 Shehla Pervin
Shehla Pervin Srinivasa T. Reddy3,4
Srinivasa T. Reddy3,4 Rajan Singh
Rajan Singh- 1Department of Obstetrics and Gynecology, David Geffen School of Medicine at University of California Los Angeles (UCLA), Los Angeles, CA, United States
- 2Division of Endocrinology and Metabolism, Charles R. Drew University of Medicine and Science, Los Angeles, CA, United States
- 3Department of Molecular and Medical Pharmacology, David Geffen School of Medicine at UCLA, Los Angeles, CA, United States
- 4Department of Medicine, Division of Cardiology, David Geffen School of Medicine, University of California Los Angeles, Los Angeles, CA, United States
- 5Department of Endocrinology, Men’s Health: Aging and Metabolism, Brigham and Women’s Hospital, Boston, MA, United States
Obesity is a global health problem and a major risk factor for several metabolic conditions including dyslipidemia, diabetes, insulin resistance and cardiovascular diseases. Obesity develops from chronic imbalance between energy intake and energy expenditure. Stimulation of cellular energy burning process has the potential to dissipate excess calories in the form of heat via the activation of uncoupling protein-1 (UCP1) in white and brown adipose tissues. Recent studies have shown that activation of transforming growth factor-β (TGF-β) signaling pathway significantly contributes to the development of obesity, and blockade or inhibition is reported to protect from obesity by promoting white adipose browning and increasing mitochondrial biogenesis. Identification of novel compounds that activate beige/brown adipose characteristics to burn surplus calories and reduce excess storage of fat are actively sought in the fight against obesity. In this review, we present recent developments in our understanding of key modulators of TGF-β signaling pathways including follistatin (FST) and myostatin (MST) in regulating adipose browning and brown adipose mass and activity. While MST is a key ligand for TGF-β family, FST can bind and regulate biological activity of several TGF-β superfamily members including activins, bone morphogenic proteins (BMP) and inhibins. Here, we review the literature supporting the critical roles for FST, MST and other proteins in modulating TGF-β signaling to influence beige and brown adipose characteristics. We further review the potential therapeutic utility of FST for the treatment of obesity and related metabolic disorders.
Introduction
The obesity epidemic significantly affects every region and demographical group worldwide with no signs of abatement. Obesity substantially increases the risk for several chronic diseases including cardiovascular diseases, fatty liver, diabetes, insulin resistance and cancer. It has been estimated that by 2030 approximately 2.16 billion individuals will be overweight and 1.12 billion individuals will be obese as defined by body mass index (BMI) of 30 or higher (1). The economic impact of obesity and its related complications on United States has been estimated between 4-8% of gross domestic product and comparable to 2018 defense budget ($643 billion) and Medicare ($588 billion) (2), and significantly impacts low-income and economically disadvantaged populations. The strategies, to date, to combat the obesity epidemic have not been successful and there is an unmet need for the development of novel therapies to prevent and treat obesity and related metabolic complications. As obesity develops from surplus energy stored in adipose tissues, therapeutic approaches to reduce energy intake, increase energy expenditure, or both would provide attractive avenues for the fight against obesity and related diseases. Although thermogenic adipocytes and their precursors are composed of various distinct cell populations (3, 4), adipose tissue mass is composed mostly of white adipose tissue (WAT) and brown adipose tissue (BAT), which metabolically play opposing roles in regulating energy balance.
Recent clinical cross-sectional studies using [18F] FDG-PET/CT, suggest a clear decline in BAT activity and mass during aging that coincides with the development of obesity and insulin resistance. Several laboratories have presented evidence for expression of the thermogenic molecule UCP1 as well as its energy dissipating capacity in human BAT and contribute towards improved metabolic profiles (5–7). WAT, which is specialized for storage of energy could be manipulated via genetic or pharmacological means to promote browning. Such browning, also called as “beige” or “brite” (brown in white), is associated with increased expression of mitochondrial uncoupling protein-1 (UCP1) expression in response to external stimuli including chronic cold exposure, treatment with β-adrenergic agonists CL 316,243, exercise, and endocrine factors (8, 9). This type of adipose browning is also associated with increased thermogenic capacity of the cells since activation of UCP1 that uncouples mitochondrial respiration from ATP production provides significant metabolic benefits that are comparable to BAT (8, 9). Experimental mice with selective ablation of beige adipose cells are prone to obesity and metabolic dysfunction probably by reducing lipogenic capacity and energy expenditure as well as by modulating the inflammatory environment inside the WAT (10). Overexpression of Prdm16 (PR-domain containing 16) resulted in abundant beige adipocytes in subcutaneous adipose depots, associated with significantly increased energy expenditure and were resistant to weight gain in response to a high fat diet (HFD) (11). Similarly, CRISPR/CAS9-mediated reconstitution of UCP1 in WAT of pigs led to significantly decreased fat mass and improved energy expenditure (12), suggesting the therapeutic potential of modulating WAT phenotype by activation of key thermogenic genes for fighting obesity and metabolic syndrome. While implementation of findings from animal studies remains challenging to apply in humans, such studies have yielded novel insights into the molecular mechanisms underlying thermogenic regulation of brown and beige adipocytes and highlight their ability to reduce obesity and related metabolic disorders. Thus, further studies directed to translate the proofs of concept generated in animal models are crucial.
Transforming growth factor-beta (TGF-β) signaling has been shown to regulate glucose and energy homeostasis (13). TGF-β levels are reported to increase with adiposity in overweight (BMI between 25-29.9 kg/m2) and obese (BMI ≥ 30 kg/m2) subjects compared to the normal subjects with BMI less than 24.9 kg/m2), and systemic blockade of TGF-β/SMAD3 signaling resulted in protection against diet-induced obesity in experimental mice (13). This effect was associated with acquisition of energy dissipating brown adipocyte phenotype in WAT. In this review, we will discuss the evidence for the novel role of FST, MST and other related proteins in modulating TGF-β signaling and adipocyte browning to explore possible therapeutic avenues for the treatment of obesity and associated metabolic disorders.
Beige and Brown Adipocyte Developmental Origin and Molecular Signatures
White, beige and brown adipocytes are three major types of adipocytes that have distinctly different fat morphology and differ in their developmental origin as well as function. During embryogenesis, BAT development precedes the formation of WAT, where it primarily contributes to non-shivering thermogenesis and maintain body temperature in newborns. Interscapular BAT mainly contributes to the temperature regulation during early stages of life and its levels slowly regress with age (14–16). Lineage-tracing studies demonstrated that classical brown adipocytes present in BAT depots originate from a sub-population of dermomyotome expressing specific transcription factors, including Pax7, engrailed 1, and Myf5 (17–20). Previously, these Myf5-expressing (Myf5+) precursors were assumed to be present exclusively in skeletal muscle precursors and absent in both in WAT and beige adipocytes (20, 21). Beige adipocytes present in the inguinal white adipose depots are reported to be derived from Myf5 negative (Myf5-) precursor pool (22). However, more recent lineage tracing studies have identified subsets of white adipocytes that are derived from both Myf5+ and Myf5- precursors (23–25), and beige adipocytes are derived from progenitor populations expressing Sma, Myh11 (a selective marker for smooth muscle cells), platelet-derived growth factor receptor (PDGFR)-α, or PDGFR-β in mice (26–29). Retinoic acid (RA)-induced adipose browning in endothelial cells and capillaries has also been reported via the activation of vascular endothelial growth factor (VEGF) A/VEGFR2 signaling that facilitate PDGFR-α-expressing adipocyte precursors (18). Beige cells could also appear as a result of trans-differentiation of mature white adipocytes (15). Trans-differentiation of beige adipocytes to WAT has been reported during warm adaptations and aging (30, 31). Collectively, the above studies strongly suggest that beige adipocytes that emerge in WAT depots appear to have multiple origins compared to the brown adipocytes.
PGC-1α is a master regulator of adaptive thermogenesis that binds to PPAR-γ and coactivates PPAR-γ to stimulate the transcription of genes involved in the brown adipocyte differentiation process and acquisition of morphological and molecular features of brown and beige fat (32). PGC-1α expression is rapidly induced by cold exposure that turns on several key components of the adaptive thermogenic program including fatty acid oxidation, mitochondrial biogenesis, and increased oxygen consumption (33). The transcriptional factor PR domain zinc finger 16 (PRDM16) is selectively expressed in brown/beige compared to the visceral white fat cells and plays an important role in controlling the differentiation-linked brown adipose/skeletal muscle fate determination and gene expression program (34). Gain and loss-of-function studies of PRDM16 in various cell systems have clearly established its major role in brown adipose/skeletal muscle cell fate determination (35). Using analysis of clonal cell lines, Wu et al. also suggested that beige and brown adipose cells express related but distinctly different gene expression profiles (36). Beige cells are selectively enriched in Tmem26, Tbx1, and CD137 expression (36). The same study identified additional beige selective genes including Ear2, CD40, Sp100, Klh113, and Slc27a from interscapular BAT and inguinal fat. Wang et al. identified early B-cell factor 2 (Ebf2) as one of the most selective markers for brown and beige adipogenic precursor cells (37). More beige-selective genes including HoxC8, HoxC9, Cited1, and Shox2 were identified using molecular profiling of human BAT (38, 39). On the other hand, epithelial V-like antigen (Eva1), Lhx1, Zic1, and Epsti1 are selectively expressed in classical brown adipocytes (36–41). Additionally, Ebf3, Pdk4, Fbxo31, Oplah, and Hsbp7 were also found to be highly enriched in interscapular BAT of 129SVE mice (36). Ussar et al. have reported few selective cell surface markers for white, beige and brown adipocytes that could provide unique tools to identify various adipocyte populations in both humans and rodents and potentially target them for therapy in vivo (42). The authors identified amino acid transporter Asc1, encoded by the SLC7A10 gene as a white adipocyte-specific cell surface protein, which was barely expressed in brown adipocytes (42). Expression level of purigenic receptor P2RX5, part of a seven-member family of ATP gated ion channels, was highest in brown adipocytes. Proton coupled amino acid transporter PAT2, another cell surface protein, show highest specificity for adipose tissue among all three markers identified in this study with significantly higher expression in brown fat compared to white fat. Better understanding of the gene expression pattern of such adipose-specific cell surface markers should provide novel tools to selectively mark and access intact white and brown adipocytes and could be used for diagnostic and therapeutic purposes. Analysis of microRNA (miRNA) between beige and brown fat have provided clear differences in their expression profile. Several miRNAs including miRNA-30, miRNA-182, and miRNA-203 are reported to positively regulate both beige and brown adipocytes (43, 44). On the other hand, miRNA-27 and miRNA-34a negatively regulator beige and brown adipogenesis (45, 46). Recent studies have also highlighted some specific miRNAs including miRNA-196b and miRNA-26 that positively and negatively regulate beige adipogenesis respectively (47, 48).
Transforming Growth Factor-Beta (TGF-β), Adipose Browning and Obesity
The TGF-β superfamily consists of several members including TGFβ1, TGFβ2, and TGFβ3, bone morphogenetic proteins (BMPs), growth differentiation factors (GDFs), and activins that regulate diverse biological processes during embryogenesis, adult tissue homeostasis, and function of several cell types including adipocytes (49, 50). The pleiotropic effects of TGF-β/Smad3 signaling on cell metabolism and energy homeostasis plays an important part in the progression of obesity-linked diabetes; these include adipocyte differentiation, adipose browning, inflammation and regulation of insulin signaling amongst others. Members of TGF-β superfamily transmit their signals via dual serine/threonine kinase receptors and transcription factors called Smads. Recent studies have clearly established an essential role of TGF-β/Smad3 signaling in the pathogenesis of obesity and type 2 diabetes. Elevated levels of TGF-β has been reported in mice and human adipose tissue during hypertension and other cardiovascular diseases as well as in morbid obesity and diabetic neuropathy (51–53). Increased TGF-β levels have also been associated with a higher risk for type 2 diabetes in a prospective case-cohort study (54). Perry et al. identified Samd3 gene in a type2 diabetes genome-wide association study (55). Smad3 is known to bind to the PGC-1α promoter to repress its transcription (13). As PGC-1α is an important transcriptional coactivator for UCP1 gene induction, mitochondrial biogenesis, and fatty acid oxidation, it is not surprising that TGF-β/Smad3 signaling would inhibit beige/brown adipocyte differentiation and their thermogenic action. The discovery of TGF-β/Smad3 signaling as novel modifiers of beige adipocyte phenotype and metabolic characteristics by Yadav et al. has opened therapeutic avenues for identifying potent inhibitors of this signaling pathway for the treatment of obesity related complications (13). The authors observed significant positive correlation between TGF-β1 levels and adiposity in both rodents and human subjects. Smad3−/− mice displayed protection against diet-induced obesity and related metabolic syndromes. These effects were associated with significant induction of white to brown phenotype and increased mitochondrial biogenesis. Examination of a group of nondiabetic human subjects from diverse ethnic groups, the authors found direct relationship between circulating TGF-β1 levels and BMI, fat mass, and oxygen consumption. The same group assessed the effect of blocking TGF-β/Smad3 signaling in two well-characterized mouse models of obesity and type 2 diabetes. Treatment with anti-TGF-β neutralizing antibody 1D11 resulted in significantly reduced body weight, improved glucose and insulin tolerance, as well as fasting glucose and insulin levels. These beneficial effects were associated with elevated expression of BAT and mitochondria-specific proteins including UCP1, COX-1 and PGC-1α as well as decreased phosphorylation of Smad3 in white adipose tissues. Such links between TGF-β signaling and mitochondrial energy metabolism pathway have also been reported by other laboratories (56). In addition, several studies have demonstrated extensive interaction between TGF-β and key energy sensors including adenosine monophosphate protein kinase (AMPK) and sirtuin family members (57, 58). Inhibition of activin receptor IIB (ActRIIB) responsible for integrating actions of TGF-β ligands promotes differentiation of primary brown adipocytes in-vitro and increases brown fat mass, but not white fat mass in mouse (59). Furthermore, inhibition of ActRIIB via a decoy receptor containing extracellular domain of ActRIIB fused with human Fc (ActRIIB-Fc) resulted in suppression of diet-induced obesity and related metabolic complications in mice (60). This blockade of ActRIIB was associated with increased browning and robust upregulation of UCP1 and PGC-1α expression in the epididymal white adipose fat and led to increased energy expenditure under ambient or cold temperature. Gene signature induced as result of ActRIIB inhibition, displayed an interesting similarity with PGC-1α overexpression in-vivo. Combined together, these studies provide significant insights into the role of TGF-β signaling in suppressing adipose browning program within white fat tissues in both mouse models and human subjects, suggesting that blockade of TGF-β activity could serve as an effective treatment strategy for obesity and diabetes.
