 Chelsie Villanueva-Hayes
Chelsie Villanueva-Hayes Steven J. Millership
Steven J. Millership- Section of Cell Biology and Functional Genomics, Department of Metabolism, Digestion and Reproduction, Imperial College London, London, United Kingdom
Beta cell failure lies at the centre of the aetiology and pathogenesis of type 2 diabetes and the epigenetic control of the expression of critical beta cell genes appears to play a major role in this decline. One such group of epigenetically-controlled genes, termed ‘imprinted’ genes, are characterised by transgenerational monoallelic expression due to differential allelic DNA methylation and play key functional roles within beta cells. Here, we review the evidence for this functional importance of imprinted genes in beta cells as well as their nutritional regulation by the diet and their altered methylation and/or expression in rodent models of diabetes and in type 2 diabetic islets. We also discuss imprinted genes in the context of the next generation, where dietary overnutrition in the parents can lead to their deregulation in the offspring, alongside beta cell dysfunction and defective glucose handling. Both the modulation of imprinted gene expression and the likelihood of developing type 2 diabetes in adulthood are susceptible to the impact of nutritional status in early life. Imprinted loci, therefore, represent an excellent opportunity with which to assess epigenomic changes in beta cells due to the diet in both the current and next generation.
Introduction
The term “epigenetics” has been redefined frequently since the 1940s, and therefore, we will use this term to define “the study of molecules and mechanisms that can perpetuate alternative gene activity states in the context of the same DNA sequence” (1). Epigenetic mechanisms control genetic information whilst unaltering the underlying DNA sequence (2) and include DNA methylation, chromatin remodelling, histone modifications, and gene regulation by non-coding RNA. Moreover, these epigenetic pathways modulate expression of target genes, and therefore, have a significant role in the establishment, maintenance and dynamic changes in the cell (3). In mammalian genomes, DNA methylation usually refers to methylation of 5’-cytosines within CpG dinucleotides (4) and is the major pathway controlling several epigenetic phenomena, including genomic imprinting, X chromosome inactivation and repression of transposable elements (3). CpG methylation is carried out by a family of DNA methyltransferases (DNMTs) [reviewed in (5, 6)] at key regulatory genomic regions, e.g. promoters, and is associated with activation and repression of gene expression (7–13).
The epigenetic phenomenon of genomic imprinting results in monoallelic and parent-of-origin-specific gene expression in a select group of genes (14, 15). The discovery of inconsistencies in the level of DNA methylation at the same locus between paternal and maternal alleles revealed the involvement of epigenetic alterations in the regulation and conservation of monoallelic silencing in genomic imprinting (16–18). Nuclear transplantation experiments showed that embryos containing one set of parental chromosomes (uniparental disomy; UPD) did not survive beyond early gestation, demonstrating that the parental genomes were not functionally equal (19–23). Imprinting is a highly conserved process in mammals and to date, approximately 150 imprinted genes are known in mice and around 100 in humans. Imprinted genes are generally found in clusters throughout the genome and have been found to be regulated by discrete DNA elements called imprinting control regions (ICR), which are a differentially methylated region (DMR) (14, 17). Parental allele-specific imprinting marks are preserved during the lifespan (24–26), [reviewed in (27, 28)] and are reset and re-established transgenerationally (29). Secondary ‘somatic’ imprints can also be established post-fertilisation and are believed to reinforce the allele-specific gene repression at imprinted loci (30).
Imprinted Genes Play Key Functional Roles in Pancreatic Beta Cells
Imprinted genes are highly expressed in metabolic systems where they play a central role in controlling growth, development and metabolism (31). Pancreatic beta cells express a number of imprinted genes that are critical for beta cell function. Demonstrating the importance of maintaining the correct dosage of imprinted gene expression are the presence of several human imprinting syndromes (Prader-Willi, Angelman, Beckwith-Wiedemann and Silver-Russell) that result in severe developmental and metabolic abnormalities, due to altered imprinted gene expression at imprinted loci (15, 32, 33) [reviewed in (34)], including a transient form of neonatal diabetes caused by paternal UPD of the 6q24 region (35–37). Here, we discuss several imprinted genes with known function in beta cells (Figure 1), primarily through their characterisation in cellular or mutant mouse models. As a significant decline in beta cell function often coincides with a reduction in whole-body glucose homeostasis, we also discuss imprinted genes in the context of their nutritional regulation by the diet and the evidence for altered imprinted gene expression in type 2 diabetes (T2D). Finally, we explore what is known regarding susceptibility to diabetes in the next generation via epigenetic changes in the offspring due to parental under- or overnutrition.
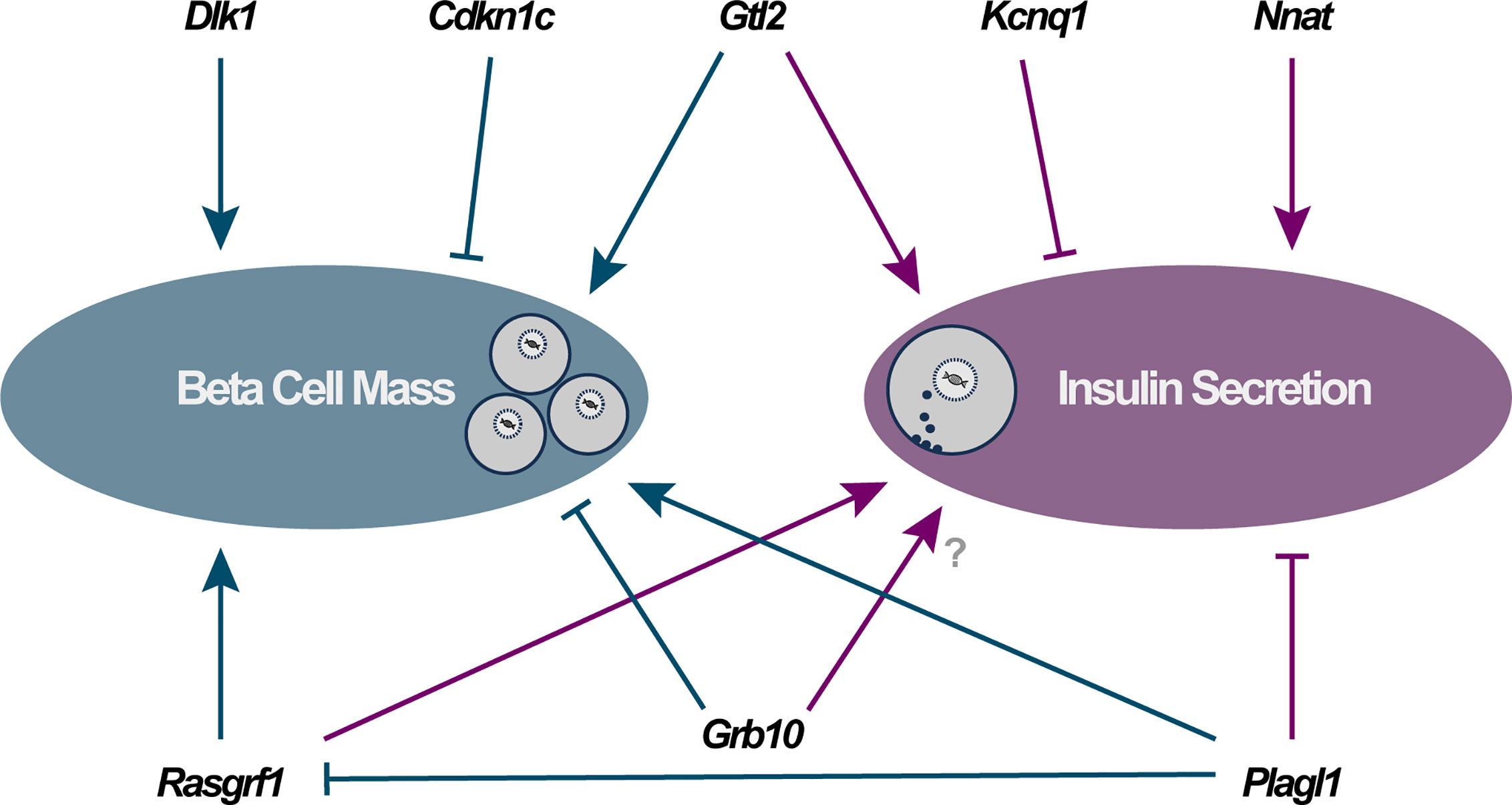
Figure 1 Direct functional importance of imprinted genes in pancreatic beta cells falls into two major categories: modulators of beta cell mass (via changes in cellular proliferation, apoptosis and/or differentiation) and alterations to specific components of the insulin secretory apparatus. Arrowheads and blocked lines represent stimulatory and inhibitory actions on these cellular pathways, respectively.
