 Sravya Jannapureddy
Sravya Jannapureddy Mira Sharma
Mira Sharma Gautham Yepuri
Gautham Yepuri Ann Marie Schmidt
Ann Marie Schmidt Ravichandran Ramasamy
Ravichandran Ramasamy- Diabetes Research Program, Division of Endocrinology, Diabetes and Metabolism, Department of Medicine, NYU Grossman School of Medicine, New York, NY, United States
Diabetes is a leading cause of cardiovascular morbidity and mortality. Despite numerous treatments for cardiovascular disease (CVD), for patients with diabetes, these therapies provide less benefit for protection from CVD. These considerations spur the concept that diabetes-specific, disease-modifying therapies are essential to identify especially as the diabetes epidemic continues to expand. In this context, high levels of blood glucose stimulate the flux via aldose reductase (AR) pathway leading to metabolic and signaling changes in cells of the cardiovascular system. In animal models flux via AR in hearts is increased by diabetes and ischemia and its inhibition protects diabetic and non-diabetic hearts from ischemia-reperfusion injury. In mouse models of diabetic atherosclerosis, human AR expression accelerates progression and impairs regression of atherosclerotic plaques. Genetic studies have revealed that single nucleotide polymorphisms (SNPs) of the ALD2 (human AR gene) is associated with diabetic complications, including cardiorenal complications. This Review presents current knowledge regarding the roles for AR in the causes and consequences of diabetic cardiovascular disease and the status of AR inhibitors in clinical trials. Studies from both human subjects and animal models are presented to highlight the breadth of evidence linking AR to the cardiovascular consequences of diabetes.
Introduction
Diabetes prevalence worldwide has been increasing at an alarming rate. World Health organization estimates that currently greater than 400 million people live with diabetes (https://www.who.int/health-topics/diabetes). As the number of people with diabetes has increased, consequent increases in diabetic complications has been observed (1, 2). Among the various diabetic complications, cardiovascular disease (CVD) is the leading cause of morbidity and mortality in patients with diabetes mellitus (3). CVD entities include increased sensitivity of diabetic myocardium to ischemic episodes (4) and diabetic cardiomyopathy, manifested as a subnormal functional response of the diabetic heart independent of coronary artery disease (5, 6). Macrovascular disease in patients with diabetes, includes, atherosclerosis (7), coronary artery disease (CAD), peripheral vascular disease (PVD) and stroke (8), and restenosis of large vessels (9–11). The United Kingdom Prospective Diabetes Study (UKPDS) demonstrated that despite significant reductions in HbA1c, diabetes related mortality and myocardial infarction (MI) events were not reduced (12).
CVD significantly reduces the median life expectancy for diabetic adults in the 55–64 age group (13, 14). This is likely due to diabetes specific cardiovascular disorders. Key among them is the accelerated atherosclerosis in diabetes, with greater infiltration of inflammatory cells, and larger necrotic core size (15). While the deaths due to CAD have declined in the general population, the reduction in deaths due to CAD has been much less dramatic in diabetic patients (16). Another factor contributing to CV death in diabetics is heart failure. Prevalence of diastolic heart failure with preserved ejection fraction (HFpEF) and systolic heart failure with reduced ejection fraction (HFrEF) are higher in patients with diabetes compared to those without diabetes. While the precise mechanisms by which diabetes mediates heart failure are unknown, contributors include impaired endothelial dysfunction, pathways driving fibrosis, cardiomyocyte dysfunction, and defective remodeling after myocardial infarction (17, 18). Similarly, the Framingham Heart Study showed that diabetes independently increases the risk of heart failure (19–21). Recently meta-analysis of sixteen CV outcome trials by Sacre et al. (22) found that hospitalization for heart failure and myocardial infarction are the most frequent CV events in clinical trials in Type 2 diabetes.
High rate of mortality, post-MI, has been observed in people with diabetes vs those without diabetes (21, 23, 24), presumably due to ventricular arrhythmia (25). Mechanisms causing arrhythmias in diabetes include calcium channel function changes driven by downregulation of SERCA2a and increased phosphorylation of the ryanodine receptor (26, 27), oxidative stress (28, 29), AGEs-RAGE axis (29–31). Cardiac autonomic neuropathy prevalent in patients with diabetes, has been linked to increased risk for fatal cardiac arrhythmias (32).
In the recent outbreak of the coronavirus disease 2019 (COVID-19), diabetes and cardiovascular disease are risk factors for severe adverse clinical outcome in COVID19 patients (33). Emerging data reveal that diabetes and obesity are among the strong predictors for hospitalization among COVID-19 patients and risk factor for severe COVID-19 morbidity and mortality (34–38). In these hospitalized COVID-19 patients, myocardial infarction with or without obstructive coronary lesions (33, 39) were observed. These and the above findings strongly highlight the urgent need for focused therapies for alleviating the devastating impact of cardiovascular complications induced by diabetes. Global efforts are underway to find more effective strategies to mitigate and or attenuate the devastating consequences of diabetic cardiovascular complications.
In this review, we will focus on aldose reductase (AR), its possible link to the cardiovascular complications of diabetes mellitus and the potential impact of pharmacological inhibition of AR on cardiovascular complications of diabetes.
Hyperglycemia in Cardiovascular Cells
One of the key mechanisms by which chronic hyperglycemia(CH) exerts its deleterious effects on CV tissue involves nonenzymatic glycation reactions of reducing sugars with free amino groups of proteins, DNA, and lipids. Amadori products formed by this reaction leads to the formation of advanced glycation end products (AGEs) (40–48). These derivatives can bind to pre-existing cell surface receptors of AGEs and such interactions often lead to generation of reactive oxygen species through perturbation of NADPH oxidase (33–38, 49). CV tissue is less dependent on insulin for glucose uptake from extracellular environment due to abundance of GLUT1, an insulin independent glucose transporter in the plasma membrane (50, 51). During CH, there is chronic and abnormal influx of extracellular glucose due to down regulation of GLUTs altering the biochemical homeostasis of cardiovascular cells (52, 53). Consequently, changes, in flux via the polyol pathway, cytoplasmic redox state, activity of specific isoforms of protein kinase C, in the glucosamine biosynthesis pathway, and production of glycating species are observed (9, 53–61).
The present review mainly focuses on aldose reductase (AR), the first enzyme of the polyol pathway that regulates the uptake of excess glucose by the cardiovascular cells.
Properties of AR and Its Gene, ALD2
Aldose reductase (E.C. 1.1.1.21; AKR1B1, ALD2, or AR), a monomeric enzyme of ~35,900 Daltons, belongs to the aldo-keto reductase superfamily (62–67). The enzyme reversibly binds NADPH when it reduces an aldehydic substrate to the corresponding alcohol, e.g., glucose to sorbitol.
AR reduces a variety of aldehydic substrates with differing affinities (68, 69). The enzyme efficiently catalyzes reduction of glyceraldehyde, 4-hydorxynonenal (4-HNE), 2-methylpentanal, methylglyoxal, retinoids and host of other aldehydes (69–71). These AR studies determined that the Km values for the above substrates are in the range of 8 to 50 µmol/L. For glucose, Inagaki et al. (72) and Grimshaw (73) showed for AR a Km for the open chain of glucose of 0.66 µmol/L.
Oxidation of cysteine residue, Cys 298 causes AR to exhibit altered activity and inhibitor sensitivity (74). AR activity is altered by S−nitrosothiols (75), activated by nitric oxide (NO) under ischemic/acidic conditions (76) or inhibited by elevated NO levels in non-acidic conditions (77). In human tissues AR occurs mostly in the reduced enzyme form (78).
The human AR gene (ALD2 or AKR1B1), approximately 18 kilobases (kb) long and includes ten exons coding for 316 amino acids, has been mapped to locus q35 on human chromosome 7 (79, 80). The TATA box (at -37), a CCAAT box (−104), and an androgen-like response element (−396–382) are in the ALD2 promoter region of (81). The region containing three osmotic response elements: OreA, OreB and OreC reside upstream of the transcription start site (82).
ALD2 gene polymorphisms have been found to be associated with most diabetic complications (83, 84). Microsatellite polymorphism in (AC)n repeat region located ~2.1 kb upstream of the transcription start site was first identified in patients with diabetic retinopathy (85). Subsequent studies detected single nucleotide polymorphisms C(-106)T (86) and C(−12)G (87) in the basal promoter region of the ALD2 gene. An intragenic polymorphism in the BamHI site consisting of an A to C substitution associated with diabetic retinopathy, was also identified (88). Studies by Demaine et al. and Moczulski et al. (89, 90) showed that the (AC)n and C(-106)T polymorphisms are closely linked. Majority of studies have demonstrated an association between polymorphisms in the ALD2 gene and the increased risk for rapid onset or increased prevalence of diabetic complications. The “Z-2” (AC)23 microsatellite polymorphism has been associated with high expression levels of AR (91) and with diabetic retinopathy (85, 89), diabetic nephropathy (90–93). The link between Z-2 allele and diabetic neuropathy is rather modest (94).
In some studies an association between ALD2 alleles and complications risk has not been detected (95, 96). In one study of Type 2 diabetic patients, although no association of Z−2 with proteinuria was found, a statistically significant association of erythrocyte AR concentration with proteinuria was found (97). ALD2 gene polymorphism has been detected in Type 2 diabetic patients with cardiorenal complications (98) and microangiopathy (99). It is important to note that most studies across the globe has demonstrated link between diabetic complications and AL2 polymorphisms (100–104).
Polyol Pathway and the Osmotic Hypothesis
Based on thee replicated genetic links, the impact of chronically elevated glucose metabolism via the AR pathway aka polyol pathway has received considerable attention in the study of diabetic complications. In this pathway (105), AR in the presence of NADPH reduces glucose to sorbitol, while sorbitol dehydrogenase (SDH) uses NAD+ to oxidize sorbitol to fructose. The pioneering studies of Kinoshita, Gabbay, Dvornik and colleagues (106) demonstrated the presence of elevated polyol pathway intermediates in diabetic rat tissues and suggested a pathogenic link to diabetic complications. In the seminal “Osmotic Hypothesis” paradigm, high levels of glucose are metabolized through AR and SDH to sorbitol and fructose. Accumulation of sorbitol in tissues like eye lens induces a osmosis driven cascade of altered ion and metabolite homeostasis, culminating in the formation of the sugar cataract (106). Data demonstrating an accelerated rate of sorbitol accumulation and cataract formation in human AR transgenic, and SDH-deficient mice (107) provides clear confirmation of this mechanism for sugar cataract formation.
Polyol Pathway and Metabolic Flux Hypothesis
The past several decades of research have reemphasized that in many tissues/cells the polyol pathway is integrally linked via its coenzymes to various metabolic and signaling pathways (108–111). In studies involving the lens tissue, it was shown that increased flux via the polyol pathway, increased turnover of NADPH (112) and that AR and antioxidant defense enzyme glutathione reductase compete for the same pool of cytoplasmic NADPH. Another study showed that increased metabolic flux via the polyol pathway impairs the glycolysis in diabetic hearts, resulting from competition between SDH and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) for cytosolic NAD+ (58). Furthermore, studies by Williamson and his team have demonstrated increased polyol pathway flux modulates the ratio of free cytosolic NADH to NAD+ and consequently impairs neural and vascular function (113–117).
Realization that in conjunction with possible osmotic stress in vascular tissue, excess metabolic flux of glucose through AR affects key pathways linked to diabetic complications via its ability to generate precursors/intermediates/activators, has heightened interest in AR and the polyol pathway.
The presence and levels of AR vary in tissues and cells (118), with the inner medulla of kidney expressing the highest amount of AR (119). While sciatic nerve, lens, testis, heart, and cornea, express high levels of AR, organs/tissues such as liver, renal cortex, stomach, spleen, lung, small intestine, and colon express low levels of AR (119). AR is present in cells such as cardiomyocytes, endothelial cells, smooth muscle cells, and fibroblasts. In this review, we summarize the key data on AR and evidence linking AR to cardiovascular complications in diabetes.
AR and Its Physiological Role
To date, the basic physiological function of AR remains elusive (120). By synthesizing intracellular sorbitol, AR forms parts of a multi-tiered renal osmolyte system that helps protect cells in the renal inner medulla from the locally high osmotic stress (121, 122). Interestingly, AR inhibiton results in upregulation of sorbitol compensatory pathways in the renal osmolyte system (123).
Several potential physiological roles have been proposed, they include (a) generation of intermediates to facilitate production of advanced glycation end product precursors (124–127), (b) process to divert glucose from glycolysis and glucose oxidation (53), (c) participation in the metabolism of steroids (128), norepinephrine intermediates (129), detoxification of aldehydes, e.g., (130), or of their glutathionylated derivatives (131). Like AR, aldehyde dehydrogenase 2 (ALDH2) has been shown to detoxify 4-hydroxynonenal (4-HNE) and is expressed in cardiovascular cells (132, 133). Furthermore, studies have shown that 4HNE is a substrate ALDH2, with Km and Vmax values of 14.3 μM and 3.5 nmol min-1 mg protein-1, respectively (134, 135). The fact that AR, aldehyde reductases, and aldehyde dehydrogenases can essentially compete for various aldehydes (136, 137), makes it challenging to determine specific physiological role for AR.
