 Suyan Duan
Suyan Duan Fang Lu
Fang Lu Dandan Song
Dandan Song Chengning Zhang
Chengning Zhang Bo Zhang
Bo Zhang Changying Xing
Changying Xing Yanggang Yuan
Yanggang Yuan- Department of Nephrology, The First Affiliated Hospital of Nanjing Medical University, Nanjing Medical University, Nanjing, China
Over decades, substantial progress has been achieved in understanding the pathogenesis of proteinuria in diabetic kidney disease (DKD), biomarkers for DKD screening, diagnosis, and prognosis, as well as novel hypoglycemia agents in clinical trials, thereby rendering more attention focused on the role of renal tubules in DKD. Previous studies have demonstrated that morphological and functional changes in renal tubules are highly involved in the occurrence and development of DKD. Novel tubular biomarkers have shown some clinical importance. However, there are many challenges to transition into personalized diagnosis and guidance for individual therapy in clinical practice. Large-scale clinical trials suggested the clinical relevance of increased proximal reabsorption and hyperfiltration by sodium-glucose cotransporter-2 (SGLT2) to improve renal outcomes in patients with diabetes, further promoting the emergence of renal tubulocentric research. Therefore, this review summarized the recent progress in the pathophysiology associated with involved mechanisms of renal tubules, potential tubular biomarkers with clinical application, and renal tubular factors in DKD management. The mechanism of kidney protection and impressive results from clinical trials of SGLT2 inhibitors were summarized and discussed, offering a comprehensive update on therapeutic strategies targeting renal tubules.
Introduction
Along with the disease spectrum that evolved around the world over the past 30 years, diabetic kidney disease (DKD) has become the leading cause of end-stage kidney disease (ESKD) at daunting rates in both developed and developing countries (1, 2). Due to the increased risk of morbidity and mortality of DKD, the number of DKD related studies rapidly increased over the past two decades, with more than 27,500 papers published from 2000 to 2017 (3). Growing evidence suggests the underlying pathogenesis of DKD involves the renal proximal tubular epithelial cell dysfunction in a high glucose environment, oxidative stress, inflammation, fibrosis, and apoptosis (4). In addition, a large number of tubular biomarkers for DKD screening, diagnosis, and prognosis are tightly associated with the prognosis of the kidney in DKD, providing evidence for potential shifting of the paradigm from glomerulocentric to tubulocentric theory (5). It has been repeatedly shown that compared with the glomerular lesions, the extent of tubulointerstitial lesions correlates well with renal function, and the associated biomarkers have been identified (6). Urinary tubular injury markers may increase in patients with diabetes even before the onset of microalbuminuria (7, 8). Plasma tubular markers, which may reflect inflammatory and fibrotic responses, oxidative stress, and capacity of reabsorption in DKD, were also reported to be associated with early renal function decline and DKD progression (9, 10). Moreover, the biomarkers of tubulointerstitial function and structural changes were ultimately proven to be better predictors of disease progression and long-term prognosis than the current markers (11). Current prognostic markers of DKD have certain limitations. Estimated glomerular filtration rate (eGFR) and albuminuria are only modestly useful for risk prediction in type 2 diabetes mellitus (T2DM) patients with preserved renal function, and DKD progresses even in the absence of albuminuria (12, 13). Most importantly, inhibition of proximal tubule glucose transport via sodium-glucose cotransporter-2 (SGLT-2) has shown nephroprotective effects in a variety of large-sample, multicenter, placebo-controlled, and randomized clinical trials. By investigating the mechanism of the newest disease-modifying treatments for DKD, an accumulating body of research had documented the vital role of tubule function in regulating glomerular filtration through tubuloglomerular feedback. Moreover, the growth of the proximal tubule in the diabetic context supplies muscular strength to the established status of renal tubules in DKD (14, 15). The tubuloglomerular feedback mechanism begins with the theory that diabetic hyperfiltration and glomerular capillary hypertension are significant treatable stressors contributing to the progression of DKD (16–19). In diabetic conditions, the increased filtered load of glucose results in an increase in sodium-coupled glucose reabsorption by the proximal tubule. Decreased sodium delivery to the macula densa subsequently inhibits adenosine-triphosphate (ATP) conversion into adenosine, which results in the vasodilation of the afferent arteriole and the intrarenal activation of the renin–angiotensin–aldosterone system (RAAS), ultimately inducing glomerular hypertension and kidney hyperfiltration (15, 16). Hence, hyperreabsorption of water and solutes has a central role in the regulation of eGFR, highlighting the importance of alteration in the tubuloglomerular feedback for the development of DKD.
This review aimed to summarize the latest updates on the pathogenesis of renal tubular dysfunction in DKD, potential applications of tubular biomarkers, and renal tubule-targeting therapeutics based on evidence from recent trials in DKD.
New Insights Into the Pathophysiology of Renal Tubules in DKD
Morphological Changes
Recently, there has been a growing consensus that tubular abnormalities, a consistent feature of DKD, are not the aftermath of glomerular damage. Tubular cells have the potential to be the primary targets for diverse pathophysiological influences (20). The shift has been suggested from the traditional paradigm of glomerulus-centered pathophysiology extended to the tubule-interstitium (21, 22). Morphological changes of tubulointerstitial lesions in DKD include thickening of the tubular basement membrane, tubular atrophy, interstitial inflammation, interstitial fibrosis, and vascular abnormalities (23). The association of tubulointerstitial lesions with DKD progression has been validated in several reports. A study in a Chinese population with an early stage of biopsy-proven DKD suggested that interstitial lesions and glomerular injuries were independently predictive of the time to ESKD (24). Another study from the United States population at relatively late stages of biopsy-proven DKD showed that interstitial fibrosis and tubular atrophy were of univariate significance for their ability to predict clinical prognosis (25). Moreover, the association of histological lesions with renal progression may differ in type 1 and type 2 DKD. In type 1 diabetes mellitus (T1DM), glomerular damage was observed through all stages. Nevertheless, minimal or no glomerular lesions but notable tubulointerstitial and/or arteriolar abnormalities were observed in type 2 DKD patients with microalbuminuria or overt proteinuria (26–28). Additionally, tubulointerstitial lesions were observed mainly in advanced disease and might contribute to the progression to ESKD in patients with T1DM (28, 29). The pathological disparity in different types of DM may be attributed to various other diabetogenic stimuli other than high glucose, including insulin resistance and growth factors and cytokines, which activate inflammatory, apoptosis, ischemic, pro-oxidant, and fibrotic pathways. A growing number of studies have proven that genes associated with pathological features of DKD are regulated not only by classical signaling pathways but also by epigenetic mechanisms involving chromatin histone modifications, deoxyribonucleic acid (DNA) methylation, and non-coding ribonucleic acid (RNA) (30).
Functional Changes
Tubular functional changes in DKD mainly correspond to the modulation of high-glucose, oxygen metabolic disorder, inflammation, fibrosis, and apoptosis (31). Figure 1 displays the primary mechanism of tubular damage in DKD. Hyperglycemia directly destroys renal tubular cells, resulting in a wide range of cellular and metabolic dysfunctions. Three interrelated and cardinal pathways, including overproduction of reactive oxygen species (ROS), initiation of autophagy, and activation of the apoptotic pathway, are triggered by high glucose and are associated with the progression of DKD (32, 33). Oxidative stress is a state of imbalance in the production of ROS and antioxidant activity in the body, resulting in the activation of downstream inflammation (34) and tubulointerstitial fibrosis-related genes such as transforming growth factor (TGF)-β1 and RAAS-related genes (35). Nitric oxide (NO) synthase, xanthine oxidase, nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidase enzymes, and the mitochondrial respiratory chain contribute to kidney ROS generation in a physiological context (36). The pro-oxidant nitrogen oxide (Nox) family members, especially Nox4 and Nox5 isoforms, have been reported to have an important role in the generation of renal ROS in diabetes. Thallas-Bonke V et al. indicated that targeted deletion of NADPH oxidase Nox4 from proximal tubules was dispensable for DKD development (36, 37).
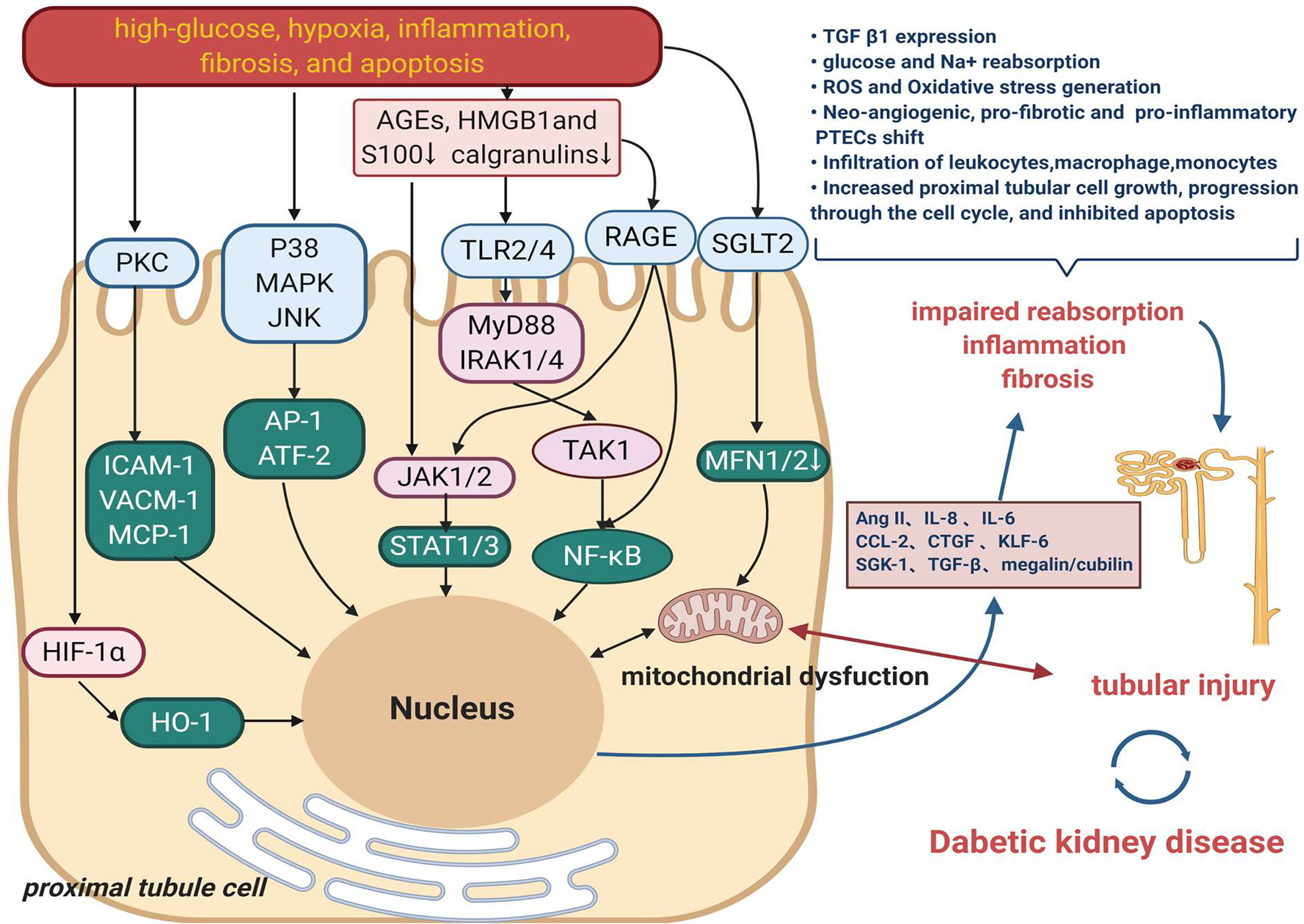
Figure 1 The main mechanism of tubular damage in DKD. Diabetogenic stimuli including high-glucose, oxygen metabolic disorder, inflammation, fibrosis, and apoptosis result in a wide range of injured pathway such as MAPK, PKC signaling. High-mobility group box 1 (HMGB1), s100/calgranulins and advanced glycation end products (AGEs) are danger-associated molecular patterns (DAMPs) that activate cell surface pattern recognition receptors (PRRs), induce signaling events to promote the development of inflammation in DKD. Another mechanism that also might contribute to tubular damage is the increased renal content of HIF1-α. Multiple effects on proximal tubule ultimately result in impaired reabsorption, inflammation and fibrosis, which contribute to tubule injury and therefore DKD.
Recent studies stress that the oxygen metabolic disorder which leads to oxidative stress, advanced glycation, hypoxia, and other harmful effects, plays a vital role in renal tubules injury (38, 39). Production and utilization of ATP by the proximal tubular cells are balanced by kidney blood flow, oxygen, and metabolite reabsorption, delivery, and consumption. This balance is now believed to the principal mechanism for regulating tubuloglomerular feedback and maintaining kidney function in diabetes (5, 33). A lately report found that hypoxia-inducible factor-1α (HIF-1α) activation in tubular cells played an important protective role against diabetic kidney injury by modulation of mitochondrial dynamics through heme oxygenase-1 (HO-1) upregulation, highlighting the potential mechanism and target in DKD (40).
Tubular inflammation is a hallmark of the progression of kidney disease in patients with DM (4). DKD inflammation produces several chemokines, which promote a pro-inflammatory microenvironment and amplify renal injury (41, 42). The majority of the pro-inflammatory responses observed in diabetic kidneys involve the activation of the transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). The activation of NF-κB and the transcription of certain pro-inflammatory chemokines in tubular epithelial cells are the markers of progressive DKD (43). Gene expression profiling of the tubulointerstitial compartment of patient biopsies has also identified 54 upregulated NF-κB target genes in progressive DKD (44). These studies showed that NF-κB activation stimulated macrophage recruitment and production of inflammatory cytokines [monocyte chemotactic protein-1 (MCP-1)], tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6) in diabetic kidneys, which were associated with the progression of the disease (45, 46).