Nnat
Nnat is a paternally expressed imprinted gene which is highly enriched in neuroendocrine systems, including pancreatic beta cells (38–40). Early in vitro work reported a potential role in glucose-stimulated insulin secretion (GSIS) in two different stable mouse pancreatic beta cell lines and expression of both known isoforms of neuronatin, Nnat-α and Nnat-β, found predominantly in the endoplasmic reticulum (ER), were increased after acute stimulation with high glucose (41, 42). It has been postulated that NNAT plays a role in the regulation of the intracellular calcium dynamics in several cell types (42–44); however, primary islets from Nnat-null mutant mice displayed unaltered Ca2+ signalling (45). Both global and beta cell-specific Nnat deficient mice demonstrate impaired GSIS due to reduced beta cell insulin content (45). Furthermore, NNAT was shown to interact with the signal peptidase complex (SPC) and facilitates the translocation of nascent preproinsulin into the ER (45). Nnat expression is also regulated by nutrient status in pancreatic beta cells both in vitro and in vivo (42, 45) and in rodent models of diabetes (42, 45, 46).
Plagl1
Plagl1 (also known as Zac1) is a zinc finger transcription factor that is implicated in anti-proliferative activities such as the regulation of cell cycle arrest and apoptosis (47–51). PLAGL1 is a paternally expressed imprinted gene on chromosome 6q24, a region where paternal duplication or loss of methylation at the PLAGL1 DMR causes transient neonatal diabetes mellitus (TNDM) locus owing to PLAGL1 overexpression (35–37, 49, 52, 53). PLAGL1 overexpression appears to reduce beta cell mass in neonates via its apoptotic and/or anti-proliferative capabilities (51). This is potentially due to PLAGL1-mediated modulation of PPARG and PACAP1-R expression, two key regulators of beta cell proliferation and insulin secretion, respectively (54). Additionally, high glucose levels reduced Plagl1 expression in rodent beta cell lines and in primary mouse islets (55). Plagl1 overexpression in several rodent beta cell lines impaired insulin secretion (56) and overexpression in mice recapitulates the early-onset diabetes observed in TNDM patients (57). Furthermore, induced Plagl1 expression resulted in a decrease of glucose-stimulated proinsulin biosynthesis, despite an increase in insulin mRNA (55) suggesting that Plagl1 can also negatively regulate the translational apparatus and ultimately the efficiency of insulin mRNA translation.
Rasgrf1
Rasgrf1 is a paternally expressed imprinted gene that functions as a guanine nucleotide exchange factor for Ras GTPases (58). Pancreatic islets from the db/db diabetic mouse model had a significant reduction in Rasgrf1 expression (59). Mice with deletion of Rasgrf1 showed reduced beta cell proliferation and neogenesis, and thus decreased beta cell mass, resulting in hypoinsulinaemia and impaired glucose tolerance (60). Interestingly, it was found that Rasgrf1 is a direct Plagl1 target gene in multiple rodent beta cell lines and in mouse islets (56). Moreover, a two-fold overexpression of Plagl1 in beta cells resulted in repression of Rasgrf1 expression and impaired insulin secretion, which could be rescued by restoring Rasgrf1 expression (56).
Cdkn1c and Kcnq1
Cdkn1c is expressed solely from the maternal allele (61, 62) and regulates cell proliferation and differentiation (63–65). Indeed, suppression of CDKN1C expression via viral delivery of shRNAs into isolated human islets provoked a 3-fold increase in beta cell proliferation and was sufficient to rescue hyperglycaemia when transplanted into diabetic mice (66). Importantly, newly-replicated beta cells retained the characteristics of mature beta cells, with expression of key functional markers (insulin, PDX1 and NKX6.1) and a robust response to high glucose in terms of calcium dynamics (66). Also found at the CDKN1C-containing 11p15/ICR2 imprinted region, the KCNQ1 gene encodes a voltage-gated potassium channel, and overexpression of this protein in mouse MIN6 beta cells causes impaired insulin secretion (67). Furthermore, administration of a KCNQ1 inhibitor enhanced insulin secretion in isolated islets and in mice (68). Genetic disruption of the Kcnq1 gene in mice caused a reduction in beta cell mass and subsequent glucose intolerance, although this was likely due to a subsequent upregulation of Cdkn1c expression (69).
The incidence of hypoglycaemia is approximately 50% in patients with Beckwith-Wiedemann Syndrome (caused by genetic disruption at 11p15) and is associated with beta cell hyperplasia and subsequent hyperinsulinaemia in affected individuals (70–74) [reviewed in (75)]. In many Beckwith-Wiedemann Syndrome patients, the ICR2 region at 11p15 is hypomethylated on both alleles (76), causing loss of expression of CDKN1C (77) and is linked with an increase in proliferation of beta cells (66, 76, 78, 79). Furthermore, targeted demethylation of ICR2 using a methylcytosine dioxygenase 1 (TET1)-based approach repressed CDKN1C expression in human islets, with subsequent increased levels of Ki-67 and significant beta cell proliferation (80). Interestingly, a point mutation in the CDKN1C gene was found in a family with several features consistent with IMAGe syndrome (81), a growth and developmental disorder similar to Beckwith-Wiedemann Syndrome (82, 83), as well as an early-adult-onset form of diabetes (81). It remains to be determined whether IMAGe patients also develop diabetes at a later stage in life; however, these findings suggest that mutation of CDKN1C alone may be sufficient to drive a monogenic form of diabetes.
Dlk1 and Gtl2/MEG3
The imprinting region on human chromosome 14q32 carries a cluster of imprinted genes, including the paternally expressed gene DLK1 and the maternally expressed long non-coding RNA (lncRNA) MEG3 (Gtl2 in rodents) (84–86). Overexpression of Dlk1 in mice improves glucose tolerance and whole-body insulin sensitivity (87), potentially by promoting proliferation and differentiation of beta cells (88). Transgenic mice overexpressing Dlk1 in pancreatic beta cells demonstrate an increase in islet mass with higher proportion of larger islets, whereas Dlk1 null mice showed the opposite trend (89). Transgenic mice, therefore, had increased insulin secretion and improved glucose tolerance (89), although conversely, a different group has demonstrated increased proliferation (and size) of pancreatic islets upon Dlk1 ablation in mice (90). At the same locus, constituent deletion of Gtl2 and its associated promoter in mice led to severe parent-of-origin-dependent peri-/postnatal developmental defects and early lethality (91). Increased methylation at the Gtl2 promoter DMR in the mouse beta cell line, βTC6, resulted in decreased Gtl2 expression and increased beta cell sensitivity to cytokine-mediated oxidative stress (92). Gtl2 has also been shown to maintain the expression of Mafa, a critical beta cell transcription factor that positively influences insulin synthesis and secretion (93). Gtl2 expression is also decreased in islets in the db/db diabetic mouse model and its expression is glucose-regulated in both the MIN6 mouse beta cell line and in primary mouse islets (94). Knockdown of Gtl2 using siRNA in both MIN6 beta cells and primary islets impaired insulin synthesis and secretion and caused beta cell apoptosis (94). Furthermore, knockdown of Gtl2 in vivo resulted in impaired glucose tolerance and insulin secretion in mice, likely due to a reduction in beta cell mass (94).
Grb10
Grb10 functions via intracellular signalling pathways regulating growth and metabolism (95) and has been implicated in binding to, and negatively regulating signals from, the insulin receptor (IR) and insulin-like growth factor 1 receptor (IGF1R) (96, 97). Differential transcriptome analysis of mouse islets from diabetes-resistant (ob/ob) vs diabetes-sensitive (New Zealand obese, NZO) mouse strains revealed a number of human diabetes candidate genes, including Grb10 (98). Several studies have also uncovered a role for Grb10 in the regulation of glucose handling; however, this has required picking apart the relative contribution from Grb10 expression in the beta cell and other peripheral, insulin target, tissues (99–101). Disruption of Grb10 expression in mice is associated with postnatal overgrowth and enhanced insulin secretion and sensitivity and improved glucose tolerance (99–103). Conversely, Grb10 overexpression in mice caused postnatal growth retardation, accompanied by severe insulin resistance and worsened glucose intolerance (104, 105). Moreover, pancreatic-specific Grb10 deletion resulted in increased beta cell proliferation and a subsequent increase in insulin content and secretion, and improved glucose tolerance (106). However, Prokopenko et al. found that shRNA-mediated knockdown of GRB10 in isolated human islets led to a reduction in insulin secretion (107).