In addition to the enzymatic activity, two recent studies have revealed other functions for AR. First, our studies (138) showed that the interaction of AR with deacetylation domain (DAD) of the nuclear corepressors, silencing mediator of retinoic and thyroid receptor (SMRT) and nuclear corepressor 1 (NCOR1), could lead to histone deacetylase 3 (HDAC3) degradation (Figure 1). HDAC3 binds to the DAD of either (SMRT) or (NCOR1) and protects itself from degradation (139). We observed that interaction of AR with DAD of SMRT/NCOR1 in hearts of ischemic, diabetic, and aged mice drives HDAC3 degradation, consequently leading to PPARγ activation and lipid accumulation in the hearts (138). These findings revealed a novel role for AR in modulating lipid metabolism via its ability to regulate HDAC3 degradation and consequent activation of PPARγ.
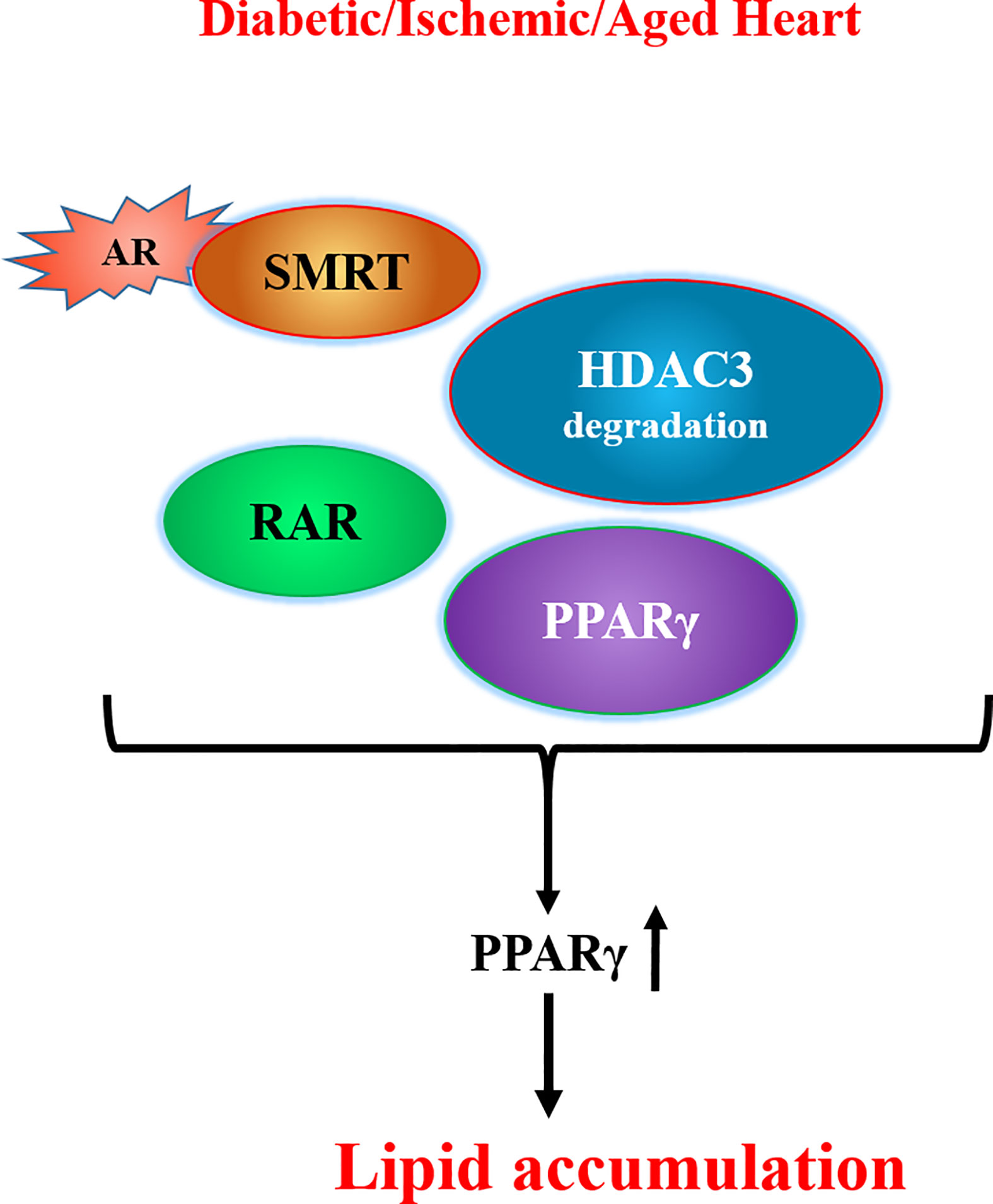
Figure 1 Scheme showing competition between AR and HDAC3 for the DAD of SMRT/NCOR1 and consequent transcriptional changes leading to lipid accumulation. [adapted from (128)]. AR denotes aldose reductase; DAD refers to deacetylation domain of the nuclear corepressors, SMRT refers to silencing mediator of retinoic and thyroid receptor, NCOR1 denotes nuclear corepressor 1, RAR denotes retinoic acid receptor, HDAC3 denotes histone deacetylase 3.
Second, AR actions independent of its enzymatic activity were revealed in a study by Shimizu et al. (140). Using phosphoproteome analysis and molecular studies, they showed that AR phosphorylation/dephosphorylation is essential for the transduction of T cell receptor-mediated T-cell stimulatory signals. Notably, they showed that AR expression in T cells was unaffected by TCR stimulation or by the presence of suppressor signals from immunosuppressive macrophages. Importantly, upregulation of ERK1/2-mediated signaling pathways in T lymphocytes was linked to AR phosphorylation driven events. Shimizu et al. (140) concluded that AR mediates intracellular transmission of the suppressor signal of immunosuppressive macrophages toward downstream ERK1/2 pathways, possibly through its direct interaction with acceptor proteins.
Adding to the AR functional conundrum are data from mice devoid of AR (141). These AR null mice, otherwise normal from structural, biochemical, reproductive and physiological standpoint, display mild polyuria, and mild polydipsia (141), and moderately altered divalent cation levels (142). Nerve conduction velocity (NCV) is unaffected by the overexpression of AR; however, in a diabetic setting, in marked contrast to the fall in NCV in wild type mice, NCV is normal in the AR knockout mouse (141). Similarly, cardiac contractile function is unaffected by pharmacological inhibition of AR (143–145).
Though the physiological function of AR in normal cellular and organ physiology is unclear, the pathogenic role of AR as a key player mediating diabetic complications is well established. This review will focus on the pathogenic role of AR in diabetic cardiovascular complications.
Diabetic Cardiac Ischemia and AR
The presence and activity of AR in cardiac myocytes of rats and rabbits has been demonstrated in several studies, e.g., (76, 143–147), and cardiac sorbitol and fructose tissue concentrations were shown to be significantly increased in diabetic rats compared to control rats (146). Studies have shown that diabetes and ischemia increase AR activity in hearts (76, 148) and that blockade of AR with ARI zopolrestat or sorbinil was found to improve cardiac glucose metabolism and to dramatically reduce acute ischemia-reperfusion-induced cardiac damage in diabetic rat hearts and in non-diabetic rat and rabbit hearts (76, 143–145).
Humans have much greater activity of AR than mice. For this reason, we used a transgenic mouse line in which human AR (hAR) was expressed via a histocompatibility gene promoter (149). These transgenic mice have tissue levels of AR activity comparable to those of humans (108). These hAR transgenic mice have been invaluable in recapitulating human diabetic cardiovascular disease. When subjected to ischemia/reperfusion (I/R), hearts from hAR transgenic mice exhibited greater injury, reduced ATP levels, and impaired functional recovery than wild-type mice (148). AR inhibitor zopolrestat attenuated I/R injury and improved functional recovery in these hAR transgenic mice (148). Studies in hAR transgenic mice addressing potential mechanisms revealed that opening the mitochondrial permeability transition pore (MPTP) (Figure 2) is linked to increased I/R injury (150). Increased generation of hydrogen peroxide, and reduced levels of antioxidant glutathione were key to MPTP opening in these hAR mice undergoing I/R (150). Attenuation of reactive oxygen species generation either by antioxidants or by ARIs reduced MPTP opening and reduced I/R injury in hAR transgenic mice hearts (150). Since MPTP opening is linked to phosphorylation of glycogen synthase kinase 3 β (GSK3β), subsequent studies in hAR mice and AR null mice, revealed that flux via AR reduces phosphorylation GSK3β via the Akt pathway in I/R hearts (151). These studies linked key signaling mechanisms by which AR impairs MPTP opening in I/R hearts.
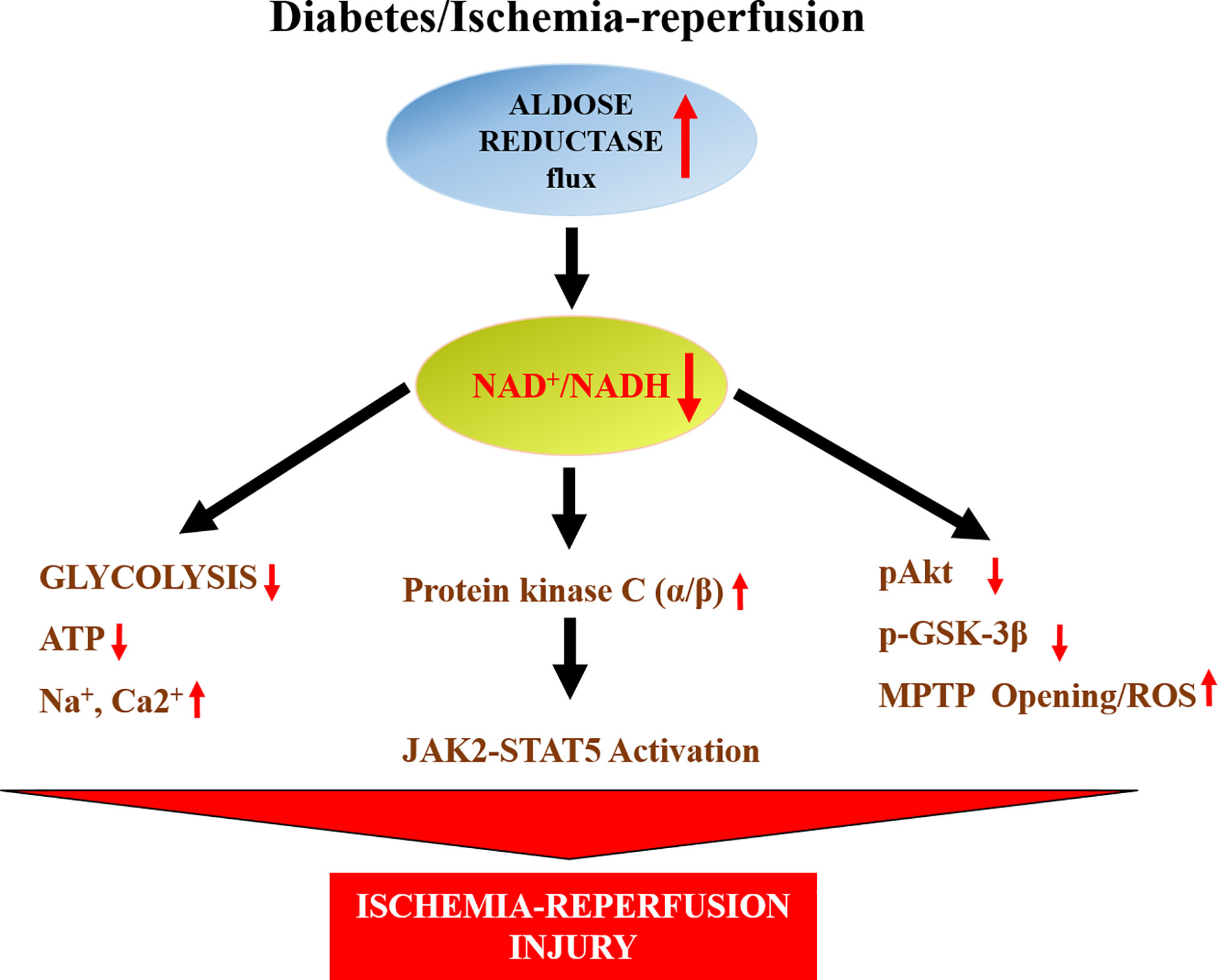
Figure 2 Scheme displays the impact of AR on changes in NAD+/NADH and consequent changes in glycolysis, mitochondrial properties and key signaling pathways leading to ischemic injury in hearts. ATP- adenosine triphosphate; JAK2- Janus activated kinase 2; MPTP-mitochondrial permeability transition pore, Akt- a serine/threonine-specific protein also known as Protein kinase B (PKB), pAkt- phosphorylated Akt, GSK3β-Glycogen synthase kinase 3 beta, STAT5- Signal transducer and activator of transcription 5.