Since bone morphogenic proteins (BMPs) belong to the same superfamily of growth factors as TGF-β, and regulate various aspects of white and brown adipocyte differentiation, we discussed below briefly their biological functions in modulating adipose tissue functions (61–66). BMP4 has been shown to promote differentiation of human adipose stem cells into beige adipocytes (61, 62). BMP4 overexpressing transgenic mice display reduced adiposity, improved insulin sensitivity, and induction of brown adipocytes within inguinal subcutaneous fat depots (63, 64). Interestingly, these transgenic mice display decreased expression of brown adipocyte markers including UCP1 and PGC-1α in the BAT (62). In spite of reduced BAT activity, these BMP4 overexpressing mice are protected from diet-induced obesity and insulin resistance perhaps due to increased WAT browning (63). It, therefore, appears that BMP4 may have opposite effects on the development of brown adipocytes in BAT and beige adipocytes in WAT in-vivo. BMP7 promotes the commitment of mesenchymal progenitor cells to a brown adipocyte lineage while it prevents osteogenesis by inhibiting the expression of runt-related transcription factor 2 (Runx2) (67). In C3H/10T1/2 cells, pretreatment with BMP7 results in brown adipogenesis with lipid accrual and expression of Ucp1 (67). Tail vein injection of adenovirus expressing BMP7 increases BAT, without affecting the mass of WAT (67). Although BMP7 increases Prdm16 and Ucp1 expression in brown adipose, there are no changes in the expression of genes involved in energy metabolism in white adipose, muscle, or liver. The increase in BAT mass results in increased energy expenditure, higher basal body temperature, and decreased body weight attributes that clearly link BMP7 signaling to energy balance. BMP7 knockout mice show significant reduction of brown fat mass (67). Conversely, adenoviral-mediated expression of BMP7 in mice results in significant increase in brown fat mass, increased energy expenditure and reduction in weight gain and subcutaneous implantation of BMP7-treated MSCs into athymic nude mice results in ectopic brown adipose tissue formation (67). BMP8b promotes brown adipose tissue thermogenesis through both central and peripheral actions (65). This thermogenic effect of BMP8a is observed only in female mice and is thought to be mediated by estrogens (66). The molecular mechanisms responsible for such differential regulation of WAT, beige and BAT by various BMP members remains largely unknown.
Myostatin, Irisin, Adipose Browning and Energy Metabolism
Myostatin (MST), also referred to as growth and differentiation factor 8 (GDF8), is a member of TGF-β superfamily. MST is synthesized as a precursor protein, which consists of a N-terminal propeptide domain that contains the signal sequence and a C-terminal domain that forms a disulfide-linked dimer and functions as the active ligand (68). MST requires release from the propeptide to be biologically active (69). MST binding to ActRIIB leads to the phosphorylation of Smad3 (70). Phosphorylated Smad3 can bind other Smad proteins and these complexes translocate into the nucleus, where they regulate the transcription of target genes (70). It is mainly expressed in skeletal muscle but is also detectable in cardiac muscle, blood, and to a limited extent in adipose cells. MST is known as the potent negative regulator of muscle mass as inactivation of Mst gene significantly accelerates muscle growth in cattle, sheep, fish and humans (71–75).
Recent studies from several laboratories have provided conclusive evidence that the effect of MST extends beyond its role in skeletal muscle, and it plays a significant role in the regulation of body fat and overall energy metabolism. Mst-knockout (Mst-KO) mice show significantly increased muscle mass, decreased fat mass, improved insulin sensitivity and resistance to diet-induced obesity (76, 77). On the other hand, overexpression of MST in mice has been shown to promote catabolic conditions and result in muscle wasting and cause insulin resistance (78). Since MST is expressed in very low amounts in fat tissues, it is not clear how lack of MST can suppress fat accumulation in Mst-KO mice. Significantly increased energy expenditure and leptin sensitivity was observed in Mst-KO mice that could potentially explain reduced fat mass in these mice when compared to the WT mice (79). In primary cultures of mouse preadipocyte cells, Kim et al. reported decreased expression of key thermogenic genes Ucp1, Prdm16, and Pgc-1a and significant inhibition of brown adipogenic differentiation following treatment of the cells with recombinant MST protein (80). Using differentiating primary cultures of mouse embryonic fibroblast (MEF) isolated from Mst-KO and WT embryos, Braga et al. reported significant upregulation of key thermogenic markers in differentiating cultures of Mst-KO group compared to the WT group (81). In the same study, treatment with recombinant MST protein led to a significant decrease in Oil-Red O stained adipocytes and expression of key thermogenic genes in both WT and Mst-KO groups. Comparative analyses of epididymal (Epi) and subcutaneous (SC) adipose tissues isolated from WT and Mst-KO mice show clear induction of thermogenic proteins including UCP1, and PRDM16 along with C/EBPα. Gene expression analyses further confirmed significant upregulation of key adipogenic differentiation markers Cebpα and Pparγ, as well as key thermogenic genes including Prdm16, Ucp1, Bmp7, PGC-1a/b and Cidea, suggesting that loss of MST significantly promotes brown adipose-related markers in two main adipose depots in Mst-KO mice (81). Similar comparative analyses of muscle tissues from androgen-dependent (levator ani, LA) and independent (gastrocnemius, Gas) muscle tissues show upregulation of UCP1 and PRDM16 protein and several genes involved in the regulation of overall thermogenic program. These combined in-vitro and in-vivo approaches using differentiating MEF cultures, as well as Mst-KO and their WT littermates show that MST inhibition could not only promote white adipocyte browning in adipose depots but could also promote the conversion of inter-muscular white adipocytes into beige/brown adipocytes. Furthermore, protein expression analysis of energy-sensing adenosine monophosphate (AMP)-activated protein kinase (AMPK), a critical regulator of mitochondrial biogenesis that controls energy metabolism by acting in co-ordination with NAD+-dependent type III deacetylase sirtuin1 (Sirt1) was found to be significantly upregulated in differentiating Mst-KO MEF primary cultures compared to the WT group (81). Protein expression of adiponectin, a key protein secreted from adipocytes and regulator of adipocyte energy metabolism was also found to be upregulated in differentiating MEF cultures isolated from Mst-KO group compared to the WT (81). Adiponectin is reported to limit triglyceride (TG) accumulation in liver (82), increase glucose clearance and improve hepatic insulin action in adiponectin transgenic mice (82, 83). Zhang et al. also reported that inhibition of MST leads to increased skeletal muscle mass, slows down fat accumulation, lowers body weight and circulating levels of triacylglycerol in mice on high-fat diet (84). The authors reported that white adipose tissue of Mst-KO mouse express significantly higher levels of genes involved in lipid transport, synthesis, oxidation and hydrolysis. In addition, adipose tissues isolated from Mst-KO mice show increased expression of UCP1 and upregulation of AMPK signaling pathway when compared to the WT mice (84). Histological analysis of WAT isolated from Mst-KO revealed BAT-like cells filled with multilocular smaller lipid droplets and immunopositive for UCP1 (81), suggesting that MST deletion induced brown-like phenotype. Genetic loss of MST has also been reported to promote white adipose browning and improve insulin sensitivity by several other laboratories (85, 86). Shan et al. performed a thorough analysis of various muscle-derived circulatory factors to identify possible mediators of adipose browning phenotype in these Mst-KO mice (86). The authors reported that skeletal muscle derived irisin (encoded by Fndc5 gene) plays a central role in promoting adipose browning in Mst-KO mice by activating AMPK-PGC-1α-Fndc5 signaling, providing an interesting involvement of muscle-adipose cross talk during adipose browning (86). Dong et al. also reported the involvement of Fndc5/irisin-mediated white adipose browning and improvement in insulin signaling in Mst-KO mice (87). Mst-KO Meishan pigs with functional deletion of Mst show increased insulin sensitivity, adipose browning and upregulation of several browning-like gene signature including Ucp1, Prdm16, Pgc-1α, Cidea, Cd137 and Tmem26 (85). Protein expression analysis of skeletal muscle in these Mst-KO pigs shows significantly increased levels of insulin receptor (IR) and insulin receptor substrate (IRS). Skeletal muscle protein expression of irisin precursor protein Fndc5 as well the serum irisin levels were significantly higher in Meishan Mst-KO pigs compared to the WT pigs. Activation of insulin signaling pathway could not be blocked via inhibition of irisin in this study, suggesting possible irisin independent activation of insulin signaling in MST deficient skeletal muscle (85). Reduction of interferon regulatory factor 4 (IRF4) leads to significantly reduced exercise capacity, mitochondrial function and ribosomal protein synthesis in brown fat, an effect that was associated with induction of MST levels (88). On the other hand, overexpression of IRF4 led to significantly reduced levels of serum MST and increased exercise capacity in muscle. IRF4 was shown to physically interact with PGC-1α and promote the thermogenic program by upregulating the transcription of UCP1 gene and driving mitochondrial biogenesis in BAT. In addition, IRF4 levels in BAT was found to be significantly induced following cold exposure and β3-adrenergic receptor (AR) agonist (88). These findings, therefore, suggest that IRF4 is a novel inducer of overall thermogenic program with the potential to inactivate MST bioactivity. Guo et al. reported additional role of MST regulation in the development of proatherogenic dyslipidemia, insulin-mediated glucose disposal as well as protection against hepatic steatosis (89). The authors show that administration of adeno-associated virus 9 (AAV9)-mediated MST pro-peptide significantly blocked the progression of atherosclerosis and development of hepatosteatosis in LDLR-/- mice on western diet. In this study, the beneficial effects of both Mst genetic ablation as well as its inactivation by MST pro-peptide were attributed to result from the enlarged muscle mass although the authors did not study its effect on adipose browning and brown fat activation. Several laboratories provided compelling evidence to support the notion that brown fat activation could reduce hypercholesterolemia and elicit protection form atherosclerosis development (90–92). Most recently, Pydi et al. demonstrated that increased plasma MST levels in mice lacking β-arrestin 1 (barr1) (adipo-baar1-KO) led to impaired insulin signaling in multiple peripheral tissues (93). On the other hand, overexpression of baar1 in adipo-baar1-OE mice on high fat diet displayed pronounced improvements in glucose tolerance, insulin sensitivity, and displayed significant reduction in MST levels, suggesting that overexpression of baar1 in adipocytes protects mice from obesity-associated metabolic disorders. Collectively, these data provide strong evidence that inhibition of MST could provide justification not only for increased muscle mass but could also be beneficial for the treatment of obesity and associated metabolic disorders through activation of adipose browning.