Evidence of Altered Imprinted Gene Expression in T2D
T2D is predominantly a consequence of beta cell failure (108, 109) and the majority of the genes associated with T2D pathogenesis encode modulators of beta cell function (110). The studies described above demonstrate that careful control of imprinted gene expression is required in order to maintain normal beta cell function and glucose homeostasis. Interestingly, several studies have shown that the expression and methylation patterns of several imprinted genes show notable differences in T2D vs non-diabetic islets (86, 107, 111–114) (summarised in Table 1). Specifically, Dlk1 expression was found to be elevated in beta cells from patients with T2D (113) and has been posited as a biomarker for identifying women at high risk of developing diabetes (119). MEG3 expression was also found to be downregulated in islets from T2D donors as a result of hypermethylation at the MEG3 DMR (86). Additionally, a study consisting of patients with gestational diabetes mellitus showed that DNA methylation at the MEG3-DMR was positively correlated with maternal glycaemia and foetal growth (120). Using a genome-wide association study (GWAS) based on assessment of GSIS, Prokopenko et al. (107) demonstrated that inheriting variants of GRB10 were associated with reduced GSIS and an increased risk of T2D when inherited from the father, but improved glycaemia when inherited from the mother, which may be due to the different parent-of-origin tissue expression patterns of Grb10 (121, 122). Several studies have also identified single nucleotide polymorphisms (SNPs) at multiple imprinted loci associated with T2D and impaired glucose tolerance including those at the CDKN1C locus (amongst others) (115), KCNQ1 (116, 117) and GRB10 (118) (Table 1). Indeed, several imprinted genes including Plagl1, Dlk1, Gtl2 and Nnat were differentially expressed between a ‘responder’ subclone of mouse MIN6 beta cells (based on their sustained GSIS capacity) vs. ‘non-responder’ beta cells (123). In the above scenarios, where the diabetic state is associated with altered DNA methylation and misexpression of imprinted genes in pancreatic beta cells, a major question centres around the temporal nature of these events. Are the observed changes in DNA methylation at key regulatory genomic regions acting as a primary driver of imprinted gene misexpression (and therefore functional changes) in these beta cells? Or do changes in nutrient status lead to misexpression of imprinted genes via other mechanisms (e.g. nutrient-specific transcription factors) that are later reinforced by long term epigenetic changes such as DNA methylation? A recent study has shown that even mild hyperglycaemia in rodents is sufficient to evoke deregulation of critical genes for beta cell identity, including Nnat (46), and it will be interesting to further explore this model in terms of epigenetic alterations longitudinally over periods of chronic, albeit mild, hyperglycaemia.

Table 1 Imprinted gene candidates for conferring susceptibility to type 2 diabetes.
Metabolic Programming and T2D in the Next Generation
It is becoming increasingly apparent that individuals can be predisposed to adult-onset metabolic diseases, such as T2D, due to the direct effect of their nutritional status in early development, either in utero or in the first few years of life (124, 125). The developmental origin of health and disease (DOHaD) hypothesis, first put forward by Barker (126, 127) suggested that exposure to environmental factors during vulnerable periods of foetal development or early childhood might increase an individual’s risk to metabolic disease in later life; this has since been linked with possible mediation by epigenetic factors (1, 128–133). Indeed, intrauterine growth restriction (IUGR) or parental overnutrition in rodents and humans results in impaired glucose homeostasis in adulthood (134–140). One of the first examples of this phenomenon was the finding that children born during the Dutch Winter Famine of 1944-45, who were exposed to maternal undernutrition in utero, went on to develop diabetes in later life (141) with evidence for altered DNA methylation at imprinted loci (142, 143). Moreover, it has been suggested that the transfer of epigenetic changes to the next generation are not limited to exposure to the developing foetus (i.e. in utero nutrition), but also directly to gametic cells, with evidence for altered DNA methylation at imprinted loci in oocytes from diabetic female mice (144). Furthermore, chronic paternal high-fat diet feeding, prior to conception, in rodents leads to impaired insulin secretion and glucose tolerance in their offspring, including altered expression of imprinted genes (145), indicating that both these epigenetic changes and beta cell dysfunction can be passed on to the next generation via the male germline. Similar findings have also been documented in children who were conceived by obese fathers, with evidence for altered expression (and methylation) at imprinted loci (IGF2, PEG3, MEG3, PLAGL1 and NNAT) in F1 offspring (146, 147) (Figure 2).
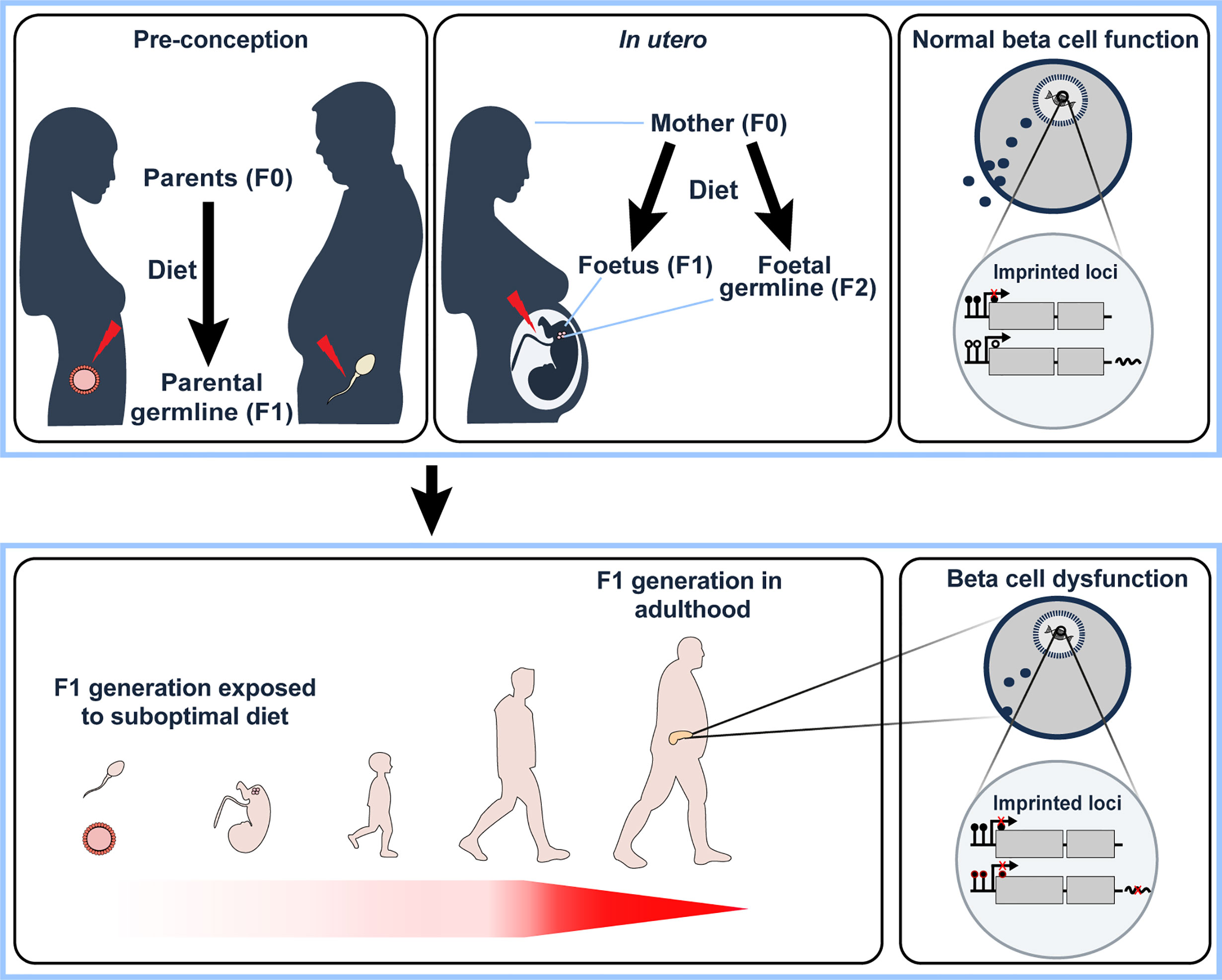
Figure 2 Under- or overnutrition influences imprinted gene expression not only in the individual (F0 generation) but also in the next generation (F1). This may occur indirectly via pre-conceptual changes in the germ cells (upper left panel) or via direct exposure in utero (and potentially the subsequent (F2) generation via direct exposure of foetal germ cells to nutritional alterations in utero, upper middle panel). F1 offspring that have been directly or indirectly exposed to a suboptimal nutritional status in early development have been shown to develop beta cell dysfunction in adulthood (lower left panel). In the F0 generation, overnutrition has been shown to alter imprinted gene expression in pancreatic beta cells via changes in DNA methylation at key regulatory genomic regions. We therefore hypothesise that changes in nutritional status affects the monoallelic expression of imprinted genes that is observed in ‘normal’ conditions (upper right panel) via alterations to CpG methylation, with an example illustrated in the lower right panel (closed circles – methylated CpGs, open circles – unmethylated CpGs). With their known functional role in beta cells, deregulation of imprinted gene expression via the diet would, therefore, lead to beta cell dysfunction. It will be interesting to determine the relative contribution of imprinted gene deregulation on the observed beta cell dysfunction in the F1 generation due to nutritional status in the F0 generation.