Studies by Hwang et al. (152), in isolated perfused rat and mice hearts, revealed that ischemia drives JAK2 phosphorylation followed by STAT5 activation and that inhibition of AR or SDH blocked JAK2 and STAT5 activation (Figure 2). Furthermore, using pharmacological strategies they showed that the activation of JAK2-STAT5 pathway during ischemia in hAR mice was dependent on lowering of cytosolic NAD+/NADH and increased protein kinase C α/β activity. These data (152) (Figure 2), showed that AR mediates myocardial ischemic injury by modulating NAD+/NADH/protein kinase C α/β/JAK-STAT signaling.
To determine if AR actions in the heart are specifically in cardiomyocytes, we generated mice with cardiac specific expression of human AR (hAR) using the α-myosin heavy chain (MHC) promoter (153). Cardiomyocyte specific hAR transgenic expression did not alter cardiac function or glucose and fatty acid (FA) oxidation gene expression in young mice, whereas cardiac dysfunction was observed in older mice. Like the global hAR transgenic mice, these cardiac specific hAR mice also had greater infarct area and reduced functional recovery than non-transgenic littermates. In these studies, when the hAR transgene was crossed onto the PPAR alpha knockout background, hAR expressing mice had increased heart fructose content, cardiac fibrosis, reactive oxygen species (ROS), and apoptosis. These studies informed us that cardiomyocyte specific overexpression of hAR leads to cardiac dysfunction with aging and in the setting of reduced FA oxidation and increased glucose metabolism (153).
Studies addressing the short term and long term remodeling consequences of in vivo I/R injury model revealed that AR null mice was protected, in part, due to short term activation of the β-catenin pathway and subsequent increases in mesenchymal markers and fibrosis provoking genes (154). The increased activity of the β-catenin pathway and its downstream target genes in AR null mice was observed at early time points (48 h) of recovery after ligation of the descending coronary artery. At later time points of recovery (28 days), these changes in β-catenin activity were not observed in the AR null mice hearts. Thus, these data demonstrated that long term protection in AR null mice hearts was independent of β-catenin pathway.
In vitro and cellular studies described in the earlier sections of this review indicate that AR can detoxify aldehydes, such as 4-HNE, that accumulate during I/R. Studies in hAR expressing mice hearts have demonstrated increased injury and poor functional recovery after I/R (148, 155), along with increased oxidative stress. Furthermore, studies in AR-null mice hearts revealed reduced oxidative stress and reduced I/R injury (156). Similar findings linking increased AR activity and flux to increased oxidative stress has been demonstrated in rat hearts (157–161). It is possible that activation of aldehyde dehydrogenase 2 (ALDH2) reduces 4-HNE accumulation and protects hearts from I/R injury (162). Furthermore, it is possible that as shown in some studies, glutathione adduct of 4-HNE (GS-HNE) is converted by AR to its dihydroxynonane form (GS-DHN) and that inhibition of AR reduces GS-DHN and mitigates adverse signaling mechanisms driving inflammation and injury (163–165). In the context of diabetes, ALDH2 activity is known to be reduced in multiple tissue, including the heart (132, 133). Could the accumulation of 4-HNE observed in the diabetic hearts (especially during I/R) be due to lack of ALDH2? Comprehensive murine studies are warranted to establish the precise role of AR vs ALDH2 in modulating 4-HNE metabolism cascade in I/R hearts.
Like in I/R hearts, flux via AR is also increased in diabetic cardiomyopathy and heart failure. AR and SDH protein expression, activities and substrate flux were increased in hearts of Type 2 BBZDR diabetic rat hearts along with functional changes (166). AR expression was attenuated in pacing induced canine model of heart failure (167). Hearts tissue samples from patients with ischemic cardiomyopathy and diabetic cardiomyopathy exhibited elevated AR expression (168). These observations provide rationale for addressing the role of AR in mediating cardiac dysfunction and heart failure, both in diabetic and non-diabetic models.
In summary, the studies discussed establish AR as a key driver of functional and metabolic impairment in diabetic and ischemic hearts and that blockade of AR presents a therapeutic target for protection of these stressed hearts
Atherosclerosis in Diabetes and AR
Patients with diabetes are at increased risk for CAD (5, 6). Gleissner et al. showed that AR is expressed in CD68+ cells (monocytes/macrophages) from human atherosclerotic plaques (108, 169), and that patients with diabetes had significantly greater CD68+AR+ macrophages in the plaques than patients without diabetes (170).
As discussed earlier, hAR transgenic mice (149) exhibit AR levels similar to those observed in humans. Previously, we reported that overexpression of hAR in LDL receptor knockout (Ldlr−/−) (171) and apolipoprotein E null (Apoe−/−) (172) mice promoted atherosclerosis under hyperglycemic conditions and that pharmacological inhibition of AR reduced lesion size (172). Subsequent studies by us probed the mechanisms by which AR promoted atherosclerosis in hyperglycemic conditions.
Early events in atherosclerosis progression include endothelial dysfunction and upregulation of VCAM-1 (173). Vedantham et al. showed that, in both diabetic Apoe−/− mice and in human atherosclerotic carotid artery, AR is expressed in endothelial cells and this endothelial AR leads to endothelial dysfunction and increased expression of VCAM-1 and MMP-2 (148). Importantly, this study showed that AR inhibition improved endothelial function and was linked to attenuated VCAM-1 and MMP-2 expression (148) and that these findings were similar to those observed by blockade of RAGE in atherosclerotic Apoe−/− mice (174). Studies in cells linked AR to AGE and RAGE activation and consequent changes in intercellular adhesion molecule-1 and monocyte chemoattractant protein-1, migration, and monocyte adhesion (175) and that ARI or AR antisense oligonucleotides (176) blocked these changes, suggesting that, AR may promote progression of atherosclerotic plaques via AGE-RAGE axis. Pharmacological studies have shown that AR inhibitor treatments improves endothelium-dependent relaxation to acetylcholine of aortas from diabetic rabbits (176), diabetic rats (177), and galactosemic rats (177). Taken together, these findings establish a central role for AR pathway as a key mediator of impaired endothelium-dependent relaxation, endothelial dysfunction, cell adhesion, and inflammatory events in diabetic blood vessels.
Mechanisms probing cellular and in vivo studies to address link between AR and inflammation, revealed that flux via AR impaired drives inflammatory gene changes via Egr-1. Specifically, changes in AR activity and flux reduces NAD+ levels triggering reduced activity of NAD+-dependent deacetylase Sirt-1 and consequent acetylation and prolonged expression of Egr-1 in hyperglycemic conditions (Figure 3) (178). These data established a novel AR-SIRT1-EGR1 mechanism by which glucose may lead to proinflammatory and prothrombotic responses in diabetic atherosclerosis.
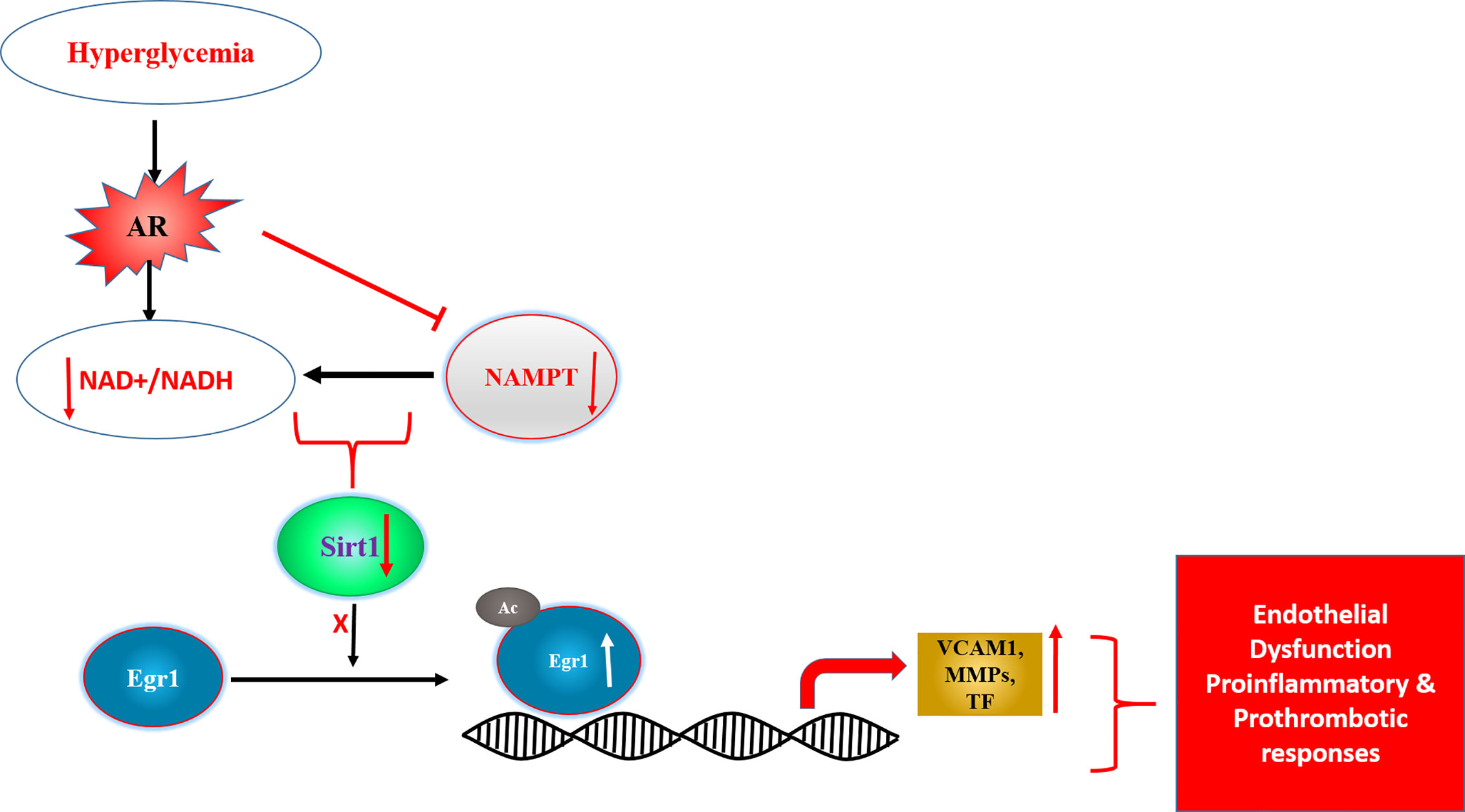
Figure 3 Scheme displaying AR driven changes in NAD+/NADH and SIRT1 activity as key driver of transcription factor Egr1 acetylation and consequent induction of proinflammatory and prothrombotic genes. [adapted from (169)]. Egr1- early growth response 1, SIRT1- NAD+ dependent Sirtuin1, Ac- acetylation, NAMPT-Nicotinamide phosphoribosyltransferase, VCAM1- vascular cell adhesion molecule 1, MMPs- matrix metalloproteinases, TF-tissue factor.
While the studies in hAR overexpressing mice revealed that AR promotes atherosclerosis progression in diabetes, an unexpected finding of increased early lesion size was observed in diabetic Apoe−/−mice devoid of AR (179). In this study lesion size positively correlated with 4-HNE in mice devoid of AR, which the authors postulated was likely to reduced metabolism of toxic aldehydes. Given the potential impact of ALDH-2 in detoxifying 4-HNE (162) and recent studies by Singh et al. showing that macrophages from AR null mice exhibit higher basal and lipopolysaccharide stimulated phagocytic activity (180), additional studies are warranted to understand the mechanisms in play when AR is deleted in Apoe−/−mice. Human AR expression does appear to recapitulate human diabetic atherosclerosis more closely in Ldlr−/− and Apoe−/− mice models, suggesting that, AR deletion may have unintended consequences, including compensatory regulation influencing vascular properties.
Diabetic patients demonstrate impaired atherosclerosis regression and persistent absolute risk level of a cardiovascular event following lipid lowering drugs compared to nondiabetic patients. Murine studies, in atherosclerosis regression models, attributed the impairment to hyperglycemia-induced monocytosis and recruitment of these macrophages to plaques (181). Yuan et al. (182), using Type 1 diabetic Akita mice with and without hAR overexpression and aortic transplantation model, addressed the role of AR in impaired atherosclerosis regression in diabetes. In the surgical model of atherosclerosis regression, the donor aortic arch containing the preformed atherosclerotic plaques are transplanted into a recipient mice that are kept on normal chow diet. Yuan et al. transplanted donor aorta into the following recipient mice; either Ldlr−/−, non-diabetic wild type, Akita, hAR transgenic, or Akita/hAR mouse. In the recipient Type 1 diabetic mice, hyperglycemia significantly impaired the decrease in percent of CD68+ lesion area, even after hyperlipidemia was attenuated. The combination of Akita with overexpression of hAR significantly increased the percent of lesion macrophage content in the plaques, suggesting continued atherosclerosis progression. Plaque CD68+ cells from the Akita+/-/hAR mice demonstrated increased oxidant stress as measured by DHE fluorescence. They also exhibited higher expression of genes linked to pro-inflammation and reduced expression of anti-inflammatory genes. This study demonstrated that hAR expression amplifies impaired atherosclerosis regression in Type 1 diabetic mice. Taken together, the atherosclerosis progression and regression studies in diabetes demonstrate a key pathogenic role for AR and that interventions to block AR may be beneficial in diabetic atherosclerosis.
