
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 20 May 2021
Sec. Gut Endocrinology
Volume 12 - 2021 | https://doi.org/10.3389/fendo.2021.636175
This article is part of the Research TopicEmerging Roles of the Gut Microbiota in the Pathogenesis of Metabolic DisordersView all 11 articles
 Qing Fang1,2,3,4,5
Qing Fang1,2,3,4,5 Na Liu1,2,3,4,5Binjie Zheng1,2,3,4,5Fei Guo1,2,3,4,5Xiangchang Zeng1,2,3,4,5Xinyi Huang1,2,3,4,5
Na Liu1,2,3,4,5Binjie Zheng1,2,3,4,5Fei Guo1,2,3,4,5Xiangchang Zeng1,2,3,4,5Xinyi Huang1,2,3,4,5 Dongsheng Ouyang1,2,3,4,5*
Dongsheng Ouyang1,2,3,4,5*Diabetes is a highly prevalent metabolic disease that has emerged as a global challenge due to its increasing prevalence and lack of sustainable treatment. Diabetic kidney disease (DKD), which is one of the most frequent and severe microvascular complications of diabetes, is difficult to treat with contemporary glucose-lowering medications. The gut microbiota plays an important role in human health and disease, and its metabolites have both beneficial and harmful effects on vital physiological processes. In this review, we summarize the current findings regarding the role of gut microbial metabolites in the development and progression of DKD, which will help us better understand the possible mechanisms of DKD and explore potential therapeutic approaches for DKD.
With the continuous improvement of people’s living standards, changes in lifestyle, and environmental factors, the incidence of diabetes mellitus (DM), which is a metabolic disorder, has been increasing year by year. According to data released by the International Diabetes Federation (IDF), about 463 million people worldwide were living with DM in 2019. The global prevalence of DM is expected to rise to rise to 10.2% (578 million people) by 2030 and to 10.9% (700 million people) by 2045 (1). Patients with DM are more prone to serious complications that contribute to increased mortality and reduced quality of life. About 30-40% of patients with DM develop diabetic kidney disease (DKD), which is one of the major complications of DM, and most cases progress to end-stage renal disease (2).
The hyperglycemic condition initiates multiple events that damage the kidney structurally and functionally, such as glomerular hyperfiltration, proteinuria, thickening of the glomerular basement membrane, mesangial matrix accumulation, podocyte damage and glomerulosclerosis (3). Renal hemodynamics changes, the renin-angiotensin-aldosterone system, oxidative stress, inflammatory responses and fibrosis are also major factors in the pathogenesis of DKD (4). Current therapy for DKD includes antihypertensive and antiproteinuric means, as well as the use of angiotensin receptor blockers and angiotensin converting enzyme inhibitors. In addition, sodium glucose cotransporter 2 (SGLT-2) inhibitors and glucagon-like peptide 1 (GLP-1) receptor agonists are novel diabetes medications that prevent kidney failure (5). However, these means have limited efficacy in preventing the progression of DKD (6). The complicated pathogenesis of DKD has not yet been elucidated. Understanding the pathophysiology of DKD is crucial for its prevention and treatment.
Recent advances in high-throughput metagenomic sequencing technologies have increased our knowledge of the symbiotic relationship between the gut microbiota and its host (7). The gut microbiota, as an important environmental factor, has emerged as a crucial regulator of human health and disease (8–10). The metabolites produced by the gut microbiota may also have pathogenic or beneficial effects on the host. These metabolites and their end products may play key roles in the host’s metabolic network (11), immune processes (12) and neurobiological processes (13).
Recently, metabolomic studies have found that the microbial metabolite profile is altered in patients with type 2 diabetes (T2D) (14) and DKD (15). Mounting evidence supports the critical role of the gut microbiota as a factor (either beneficial or harmful) in the development of T2D (11) and DKD (16). Moreover, a number of metabolites are produced by the gut microbiota, using dietary nutrients as precursors, suggesting that diet has an important impact on gut microbial metabolites. In this review, we offer a summary of short-chain fatty acids (SCFAs), trimethylamine-N-oxide (TMAO), bile acids (BAs), polyphenols, tryptophan-derived metabolites, branched-chain amino acids (BCAAs) and other metabolites that play important roles in the pathogenesis and progression of DKD. We also discuss the potential mechanisms of microbial metabolites and DKD.