In diabetic kidneys, excessive amount of plasma proteins, including albumin, filtered through the damaged glomerulus appears in the glomerular filtrate. Conventional perspectives have emphasized the role of glomerular hypertension and hyperfiltration in the early stage of DKD, which induce the increase in serum creatinine and urinary albumin excretion (47). However, more recent studies have focused on an unchanged glomerular albumin filtration and reduced tubular albumin reabsorption (7, 48). A membrane-associated endocytic receptor megalin (low-density lipoprotein receptor-related protein 2; LRP2) drives the reabsorption of nearly all filtered plasma proteins in cooperation with the receptor protein cubilin (49–51). Protein-overloaded condition occurs in the proximal tubular epithelial cells of the diabetic kidney. Several experimental studies have indicated that protein overload induces proximal tubular cell apoptosis (52), oxidative stress (53), inflammation, and tubulointerstitial fibrosis (54–56). The clinical relevance of increased proximal reabsorption and hyperfiltration in diabetes has been demonstrated by the ability of SGLT2 inhibitors (SGLT2is) to improve renal outcomes in patients with diabetes in large-scale clinical trials, promoting the emergence of the renal tubulocentric hypothesis (15).
A Link of Diabetogenic Stimuli to Morphological and Functional Changes in Tubules
High levels of glucose-induced oxidative stress contribute to cell death in tubule injury and tubulointerstitial fibrosis in DKD (57). In addition, persistently high levels of glucose can cause abnormal activation of mitochondrial and endoplasmic reticulum stress and intracellular signal transduction pathways, leading to further activation of downstream inflammatory factors and induction of innate immune response (58). The innate immunity in native kidney cells is upregulated at the stage of diabetic microalbuminuria, while tubulointerstitial kidney cell infiltration is associated with albuminuria and fibrosis at a more advanced stage (59). Moreover, it was shown that macrophage accumulation in the interstitium, but not glomeruli, was associated with albuminuria and renal function loss (58). Clustered renal neutrophils were mostly observed in the peritubular space and were associated with accelerated progression and eventual kidney function loss (60). Mast cell accumulation and degranulation were observed in patients with T2DM at varying stages in the periglomerular, peritubular, and perivascular regions of the interstitium. Their presence correlated with tubulointerstitial injury and disease progression (61). These studies suggested that renal tubulointerstitial infiltration by inflammatory cells could accelerate tissue damage. Besides, the components of the glomerular filtrate, such as albumin, advanced glycation end products, growth hormones, etc., interacted with the tubular system and contributed to increased energy consumption, renal oxidative stress, cortical interstitial inflammation, impairment of autophagy, stimulation of hypoxia, and tubulointerstitial fibrosis in DKD (6, 62–64). More convincingly, Vallon et al. illustrated that several diabetogenic stimuli (oxidative stress, tubular renin–angiotensin system, enhanced filtration, and tubular expression of growth factors) induced the growth of the proximal tubules and enhanced tubule reabsorptive capacity, resulting in inflammation, fibrosis, scarring, and impairment of renal function in the diabetic kidney (15).
Challenges and Progress in the Application of Novel Tubular Biomarkers
In clinical practice, therapeutic strategies for early identification of the kidney lesions in diabetic conditions and consequent slowing of the progression of DKD are still limited and currently mostly rely upon conventional biomarkers. The urine albumin-to-creatinine ratio (uACR) and eGFR are well-standardized and widely used biomarkers for evaluating kidney function and determining different stages of kidney disease in clinical practice. Although carrying prognostic information, eGFR is subject to variation owing to the analytical error of the creatinine measurement and biological variation derived from serum creatinine, patient’s age, and gender (65, 66). ACR, a tubuloglomerular-centric marker, has been recognized as the hallmark of DKD and precedes renal function loss in years. It not only reflects the capacity of glomerular permeability but is also a valuable indicator of tubular damage or dysfunction. The increase in albuminuria followed by glomerular hyperfiltration places a burden on the proximal tubule and elicits an inflammatory response leading to tubulointerstitial damage (67). Nevertheless, a substantial proportion of patients with T1DM or T2DM have renal function impairment without proteinuria, which is known as non-proteinuric DKD (68–70). The data on clinicopathological characteristics, renal prognosis, and all-cause mortality are limited to a handful of clinical trials and longitudinal studies focused on this phenotype. In 2018, the Chronic Renal Insufficiency Cohort (CRIC) study showed that the absence of albuminuria or proteinuria was common and carried a much lower risk for ESKD, chronic kidney disease (CKD) progression, or rapid decline in eGFR than those with albuminuria or proteinuria did (71). In line with this, another propensity score-matched analysis of a nationwide, biopsy-based cohort reported that non-proteinuric DKD patients presented better-controlled blood pressure and fewer typical morphological changes. They were also at a lower risk of CKD progression and all-cause mortality (72). The possible mechanism of developing non-proteinuric DKD may rely on racial/ethnic differences, aging, and response to RAAS inhibitors or other glomerulus-protective drugs before the diagnosis of DKD (68, 73). Therefore, there is still a compelling need to discover potential novel biomarkers for early diagnosis and timely risk stratification in DKD.
Recent advancements in omics-based biomarkers including proteomics, metabolomics, genome, transcriptome, or lipidome and the integration of these different approaches continue to unveil new potential biomarkers (74). Urinary novel proteomics, peptidomics markers may be associated with impaired proximal tubular reabsorption that almost all of these filtered proteins are reabsorbed into the proximal tubules through megalin/cubilin-mediated endocytosis (75). One study also demonstrated that empagliflozin, the SGLT2i, significantly impacts urinary peptides (76). However, their detection is relatively expensive and still needs time to promote clinical use. Rigorous technical and clinical validation studies are demanded to clarify the specific role and the underlying mechanism. Future research in DKD should attempt to explain how the novel biomarkers can be combined with traditional clinical and biochemical biomarkers in clinical practice to guide screening programs, improve risk stratification, predict response to treatment, and provide a method of monitoring response to treatment. The tubular biomarkers in DKD are summarized in Figure 2, which outlines three main classes of the principal tubular biomarkers that may be helpful in early detection and risk-stratification of DKD. The potential applications of these biomarkers in DKD were shown in Table 1.
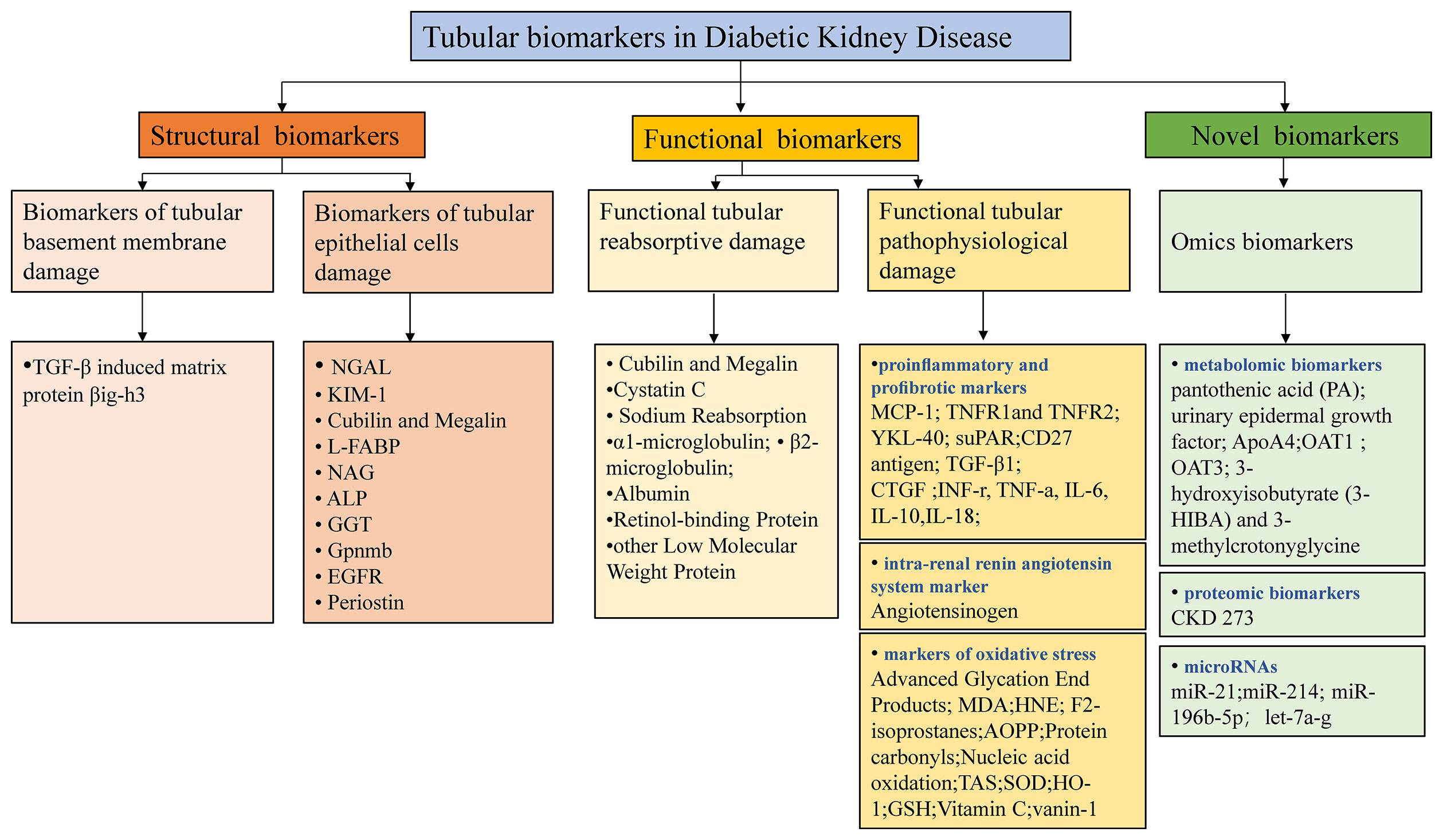
Figure 2 Potential tubular biomarkers in DKD. TGF-β, transforming growth factor-β; NGAL, neutrophil gelatinase-associated apolipoprotein; KIM-1, kidney injury molecule 1; YKL-40, chitinase-3-like protein 1; MCP-1, monocyte chemoattractant protein-1; L-FABP, liver-type fatty acid binding protein; NAG, N-acetyl-β-D-glucosidase; ALP, alkaline phosphatase; GGT, gamma-glutamyl transpeptidase; Gpnmb, glycoprotein Nmb; EGFR, epidermal growth factor receptor; TNFR1/2, tumor necrosis factor receptor 1/2; suPAR, soluble urokinase receptor; CTGF, connective tissue growth factor; INF-γ, interferon-γ; TNF-α, tumor necrosis factor-α; IL-6/10/18, interleukin 6/10/18; MDA, malondialdehyde; AOPP, advanced oxidation protein products; SOD, superoxide dismutase; HO-1, hemeoxygenase-1; GSH, glutathione; PA, pantothenic acid; OAT1/3, organic anion transporter1/3; 3-HIBA, 3-hydroxyisobutyrate; CKD, chronic kidney disease; TAS, total antioxidant status.
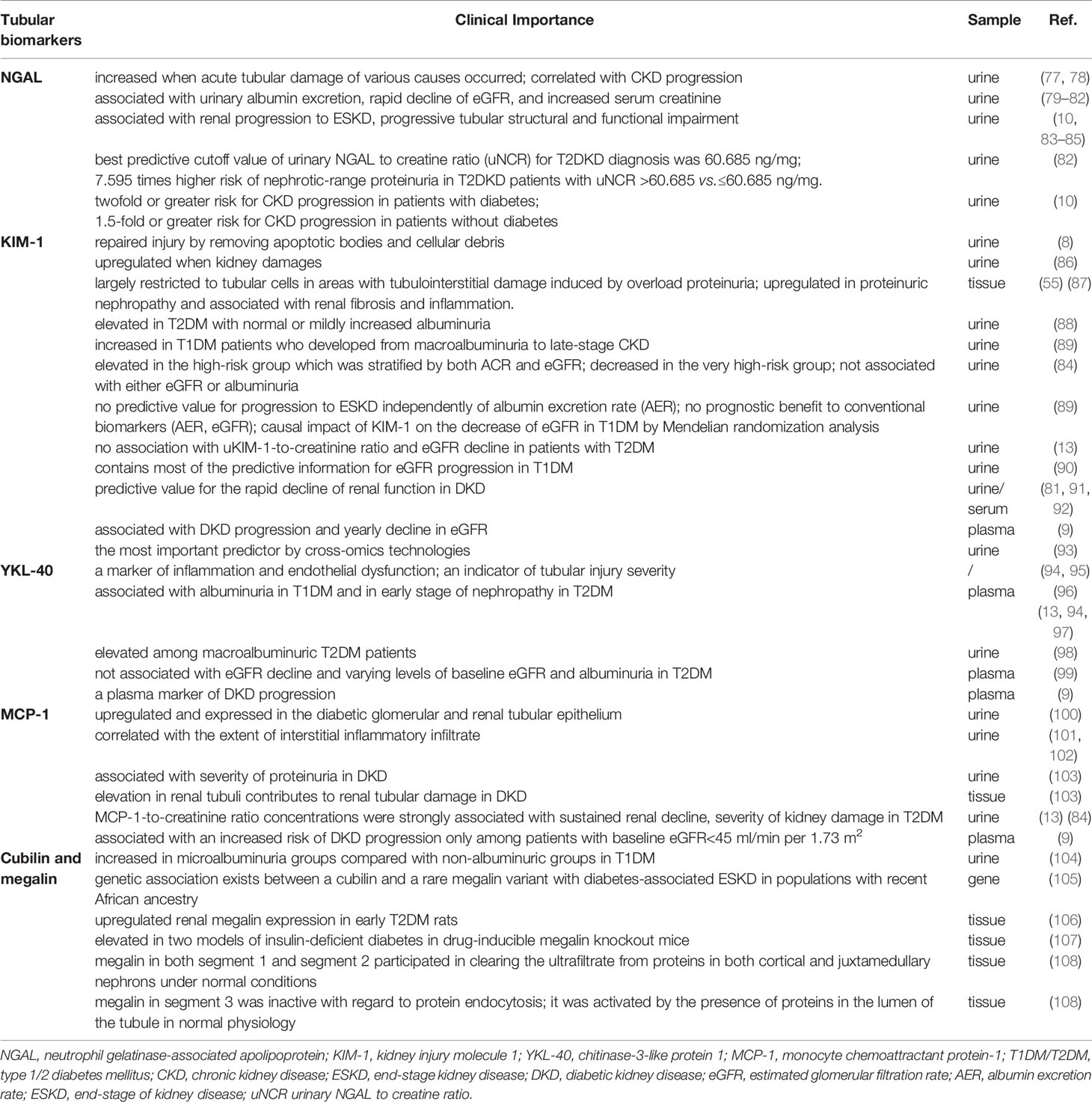
Table 1 Summary of principal tubular biomarkers of DKD in clinical use.