AR and platelets in diabetes
Platelet abnormalities, one of the hallmarks of diabetes, contributes to the pathogenesis of atherosclerosis and thrombosis. Studies by Tang et al. (183), in human platelets, demonstrated that AR plays a key role in mediating thromboxane release, increased cell surface thromboxane receptor expression, and enhanced platelet activity in human platelets treated with hyperglycemic conditions and/or collagen. Importantly, they linked these changes to increased oxidative stress and the activation of PLCγ2, PKCβII, PKCδ, and p38α MAPK (183). Furthermore, studies in diabetic subjects and humanized AR transgenic mice rendered diabetic with STZ revealed that hyperglycemia driven AR activation and subsequent increases in oxidative stress leads to increased p53 phosphorylation, followed by mitochondrial dysfunction, damage, and rupture of platelets by sequestration of the antiapoptotic protein Bcl-xL (184). Taken together, these human and animal studies established that AR is key mediator of abnormal platelet activity in diabetes, thus adding to the multiple processes that contribute to the pathogenesis of diabetic cardiovascular complications.
AR and Vascular Injury
Diabetes is known to cause increased restenosis after angioplasty. AR plays a central role in smooth muscle cell (SMC) proliferation caused by balloon injury in animal models of restenosis. Studies have shown that AR inhibition prevents SMC growth in in animal models of restenosis (185–190). Studies in cells and tissue demonstrated that high glucose flux via the AR pathway leads to diacylglycerol accumulation and consequent protein kinase C activation (186). In addition, AR was shown to modulate hyperglycemia and TNF-α driven increases in the extracellular signal–related kinase/mitogen-activated protein kinase and phosphatidylinositol 3-kinase, (187), as well as activation of nuclear factor κB (188), and G1/S-phase proteins E2F-1, cdks, and cyclins (191). These signaling changes lead to upregulation of SMC chemotaxis, vascular inflammation, and cell adhesion. AR inhibition attenuated the above signaling events and arrested proliferation and migration of SMCs. Findings from these cellular and animal studies provide a compelling rationale for testing AR inhibitors for safety and efficacy in diabetic patients undergoing angioplasty and at risk for restenosis (185–190).
AR Inhibitors and Properties
AR inhibitors (ARIs) have been extensively reviewed in the literature, e.g., (53, 83, 192). At this time, epalrestat is the only ARI that is being used, in Japan, India, and China, to treat patients with diabetic neuropathy (98, 192). X−ray crystallographic studies of ARIs revealed that they bind in the active site of AR. Most ARIs that have been tested in human trials belong to the chemical classes of spirohydantions or carboxylic acids (192, 193). ARIs of the carboxylic acid class are quite selective for AR vs. aldehyde reductase (192, 194). One efficacy challenge of the carboxylic acid class of ARIs is that they are highly protein bound in vivo. Hydantoin class of ARIs inhibit both aldehyde and AR with comparable efficacies (192, 195), thus likely to cause to off target effects. Another strategic approach that is under active consideration is the design and use of inhibitors to preferentially inhibit glucose reduction while preserving the detoxifying ability of AR toward toxic aldehydes (196, 197).
The number of patents filed over the last 5 years demonstrates that, after decades of uncertainty, scientific interest in AR and its inhibitors has resurged. During the last 5 years, a number of synthetic compounds have been designed and patented as AR inhibitors, mainly belonging to the carboxylic-type class. Inspired by the well-known inhibitor zopolrestat, Mylari and co-workers designed a novel class of carboxylic acid inhibitors and its water-soluble formulations (198–201) to overcome some of the limitations of this class of ARIs. Shendelman recently patented two novel series of phthalazino and pyrazinopyridazino derivatives (202). Although these compounds are closely related to ones described by Mylari and coworkers, in their heterocyclic portion, the novel derivatives possess a boronic residue that replaces the carboxylic acid moiety. This gives the ARI field a rather new chemical approach for developing active inhibitors. These newly developed ARIs are actively being tested for its efficacy in animal and human diabetic cardiovascular disorders.
Clinical Applications of ARIs in Humans
Initial studies on AR inhibition attenuating injury and improving functional recovery after I/R in both diabetic and nondiabetic hearts generated considerable interest toward testing these molecules for diabetic heart disease (143, 144, 148, 203, 204), and for development of new ARIs (193, 194). In clinical studies, AR inhibitor, zopolrestat, treated diabetic subjects displayed increased left ventricular ejection fraction (LVEF), cardiac output, left ventricle stroke volume and exercise LVEF (205) whereas, placebo-treated diabetic subjects exhibited decreased exercise cardiac output, stroke volume and end diastolic volume (205). Didangelos et al. showed that AR inhibition beneficially altered heart rate variability in patients with severe or moderate diabetic autonomic neuropathy (206). These promising studies in human subjects with established diabetic complications paved the way for the development and use of new ARIs, such as AT-001, currently in clinical trials. Multicenter, randomized, placebo-controlled, 2-part study to evaluate the safety and efficacy of AT-001, a novel AR inhibitor, in adult patients with diabetic cardiomyopathy at high risk of progression to overt heart failure, is currently in progress (NCT04083339). AT-001 treatment for 28 days was shown to reduce blood levels of sorbitol and N-terminal pro-B-type natriuretic peptide levels in diabetic patients (207). In addition to its role in mediating cardiac dysfunction and injury, preclinical studies have shown that AR exacerbates lung inflammation (204). Taken together these studies formed the basis for the current testing of ARI in COVID19 patients [see review by Kadosh et al. (208)]. Currently, AR inhibitor, AT-001 is undergoing trials to assess safety and efficacy in reducing inflammation and cardiac injury in COVID-19 diabetic patients with heart disease (NCT04365699). Taken together, these findings from animal and human studies strongly suggest that AR promotes diabetic cardiovascular complications. Current large randomized multicenter human trials using the newly developed potent ARIs are likely to establish its therapeutic potential in diabetic cardiovascular complications.
Conclusions
The DCCT, UKPDS, and prior ARI studies have indicated that relatively long clinical trials with focused recruitment strategies and end points will be needed to demonstrate efficacy on microvascular and macrovascular complications of diabetes. Focus on prevention or slowing of cardiovascular disease progression in diabetic patients should be the primary goal, not rapid reversal of disease endpoints. Despite developmental setbacks over the last two decades, preclinical and clinical evidence linking progression of diabetic cardiovascular complications and elevated flux via AR unambiguously confirm pathogenic role for AR in mediating diabetic complications. Importantly, data on genetic polymorphisms of ALD2 from around the globe indicate an association between “high AR” alleles and diabetic complications. The preclinical and clinical data reviewed above indicate that inhibiting AR could play a key role in our therapeutic strategy to prevent/arrest progression of diabetic cardiovascular complications. Recent development of potent AR inhibitors and new formulations has set the stage for successful clinical testing of these molecules in patients with diabetic cardiovascular complications.
Author Contributions
SJ, MS, and RR wrote the first draft of the manuscript and completed all of the editing. GY and RR prepared figures. SJ, MS, GY, AS, and RR provided critical comments on the manuscript and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
AS and RR are supported, in part, by funds from the Diabetes Research Program, NYU Grossman School of Medicine and grants from the U.S. Public Health Service (P01HL143697, R01HL132516, and R01DK109675 (to AS and RR). SJ was supported by a training grant for medical students [T35 DK007421, RR (PI)]. RR is a consultant for Applied Therapeutics and receives funding from them for ARI studies.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Ms. Latoya Woods for assistance in the preparation of this manuscript. AS and RR are supported, in part, by funds from the Diabetes Research Program, NYU Grossman School of Medicine and grants from the National Institutes of Health [P01HL143697, R01HL132516, and R01DK109675 to AS and RR].
References
1. Demir Y, Taslimi P, Koçyiğit ÜM, Akkuş M, Özaslan MS, Duran HE, et al. Determination of the inhibition profiles of pyrazolyl-thiazole derivatives against aldose reductase and α-glycosidase and molecular docking studies. Archiv der Pharm (2020) 353(12):e2000118. doi: 10.1002/ardp.202000118
2. Taslimi P, Aslan HE, Demir Y, Oztaskin N, Maraş A, Gulçin İ, et al. Diarylmethanon, bromophenol and diarylmethane compounds: Discovery of potent aldose reductase, α-amylase and α-glycosidase inhibitors as new therapeutic approach in diabetes and functional hyperglycemia. Int J Biol Macromol (2018) 119:857–63. doi: 10.1016/j.ijbiomac.2018.08.004
3. Jandeleit-Dahm K, Cooper ME. Hypertension and diabetes. Curr Opin Nephrol Hypertens (2002) 11(2):221–8. doi: 10.1097/00041552-200203000-00014
4. Lopaschuk GD. Metabolic abnormalities in the diabetic heart. Heart Fail Rev (2002) 7(2):149–59. doi: 10.1023/A:1015328625394
5. Zarich SW, Nesto RW. Diabetic cardiomyopathy. Am Heart J (1989) 118(5 Pt 1):1000–12. doi: 10.1016/0002-8703(89)90236-6
6. Sowers JR, Epstein M, Frohlich ED. Diabetes, hypertension, and cardiovascular disease: an update. Hypertens (Dallas Tex 1979) (2001) 37(4):1053–9. doi: 10.1161/01.HYP.37.4.1053
7. Beckman JA, Creager MA, Libby P. Diabetes and atherosclerosis: epidemiology, pathophysiology, and management. Jama (2002) 287(19):2570–81. doi: 10.1001/jama.287.19.2570
8. Weinberger J. Prevention of Ischemic Stroke. Curr Treat Options Cardiovasc Med (2002) 4(5):393–403. doi: 10.1007/s11936-002-0019-z
9. Wendt T, Bucciarelli L, Qu W, Lu Y, Yan SF, Stern DM, et al. Receptor for advanced glycation endproducts (RAGE) and vascular inflammation: insights into the pathogenesis of macrovascular complications in diabetes. Curr Atheroscl Rep (2002) 4(3):228–37. doi: 10.1007/s11883-002-0024-4
10. Malloy MJ, Kane JP. A risk factor for atherosclerosis: triglyceride-rich lipoproteins. Adv Internal Med (2001) 47:111–36.