Conclusions and Final Perspectives
The expression of imprinted genes is heavily influenced by epigenetic mechanisms such as DNA methylation. Multiple lines of evidence demonstrate that imprinted genes are critical for beta cell function and that they are nutritionally regulated in these cells. Misexpression of imprinted genes is associated with both rodent models of diabetes and T2D islets, with evidence that altered methylation and/or expression at these loci by the diet can be passed on to the next generation either in utero or via gametic cells. Using imprinted gene loci, with their well understood epigenetic control and functional importance in beta cells, will help us to understand the type and genomic distribution of epigenetic marks that are established in response to overnutrition. Indeed, the plasticity of the epigenome enables both a flexibility in response to environmental factors (e.g. diet) and also a potential target for epigenetic-modifying drugs that may be used to enhance insulin secretion. Epigenetic editing at imprinted loci has already been shown to be a promising tool to promote beta cell expansion (80) and epidrugs directed as specific molecular targets e.g. methyltransferases, that preserve beta cell functional identity during periods of suboptimal nutritional status, represent an exciting therapeutic possibility for T2D. We therefore need a better understanding of the diet-induced epigenomic changes responsible for misexpression of imprinted (and non-imprinted) genes that negatively impact beta cell function. This would enable us to test the ability of specific epidrugs to target and inhibit these pathways in beta cells in the face of nutrient excess. Indeed, if modification of epigenetic status at imprinted loci is proven to be a reliable biomarker for reduced beta cell function, this approach could be employed to assess the effect of specific dietary components/macromolecule content on insulin secretion in model systems. A key question for the future is also whether any epigenomic changes observed at the beta cell level are preserved in other cells (e.g. blood cells, subcutaneous adipose tissue) that can easily be sampled from patients. In this scenario, we could harness these molecular alterations to better predict future diabetic outcomes in patients and intervene in disease progression prior to long term hyperglycaemia and beta cell failure.
Author Contributions
All authors contributed to the article and approved the submitted version. Writing – Original Draft Preparation, CV-H and SM. Writing – Review and Editing, CV-H and SM.
Funding
SM was supported by a Wellcome Trust/Imperial College ISSF Fellowship (PS3619_WREC).
Conflict of Interest
SM has received a speaker honorarium from the Society for Endocrinology.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are extremely grateful to the Frontiers Fee Support Programme for providing a portion of the publishing fees for this article.
References
1. Cavalli G, Heard E. Advances in Epigenetics Link Genetics to the Environment and Disease. Nature (2019) 571:489–99. doi: 10.1038/s41586-019-1411-0
2. Cedar H, Bergman Y. Linking DNA Methylation and Histone Modification: Patterns and Paradigms. Nat Rev Genet (2009) 10:295–304. doi: 10.1038/nrg2540
3. Adalsteinsson BT, Ferguson-Smith AC. Epigenetic Control of the Genome-Lessons From Genomic Imprinting. Genes (Basel) (2014) 5:635–55. doi: 10.3390/genes5030635
4. Li E, Zhang Y. DNA Methylation in Mammals. Cold Spring Harb Perspect Biol (2014) 6:a019133. doi: 10.1101/cshperspect.a019133
5. Goll MG, Bestor TH. Eukaryotic Cytosine Methyltransferases. Annu Rev Biochem (2005) 74:481–514. doi: 10.1146/annurev.biochem.74.010904.153721
6. Lyko F. The DNA Methyltransferase Family: A Versatile Toolkit for Epigenetic Regulation. Nat Rev Genet (2018) 19:81–92. doi: 10.1038/nrg.2017.80
7. Bird AP, Taggart MH, Smith BA. Methylated and Unmethylated DNA Compartments in the Sea Urchin Genome. Cell (1979) 17:889–901. doi: 10.1016/0092-8674(79)90329-5
8. Bird A, Taggart M, Macleod D. Loss of rDNA Methylation Accompanies the Onset of Ribosomal Gene Activity in Early Development of X. Laevis. Cell (1981) 26:381–90. doi: 10.1016/0092-8674(81)90207-5
9. Kuo MT, Mandel JL, Chambon P. DNA Methylation: Correlation With Dnase I Sensitivity of Chicken Ovalbumin and Conalbumin Chromatin. Nucleic Acids Res (1979) 7:2105–13. doi: 10.1093/nar/7.8.2105
10. Mandel JL, Chambon P. DNA Methylation: Organ Specific Variations in the Methylation Pattern Within and Around Ovalbumin and Other Chicken Genes. Nucleic Acids Res (1979) 7:2081–103. doi: 10.1093/nar/7.8.2081
11. McGhee JD, Ginder GD. Specific DNA Methylation Sites in the Vicinity of the Chicken Beta-Globin Genes. Nature (1979) 280:419–20. doi: 10.1038/280419a0
12. Trowbridge JJ, Orkin SH. DNA Methylation in Adult Stem Cells: New Insights Into Self-Renewal. Epigenetics (2010) 5:189–93. doi: 10.4161/epi.5.3.11374
13. van der Ploeg LH, Flavell RA. DNA Methylation in the Human Gamma Delta Beta-Globin Locus in Erythroid and Nonerythroid Tissues. Cell (1980) 19:947–58. doi: 10.1016/0092-8674(80)90086-0
14. Ferguson-Smith AC. Genomic Imprinting: The Emergence of an Epigenetic Paradigm. Nat Rev Genet (2011) 12:565–75. doi: 10.1038/nrg3032
15. Monk D, Mackay DJG, Eggermann T, Maher ER, Riccio A. Genomic Imprinting Disorders: Lessons on How Genome, Epigenome and Environment Interact. Nat Rev Genet (2019) 20:235–48. doi: 10.1038/s41576-018-0092-0
16. Ferguson-Smith AC, Sasaki H, Cattanach BM, Surani MA. Parental-Origin-Specific Epigenetic Modification of the Mouse H19 Gene. Nature (1993) 362:751–5. doi: 10.1038/362751a0
17. Reik W, Walter J. Genomic Imprinting: Parental Influence on the Genome. Nat Rev Genet (2001) 2:21–32. doi: 10.1038/35047554
18. Stoger R, Kubicka P, Liu CG, Kafri T, Razin A, Cedar H, et al. Maternal-Specific Methylation of the Imprinted Mouse Igf2r Locus Identifies the Expressed Locus as Carrying the Imprinting Signal. Cell (1993) 73:61–71. doi: 10.1016/0092-8674(93)90160-R
19. McGrath J, Solter D. Nuclear Transplantation in Mouse Embryos. J Exp Zool (1983) 228:355–62. doi: 10.1002/jez.1402280218
20. McGrath J, Solter D. Nuclear Transplantation in the Mouse Embryo by Microsurgery and Cell Fusion. Science (1983) 220:1300–2. doi: 10.1126/science.6857250
21. McGrath J, Solter D. Completion of Mouse Embryogenesis Requires Both the Maternal and Paternal Genomes. Cell (1984) 37:179–83. doi: 10.1016/0092-8674(84)90313-1
22. Surani MA, Barton SC. Development of Gynogenetic Eggs in the Mouse: Implications for Parthenogenetic Embryos. Science (1983) 222:1034–6. doi: 10.1126/science.6648518
23. Surani MA, Barton SC, Norris ML. Development of Reconstituted Mouse Eggs Suggests Imprinting of the Genome During Gametogenesis. Nature (1984) 308:548–50. doi: 10.1038/308548a0
24. Bourc’his D, Xu GL, Lin CS, Bollman B, Bestor TH. Dnmt3L and the Establishment of Maternal Genomic Imprints. Science (2001) 294:2536–9. doi: 10.1126/science.1065848