Neutrophil Gelatinase-Associated Lipocalin
NGAL is a 24 kDa secreted glycoprotein that belongs to the lipocalin protein family. As mainly released by neutrophils and distal tubular cells, it rapidly increases when acute tubular damage of various causes occurs (109). Following the discovery that NGAL levels are also raised in the CKD setting, this marker has been suggested to correlate with CKD progression (77, 78). More importantly, a great number of studies have demonstrated the important role of NGAL in predicting the evolution of DKD. In a study of T2DM patients and healthy controls, Fu et al. reported that NGAL increased across the four groups from controls to normoalbuminuric, microalbuminuric, and macroalbuminuric patients (79). In several observational single-center follow-up studies, elevated urine NGAL level was shown to be associated with urinary albumin excretion (80), the rapid decline in eGFR and increased serum creatinine (81), renal progression to ESKD (83), and progressive tubular structural and functional impairment (84). Consistently, our cohort study found that the best predictive cutoff value of urinary NGAL to creatine ratio (uNCR) for DKD diagnosis was 60.685 ng/mg, and T2DM patients with the increased level of uNCR had a higher risk of nephrotic-range proteinuria and worse renal outcome (82). Furthermore, a more recent report from the CRIC study conducted at seven US clinical centers provided solid evidence that higher urinary NGAL levels were not only strongly associated with cardiac markers, but were also linked to an approximately twofold or greater risk of CKD progression in patients with DM (10). It has been postulated that NGAL captures some of the variability in the rate of kidney function decline independently of albuminuria or other risk factors and reflects tubular injury and inflammation in the setting of DKD (10, 85).
Kidney Injury Molecule 1
KIM-1 is a transmembrane protein expressed on the apical membrane of proximal tubule cells (110). KIM-1 facilitates the repair of the injury by removing apoptotic bodies and cellular debris from the damaged tubulointerstitial compartment (8). Han et al. reported that urinary KIM-1 was not detectable in normal kidneys while its levels were upregulated with the occurrence of kidney injury (86). Consistently, renal KIM-1 expression was largely restricted to tubular cells in areas with tubulointerstitial damage in an experimental model of tubulointerstitial damage induced by overload proteinuria (55), and it was also upregulated in patients with proteinuric nephropathy (87). Hence, KIM-1 was suggested to be a specific and sensitive biomarker of proximal tubular damage. However, there has been a controversy about the changes in its serum and urine levels, as well as its association with kidney progression in DKD. In several studies, urine KIM-1 was elevated in T2DM patients with normal or mildly increased albuminuria (88) and in T1DM patients who developed from macroalbuminuria to late-stage CKD (89). However, Siddiqui et al. found that urinary KIM-1 was elevated in the high-risk group (stratified by both ACR and eGFR) and reduced in the very high-risk group. Also, it was not found to be associated with either eGFR or albuminuria (84). The disparity of those studies may be due to the limited sample sizes and selected population. In a large-sample randomized-controlled trial in T1DM conducted by Panduru et al., KIM-1 had no predictive value for progression to ESKD independently of albumin excretion rate (AER) and added no prognostic benefit to conventional biomarkers (AER, eGFR). However, the causal impact of KIM-1 on the decrease of eGFR in T1DM was confirmed by Mendelian randomization analysis (89). Nadkarni et al. did not find any association with uKIM-1-to-creatinine ratio and eGFR decline in patients with T2DM and preserved renal function from the ACCORD Trial population (13). Another recent report in T1DM patients from the Scottish Diabetes Research Network Type 1 Bioresource (SDRNT1BIO) and the Finnish Diabetic Nephropathy (FinnDiane) study showed that just the serum KIM-1, as well as CD27, contained most of the predictive information for eGFR progression among a large set of associated biomarkers evaluated with the Luminex platform and LC electrospray tandem MS (LC-MS/MS) (90). More recent evidence still emphasizes the important role of KIM-1 in DKD. In 2020, a multicenter and prospective cohort within the CRIC Study suggested that higher plasma KIM-1 levels were associated with DKD progression and yearly decline in eGFR (9). Kammer et al. reported that the discrimination of eGFR trajectories in individuals with the incident or early DKD and maintained baseline eGFR was modest, and KIM-1 was the most critical predictor by cross-omics technologies (93).
YKL-40
YKL-40, which is composed of three N terminal amino acids tyrosine (Y), lysine (K), and leucine (L), is a low-molecular-weight (40 kDa) heparin- and chitin-binding glycoprotein. Also known as cartilage glycoprotein-39 or chitinase 3-like protein 1 (CHI3L1), YKL-40 is a product of the chitinase 3-like 1 gene and a growth factor for several cell types. It has an established role in extracellular matrix remodeling and angiogenesis (111). Moreover, YKL-40 acts as a marker of inflammation and endothelial dysfunction. It is secreted by various cells such as neutrophils and activated macrophages in different inflamed tissues and vascular smooth muscle cells (94). Increasing evidence stressed the role of YKL-40 in kidney disease. YKL-40 was demonstrated to be an indicator of tubular injury severity, and it was upregulated in kidney macrophages after ischemia–reperfusion injury (95). It played a role in limiting tubular cell apoptosis during the repair phase of acute kidney injury (AKI) (95). The association of YKL-40 with DKD has also been suggested. Several studies have suggested that urine YKL-40 has a limited role. In contrast, plasma YKL-40 was independently associated with albuminuria in T1DM and in the early stage of nephropathy in T2DM patients (13, 94, 96, 97). However, one study documented that urinary excretion of YKL-40 was significantly elevated among macroalbuminuric T2DM patients (98), while another study reported that plasma YKL-40 was not associated with eGFR decline in participants with type 2 diabetes and varying levels of baseline eGFR (mean eGFR 78 ml/min per 1.73 m2) and albuminuria (99). More convincing results were obtained from a multicenter, prospective, large-sample cohort within the CRIC Study, providing new insights on YKL-40 as a plasma marker of DKD progression. Increased plasma YKL-40 concentrations were associated with DKD progression and decline in eGFR over time, even after adjustment for potential confounders and other plasma biomarkers (9).
Monocyte Chemoattractant Protein-1
MCP-1 (or C-C chemokine ligand 2) is a member of the C-C chemokine family, recruiting monocytes and influencing macrophage accumulation (112, 113). As an inflammatory biomarker, MCP-1 is highly upregulated in the diabetic glomerular and tubular epithelium (100). Previous studies have documented that urinary MCP-1 levels not only correlate with the extent of interstitial inflammatory infiltration but also are associated with the development of albuminuria and renal damage (101, 114). Morii et al. found that MCP-1 was produced in renal tubular cells and released into the urine in proportion to the degree of albuminuria. Increased renal tubular MCP-1 expression contributed to tubular damage in DKD (103). The ACCORD trial enrolled 10,251 T2DM patients with preserved renal function and examined the association of four biomarker-to-creatinine ratio levels; only MCP-1-to-creatinine ratio concentrations were strongly associated with the sustained renal decline (13). Siddiqui et al. also found that elevated urinary MCP-1 was related to the severity of kidney damage, and it was expressed more in progressive renal impairment in T2DM (84). The 2020 CRIC Study first reported an association of plasma MCP-1 concentrations and DKD progression among individuals with moderate to severe kidney disease. Higher plasma MCP-1 levels were associated with an increased risk of DKD progression only among patients with baseline eGFR<45 ml/min per 1.73 m2 (9).
Cubilin and Megalin
In physiological conditions, proximal tubule epithelial cells have the capacity of reabsorbing nearly all low-molecular-weight serum proteins and ultrafiltrated albumin, along with glucose, phosphate, amino acids, and various ions. The key contributor for the uptake ability of the epithelial cells essentially relies on the collective effort of two apical membrane receptors cubilin (CUBN) and megalin (LRP2), which form a complex expressed at the brush border (115). Both cubilin and megalin are huge multiligand receptors (460 and 600 kDa, respectively), each of which could independently bind to an amount of identified substrates including albumin and vitamin D binding protein (VDBP) (49). After ligand binding, cubilin/megalin ligands interact and are internalized to proximal tubular epithelial cells’ (PTECs) endosomes and lysosomes for catabolic degradation and receptor recycling (116). Using a GeLC/MS platform proteomics approach, Thrailkill et al. first propose that enhanced cubilin and megalin excretion might serve as important markers of DKD, considering that urinary cubilin and megalin were significantly higher in microalbuminuria groups than in non-albuminuric groups in T1DM patients (104). Both album infiltration and reabsorption were observed elevated in two models of insulin-deficient diabetes and drug-inducible megalin knockout mice (107). A study published in 2020 explained that megalin in both segment 1 and segment 2 participated in clearing the ultrafiltrate from proteins in both cortical and juxtamedullary nephrons under normal conditions. Although megalin in segment 3 was inactive concerning protein endocytosis, it was activated by the presence of proteins in the lumen of the tubule in normal physiological conditions (108). These studies provided a theoretical rationale and backbone for early treatment to improve the capacity of proximal tubule to avoid the development of proteinuria.
Renal Tubule-Targeting Therapeutics: A New Era for DKD Management
In addition to the new tubulocentric insights for DKD mentioned above, the emergence of new anti-hyperglycemic agents has considerably altered the therapeutic landscape of DKD. For decades, the cornerstone of DKD therapeutics relied on lifestyle interventions, strategies for hyperglycemia and hypertension in combination with the use of angiotensin-converting enzyme inhibitors (ACEIs) or angiotensin receptor blockers (ARBs) (117). Recent advances in studies on novel glucose-lowering agents promote the new era in the advanced glycemic control and concurrently promise cardiorenal protection in DKD management. Figure 3 depicts the current high-profile classes of potential novel anti-hyperglycemic agents for DKD, mainly grouped into renal tubule-targeting therapies, incretin therapies, and energy pathways-targeting therapies (117, 118). The tubule-targeting medicine, SGLT2i also affects the energy pathway associated with enhanced sirtuin1 and hypoxia-inducible factor (HIF)-2a signaling (119). In addition to SGLT2i, incretin drugs include glucagon-like peptide 1 receptor (GLP1R) agonists and dipeptidyl peptidase 4 (DPP4) inhibitors, which also have the potential to improve tubulointerstitial function. GLP1R expression was detected in macrophages, endothelial cells, juxtaglomerular cells, and proximal tubules within the kidney in various animal models and human tissue (117). Endogenous GLP1R signaling exerts a natriuretic action in DKD. Direct GLP1R-stimulation induces diuresis and natriuresis by increasing GFR and inhibiting the activity of the sodium-hydrogen exchanger isoform 3 (NHE3) in the proximal tubule (120, 121). Nevertheless, DPP4 inhibitors demonstrate modest kidney-protective effects. Compared with the GLP1R agonists, they mainly attenuate albuminuria without an impact on eGFR decline. DPP4 inhibitors indirectly modulate glucose-dependent insulin secretion and suppress glucagon secretion from pancreatic α-cells by elevating endogenous GLP1 levels (122). Linagliptin, the only available DPP4 inhibitor, showed a significant improvement in albuminuria progression but not in kidney outcomes in the Cardiovascular and Renal Microvascular Outcome Study with Linagliptin (CARMELINA) trial (123). No significant placebo-adjusted changes in eGFR or albuminuria with linagliptin therapy were observed in the Modification of Albuminuria in T2D and CKD with the LINAgliptin (MARLINA-T2D™) study (124).