11. Gotto AM Jr. Management of dyslipidemia. Am J Med (2002) 112(Suppl 8A):10s–8s. doi: 10.1016/S0002-9343(02)01085-9
12. Group UPDSU. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet (London England) (1998) 352(9131):837–53. doi: 10.1016/S0140-6736(98)07019-6
13. Gu K, Cowie CC, Harris MI. Mortality in adults with and without diabetes in a national cohort of the U.S. Popul 1971-1993 Diabetes Care (1998) 21(7):1138–45. doi: 10.2337/diacare.21.7.1138
14. Reaven GM. Multiple CHD risk factors in type 2 diabetes: beyond hyperglycaemia. Diabetes Obes Metab (2002) 4(Suppl 1):S13–8. doi: 10.1046/j.1462-8902.2001.00037.x
15. Virmani R, Burke AP, Kolodgie F. Morphological characteristics of coronary atherosclerosis in diabetes mellitus. Can J Cardiol (2006) 22(Suppl B):81b–4b. doi: 10.1016/S0828-282X(06)70991-6
16. Gu K, Cowie CC, Harris MI. Diabetes and decline in heart disease mortality in US adults. Jama (1999) 281(14):1291–7. doi: 10.1001/jama.281.14.1291
17. Shah MS, Brownlee M. Molecular and Cellular Mechanisms of Cardiovascular Disorders in Diabetes. Circ Res (2016) 118(11):1808–29. doi: 10.1161/CIRCRESAHA.116.306923
18. Burchfield JS, Xie M, Hill JA. Pathological ventricular remodeling: mechanisms: part 1 of 2. Circulation (2013) 128(4):388–400. doi: 10.1161/CIRCULATIONAHA.113.001878
19. Dei Cas A, Khan SS, Butler J, Mentz RJ, Bonow RO, Avogaro A, et al. Impact of diabetes on epidemiology, treatment, and outcomes of patients with heart failure. JACC Heart Fail (2015) 3(2):136–45. doi: 10.1016/j.jchf.2014.08.004
20. Nichols GA, Hillier TA, Erbey JR, Brown JB. Congestive heart failure in type 2 diabetes: prevalence, incidence, and risk factors. Diabetes Care (2001) 24(9):1614–9. doi: 10.2337/diacare.24.9.1614
21. Kenny HC, Abel ED. Heart Failure in Type 2 Diabetes Mellitus. Circ Res (2019) 124(1):121–41. doi: 10.1161/CIRCRESAHA.118.311371
22. Sacre JW, Magliano DJ, Shaw JE. Incidence of Hospitalization for Heart Failure Relative to Major Atherosclerotic Events in Type 2 Diabetes: A Meta-analysis of Cardiovascular Outcomes Trials. Diabetes Care (2020) 43(10):2614–23. doi: 10.2337/dc20-0654
23. Donahoe SM, Stewart GC, McCabe CH, Mohanavelu S, Murphy SA, Cannon CP, et al. Diabetes and mortality following acute coronary syndromes. Jama (2007) 298(7):765–75. doi: 10.1001/jama.298.7.765
24. Shan J, Xie W, Betzenhauser M, Reiken S, Chen BX, Wronska A, et al. Calcium leak through ryanodine receptors leads to atrial fibrillation in 3 mouse models of catecholaminergic polymorphic ventricular tachycardia. Circ Res (2012) 111(6):708–17. doi: 10.1161/CIRCRESAHA.112.273342
25. Wang A, Green JB, Halperin JL, Piccini JP Sr. Atrial Fibrillation and Diabetes Mellitus: JACC Review Topic of the Week. J Am Coll Cardiol (2019) 74(8):1107–15. doi: 10.1016/j.jacc.2019.07.020
26. Banerjee PS, Ma J, Hart GW. Diabetes-associated dysregulation of O-GlcNAcylation in rat cardiac mitochondria. Proc Natl Acad Sci USA (2015) 112(19):6050–5. doi: 10.1073/pnas.1424017112
27. Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, et al. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature (2013) 502(7471):372–6. doi: 10.1038/nature12537
28. Joseph LC, Barca E, Subramanyam P, Komrowski M, Pajvani U, Colecraft HM, et al. Inhibition of NAPDH Oxidase 2 (NOX2) Prevents Oxidative Stress and Mitochondrial Abnormalities Caused by Saturated Fat in Cardiomyocytes. PloS One (2016) 11(1):e0145750. doi: 10.1371/journal.pone.0145750
29. Karam BS, Chavez-Moreno A, Koh W, Akar JG, Akar FG. Oxidative stress and inflammation as central mediators of atrial fibrillation in obesity and diabetes. Cardiovasc Diabetol (2017) 16(1):120. doi: 10.1186/s12933-017-0604-9
30. Bell DSH, Goncalves E. Atrial fibrillation and type 2 diabetes: Prevalence, etiology, pathophysiology and effect of anti-diabetic therapies. Diabetes Obes Metab (2019) 21(2):210–7. doi: 10.1111/dom.13512
31. Kato T, Yamashita T, Sekiguchi A, Tsuneda T, Sagara K, Takamura M, et al. AGEs-RAGE system mediates atrial structural remodeling in the diabetic rat. J Cardiovasc Electrophysiol (2008) 19(4):415–20. doi: 10.1111/j.1540-8167.2007.01037.x
32. Vinik AI, Ziegler D. Diabetic cardiovascular autonomic neuropathy. Circulation (2007) 115(3):387–97. doi: 10.1161/CIRCULATIONAHA.106.634949
33. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet (2020) 395(10229):1054–62. doi: 10.1016/S0140-6736(20)30566-3
34. Cummings MJ, Baldwin MR, Abrams D, Jacobson SD, Meyer BJ, Balough EM, et al. Epidemiology, clinical course, and outcomes of critically ill adults with COVID-19 in New York City: a prospective cohort study. Lancet (London England) (2020) 395(10239):1763–70. doi: 10.1016/S0140-6736(20)31189-2
35. Petrilli CM, Jones SA, Yang J, Rajagopalan H, O’Donnell L, Chernyak Y, et al. Factors associated with hospital admission and critical illness among 5279 people with coronavirus disease 2019 in New York City: prospective cohort study. BMJ (Clin Res ed) (2020) 369:m1966. doi: 10.1136/bmj.m1966
36. Stokes EK, Zambrano LD, Anderson KN, Marder EP, Raz KM, El Burai Felix S, et al. Coronavirus Disease 2019 Case Surveillance - United States, January 22-May 30, 2020. MMWR Morb Mortal Wkly Rep (2020) 69(24):759–65. doi: 10.15585/mmwr.mm6924e2
37. de Lusignan S, Dorward J, Correa A, Jones N, Akinyemi O, Amirthalingam G, et al. Risk factors for SARS-CoV-2 among patients in the Oxford Royal College of General Practitioners Research and Surveillance Centre primary care network: a cross-sectional study. Lancet Infect Dis (2020) 20(9):1034–42. doi: 10.1016/S1473-3099(20)30371-6
38. Jordan RE, Adab P. Who is most likely to be infected with SARS-CoV-2? Lancet Infect Dis (2020) 20(9):995–6. doi: 10.1016/S1473-3099(20)30395-9
39. Bangalore S, Sharma A, Slotwiner A, Yatskar L, Harari R, Shah B, et al. ST-Segment Elevation in Patients with Covid-19 - A Case Series. New Engl J Med (2020) 382(25):2478–80. doi: 10.1056/NEJMc2009020
40. Litwinoff E, Hurtado Del Pozo C, Ramasamy R, Schmidt AM. Emerging Targets for Therapeutic Development in Diabetes and Its Complications: The RAGE Signaling Pathway. Clin Pharmacol Ther (2015) 98(2):135–44. doi: 10.1002/cpt.148
41. Ramasamy R, Shekhtman A, Schmidt AM. The multiple faces of RAGE–opportunities for therapeutic intervention in aging and chronic disease. Expert Opin Ther Targets (2016) 20(4):431–46. doi: 10.1517/14728222.2016.1111873
42. López-Díez R, Shekhtman A, Ramasamy R, Schmidt AM. Cellular mechanisms and consequences of glycation in atherosclerosis and obesity. Biochim Biophys Acta (2016) 1862(12):2244–52. doi: 10.1016/j.bbadis.2016.05.005
43. Ramasamy R, Friedman RA, Shekhtman A, Schmidt AM. The receptor for advanced glycation end products (RAGE) and DIAPH1: unique mechanisms and healing the wounded vascular system. Expert Rev Proteomics (2019) 16(6):471–4. doi: 10.1080/14789450.2018.1536551
44. Ruiz HH, Ramasamy R, Schmidt AM. Advanced Glycation End Products: Building on the Concept of the “Common Soil” in Metabolic Disease. Endocrinology (2020) 161(1):1–10. doi: 10.1210/endocr/bqz006
45. Egaña-Gorroño L, López-Díez R, Yepuri G, Ramirez LS, Reverdatto S, Gugger PF, et al. Receptor for Advanced Glycation End Products (RAGE) and Mechanisms and Therapeutic Opportunities in Diabetes and Cardiovascular Disease: Insights From Human Subjects and Animal Models. Front Cardiovasc Med (2020) 7:37. doi: 10.3389/fcvm.2020.00037
46. Ahmed N, Thornalley PJ. Advanced glycation endproducts: what is their relevance to diabetic complications? Diabetes Obes Metab (2007) 9(3):233–45. doi: 10.1111/j.1463-1326.2006.00595.x
47. Ahmed N, Thornalley PJ. Advanced glycation endproducts: what is their relevance to diabetic complications? Diabetes Obes Metab (2007) 9(3)233–45. doi: 10.1111/j.1463-1326.2006.00595.x
48. Schmidt AM, Yan SD, Yan SF, Stern DM. The biology of the receptor for advanced glycation end products and its ligands. Biochim Biophys Acta (2000) 1498(2–3):99–111. doi: 10.1016/S0167-4889(00)00087-2
49. Wautier MP, Chappey O, Corda S, Stern DM, Schmidt AM, Wautier JL. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am J Physiol Endocrinol Metab (2001) 280(5):E685–94. doi: 10.1152/ajpendo.2001.280.5.E685
50. Ramasamy R, Hwang YC, Whang J, Bergmann SR. Protection of ischemic hearts by high glucose is mediated, in part, by GLUT-4. Am J Physiol Heart Circulatory Physiol (2001) 281(1):H290–7. doi: 10.1152/ajpheart.2001.281.1.H290
51. Depre C, Vanoverschelde JL, Taegtmeyer H. Glucose for the heart. Circulation (1999) 99(4):578–88. doi: 10.1161/01.CIR.99.4.578
52. Heilig CW, Brosius FC3, Henry DN. Glucose transporters of the glomerulus and the implications for diabetic nephropathy. Kidney Int Suppl (1997) 60:S91–9.
53. Oates PJ. Polyol pathway and diabetic peripheral neuropathy. Int Rev Neurobiol (2002) 50:325–92. doi: 10.1016/S0074-7742(02)50082-9
54. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature (2001) 414(6865):813–20. doi: 10.1038/414813a
55. Ishii H, Tada H, Isogai S. An aldose reductase inhibitor prevents glucose-induced increase in transforming growth factor-beta and protein kinase C activity in cultured mesangial cells. Diabetologia (1998) 41(3):362–4. doi: 10.1007/s001250050916
56. Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature (2000) 404(6779):787–90. doi: 10.1038/35008121
57. Tilton RG. Diabetic vascular dysfunction: links to glucose-induced reductive stress and VEGF. Microsc Res Tech (2002) 57(5):390–407. doi: 10.1002/jemt.10092
58. Trueblood N, Ramasamy R. Aldose reductase inhibition improves altered glucose metabolism of isolated diabetic rat hearts. Am J Physiol (1998) 275(1):H75–83. doi: 10.1152/ajpheart.1998.275.1.H75
59. Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res (2010) 106(8):1319–31. doi: 10.1161/CIRCRESAHA.110.217117
60. Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res (2010) 107(9):1058–70. doi: 10.1161/CIRCRESAHA.110.223545
61. Kitada M, Zhang Z, Mima A, King GL. Molecular mechanisms of diabetic vascular complications. J Diabetes Invest (2010) 1(3):77–89. doi: 10.1111/j.2040-1124.2010.00018.x
62. Jez JM, Penning TM. The aldo-keto reductase (AKR) superfamily: an update. Chem-Biol Interact (2001) 130-132(1-3):499–525. doi: 10.1016/S0009-2797(00)00295-7
63. Petrash JM, Tarle I, Wilson DK, Quiocho FA. Aldose reductase catalysis and crystallography. Insights from recent advances in enzyme structure and function. Diabetes (1994) 43(8):955–9. doi: 10.2337/diabetes.43.8.955
64. Grimshaw CE, Bohren KM, Lai CJ, Gabbay KH. Human aldose reductase: rate constants for a mechanism including interconversion of ternary complexes by recombinant wild-type enzyme. Biochemistry (1995) 34(44):14356–65. doi: 10.1021/bi00044a012
65. Clements RS Jr., Weaver JP, Winegrad AI. The distribution of polyol: NADP oxidoreductase in mammalian tissues. Biochem Biophys Res Commun (1969) 37(2):347–53. doi: 10.1016/0006-291X(69)90741-4
66. Ludvigson MA, Sorenson RL. Immunohistochemical localization of aldose reductase. I. Enzyme purification and antibody preparation–localization in peripheral nerve, artery, and testis. Diabetes (1980) 29(6):438–49. doi: 10.2337/diabetes.29.6.438
67. Rondeau JM, Tête-Favier F, Podjarny A, Reymann JM, Barth P, Biellmann JF, et al. Novel NADPH-binding domain revealed by the crystal structure of aldose reductase. Nature (1992) 355(6359):469–72. doi: 10.1038/355469a0
68. Hers HG. [Aldose reductase]. Biochim Biophys Acta (1960) 37:120–6. doi: 10.1016/0006-3002(60)90085-8
69. Srivastava S, Watowich SJ, Petrash JM, Srivastava SK, Bhatnagar A. Structural and kinetic determinants of aldehyde reduction by aldose reductase. Biochemistry (1999) 38(1):42–54. doi: 10.1021/bi981794l