25. Bourc’his D, Bestor TH. Meiotic Catastrophe and Retrotransposon Reactivation in Male Germ Cells Lacking Dnmt3L. Nature (2004) 431:96–9. doi: 10.1038/nature02886
26. Kaneda M, Okano M, Hata K, Sado T, Tsujimoto N, Li E, et al. Essential Role for De Novo DNA Methyltransferase Dnmt3a in Paternal and Maternal Imprinting. Nature (2004) 429:900–3. doi: 10.1038/nature02633
27. Arnaud P. Genomic Imprinting in Germ Cells: Imprints are Under Control. Reproduction (2010) 140:411–23. doi: 10.1530/REP-10-0173
28. Wood AJ, Oakey RJ. Genomic Imprinting in Mammals: Emerging Themes and Established Theories. PloS Genet (2006) 2:e147. doi: 10.1371/journal.pgen.0020147
29. Cedar H, Bergman Y. Programming of DNA Methylation Patterns. Annu Rev Biochem (2012) 81:97–117. doi: 10.1146/annurev-biochem-052610-091920
30. John RM, Lefebvre L. Developmental Regulation of Somatic Imprints. Differentiation (2011) 81:270–80. doi: 10.1016/j.diff.2011.01.007
31. Millership SJ, Van de Pette M, Withers DJ. Genomic Imprinting and its Effects on Postnatal Growth and Adult Metabolism. Cell Mol Life Sci (2019) 76:4009–21. doi: 10.1007/s00018-019-03197-z
32. Brucato N, DeLisi LE, Fisher SE, Francks C. Hypomethylation of the Paternally Inherited LRRTM1 Promoter Linked to Schizophrenia. Am J Med Genet B Neuropsychiatr Genet (2014) 165B:555–63. doi: 10.1002/ajmg.b.32258
33. Eggermann T, Soellner L, Buiting K, Kotzot D. Mosaicism and Uniparental Disomy in Prenatal Diagnosis. Trends Mol Med (2015) 21:77–87. doi: 10.1016/j.molmed.2014.11.010
34. Eggermann T, Perez de Nanclares G, Maher ER, Temple IK, Tumer Z, Monk D, et al. Imprinting Disorders: A Group of Congenital Disorders With Overlapping Patterns of Molecular Changes Affecting Imprinted Loci. Clin Epigenet (2015) 7:123. doi: 10.1186/s13148-015-0143-8
35. Gardner RJ, Mackay DJ, Mungall AJ, Polychronakos C, Siebert R, Shield JP, et al. An Imprinted Locus Associated With Transient Neonatal Diabetes Mellitus. Hum Mol Genet (2000) 9:589–96. doi: 10.1093/hmg/9.4.589
36. Temple IK, James RS, Crolla JA, Sitch FL, Jacobs PA, Howell WM, et al. An Imprinted Gene(s) for Diabetes? Nat Genet (1995) 9:110–2. doi: 10.1038/ng0295-110
37. Temple IK, Gardner RJ, Robinson DO, Kibirige MS, Ferguson AW, Baum JD, et al. Further Evidence for an Imprinted Gene for Neonatal Diabetes Localised to Chromosome 6q22-Q23. Hum Mol Genet (1996) 5:1117–21. doi: 10.1093/hmg/5.8.1117
38. Arava Y, Adamsky K, Ezerzer C, Ablamunits V, Walker MD. Specific Gene Expression in Pancreatic Beta-Cells: Cloning and Characterization of Differentially Expressed Genes. Diabetes (1999) 48:552–6. doi: 10.2337/diabetes.48.3.552
39. Joseph RM. Neuronatin Gene: Imprinted and Misfolded: Studies in Lafora Disease, Diabetes and Cancer may Implicate NNAT-aggregates as a Common Downstream Participant in Neuronal Loss. Genomics (2014) 103:183–8. doi: 10.1016/j.ygeno.2013.12.001
40. Rolletschek A, Schroeder IS, Schulz H, Hummel O, Huebner N, Wobus AM. Characterization of Mouse Embryonic Stem Cell Differentiation Into the Pancreatic Lineage In Vitro by Transcriptional Profiling, Quantitative RT-PCR and Immunocytochemistry. Int J Dev Biol (2010) 54:41–54. doi: 10.1387/ijdb.082694ar
41. Chu K, Tsai MJ. Neuronatin, a Downstream Target of BETA2/NeuroD1 in the Pancreas, is Involved in Glucose-Mediated Insulin Secretion. Diabetes (2005) 54:1064–73. doi: 10.2337/diabetes.54.4.1064
42. Joe MK, Lee HJ, Suh YH, Han KL, Lim JH, Song J, et al. Crucial Roles of Neuronatin in Insulin Secretion and High Glucose-Induced Apoptosis in Pancreatic Beta-Cells. Cell Signal (2008) 20:907–15. doi: 10.1016/j.cellsig.2008.01.005
43. Lin HH, Bell E, Uwanogho D, Perfect LW, Noristani H, Bates TJ, et al. Neuronatin Promotes Neural Lineage in ESCs Via Ca(2+) Signaling. Stem Cells (2010) 28:1950–60. doi: 10.1002/stem.530
44. Suh YH, Kim WH, Moon C, Hong YH, Eun SY, Lim JH, et al. Ectopic Expression of Neuronatin Potentiates Adipogenesis Through Enhanced Phosphorylation of cAMP-response Element-Binding Protein in 3T3-L1 Cells. Biochem Biophys Res Commun (2005) 337:481–9. doi: 10.1016/j.bbrc.2005.09.078
45. Millership SJ, Da Silva Xavier G, Choudhury AI, Bertazzo S, Chabosseau P, Pedroni SM, et al. Neuronatin Regulates Pancreatic Beta Cell Insulin Content and Secretion. J Clin Invest (2018) 128:3369–81. doi: 10.1172/JCI120115
46. Ebrahimi AG, Hollister-Lock J, Sullivan BA, Tsuchida R, Bonner-Weir S, Weir GC. Beta Cell Identity Changes With Mild Hyperglycemia: Implications for Function, Growth, and Vulnerability. Mol Metab (2020) 35:100959. doi: 10.1016/j.molmet.2020.02.002
47. Pagotto U, Arzberger T, Ciani E, Lezoualc’h F, Pilon C, Journot L, et al. Inhibition of Zac1, a New Gene Differentially Expressed in the Anterior Pituitary, Increases Cell Proliferation. Endocrinology (1999) 140:987–96. doi: 10.1210/endo.140.2.6532
48. Spengler D, Villalba M, Hoffmann A, Pantaloni C, Houssami S, Bockaert J, et al. Regulation of Apoptosis and Cell Cycle Arrest by Zac1, a Novel Zinc Finger Protein Expressed in the Pituitary Gland and the Brain. EMBO J (1997) 16:2814–25. doi: 10.1093/emboj/16.10.2814
49. Temple IK, Gardner RJ, Mackay DJ, Barber JC, Robinson DO, Shield JP. Transient Neonatal Diabetes: Widening the Understanding of the Etiopathogenesis of Diabetes. Diabetes (2000) 49:1359–66. doi: 10.2337/diabetes.49.8.1359
50. Varrault A, Ciani E, Apiou F, Bilanges B, Hoffmann A, Pantaloni C, et al. hZAC Encodes a Zinc Finger Protein With Antiproliferative Properties and Maps to a Chromosomal Region Frequently Lost in Cancer. Proc Natl Acad Sci USA (1998) 95:8835–40. doi: 10.1073/pnas.95.15.8835
51. Vega-Benedetti AF, Saucedo C, Zavattari P, Vanni R, Zugaza JL, Parada LA. PLAGL1: An Important Player in Diverse Pathological Processes. J Appl Genet (2017) 58:71–8. doi: 10.1007/s13353-016-0355-4
52. Kamiya M, Judson H, Okazaki Y, Kusakabe M, Muramatsu M, Takada S, et al. The Cell Cycle Control Gene ZAC/PLAGL1 is Imprinted–a Strong Candidate Gene for Transient Neonatal Diabetes. Hum Mol Genet (2000) 9:453–60. doi: 10.1093/hmg/9.3.453
53. Varrault A, Bilanges B, Mackay DJ, Basyuk E, Ahr B, Fernandez C, et al. Characterization of the Methylation-Sensitive Promoter of the Imprinted ZAC Gene Supports its Role in Transient Neonatal Diabetes Mellitus. J Biol Chem (2001) 276:18653–6. doi: 10.1074/jbc.C100095200
54. Barz T, Hoffmann A, Panhuysen M, Spengler D. Peroxisome Proliferator-Activated Receptor Gamma is a Zac Target Gene Mediating Zac Antiproliferation. Cancer Res (2006) 66:11975–82. doi: 10.1158/0008-5472.CAN-06-1529
55. Du X, Ounissi-Benkalha H, Loder MK, Rutter GA, Polychronakos C. Overexpression of ZAC Impairs Glucose-Stimulated Insulin Translation and Secretion in Clonal Pancreatic Beta-Cells. Diabetes Metab Res Rev (2012) 28:645–53. doi: 10.1002/dmrr.2325
56. Hoffmann A, Spengler D. Transient Neonatal Diabetes Mellitus Gene Zac1 Impairs Insulin Secretion in Mice Through Rasgrf1. Mol Cell Biol (2012) 32:2549–60. doi: 10.1128/MCB.06637-11