Irisin is a key myokine and adipokine that is secreted following the proteolytic cleavage of its precursor fibronectin type III domain containing protein 5 (FNDC5). Secreted irisin exerts its major action by upregulating the expression of UCP1 and promoting browning of WAT (94). Circulating levels of irisin are regulated by various factors including diet, exercise, obesity and pharmacological agents (95). Bostrom et al. first isolated irisin from muscle tissues and performed its chemical characterization (96). Following exercise stimulation and activation of transcriptional co-activator PGC-1α, FNDC5 expression levels are increased in muscle, resulting in the secretion of irisin to induce adipose browning through activation of thermogenic genes (96). These findings provided strong evidence for the beneficial role of irisin in cardiovascular, obesity, diabetes, skeletal and other diseases. Cold activation and physical activity among several other factors are known to alter the level of circulating irisin (97, 98). Plasma irisin levels are reported to increase by 65% after 3 weeks of freewheel running, while in healthy humans irisin levels double after 10 weeks of endurance exercise (96). Several studies demonstrated that irisin improves glucose homeostasis, and its circulating levels are inversely associated with liver fat content (99–101). Based on these findings, irisin was revealed as a potential new target for the treatment of metabolic diseases. However, in contrast with the above reports, several other studies question the beneficial role of irisin and in some cases even its existence (101–104). There is also a disagreement regarding the induction of FNDC5/irisin by exercise (105, 106), and its association with markers of glucose and lipid homeostasis disturbance in obesity and metabolic syndrome (107–110). Such controversies could be explained by the fact that irisin levels increase only when muscle ATP concentration decreased in absence of physical activity during sedentary lifestyle (105). Perez-Sotelo et al. reported decreased browning capacity and increased adipogenesis of differentiating adipocytes by blocking adipose endogenous expression of FNDC5 (111). The authors reported that incubation of normal adipocytes with secreted factors from the WAT of obese patients resulted in significant reduction of FNDC5, PGC-1α and UCP1 expression. Irisin is also reported to influences glucose metabolism in skeletal muscle (112) and myocytes in-vitro via increased oxidative phosphorylation, mitochondrial biogenesis and upregulation of various genes involved in glucose transport as well as in mitochondrial uncoupling (113). Furthermore, exogenous FNDC5 induces UCP1 expression in subcutaneous white adipocytes in animal models, and FNDC5 overexpression in the liver prevented diet-induced weight gain, metabolic disturbances, and stimulation of oxygen consumption (114). Irisin administration was also found to increase the secretion of glycerol and decrease lipid accumulation via regulating the expression of hormone-sensitive lipase (HSL), adipose triglyceride lipase (ATGL) and fatty acid-binding protein 4 (FABP4) (115). Moreover, irisin was found to inhibit hepatic cholesterol synthesis through AMPK-SREBP2 signaling (116) in addition to its ability to lower plasma glucose levels and altered food intake in streptozotocin-induced diabetes mellitus model (117). Subcutaneous perfusion of irisin resulted in significantly increased energy expenditure, reduced hyperlipidemia and hyperglycemia, and improved insulin resistance (118). These beneficial effects of irisin were mediated via upregulation of cAMP/PKA/HSL-perilipin pathway (118). In a recent report, Li et al. provided supporting evidence for a critical role of irisin in mediating Fst-induced browning (119). They reported that Fst injection promoted increased secretion of irisin from the subcutaneous fat depots via AMPK-PGC1-α-irisin mediated signaling during adipose browning. In cardiomyocyte H9C2 cells, recombinant irisin (r-irisin) activated PI3K/AKT pathway, induced intracellular Ca2+ signaling, and increased cellular oxygen consumption (120). In primary adipocytes and 3T3-L1 cells, r-irisin significantly increased the expression levels of key thermogenic genes including Ucp-1, Pgc-1a, Cox7a, Ebf3, and Elovl3 and phosphorylated forms of p38 MAPK and ERK1/2. Pharmacological inhibition of p38 MAPK and ERK1/2 phosphorylation significantly lowered irisin-induced UCP-1 expression (94). Collectively, these studies provide novel beneficial role of irisin in regulating key metabolic parameters associated with perturbed lipid, cholesterol and energy metabolism.
Follistatin
Follistatin and Follistatin-Like Proteins
Follistatin (FST) was initially identified as component of the follicular fluid capable of inhibiting follicle-stimulating hormone (FSH) (121). FST is a monomeric glycosylated protein that binds and neutralizes activins with high affinity and neutralizes their bioactivity (121). FST also binds with lower affinity to several other members of the TGF-β superfamily including MST and BMPs 2, 5, 7, and 8 (122–125). These reports highlight the potential for FST to modulate the biological activities of several TGFβ superfamily, particularly at higher concentrations. Two variants of FST are generated through alternate splicing at the C-terminus of the common precursor gene (126). A third isoform of approximately 300-303 amino acids (FST300 or FST303) is also reported to be produced by proteolytic cleavage of the C-terminus of FST315 (127). The shorter isoform FST288 is capable of binding heparin-sulfated proteoglycans on the cell surface with high affinity. The longer isoform FST315 is localized primarily in the circulation and has reduced affinity for heparin as a result of masking of the heparin-binding site at the C-terminal (121). Another molecule related to FST, called follistatin-like 3 (FSTL3) has been identified (128). This protein lacks the heparin-binding sequence, but similar to Fst, it binds activin A with high affinity and activin B with relatively lower affinity (129). FSTL3 is relatively less effective in blocking endogenous activin A in various cells (130). It is, therefore, possible that the ability of FST to bind to the proteoglycan cell surface could be important for potent inhibition of activin A action. While Fst is expressed in several tissues including ovary, pituitary, muscle and adipose tissues, FSTL3 is distributed predominantly in testis, placenta, heart and pancreas (131). Moreover, unlike Fst, FSTL3 is located in the nucleus, though it is also secreted at a relatively slower rate (131). Based on the tissue distribution, subcellular localization and intracellular transport pattern of FST and FSTL3, it is evident that they are not functionally redundant. Glycosylation of these core proteins produces a number of protein variants ranging in size from 31 to 42 kDa in size. Human FST is glycosylated at two specific sites, but point mutation of these sites does not change the affinity of FS315 for activin A (132). It is important to note that FSTL3 along with GASP1 and MST-propeptide binds with MST in circulation, suggesting that FST might not be the sole physiological regulator of MST in-vivo (133–135).
Follistatin and Muscle Mass
Matzuk et al. elegantly assessed the role of FST in regulating muscle mass and reported that FST loss-of-function mutant (Fst-KO) mice show decreased diaphragm and intercostal muscles and die within hours of birth (136). Based on the role of MST in being the most potent negative regulator of muscle mass to date and its inhibition by FST, Amthor et al. explored the role of possible interaction between FST and MST during chick development using yeast and mammalian two-hybrid system (123). The authors demonstrated that FST and MST interact directly with a high affinity of 5.84 × 10−10 M, and are expressed in the overlapping domains during muscle development. Moreover, MST-induced decrease in the expression levels of key myogenic proteins Pax3 and MyoD was significantly blocked in the presence of FST, suggesting an important role of FST in antagonizing the inhibitory effect on muscle development. Subsequently, it was reported that FST-induced muscle hypertrophy was associated inhibition of both MST and activin A and induction of satellite cell proliferation (137). Fst gene delivery of AAV1-FST344 in normal and dystrophic mice as well as in non-human primates led to significant increase in muscle mass and strength (138, 139). Transgenic expression of Fst in mdx mice, a popular model for Duchenne muscular dystrophy (DMD), showed amelioration of dystrophic pathology and increase in skeletal muscle mass (140). Interestingly, in a gene therapy trial Mendell et al. demonstrated beneficial effects of FST344 direct delivery into intramuscular quadriceps in patients suffering from Becker Muscular Dystrophy without any apparent side effects (141). Initially, FST was identified as a direct downstream target of testosterone action during its pro-myogenic action in both mouse models (142) and cell-culture studies (143). Protein and gene expression of FST was significantly upregulated in mouse mesenchymal pluripotent C3H 10T1/2 cells following testosterone treatment (142). This upregulation of FST was associated with a parallel increase in key myogenic markers MyoD and myosin heavy chain (MHC) II proteins, and co-treatment of the testosterone treated C3H 10T1/2 cells with anti-FST antibody abolished the myogenic action of testosterone. Furthermore, castration-induced decrease in FST expression was normalized to basal levels following testosterone supplementation, suggesting an intermediate role of FST in mediating testosterone’s promyogenic action on muscle mass (142). Subsequently, Braga et al. reported for the first time that FST is expressed in primary cultures of muscle satellite cells and respond to the myogenic action of testosterone (143). FST significantly antagonized the TGF-β-induced inhibition of MHC II expression and phosphorylation of Smad2/3 in satellite cells (143). Combined together, these findings provide conclusive support for a central role of FST in promoting muscle mass and function, and its potential therapeutic use for the treatment of muscle wasting cachexic conditions often associated with aging, HIV, and cancer.
Follistatin and Adipose Browning
Although the role of FST in regulating skeletal muscle mass has been supported by abundant literature, its potential role in lipid metabolism has not been thoroughly investigated. Based on the established role of FST in inhibiting TGF-β/MST signaling pathway known to inhibit adipose browning and thermogenic program, it is logical to hypothesize that FST may promote brown adipose characteristics and favorably alter overall lipid and energy metabolism (144). Since both skeletal muscle and brown adipose tissue share common Myf5+ precursor population, there is a possibility that severe musculoskeletal defects and death of Fst-KO newborn pups could also be complicated by their concomitant decrease of BAT mass and activity, resulting in their inability to maintain proper body temperature especially during the early neonatal life. Braga et al. provided the first evidence for a potential role of FST in regulating brown adipose metabolic characteristics and thermogenesis (144). First insight regarding a direct role for FST in adipose tissues was obtained from analysis of Fst gene expression of a tissue panel from C57BL6/J mice that included WAT (inguinal subcutaneous and epididymal) and BAT depots, as well as several other metabolic tissues including brain, heart, intestine, liver, skeletal muscle, and testis (144). Interestingly, Fst gene expression was highest in BAT and skeletal muscle, and at substantial levels in inguinal WAT and liver compared to other tissues where its expression was significantly low. This finding, therefore, suggested a possible novel role of FST in regulating WAT and BAT metabolic characteristics. Differentiated mouse brown preadipocyte primary cultures show significant upregulation of FST expression along with key thermogenic markers UCP1 and PRDM16, compared to the undifferentiated cells. Interestingly, Fst gene expression dramatically increased in mouse BAT following cold-exposure, suggesting a possible functional role of FST during brown adipocyte differentiation and regulation of thermogenesis. Comparative analysis of mouse embryonic fibroblast (MEF) primary cultures isolated from WT and Fst-KO embryos, Braga et al. demonstrated significant inhibition of key brown adipogenic markers including PRDM16, UCP1 and PGC-1α in Fst-KO differentiating MEF cultures compared to the WT group (144). Treatment of these cells with recombinant FST protein (rFST) resulted in significant upregulation of BAT-related genes and proteins in both WT and Fst-KO MEF differentiated cultures. Comprehensive analysis of global gene expression profile revealed lipid metabolism pathways as the most significantly altered pathways between WT and Fst-KO groups. Furthermore, Fst-KO differentiating cultures displayed significantly compromised basal mitochondrial respiration compared to the WT group. Addition of exogenous rFST protein to the Fst-KO cultures rescued this respiration impairment by increasing the cellular respiration. In addition, expression level of phosphorylated adenosine monophosphate (pAMPK), a key energy sensor implicated in the regulation of cellular energy balance, was significantly down regulated in Fst-KO compared to the WT MEFs. More recently, Li et al. further confirmed FST-induced adipose browning in high fat diet (HFD)-fed obese mice (119). The authors demonstrated that intraperitoneal injection of FST increased thermogenesis, energy expenditure and browning of subcutaneous adipose fat in mice on HFD. FST injected mice had significantly higher body temperature 37.50C compared to the control group. This FST-induced thermogenesis was further confirmed by infrared imaging that demonstrated high-temperature areas in the FST injected group compared to the control group (119). In agreement with previous reports, a recent study reported that a single injection of AAV-mediated FST administration after several weeks of HFD feeding induced browning of subcutaneous WAT by upregulation of PGC-1α, PRDM16, UCP1 and beige-specific CD137, and decreased obesity-associated metabolic inflammation (145). Collectively, these data provided interesting novel insight regarding the importance of FST in modulating lipid and energy metabolism and suggest that overexpression of FST in-vivo may promote both beige and brown adipose tissue mass and activity.
Molecular Targets of FST During Adipose Browning
Using follistatin transgenic (Fst-Tg) mice (146), Singh et al. systematically analyzed the effect of FST in both WAT and interscapular classical BAT (147). These Fst-Tg mice express Fst under a muscle-specific promoter and have significantly elevated (1.5 fold) circulating levels of FST as well as interscapular BAT mass (70% higher) compared to age-matched WT control mice (147). Analysis of BAT signature genes important for differentiation (Ucp1, Prdm16, Zic1, Myf5, Lhx8), fatty acid oxidation (Ascl1, Fabp3, Cidea) and mitochondrial biogenesis and function (Pgc1α, Cox7a1, Cox8) as well key thermogenic proteins (UCP1, PRDM16, PGC1α) were significantly upregulated in the interscapular BAT of the Fst-Tg mice compared to the WT mice (147). Comparative analysis of epididymal and subcutaneous WAT between Fst-Tg and WT mice displayed similar upregulation of key BAT-related markers. These changes observed in both WAT obtained from Fst-Tg mice was associated with distinct adipose browning characteristics including increased UCP1 immunostating, and upregulation of key beige-specific Cd137 gene in both WAT depots, with greater changes observed in subcutaneous WAT compared to the epididymal WAT. These findings provided the first line of evidence that Fst promotes adipose browning in both WAT depots and increase BAT mass in-vivo.