70. Vander Jagt DL, Kolb NS, Vander Jagt TJ, Chino J, Martinez FJ, Hunsaker LA, et al. Substrate specificity of human aldose reductase: identification of 4-hydroxynonenal as an endogenous substrate. Biochim Biophys Acta (1995) 1249(2):117–26. doi: 10.1016/0167-4838(95)00021-L
71. Crosas B, Hyndman DJ, Gallego O, Martras S, Parés X, Flynn TG, et al. Human aldose reductase and human small intestine aldose reductase are efficient retinal reductases: consequences for retinoid metabolism. Biochem J (2003) 373(Pt 3):973–9. doi: 10.1042/bj20021818
72. Inagaki K, Miwa I, Okuda J. Affinity purification and glucose specificity of aldose reductase from bovine lens. Arch Biochem Biophys (1982) 216(1):337–44. doi: 10.1016/0003-9861(82)90219-3
73. Grimshaw CE. Direct measurement of the rate of ring opening of D-glucose by enzyme-catalyzed reduction. Carbohydr Res (1986) 148(2):345–8. doi: 10.1016/S0008-6215(00)90401-4
74. Petrash JM, Harter TM, Devine CS, Olins PO, Bhatnagar A, Liu S, et al. Involvement of cysteine residues in catalysis and inhibition of human aldose reductase. Site-directed mutagenesis of Cys-80, -298, and -303. J Biol Chem (1992) 267(34):24833–40. doi: 10.1016/S0021-9258(18)35839-3
75. Srivastava S, Dixit BL, Ramana KV, Chandra A, Chandra D, Zacarias A, et al. Structural and kinetic modifications of aldose reductase by S-nitrosothiols. Biochem J (2001) 358(Pt 1):111–8. doi: 10.1042/bj3580111
76. Hwang YC, Sato S, Tsai JY, Yan S, Bakr S, Zhang H, et al. Aldose reductase activation is a key component of myocardial response to ischemia. FASEB J: Off Publ Fed Am Soc Exp Biol (2002) 16(2):243–5. doi: 10.1096/fj.01-0368fje
77. Chandra D, Jackson EB, Ramana KV, Kelley R, Srivastava SK, Bhatnagar A. Nitric oxide prevents aldose reductase activation and sorbitol accumulation during diabetes. Diabetes (2002) 51(10):3095–101. doi: 10.2337/diabetes.51.10.3095
78. Robinson B, Hunsaker LA, Stangebye LA, Vander Jagt DL. Aldose and aldehyde reductases from human kidney cortex and medulla. Biochim Biophys Acta (1993) 1203(2):260–6. doi: 10.1016/0167-4838(93)90092-6
79. Graham A, Heath P, Morten JE, Markham AF. The human aldose reductase gene maps to chromosome region 7q35. Hum Genet (1991) 86(5):509–14. doi: 10.1007/BF00194644
80. Chung S, LaMendola J. Cloning and sequence determination of human placental aldose reductase gene. J Biol Chem (1989) 264(25):14775–7. doi: 10.1016/S0021-9258(18)63766-4
81. Wang K, Bohren KM, Gabbay KH. Characterization of the human aldose reductase gene promoter. J Biol Chem (1993) 268(21):16052–8. doi: 10.1016/S0021-9258(18)82356-0
82. Ko BC, Ruepp B, Bohren KM, Gabbay KH, Chung SS. Identification and characterization of multiple osmotic response sequences in the human aldose reductase gene. J Biol Chem (1997) 272(26):16431–7. doi: 10.1074/jbc.272.26.16431
83. Oates PJ, Mylari BL. Aldose reductase inhibitors: therapeutic implications for diabetic complications. Expert Opin Investig Drugs (1999) 8(12):2095–119. doi: 10.1517/13543784.8.12.2095
84. Neamat-Allah M, Feeney SA, Savage DA, Maxwell AP, Hanson RL, Knowler WC, et al. Analysis of the association between diabetic nephropathy and polymorphisms in the aldose reductase gene in Type 1 and Type 2 diabetes mellitus. Diabetic Med: J Br Diabetic Assoc (2001) 18(11):906–14. doi: 10.1046/j.0742-3071.2001.00598.x
85. Ko BC, Lam KS, Wat NM, Chung SS. An (A-C)n dinucleotide repeat polymorphic marker at the 5’ end of the aldose reductase gene is associated with early-onset diabetic retinopathy in NIDDM patients. Diabetes (1995) 44(7):727–32. doi: 10.2337/diabetes.44.7.727
86. Kao YL, Donaghue K, Chan A, Knight J, Silink M. A novel polymorphism in the aldose reductase gene promoter region is strongly associated with diabetic retinopathy in adolescents with type 1 diabetes. Diabetes (1999) 48(6):1338–40. doi: 10.2337/diabetes.48.6.1338
87. Lee SC, Wang Y, Ko GT, Critchley JA, Ng MC, Tong PC, et al. Association of retinopathy with a microsatellite at 5’ end of the aldose reductase gene in Chinese patients with late-onset Type 2 diabetes. Ophthalmic Genet (2001) 22(2):63–7. doi: 10.1076/opge.22.2.63.2230
88. Kao YL, Donaghue K, Chan A, Knight J, Silink M. An aldose reductase intragenic polymorphism associated with diabetic retinopathy. Diabetes Res Clin Pract (1999) 46(2):155–60. doi: 10.1016/S0168-8227(99)00087-X
89. Demaine A, Cross D, Millward A. Polymorphisms of the aldose reductase gene and susceptibility to retinopathy in type 1 diabetes mellitus. Invest Ophthalmol Visual Sci (2000) 41(13):4064–8.
90. Moczulski DK, Scott L, Antonellis A, Rogus JJ, Rich SS, Warram JH, et al. Aldose reductase gene polymorphisms and susceptibility to diabetic nephropathy in Type 1 diabetes mellitus. Diabetic Med: J Br Diabetic Assoc (2000) 17(2):111–8. doi: 10.1046/j.1464-5491.2000.00225.x
91. Shah VO, Scavini M, Nikolic J, Sun Y, Vai S, Griffith JK, et al. Z-2 microsatellite allele is linked to increased expression of the aldose reductase gene in diabetic nephropathy. J Clin Endocrinol Metab (1998) 83(8):2886–91. doi: 10.1210/jc.83.8.2886
92. Hodgkinson AD, Millward BA, Demaine AG. Polymorphisms of the glucose transporter (GLUT1) gene are associated with diabetic nephropathy. Kidney Int (2001) 59(3):985–9. doi: 10.1046/j.1523-1755.2001.00581.x
93. Heesom AE, Hibberd ML, Millward A, Demaine AG. Polymorphism in the 5’-end of the aldose reductase gene is strongly associated with the development of diabetic nephropathy in type I diabetes. Diabetes (1997) 46(2):287–91. doi: 10.2337/diabetes.46.2.287
94. Heesom AE, Millward A, Demaine AG. Susceptibility to diabetic neuropathy in patients with insulin dependent diabetes mellitus is associated with a polymorphism at the 5’ end of the aldose reductase gene. J Neurol Neurosurg Psychiatry (1998) 64(2):213–6. doi: 10.1136/jnnp.64.2.213
95. Moczulski DK, Burak W, Doria A, Zychma M, Zukowska-Szczechowska E, Warram JH, et al. The role of aldose reductase gene in the susceptibility to diabetic nephropathy in Type II (non-insulin-dependent) diabetes mellitus. Diabetologia (1999) 42(1):94–7. doi: 10.1007/s001250051119
96. Ng DP, Conn J, Chung SS, Larkins RG. Aldose reductase (AC)(n) microsatellite polymorphism and diabetic microvascular complications in Caucasian Type 1 diabetes mellitus. Diabetes Res Clin Pract (2001) 52(1):21–7. doi: 10.1016/S0168-8227(00)00239-4
97. Maeda S, Haneda M, Yasuda H, Tachikawa T, Isshiki K, Koya D, et al. Diabetic nephropathy is not associated with the dinucleotide repeat polymorphism upstream of the aldose reductase (ALR2) gene but with erythrocyte aldose reductase content in Japanese subjects with type 2 diabetes. Diabetes (1999) 48(2):420–2. doi: 10.2337/diabetes.48.2.420
98. So WY, Wang Y, Ng MC, Yang X, Ma RC, Lam V, et al. Aldose reductase genotypes and cardiorenal complications: an 8-year prospective analysis of 1,074 type 2 diabetic patients. Diabetes Care (2008) 31(11):2148–53. doi: 10.2337/dc08-0712
99. Watarai A, Nakashima E, Hamada Y, Watanabe G, Naruse K, Miwa K, et al. Aldose reductase gene is associated with diabetic macroangiopathy in Japanese Type 2 diabetic patients. Diabetic Med: J Br Diabetic Assoc (2006) 23(8):894–9. doi: 10.1111/j.1464-5491.2006.01946.x
100. Rojas-Carranza CA, Bustos-Cruz RH, Pino-Pinzon CJ, Ariza-Marquez YV, Gomez-Bello RM, Canadas-Garre M. Diabetes-Related Neurological Implications and Pharmacogenomics. Curr Pharm Des (2018) 24(15):1695–710. doi: 10.2174/1381612823666170317165350
101. Hampton BM, Schwartz SG, Brantley MA Jr., Flynn HW Jr. Update on genetics and diabetic retinopathy. Clin Ophthalmol (Auckland NZ) (2015) 9:2175–93. doi: 10.2147/OPTH.S94508
102. Zhou M, Zhang P, Xu X, Sun X. The Relationship Between Aldose Reductase C106T Polymorphism and Diabetic Retinopathy: An Updated Meta-Analysis. Invest Ophthalmol Visual Sci (2015) 56(4):2279–89. doi: 10.1167/iovs.14-16279
103. Demaine AG. Polymorphisms of the aldose reductase gene and susceptibility to diabetic microvascular complications. Curr Med Chem (2003) 10(15):1389–98. doi: 10.2174/0929867033457359
104. Chung SS, Chung SK. Genetic analysis of aldose reductase in diabetic complications. Curr Med Chem (2003) 10(15):1375–87. doi: 10.2174/0929867033457322
105. Hers HG. The mechanism of the transformation of glucose in fructose in the seminal vesicles. Biochim Biophys Acta (1956) 22(1):202–3. doi: 10.1016/0006-3002(56)90247-5
106. Kinoshita JH. A thirty year journey in the polyol pathway. Exp Eye Res (1990) 50(6):567–73. doi: 10.1016/0014-4835(90)90096-D
107. Lee AY, Chung SK, Chung SS. Demonstration that polyol accumulation is responsible for diabetic cataract by the use of transgenic mice expressing the aldose reductase gene in the lens. Proc Natl Acad Sci USA (1995) 92(7):2780–4. doi: 10.1073/pnas.92.7.2780
108. Ramasamy R, Goldberg IJ. Aldose reductase and cardiovascular diseases, creating human-like diabetic complications in an experimental model. Circ Res (2010) 106(9):1449–58. doi: 10.1161/CIRCRESAHA.109.213447
109. Yadav UC, Srivastava SK, Ramana KV. Understanding the role of aldose reductase in ocular inflammation. Curr Mol Med (2010) 10(6):540–9. doi: 10.2174/156652410792231303
110. Pandey S, Srivastava SK, Ramana KV. A potential therapeutic role for aldose reductase inhibitors in the treatment of endotoxin-related inflammatory diseases. Expert Opin Investig Drugs (2012) 21(3):329–39. doi: 10.1517/13543784.2012.656198
111. Srivastava SK, Yadav UC, Reddy AB, Saxena A, Tammali R, Shoeb M, et al. Aldose reductase inhibition suppresses oxidative stress-induced inflammatory disorders. Chem-Biol Interact (2011) 191(1-3):330–8. doi: 10.1016/j.cbi.2011.02.023
112. Cheng HM, González RG. The effect of high glucose and oxidative stress on lens metabolism, aldose reductase, and senile cataractogenesis. Metabol: Clin Exp (1986) 35(4 Suppl 1):10–4. doi: 10.1016/0026-0495(86)90180-0
113. Ido Y, Chang K, Woolsey TA, Williamson JR. NADH: sensor of blood flow need in brain, muscle, and other tissues. FASEB J: Off Publ Fed Am Soc Exp Biol (2001) 15(8):1419–21. doi: 10.1096/fj.00-0652fje
114. Williamson JR, Chang K, Frangos M, Hasan KS, Ido Y, Kawamura T, et al. Hyperglycemic pseudohypoxia and diabetic complications. Diabetes (1993) 42(6):801–13. doi: 10.2337/diabetes.42.6.801
115. Tilton RG, Kawamura T, Chang KC, Ido Y, Bjercke RJ, Stephan CC, et al. Vascular dysfunction induced by elevated glucose levels in rats is mediated by vascular endothelial growth factor. J Clin Invest (1997) 99(9):2192–202. doi: 10.1172/JCI119392
116. Ido Y, Nyengaard JR, Chang K, Tilton RG, Kilo C, Mylari BL, et al. Early neural and vascular dysfunctions in diabetic rats are largely sequelae of increased sorbitol oxidation. Antioxid Redox Signaling (2010) 12(1):39–51. doi: 10.1089/ars.2009.2502
117. Ido Y, Chang K, Williamson JR. NADH augments blood flow in physiologically activated retina and visual cortex. Proc Natl Acad Sci USA (2004) 101(2):653–8. doi: 10.1073/pnas.0307458100
118. Markus HB, Raducha M, Harris H. Tissue distribution of mammalian aldose reductase and related enzymes. Biochem Med (1983) 29(1):31–45. doi: 10.1016/0006-2944(83)90051-0
119. Tanimoto T, Maekawa K, Okada S, Yabe-Nishimura C. Clinical analysis of aldose reductase for differential diagnosis of the pathogenesis of diabetic complication. Anal Chim Acta (1998) 365(1):285–92. doi: 10.1016/S0003-2670(97)00649-1
120. Yabe-Nishimura C. Aldose reductase in glucose toxicity: a potential target for the prevention of diabetic complications. Pharmacol Rev (1998) 50(1):21–33.
