doi: 10.1038/s41374-020-00513-1
23. Patiño LC, Beau I, Carlosama C, Buitrago JC, González R, Suárez CF, et al. New Mutations in Non-Syndromic Primary Ovarian Insufficiency Patients Identified via Whole-Exome Sequencing. Hum Reprod (2017) 32:1512–20. doi: 10.1093/humrep/dex089
24. Yang H, Wang K. Genomic Variant Annotation and Prioritization With ANNOVAR and wANNOVAR. Nat Protoc (2015) 10:1556–66. doi: 10.1038/nprot.2015.105
25. Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Aguilera MA, Meyer R, et al. VarSome: The Human Genomic Variant Search Engine. Bioinformatics (2019) 35:1978–80. doi: 10.1093/bioinformatics/bty897
26. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med (2015) 17:405–24. doi: 10.1038/gim.2015.30
27. Huang da W, Sherman BT, Lempicki RA. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat Protoc (2009) 4:44–57. doi: 10.1038/nprot.2008.211
28. Jassal B, Matthews L, Viteri G, Gong C, Lorente P, Fabregat A, et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res (2020) 48:D498–503. doi: 10.1093/nar/gkz1031
29. França MM, Funari MFA, Lerario AM, Santos MG, Nishi MY, Domenice S, et al. Screening of Targeted Panel Genes in Brazilian Patients With Primary Ovarian Insufficiency. PLoS One (2020) 23:15e0240795. doi: 10.1371/journal.pone.0240795
30. Pacheco S, Garcia-Caldés M, Roig I. ATR Function Is Indispensable to Allow Proper Mammalian Follicle Development. Chromosoma (2019) 128:489–500. doi: 10.1007/s00412-019-00723-7
31. Rossetti R, Pasquale ED, Marozzi A, Bione S, Toniolo D, Grammatico P, et al. BMP15 Mutations Associated With Primary Ovarian Insufficiency Cause a Defective Production of Bioactive Protein. Hum Mutat (2009) 30:804–10. doi: 10.1002/humu.20961
32. Wang J, Tian GG, Zheng Z, Li B, Xing Q, Wu J. Comprehensive Transcriptomic Analysis of Mouse Gonadal Development Involving Sexual Differentiation, Meiosis and Gametogenesis. Biol Procedures Online (2019) 21. doi: 10.1186/s12575-019-0108-y
33. Aittomäki K, Lucena JD, Pakarinen P, Sistonen P, Tapanainen J, Gromoll J, et al. Mutation in the Follicle-Stimulating Hormone Receptor Gene Causes Hereditary Hypergonadotropic Ovarian Failure. Cell (1995) 82:959–68. doi: 10.1016/0092-8674(95)90275-9
34. Palmer JS, Zhao ZZ, Hoekstra C, Hayward NK, Webb PM, Whiteman DC, et al. Novel Variants in Growth Differentiation Factor 9 in Mothers of Dizygotic Twins. J Clin Endocrinol Metab (2006) 91:4713–6. doi: 10.1210/jc.2006-0970
35. Tiosano D, Mears JA, Buchner DA. Mitochondrial Dysfunction in Primary Ovarian Insufficiency. Endocrinology (2019) 160:2353–66. doi: 10.1210/en.2019-00441
36. Cattoni A, Spano A, Tulone A, Boneschi A, Masera N, Maitz S, et al. The Potential Synergic Effect of a Complex Pattern of Multiple Inherited Genetic Variants as a Pathogenic Factor for Ovarian Dysgenesis: A Case Report. Front Endocrinol (2020) 11:540683. doi: 10.3389/fendo.2020.540683