SCFAs, including acetate, propionate, and butyrate, are produced by the microbial community through the fermentation of non-digestible carbohydrates. Acetate and propionate are mainly produced by Bacteroidetes, whereas butyrate is primarily generated by Firmicutes (17). SCFAs can be used by the host for the biosynthesis of lipids, cholesterol, and proteins or as an energy source by gut mucosal cells (18). The effects of SCFAs are in part mediated by G-protein coupled receptors (GPR41, GPR43, and GPR109A) and histone deacetylase (HDAC), which are related to oxidative stress, immune, and inflammatory responses (19–22).
SCFAs have been reported to have multiple beneficial regulatory roles in both type 1 diabetes (T1D) (23, 24) and T2D (25). Several studies have also indicated their important roles in DKD (Table 1). SCFAs, especially acetate and butyrate, inhibit oxidative stress and inflammation in mouse glomerular mesangial cells that have been induced by high glucose and lipopolysaccharides (26). Treatment with a high-fiber diet or directly treatment with SCFAs can both protect against the development of DKD in mice by regulating the key pathways and genes involved in innate immunity, inflammation, and macrophage recruitment. GPR43 and GPR109A are critical to SCFAs-mediated protection against DKD (31). Exogenous sodium butyrate administration can improve DKD by reducing inflammation and oxidative stress, and by ameliorating fibrosis, apoptosis, and DNA damage, and this has been proven in different animal models (19, 22, 31). Exogenous sodium butyrate can also could protect human glomerular mesangial cells against high glucose-induced pyroptosis (32). These studies suggest that SCFAs, especially butyrate, may act as potential therapeutic targets for DKD.
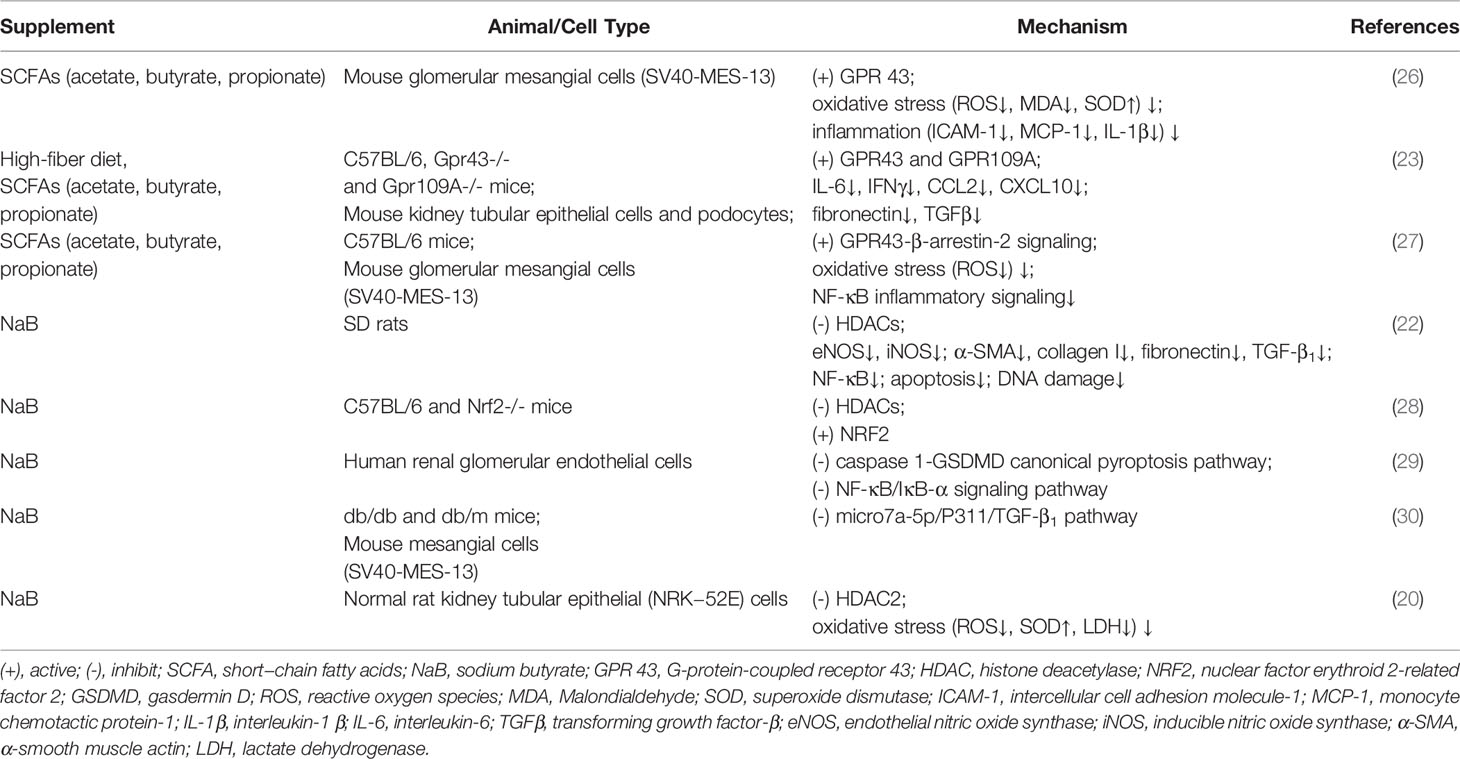
Table 1 Beneficial and Harmful effect of SCFAs on DKD in vivo and vitro.
Although much evidence suggests that increased butyrate production benefits the host through antidiabetic effects, some studies have suggested that acetate may exacerbate DKD. In some animal models, plasma acetate levels have been reported to be positively correlated with the intrarenal angiotensin II protein, which has long been considered to be one of the initiators of DKD. Acetate might also be involved in the kidney injury of early DKD (33). In addition, acetate has been reported to dysregulate cholesterol homeostasis through activating GPR43, thereby contributing to the tubulointerstitial injury of DKD (34). Whether SCFAs production is beneficial or harmful when it comes to DKD remains to be further studied.