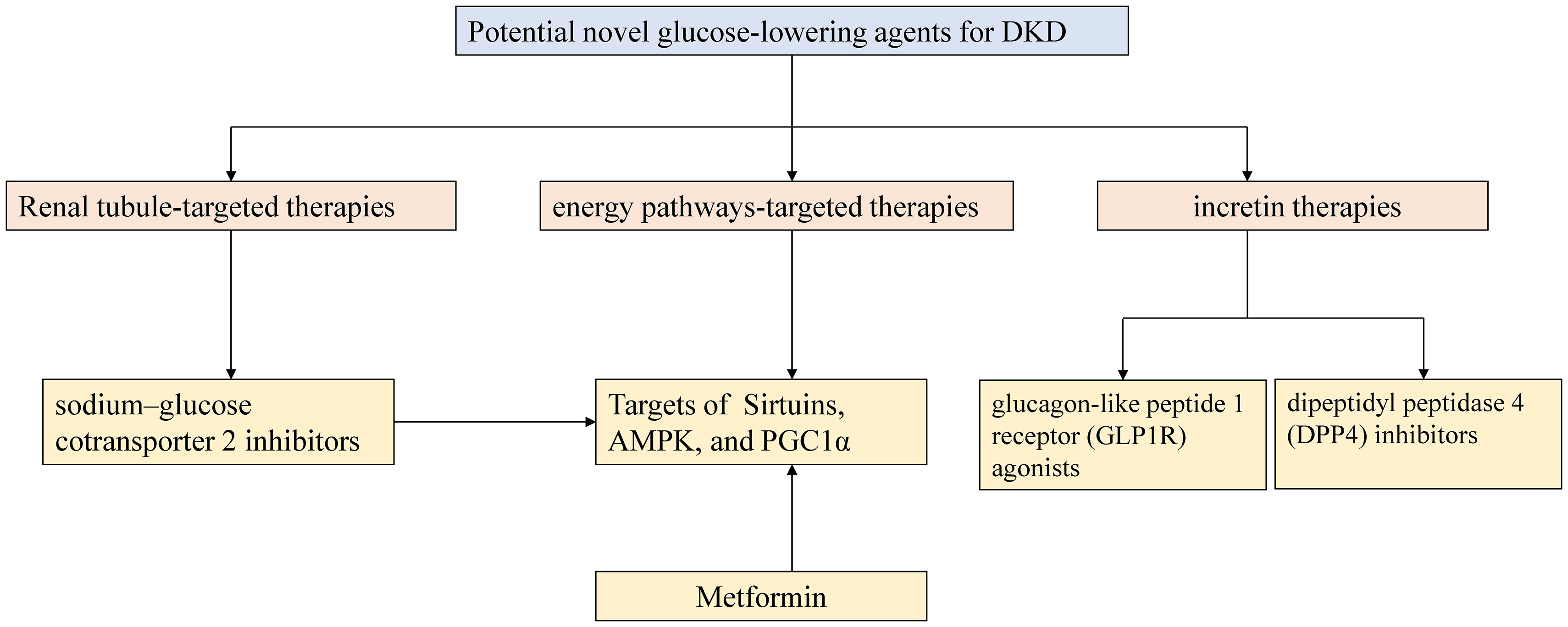
Figure 3 Outlines of potential novel glucose-lowering agents for DKD. AMPK 5-AMP-activated protein kinase; PGC-1α peroxisome proliferator-activated receptor γ coactivator-1 alpha.
Among diabetic medications, SGLT2i attracts considerable attention for their pleiotropic effects on glycemic control, renal protection, cardiovascular benefits, blood pressure control, and attenuation of lipid levels. SGLT2 is a low-capacity and high-affinity glucose transporter with 1:1 Na+/glucose stoichiometry. It is located in the S1–2 segment of the proximal convoluted tubules and is responsible for reabsorption of 90% of glucose filtered through the glomerulus (125). Multiple mechanisms are explored involving the kidney protection of SGLT2 inhibition, mainly characterized into (1) attenuation of proximal tubular oxidative stress, mitochondrial morphology, modulation of key metabolism and reabsorptive proteins, pro-inflammatory and profibrotic cytokines, and improvement of tubulointerstitial fibrosis; (2) through activation of tubuloglomerular feedback to regulate glomerular hemodynamic stability and metabolic effects (126, 127). Table 2 summarizes the underlying mechanism of kidney protection by SGLT2 inhibition in DM reported in recent years.

Table 2 Proposed hypotheses for the kidney protective mechanisms of SGLT2 inhibitors in DKD.
There is increasing evidence suggesting that SGLT2i has renal protective effects in addition to cardiovascular protection, as reported by diverse clinical trials (summarized in Table 3). The first clues involving the potential nephroprotection with SGLT-2 inhibitors originated from glucose-lowering trials that set albuminuria as a secondary outcome (167). In the Empagliflozin Cardiovascular Outcome Event (EMPA-REG OUTCOME) trial, the treatment of empagliflozin significantly reduced the primary end points which were defined as progression to macroalbuminuria, doubling of the serum creatinine level (D-Scr), initiation of kidney replacement therapy, or renal death, and incident albuminuria (159) (Table 3). In addition, all individual renal end points showed notable attenuation (31, 168). In the subsequent published Canagliflozin Cardiovascular Assessment Study (CANVAS) and CANVAS-Renal (R) program studies, clear renal protective effects were also noted (160, 161). Kidney function declined in a relatively stable manner, and urine albumin loss decreased in participants who received canagliflozin vs. placebo. Regarding the Dapagliflozin Effect on Cardiovascular Events-Thrombolysis in Myocardial Infarction 58 (DECLARE-TIMI 58) trials, although treatment with dapagliflozin showed a non-inferior rate of major adverse cardiovascular events (MACEs) than placebo, a possible lower rate of adverse renal outcomes in the dapagliflozin group than in the placebo group was observed (163). Although the above cardiovascular trials indicated nephroprotective effects of SGLT2i, it should be noted that the recruitment of participants was biased, considering that the selected patients had a high risk of cardiovascular events and mostly normal kidney function (169). Canagliflozin and Renal Events in Diabetes with Established Nephropathy Clinical Evaluation(CREDENCE) was the first dedicated renal outcomes trial of an SGLT2i canagliflozin, the recruitment of which was randomized in 4,401 T2DM patients with CKD, severely elevated albuminuria, and already ACEIs or ARBs receivers (162). The incidence rates of primary composite outcomes (D-Scr, ESKD or renal/CV death) and the renal-specific composite outcomes (D-Scr, ESKD or renal death) were significantly lower in the canagliflozin group than in the placebo group. Subsequently, two trials embarked on investigating the kidney effects of SGLT-2 inhibitors in CKD patients with or without DM (169). The Dapagliflozin and Prevention of Adverse Outcomes in Chronic Kidney Disease (DAPA-CKD) trial enrolled 4,304 CKD patients with an eGFR ranging from 25 to 75 ml/min/1.73 m², and uACR range from 200 to 5,000 mg/g (164). The trial aimed to evaluate the effect of dapagliflozin 10 mg once daily compared with placebo in addition to a maximum tolerated labeled dose of an ACEI or ARB. Reductions of the same magnitude in the primary outcomes (a composite of a sustained decline in the estimated GFR of at least 50%, ESKD, or renal/CV death) and renal-specific composite outcomes (D-Scr, ESKD, or renal death) were noted. The benefit was comparable for patients with diabetic and non-diabetic CKD. The Heart and Kidney Protection with Empagliflozin (EMPA-KIDNEY) trial commenced in November 2018, with a plan to recruit 5,000 participants and to be completed in June 2022 (170). The empagliflozin on estimated extracellular volume, estimated plasma volume, and measured glomerular filtration rate in patients with heart failure (Empire HF Renal) trial focused on the effects of empagliflozin in both heart failure and CKD patients. It enrolled 391 patients with left ventricular ejection fraction (LVEF) ≤40% and eGFR >30 ml/min/1.73 m². The results showed that empagliflozin reduced estimated extracellular volume, estimated plasma volume, and measured GFR after 12 weeks, implying that fluid volume changes might be an important mechanism underlying the beneficial clinical effects of SGLT2i (165). However, the recent Evaluation of Ertugliflozin Efficacy and Safety Cardiovascular Outcomes Trial (VERTIS CV) reported no significant benefit of ertugliflozin for the renal composite outcomes (death from renal causes, renal replacement therapy, or D-Scr) (166). Further analyses in the trial using renal different end points are underway and may give more clues. To sum up, both in the cardiovascular outcomes trials, which set different definitions of renal outcomes as secondary end points, and in the dedicated trials in CKD patients in which cardiorenal composite outcomes were primary end points, SGLT2i mostly displayed a convincing significant hindering of kidney progression.
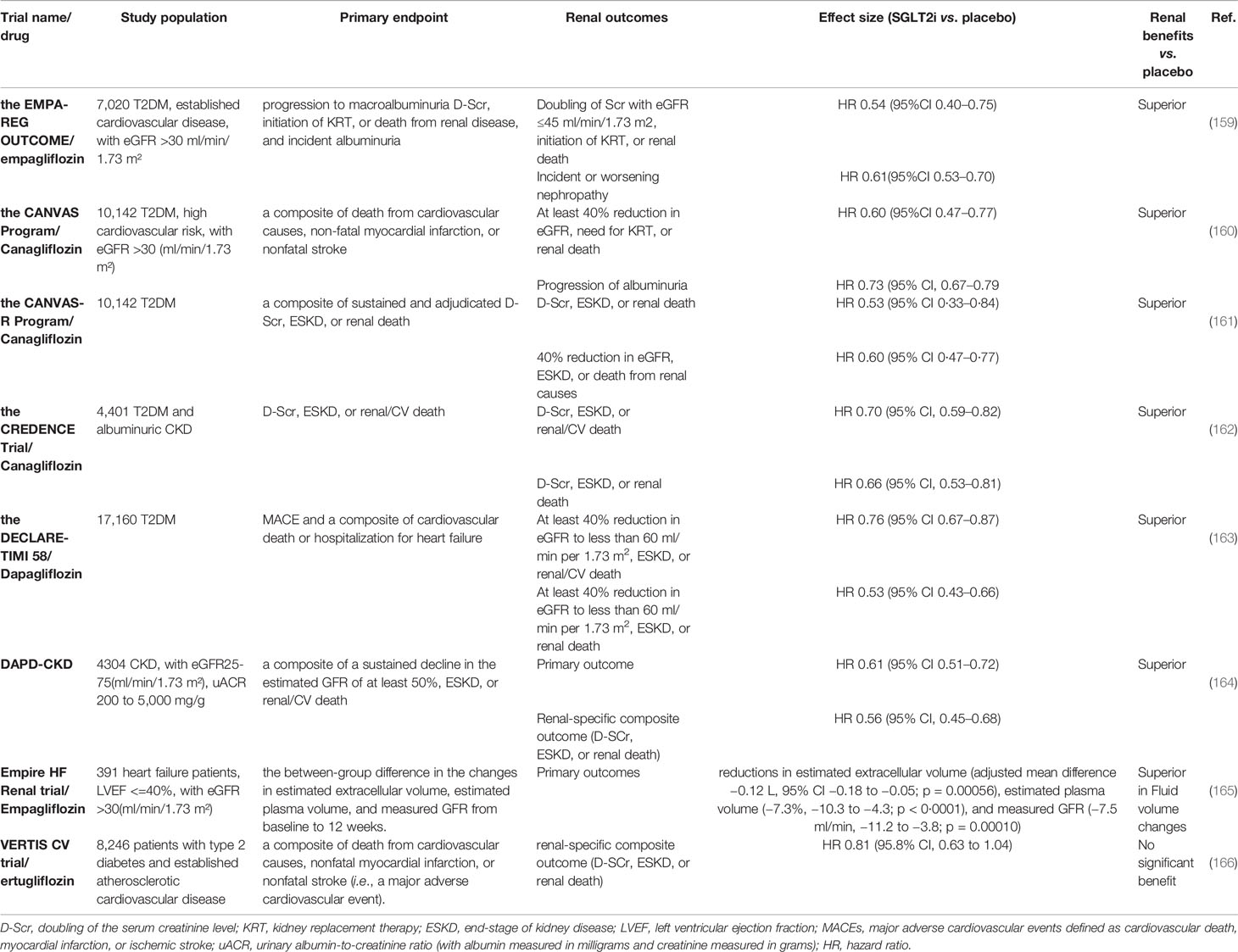
Table 3 Summary of the main renal outcomes of the SGLT2 inhibitors trials.
These impressive clinical trials and mechanistic studies of SGLT2i promoted the clinical guidelines and recommendations to update the optimal approaches for the prevention and management of DKD. In 2019, the American Diabetes Association (ADA), European Association for the Study of Diabetes (EASD), and European Society of Cardiology (ESC) published updated recommendations for the management of patients with T2DM and a high cardiovascular risk, highlighting the cardiorenal benefits of SGLT2i and glucagon-like peptide-1 receptor agonists (GLP-1 RA) (171–174). The ESC guidelines suggest that SGLT2i or GLP1 receptor agonists should have priority when patients coexist with cardiovascular disease and those at high or very high cardiovascular risk. Likewise, the ADA-EASD consensus report indicates that patients at high risk of cardiorenal disease are recommended to be treated with SGLT2i or GLP1 receptor agonists, independent of glycosylated hemoglobin (HbA1c) levels. Additionally, SGLT2i, as well as metformin, was recommended as first-line glycemic management for patients with T2D and CKD according to the 2020 Kidney Disease Improving Global Outcomes (KDIGO) guideline for diabetes management in CKD, in light of the kidney benefits for most patients with eGFR ≥30 ml/min per 1.73 m2 (175). Empagliflozin and canagliflozin are FDA-approved for use in patients with eGFR ≥45 ml/min/1.73 m2, and ertugliflozin and dapagliflozin are used for those with eGFR ≥60 ml/min/1.73 m2 (166, 176).
Future Perspectives
Great research progress in understanding the pathogenesis of tubular damage and novel biomarkers and treatments has been made, promoting us the transition into a new era of personalized diagnosis and therapy in DKD. As a complex and major complication of metabolism disease, diabetic tubular dysfunction should be regarded with close interconnection with glomerular changes and compact interrelation with systemic metabolic changes. The major current challenges in discovered biomarkers in DKD include the integration of clinical and biochemical biomarkers and omic biomarkers and translation into the pathophysiology, differential diagnosis, risk stratification, prognosis, and individual therapy in clinical practice. The ongoing progress with new anti-hyperglycemic agents provides invaluable and novel insights into the pathophysiology and potential biomarkers of renal tubules in DKD, the combination of which will shed light on better clinical management of DKD.