121. Burg MB, Kwon ED, Kültz D. Regulation of gene expression by hypertonicity. Annu Rev Physiol (1997) 59:437–55. doi: 10.1146/annurev.physiol.59.1.437
123. Burg MB. Coordinate regulation of organic osmolytes in renal cells. Kidney Int (1996) 49(6):1684–5. doi: 10.1038/ki.1996.247
124. Srivastava SK, Ansari NH, Brown JH, Petrash JM. Formation of sorbitol 6-phosphate by bovine and human lens aldose reductase, sorbitol dehydrogenase and sorbitol kinase. Biochim Biophys Acta (1982) 717(2):210–4. doi: 10.1016/0304-4165(82)90171-4
125. Szwergold BS, Kappler F, Brown TR, Pfeffer P, Osman SF. Identification of D-sorbitol 3-phosphate in the normal and diabetic mammalian lens. J Biol Chem (1989) 264(16):9278–82. doi: 10.1016/S0021-9258(18)60526-5
126. Szwergold BS, Kappler F, Brown TR. Identification of fructose 3-phosphate in the lens of diabetic rats. Sci (New York NY) (1990) 247(4941):451–4. doi: 10.1126/science.2300805
127. Hallam KM, Li Q, Ananthakrishnan R, Kalea A, Zou YS, Vedantham S, et al. Aldose reductase and AGE-RAGE pathways: central roles in the pathogenesis of vascular dysfunction in aging rats. Aging Cell (2010) 9(5):776–84. doi: 10.1111/j.1474-9726.2010.00606.x
128. Petrash JM, Harter TM, Murdock GL. A potential role for aldose reductase in steroid metabolism. Adv Exp Med Biol (1997) 414:465–73. doi: 10.1007/978-1-4615-5871-2_53
129. Kawamura M, Kopin IJ, Kador PF, Sato S, Tjurmina O, Eisenhofer G. Effects of aldehyde/aldose reductase inhibition on neuronal metabolism of norepinephrine. J Auton Nerv Syst (1997) 66(3):145–8. doi: 10.1016/S0165-1838(97)00086-6
130. Grimshaw CE. Aldose reductase: model for a new paradigm of enzymic perfection in detoxification catalysts. Biochemistry (1992) 31(42):10139–45. doi: 10.1021/bi00157a001
131. Dixit BL, Balendiran GK, Watowich SJ, Srivastava S, Ramana KV, Petrash JM, et al. Kinetic and structural characterization of the glutathione-binding site of aldose reductase. J Biol Chem (2000) 275(28):21587–95. doi: 10.1074/jbc.M909235199
132. Chen CH, Ferreira JCB, Mochly-Rosen D. ALDH2 and Cardiovascular Disease. Adv Exp Med Biol (2019) 1193:53–67. doi: 10.1007/978-981-13-6260-6_3
133. Munukutla S, Pan G, Palaniyandi SS. Aldehyde Dehydrogenase (ALDH) 2 in Diabetic Heart Diseases. Adv Exp Med Biol (2019) 1193:155–74. doi: 10.1007/978-981-13-6260-6_9
134. Lee SH, Takahashi R, Goto T, Oe T. Mass Spectrometric Characterization of Modifications to Angiotensin II by Lipid Peroxidation Products, 4-Oxo-2(E)-nonenal and 4-Hydroxy-2(E)-nonenal. Chem Res Toxicol (2010) 23(11):1771–85. doi: 10.1021/tx100228q
135. Allen EMG, Anderson DGR, Florang VR, Khanna M, Hurley TD, Doorn JA. Relative Inhibitory Potency of Molinate and Metabolites with Aldehyde Dehydrogenase 2: Implications for the Mechanism of Enzyme Inhibition. Chem Res Toxicol (2010) 23(11):1843–50. doi: 10.1021/tx100317q
136. Rees-Milton KJ, Jia Z, Green NC, Bhatia M, El-Kabbani O, Flynn TG. Aldehyde reductase: the role of C-terminal residues in defining substrate and cofactor specificities. Arch Biochem Biophys (1998) 355(2):137–44. doi: 10.1006/abbi.1998.0721
137. Vasiliou V, Pappa A, Petersen DR. Role of aldehyde dehydrogenases in endogenous and xenobiotic metabolism. Chem-Biol Interact (2000) 129(1-2):1–19. doi: 10.1016/S0009-2797(00)00211-8
138. Thiagarajan D, Ananthakrishnan R, Zhang J, O’Shea KM, Quadri N, Li Q, et al. Aldose Reductase Acts as a Selective Derepressor of PPARγ and the Retinoic Acid Receptor. Cell Rep (2016) 15(1):181–96. doi: 10.1016/j.celrep.2016.02.086
139. Thiagarajan D, Vedantham S, Ananthakrishnan R, Schmidt AM, Ramasamy R. Mechanisms of transcription factor acetylation and consequences in hearts. Biochim Biophys Acta (BBA) - Mol Basis Dis (2016) 1862(12):2221–31. doi: 10.1016/j.bbadis.2016.08.011
140. Shimizu T, Tatano Y, Tomioka H. Aldose reductase participates in the downregulation of T cell functions due to suppressor macrophages. Sci Rep (2016) 6:21093. doi: 10.1038/srep21093
141. Ho HT, Chung SK, Law JW, Ko BC, Tam SC, Brooks HL, et al. Aldose reductase-deficient mice develop nephrogenic diabetes insipidus. Mol Cell Biol (2000) 20(16):5840–6. doi: 10.1128/MCB.20.16.5840-5846.2000
142. Aida K, Ikegishi Y, Chen J, Tawata M, Ito S, Maeda S, et al. Disruption of aldose reductase gene (Akr1b1) causes defect in urinary concentrating ability and divalent cation homeostasis. Biochem Biophys Res Commun (2000) 277(2):281–6. doi: 10.1006/bbrc.2000.3648
143. Ramasamy R, Oates PJ, Schaefer S. Aldose reductase inhibition protects diabetic and nondiabetic rat hearts from ischemic injury. Diabetes (1997) 46(2):292–300. doi: 10.2337/diabetes.46.2.292
144. Ramasamy R, Trueblood N, Schaefer S. Metabolic effects of aldose reductase inhibition during low-flow ischemia and reperfusion. Am J Physiol (1998) 275(1):H195–203. doi: 10.1152/ajpheart.1998.275.1.H195
145. Tracey WR, Magee WP, Ellery CA, MacAndrew JT, Smith AH, Knight DR, et al. Aldose reductase inhibition alone or combined with an adenosine A(3) agonist reduces ischemic myocardial injury. Am J Physiol Heart Circulatory Physiol (2000) 279(4):H1447–52. doi: 10.1152/ajpheart.2000.279.4.H1447
146. Kashiwagi A, Obata T, Suzaki M, Takagi Y, Kida Y, Ogawa T, et al. Increase in cardiac muscle fructose content in streptozotocin-induced diabetic rats. Metabol: Clin Exp (1992) 41(10):1041–6. doi: 10.1016/0026-0495(92)90283-G
147. Shinmura K, Bolli R, Liu SQ, Tang XL, Kodani E, Xuan YT, et al. Aldose reductase is an obligatory mediator of the late phase of ischemic preconditioning. Circ Res (2002) 91(3):240–6. doi: 10.1161/01.RES.0000029970.97247.57
148. Hwang YC, Kaneko M, Bakr S, Liao H, Lu Y, Lewis ER, et al. Central role for aldose reductase pathway in myocardial ischemic injury. FASEB J: Off Publ Fed Am Soc Exp Biol (2004) 18(11):1192–9. doi: 10.1096/fj.03-1400com
149. Yamaoka T, Nishimura C, Yamashita K, Itakura M, Yamada T, Fujimoto J, et al. Acute onset of diabetic pathological changes in transgenic mice with human aldose reductase cDNA. Diabetologia (1995) 38(3):255–61. doi: 10.1007/s001250050278
150. Ananthakrishnan R, Kaneko M, Hwang YC, Quadri N, Gomez T, Li Q, et al. Aldose reductase mediates myocardial ischemia-reperfusion injury in part by opening mitochondrial permeability transition pore. Am J Physiol Heart Circulatory Physiol (2009) 296(2):H333–41. doi: 10.1152/ajpheart.01012.2008
151. Abdillahi M, Ananthakrishnan R, Vedantham S, Shang L, Zhu Z, Rosario R, et al. Aldose reductase modulates cardiac glycogen synthase kinase-3β phosphorylation during ischemia-reperfusion. Am J Physiol Heart Circulatory Physiol (2012) 303(3):H297–308. doi: 10.1152/ajpheart.00999.2011
152. Hwang YC, Shaw S, Kaneko M, Redd H, Marrero MB, Ramasamy R. Aldose reductase pathway mediates JAK-STAT signaling: a novel axis in myocardial ischemic injury. FASEB J: Off Publ Fed Am Soc Exp Biol (2005) 19(7):795–7. doi: 10.1096/fj.04-2780fje
153. Son NH, Ananthakrishnan R, Yu S, Khan RS, Jiang H, Ji R, et al. Cardiomyocyte aldose reductase causes heart failure and impairs recovery from ischemia. PloS One (2012) 7(9):e46549. doi: 10.1371/journal.pone.0046549
154. Thiagarajan D, OS K, Sreejit G, Ananthakrishnan R, Quadri N, Li Q, et al. Aldose reductase modulates acute activation of mesenchymal markers via the β-catenin pathway during cardiac ischemia-reperfusion. PloS One (2017) 12(11):e0188981. doi: 10.1371/journal.pone.0188981
155. Iwata K, Matsuno K, Nishinaka T, Persson C, Yabe-Nishimura C. Aldose reductase inhibitors improve myocardial reperfusion injury in mice by a dual mechanism. J Pharmacol Sci (2006) 102(1):37–46. doi: 10.1254/jphs.FP0060218
156. Lo AC, Cheung AK, Hung VK, Yeung CM, He QY, Chiu JF, et al. Deletion of aldose reductase leads to protection against cerebral ischemic injury. J Cereb Blood Flow Metabol: Off J Int Soc Cereb Blood Flow Metab (2007) 27(8):1496–509. doi: 10.1038/sj.jcbfm.9600452
157. Tang WH, Wu S, Wong TM, Chung SK, Chung SS. Polyol pathway mediates iron-induced oxidative injury in ischemic-reperfused rat heart. Free Radical Biol Med (2008) 45(5):602–10. doi: 10.1016/j.freeradbiomed.2008.05.003
158. Lee AY, Chung SS. Contributions of polyol pathway to oxidative stress in diabetic cataract. FASEB J: Off Publ Fed Am Soc Exp Biol (1999) 13(1):23–30. doi: 10.1096/fasebj.13.1.23
159. Chung SS, Ho EC, Lam KS, Chung SK. Contribution of polyol pathway to diabetes-induced oxidative stress. J Am Soc Nephrol: JASN (2003) 14(8 Suppl 3):S233–6. doi: 10.1097/01.ASN.0000077408.15865.06
160. Ho EC, Lam KS, Chen YS, Yip JC, Arvindakshan M, Yamagishi S, et al. Aldose reductase-deficient mice are protected from delayed motor nerve conduction velocity, increased c-Jun NH2-terminal kinase activation, depletion of reduced glutathione, increased superoxide accumulation, and DNA damage. Diabetes (2006) 55(7):1946–53. doi: 10.2337/db05-1497
161. Obrosova IG, Minchenko AG, Vasupuram R, White L, Abatan OI, Kumagai AK, et al. Aldose reductase inhibitor fidarestat prevents retinal oxidative stress and vascular endothelial growth factor overexpression in streptozotocin-diabetic rats. Diabetes (2003) 52(3):864–71. doi: 10.2337/diabetes.52.3.864
162. Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Sci (New York NY) (2008) 321(5895):1493–5. doi: 10.1126/science.1158554
163. Srivastava S, Dixit BL, Cai J, Sharma S, Hurst HE, Bhatnagar A, et al. Metabolism of lipid peroxidation product, 4-hydroxynonenal (HNE) in rat erythrocytes: role of aldose reductase. Free Radical Biol Med (2000) 29(7):642–51. doi: 10.1016/S0891-5849(00)00351-8
164. Ramana KV, Dixit BL, Srivastava S, Balendiran GK, Srivastava SK, Bhatnagar A. Selective recognition of glutathiolated aldehydes by aldose reductase. Biochemistry (2000) 39(40):12172–80. doi: 10.1021/bi000796e
165. Shoeb M, Ansari NH, Srivastava SK, Ramana KV. 4-Hydroxynonenal in the pathogenesis and progression of human diseases. Curr Med Chem (2014) 21(2):230–7. doi: 10.2174/09298673113209990181
166. Li Q, Hwang YC, Ananthakrishnan R, Oates PJ, Guberski D, Ramasamy R. Polyol pathway and modulation of ischemia-reperfusion injury in Type 2 diabetic BBZ rat hearts. Cardiovasc Diabetol (2008) 7:33. doi: 10.1186/1475-2840-7-33
167. Srivastava S, Chandrasekar B, Bhatnagar A, Prabhu SD. Lipid peroxidation-derived aldehydes and oxidative stress in the failing heart: role of aldose reductase. Am J Physiol Heart Circulatory Physiol (2002) 283(6):H2612–9. doi: 10.1152/ajpheart.00592.2002