Analysis of molecular targets of FST in these two adipose depots identified two distinctly different mechanisms. While FST increased phosphorylation of p38 MAPK and ERK1/2 in both WAT depots, it increased Myf5 expression in BAT of Fst-Tg mice (147). The authors also utilized in- vitro model of differentiating 3T3-L1 cultures to confirm that recombinant FST (rFST) treatment led to significant upregulation of UCP1 and beige-specific CD137 protein and beige-selective genes Cd137, Tbx1, and Tmem26. This rFST-induced increase in beige-selective markers in 3T3-L1 cells was also associated with concomitant increase in p38MAPK and ERK1/2 phosphorylation. Furthermore, pharmacological inhibition of their phosphorylation in these cells by either SB023580 or PD98059 resulted in abrogation of rFst-induced upregulation of UCP1 protein expression, suggesting that FST stimulates adipose browning via p38MAPK/ERK1/2 pathway. Since FST overexpression in Fst-Tg mice displayed distinctly different targets in WAT and BAT tissues, Singh et al. analyzed differential expression of key TGF-β signaling components Smad3/pSmad3 and activin receptor type IIB (Act RIIB) in adipose tissues obtained from WT and Fst-Tg mice. FST overexpression in these Fst-Tg mice led to significant inhibition of Smad3/pSmad3 as well as Act RIIB expression in both SC and Epi WAT as well as in BAT (147), suggesting that inhibition of Smad3 signaling may be the common upstream target of FST action that precedes phosphorylation of p38 MAPK/ERK1/2 and activation of Myf5. FST-induced inhibition of TGF-β/Smad3 signaling has also been reported previously in satellite cells and muscle tissues (142, 143). In order to further test the effect of FST overexpression on adipocyte browning in differentiating 3T3-L1 cells, Singh et al. cloned full-length mouse Fst gene in Piggyback Transposon cargo plasmid vector to perform systematic beige/brown adipose gene expression analysis following Fst overexpression (148). Comparative gene expression analysis of Fst-overexpressing 3T3-L1 Fst cells with the parental 3T3-L1 cells displayed significantly higher levels of follistatin protein and gene in the cells and in the cell supernatant compared to the 3T3-L1 cells (148). Expression levels of key thermogenic and several adipose browning markers including CD137, Tbx1, and Tmem26 were significantly upregulated following Fst overexpression in differentiating 3T3-L1 cells (148). Table 1 and Table 2 summarizes a comprehensive list of proteins and genes respectively that are influenced by increased levels of FST in various cell culture and Fst transgenic (Fst-Tg) mouse model. Fst overexpression also led to significant induction of p38 MAPK and ERK1/2 phosphorylation in-vitro in 3T3-L1 confirming previous findings of induced phosphorylation of these proteins in both WAT depots of Fst-Tg mice. Previous reports have suggested an essential role for p38MAPK in promoting cyclic-AMP-dependent activation of protein kinase A (PKA) and activation of UCP1 transcription (149, 150). Phosphorylation of p38 MAPK following stimulation of beta-adrenergic receptor (β-AR) results in phosphorylation and recruitment of ATF2 and PGC-1α to PPRE and CRF2 motifs within the UCP1 enhancer following their interactions with PPARγ and RXRα to activate the brown adipose thermogenic program (151). Phosphorylation of p38MAPK has also been shown to stimulate adipose browning via induction of irisin, a key myokine that can be significantly induced by exercise and PGC1-α (117). Recent data suggest that Fndc5 is also secreted from WAT (152). Since FST is known to induce irisin encoded Fndc5 gene in mouse muscle cells (86), it is possible that induced levels of irisin/Fndc5 will have contributed to increased browning via phosphorylation of p38 MAPK and ERK1/2 in Fst-Tg mice. Robust activation of FGF21/adiponectin/pAMPK signaling pathway was found in both adipose depots of Fst-Tg mice suggest a possible link between Fst overexpression and FGF21 activation (147). In order to test the possible intermediate role of β3-AR signaling during FST-induced browning, Singh et al. also tested whether treatment of β3 agonist CL316, 243 would promote BAT activation and adipose browning in Fst-Tg compared to the WT mice. The authors were able to show heightened response to β3-AR activation on UCP1 expression in both WAT depots and BAT tissues obtained from Fst-Tg mice compared to the WT mice (147).
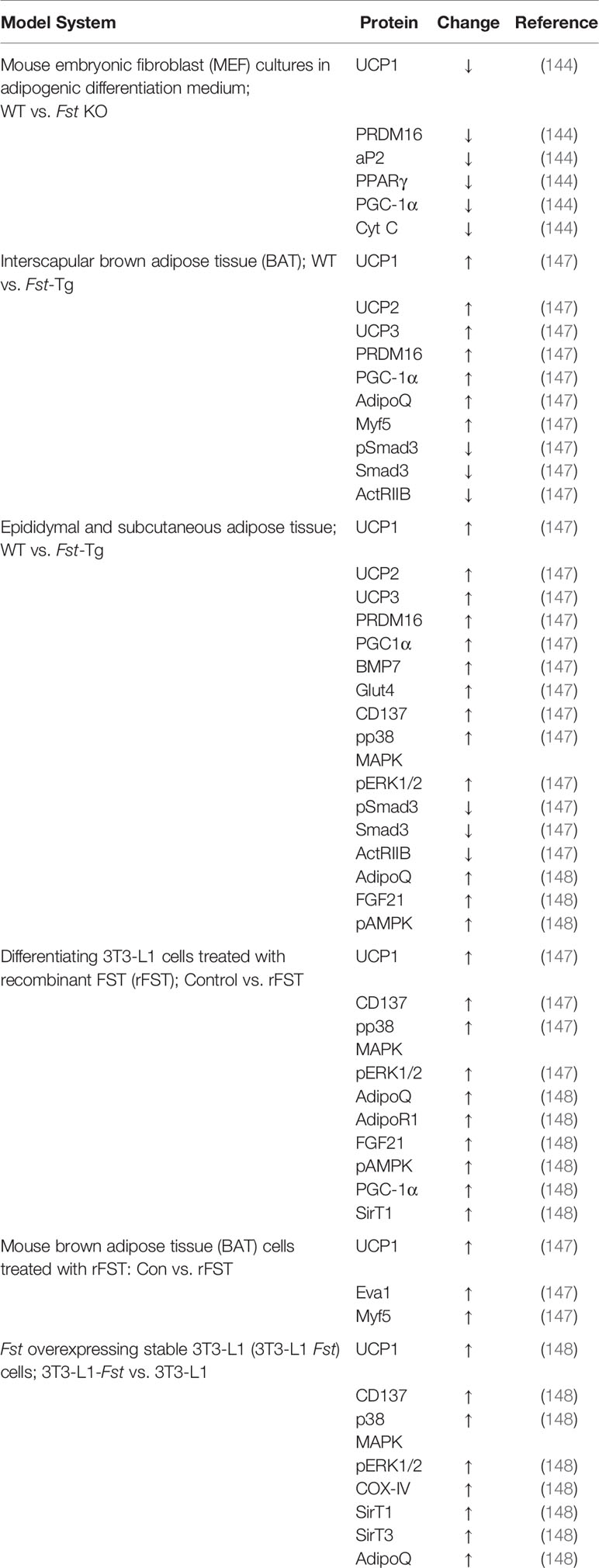
Table 1 List of proteins targeted by follistatin.

Table 2 List of genes targeted by follistatin.
In order to identify the molecular targets of FST in classical brown fat, Singh et al. also analyzed the effects of exogenous rFST on differentiating mouse brown preadipocyte BAT cultures (147). rFST treatment led to significant increase in BAT-selective UCP1, Eva1, and Myf5 protein and gene expression. They also showed that siRNA-mediated knockdown of mouse Myf5 expression led to significant blockade of FST-induced UCP1 protein and gene expression and two key BAT-selective genes Lhx8 and Zic1. Furthermore, Fst-KO embryo sections show decreased Myf5 immunostating compared to the WT, and treatment of differentiating MEF cultures derived from Fst-KO embryo with rFST was able to rescue Myf5 protein expression (147). Collectively, these findings obtained from differentiating BAT cells and Fst KO primary cultures provide strong evidence that Myf5 acts as an obligatory target of FST in promoting brown adipose characteristics. It appears however, that major action of FST on adipose browning is primarily due to the blocking of TGF-β ligands to inhibit Smad3 signaling as shown in Figure 1. A comprehensive list of proteins and genes targeted by FST during adipose browning are also summarized in Tables 1 and 2 respectively.
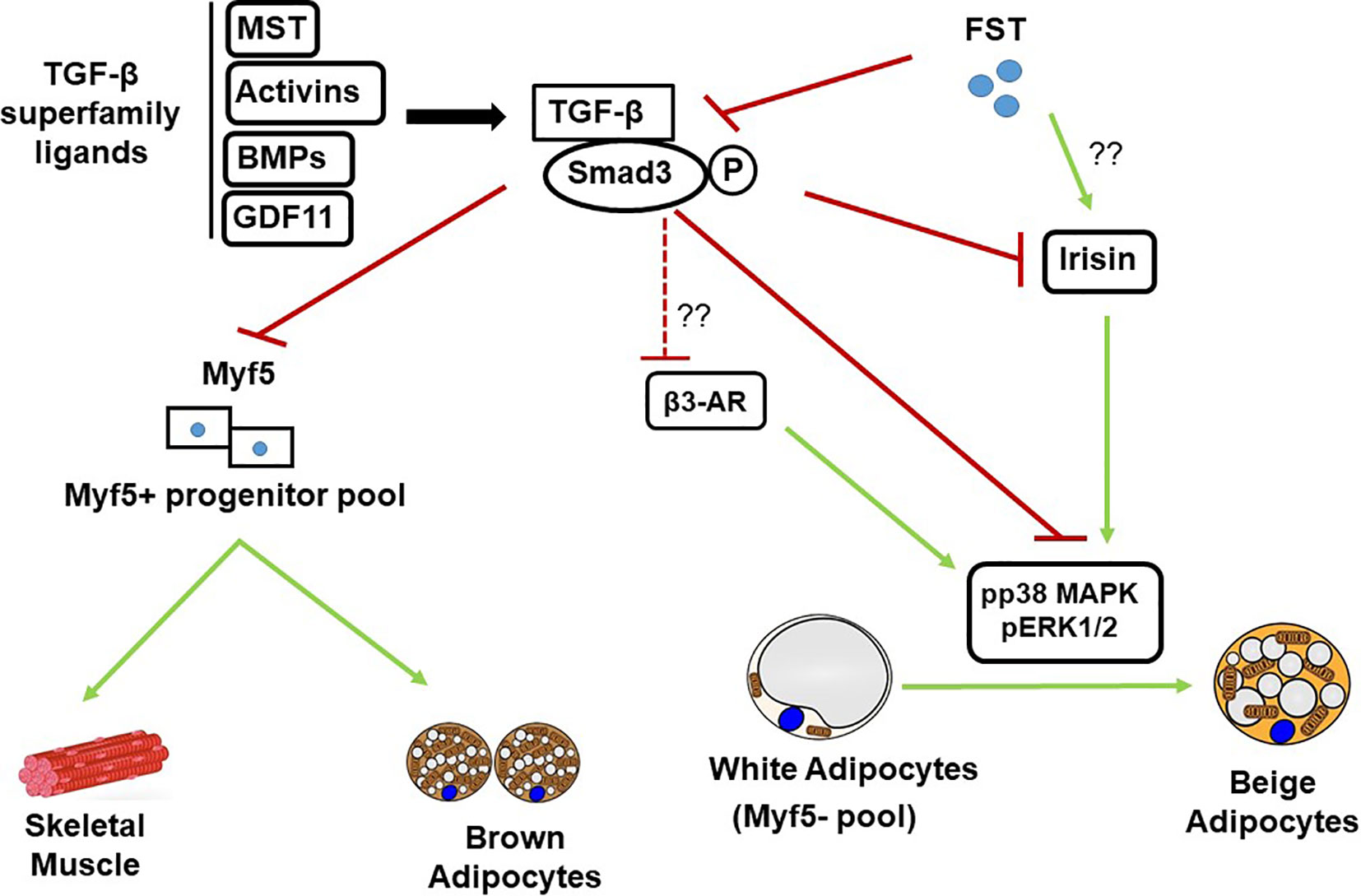
Figure 1 Schematic diagram showing FST modulation of TGF-β/Samd 3 signaling pathway during adipose browning. MST, Myostatin; BMPs, Bone morphogenic proteins; GDF11, Growth and differentiation factor 11; TGF-β, Transforming growth factor beta.