57. Ma D, Shield JP, Dean W, Leclerc I, Knauf C, Burcelin RR, et al. Impaired Glucose Homeostasis in Transgenic Mice Expressing the Human Transient Neonatal Diabetes Mellitus Locus, TNDM. J Clin Invest (2004) 114:339–48. doi: 10.1172/JCI200419876
58. Plass C, Shibata H, Kalcheva I, Mullins L, Kotelevtseva N, Mullins J, et al. Identification of Grf1 on Mouse Chromosome 9 as an Imprinted Gene by RLGS-M. Nat Genet (1996) 14:106–9. doi: 10.1038/ng0996-106
59. Neelankal John A, Ram R, Jiang FX. Rna-Seq Analysis of Islets to Characterise the Dedifferentiation in Type 2 Diabetes Model Mice Db/Db. Endocr Pathol (2018) 29:207–21. doi: 10.1007/s12022-018-9523-x
60. Font de Mora J, Esteban LM, Burks DJ, Nunez A, Garces C, Garcia-Barrado MJ, et al. Ras-GRF1 Signaling is Required for Normal Beta-Cell Development and Glucose Homeostasis. EMBO J (2003) 22:3039–49. doi: 10.1093/emboj/cdg280
61. Hatada I, Inazawa J, Abe T, Nakayama M, Kaneko Y, Jinno Y, et al. Genomic Imprinting of Human p57KIP2 and its Reduced Expression in Wilms’ Tumors. Hum Mol Genet (1996) 5:783–8. doi: 10.1093/hmg/5.6.783
62. Hatada I, Mukai T. Genomic Imprinting of p57KIP2, a Cyclin-Dependent Kinase Inhibitor, in Mouse. Nat Genet (1995) 11:204–6. doi: 10.1038/ng1095-204
63. Lee MH, Reynisdottir I, Massague J. Cloning of p57KIP2, a Cyclin-Dependent Kinase Inhibitor With Unique Domain Structure and Tissue Distribution. Genes Dev (1995) 9:639–49. doi: 10.1101/gad.9.6.639
64. Matsuoka S, Edwards MC, Bai C, Parker S, Zhang P, Baldini A, et al. p57KIP2, a Structurally Distinct Member of the P21cip1 Cdk Inhibitor Family, is a Candidate Tumor Suppressor Gene. Genes Dev (1995) 9:650–62. doi: 10.1101/gad.9.6.650
65. Yan Y, Frisen J, Lee MH, Massague J, Barbacid M. Ablation of the CDK Inhibitor p57Kip2 Results in Increased Apoptosis and Delayed Differentiation During Mouse Development. Genes Dev (1997) 11:973–83. doi: 10.1101/gad.11.8.973
66. Avrahami D, Li C, Yu M, Jiao Y, Zhang J, Naji A, et al. Targeting the Cell Cycle Inhibitor p57Kip2 Promotes Adult Human Beta Cell Replication. J Clin Invest (2014) 124:670–4. doi: 10.1172/JCI69519
67. Yamagata K, Senokuchi T, Lu M, Takemoto M, Fazlul Karim M, Go C, et al. Voltage-Gated K+ Channel KCNQ1 Regulates Insulin Secretion in MIN6 Beta-Cell Line. Biochem Biophys Res Commun (2011) 407:620–5. doi: 10.1016/j.bbrc.2011.03.083
68. Liu L, Wang F, Lu H, Ren X, Zou J. Chromanol 293B, an Inhibitor of KCNQ1 Channels, Enhances Glucose-Stimulated Insulin Secretion and Increases Glucagon-Like Peptide-1 Level in Mice. Islets (2014) 6:e962386. doi: 10.4161/19382014.2014.962386
69. Asahara S, Etoh H, Inoue H, Teruyama K, Shibutani Y, Ihara Y, et al. Paternal Allelic Mutation At the Kcnq1 Locus Reduces Pancreatic Beta-Cell Mass by Epigenetic Modification of Cdkn1c. Proc Natl Acad Sci USA (2015) 112:8332–7. doi: 10.1073/pnas.1422104112
70. Elliott M, Bayly R, Cole T, Temple IK, Maher ER. Clinical Features and Natural History of Beckwith-Wiedemann Syndrome: Presentation of 74 New Cases. Clin Genet (1994) 46:168–74. doi: 10.1111/j.1399-0004.1994.tb04219.x
71. Lteif AN, Schwenk WF. Hypoglycemia in Infants and Children. Endocrinol Metab Clin North Am (1999) 28:619–46, vii. doi: 10.1016/S0889-8529(05)70091-8
72. Martinez y Martinez R. Clinical Features in the Wiedemann-Beckwith Syndrome. Clin Genet (1996) 50:272–4. doi: 10.1111/j.1399-0004.1996.tb02644.x
73. Shepherd RM, Cosgrove KE, O’Brien RE, Barnes PD, Ammala C, Dunne MJ. Hyperinsulinism of Infancy: Towards an Understanding of Unregulated Insulin Release. European Network for Research Into Hyperinsulinism in Infancy. Arch Dis Child Fetal Neonatal Ed (2000) 82:F87–97. doi: 10.1136/fn.82.2.F87
74. Stanley CA. Hyperinsulinism in Infants and Children. Pediatr Clin North Am (1997) 44:363–74. doi: 10.1016/S0031-3955(05)70481-8
75. DeBaun MR, King AA, White N. Hypoglycemia in Beckwith-Wiedemann Syndrome. Semin Perinatol (2000) 24:164–71. doi: 10.1053/sp.2000.6366
76. Calvello M, Tabano S, Colapietro P, Maitz S, Pansa A, Augello C, et al. Quantitative DNA Methylation Analysis Improves Epigenotype-Phenotype Correlations in Beckwith-Wiedemann Syndrome. Epigenetics (2013) 8:1053–60. doi: 10.4161/epi.25812
77. Diaz-Meyer N, Day CD, Khatod K, Maher ER, Cooper W, Reik W, et al. Silencing of CDKN1C (p57KIP2) is Associated With Hypomethylation At KvDMR1 in Beckwith-Wiedemann Syndrome. J Med Genet (2003) 40:797–801. doi: 10.1136/jmg.40.11.797
78. Borges KS, Arboleda VA, Vilain E. Mutations in the PCNA-binding Site of CDKN1C Inhibit Cell Proliferation by Impairing the Entry Into S Phase. Cell Div (2015) 10:2. doi: 10.1186/s13008-015-0008-8
79. Ueberberg S, Tannapfel A, Schenker P, Viebahn R, Uhl W, Schneider S, et al. Differential Expression of Cell-Cycle Regulators in Human Beta-Cells Derived From Insulinoma Tissue. Metabolism (2016) 65:736–46. doi: 10.1016/j.metabol.2016.02.007
80. Ou K, Yu M, Moss NG, Wang YJ, Wang AW, Nguyen SC, et al. Targeted Demethylation At the CDKN1C/p57 Locus Induces Human Beta Cell Replication. J Clin Invest (2019) 129:209–14. doi: 10.1172/JCI99170
81. Kerns SL, Guevara-Aguirre J, Andrew S, Geng J, Guevara C, Guevara-Aguirre M, et al. A Novel Variant in CDKN1C is Associated With Intrauterine Growth Restriction, Short Stature, and Early-Adulthood-Onset Diabetes. J Clin Endocrinol Metab (2014) 99:E2117–22. doi: 10.1210/jc.2014-1949
82. Arboleda VA, Lee H, Parnaik R, Fleming A, Banerjee A, Ferraz-de-Souza B, et al. Mutations in the PCNA-binding Domain of CDKN1C Cause IMAGe Syndrome. Nat Genet (2012) 44:788–92. doi: 10.1038/ng.2275
83. Hamajima N, Johmura Y, Suzuki S, Nakanishi M, Saitoh S. Increased Protein Stability of CDKN1C Causes a Gain-of-Function Phenotype in Patients With IMAGe Syndrome. PloS One (2013) 8:e75137. doi: 10.1371/journal.pone.0075137
84. Cavaille J, Seitz H, Paulsen M, Ferguson-Smith AC, Bachellerie JP. Identification of Tandemly-Repeated C/D snoRNA Genes At the Imprinted Human 14q32 Domain Reminiscent of Those At the Prader-Willi/Angelman Syndrome Region. Hum Mol Genet (2002) 11:1527–38. doi: 10.1093/hmg/11.13.1527
85. da Rocha ST, Edwards CA, Ito M, Ogata T, Ferguson-Smith AC. Genomic Imprinting At the Mammalian Dlk1-Dio3 Domain. Trends Genet (2008) 24:306–16. doi: 10.1016/j.tig.2008.03.011
86. Kameswaran V, Bramswig NC, McKenna LB, Penn M, Schug J, Hand NJ, et al. Epigenetic Regulation of the DLK1-MEG3 microRNA Cluster in Human Type 2 Diabetic Islets. Cell Metab (2014) 19:135–45. doi: 10.1016/j.cmet.2013.11.016
87. Charalambous M, Da Rocha ST, Radford EJ, Medina-Gomez G, Curran S, Pinnock SB, et al. DLK1/PREF1 Regulates Nutrient Metabolism and Protects From Steatosis. Proc Natl Acad Sci USA (2014) 111:16088–93. doi: 10.1073/pnas.1406119111
88. Rhee M, Lee SH, Kim JW, Ham DS, Park HS, Yang HK, et al. Preadipocyte Factor 1 Induces Pancreatic Ductal Cell Differentiation Into Insulin-Producing Cells. Sci Rep (2016) 6:23960. doi: 10.1038/srep23960