Dietary choline, phosphatidylcholine, and L-carnitine are metabolized into trimethylamine (TMA) by intestinal commensal bacteria. A choline-utilization gene cluster (Cut) responsible for anaerobic conversion of choline to TMA was identified in the sulfate-reducing bacterium Desulfovibrio desulfuricans. CutC and CutD, which are crucial genes in the cluster, encoding for choline TMA-lyase and its activating protein. Moreover, in some bacteria of the gender Acinetobacter and Serratia, CntA and CntB gens encode the two subunits of the oxidoreductase enzyme necessary to convert L-carnitine into TMA (35). On the other hand, a YeaW/YeaX gene pair encodes some oxygenase and oxidoreductase enzymes with substrate promiscuity for betaine, γ-butyrobetaine, choline and L-carnitine. These genes, not only CntA/CntB and YeaW/YeaX, but also orthologs and homologs of them, can be found in a wide range of gut microbiota: Actinobacteria, Betaproteobacteria (Achromobacter), Firmicutes (Sporosarcina), and Gammaproteobacteria (Citrobacter, E.coli, Klebsiella pneumoniae, Providencia, and Shigella) (36).
Once TMA is produced, most TMA is oxidized into TMAO in the liver predominantly by the enzyme flavin-containing monooxygenase 3 (37). Some clinical studies have investigated the role of TMAO in predicting the prognostic outcomes and mortality of diverse diseases (38–40). In addition to its predictive value, TMAO has been implicated in the pathogenesis of various human diseases, including cardiovascular (41), kidney (42), metabolic (43) and neurological (44) disorders.
TMAO is linked to the pathogenesis of many diseases by activating inflammatory pathways, such as nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 (NLRP3) inflammasome signals (45–48) and nuclear factor-κB signals (48–50), resulting in the release of inflammatory cytokines. Moreover, some studies have indicated that TMAO and its precursor can contribute to the pathogenesis of cardiovascular diseases by inducing endothelial dysfunction (51–54). In addition, both inflammation and endothelial dysfunction play important roles in the pathogenesis of DKD.