37. Bouilly J, Roucher-Boulez F, Gompel A, Bry-Gauillard H, Azibi K, Beldjord C, et al. New NOBOX Mutations Identified in a Large Cohort of Women With Primary Ovarian Insufficiency Decrease KIT-L Expression. J Clin Endocrinol Metab (2015) 100:994–1001. doi: 10.1210/jc.2014-2761
38. Ferrari I, Bouilly J, Beau I, Guizzardi F, Ferlin A, Pollazzon M, et al. Impaired Protein Stability and Nuclear Localization of NOBOX Variants Associated With Premature Ovarian Insufficiency. Hum Mol Genet (2016) 25(23):5223–33. doi: 10.1093/hmg/ddw342
40. Di Fonzo A, Bordoni A, Crimi M, Sara G, Bo RD, Bresolin N, et al. POLG Mutations in Sporadic Mitochondrial Disorders With Multiple mtDNA Deletions. Hum Mutat (2003) 22:498–9. doi: 10.1002/humu.9203
41. Xiao W-J, He W-B, Zhang Y-X, Meng L-L, Lu G-X, Lin G, et al. In-Frame Variants in STAG3 Gene Cause Premature Ovarian Insufficiency. Front Genet (2019) 10:1016. doi: 10.3389/fgene.2019.01016
42. Mathorne SW, Ravn P, Hansen D, Beck-Nielsen SS, Gjørup H, Sørensen KP, et al. Novel Phenotype of Syndromic Premature Ovarian Insufficiency Associated With TP63 Molecular Defect. Clin Genet (2020) 97:779–84. doi: 10.1111/cge.13725
43. Al-Jaroudi D, Enabi S, Althagafi MS. Perrault Syndrome With Amenorrhea, Infertility, Tarlov Cyst, and Degenerative Disc. Gynecol Endocrinol (2019) 35:1037–9. doi: 10.1080/09513590.2019.1637407
44. Karaca E, Posey JE, Coban Akdemir Z, Pehlivan D, Harel T, Jhangiani SN, et al. Phenotypic Expansion Illuminates Multilocus Pathogenic Variation. Genet Med (2018) 20:1528–37. doi: 10.1038/gim.2018.33
45. Kumar S, Cunningham TJ, Duester G. Nuclear Receptor Corepressors Ncor1 and Ncor2 (Smrt) Are Required for Retinoic Acid-Dependent Repression of Fgf8 During Somitogenesis. Dev Biol (2016) 418:204–15. doi: 10.1016/j.ydbio.2016.08.005
46. Endo T, Mikedis MM, Nicholls PK, Page DC, de Rooij DG. Retinoic Acid and Germ Cell Development in the Ovary and Testis. Biomolecules (2019) 9:775. doi: 10.3390/biom9120775
47. Sugiura K, Su YQ, Diaz FJ, et al. Oocyte-Derived BMP15 and FGFs Cooperate to Promote Glycolysis in Cumulus Cells. (2007) 134:2593–603. doi: 10.1242/dev.006882
48. Baudat F, Imai Y, de Massy B. Meiotic Recombination in Mammals: Localization and Regulation. Nat Rev Genet (2013) 14:794–806. doi: 10.1038/nrg3573
49. Winship AL, Stringer JM, Liew SH, Hutt KJ. The Importance of DNA Repair for Maintaining Oocyte Quality in Response to Anti-Cancer Treatments, Environmental Toxins and Maternal Ageing. Hum Reprod Update (2018) 24:119–34. doi: 10.1093/humupd/dmy002
50. Veitia RA. Primary Ovarian Insufficiency, Meiosis and DNA Repair. BioMed J (2020) 43:115–23. doi: 10.1016/j.bj.2020.03.005
51. Zhou J, Peng X, Mei S. Autophagy in Ovarian Follicular Development and Atresia. Int J Biol Sci (2019) 15:726–37. doi: 10.7150/ijbs.30369
52. Witchel SF. Disorders of Sex Development. Best Pract Res Clin Obstet Gynaecol (2018) 48:90–102. doi: 10.1016/j.bpobgyn.2017.11.005