Author Contributions
SD drafted the manuscript, designed the figures and tables. FL and DS corrected the figures and tables. CZ and BZ reviewed the draft. CX was responsible for the final substance. YY was the guarantor and supervised the review and edited the review. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 81870469, 81670628, 81300573), the Natural Science Foundation of Jiangsu Province (No. BK20131030 to Yanggang Yuan, BK20191075 to Suyan Duan), the China Scholarship Council (CSC, File No. 201608320124), Chinese Society of Nephrology (17010060675 to Yanggang Yuan, 17010090678 to Suyan Duan), the Clinic Research Center of Jiangsu Province (No. BL2014080) and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Afkarian M, Zelnick LR, Hall YN, Heagerty PJ, Tuttle K, Weiss NS, et al. Clinical Manifestations of Kidney Disease Among Us Adults With Diabetes, 1988-2014. Jama (2016) 316:602–10. doi: 10.1001/jama.2016.10924
2. Fu H, Liu S, Bastacky SI, Wang X, Tian XJ, Zhou D. Diabetic Kidney Diseases Revisited: A New Perspective for a New Era. Mol Metab (2019) 30:250–63. doi: 10.1016/j.molmet.2019.10.005
3. Zou LX, Sun L. Global Diabetic Kidney Disease Research From 2000 to 2017: A Bibliometric Analysis. Medicine (2019) 98:e14394. doi: 10.1097/MD.0000000000014394
4. Tang SC, Lai KN. The Pathogenic Role of the Renal Proximal Tubular Cell in Diabetic Nephropathy. Nephrol Dial Transplant (2012) 27:3049–56. doi: 10.1093/ndt/gfs260
5. Gilbert RE. Proximal Tubulopathy: Prime Mover and Key Therapeutic Target in Diabetic Kidney Disease. Diabetes (2017) 66:791–800. doi: 10.2337/db16-0796
6. Thomas MC, Burns WC, Cooper ME. Tubular Changes in Early Diabetic Nephropathy. Adv Chronic Kidney Dis (2005) 12:177–86. doi: 10.1053/j.ackd.2005.01.008
7. Russo LM, Sandoval RM, Campos SB, Molitoris BA, Comper WD, Brown D. Impaired Tubular Uptake Explains Albuminuria in Early Diabetic Nephropathy. J Am Soc Nephrol JASN (2009) 20:489–94. doi: 10.1681/ASN.2008050503
8. Zeni L, Norden AGW, Cancarini G, Unwin RJ. A More Tubulocentric View of Diabetic Kidney Disease. J Nephrol (2017) 30:701–17. doi: 10.1007/s40620-017-0423-9
9. Schrauben SJ, Shou H, Zhang X, Anderson AH, Bonventre JV, Chen J, et al. Association of Multiple Plasma Biomarker Concentrations With Progression of Prevalent Diabetic Kidney Disease: Findings From the Chronic Renal Insufficiency Cohort (Cric) Study. J Am Soc Nephrol JASN (2021) 32:115–26. doi: 10.1681/ASN.2020040487
10. Anderson AH, Xie D, Wang X, Baudier RL, Orlandi P, Appel LJ, et al. Novel Risk Factors for Progression of Diabetic and Nondiabetic Ckd: Findings From the Chronic Renal Insufficiency Cohort (Cric) Study. Am J Kidney Dis Off J Natl Kidney Foundation (2021) 77:56–73.e1. doi: 10.1053/j.ajkd.2020.07.011
11. Satirapoj B, Nast CC, Adler SG. Novel Insights Into the Relationship Between Glomerular Pathology and Progressive Kidney Disease. Adv Chronic Kidney Dis (2012) 19:93–100. doi: 10.1053/j.ackd.2011.12.001
12. Dunkler D, Gao P, Lee SF, Heinze G, Clase CM, Tobe S, et al. Risk Prediction for Early CKD in Type 2 Diabetes. Clin J Am Soc Nephrol CJASN (2015) 10:1371–9. doi: 10.2215/CJN.10321014
13. Nadkarni GN, Rao V, Ismail-Beigi F, Fonseca VA, Shah SV, Simonson MS, et al. Association of Urinary Biomarkers of Inflammation, Injury, and Fibrosis With Renal Function Decline: The ACCORD Trial. Clin J Am Soc Nephrol CJASN (2016) 11:1343–52. doi: 10.2215/CJN.12051115
14. Vallon V, Thomson SC. Renal Function in Diabetic Disease Models: The Tubular System in the Pathophysiology of the Diabetic Kidney. Annu Rev Physiol (2012) 74:351–75. doi: 10.1146/annurev-physiol-020911-153333
15. Vallon V, Thomson SC. The Tubular Hypothesis of Nephron Filtration and Diabetic Kidney Disease. Nat Rev Nephrol (2020) 16:317–36. doi: 10.1038/s41581-020-0256-y
16. DeFronzo RA, Reeves WB. Pathophysiology of Diabetic Kidney Disease: Impact of SGLT2 Inhibitors. Nat Rev Nephrol (2021) 17:319–34. doi: 10.1038/s41581-021-00393-8
17. Thomson SC, Vallon V. Effects of SGLT2 Inhibitor and Dietary NaCl on Glomerular Hemodynamics Assessed by Micropuncture in Diabetic Rats. Am J Physiol Renal Physiol (2021) 320:F761–71. doi: 10.1152/ajprenal.00552.2020
18. Brenner BM. Hemodynamically Mediated Glomerular Injury and the Progressive Nature of Kidney Disease. Kidney Int (1983) 23:647–55. doi: 10.1038/ki.1983.72
19. Thomson SC, Blantz RC. Glomerulotubular Balance, Tubuloglomerular Feedback, and Salt Homeostasis. J Am Soc Nephrol JASN (2008) 19:2272–5. doi: 10.1681/ASN.2007121326
20. Akhtar M, Taha NM, Nauman A, Mujeeb IB, Al-Nabet A. Diabetic Kidney Disease: Past and Present. Adv Anatomic Pathol (2020) 27:87–97. doi: 10.1097/PAP.0000000000000257
21. Yu SM, Bonventre JV. Acute Kidney Injury and Progression of Diabetic Kidney Disease. Adv Chronic Kidney Dis (2018) 25:166–80. doi: 10.1053/j.ackd.2017.12.005
22. Chen Y, Lee K, Ni Z, He JC. Diabetic Kidney Disease: Challenges, Advances, and Opportunities. Kidney Dis (Basel Switzerland) (2020) 6:215–25. doi: 10.1159/000506634
23. Tervaert TW, Mooyaart AL, Amann K, Cohen AH, Cook HT, Drachenberg CB, et al. Pathologic Classification of Diabetic Nephropathy. J Am Soc Nephrol JASN (2010) 21:556–63. doi: 10.1681/ASN.2010010010
24. An Y, Xu F, Le W, Ge Y, Zhou M, Chen H, et al. Renal Histologic Changes and the Outcome in Patients With Diabetic Nephropathy. Nephrol Dial Transplant (2015) 30:257–66. doi: 10.1093/ndt/gfu250
25. Mottl AK, Gasim A, Schober FP, Hu Y, Dunnon AK. Segmental Sclerosis and Extracapillary Hypercellularity Predict Diabetic Esrd. J Am Soc Nephrol JASN (2018) 29:694–703. doi: 10.1681/ASN.2017020192
26. Fioretto P, Mauer M, Brocco E, Velussi M, Frigato F, Muollo B, et al. Patterns of Renal Injury in NIDDM Patients With Microalbuminuria. Diabetologia (1996) 39:1569–76. doi: 10.1007/s001250050616
27. Fioretto P, Caramori ML, Mauer M. The Kidney in Diabetes: Dynamic Pathways of Injury and Repair. The Camillo Golgi Lecture 2007. Diabetologia (2008) 51:1347–55. doi: 10.1007/s00125-008-1051-7
28. Di Vincenzo A, Bettini S, Russo L, Mazzocut S, Mauer M, Fioretto P. Renal Structure in Type 2 Diabetes: Facts and Misconceptions. J Nephrol (2020) 33:901–7. doi: 10.1007/s40620-020-00797-y
29. Lane PH, Steffes MW, Fioretto P, Mauer SM. Renal Interstitial Expansion in Insulin-Dependent Diabetes Mellitus. Kidney Int (1993) 43:661–7. doi: 10.1038/ki.1993.95
30. Kato M, Natarajan R. Epigenetics and Epigenomics in Diabetic Kidney Disease and Metabolic Memory. Nat Rev Nephrol (2019) 15:327–45. doi: 10.1038/s41581-019-0135-6
31. Jaikumkao K, Pongchaidecha A, Chatsudthipong V, Chattipakorn SC, Chattipakorn N, Lungkaphin A. The Roles of Sodium-Glucose Cotransporter 2 Inhibitors in Preventing Kidney Injury in Diabetes. Biomed Pharmacother (2017) 94:176–87. doi: 10.1016/j.biopha.2017.07.095
32. Zha D, Cheng H, Li W, Wu Y, Li X, Zhang L, et al. High Glucose Instigates Tubulointerstitial Injury by Stimulating Hetero-Dimerization of Adiponectin and Angiotensin II Receptors. Biochem Biophys Res Commun (2017) 493:840–6. doi: 10.1016/j.bbrc.2017.08.047
33. Wei PZ, Szeto CC. Mitochondrial Dysfunction in Diabetic Kidney Disease. Clin Chim Acta; Int J Clin Chem (2019) 496:108–16. doi: 10.1016/j.cca.2019.07.005
34. Pickering TG. Stress, Inflammation, and Hypertension. J Clin Hypertension (2007) 9:567–71. doi: 10.1111/j.1524-6175.2007.06301.x
35. Wei L, Xiao Y, Li L, Xiong X, Han Y, Zhu X, et al. The Susceptibility Genes in Diabetic Nephropathy. Kidney Dis (Basel Switzerland) (2018) 4:226–37. doi: 10.1159/000492633
36. Jha JC, Banal C, Chow BS, Cooper ME, Jandeleit-Dahm K. Diabetes and Kidney Disease: Role of Oxidative Stress. Antioxid Redox Signaling (2016) 25:657–84. doi: 10.1089/ars.2016.6664
37. Thallas-Bonke V, Tan SM, Lindblom RS, Snelson M, Granata C, Jha JC, et al. Targeted Deletion of NADPH-Oxidase Nox4 From Proximal Tubules is Dispensable for Diabetic Kidney Disease Development. Nephrol Dial Transplant (2020) 36:988–97. doi: 10.1093/ndt/gfaa376
38. Blantz RC. Phenotypic Characteristics of Diabetic Kidney Involvement. Kidney Int (2014) 86:7–9. doi: 10.1038/ki.2013.552
39. Miyata T, Suzuki N, van Ypersele de Strihou C. Diabetic Nephropathy: Are There New and Potentially Promising Therapies Targeting Oxygen Biology? Kidney Int (2013) 84:693–702. doi: 10.1038/ki.2013.74
40. Jiang N, Zhao H, Han Y, Li L, Xiong S, Zeng L, et al. Hif-1α Ameliorates Tubular Injury in Diabetic Nephropathy Via HO-1-Mediated Control of Mitochondrial Dynamics. Cell Proliferation (2020) 53:e12909. doi: 10.1111/cpr.12909
41. Zheng Z, Zheng F. Immune Cells and Inflammation in Diabetic Nephropathy. J Diabetes Res (2016) 2016:1841690. doi: 10.1155/2016/1841690
42. Lim AK, Tesch GH. Inflammation in Diabetic Nephropathy. Mediators Inflammation (2012) 2012:146154. doi: 10.1155/2012/146154
43. Mezzano S, Aros C, Droguett A, Burgos ME, Ardiles L, Flores C, et al. NF-Kappab Activation and Overexpression of Regulated Genes in Human Diabetic Nephropathy. Nephrol Dial Transplant (2004) 19:2505–12. doi: 10.1093/ndt/gfh207
44. Schmid H, Boucherot A, Yasuda Y, Henger A, Brunner B, Eichinger F, et al. Modular Activation of Nuclear Factor-Kappab Transcriptional Programs in Human Diabetic Nephropathy. Diabetes (2006) 55:2993–3003. doi: 10.2337/db06-0477
45. Lee FT, Cao Z, Long DM, Panagiotopoulos S, Jerums G, Cooper ME, et al. Interactions Between Angiotensin II and NF-KappaB-Dependent Pathways in Modulating Macrophage Infiltration in Experimental Diabetic Nephropathy. J Am Soc Nephrol JASN (2004) 15:2139–51. doi: 10.1097/01.ASN.0000135055.61833.A8
46. Kolati SR, Kasala ER, Bodduluru LN, Mahareddy JR, Uppulapu SK, Gogoi R, et al. Bay 11-7082 Ameliorates Diabetic Nephropathy by Attenuating Hyperglycemia-Mediated Oxidative Stress and Renal Inflammation Via NF-κb Pathway. Environ Toxicol Pharmacol (2015) 39:690–9. doi: 10.1016/j.etap.2015.01.019
47. Brenner BM, Lawler EV, Mackenzie HS. The Hyperfiltration Theory: A Paradigm Shift in Nephrology. Kidney Int (1996) 49:1774–7. doi: 10.1038/ki.1996.265
48. Tojo A, Onozato ML, Ha H, Kurihara H, Sakai T, Goto A, et al. Reduced Albumin Reabsorption in the Proximal Tubule of Early-Stage Diabetic Rats. Histochem Cell Biol (2001) 116:269–76. doi: 10.1007/s004180100317
49. Nielsen R, Christensen EI, Birn H. Megalin and Cubilin in Proximal Tubule Protein Reabsorption: From Experimental Models to Human Disease. Kidney Int (2016) 89:58–67. doi: 10.1016/j.kint.2015.11.007