Genetic Manipulation of Follistatin Expression and its Relevance to Obesity Related Metabolic Diseases
As adipose tissues are the primary site of energy storage and its mobilization, activation of adipose browning has the potential to positively regulate overall systemic metabolism (153, 154). Adipose browning-induced biochemical changes are implicated in alterations of several key metabolic pathways that regulate plasma glucose levels and triglyceride metabolism in mice. Beige and brown adipose tissues consume and metabolize nutrients in a specialized way to facilitate weight loss, amelioration of insulin resistance and protection from hyperlipidemia and obesity related metabolic syndromes (155, 156). In order to assess the metabolic consequences of FST-induced adipose browning, Singh et al. performed quantitative analysis of abdominal fat volume, glucose clearance and comprehensive analysis of serum lipid profiles of Fst-Tg mice (148). Computerized tomography (CT) scan analysis of Fst-Tg mice revealed significantly lower percentage of abdominal fat mass and increased glucose disposal rate compared to the WT mice (148). Also, serum levels of triglycerides (TG), free fatty acid (FFA), and glucose levels were significantly lower in Fst-Tg mice compared to the WT mice without any significant changes in total cholesterol (TC) and high-density lipoprotein (HDL) levels. Major urinary protein 1 (Mup1), a key regulator of glucose and lipid metabolism (157), and energy expenditure (158) was significantly upregulated in liver and both WAT depots of Fst-Tg mice compared to the WT (148). Braga et al. previously reported a significant decrease in Mup1 gene expression in Fst-KO MEF differentiating cultures compared to the WT (144). In another recent study, Davey et al. reported that intravascular gene delivery via rAAV6-FST 317 to prediabetic db/db mice ameliorates progression of hyperglycemia, maintains insulinemia, promote abundance of insulin producing beta cell population, and reduced number of α‐like cells (159). The authors also reported that Fst gene delivery to older mice with hyperglycemia and declining insulinemia led to significant restoration of serum insulin concentration. Diabetic db/db mice display compromised β-cell function and reduced insulin content. Overexpression of FST in pancreatic-β cells has previously been reported to counter insulin insufficiency and extend the life span of db/db mice mainly by inhibition of SMAD pathway and activation of the PI3-kinase/Akt pathway (160). Most recently, Tang et al. also reported significant decrease in body fat percentage in mice on normal diet, and ameliorated the increase in body fat after HFD following AAV-Fst mediated gene delivery (145). In this study, Fst gene delivery in the HFD group significantly decreased serum levels of insulin, leptin, resistin, and C-peptide as well as serum glucose, triglycerides, cholesterol, and free FFAs as compared to control group. AAV-Fst gene delivery also significantly increased circulating levels of vascular endothelial growth factor (VEGF) and lowered serum levels of inflammatory cytokine IL-1α. In addition, reduced levels of mitochondrial oxidative phosphorylation (OXPHOS) complex subunits in subcutaneous WAT of mice on HFD was normalized following Fst overexpression via increased expression of PGC-1α. Although Fst has been reported to promote PGC-1α expression in previous studies (144, 147), the precise mechanism responsible for Fst-induced upregulation of PGC-1α remains unknown. Collectively, these findings provide exciting supporting evidence that Fst gene therapy could elicit beneficial metabolic effects and mitigate HFD-induced obesity. In order to test the effect of FST overexpression on overall lipidomic profiles in differentiating 3T3-L1 cells, Singh et al. performed comparative metabolic profiling of basal 3T3-L1 and Fst overexpressing 3T3-L1 Fst cells (148). Increased mitochondrial biogenesis in differentiated 3T3-L1 Fst cultures was also confirmed by significantly increased maximal oxygen consumption rate (OCR) (148). Analysis of endogenous lipid metabolites displayed a general reduction in diglycerides (DG), triglycerides (TG), ceramide, FA, phosphatidylcholine (PC), phosphatidylethanolamine (PE), and lysophosphatidylethanolamine (LPE) in FST overexpressing 3T3-L1 Fst cells compared to the basal 3T3-L1 cells (148). On the other hand, levels of several lyosophosphatidylcholines (LPL) such as LPC (16.0), LPC (18.0), and LPC (18.1) were significantly increased in 3T3-L1 Fst cells in comparison with the 3T3-L1 cells (148). These in- vitro data, thus, provide supporting evidence that genetic manipulation of FST could favorably alter overall lipid metabolites known to be associated with fat mass and promote obesity and associated metabolic conditions (161, 162). In-vivo analysis of adipose tissues from Fst-Tg mice also show significant differences in several amino acids including leucine, isoleucine, and valine also collectively referred to as branched-chain amino acids (BCAA), key components of urea cycle and arginine metabolism, and components of the Kreb’s cycle including citrate, succinylcarnitine, and fumarate were significantly lower compared to the WT tissues. FST overexpression in Fst-Tg mice was associated with significant upregulation of two key BCAA catabolic proteins BCAT2 and BCKDHA in epididymal WAT (148). Several recent studies have provided convincing evidence in support of a positive association between BCAA levels and insulin resistance and type 2 diabetes as their levels are significantly induced in obese subjects compared to the lean humans (163, 164). Levels of ω-3 polyunsaturated fatty acids (PUFAs), reported to improve obesity-associated chronic inflammation, insulin resistance and dyslipidemia (165), and regulate several aspects of energy and lipid metabolism (166) were significantly increased in the subcutaneous WAT of Fst-Tg mice. Levels of key lysolipids, known metabolic regulators of childhood obesity (167) were significantly elevated in the Epi WAT of Fst-Tg compared to the WT mice. Combined together, these findings obtained from comprehensive metabolomic profiling of Fst transgenic mice provide compelling evidence that genetic manipulation of Fst in-vivo favorably alters the levels of key metabolites known to influence various aspects of metabolic conditions, and warrant future studies for the use of FST based therapeutic interventions to combat obesity and related diseases (168).
Conclusion
Obesity and associated comorbidities resulting from accumulation of dysfunctional white adipose tissues and chronic imbalance between energy intake and energy expenditure represent a growing worldwide problem. Activation of adipose browning characteristics leads to the dissipation of excess stored energy and provide metabolic benefits to combat the burden of obesity and related abnormalities including insulin resistance, hyperlipidemia, type 2 diabetes and cardiovascular diseases. Adipose browning phenomenon in humans has been confirmed based on both morphological, and functional studies (5, 6, 16). Accordingly, new strategies are being explored to identify novel compounds that can promote adipose browning and reduce the development of obesity and associated conditions. Recent reports from several laboratories provide convincing evidence that inhibition of TGF-β signaling pathway provides metabolic protection from obesity and diabetes by regulating glucose and energy homeostasis via activation of white adipose browning (13, 59). Genetic inactivation of MST, a key member of TGF-β superfamily not only results in increased muscle mass but also promotes activation of adipose browning and favorably alters several metabolic parameters implicated in the development of metabolic complications (86, 87). Since Fst is a known inhibitor of MST and reported to antagonize overall TGF-β signaling, it is logical to explore the therapeutic potential of FST in regulating key metabolic functions in both adipose depots besides its established role in promoting muscle mass. Recent findings by Braga et al. provided the first evidence that FST enhances the acquisition of beige and brown adipose characteristics by directly targeting Myf5- and Myf5+ populations to promote beige and brown adipose characteristics respectively (142). Since Myf5+ precursor population gives rise to both skeletal muscle and brown fat (20), it is not surprising that FST could selectively target these populations to promote both muscle and BAT mass (147, 169). Additionally, identification of key molecular and cellular targets responsible for FST-induced adipose browning is necessary to develop therapeutic strategies for the treatment of obesity and related diseases. Although activation of p38MAPK and ERK1/2 signaling is necessary for FST-induced adipose browning in both adipose depots (147), it is important to explore the possible role of irisin/Fndc5 during the process as secretion of irisin and subsequent activation of p38 MAPK and ERK1/2 has been reported during exercise (94). Since FST secretion is also induced following exercise (169, 170) and rFST treatment leads to elevated Fndc5 gene expression in muscle (86), it is possible that FST will indirectly affect p38MAPK and ERK1/2 activation via increased secretion of irisin. β3-AR signaling has been shown to promote p38 MAPK activation and induce browning of WAT and nonshivering thermogenesis in BAT (150, 171, 172). It is, therefore, possible that FST activates β3-AR signaling to promote p38 MAPK phosphorylation during adipose browning as β3 agonist CL 316,243 treatment elicited additive response in UCP1 levels in both WAT depots as well as in BAT (147). FGF21, another key regulator of adipose browning and a downstream target of β3-AR signaling (173, 174) is upregulated in WAT of Fst transgenic mice, suggesting a possible link between FST and FGF21 signaling during adipose browning. Based on available data, it appears that the beneficial effects of FST on adipose browning, obesity, and related metabolic conditions are mainly due to blocking of TGF-β ligands including MST and inhibition of Smad3 signaling as summarized in Figure 1. Finally, data obtained from Fst gene therapy studies in both human and nonhuman primates did not indicate apparent structural or functional aberration in various tissues, suggesting that FST may have therapeutic potential in clinical settings for the treatment of obesity and related diseases.
Author Contributions
RS and SP organized and wrote the manuscript. SR edited the manuscript and provided constructive comments. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Institute of Health grant numbers SC1AG049682 (RS), SC1CA232319 (SP), TRDRP grant number T31IP1551 (RS, SR), Boston Pepper Center P30 AG031679 (RS), UHI NIMHD S21MD000103, and Accelerating Excellence in Translational Sciences (AXIS) Center U54MD007598 to Charles R. Drew University of Medicine and Science.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Azra Tezein Devonshire for language editing.
References
1. Tam CS, Lecoultre V, Ravussin E. Brown adipose tissue: mechanisms and potential therapeutic targets. . Circulation (2012) 125:2782–91. doi: 10.1161/CIRCULATIONAHA.111.042929
2. The Toll of America’s Obesity. The New York Times (2018). Available at: https://www.nytimes.com/2018/08/09/opinion/cost-diabetes-obesity-budget.html.
3. Chen Y, Ikeda K, Yoneshiro T, Scaramozza A, Tajima K, Wang Q, et al. Thermal stress induces glycolytic beige fat formation via a myogenic state. Nature (2019) 565:180–5. doi: 10.1038/s41586-018-0801-z
4. Oguri Y, Shinoda K, Kim H, Alba DL, Bolus WR, Wang Q, et al. CD81 controls beige fat progenitor cell growth and energy balance via FAK signaling. Cell (2020) 182:563–77. doi: 10.1016/j.cell.2020.06.021
5. Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med (2009) 360(15):1509–17. doi: 10.1056/NEJMoa0810780
6. van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, et al. Cold-activated brown adipose tissue in healthy men. N Engl J Med (2009) 360(15):1500–8. doi: 10.1056/NEJMoa0808718