53. Tsai CL, Tsai CN, Lee YS, Wang HS, Lee LY, Lin CY, et al. Genetic Analysis of a Taiwanese Family Identifies a DMRT3-OAS3 Interaction That Is Involved in Human Sexual Differentiation Through the Regulation of ESR1 Expression. Fertil Steril (2020) 114:133–43. doi: 10.1016/j.fertnstert.2020.03.008
54. Diggle CP, Martinez-Garay I, Molnar Z, Brinkworth MH, White E, Fowler E, et al. A Tubulin Alpha 8 Mouse Knockout Model Indicates a Likely Role in Spermatogenesis But Not in Brain Development. PLoS One (2017) 12:e0174264. doi: 10.1371/journal.pone.0174264
55. Bhagwat S, Dalvi V, Chandrasekhar D, Matthew T, Acharya K, Gajbhiye R, et al. Acetylated α-Tubulin Is Reduced in Individuals With Poor Sperm Motility. Fertil Steril (2014) 101:95–104.e3. doi: 10.1016/j.fertnstert.2013.09.016
56. Clarke TR, Hoshiya Y, Yi SE, Liu X, Lyons KM, Donahoe PK. Müllerian Inhibiting Substance Signaling Uses a Bone Morphogenetic Protein (BMP)-Like Pathway Mediated by ALK2 and Induces SMAD6 Expression. Mol Endocrinol (2001) 15:946–59. doi: 10.1210/mend.15.6.0664
57. Li L, Zhou X, Wang X, Wang J, Zhang W, Wang B, et al. A Dominant Negative Mutation at the ATP Binding Domain of AMHR2 Is Associated With a Defective Anti-Müllerian Hormone Signaling Pathway. Mol Hum Reprod (2016) 22:669–78. doi: 10.1093/molehr/gaw040
58. Bögershausen N, Wollnik B. Unmasking Kabuki Syndrome. Clin Genet (2013) 83:201–11. doi: 10.1111/cge.12051
59. Berkholtz CB, Shea LD, Woodruff TK. Extracellular Matrix Functions in Follicle Maturation. Semin Reprod Med (2006) 24(4):262–9. doi: 10.1055/s-2006-948555
60. Vanorny DA, Mayo KE. The Role of Notch Signaling in the Mammalian Ovary. Reproduction (2017) 153:R187–204. doi: 10.1530/REP-16-0689
61. Patiño LC, Beau I, Morel A, Delemer B, Young J, Binart N, et al. Functional Evidence Implicating NOTCH2 Missense Mutations in Primary Ovarian Insufficiency Etiology. Hum Mutat (2019) 40:25–30. doi: 10.1002/humu.23667
62. Wyatt E, Wu R, Rabeh W, Park HW, Ghanefar M, Ardehali H. Regulation and Cytoprotective Role of Hexokinase III. PLoS One (2010) 5:e13823. doi: 10.1371/journal.pone.0013823
63. Chen CT, Liu CT, Chen GK, Andrews JS, Arnold AM, Dreyfus J, et al. Meta-Analysis of Loci Associated With Age at Natural Menopause in African-American Women. Hum Mol Genet (2014) 23:3327–42. doi: 10.1093/hmg/ddu041
64. Wang S, Zheng Y, Li J, Yu Y, Zhang W, Song M, et al. Single-Cell Transcriptomic Atlas of Primate Ovarian Aging. Cell (2020) 180:585–600.e19. doi: 10.1016/j.cell.2020.01.009
65. Cohen BH, Naviaux RK. The Clinical Diagnosis of POLG Disease and Other Mitochondrial DNA Depletion Disorders. Methods (2010) 51:364–73. doi: 10.1016/j.ymeth.2010.05.008
66. Pierce SB, Gersak K, Michaelson-Cohen R, Walsh T, Lee MK, Malach D, et al. Mutations in LARS2, Encoding Mitochondrial leucyl-tRNA Synthetase, Lead to Premature Ovarian Failure and Hearing Loss in Perrault Syndrome. Am J Hum Genet (2013) 92:614–20. doi: 10.1016/j.ajhg.2013.03.007
67. Li J, Li C, Li Q, Li G, Li W, Li H, et al. Novel Regulatory Factors in the Hypothalamic-Pituitary-Ovarian Axis of Hens at Four Developmental Stages. Front Genet (2020) 11:591672. doi: 10.3389/fgene.2020.591672