50. De S, Kuwahara S, Saito A. The Endocytic Receptor Megalin and Its Associated Proteins in Proximal Tubule Epithelial Cells. Membranes (2014) 4:333–55. doi: 10.3390/membranes4030333
51. Amsellem S, Gburek J, Hamard G, Nielsen R, Willnow TE, Devuyst O, et al. Cubilin Is Essential for Albumin Reabsorption in the Renal Proximal Tubule. J Am Soc Nephrol JASN (2010) 21:1859–67. doi: 10.1681/ASN.2010050492
52. Zhuang Y, Yasinta M, Hu C, Zhao M, Ding G, Bai M, et al. Mitochondrial Dysfunction Confers Albumin-Induced NLRP3 Inflammasome Activation and Renal Tubular Injury. Am J Physiol Renal Physiol (2015) 308:F857–66. doi: 10.1152/ajprenal.00203.2014
53. Nishi Y, Satoh M, Nagasu H, Kadoya H, Ihoriya C, Kidokoro K, et al. Selective Estrogen Receptor Modulation Attenuates Proteinuria-Induced Renal Tubular Damage by Modulating Mitochondrial Oxidative Status. Kidney Int (2013) 83:662–73. doi: 10.1038/ki.2012.475
54. Takagaki Y, Shi S, Katoh M, Kitada M, Kanasaki K. Dipeptidyl Peptidase-4 Plays a Pathogenic Role in BSA-Induced Kidney Injury in Diabetic Mice. Sci Rep (2019) 9:7519. doi: 10.1038/s41598-019-43730-5
55. van Timmeren MM, Bakker SJ, Vaidya VS, Bailly V, Schuurs TA, Damman J, et al. Tubular Kidney Injury Molecule-1 in Protein-Overload Nephropathy. Am J Physiol Renal Physiol (2006) 291:F456–64. doi: 10.1152/ajprenal.00403.2005
56. Otomo H, Nara M, Kato S, Shimizu T, Suganuma Y, Sato T, et al. Sodium-Glucose Cotransporter 2 Inhibition Attenuates Protein Overload in Renal Proximal Tubule Via Suppression of Megalin O-GlcNacylation in Progressive Diabetic Nephropathy. Metabol: Clin Exp (2020) 113:154405. doi: 10.1016/j.metabol.2020.154405
57. Chang J, Yan J, Li X, Liu N, Zheng R, Zhong Y. Update on the Mechanisms of Tubular Cell Injury in Diabetic Kidney Disease. Front Med (2021) 8:661076. doi: 10.3389/fmed.2021.661076
58. Tesch GH. Diabetic Nephropathy - Is This an Immune Disorder? Clin Sci (2017) 131:2183–99. doi: 10.1042/CS20160636
59. Verzola D, Cappuccino L, D’Amato E, Villaggio B, Gianiorio F, Mij M, et al. Enhanced Glomerular Toll-Like Receptor 4 Expression and Signaling in Patients With Type 2 Diabetic Nephropathy and Microalbuminuria. Kidney Int (2014) 86:1229–43. doi: 10.1038/ki.2014.116
60. Kelly KJ, Dominguez JH. Rapid Progression of Diabetic Nephropathy is Linked to Inflammation and Episodes of Acute Renal Failure. Am J Nephrol (2010) 32:469–75. doi: 10.1159/000320749
61. Zheng JM, Yao GH, Cheng Z, Wang R, Liu ZH. Pathogenic Role of Mast Cells in the Development of Diabetic Nephropathy: A Study of Patients at Different Stages of the Disease. Diabetologia (2012) 55:801–11. doi: 10.1007/s00125-011-2391-2
62. Magri CJ, Fava S. The Role of Tubular Injury in Diabetic Nephropathy. Eur J Internal Med (2009) 20:551–5. doi: 10.1016/j.ejim.2008.12.012
63. Singh DK, Winocour P, Farrington K. Mechanisms of Disease: The Hypoxic Tubular Hypothesis of Diabetic Nephropathy. Nat Clin Practice Nephrol (2008) 4:216–26. doi: 10.1038/ncpneph0757
64. Vallon V, Komers R. Pathophysiology of the Diabetic Kidney. Compr Physiol (2011) 1:1175–232. doi: 10.1002/cphy.c100049
65. Badrick T, Turner P. The Uncertainty of the Egfr. Indian J Clin Biochem IJCB (2013) 28:242–7. doi: 10.1007/s12291-012-0280-1
66. Porrini E, Ruggenenti P, Luis-Lima S, Carrara F, Jiménez A, de Vries APJ, et al. Time for a Critical Appraisal. Nat Rev Nephrol (2019) 15:177–90. doi: 10.1038/s41581-018-0080-9
67. Abbate M, Zoja C, Remuzzi G. How Does Proteinuria Cause Progressive Renal Damage? J Am Soc Nephrol JASN (2006) 17:2974–84. doi: 10.1681/ASN.2006040377
68. Yamanouchi M, Furuichi K, Hoshino J, Ubara Y, Wada T. Nonproteinuric Diabetic Kidney Disease. Clin Exp Nephrol (2020) 24:573–81. doi: 10.1007/s10157-020-01881-0
69. Molitch ME, Steffes M, Sun W, Rutledge B, Cleary P, de Boer IH, et al. Development and Progression of Renal Insufficiency With and Without Albuminuria in Adults With Type 1 Diabetes in the Diabetes Control and Complications Trial and the Epidemiology of Diabetes Interventions and Complications Study. Diabetes Care (2010) 33:1536–43. doi: 10.2337/dc09-1098
70. Porrini E, Ruggenenti P, Mogensen CE, Barlovic DP, Praga M, Cruzado JM, et al. Non-Proteinuric Pathways in Loss of Renal Function in Patients With Type 2 Diabetes. Lancet Diabetes Endocrinol (2015) 3:382–91. doi: 10.1016/S2213-8587(15)00094-7
71. Koye DN, Magliano DJ, Reid CM, Jepson C, Feldman HI, Herman WH, et al. Risk of Progression of Nonalbuminuric CKD to End-Stage Kidney Disease in People With Diabetes: The Cric (Chronic Renal Insufficiency Cohort) Study. Am J Kidney Dis (2018) 72:653–61. doi: 10.1053/j.ajkd.2018.02.364
72. Yamanouchi M, Furuichi K, Hoshino J. Nonproteinuric Versus Proteinuric Phenotypes in Diabetic Kidney Disease: A Propensity Score-Matched Analysis of a Nationwide, Biopsy-Based Cohort Study. Diabetes Care (2019) 42:891–902. doi: 10.2337/dc18-1320
73. Bhalla V, Zhao B, Azar KM, Wang EJ, Choi S, Wong EC, et al. Racial/Ethnic Differences in the Prevalence of Proteinuric and Nonproteinuric Diabetic Kidney Disease. Diabetes Care (2013) 36:1215–21. doi: 10.2337/dc12-0951
74. Gan WZ, Ramachandran V, Lim CSY, Koh RY. Omics-Based Biomarkers in the Diagnosis of Diabetes. J Basic Clin Physiol Pharmacol (2019) 31(2):20190120. doi: 10.1515/jbcpp-2019-0120
75. Van JA, Scholey JW, Konvalinka A. Insights Into Diabetic Kidney Disease Using Urinary Proteomics and Bioinformatics. J Am Soc Nephrol JASN (2017) 28:1050–61. doi: 10.1681/ASN.2016091018
76. Cherney D, Perkins BA, Lytvyn Y, Heerspink H, Rodríguez-Ortiz ME, Mischak H. The Effect of Sodium/Glucose Cotransporter 2 (SGLT2) Inhibition on the Urinary Proteome. PloS One (2017) 12:e0186910. doi: 10.1371/journal.pone.0186910
77. Liu KD, Yang W, Go AS, Anderson AH, Feldman HI, Fischer MJ, et al. Urine Neutrophil Gelatinase-Associated Lipocalin and Risk of Cardiovascular Disease and Death in CKD: Results From the Chronic Renal Insufficiency Cohort (Cric) Study. Am J Kidney Dis (2015) 65:267–74. doi: 10.1053/j.ajkd.2014.07.025
78. Smith ER, Lee D, Cai MM, Tomlinson LA, Ford ML, McMahon LP, et al. Urinary Neutrophil Gelatinase-Associated Lipocalin may Aid Prediction of Renal Decline in Patients With Non-Proteinuric Stages 3 and 4 Chronic Kidney Disease (CKD). Nephrol Dial Transplant (2013) 28:1569–79. doi: 10.1093/ndt/gfs586
79. Fu WJ, Xiong SL, Fang YG, Wen S, Chen ML, Deng RT, et al. Urinary Tubular Biomarkers in Short-Term Type 2 Diabetes Mellitus Patients: A Cross-Sectional Study. Endocrine (2012) 41:82–8. doi: 10.1007/s12020-011-9509-7
80. Assal HS, Tawfeek S, Rasheed EA, El-Lebedy D, Thabet EH. Serum Cystatin C and Tubular Urinary Enzymes as Biomarkers of Renal Dysfunction in Type 2 Diabetes Mellitus. Clin Med Insights Endocrinol Diabetes (2013) 6:7–13. doi: 10.4137/CMED.S12633
81. Satirapoj B, Aramsaowapak K, Tangwonglert T, Supasyndh O. Novel Tubular Biomarkers Predict Renal Progression in Type 2 Diabetes Mellitus: A Prospective Cohort Study. J Diabetes Res (2016) 2016:3102962. doi: 10.1155/2016/3102962
82. Duan S, Chen J, Wu L, Nie G, Sun L, Zhang C, et al. Assessment of Urinary NGAL for Differential Diagnosis and Progression of Diabetic Kidney Disease. J Diabetes its Complications (2020) 34:107665. doi: 10.1016/j.jdiacomp.2020.107665
83. Yang YH, He XJ, Chen SR, Wang L, Li EM, Xu LY. Changes of Serum and Urine Neutrophil Gelatinase-Associated Lipocalin in Type-2 Diabetic Patients With Nephropathy: One Year Observational Follow-Up Study. Endocrine (2009) 36:45–51. doi: 10.1007/s12020-009-9187-x
84. Siddiqui K, Joy SS. Potential Role and Excretion Level of Urinary Transferrin, KIM-1, Rbp, MCP-1 and NGAL Markers in Diabetic Nephropathy. Diabetes Metab Syndrome Obes Targets Ther (2020) 13:5103–11. doi: 10.2147/DMSO.S282166
85. Satirapoj B. Tubulointerstitial Biomarkers for Diabetic Nephropathy. J Diabetes Res (2018) 2018:2852398. doi: 10.1155/2018/2852398
86. Han WK, Bailly V, Abichandani R, Thadhani R, Bonventre JV. Kidney Injury Molecule-1 (Kim-1): A Novel Biomarker for Human Renal Proximal Tubule Injury. Kidney Int (2002) 62:237–44. doi: 10.1046/j.1523-1755.2002.00433.x
87. van Timmeren MM, van den Heuvel MC, Bailly V, Bakker SJ, van Goor H, Stegeman CA. Tubular Kidney Injury Molecule-1 (KIM-1) in Human Renal Disease. J Pathol (2007) 212:209–17. doi: 10.1002/path.2175
88. de Carvalho JA, Tatsch E, Hausen BS, Bollick YS, Moretto MB, Duarte T, et al. Urinary Kidney Injury Molecule-1 and Neutrophil Gelatinase-Associated Lipocalin as Indicators of Tubular Damage in Normoalbuminuric Patients With Type 2 Diabetes. Clin Biochem (2016) 49:232–6. doi: 10.1016/j.clinbiochem.2015.10.016
89. Panduru NM, Sandholm N, Forsblom C, Saraheimo M, Dahlström EH, Thorn LM, et al. Kidney Injury Molecule-1 and the Loss of Kidney Function in Diabetic Nephropathy: A Likely Causal Link in Patients With Type 1 Diabetes. Diabetes Care (2015) 38:1130–7. doi: 10.2337/dc14-2330
90. Colombo M, Valo E, McGurnaghan SJ, Sandholm N, Blackbourn LAK, Dalton RN, et al. Biomarker Panels Associated With Progression of Renal Disease in Type 1 Diabetes. Diabetologia (2019) 62:1616–27. doi: 10.1007/s00125-019-4915-0
91. Nielsen SE, Reinhard H, Zdunek D, Hess G, Gutiérrez OM, Wolf M, et al. Tubular Markers Are Associated With Decline in Kidney Function in Proteinuric Type 2 Diabetic Patients. Diabetes Res Clin Pract (2012) 97:71–6. doi: 10.1016/j.diabres.2012.02.007
92. Satirapoj B, Pooluea P, Nata N, Supasyndh O. Urinary Biomarkers of Tubular Injury to Predict Renal Progression and End Stage Renal Disease in Type 2 Diabetes Mellitus With Advanced Nephropathy: A Prospective Cohort Study. J Diabetes Complications (2019) 33:675–81. doi: 10.1016/j.jdiacomp.2019.05.013
93. Kammer M, Heinzel A, Willency JA, Duffin KL, Mayer G, Simons K, et al. Integrative Analysis of Prognostic Biomarkers Derived From Multiomics Panels Helps Discrimination of Chronic Kidney Disease Trajectories in People With Type 2 Diabetes. Kidney Int (2019) 96:1381–8. doi: 10.1016/j.kint.2019.07.025
94. Umapathy D, Dornadula S, Krishnamoorthy E, Mariappanadar V, Viswanathan V, Ramkumar KM. Ykl-40: A Biomarker for Early Nephropathy in Type 2 Diabetic Patients and Its Association With Inflammatory Cytokines. Immunobiology (2018) 223:718–27. doi: 10.1016/j.imbio.2018.07.020