89. Wang Y, Lee K, Moon YS, Ahmadian M, Kim KH, Roder K, et al. Overexpression of Pref-1 in Pancreatic Islet Beta-Cells in Mice Causes Hyperinsulinemia With Increased Islet Mass and Insulin Secretion. Biochem Biophys Res Commun (2015) 461:630–5. doi: 10.1016/j.bbrc.2015.04.078
90. Abdallah BM, Ditzel N, Laborda J, Karsenty G, Kassem M. Dlk1 Regulates Whole-Body Glucose Metabolism: A Negative Feedback Regulation of the Osteocalcin-Insulin Loop. Diabetes (2015) 64:3069–80. doi: 10.2337/db14-1642
91. Takahashi N, Okamoto A, Kobayashi R, Shirai M, Obata Y, Ogawa H, et al. Deletion of Gtl2, Imprinted non-Coding RNA, With its Differentially Methylated Region Induces Lethal Parent-Origin-Dependent Defects in Mice. Hum Mol Genet (2009) 18:1879–88. doi: 10.1093/hmg/ddp108
92. Kameswaran V, Golson ML, Ramos-Rodriguez M, Ou K, Wang YJ, Zhang J, et al. The Dysregulation of the DLK1-MEG3 Locus in Islets From Patients With Type 2 Diabetes is Mimicked by Targeted Epimutation of Its Promoter With Tale-DNMT Constructs. Diabetes (2018) 67:1807–15. doi: 10.2337/db17-0682
93. Wang N, Zhu Y, Xie M, Wang L, Jin F, Li Y, et al. Long Noncoding Rna Meg3 Regulates Mafa Expression in Mouse Beta Cells by Inactivating Rad21, Smc3 or Sin3alpha. Cell Physiol Biochem (2018) 45:2031–43. doi: 10.1159/000487983
94. You L, Wang N, Yin D, Wang L, Jin F, Zhu Y, et al. Downregulation of Long Noncoding RNA Meg3 Affects Insulin Synthesis and Secretion in Mouse Pancreatic Beta Cells. J Cell Physiol (2016) 231:852–62. doi: 10.1002/jcp.25175
95. Holt LJ, Siddle K. Grb10 and Grb14: Enigmatic Regulators of Insulin Action–and More? Biochem J (2005) 388:393–406. doi: 10.1042/BJ20050216
96. Dufresne AM, Smith RJ. The Adapter Protein GRB10 is an Endogenous Negative Regulator of Insulin-Like Growth Factor Signaling. Endocrinology (2005) 146:4399–409. doi: 10.1210/en.2005-0150
97. O’Neill TJ, Rose DW, Pillay TS, Hotta K, Olefsky JM, Gustafson TA. Interaction of a GRB-IR Splice Variant (a Human GRB10 Homolog) With the Insulin and Insulin-Like Growth Factor I Receptors. Evidence for a Role in Mitogenic Signaling. J Biol Chem (1996) 271:22506–13. doi: 10.1074/jbc.271.37.22506
98. Kluth O, Matzke D, Schulze G, Schwenk RW, Joost HG, Schurmann A. Differential Transcriptome Analysis of Diabetes-Resistant and -Sensitive Mouse Islets Reveals Significant Overlap With Human Diabetes Susceptibility Genes. Diabetes (2014) 63:4230–8. doi: 10.2337/db14-0425
99. Holt LJ, Lyons RJ, Ryan AS, Beale SM, Ward A, Cooney GJ, et al. Dual Ablation of Grb10 and Grb14 in Mice Reveals Their Combined Role in Regulation of Insulin Signaling and Glucose Homeostasis. Mol Endocrinol (2009) 23:1406–14. doi: 10.1210/me.2008-0386
100. Smith FM, Holt LJ, Garfield AS, Charalambous M, Koumanov F, Perry M, et al. Mice With a Disruption of the Imprinted Grb10 Gene Exhibit Altered Body Composition, Glucose Homeostasis, and Insulin Signaling During Postnatal Life. Mol Cell Biol (2007) 27:5871–86. doi: 10.1128/MCB.02087-06
101. Wang L, Balas B, Christ-Roberts CY, Kim RY, Ramos FJ, Kikani CK, et al. Peripheral Disruption of the Grb10 Gene Enhances Insulin Signaling and Sensitivity In Vivo. Mol Cell Biol (2007) 27:6497–505. doi: 10.1128/MCB.00679-07
102. Charalambous M, Smith FM, Bennett WR, Crew TE, Mackenzie F, Ward A. Disruption of the Imprinted Grb10 Gene Leads to Disproportionate Overgrowth by an Igf2-independent Mechanism. Proc Natl Acad Sci USA (2003) 100:8292–7. doi: 10.1073/pnas.1532175100
103. Shiura H, Nakamura K, Hikichi T, Hino T, Oda K, Suzuki-Migishima R, et al. Paternal Deletion of Meg1/Grb10 DMR Causes Maternalization of the Meg1/Grb10 Cluster in Mouse Proximal Chromosome 11 Leading to Severe Pre- and Postnatal Growth Retardation. Hum Mol Genet (2009) 18:1424–38. doi: 10.1093/hmg/ddp049
104. Shiura H, Miyoshi N, Konishi A, Wakisaka-Saito N, Suzuki R, Muguruma K, et al. Meg1/Grb10 Overexpression Causes Postnatal Growth Retardation and Insulin Resistance Via Negative Modulation of the IGF1R and IR Cascades. Biochem Biophys Res Commun (2005) 329:909–16. doi: 10.1016/j.bbrc.2005.02.047
105. Yamamoto Y, Ishino F, Kaneko-Ishino T, Shiura H, Uchio-Yamada K, Matsuda J, et al. Type 2 Diabetes Mellitus in a non-Obese Mouse Model Induced by Meg1/Grb10 Overexpression. Exp Anim (2008) 57:385–95. doi: 10.1538/expanim.57.385
106. Zhang J, Zhang N, Liu M, Li X, Zhou L, Huang W, et al. Disruption of Growth Factor Receptor-Binding Protein 10 in the Pancreas Enhances Beta-Cell Proliferation and Protects Mice From Streptozotocin-Induced Beta-Cell Apoptosis. Diabetes (2012) 61:3189–98. doi: 10.2337/db12-0249
107. Prokopenko I, Poon W, Magi R, Prasad BR, Salehi SA, Almgren P, et al. A Central Role for GRB10 in Regulation of Islet Function in Man. PloS Genet (2014) 10:e1004235. doi: 10.1371/journal.pgen.1004235
108. Gilbert ER, Liu D. Epigenetics: The Missing Link to Understanding Beta-Cell Dysfunction in the Pathogenesis of Type 2 Diabetes. Epigenetics (2012) 7:841–52. doi: 10.4161/epi.21238
109. Rutter GA, Pullen TJ, Hodson DJ, Martinez-Sanchez A. Pancreatic Beta-Cell Identity, Glucose Sensing and the Control of Insulin Secretion. Biochem J (2015) 466:203–18. doi: 10.1042/BJ20141384
110. Petrie JR, Pearson ER, Sutherland C. Implications of Genome Wide Association Studies for the Understanding of Type 2 Diabetes Pathophysiology. Biochem Pharmacol (2011) 81:471–7. doi: 10.1016/j.bcp.2010.11.010
111. Dayeh T, Volkov P, Salo S, Hall E, Nilsson E, Olsson AH, et al. Genome-Wide DNA Methylation Analysis of Human Pancreatic Islets From Type 2 Diabetic and non-Diabetic Donors Identifies Candidate Genes That Influence Insulin Secretion. PloS Genet (2014) 10:e1004160. doi: 10.1371/journal.pgen.1004160
112. Fadista J, Vikman P, Laakso EO, Mollet IG, Esguerra JL, Taneera J, et al. Global Genomic and Transcriptomic Analysis of Human Pancreatic Islets Reveals Novel Genes Influencing Glucose Metabolism. Proc Natl Acad Sci USA (2014) 111:13924–9. doi: 10.1073/pnas.1402665111
113. Lawlor N, George J, Bolisetty M, Kursawe R, Sun L, Sivakamasundari V, et al. Single-Cell Transcriptomes Identify Human Islet Cell Signatures and Reveal Cell-Type-Specific Expression Changes in Type 2 Diabetes. Genome Res (2017) 27:208–22. doi: 10.1101/gr.212720.116
114. Olsson AH, Volkov P, Bacos K, Dayeh T, Hall E, Nilsson EA, et al. Genome-Wide Associations Between Genetic and Epigenetic Variation Influence mRNA Expression and Insulin Secretion in Human Pancreatic Islets. PloS Genet (2014) 10:e1004735. doi: 10.1371/journal.pgen.1004735
115. Kong A, Steinthorsdottir V, Masson G, Thorleifsson G, Sulem P, Besenbacher S, et al. Parental Origin of Sequence Variants Associated With Complex Diseases. Nature (2009) 462:868–74. doi: 10.1038/nature08625
116. Yasuda K, Miyake K, Horikawa Y, Hara K, Osawa H, Furuta H, et al. Variants in KCNQ1 are Associated With Susceptibility to Type 2 Diabetes Mellitus. Nat Genet (2008) 40:1092–7. doi: 10.1038/ng.207
117. Unoki H, Takahashi A, Kawaguchi T, Hara K, Horikoshi M, Andersen G, et al. Snps in KCNQ1 are Associated With Susceptibility to Type 2 Diabetes in East Asian and European Populations. Nat Genet (2008) 40:1098–102. doi: 10.1038/ng.208