Recently, it has been widely accepted that increased levels of circulating TMAO directly contribute to renal dysfunction by promoting inflammation, oxidative stress, and fibrosis. Both TMAO-supplemented and choline-supplemented mice have shown elevated TMAO levels, which were associated with increases in tubulointerstitial fibrosis and collagen deposition (42). In a mouse model of high-fat-diet-induced obesity, elevated TMAO levels were found to promote renal oxidative stress and inflammation, subsequently contributing to renal interstitial fibrosis and dysfunction (55). In rats with chronic kidney disease (CKD), elevated TMAO levels promote vascular oxidative stress and inflammation, contributing to endothelial dysfunction (56). In a CKD mouse model, dietary supplementation with either choline or TMAO was found to significantly augment multiple indices of renal functional impairment and fibrosis (57). Supplementation with 3,3-dimethyl-1-butanol (an inhibitor of trimethylamine formation) or iodomethylcholine (an inhibitor of prototypic mechanism-based gut microbial choline TMA-lyase) can reduce plasma TMAO levels and prevent adverse renal structural and functional alterations in animal models (55–57). Therefore, high TMAO levels may exacerbate DKD, and TMAO inhibitors may have therapeutic potential to ameliorate DKD (Figure 1).
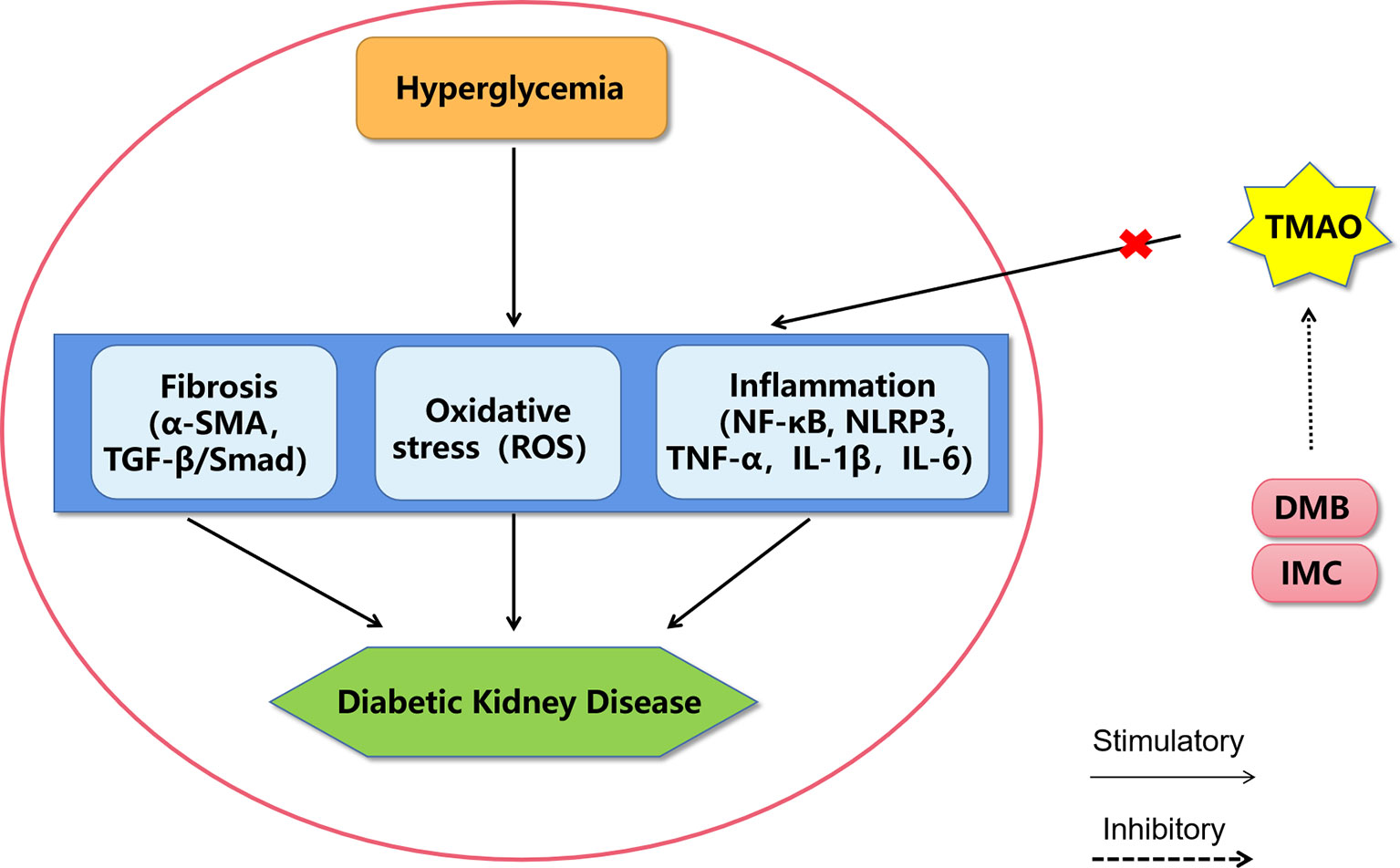
Figure 1 A proposed model of diabetic kidney disease mediated by TMAO. The mechanism of TMAO promoting the progression of diabetic kidney disease may be through promoting inflammation, oxidative stress and fibrosis in renal system. The choline TMA lyase inhibitor DMB and IMC may improve diabetic kidney disease by inhibiting TMAO levels. DMB, 3,3-dimethyl-1-butanol; IMC, iodomethylcholine; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NLRP3, Nod-like receptor pyrin domain 3 inflammasome, TNF-α, tumor necrosis factor α; IL-1β, interleukin-1β; IL-6, interleukin-6; ROS, reactive oxygen species; α-SMA, alpha sarcomeric actin; TGF-β, transforming growth factor-β.
The regulation of TMAO in the DKD still warrants more investigation. Despite growing interest in TMAO biology, the receptor for TMAO is not yet known. Recent evidence has shown that TMAO directly bounds to and activates the protein kinase R-like endoplasmic reticulum kinase, which is an endoplasmic reticulum stress kinase (43). The next milestone is to identify the direct targets of TMAO.