95. Schmidt IM, Hall IE, Kale S, Lee S, He CH, Lee Y, et al. Chitinase-Like Protein Brp-39/YKL-40 Modulates the Renal Response to Ischemic Injury and Predicts Delayed Allograft Function. J Am Soc Nephrol JASN (2013) 24:309–19. doi: 10.1681/ASN.2012060579
96. Lee JH, Kim SS, Kim IJ, Song SH, Kim YK, In Kim J, et al. Clinical Implication of Plasma and Urine YKL-40, as a Proinflammatory Biomarker, on Early Stage of Nephropathy in Type 2 Diabetic Patients. J Diabetes Complications (2012) 26:308–12. doi: 10.1016/j.jdiacomp.2012.04.012
97. Rathcke CN, Persson F, Tarnow L, Rossing P, Vestergaard H. Ykl-40, a Marker of Inflammation and Endothelial Dysfunction, Is Elevated in Patients With Type 1 Diabetes and Increases With Levels of Albuminuria. Diabetes Care (2009) 32:323–8. doi: 10.2337/dc08-1144
98. Al-Rubeaan K, Siddiqui K. Assessment of the Diagnostic Value of Different Biomarkers in Relation to Various Stages of Diabetic Nephropathy in Type 2 Diabetic Patients. Sci Rep (2017) 7:2684. doi: 10.1038/s41598-017-02421-9
99. Pena MJ, Heinzel A, Heinze G, Alkhalaf A, Bakker SJ, Nguyen TQ, et al. A Panel of Novel Biomarkers Representing Different Disease Pathways Improves Prediction of Renal Function Decline in Type 2 Diabetes. PloS One (2015) 10:e0120995. doi: 10.1371/journal.pone.0120995
100. Banba N, Nakamura T, Matsumura M, Kuroda H, Hattori Y, Kasai K. Possible Relationship of Monocyte Chemoattractant Protein-1 With Diabetic Nephropathy. Kidney Int (2000) 58:684–90. doi: 10.1046/j.1523-1755.2000.00214.x
101. Grandaliano G, Gesualdo L, Ranieri E, Monno R, Montinaro V, Marra F, et al. Monocyte Chemotactic Peptide-1 Expression in Acute and Chronic Human Nephritides: A Pathogenetic Role in Interstitial Monocytes Recruitment. J Am Soc Nephrol JASN (1996) 7:906–13. doi: 10.1681/ASN.V76906
102. Rovin BH, Doe N, Tan LC. Monocyte Chemoattractant Protein-1 Levels in Patients With Glomerular Disease. Am J Kidney Dis (1996) 27:640–6. doi: 10.1016/S0272-6386(96)90097-9
103. Morii T, Fujita H, Narita T, Shimotomai T, Fujishima H, Yoshioka N, et al. Association of Monocyte Chemoattractant Protein-1 With Renal Tubular Damage in Diabetic Nephropathy. J Diabetes its Complications (2003) 17:11–5. doi: 10.1016/S1056-8727(02)00176-9
104. Thrailkill KM, Nimmo T, Bunn RC, Cockrell GE, Moreau CS, Mackintosh S, et al. Microalbuminuria in Type 1 Diabetes Is Associated With Enhanced Excretion of the Endocytic Multiligand Receptors Megalin and Cubilin. Diabetes Care (2009) 32:1266–8. doi: 10.2337/dc09-0112
105. Ma J, Guan M, Bowden DW, Ng MC, Hicks PJ, Lea JP, et al. Association Analysis of the Cubilin (CUBN) and Megalin (Lrp2) Genes With ESRD in African Americans. Clin J Am Soc Nephrol CJASN (2016) 11:1034–43. doi: 10.2215/CJN.12971215
106. Bryniarski MA, Yee BM, Jaffri I, Chaves LD, Yu JA, Guan X, et al. Increased Megalin Expression in Early Type 2 Diabetes: Role of Insulin-Signaling Pathways. Am J Physiol Renal Physiol (2018) 315:F1191–207. doi: 10.1152/ajprenal.00210.2018
107. Mori KP, Yokoi H, Kasahara M, Imamaki H, Ishii A, Kuwabara T, et al. Increase of Total Nephron Albumin Filtration and Reabsorption in Diabetic Nephropathy. J Am Soc Nephrol JASN (2017) 28:278–89. doi: 10.1681/ASN.2015101168
108. Christensen EI, Kristoffersen IB, Grann B, Thomsen JS, Andreasen A, Nielsen R. A Well-Developed Endolysosomal System Reflects Protein Reabsorption in Segment 1 and 2 of Rat Proximal Tubules. Kidney Int (2020) 99:841–53. doi: 10.1016/j.kint.2020.11.015
109. Mori K, Nakao K. Neutrophil Gelatinase-Associated Lipocalin as the Real-Time Indicator of Active Kidney Damage. Kidney Int (2007) 71:967–70. doi: 10.1038/sj.ki.5002165
110. Bangstad HJ, Seljeflot I, Berg TJ, Hanssen KF. Renal Tubulointerstitial Expansion Is Associated With Endothelial Dysfunction and Inflammation in Type 1 Diabetes. Scandinavian J Clin Lab Invest (2009) 69:138–44. doi: 10.1080/00365510802444080
111. Malinda KM, Ponce L, Kleinman HK, Shackelton LM, Millis AJ. Gp38k, a Protein Synthesized by Vascular Smooth Muscle Cells, Stimulates Directional Migration of Human Umbilical Vein Endothelial Cells. Exp Cell Res (1999) 250:168–73. doi: 10.1006/excr.1999.4511
112. Segerer S, Nelson PJ, Schlöndorff D. Chemokines, Chemokine Receptors, and Renal Disease: From Basic Science to Pathophysiologic and Therapeutic Studies. J Am Soc Nephrol JASN (2000) 11:152–76. doi: 10.1681/ASN.V111152
113. Wada T, Furuichi K, Sakai N, Iwata Y, Yoshimoto K, Shimizu M, et al. Up-Regulation of Monocyte Chemoattractant Protein-1 in Tubulointerstitial Lesions of Human Diabetic Nephropathy. Kidney Int (2000) 58:1492–9. doi: 10.1046/j.1523-1755.2000.00311.x
114. Ibrahim S, Rashed L. Correlation of Urinary Monocyte Chemo-Attractant Protein-1 With Other Parameters of Renal Injury in Type-II Diabetes Mellitus. Saudi J Kidney Dis Transplant (2008) 19:911–7.
115. Christensen EI, Birn H. Megalin and Cubilin: Multifunctional Endocytic Receptors. Nat Rev Mol Cell Biol (2002) 3:256–66. doi: 10.1038/nrm778
116. Terryn S, Tanaka K, Lengelé JP, Olinger E, Dubois-Laforgue D, Garbay S, et al. Tubular Proteinuria in Patients With HNF1α Mutations: HNF1α Drives Endocytosis in the Proximal Tubule. Kidney Int (2016) 89:1075–89. doi: 10.1016/j.kint.2016.01.027
117. Alicic RZ, Cox EJ, Neumiller JJ, Tuttle KR. Incretin Drugs in Diabetic Kidney Disease: Biological Mechanisms and Clinical Evidence. Nat Rev Nephrol (2020) 17(4):227–44. doi: 10.1038/s41581-020-00367-2
118. Clark AJ, Parikh SM. Targeting Energy Pathways in Kidney Disease: The Roles of Sirtuins, AMPK, and PGC1α. Kidney Int (2020) 99:828–40. doi: 10.1016/j.kint.2020.09.037.
119. Packer M. Role of Impaired Nutrient and Oxygen Deprivation Signaling and Deficient Autophagic Flux in Diabetic Ckd Development: Implications for Understanding the Effects of Sodium-Glucose Cotransporter 2-Inhibitors. J Am Soc Nephrol JASN (2020) 31:907–19. doi: 10.1681/ASN.2020010010
120. Tonneijck L, Muskiet MHA, Blijdorp CJ, Smits MM, Twisk JW, Kramer MHH, et al. Renal Tubular Effects of Prolonged Therapy With the GLP-1 Receptor Agonist Lixisenatide in Patients With Type 2 Diabetes Mellitus. Am J Physiol Renal Physiol (2019) 316:F231–40. doi: 10.1152/ajprenal.00432.2018
121. Martins FL, Bailey MA, Girardi ACC. Endogenous Activation of Glucagon-Like Peptide-1 Receptor Contributes to Blood Pressure Control: Role of Proximal Tubule Na(+)/H(+) Exchanger Isoform 3, Renal Angiotensin II, and Insulin Sensitivity. Hypertension (2020) 76:839–48. doi: 10.1161/HYPERTENSIONAHA.120.14868
122. Scheen AJ. Pharmacokinetics of Dipeptidylpeptidase-4 Inhibitors. Diabetes Obes Metab (2010) 12:648–58. doi: 10.1111/j.1463-1326.2010.01212.x
123. Rosenstock J, Perkovic V, Johansen OE, Cooper ME, Kahn SE, Marx N, et al. Effect of Linagliptin vs Placebo on Major Cardiovascular Events in Adults With Type 2 Diabetes and High Cardiovascular and Renal Risk: The Carmelina Randomized Clinical Trial. Jama (2019) 321:69–79. doi: 10.1001/jama.2018.18269
124. Groop PH, Cooper ME, Perkovic V, Hocher B, Kanasaki K, Haneda M, et al. Linagliptin and Its Effects on Hyperglycaemia and Albuminuria in Patients With Type 2 Diabetes and Renal Dysfunction: The Randomized MARLINA-T2D Trial. Diabetes Obes Metab (2017) 19:1610–9. doi: 10.1111/dom.13041
125. Alicic RZ, Neumiller JJ. Sodium-Glucose Cotransporter 2 Inhibition and Diabetic Kidney Disease. Diabetes (2019) 68:248–57. doi: 10.2337/dbi18-0007
126. Hou YC, Zheng CM. Molecular Mechanisms of SGLT2 Inhibitor on Cardiorenal Protection. Int J Mol Sci 21 (2020) 21:7833. doi: 10.3390/ijms21217833
127. Castañeda AM, Dutra-Rufato A, Juarez MJ, Grosembacher L, Gonzalez-Torres H, Musso CG. Sodium-Glucose Cotransporter 2 Inhibitors (SGLT2i): Renal Implications. Int Urol Nephrology (2020) 53:291–9. doi: 10.1007/s11255-020-02585-w
128. Vallon V, Schwark JR, Richter K, Hropot M. Role of Na(+)/H(+) Exchanger NHE3 in Nephron Function: Micropuncture Studies With S3226, an Inhibitor of NHE3. Am J Physiol Renal Physiol (2000) 278:F375–9. doi: 10.1152/ajprenal.2000.278.3.F375
129. Pessoa TD, Campos LC, Carraro-Lacroix L, Girardi AC, Malnic G. Functional Role of Glucose Metabolism, Osmotic Stress, and Sodium-Glucose Cotransporter Isoform-Mediated Transport on Na+/H+ Exchanger Isoform 3 Activity in the Renal Proximal Tubule. J Am Soc Nephrol JASN (2014) 25:2028–39. doi: 10.1681/ASN.2013060588
130. Onishi A, Fu Y, Darshi M, Crespo-Masip M, Huang W, Song P, et al. Effect of Renal Tubule-Specific Knockdown of the Na(+)/H(+) Exchanger NHE3 in Akita Diabetic Mice. Am J Physiol Renal Physiol (2019) 317:F419–f434. doi: 10.1152/ajprenal.00497.2018
131. Silva Dos Santos D, Polidoro JZ, Borges-Júnior FA, Girardi ACC. Cardioprotection Conferred by Sodium-Glucose Cotransporter 2 Inhibitors: A Renal Proximal Tubule Perspective. Am J Physiol Cell Physiol (2020) 318:C328–c336. doi: 10.1152/ajpcell.00275.2019
132. Chung S, Kim S, Son M, Kim M, Koh ES, Shin SJ, et al. Empagliflozin Contributes to Polyuria Via Regulation of Sodium Transporters and Water Channels in Diabetic Rat Kidneys. Front Physiol (2019) 10:271. doi: 10.3389/fphys.2019.00271
133. Onishi A, Fu Y, Patel R, Darshi M, Crespo-Masip M, Huang W, et al. A Role for Tubular Na(+)/H(+) Exchanger NHE3 in the Natriuretic Effect of the SGLT2 Inhibitor Empagliflozin. Am J Physiol Renal Physiol (2020) 319:F712–f728. doi: 10.1152/ajprenal.00264.2020
134. Shin SJ, Chung S, Kim SJ, Lee EM, Yoo YH, Kim JW, et al. Effect of Sodium-Glucose Co-Transporter 2 Inhibitor, Dapagliflozin, on Renal Renin-Angiotensin System in an Animal Model of Type 2 Diabetes. PloS One (2016) 11:e0165703. doi: 10.1371/journal.pone.0165703
135. Ansary TM, Nakano D, Nishiyama A. Diuretic Effects of Sodium Glucose Cotransporter 2 Inhibitors and Their Influence on the Renin-Angiotensin System. Int J Mol Sci (2019) 20:629. doi: 10.3390/ijms20030629
136. Yoshimoto T, Furuki T, Kobori H, Miyakawa M, Imachi H, Murao K, et al. Effects of Sodium-Glucose Cotransporter 2 Inhibitors on Urinary Excretion of Intact and Total Angiotensinogen in Patients With Type 2 Diabetes. J Invest Med (2017) 65:1057–61. doi: 10.1136/jim-2017-000445
137. Cherney DZ, Perkins BA, Soleymanlou N, Maione M, Lai V, Lee A, et al. Renal Hemodynamic Effect of Sodium-Glucose Cotransporter 2 Inhibition in Patients With Type 1 Diabetes Mellitus. Circulation (2014) 129:587–97. doi: 10.1161/CIRCULATIONAHA.113.005081
138. Cherney DZ, Perkins BA, Soleymanlou N, Xiao F, Zimpelmann J, Woerle HJ, et al. Sodium Glucose Cotransport-2 Inhibition and Intrarenal RAS Activity in People With Type 1 Diabetes. Kidney Int (2014) 86:1057–8. doi: 10.1038/ki.2014.246