7. Cao L, Choi EY, Liu X, Martin A, Wang C, Xu X, et al. White to brown fat phenotypic switch induced by genetic and environmental activation of a hypothalamic-adipocyte axis. Cell Metab (2011) 14:324–38. doi: 10.1016/j.cmet.2011.06.020
8. Bartelt A, Heeren J. Adipose tissue browning and metabolic health. Nat Rev Endocrinol (2014) 10:24–36. doi: 10.1038/nrendo.2013.204
9. Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev (2004) 84:277–359. doi: 10.1152/physrev.00015.2003
10. Cohen P, Levy JD, Zhang Y, Frontini A, Kolodin DP, Svensson KJ, et al. Ablation of PRDM16 and beige adipose causes metabolic dysfunction and a subcutaneous to visceral fat switch. Cell (2014) 156:304–16. doi: 10.1016/j.cell.2013.12.021
11. Seale P, Conroe HM, Estall J, Kajimura S, Frontini A, Ishibashi J, et al. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J Clin Invest (2011) 121(1):96–105. doi: 10.1172/JCI44271
12. Zheng Q, Lin J, Huang J, Zhang H, Zhang R, Zhang X, et al. Reconstitution of UCP1 using CRISPR/Cas9 in the white adipose tissue of pigs decreases fat deposition and improves thermogenic capacity. Proc Natl Acad Sci USA (2017) 114(45):E9474–82. doi: 10.1073/pnas.1707853114
13. Yadav H, Quijano C, Kamaraju AK, Gavrilova O, Malek R, Chen W, et al. Protection from obesity and diabetes by blockade of TGF-β/Smad3 signaling. Cell Metab (2011) 14:67–79. doi: 10.1016/j.cmet.2011.04.013
14. Gilsanz V, Hu HH, Kajimura S. Relevance of brown adipose tissue in infancy and adolescence. Pediatr Res (2013) 73:3–9. doi: 10.1038/pr.2012.141
15. Palmer AK, Kirkland JL. Aging and adipose tissue: potential interventions for diabetes and regenerative medicine. Exp Gerontol (2016) 86:97–105. doi: 10.1016/j.exger.2016.02.013
16. Sidossis L, Kajimura S. Brown and beige fat in humans: thermogenic adipocytes that control energy and glucose homeostasis. J Clin Invest (2015) 125:478–86. doi: 10.1172/JCI78362
17. Atit R, Sgaier SK, Mohamed OA, Taketo MM, Dufort D, Joyner AL, et al. Beta-catenin activation is necessary and sufficient to specify the dorsal dermal fate in the mouse. Dev Biol (2006) 296:164–76. doi: 10.1016/j.ydbio.2006.04.449
18. Lepper C, Fan CM. Inducible lineage tracing of Pax7-descendant cells reveals embryonic origin of adult satellite cells. Genesis (2010) 48:424–36. doi: 10.1002/dvg.20630
19. Sanchez-Gurmaches J, Hung CM, Sparks CA, Tang Y, Li H, Guertin DA. PTEN loss in the Myf5 lineage redistributes body fat and reveals subsets of white adipocytes that arise from Myf5 precursors. Cell Metab (2012) 16:348–62. doi: 10.1016/j.cmet.2012.08.003
20. Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature (2008) 454:961–7. doi: 10.1038/nature07182
21. Kajimura S, Seale P, Spiegelman BM. Transcriptional control of brown fat development. Cell Metab (2010) 11:257–62. doi: 10.1016/j.cmet.2010.03.005
22. Harms M, Seale P. Brown and beige fat: development, function and therapeutic potential. Nat Med (2013) 19:1252–63. doi: 10.1038/nm.3361
23. Porter C, Chondronikola M, Sidossis LS. The therapeutic potential of brown adipocytes in humans. Front Endocrinol (Lausanne) (2015) 6:156. doi: 10.3389/fendo.2015.00156
24. Rodríguez A, Becerril S, Ezquerro S, Méndez-Giménez L, Frühbeck G. Crosstalk between adipokines and myokines in fat browning. Acta Physiol (Oxf) (2017) 219:362–81. doi: 10.1111/apha.12686
25. Abdullahi A, Jeschke MG. White adipose tissue browning: a double-edged sword. Trends Endocrinol Metab (2016) 27:542–52. doi: 10.1016/j.tem.2016.06.006
26. Long JZ, Svensson KJ, Tsai L, Zeng X, Roh HC, Kong X, et al. A smooth muscle-like origin for beige adipocytes. Cell Metab (2014) 19:810–20. doi: 10.1016/j.cmet.2014.03.025
27. Vishvanath L, MacPherson KA, Hepler C, Wang QA, Shao M, Spurgin SB, et al. Pdgfrβ+ mural preadipocytes con-tribute to adipocyte hyperplasia induced by high-fat-diet feeding and prolonged cold exposure in adult mice. Cell Metab (2016) 23:350–9. doi: 10.1016/j.cmet.2015.10.018
28. Berry DC, Jiang Y, Graff JM. Mouse strains to study cold-inducible beige progenitors and beige adipocyte formation and function. Nat Commun (2016) 7:10184. doi: 10.1038/ncomms10184
29. Lee YH, Petkova AP, Mottillo EP, Granneman JG. In vivo identification of bipotential adipocyte progenitors recruited by beta3-adrenoceptor activation and high-fat feeding. Cell Metab (2012) 15:480–91. doi: 10.1016/j.cmet.2012.03.009
30. Frontini A, Cinti S. Distribution and development of brown adipocytes in the murine and human adipose organ. Cell Metab (2010) 11:253–6. doi: 10.1016/j.cmet.2010.03.004
31. Rosenwald M, Perdikari A, Rülicke T, Wolfrum C. Bi-directional interconversion of brite and white adipocytes. Nat Cell Biol (2013) 15:659–67. doi: 10.1038/ncb2740
32. Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell (1998) 92:829–39. doi: 10.1016/s0092-8674(00)81410-5
33. Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell (1999) 98:115–24. doi: 10.1016/S0092-8674(00)80611-X
34. Harms MJ, Ishibashi J, Wang W, Lim HW, Goyama S, Sato T, et al. Prdm16 is required for the maintenance of brown adipocyte identity and function in adult mice. Cell Metab (2014) 19(4):593–604. doi: 10.1016/j.cmet.2014.03.007
35. Seale P, Kajimura S, Yang W, Chin S, Rohas LM, Uldry M, et al. Transcriptional control of brown fat determination by PRDM16. Cell Metab (2007) 6:38–54. doi: 10.1016/j.cmet.2007.06.001
36. Wu J, Boström P, Sparks LM, Ye L, Choi JH, Giang AH, et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell (2012) 150:366–76. doi: 10.1016/j.cell.2012.05.016
37. Wang W, Kissig M, Rajakumari S, Huang L, Lim HW, Won KJ, et al. Ebf2 is a selective marker of brown and beige adipogenic precursor cells. Proc Nat Acad Sci USA (2014) 111:14466–71. doi: 10.1073/pnas.1412685111
38. Lidell ME, Betz MJ, Leinhard OD, Heglind M, Elander L, Slawik M, et al. Evidence for two types of brown adipose tissue in humans. Nat Med (2013) 19:631–4. doi: 10.1038/nm.3017
39. Sharp LZ, Shinoda K, Ohno H, Scheel DW, Tomoda E, Ruiz L, et al. Human BAT possesses molecular signatures that resemble beige/brite cells. PloS One (2012) 7:e49452. doi: 10.1371/journal.pone.0049452
40. Petrovic N, Walden TB, Shabalina IG, Timmons JA, Cannon B, Nedergaard J. Chronic peroxisome proliferator activated receptor gamma (PPARgamma) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1- containing adipocytes molecularly distinct from classic brown adipocytes. J Biol Chem (2010) 285:7153–64. doi: 10.1074/jbc.M109.053942
41. Jespersen NZ, Larsen TJ, Peijs L, Daugaard S, Homøe P, Loft A, et al. A classical brown adipose tissue mRNA signature partly overlaps with brite in the supraclavicular region of adult humans. Cell Metab (2013) 17:798–805. doi: 10.1016/j.cmet.2013.04.011
42. Ussar S, Lee KY, Dankel SN, Boucher J, Haering MF, Kleinridders A, et al. ASC-1, PAT2, and P2RX5 are cell surface markers for white, beige, and brown adipocytes. Sci Trans Med (2014) 6:247ra103. doi: 10.1126/scitranslmed.3008490
43. Kim HJ, Cho H, Alexander R, Patterson HC, Gu M, Lo KA, et al. MicroRNAs are required for the feature maintenance and differentiation of brown adipocytes. Diabetes (2014) 63:4045–56. doi: 10.2337/db14-0466
44. Hu F, Wang M, Xiao T, Yin B, He L, Meng W, et al. miR-30 promotes thermogenesis and the development of beige fat by targeting RIP140. Diabetes (2015) 64:2056–68. doi: 10.2337/db14-1117
45. Sun L, Trajkovski M. MiR-27 orchestrates the transcriptional regulation of brown adipogenesis. Metabolism (2014) 63:272–82. doi: 10.1016/j.metabol.2013.10.004
46. Fu T, Seok S, Choi S, Huang Z, Suino-Powell K, Xu HE, et al. MicroRNA 34a inhibits beige and brown fat formation in obesity in part by suppressing adipocyte fibroblast growth factor 21 signaling and SIRT1 function. Mol Cell Biol (2014) 34:4130–42. doi: 10.1128/MCB.00596-14
47. Mori M, Nakagami H, Rodriguez-Araujo G, Nimura K, Kaneda Y. Essential role for miR-196a in brown adipogenesis of white fat progenitor cells. PloS Biol (2012) 10:e1001314. doi: 10.1371/journal.pbio.1001314
48. Liu W, Bi P, Shan T, Yang X, Yin H, Wang YX, et al. miR-133a regulates adipocyte browning in vivo. PloS Genet (2013) 9:e1003626. doi: 10.1371/journal.pgen.1003626
49. Budi EH, Duan D, Derynck R. Transforming growth factor-β receptors and Smads: Regulatory complexity and functional versatility. Trends Cell Biol (2017) 27:658–72. doi: 10.1016/j.tcb.2017.04.005
50. Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell (2003) 113:685–700. doi: 10.1016/s0092-8674(03)00432-x
51. Fain JN, Tichansky DS, Madan AK. Transforming growth factor beta1 release by human adipose tissue is enhanced in obesity. Metabolism (2005) 54(11):1546–51. doi: 10.1016/j.metabol.2005.05.024
52. Alessi MC, Bastelica D, Morange P, Berthet B, Leduc I, Verdier M, et al. Plasminogen activator inhibitor 1, transforming growth factor-beta 1, and BMI are closely associated in human adipose tissue during morbid obesity. Diabetes (2000) 49:1374–80. doi: 10.2337/diabetes.49.8.1374
53. Gordon KJ, Blobe GC. Role of transforming growth factor-beta superfamily. Signaling pathways in human disease. Biochim Biophys Acta (2008) 1782:197–228. doi: 10.1016/j.bbadis.2008.01.006
54. Herder C, Zierer A, Koenig W, Roden M, Meisinger C, Thorand B. Transforming growth factor-beta1 and incident type 2 diabetes: results from the MONICA/KORA case-cohort study, 1984-2002. Diabetes Care (2009) 32:1921–3. doi: 10.2337/dc09-0476
55. Perry JR, McCarthy MI, Hattersley AT, Zeggini E, Wellcome Trust Case Control Consortium, Weedon MN, et al. Interrogating type 2 diabetes genome-wide association data using a biological pathway-based approach. Diabetes (2009) 58(6):1463–7. doi: 10.2337/db08-1378
56. Casalena G, Daehn I, Bottinger E. Transforming growth factor-β, bioenergetics, and mitochondria in renal disease. Semin Nephrol (2012) 32:295–303. doi: 10.1016/j.semnephrol.2012.04.009
57. Mishra R, Cool BL, Laderoute KR, Foretz M, Viollet B, Simonson MS. AMP-activated protein kinase inhibits transforming growth factor-beta-induced Smad3-dependent transcription and myofibroblast trans-differentiation. J Biol Chem (2008) 283:10461–9. doi: 10.1074/jbc.M800902200
58. Verdin E, Hirschey MD, Finley LW, Haigis MC. Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends Biochem Sci (2010) 35:669–75. doi: 10.1016/j.tibs.2010.07.003
59. Fournier B, Murray B, Gutzwiller S, Marcaletti S, Marcellin D, Bergling S, et al. Blockade of the activin receptor IIb activates functional brown adi-pogenesis and thermogenesis by inducing mitochondrial oxidative metabolism. Mol Cell Biol (2012) 32:2871–9. doi: 10.1128/MCB.06575-11
60. Koncarevic A, Kajimura S, Cornwall-Brady M, Andreucci A, Pullen A, Sako D, et al. A novel therapeutic approach to treating obesity through modulation of TGFβ signaling. Endocrinology (2012) 153(7):3133–46. doi: 10.1210/en.2012-1016
61. Elsen MS, Raschke N, Tennagels U, Schwahn T, Jelenik M, Roden T, et al. BMP4 and BMP7 induce the white-to-brown transition of primary human adipose stem cells. Am J Phys Cell Physiol (2014) 306:C431–C44. doi: 10.1152/ajpcell.00290.2013
62. Gustafson A, Hammarstedt S, Hedjazifar JM, Hoffmann PA, Svensson J, Grimsby C, et al. BMP4 and BMP antagonists regulate human white and beige adipogenesis. Diabetes (2015) 64(5):1670–81. doi: 10.2337/db14-1127
63. Qian SW, Tang Y, Li X, Liu Y, Zhang YY, Huang HY, et al. BMP4-mediated brown fat-like changes in white adipose tissue alter glucose and energy homeostasis. Proc Natl Acad Sci USA (2013) 110:E798–807. doi: 10.1073/pnas.1215236110
64. Tang Y, Qian SW, Wu MY, Wang J, Lu P, Li XB, et al. BMP4 mediates the interplay between adipogenesis and angiogenesis during expansion of subcutaneous white adipose tissue. J Mol Cell Biol (2016) 8:302–12. doi: 10.1093/jmcb/mjw019
65. Whittle AJ, Carobbio S, Martins L, Slawik M, Hondares E, Vazquez MJ, et al. BMP8B increases brown adipose tissue thermogenesis through both central and peripheral actions. Cell (2012) 149:871–85. doi: 10.1016/j.cell.2012.02.066
66. Martins LP, Seoane-Collazo C, Contreras I, Gonzalez-Garcia N, Martinez-Sanchez F, Gonzalez JZ, et al. A functional link between AMPK and orexin mediates the effect of BMP8B on energy balance. Cell Rep (2016) 16:2231–42. doi: 10.1016/j.celrep.2016.07.045