BAs are host-microbial co-metabolites and important signaling molecules. The primary BAs produced by the host can be metabolized by the gut microbiota into a secondary BAs (58). When a small portion of unabsorbed BAs enter the distal ileum, cecum, and colon, they undergo various reactions via microbiota: deconjugation, dehydroxylation, oxidation and epimerization reactions (59, 60). BA deconjugation is driven by bile salt hydrolase, which have been identified in Bacteroides, Bifidobacterium, Clostridium, Enterococcus, and Lactobacillus (61). Dehydroxylation occurs after deconjugation, and is catalyzed by members of the Firmicutes phylum, including Clostridium (C. scindens or C. hylemonae) and Eubacterium. Oxidation and epimerization require BA hydroxysteroid dehydrogenases produced by intestinal Bacteroides, Firmicutes (including Clostridium, Eubacterium, and Ruminococcus), and Escherichia (60).
BAs are endocrine signaling molecules that affect host physiology via the activation of BA receptors. The two major BA receptors that regulate the host metabolism are the nuclear farnesoid X receptor (FXR) and the membrane-bound Takeda G protein-coupled receptor 5 (TGR5) (62). Both FXR and TGR5 have protective roles in DKD (63).
Renal expression of FXR is predominantly tubular and less prominently glomerular, mesangial, and podocytal (64). FXR expression is decreased in people with diabetes- and obesity-related kidney disease. In a series of rodent models of diabetes, the expression levels of FXR and its target genes were found to be downregulated in the kidney (65). Supplementation with FXR agonists, such as tauroursodeoxycholic acid, has been shown to attenuate glomerular and tubular injury in db/db mice and diabetic endothelial nitric oxide synthase-deficient mice (66). Moreover, the FXR agonist GW4064 can improve the functional and structural changes in the kidney of db/db mice (67).
TGR5 expression and activity is impaired in the kidneys of humans and rodents with obesity and diabetes (68). TGR5 activation reduces the renal inflammatory reactions in diabetic mice, thereby improving renal fibrosis (69). In high glucose-treated glomerular mesangial cells, TGR5 activation was found to significantly decrease the expression levels of transforming growth factor beta 1 and fibronectin, which can both accelerate renal fibrosis (70, 71).
The BA signaling pathway plays an extremely important role in T2D and DKD, and it is an important target for drug intervention (Figure 2). BAs have been used directly to treat diabetes and obesity. Metformin, which is a first-line antidiabetic drug, acts in part through the intestinal FXR axis to improve T2D. Oral synthetic FXR antagonists may be of potential translational value in the clinical treatment of T2D (72) Moreover, both BA sequestrants and apical sodium-dependent BA transporter inhibitors can reduce BA absorption and have a therapeutic effect on T2D by activating FXR (73). In the future, semisynthetic BA analogues for the treatment of T2D and DKD need more focus.