118. Rampersaud E, Damcott CM, Fu M, Shen H, McArdle P, Shi X, et al. Identification of Novel Candidate Genes for Type 2 Diabetes From a Genome-Wide Association Scan in the Old Order Amish: Evidence for Replication From Diabetes-Related Quantitative Traits and From Independent Populations. Diabetes (2007) 56:3053–62. doi: 10.2337/db07-0457
119. Lee SH, Rhee M, Yang HK, Ha HS, Lee JH, Kwon HS, et al. Serum Preadipocyte Factor 1 Concentrations and Risk of Developing Diabetes: A Nested Case-Control Study. Diabetes Med (2016) 33:631–8. doi: 10.1111/dme.12871
120. Cheng C, Jiang Y, Yan T, Chen Y, Yang M, Lv M, et al. Placental MEG3-DMR Methylation Profile is Associated With Maternal Glucose Concentration and Newborn Birth Weight. J Diabetes Investig (2020). doi: 10.1111/jdi.13432
121. Blagitko N, Mergenthaler S, Schulz U, Wollmann HA, Craigen W, Eggermann T, et al. Human GRB10 is Imprinted and Expressed From the Paternal and Maternal Allele in a Highly Tissue- and Isoform-Specific Fashion. Hum Mol Genet (2000) 9:1587–95. doi: 10.1093/hmg/9.11.1587
122. Garfield AS, Cowley M, Smith FM, Moorwood K, Stewart-Cox JE, Gilroy K, et al. Distinct Physiological and Behavioural Functions for Parental Alleles of Imprinted Grb10. Nature (2011) 469:534–8. doi: 10.1038/nature09651
123. Yamato E, Tashiro F, Miyazaki J. Microarray Analysis of Novel Candidate Genes Responsible for Glucose-Stimulated Insulin Secretion in Mouse Pancreatic Beta Cell Line MIN6. PloS One (2013) 8:e61211. doi: 10.1371/journal.pone.0061211
124. Aerts L, Van Assche FA. Animal Evidence for the Transgenerational Development of Diabetes Mellitus. Int J Biochem Cell Biol (2006) 38:894–903. doi: 10.1016/j.biocel.2005.07.006
125. Fernandez-Twinn DS, Hjort L, Novakovic B, Ozanne SE, Saffery R. Intrauterine Programming of Obesity and Type 2 Diabetes. Diabetologia (2019) 62:1789–801. doi: 10.1007/s00125-019-4951-9
126. Barker DJ. The Fetal and Infant Origins of Adult Disease. BMJ (1990) 301:1111. doi: 10.1136/bmj.301.6761.1111
127. Hales CN, Barker DJ. Type 2 (non-Insulin-Dependent) Diabetes Mellitus: The Thrifty Phenotype Hypothesis. Diabetologia (1992) 35:595–601. doi: 10.1007/BF00400248
128. Calkins K, Devaskar SU. Fetal Origins of Adult Disease. Curr Probl Pediatr Adolesc Health Care (2011) 41:158–76. doi: 10.1016/j.cppeds.201101.001
129. Feinberg AP. Phenotypic Plasticity and the Epigenetics of Human Disease. Nature (2007) 447:433–40. doi: 10.1038/nature05919
130. Gluckman PD, Hanson MA, Buklijas T, Low FM, Beedle AS. Epigenetic Mechanisms That Underpin Metabolic and Cardiovascular Diseases. Nat Rev Endocrinol (2009) 5:401–8. doi: 10.1038/nrendo.2009.102
131. Greco EA, Lenzi A, Migliaccio S, Gessani S. Epigenetic Modifications Induced by Nutrients in Early Life Phases: Gender Differences in Metabolic Alteration in Adulthood. Front Genet (2019) 10:795. doi: 10.3389/fgene.2019.00795
132. Heindel JJ, Vandenberg LN. Developmental Origins of Health and Disease: A Paradigm for Understanding Disease Cause and Prevention. Curr Opin Pediatr (2015) 27:248–53. doi: 10.1097/MOP.0000000000000191
133. Jirtle RL, Skinner MK. Environmental Epigenomics and Disease Susceptibility. Nat Rev Genet (2007) 8:253–62. doi: 10.1038/nrg2045
134. Boney CM, Verma A, Tucker R, Vohr BR. Metabolic Syndrome in Childhood: Association With Birth Weight, Maternal Obesity, and Gestational Diabetes Mellitus. Pediatrics (2005) 115:e290–296. doi: 10.1542/peds.2004-1808
135. Forsen T, Eriksson J, Tuomilehto J, Reunanen A, Osmond C, Barker D. The Fetal and Childhood Growth of Persons Who Develop Type 2 Diabetes. Ann Intern Med (2000) 133:176–82. doi: 10.7326/0003-4819-133-3-200008010-00008
136. Hales CN, Barker DJ, Clark PM, Cox LJ, Fall C, Osmond C, et al. Fetal and Infant Growth and Impaired Glucose Tolerance At Age 64. BMJ (1991) 303:1019–22. doi: 10.1136/bmj.303.6809.1019
137. Kaati G, Bygren LO, Edvinsson S. Cardiovascular and Diabetes Mortality Determined by Nutrition During Parents’ and Grandparents’ Slow Growth Period. Eur J Hum Genet (2002) 10:682–8. doi: 10.1038/sj.ejhg.5200859
138. Ravelli AC, van der Meulen JH, Michels RP, Osmond C, Barker DJ, Hales CN, et al. Glucose Tolerance in Adults After Prenatal Exposure to Famine. Lancet (1998) 351:173–7. doi: 10.1016/S0140-6736(97)07244-9
139. Simmons RA, Templeton LJ, Gertz SJ. Intrauterine Growth Retardation Leads to the Development of Type 2 Diabetes in the Rat. Diabetes (2001) 50:2279–86. doi: 10.2337/diabetes.50.10.2279
140. Taylor PD, McConnell J, Khan IY, Holemans K, Lawrence KM, Asare-Anane H, et al. Impaired Glucose Homeostasis and Mitochondrial Abnormalities in Offspring of Rats Fed a Fat-Rich Diet in Pregnancy. Am J Physiol Regul Integr Comp Physiol (2005) 288:R134–9. doi: 10.1152/ajpregu.00355.2004
141. Ravelli GP, Stein ZA, Susser MW. Obesity in Young Men After Famine Exposure In Utero and Early Infancy. N Engl J Med (1976) 295:349–53. doi: 10.1056/NEJM197608122950701
142. Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, et al. Persistent Epigenetic Differences Associated With Prenatal Exposure to Famine in Humans. Proc Natl Acad Sci USA (2008) 105:17046–9. doi: 10.1073/pnas.0806560105
143. Tobi EW, Lumey LH, Talens RP, Kremer D, Putter H, Stein AD, et al. DNA Methylation Differences After Exposure to Prenatal Famine are Common and Timing- and Sex-Specific. Hum Mol Genet (2009) 18:4046–53. doi: 10.1093/hmg/ddp353
144. Ge ZJ, Liang XW, Guo L, Liang QX, Luo SM, Wang YP, et al. Maternal Diabetes Causes Alterations of DNA Methylation Statuses of Some Imprinted Genes in Murine Oocytes. Biol Reprod (2013) 88:117. doi: 10.1095/biolreprod.112.105981
145. Ng SF, Lin RC, Laybutt DR, Barres R, Owens JA, Morris MJ. Chronic High-Fat Diet in Fathers Programs Beta-Cell Dysfunction in Female Rat Offspring. Nature (2010) 467:963–6. doi: 10.1038/nature09491
146. Soubry A, Murphy SK, Wang F, Huang Z, Vidal AC, Fuemmeler BF, et al. Newborns of Obese Parents Have Altered DNA Methylation Patterns At Imprinted Genes. Int J Obes (Lond) (2015) 39:650–7. doi: 10.1038/ijo.2013.193
Keywords: genomic imprinting, methylation, beta cell function, type 2 diabetes, diet, nutritional regulation, pancreatic islets
Citation: Villanueva-Hayes C and Millership SJ (2021) Imprinted Genes Impact Upon Beta Cell Function in the Current (and Potentially Next) Generation. Front. Endocrinol. 12:660532. doi: 10.3389/fendo.2021.660532
Received: 29 January 2021; Accepted: 01 April 2021;
Published: 27 April 2021.
Edited by:
Senta Georgia, Children’s Hospital of Los Angeles, United StatesReviewed by:
Christine Doucette, University of Manitoba, CanadaHung Ping Shih, Beckman Research Institute, City of Hope, United States
Copyright © 2021 Villanueva-Hayes and Millership. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Steven J. Millership, cy5taWxsZXJzaGlwQGltcGVyaWFsLmFjLnVr
†Present address: Chelsie Villanueva-Hayes, Cardiovascular Disease Mechanisms Laboratory, MRC London Institute of Medical Sciences, London, United Kingdom