139. Chen L, LaRocque LM, Efe O, Wang J, Sands JM, Klein JD. Effect of Dapagliflozin Treatment on Fluid and Electrolyte Balance in Diabetic Rats. Am J Med Sci (2016) 352:517–23. doi: 10.1016/j.amjms.2016.08.015
140. Masuda T, Muto S, Fukuda K, Watanabe M, Ohara K, Koepsell H, et al. Osmotic Diuresis by SGLT2 Inhibition Stimulates Vasopressin-Induced Water Reabsorption to Maintain Body Fluid Volume. Physiol Rep (2020) 8:e14360. doi: 10.14814/phy2.14360
141. Nilsson LM, Zhang L, Bondar A, Svensson D, Wernerson A, Brismar H, et al. Prompt Apoptotic Response to High Glucose in SGLT-Expressing Renal Cells. Am J Physiol Renal Physiol (2019) 316:F1078–f1089. doi: 10.1152/ajprenal.00615.2018
142. Vallon V, Rose M, Gerasimova M, Satriano J, Platt KA, Koepsell H, et al. Knockout of Na-glucose Transporter SGLT2 Attenuates Hyperglycemia and Glomerular Hyperfiltration But Not Kidney Growth or Injury in Diabetes Mellitus. Am J Physiol Renal Physiol (2013) 304:F156–67. doi: 10.1152/ajprenal.00409.2012
143. Gembardt F, Bartaun C, Jarzebska N, Mayoux E, Todorov VT, Hohenstein B, et al. The SGLT2 Inhibitor Empagliflozin Ameliorates Early Features of Diabetic Nephropathy in BTBR Ob/Ob Type 2 Diabetic Mice With and Without Hypertension. Am J Physiol Renal Physiol (2014) 307:F317–25. doi: 10.1152/ajprenal.00145.2014
144. Oraby MA, El-Yamany MF, Safar MM, Assaf N, Ghoneim HA. Dapagliflozin Attenuates Early Markers of Diabetic Nephropathy in Fructose-Streptozotocin-Induced Diabetes in Rats. Biomed Pharmacother (2019) 109:910–20. doi: 10.1016/j.biopha.2018.10.100
145. Das NA, Carpenter AJ, Belenchia A, Aroor AR, Noda M, Siebenlist U, et al. Empagliflozin Reduces High Glucose-Induced Oxidative Stress and Mir-21-Dependent TRAF3IP2 Induction and RECK Suppression, and Inhibits Human Renal Proximal Tubular Epithelial Cell Migration and Epithelial-to-Mesenchymal Transition. Cell Signalling (2020) 68:109506. doi: 10.1016/j.cellsig.2019.109506
146. Wang XX, Levi J, Luo Y, Myakala K, Herman-Edelstein M, Qiu L, et al. Sglt2 Protein Expression Is Increased in Human Diabetic Nephropathy: Sglt2 PROTEIN Inhibition DECREASES Renal LIPID Accumulation, INFLAMMATION, and THE Development OF Nephropathy IN Diabetic Mice. J Biol Chem (2017) 292:5335–48. doi: 10.1074/jbc.M117.779520
147. Korbut AI, Taskaeva IS, Bgatova NP, Muraleva NA, Orlov NB, Dashkin MV, et al. Sglt2 Inhibitor Empagliflozin and DPP4 Inhibitor Linagliptin Reactivate Glomerular Autophagy in Db/Db Mice, a Model of Type 2 Diabetes. Int J Mol Sci (2020) 21:2987. doi: 10.3390/ijms21082987
148. Han E, Shin E, Kim G, Lee JY, Lee YH, Lee BW, et al. Combining SGLT2 Inhibition With a Thiazolidinedione Additively Attenuate the Very Early Phase of Diabetic Nephropathy Progression in Type 2 Diabetes Mellitus. Front Endocrinol (2018) 9:412. doi: 10.3389/fendo.2018.00412
149. Bonnet F, Scheen AJ. Effects of SGLT2 Inhibitors on Systemic and Tissue Low-Grade Inflammation: The Potential Contribution to Diabetes Complications and Cardiovascular Disease. Diabetes Metab (2018) 44:457–64. doi: 10.1016/j.diabet.2018.09.005
150. Xu C, Wang W, Zhong J, Lei F, Xu N, Zhang Y, et al. Canagliflozin Exerts Anti-Inflammatory Effects by Inhibiting Intracellular Glucose Metabolism and Promoting Autophagy in Immune Cells. Biochem Pharmacol (2018) 152:45–59. doi: 10.1016/j.bcp.2018.03.013
151. O’Neill J, Fasching A, Pihl L, Patinha D, Franzén S, Palm F. Acute SGLT Inhibition Normalizes O2 Tension in the Renal Cortex But Causes Hypoxia in the Renal Medulla in Anaesthetized Control and Diabetic Rats. Am J Physiol Renal Physiol (2015) 309:F227–34. doi: 10.1152/ajprenal.00689.2014
152. Sano M, Takei M, Shiraishi Y, Suzuki Y. Increased Hematocrit During Sodium-Glucose Cotransporter 2 Inhibitor Therapy Indicates Recovery of Tubulointerstitial Function in Diabetic Kidneys. J Clin Med Res (2016) 8:844–7. doi: 10.14740/jocmr2760w
153. Sano M. Inter-Organ Communication Pathway Manifested by Non-physiological Stress to the Kidney in Type Ii Diabetic Patients -Why Are Diabetic Patients Prone to Develop Heart Failure? Internal Med (Tokyo Japan) (2020) 59:1–5. doi: 10.2169/internalmedicine.2870-19
154. Lambers Heerspink HJ, de Zeeuw D, Wie L, Leslie B, List J. Dapagliflozin a Glucose-Regulating Drug With Diuretic Properties in Subjects With Type 2 Diabetes. Diabetes Obes Metab (2013) 15:853–62. doi: 10.1111/dom.12127
155. Cai T, Ke Q, Fang Y, Wen P, Chen H, Yuan Q, et al. Sodium-Glucose Cotransporter 2 Inhibition Suppresses HIF-1α-Mediated Metabolic Switch From Lipid Oxidation to Glycolysis in Kidney Tubule Cells of Diabetic Mice. Cell Death Dis (2020) 11:390. doi: 10.1038/s41419-020-2544-7
156. Shirakawa K, Sano M. Sodium-Glucose Co-Transporter 2 Inhibitors Correct Metabolic Maladaptation of Proximal Tubular Epithelial Cells in High-Glucose Conditions. Int J Mol Sci (2020) 21:7676. doi: 10.3390/ijms21207676
157. Tomita I, Kume S, Sugahara S, Osawa N, Yamahara K, Yasuda-Yamahara M, et al. Sglt2 Inhibition Mediates Protection From Diabetic Kidney Disease by Promoting Ketone Body-Induced mTORC1 Inhibition. Cell Metab (2020) 32:404–19.e6. doi: 10.1016/j.cmet.2020.06.020
158. Ndibalema AR, Kabuye D, Wen S. Empagliflozin Protects Against Proximal Renal Tubular Cell Injury Induced by High Glucose Via Regulation of Hypoxia-Inducible Factor 1-Alpha. Diabetes Metab Syndrome Obesity: Targets Ther (2020) 13:1953–67. doi: 10.2147/DMSO.S243170
159. Wanner C, Inzucchi SE, Lachin JM, Fitchett D, von Eynatten M, Mattheus M, et al. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. New Engl J Med (2016) 375:323–34. doi: 10.1056/NEJMoa1515920
160. Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. New Engl J Med (2017) 377:644–57. doi: 10.1056/NEJMoa1611925
161. Perkovic V, de Zeeuw D, Mahaffey KW, Fulcher G, Erondu N, Shaw W, et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes: Results From the CANVAS Program Randomised Clinical Trials. Lancet Diabetes Endocrinol (2018) 6:691–704. doi: 10.1016/S2213-8587(18)30141-4
162. Perkovic V, Jardine MJ, Neal B, Bompoint S, Heerspink HJL, Charytan DM, et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. New Engl J Med (2019) 380:2295–306. doi: 10.1056/NEJMoa1811744
163. Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. New Engl J Med (2019) 380:347–57. doi: 10.1056/NEJMoa1812389
164. Heerspink HJL, Stefánsson BV, Correa-Rotter R, Chertow GM, Greene T, Hou FF, et al. Dapagliflozin in Patients With Chronic Kidney Disease. New Engl J Med (2020) 383:1436–46. doi: 10.1056/NEJMoa2024816
165. Jensen J, Omar M, Kistorp C, Tuxen C, Gustafsson I, Køber L, et al. Effects of Empagliflozin on Estimated Extracellular Volume, Estimated Plasma Volume, and Measured Glomerular Filtration Rate in Patients With Heart Failure (Empire HF Renal): A Prespecified Substudy of a Double-Blind, Randomised, Placebo-Controlled Trial. Lancet Diabetes Endocrinol (2020) 9:106–16. doi: 10.1016/S2213-8587(20)30382-X
166. Cannon CP, Pratley R, Dagogo-Jack S, Mancuso J, Huyck S, Masiukiewicz U, et al. Cardiovascular Outcomes With Ertugliflozin in Type 2 Diabetes. N Engl J Med (2020) 383:1425–35. doi: 10.1056/NEJMoa2004967
167. Sarafidis P, Ortiz A, Ferro CJ, Halimi JM, Kreutz R, Mallamaci F, et al. Sodium–Glucose Co-Transporter-2 Inhibitors for Patients With Diabetic and Nondiabetic Chronic Kidney Disease: A New Era has Already Begun. J Hypertension (2021) 39:1090–7. doi: 10.1097/HJH.0000000000002776
168. Wanner C. Empa-Reg OUTCOME: The Nephrologist’s Point of View. Am J Cardiol (2017) 120:S59–67. doi: 10.1016/j.amjcard.2017.05.012
169. Sarafidis P, Ferro CJ, Morales E, Ortiz A, Malyszko J, Hojs R, et al. SGLT-2 Inhibitors and GLP-1 Receptor Agonists for Nephroprotection and Cardioprotection in Patients With Diabetes Mellitus and Chronic Kidney Disease. A Consensus Statement by the EURECA-m and the DIABESITY Working Groups of the ERA-EDTA. Nephrol Dial Transplant (2019) 34:208–30. doi: 10.1093/ndt/gfy407
170. Herrington WG, Preiss D, Haynes R, von Eynatten M, Staplin N, Hauske SJ, et al. The Potential for Improving Cardio-Renal Outcomes by Sodium-Glucose Co-Transporter-2 Inhibition in People With Chronic Kidney Disease: A Rationale for the EMPA-KIDNEY Study. Clin Kidney J (2018) 11:749–61. doi: 10.1093/ckj/sfy090
171. Davies MJ, D’Alessio DA, Fradkin J, Kernan WN, Mathieu C. Management of Hyperglycemia in Type 2 Diabetes, 2018. A Consensus Report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (Easd). Diabetes Care (2018) 41:2669–701. doi: 10.2337/dci18-0033
172. Cosentino F, Grant PJ, Aboyans V, Bailey CJ, Ceriello A, Delgado V, et al. 2019 ESC Guidelines on Diabetes, Pre-Diabetes, and Cardiovascular Diseases Developed in Collaboration With the EASD. Eur Heart J (2020) 41:255–323. doi: 10.1093/eurheartj/ehz486
173. Buse JB, Wexler DJ. 2019 Update to: Management of Hyperglycaemia in Type 2 Diabetes, 2018. A Consensus Report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (Easd). Diabetologia (2020) 63:221–8. doi: 10.1007/s00125-019-05039-w
174. Marx N, Davies MJ, Grant PJ, Mathieu C, Petrie JR, Cosentino F, et al. Guideline Recommendations and the Positioning of Newer Drugs in Type 2 Diabetes Care. Lancet Diabetes Endocrinol (2021) 9:46–52. doi: 10.1016/S2213-8587(20)30343-0
175. Kidney Disease: Improving Global Outcomes (KDIGO) Diabetes Work GroupKdigo 2020 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease. Kidney Int (2020) 98:S1–s115. doi: 10.1016/j.kint.2020.06.019
Keywords: renal tubular dysfunction, tubular biomarkers, sodium-glucose cotransporter-2, diabetic kidney disease, therapeutic strategies
Citation: Duan S, Lu F, Song D, Zhang C, Zhang B, Xing C and Yuan Y (2021) Current Challenges and Future Perspectives of Renal Tubular Dysfunction in Diabetic Kidney Disease. Front. Endocrinol. 12:661185. doi: 10.3389/fendo.2021.661185
Received: 30 January 2021; Accepted: 21 May 2021;
Published: 10 June 2021.
Edited by:
Martina Guthoff, Tübingen University Hospital, GermanyReviewed by:
Sanjay Kalra, Independent Researcher, Karnal, IndiaDilip Sharma, National Institute of Pharmaceutical Education and Research, Ahmedabad, India
Copyright © 2021 Duan, Lu, Song, Zhang, Zhang, Xing and Yuan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanggang Yuan, eWd5dWFuQG5qbXUuZWR1LmNu; Changying Xing, Y3l4aW5nNjJAMTI2LmNvbQ==