67. Tseng YH, Kokkotou E, Schulz TJ, Huang TL, Winnay JN, Taniguchi CM, et al. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature (2008) 454(7207):1000–4. doi: 10.1038/nature07221
68. Thomas M, Langley B, Berry C, Sharma M, Kir S, Bass J, et al. Myostatin, a negative regulator of muscle growth, functions by inhibiting myoblast proliferation. J Biol Chem (2000) 275:40235–43. doi: 10.1074/jbc.M004356200
69. Zimmers TA, Davies MV, Koniaris LG, Haynes P, Esquela AF, Tomkinson KN, et al. Induction of cachexia in mice by systemically administered myostatin. Science (2002) 296:1486–8. doi: 10.1126/science.1069525
71. McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature (1997) 387(6628):83–90. doi: 10.1038/387083a0
72. McPherron AC, Lee SJ. Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci USA (1997) 94(23):12457–61. doi: 10.1073/pnas.94.23.12457
73. Kambadur R, Sharma M, Smith TP, Bass JJ. Mutations in myostatin (GDF8) in double-muscled Belgian Blue and Piedmontese cattle. Genome Res (1997) 7(9):910–6. doi: 10.1101/gr.7.9.910
74. Clop A, Marcq F, Takeda H, Pirottin D, Tordoir X, Bibe B, et al. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat Genet (2006) 38:813–8. doi: 10.1038/ng1810
75. Schuelke M, Wagner KR, Stolz LE, Hubner C, Riebel T, Komen W, et al. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med (2004) 350:2682–8. doi: 10.1056/NEJMoa040933
76. Lebrasseur NK. Building muscle, browning fat and preventing obesity by inhibiting myostatin. Diabetologia (2012) 55:13–7. doi: 10.1007/s00125-011-2361-8
77. Bernardo BL, Wachtmann TS, Cosgrove PC, Kuhn M, Opsahl AC, Judkins KM, et al. Postnatal PPAR delta activation and myostatin inhibition exert distinct yet complimentary effects on the metabolic profile of obese insulin-resistant mice. PloS One (2010), e113075. doi: 10.1371/journal.pone.0011307
78. Zhang L, Rajan V, Lin E, Hu Z, Han HQ, Zhou X, et al. Pharmacological inhibition of myostatin suppresses systemic inflammation and muscle atrophy in mice with chronic kidney disease. FASEB J (2011) 5(6):e11307. doi: 10.1096/fj.10-176917
79. Choi SJ, Yablonka-Reuveni Z, Kaiyala KJ, Ogimoto K, Schwartz MW, Wisse BEIncreased energy expenditure and leptin sensitivity account for low fat mass in myostatin-deficient mice. American Journal of Physiology. Endocrinol Metab (2011) 300:E1031–7. doi: 10.1152/ajpendo.00656.2010
80. Kim WK, Choi HR, Park SG, Ko Y, Bae KH, Lee SC. Myostatin inhibits brown adipocyte differentiation via regulation of Smad3-mediated β-catenin stabilization. Intern J Biochem Cell Biol (2012) 44:327–34. doi: 10.1016/j.biocel.2011.11.004
81. Braga M, Pervin S, Norris K, Bhasin S, Singh R. Inhibition of in vitro and in vivo brown fat differentiation program by myostatin. Obesity (2013) 21:1180–8. doi: 10.1002/oby.20117
82. Asterholm IW, Scherer PE. Enhanced metabolic flexibility associated with elevated adiponectin levels. Am J Pathol (2010) 176(3):1364–76. doi: 10.2353/ajpath.2010.090647
83. Combs TP, Pajvani UB, Berg AH, Lin Y, Jelicks LA, Laplante M, et al. A transgenic mouse with a deletion in the collagenous domain of adiponectin displays elevated circulating adiponectin and improved insulin sensitivity. Endocrinology (2004) 145:367–83. doi: 10.1210/en.2003-1068
84. Zhang C, McFarlane C, Lokireddy S, Masuda S, Ge X, Gluckman PD, et al. Inhibition of myostatin protects against diet-induced obesity by enhancing fatty acid oxidation and promoting a brown adipose phenotype in mice. Diabetologia (2012) 55:183–93. doi: 10.1007/s00125-011-2304-4
85. Cai C, Qian L, Jiang S, Sun Y, Wang Q, Ma D, et al. Loss-of-function myostatin mutation increases insulin sensitivity and browning of white fat in Meishan pigs. Oncotarget (2017) 8:34911–22. doi: 10.18632/oncotarget.16822
86. Shan T, Liang X, Bi P, Kuang S. Myostatin knockout drives browning of white adipose tissue through activating the AMPK-PGC1α-Fndc5 pathway in muscle. FASEB J (2013) 27:1981–9. doi: 10.1096/fj.12-225755
87. Dong J, Dong Y, Dong Y, Chen F, Mitch WE, Zhang L. Inhibition of myostatin in mice improves insulin sensitivity via irisin-mediated cross talk between muscle and adipose tissues. Int J Obes (Lond) (2016) 40:434–42. doi: 10.1038/ijo.2015.200
88. Kong X, Banks A, Liu T, Kazak L, Rao RR, Cohen P, et al. IRF4 is a key thermogenic transcriptional partner of PGC-1α. Cell (2014) 158:69–83. doi: 10.1016/j.cell.2014.04.049
89. Guo W, Wong S, Bhasin S. AAV-mediated administration of myostatin pro-peptide mutant in adult Ldlr null mice reduces diet-induced hepatosteatosis and arteriosclerosis. PloS One (2013) 8(8):e71017. doi: 10.1371/journal.pone.0071017
90. Berbée JF, Boon MR, Khedoe PP, Bartelt A, Schlein C, Worthmann A, et al. Brown fat activation reduces hypercholesterolaemia and protects from atherosclerosis development. Nat Commun (2015) 6:6356. doi: 10.1038/ncomms7356
91. Bartelt A, John C, Schaltenberg N, Berbée JFP, Worthmann A, Cherradi ML, et al. Thermogenic adipocytes promote HDL turnover and reverse cholesterol transport. Nat Commun (2017) 8:15010. doi: 10.1038/ncomms15010
92. Hoeke G, Kooijman S, Boon MR, Rensen PC, Berbée JF. Role of brown fat in lipoprotein metabolism and atherosclerosis. Circ Res (2016) 118:173–82. doi: 10.1161/CIRCRESAHA.115.306647
93. Pydi SP, Cui Z, He Z, Barella LF, Pham J, Cui Y, et al. Beneficial metabolic role of β-arrestin-1 expressed by AgRP neurons. Sci Adv (2020) 6(23):eaaz1341. doi: 10.1126/sciadv.aaz1341
94. Zhang Y, Li R, Meng Y, Li S, Donelan W, Zhao Y, et al. Irisin stimulates browning of white adipocytes through mitogen-activated protein kinase p38 MAP kinase and ERK MAP kinase signaling. Diabetes (2014) 63(2):514–25. doi: 10.2337/db13-1106
95. Mahgoub MO, D’Souza C, Al Darmaki RSMH, Baniyas MMYH, Adeghate E. An update on the role of irisin in the regulation of endocrine and metabolic functions. Peptides (2018) 104:15–23. doi: 10.1016/j.peptides.2018.03.018
96. Bostrom P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, et al. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature (2012) 481:463–8. doi: 10.1038/nature10777
97. Lee P, Linderman JD, Smith S, Brychta RJ, Wang J, Idelson C, et al. Irisin and FGF21 are cold-induced endocrine activators of brown fat function in humans. Cell Metab (2014) 19:302–9. doi: 10.1016/j.cmet.2013.12.017
98. Moreno-Navarrete JM, Ortega F, Serrano M, Guerra E, Pardo G, Tinahones F, et al. Irisin is expressed and produced by human muscle and adipose tissue in association with obesity and insulin resistance. J Clin Endocrinol Metab (2013) 98:E769–78. doi: 10.1210/jc.2012-2749
99. Hojlund K, Bostrom P. Irisin in obesity and type 2 diabetes. J Diabetes Complicat (2013) 27:303–4. doi: 10.1016/j.jdiacomp.2013.04.002
100. Zhang HJ, Zhang XF, Ma ZM, Pan LL, Chen Z, Han HW, et al. Irisin is inversely associated with intrahepatic triglyceride contents in obese adults. J Hepatol (2013) 59:557–62. doi: 10.1016/j.jhep.2013.04.030
101. Crujeiras AB, Pardo M, Casanueva FF. Irisin: ‘fat’ or artefact. Clin Endocrinol (2015) 82:467–74. doi: 10.1111/cen.12627
102. Raschke S, Eckel J. Adipo-myokines: two sides of the same coin–mediators of inflammation and mediators of exercise. Mediators Inflamm (2013) 2013:320724. doi: 10.1155/2013/320724
103. Albrecht E, Norheim F, Thiede B, Holen T, Ohashi T, Schering L, et al. Irisin - a myth rather than an exercise-inducible myokine. Sci Rep (2015) 5:8889. doi: 10.1038/srep08889
104. Erickson HP. Irisin and FNDC5 in retrospect: An exercise hormone or a transmembrane receptor? Adipocyte (2013) 2:289–93. doi: 10.4161/adip.26082
105. Huh JY, Panagiotou G, Mougios V, Brinkoetter M, Vamvini MT, Schneider BE, et al. FNDC5 and irisin in humans: I. Predictors of circulating concentrations in serum and plasma and II. mRNA expression and circulating concentrations in response to weight loss and exercise. Metabolism (2012) 61:1725–38. doi: 10.1016/j.metabol.2012.09.002
106. Timmons JA, Davidsen PK, Atherton PJBK. Is irisin a human exercise gene? Nature (2012) 488:E9–10. doi: 10.1038/nature11364
107. Crujeiras AB, Zulet MA, Lopez-Legarrea P, de la Iglesia R, Pardo M, Carreira MC, et al. Association between circulating irisin levels and the promotion of insulin resistance during the weight maintenance period after a dietary weight-lowering program in obese patients. Metabolism (2014) 63:520–31. doi: 10.1016/j.metabol.2013.12.007
108. de la Iglesia R, Lopez-Legarrea P, Crujeiras AB, Pardo M, Casanueva FF, Zulet MA, et al. Plasma irisin depletion under energy restriction is associated with improvements in lipid profile in metabolic syndrome patients. Clin Endocrinol (Oxf) (2014) 81:1–6. doi: 10.1111/cen.12383
109. Lopez-Legarrea P, de la Iglesia R, Crujeiras AB, Pardo M, Casanueva FF, Zulet MA, et al. Higher baseline irisin concentrations are associated with greater reductions in glycemia and insulinemia after weight loss in obese subjects. Nutr Diabetes (2014) 4:e110. doi: 10.1038/nutd.2014.7
110. Park KH, Zaichenko L, Brinkoetter M, Thakkar B, Sahin-Efe A, Joung KE, et al. Circulating irisin in relation to insulin resistance and the metabolic syndrome. J Clin Endocrinol Metab (2013) 98:4899–907. doi: 10.1210/jc.2013-2373
111. Pérez-Sotelo D, Roca-Rivada A, Baamonde I, Baltar J, Castro AI, Domínguez E, et al. Lack of Adipocyte-Fndc5/Irisin Expression and Secretion Reduces Thermogenesis and Enhances Adipogenesis. Sci Rep (2017) 7(1):16289. doi: 10.1038/s41598-017-16602-z
112. Chen N, Li Q, Liu J, Jia S. Irisin, an exercise-induced myokine as a metabolic regulator: an updated narrative review. Diabetes Metab Res Rev (2016) 32(1):51–9. doi: 10.1002/dmrr.2660
113. Vaughan RA, Gannon NP, Barberena MA, Garcia-Smith R, Bisoffi M, Mermier CM, et al. Characterization of the metabolic effects of irisin on skeletal muscle in vitro. Diabetes Obes Metab (2014) 16:711–8. doi: 10.1111/dom.12268
114. Arhire LI, Mihalache L, Covasa M. Irisin: A Hope in Understanding and Managing Obesity and Metabolic Syndrome. Front Endocrinol (Lausanne) (2019) 10:524. doi: 10.3389/fendo.2019.00524
115. Gao S, Li FL, Huang Y, Liu Y, Chen Y. Effects and molecular mechanism of GST-Irisin on lipolysis and autocrine function in 3T3-L1 adipocytes. PloS One (2016) 11:e0147480. doi: 10.1371/journal.pone.0147480
116. Tang HR, Yu S, Liu B, Huwatibieke Z, Li W, Zhang W. Irisin inhibits hepatic cholesterol synthesis via AMPK-SREBP2 signaling. EBio Med (2016) 6:139–48. doi: 10.1016/j.ebiom.2016.02.041
117. Duan H, Ma B, Ma X, Wang H, Ni Z, Wang B, et al. Anti-diabetic activity of recombinant irisin in STZ-induced insulin-deficient diabetic mice. Int J Biol Macromol (2016) 84:457–63. doi: 10.1016/j.ijbiomac.2015.04.030
118. Xiong XQ, Chen D, Sun HJ, Ding L, Wang JJ, Chen Q, et al. FNDC5 over-expression and irisin ameliorate glucose/lipid metabolic derangements and enhance lipolysis in obesity. Biochim Biophys Acta (2015) 1852:1867–75. doi: 10.1016/j.bbadis.2015.06.017
119. Li H, Zhang C, Liu J, Xie W, Xu W, Liang F, et al. Intraperitoneal administration of follistatin promotes adipocyte browning in high-fat diet-induced obese mice. PloS One (2019) 14(7):e0220310. doi: 10.1371/journal.pone.0226344
120. Xie C, Zhang Y, Tran DN, Wang H, Li S, George EV, et al. Irisin Controls Growth, Intracellular Ca2+ Signals, and Mitochondrial Thermogenesis in Cardiomyoblasts. PloS One (2015) 10(8):e0136816–e0136816. doi: 10.1371/journal.pone.0136816
121. Welt C, Sidis Y, Keutmann HT, Schneyer A. Activins, inhibins and follistatins: from endocrinology to signalling—a paradigm for the new millennium. Exp Biol Med (2002) 227:724–52. doi: 10.1177/153537020222700905
122. Sidis Y, Mukherjee A, Keutmann H, Delbaere A, Sadatsuki M, Schneyer A. Biological activity of follistatin isoforms and follistatin-like-3 is dependent on differential cell surface binding and specificity for activin, myostatin, and bone morphogenetic proteins. Endocrinology (2006) 147:3586–97. doi: 10.1210/en.2006-0089
123. Amthor H, Nicholas G, McKinnell I, Kemp CF, Sharma M, Kambadur R, et al. Follistatin complexes Myostatin and antagonises Myostatin-mediated inhibition of myogenesis. Dev Biol (2004) 270(1):19–30. doi: 10.1016/j.ydbio.2004.01.046