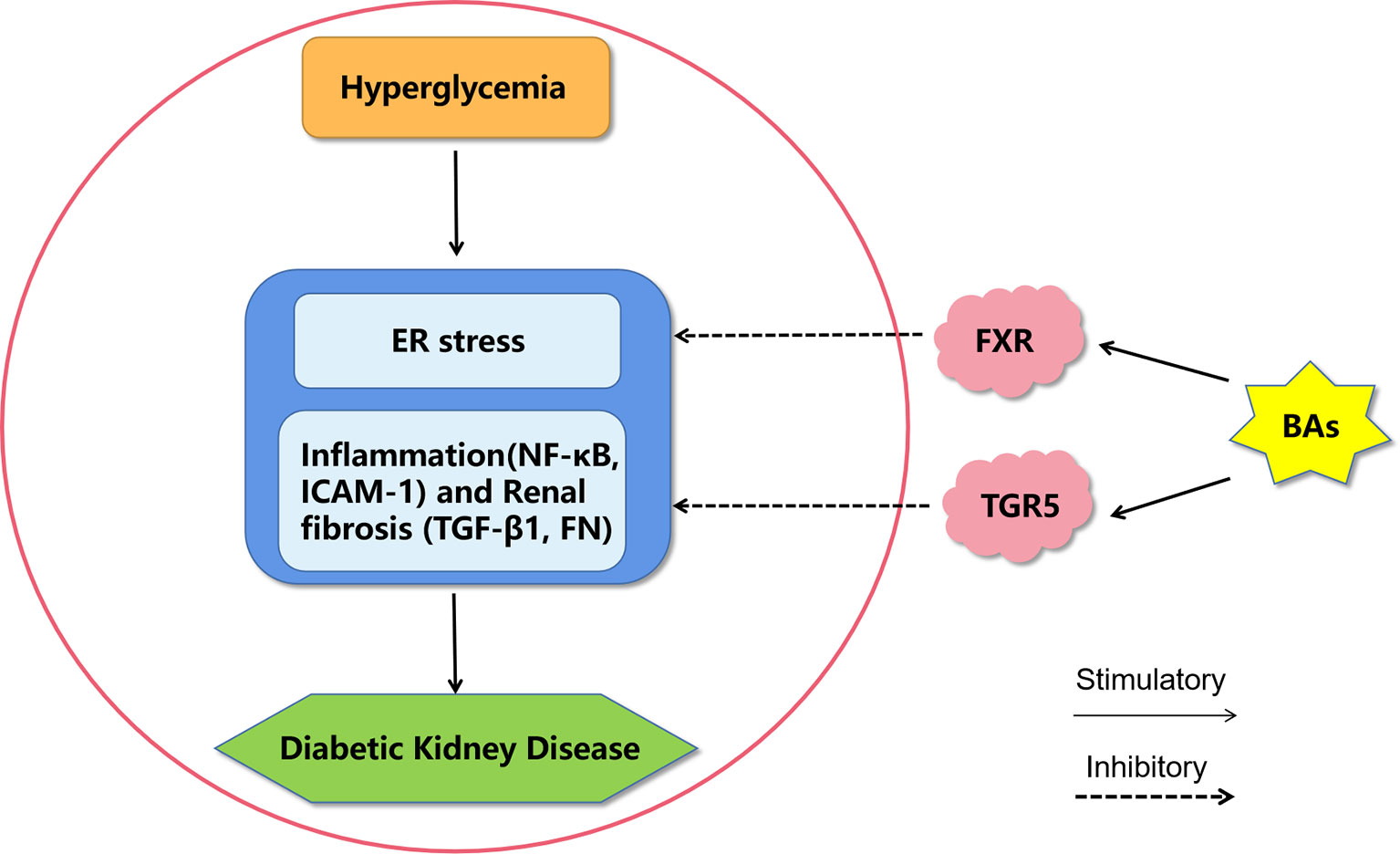
Figure 2 A proposed model of diabetic kidney disease mediated by bile acids. Bile acids may inhibit endoplasmic reticulum stress, inflammation and fibrosis by activating FXR and TGR5 to improve diabetic kidney disease. BAs, bile acids; FXR, Farnesoid X receptor; TGR5, G protein-coupled receptor 5; ER stress, endoplasmic reticulum stress; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; ICAM-1, intercellular adhesion molecule-1; TGF-β1, transforming growth factor-β1; FN, fibronectin.