68. Bujo H, Yamamoto T, Hayashi K, Hermann M, Nimpf J, Schneider WJ. Mutant Oocytic Low Density Lipoprotein Receptor Gene Family Member Causes Atherosclerosis and Female Sterility. Proc Natl Acad Sci USA (1995) 92:9905–9. doi: 10.1073/pnas.92.21.9905
69. Bestetti I, Castronovo C, Sironi A, Caslini C, Sala C, Rossetti R, et al. High-Resolution Array-CGH Analysis on 46,XX Patients Affected by Early Onset Primary Ovarian Insufficiency Discloses New Genes Involved in Ovarian Function. Hum Reprod (2019) 34:574–83. doi: 10.1093/humrep/dey389
70. Pan H, Cui H, Liu S, Qian Y, Wu H, Li L, et al. Lgr4 Gene Regulates Corpus Luteum Maturation Through Modulation of the WNT-Mediated EGFR-ERK Signaling Pathway. Endocrinology (2014) 155:3624–37. doi: 10.1210/en.2013-2183
71. Carmon KS, Gong X, Lin Q, Thomas A, Liu Q. R-Spondins Function as Ligands of the Orphan Receptors LGR4 and LGR5 to Regulate Wnt/beta-Catenin Signaling. Proc Natl Acad Sci USA (2011) 108:11452–7. doi: 10.1073/pnas.1106083108
72. Cui Y, Niziolek PJ, MacDonald BT, Zylstra CR, Alenina N, Robinson DR, et al. Lrp5 Functions in Bone to Regulate Bone Mass. Nat Med (2011) 17:684–91. doi: 10.1038/nm.2388
73. Corton M, Avila-Fernández A, Campello L, Sánchez M, Benavides B, López-Molina MI, et al. Identification of the Photoreceptor Transcriptional Co-Repressor SAMD11 as Novel Cause of Autosomal Recessive Retinitis Pigmentosa. Sci Rep (2016) 6:35370. doi: 10.1038/srep35370
74. Zhang X, Dowling JP, Zhang J. RIPK1 can Mediate Apoptosis in Addition to Necroptosis During Embryonic Development. Cell Death Dis (2019) 10(3):245. doi: 10.1038/s41419-019-1490-8
75. Park J, Kwon MS, Kim EE, Lee H, Song EJ. USP35 Regulates Mitotic Progression by Modulating the Stability of Aurora B. Nat Commun (2018) 9:688. doi: 10.1038/s41467-018-03107-0
Keywords: primary ovarian insufficiency, primary amenorrhea, secondary amenorrhea, oligogenic disease, next-generation sequencing
Citation: Rossetti R, Moleri S, Guizzardi F, Gentilini D, Libera L, Marozzi A, Moretti C, Brancati F, Bonomi M and Persani L (2021) Targeted Next-Generation Sequencing Indicates a Frequent Oligogenic Involvement in Primary Ovarian Insufficiency Onset. Front. Endocrinol. 12:664645. doi: 10.3389/fendo.2021.664645
Received: 05 February 2021; Accepted: 22 September 2021;
Published: 04 November 2021.
Edited by:
Manuela Uda, Institute of Genetic and Biomedical Research, National Research Council (CNR), ItalyReviewed by:
Andrea Angius, Institute of Genetic and Biomedical Research, National Research Council (CNR), ItalyCopyright © 2021 Rossetti, Moleri, Guizzardi, Gentilini, Libera, Marozzi, Moretti, Brancati, Bonomi and Persani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Raffaella Rossetti, ci5yb3NzZXR0aUBhdXhvbG9naWNvLml0; Luca Persani, cGVyc2FuaUBhdXhvbG9naWNvLml0
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.