124. Hedger MP, Winnall WR, Phillips DJ, de Kretser DM. The regulation and functions of activin and follistatin in inflammation and immunity. Vitam Horm (2011) 85:255–97. doi: 10.1016/B978-0-12-385961-7.00013-5
125. Iemura S, Yamamoto TS, Takagi C, Uchiyama H, Natsume T, Shimasaki S, et al. Direct binding of follistatin to a complex of bone-morphogenetic protein and its receptor inhibits ventral and epidermal cell fates in early Xenopus embryo. Proc Natl Acad Sci USA (1998) 95(16):9337–42. doi: 10.1073/pnas.95.16.9337
126. Shimasaki S, Koga M, Esch F, Cooksey K, Mercado M, Koba A, et al. Primary structure of the human follistatin precursor and its genomic organization. Proc Natl Acad Sci USA (1988) 85:4218–22. doi: 10.1073/pnas.85.12.4218
127. Inouye S, Guo Y, DePaolo L, Shimonaka M, Ling N, Shimasaki S. Recombinant expression of human follistatin with 315 and 288 amino acids: chemical and biological comparison with native porcine follistatin. Endocrinology (1991) 129:815–22. doi: 10.1210/endo-129-2-815
128. Tsuchida K, Arai KY, Kuramoto Y, Yamakawa N, Hasegawa Y, Sugino H. Identification and characterization of a novel follistatin-like protein as a binding protein for the TGF-beta family. J Biol Chem (2000) 275:40788–96. doi: 10.1074/jbc.M006114200
129. Schneyer A, Sidis Y, Xia Y, Saito S, del Re E, Lin HY, et al. Differential actions of follistatin and follistatin-like 3. Mol Cell Endocrinol (2004) 225(1-2):25–8. doi: 10.1016/j.mce.2004.02.009
130. Schneyer A, Schoen A, Quigg A, Sidis Y. Differential binding and neutralization of activins A and B by follistatin and follistatin like-3 (FSTL-3/FSRP/FLRG). Endocrinology (2003) 144:1671–4. doi: 10.1210/en.2002-0203
131. Tortoriello DV, Sidis Y, Holtzman DA, Holmes WE, Schneyer AL. Human follistatin-related protein: a structural homologue of follistatin with nuclear localization. Endocrinology (2001) 142:3426–34. doi: 10.1210/endo.142.8.8319
132. Inouye S, Guo Y, Ling N, Shimasaki S. Site-specific mutagenesis of human follistatin. Biochem Biophys Res Commun (1991) 179(1):352–8. doi: 10.1016/0006-291x(91)91377-o
133. Brown ML, Bonomi L, Ungerleider N, Zina J, Kimura F, Mukherjee A, et al. Follistatin and follistatin like-3 differentially regulate adiposity and glucose homeostasis. Obesity (Silver Spring) (2011) 19(10):1940–9. doi: 10.1038/oby.2011.97
134. Lee YS, Lee SJ. Regulation of GDF-11 and myostatin activity by GASP-1 and GASP-2. Proc Natl Acad Sci U S A (2013) 110(39):E3713–22. doi: 10.1073/pnas.1309907110
135. Lee SJ. Extracellular Regulation of Myostatin: A Molecular Rheostat for Muscle Mass. Immunol Endocr Metab Agents Med Chem (2010) 10:183–94. doi: 10.2174/187152210793663748
136. Matzuk MM, Lu N, Vogel H, Sellheyer K, Roop DR, Bradley A. Multiple defects and perinatal death in mice deficient in follistatin. Nature (1995) 374(6520):360–3. doi: 10.1038/374360a0
137. Gilson H, Schakman O, Kalista S, Lause P, Tsuchida K, Thissen JP. Follistatin induces muscle hypertrophy through satellite cell proliferation and inhibition of both myostatin and activin. Am J Physiol Endocrinol Metab (2009) 297:E157–64. doi: 10.1152/ajpendo.00193.2009
138. Haidet AM, Rizo L, Handy C, Umapathi P, Eagle A, Shilling C, et al. Long-term enhancement of skeletal muscle mass and strength by single gene administration of myostatin inhibitors. Proc Natl Acad Sci USA (2008) 105:4318–22. doi: 10.1073/pnas.0709144105
139. Kota J, Handy CR, Haidet AM, Montgomery CL, Eagle A, Rodino-Klapac LR, et al. Follistatin gene delivery enhances muscle growth and strength in nonhuman primates. Sci Transl Med (2009) 1(6):6ra15. doi: 10.1126/scitranslmed.3000112
140. Nakatani M, Takehara Y, Sugino H, Matsumoto M, Hashimoto O, Hasegawa Y, et al. Transgenic expression of a myostatin inhibitor derived from follistatin increases skeletal muscle mass and ameliorates dystrophic pathology in mdx mice. FASEB J (2008) 22:477–87. doi: 10.1096/fj.07-8673com
141. Mendell JR, Sahenk Z, Malik V, Gomez AM, Flanigan KM, Lowes LP, et al. A phase 1/2a follistatin gene therapy trial for becker muscular dystrophy. Mol Ther (2015) 23:192–201. doi: 10.1038/mt.2014.200
142. Singh R, Bhasin S, Braga M, Artaza JN, Pervin S, Taylor WE, et al. Regulation of myogenic differentiation by androgens: cross talk between androgen receptor/ beta-catenin and follistatin/transforming growth factor-beta signaling pathways. Endocrinology (2009) 150(3):1259–68. doi: 10.1210/en.2008-0858
143. Braga M, Bhasin S, Jasuja R, Pervin S, Singh R. Testosterone inhibits transforming growth factor-β signaling during myogenic differentiation and proliferation of mouse satellite cells: potential role of follistatin in mediating testosterone action. Mol Cell Endocrinol (2012) 350(1):39–52. doi: 10.1016/j.mce.2011.11.019
144. Braga M, Reddy ST, Vergnes L, Pervin S, Grijalva V, Stout D, et al. Follistatin promotes adipocyte differentiation, browning, and energy metabolism. J Lipid Res (2014) 55(3):375–84. doi: 10.1194/jlr.M039719
145. Tang R, Harasymowicz NS, Wu CL, Collins KH, Choi YR, Oswald SJ, et al. Gene therapy for follistatin mitigates systemic metabolic inflammation and post-traumatic arthritis in high-fat diet-induced obesity. Sci Adv (2020) 6(19):eaaz7492. doi: 10.1126/sciadv.aaz7492
146. Lee SJ, McPherron AC. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci USA (2001) 98(16):9306–11. doi: 10.1073/pnas.151270098
147. Singh R, Braga M, Reddy ST, Lee SJ, Parveen M, Grijalva V, et al. Follistatin Targets Distinct Pathways To Promote Brown Adipocyte Characteristics in Brown and White Adipose Tissues. Endocrinology (2017) 158(5):1217–30. doi: 10.1210/en.2016-1607
148. Singh R, Pervin S, Lee SJ, Kuo A, Grijalva V, David J, et al. Metabolic profiling of follistatin overexpression: a novel therapeutic strategy for metabolic diseases. Diabetes Metab Syndr Obes (2018) 11:65–84. doi: 10.2147/DMSO.S159315
149. Robidoux J, Cao W, Quan H, Daniel KW, Moukdar F, Bai X, et al. Selective activation of mitogen-activated protein (MAP) kinase kinase 3 and p38alpha MAP kinase is essential for cyclic AMP-dependent UCP1 expression in adipocytes. Mol Cell Biol (2005) 25(13):5466–79. doi: 10.1128/MCB.25.13.5466-5479.2005
150. Cao W, Medvedev AV, Daniel KW, Collins S. Beta-adrenergic activation of p38 MAP kinase in adipocytes: cAMP induction of the uncoupling protein 1 (UCP1) gene requires p38 MAP kinase. J Biol Chem (2001) 276(29):27077–82. doi: 10.1074/jbc.M101049200
151. Bordicchia M, Liu D, Amri EZ, Ailhaud G, Dessì-Fulgheri P, Zhang C, et al. Cardiac natriuretic peptides act via p38 MAPK to induce the brown fat thermogenic program in mouse and human adipocytes. J Clin Invest (2012) 122:1022–36. doi: 10.1172/JCI59701
152. Arturo RR, Cecilia C, Senin LL, Landrove MO, Javier B, Ana BC, et al. FNDC5/irisin is not only a myokine but also an adipokine. PloS One (2013) 8(4):e60563. doi: 10.1371/journal.pone.0060563
153. Hill BG. Insights into an adipocyte whitening program. Adipocyte (2015) 4(1):75–80. doi: 10.4161/21623945.2014.960351
154. Peirce V, Carobbio S, Vidal-Puig A. The different shades of fat. Nature (2014) 510(7503):76–83. doi: 10.1038/nature13477
155. Abdullahi A, Jeschke MG. Taming the flames: targeting white adipose tissue browning in hypermetabolic conditions. Endocr Rev (2017) 38(6):538–49. doi: 10.1210/er.2017-00163
156. Fabbiano S, Suárez-Zamorano N, Rigo D, Veyrat-Durebex C, Stevanovic DA, Colin DJ, et al. Caloric restriction leads to browning of white adipose tissue through type 2 immune signaling. Cell Metab (2016) 24:434–46. doi: 10.1016/j.cmet.2016.07.023
157. Zhou Y, Jiang L, Rui L. Identification of MUP1 as a regulator for glucose and lipid metabolism in mice. J Biol Chem (2009) 284(17):11152–9. doi: 10.1074/jbc.M900754200
158. Hui X, Zhu W, Wang Y, Lam KSL, Zhang J, Wu D, et al. Major urinary protein-1 increases energy expenditure and improves glucose intolerance through enhancing mitochondrial function in skeletal muscle of diabetic mice. J Biol Chem (2009) 284(21):14050–7. doi: 10.1074/jbc.M109.001107
159. Davey JR, Estevez E, Thomson RE, Whitham M, Watt KI, Hagg A, et al. Intravascular Follistatin gene delivery improves glycemic control in a mouse model of type 2 diabetes. FASEB J (2020) 34(4):5697–714. doi: 10.1096/fj.201802059RRR
160. Zhao C, Qioa C, Tang R-H, Jiang J, Li J, Martin CB, et al. Overcoming insulin insufficiency by forced follistatin expression in β-cells of db/db mice. Mol Ther (2015) 23:866–74. doi: 10.1038/mt.2015.29
161. Xia JY, Holland WL, Kusminski CM, Sun K, Sharma AX, Pearson MJ, et al. Targeted induction of ceramide degradation leads to improved systemic metabolism and reduced hepatic steatosis. Cell Metab (2015) 22(2):266–78. doi: 10.1016/j.cmet.2015.06.007
162. Barber MN, Risis S, Yang C, Meikle PJ, Staples M, Febbraio MA, et al. Plasma lysophosphatidylcholine levels are reduced in obesity and type 2 diabetes. PloS One (2012) 7(7):e41456. doi: 10.1371/journal.pone.0041456
163. McCormack SE, Shaham O, McCarthy MA, Deik AA, Wang TJ, Gerszten RE, et al. Circulating branched-chain amino acid concentrations are associated with obesity and future insulin resistance in children and adolescents. Pediatr Obes (2013) 8(1):52–61. doi: 10.1111/j.2047-6310.2012.00087.x
164. Lynch CJ, Adams SH. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat Rev Endocrinol (2014) 10(12):723–36. doi: 10.1038/nrendo.2014.171
165. Martínez-Fernández L, Laiglesia LM, Huerta AE, Martínez JA, Moreno-Aliaga MJ. Omega-3 fatty acids and adipose tissue function in obesity and metabolic syndrome. Prostaglandins Other Lipid Mediat (2015) 121(Pt A):24–41. doi: 10.1016/j.prostaglandins.2015.07.003
166. Kim J, Okla M, Erickson A, Carr T, Natarajan SK, Chung S. Eicosapentaenoic acid potentiates brown thermogenesis through FFAR4-dependent Up-regulation of miR-30b and miR-378. J Biol Chem (2016) 291(39):20551–62. doi: 10.1074/jbc.M116.721480
167. Butte NF, Liu Y, Zakeri IF, Mohney RP, Mehta N, Voruganti VS, et al. Global metabolomic profiling targeting childhood obesity in the Hispanic population. Am J Clin Nutr (2015) 102(2):256–67. doi: 10.3945/ajcn.115.111872
168. Singh R, Charles R. (2017). Drew University of Medicine and Science-Assignee; United States Patents, US9682093B2.
169. Li JX, Cummins CL. Getting the Skinny on Follistatin and Fat. Endocrinology (2017) 158(5):1109–12. doi: 10.1210/en.2017-00223
170. Hansen J, Brandt C, Nielsen AR, Hojman P, Whitham M, Febbraio MA, et al. Exercise induces a marked increase in plasma follistatin: evidence that follistatin is a contraction-induced hepatokine. Endocrinology (2011) 152(1):164–71. doi: 10.1210/en.2010-0868
171. Collins S, Surwit RS. The beta-adrenergic receptors and the control of adipose tissue metabolism and thermogenesis. Recent Prog Horm Res (2001) 56:309–28. doi: 10.1210/rp.56.1.309
172. Zheng Z, Liu X, Zhao Q, Zhang L, Li C, Xue Y. Regulation of UCP1 in the Browning of Epididymal Adipose Tissue by β3-Adrenergic Agonist: A Role for MicroRNAs. Int J Endocrinol (2014) 2014:530636. doi: 10.1155/2014/530636
173. Douris N, Stevanovic DM, Fisher FM, Cisu TI, Chee MJ, Nguyen NL, et al. Central Fibroblast Growth Factor 21 Browns White Fat via Sympathetic Action in Male Mice. Endocrinology (2015) 156(7):2470–81. doi: 10.1210/en.2014-2001
Keywords: follistatin, myostatin, obesity, transforming growth Factor β, UCP1, adipose tissue
Citation: Pervin S, Reddy ST and Singh R (2021) Novel Roles of Follistatin/Myostatin in Transforming Growth Factor-β Signaling and Adipose Browning: Potential for Therapeutic Intervention in Obesity Related Metabolic Disorders. Front. Endocrinol. 12:653179. doi: 10.3389/fendo.2021.653179
Received: 14 January 2021; Accepted: 19 March 2021;
Published: 09 April 2021.
Edited by:
Xinran Ma, East China Normal University, ChinaReviewed by:
Meng Dong, Institute of Zoology, Chinese Academy of Sciences (CAS), ChinaAbir Mukherjee, Royal Veterinary College (RVC), United Kingdom
Copyright © 2021 Pervin, Reddy and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rajan Singh, cmFqYW5zaW5naEBtZWRuZXQudWNsYS5lZHU=