Protein-bound uremic toxins, such as indoxyl sulfate (IS), p-cresyl sulfate (pCS), p-cresyl glucuronide (pCG), and phenyl sulfate, originate from the gut microbial metabolism of the aromatic amino acids, tyrosine, phenylalanine, and tryptophan. These uremic toxins have been associated with cardiovascular disease and mortality in CKD, and several uremic toxins have also been found to exert toxic effects in the kidney. The levels of these uremic toxins are elevated in T2D patients who progress to end-stage kidney disease (74, 75) and elevated levels of these uremic toxins increase the risk of progression to end-stage kidney disease in patients with T2D (74).
IS is derived from tryptophan metabolism. Tryptophan is digested by intestinal bacteria (E. coli, Proteus vulgaris, Paracolobactrum coliforme, Achromobacter liquefaciens, and Bacteroides spp) to indole, and it is metabolized to IS in the liver (76). Increasing levels of IS are correlated with changes in albuminuria and the estimated glomerular filtration rate, and they are associated with the progression of DKD in patients with T1D and T2D (77–79), as well as in animal models of DM (80–82). IS also can directly induce tubulointerstitial injury, renal oxidative stress and inflammation in mice that undergone nephrectomy (83, 84), as well as in human renal proximal tubular epithelial (HK-2) cells (85, 86).
Both pCS and pCG originate from the intestinal microbial fermentation of tyrosine into p-cresol, and p-cresol is subsequently conjugated to either sulfate or glucuronic acid resulting in the formation of pCS or pCG, respectively (87). The intestinal bacteria generating p-cresol mainly belong to the families Bacteroidaceae, Bifidobacteriaceae, Clostridiaceae, Enterococcaceae, Eubacteriaceae, Fusobacteriaceae, Lachnospiraceae, Lactobacillaceae, Porphyromonadaceae, Staphylococcaceae, Ruminococcaceae, and Veillonellaceae (88). The levels of pCS and pCG are elevated in patients with CKD, and pCG can cause phenotypical changes in renal proximal tubule cells (89). In addition, pCS can directly influence cell viability and induce cell death (90, 91). It has also been reported that pCS can induce reactive oxygen species and inflammatory cytokines in 5/6 nephrectomized rats and HK-2 cells (92).
Phenyl sulfate is produced by the metabolism of the amino acid tyrosine by the gut microbiota. First, phenol is synthesized in the gut and then metabolized into phenyl sulfate by the liver. The intestinal bacteria generating phenol mainly belong to the families Clostridiaceae, Enterococcaceae, Staphylococcaceae, Bacteroidaceae, Bifidobacteriaceae, and Enterobacteriaceae (93, 94). Phenyl sulfate is then secreted by proximal tubular cells through the action of SLCO4C1, which is the only organic acid transporter polypeptide in the human kidney (95). In a cohort of diabetic patients, phenyl sulfate levels were found to be statistically significantly correlated with both basal albuminuria and the 2-year progression of DKD. Further, phenyl sulfate was found to directly induce albuminuria via podocyte damage in diabetic animal models (15).
These protein-bound uremic toxins are not only markers of the risk for DKD occurrence in diabetic patients, but they are also risk factors that directly facilitate the development of DKD (Figure 3). However, the molecular mechanisms of these uremic toxins in DKD still need further study. Some drugs that target uremic toxins, such as AST-120 (Kremezin), which is an oral adsorbent, protect against the progression of both DKD (84, 96) and CKD (97, 98) by removing serum and urinary uremic toxins.