168. Yang J, Moravec CS, Sussman MA, DiPaola NR, Fu D, Hawthorn L, et al. Decreased SLIM1 expression and increased gelsolin expression in failing human hearts measured by high-density oligonucleotide arrays. Circulation (2000) 102(25):3046–52. doi: 10.1161/01.CIR.102.25.3046
169. Gleissner CA, Sanders JM, Nadler J, Ley K. Upregulation of aldose reductase during foam cell formation as possible link among diabetes, hyperlipidemia, and atherosclerosis. Arterioscler Thromb Vasc Biol (2008) 28(6):1137–43. doi: 10.1161/ATVBAHA.107.158295
170. Erbel C, Rupp G, Domschke G, Linden F, Akhavanpoor M, Doesch AO, et al. Differential regulation of aldose reductase expression during macrophage polarization depends on hyperglycemia. Innate Immun (2016) 22(3):230–7. doi: 10.1177/1753425916632053
171. Vikramadithyan RK, Hu Y, Noh HL, Liang CP, Hallam K, Tall AR, et al. Human aldose reductase expression accelerates diabetic atherosclerosis in transgenic mice. J Clin Invest (2005) 115(9):2434–43. doi: 10.1172/JCI24819
172. Vedantham S, Noh H, Ananthakrishnan R, Son N, Hallam K, Hu Y, et al. Human aldose reductase expression accelerates atherosclerosis in diabetic apolipoprotein E-/- mice. Arterioscler Thromb Vasc Biol (2011) 31(8):1805–13. doi: 10.1161/ATVBAHA.111.226902
173. Nakashima Y, Raines EW, Plump AS, Breslow JL, Ross R. Upregulation of VCAM-1 and ICAM-1 at atherosclerosis-prone sites on the endothelium in the ApoE-deficient mouse. Arterioscler Thromb Vasc Biol (1998) 18(5):842–51. doi: 10.1161/01.ATV.18.5.842
174. Harja E, Bu DX, Hudson BI, Chang JS, Shen X, Hallam K, et al. Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE-/- mice. J Clin Invest (2008) 118(1):183–94. doi: 10.1172/JCI32703
175. Dan Q, Wong R, Chung SK, Chung SS, Lam KS. Interaction between the polyol pathway and non-enzymatic glycation on aortic smooth muscle cell migration and monocyte adhesion. Life Sci (2004) 76(4):445–59. doi: 10.1016/j.lfs.2004.09.010
176. Cameron NE, Cotter MA. Impaired contraction and relaxation in aorta from streptozotocin-diabetic rats: role of polyol pathway. Diabetologia (1992) 35(11):1011–9. doi: 10.1007/BF02221675
177. Cameron NE, Cotter MA. Contraction and relaxation of aortas from galactosaemic rats and the effects of aldose reductase inhibition. Eur J Pharmacol (1993) 243(1):47–53. doi: 10.1016/0014-2999(93)90166-F
178. Vedantham S, Thiagarajan D, Ananthakrishnan R, Wang L, Rosario R, Zou YS, et al. Aldose reductase drives hyperacetylation of Egr-1 in hyperglycemia and consequent upregulation of proinflammatory and prothrombotic signals. Diabetes (2014) 63(2):761–74. doi: 10.2337/db13-0032
179. Srivastava S, Vladykovskaya E, Barski OA, Spite M, Kaiserova K, Petrash JM, et al. Aldose reductase protects against early atherosclerotic lesion formation in apolipoprotein E-null mice. Circ Res (2009) 105(8):793–802. doi: 10.1161/CIRCRESAHA.109.200568
180. Singh M, Kapoor A, McCracken J, Hill B, Bhatnagar A. Aldose reductase (AKR1B) deficiency promotes phagocytosis in bone marrow derived mouse macrophages. Chem-Biol Interact (2017) 265:16–23. doi: 10.1016/j.cbi.2017.01.012
181. 11Nagareddy PR, Murphy AJ, Stirzaker RA, Hu Y, Yu S, Miller RG, et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab (2013) 17(5):695–708. doi: 10.1016/j.cmet.2013.04.001
182. Yuan C, Hu J, Parathath S, Grauer L, Cassella CB, Bagdasarov S, et al. Human Aldose Reductase Expression Prevents Atherosclerosis Regression in Diabetic Mice. Diabetes (2018) 67(9):1880–91. doi: 10.2337/db18-0156
183. Tang WH, Stitham J, Gleim S, Di Febbo C, Porreca E, Fava C, et al. Glucose and collagen regulate human platelet activity through aldose reductase induction of thromboxane. J Clin Invest (2011) 121(11):4462–76. doi: 10.1172/JCI59291
184. Tang WH, Stitham J, Jin Y, Liu R, Lee SH, Du J, et al. Aldose reductase-mediated phosphorylation of p53 leads to mitochondrial dysfunction and damage in diabetic platelets. Circulation (2014) 129(15):1598–609. doi: 10.1161/CIRCULATIONAHA.113.005224
185. Srivastava S, Ramana KV, Tammali R, Srivastava SK, Bhatnagar A. Contribution of aldose reductase to diabetic hyperproliferation of vascular smooth muscle cells. Diabetes (2006) 55(4):901–10. doi: 10.2337/diabetes.55.04.06.db05-0932
186. Ramana KV, Friedrich B, Tammali R, West MB, Bhatnagar A, Srivastava SK. Requirement of aldose reductase for the hyperglycemic activation of protein kinase C and formation of diacylglycerol in vascular smooth muscle cells. Diabetes (2005) 54(3):818–29. doi: 10.2337/diabetes.54.3.818
187. Campbell M, Trimble ER. Modification of PI3K- and MAPK-dependent chemotaxis in aortic vascular smooth muscle cells by protein kinase CbetaII. Circ Res (2005) 96(2):197–206. doi: 10.1161/01.RES.0000152966.88353.9d
188. Ramana KV, Friedrich B, Srivastava S, Bhatnagar A, Srivastava SK. Activation of nuclear factor-kappaB by hyperglycemia in vascular smooth muscle cells is regulated by aldose reductase. Diabetes (2004) 53(11):2910–20. doi: 10.2337/diabetes.53.11.2910
189. Ramana KV, Chandra D, Srivastava S, Bhatnagar A, Aggarwal BB, Srivastava SK. Aldose reductase mediates mitogenic signaling in vascular smooth muscle cells. J Biol Chem (2002) 277(35):32063–70. doi: 10.1074/jbc.M202126200
190. Ruef J, Liu SQ, Bode C, Tocchi M, Srivastava S, Runge MS, et al. Involvement of aldose reductase in vascular smooth muscle cell growth and lesion formation after arterial injury. Arterioscler Thromb Vasc Biol (2000) 20(7):1745–52. doi: 10.1161/01.ATV.20.7.1745
191. Tammali R, Saxena A, Srivastava SK, Ramana KV. Aldose reductase regulates vascular smooth muscle cell proliferation by modulating G1/S phase transition of cell cycle. Endocrinology (2010) 151(5):2140–50. doi: 10.1210/en.2010-0160
192. Quattrini L, La Motta C. Aldose reductase inhibitors: 2013-present. Expert Opin Ther Pat (2019) 29(3):199–213. doi: 10.1080/13543776.2019.1582646
193. Maccari R, Ottanà R. Targeting aldose reductase for the treatment of diabetes complications and inflammatory diseases: new insights and future directions. J Med Chem (2015) 58(5):2047–67. doi: 10.1021/jm500907a
194. Mylari BL, Beyer TA, Siegel TW. A highly specific aldose reductase inhibitor, ethyl 1-benzyl-3-hydroxy-2(5H)-oxopyrrole-4-carboxylate, and its congeners. J Med Chem (1991) 34(3):1011–8. doi: 10.1021/jm00107a020
195. Sarges R, Oates PJ. Aldose reductase inhibitors: recent developments. Prog Drug Res Fortschr der Arzneimittelforschung Prog Des Rech Pharm (1993) 40:99–161. doi: 10.1007/978-3-0348-7147-1_5
196. Del-Corso A, Balestri F, Di Bugno E, Moschini R, Cappiello M, Sartini S, et al. Aldose Reductase Differential Inhibitors in Green Tea. PloS One (2013) 8(9):e74076. doi: 10.1371/journal.pone.0074076
197. Wasmuth A, Landry DW, Deng SX, Ramasamy R, Schmidt AM, Mylari BL, et al. Aldose Reductase Differ Inhib Green Tea. Biomol (2020) 10(7). doi: 10.3390/biom10071003
198. Wasmuth A, Landry DW, Deng SX, Ramasamy R, Schmidt AM, Mylari BL, et al. inventorsPreparation of oxodihydropyrazinopyridazine derivatives for use as aldose reductase inhibitors. U.S. Pat. Appl. Publ (2013). US20130225592, A120130829 patent U.S. Pat. Appl. Publ. US20130225592, A120130829.
199. Wasmuth A, Landry DW, Deng SX, Ramasamy R, Schmidt AM, Mylari BL. Preparation of oxodihydropyrazinopyridazine derivatives for use as aldose reductase inhibitors. USA: PCT Int. Appl (2014). WO2014113380, A120140724.
200. Mylari BL. Water soluble salts of aldose reductase inhibitors for treatment of diabetic complications. U.S. Pat. Appl. Publ (2014). US20140228319, A120140814.
201. Mylari BL. Water soluble salts of aldose reductase inhibitors for treatment of diabetic complications. PCT Int. Appl (2014). WO2014126885, A120140821.
202. Shendelman S. Aldose reductase inhibitors such as boronic acid and boronate ester compounds and uses thereof. PCT Int. Appl (2018). WO2018200258, A120181101.
203. Pal PB, Sonowal H, Shukla K, Srivastava SK, Ramana KV. Aldose Reductase Mediates NLRP3 Inflammasome-Initiated Innate Immune Response in Hyperglycemia-Induced Thp1 Monocytes and Male Mice. Endocrinology (2017) 158(10):3661–75. doi: 10.1210/en.2017-00294
204. Ravindranath TM, Mong PY, Ananthakrishnan R, Li Q, Quadri N, Schmidt AM, et al. Novel role for aldose reductase in mediating acute inflammatory responses in the lung. J Immunol (Baltimore Md: 1950) (2009) 183(12):8128–37. doi: 10.4049/jimmunol.0900720
205. Johnson BF, Nesto RW, Pfeifer MA, Slater WR, Vinik AI, Chyun DA, et al. Cardiac abnormalities in diabetic patients with neuropathy: effects of aldose reductase inhibitor administration. Diabetes Care (2004) 27(2):448–54. doi: 10.2337/diacare.27.2.448
206. Didangelos TP, Athyros VG, Karamitsos DT, Papageorgiou AA, Kourtoglou GI, Kontopoulos AG. Effect of aldose reductase inhibition on heart rate variability in patients with severe or moderate diabetic autonomic neuropathy. Clin Drug Invest (1998) 15(2):111–21. doi: 10.2165/00044011-199815020-00005
207. Perfetti R, Shendelman S. Abstract 13475: Clinical Assessment of AT-001, an Aldose Reductase Inhibitor in Development for Diabetic Cardiomyopathy: A 28-Day Proof of Concept Study. Circulation (2019) 140(Suppl_1):A13475–A.
208. Kadosh BS, Garshick MS, Gaztanaga J, Moore KJ, Newman JD, Pillinger M, et al. COVID-19 and the Heart and Vasculature: Novel Approaches to Reduce Virus-Induced Inflammation in Patients With Cardiovascular Disease. Arterioscler Thromb Vasc Biol (2020) 40(9):2045–53. doi: 10.1161/ATVBAHA.120.314513
Keywords: diabetes, cardiovascular diabetic complications, aldose reductase, polyol pathway, hyperglycemia, aldose reductase inhibitor, cardiovascular disease
Citation: Jannapureddy S, Sharma M, Yepuri G, Schmidt AM and Ramasamy R (2021) Aldose Reductase: An Emerging Target for Development of Interventions for Diabetic Cardiovascular Complications. Front. Endocrinol. 12:636267. doi: 10.3389/fendo.2021.636267
Received: 01 December 2020; Accepted: 19 January 2021;
Published: 11 March 2021.
Edited by:
Khalid Siddiqui, King Saud University, Saudi ArabiaReviewed by:
Kirtikar Shukla, Wake Forest School of Medicine, United StatesUmberto Mura, University of Pisa, Italy
Yeliz Demir, Ardahan University, Turkey
Copyright © 2021 Jannapureddy, Sharma, Yepuri, Schmidt and Ramasamy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ravichandran Ramasamy, UmF2aWNoYW5kcmFuLlJhbWFzYW15QG55dWxhbmdvbmUub3Jn
†Present address: Sravya Jannapureddy, New York Institute of Technology College of Osteopathic Medicine, Glen Head, NY, United States