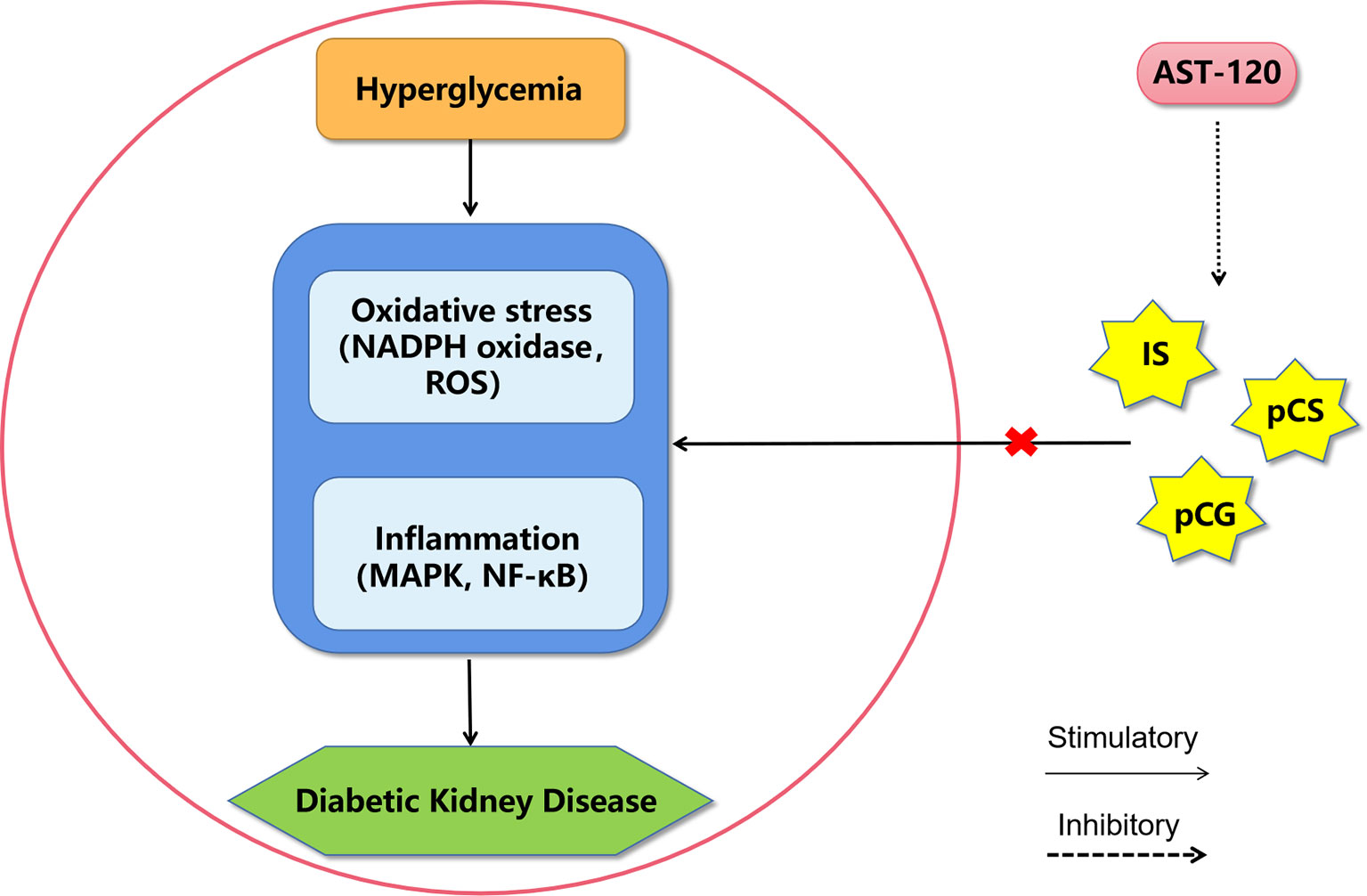
Figure 3 A proposed model of diabetic kidney disease mediated by protein-bound uremic toxins. The mechanism of protein-bound uremic toxins promoting the progression of diabetic kidney disease may be through promoting inflammation and oxidative stress in renal system. AST-120 may improve diabetic kidney disease by removing serum and urinary uremic toxins. IS, Indoxyl sulfate; pCG, p-cresyl glucuronide; pCS, p-cresyl sulfate; ROS, reactive oxygen species; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells.
Polyphenols are produced in plants and have excellent antibacterial, antifungal, antioxidant, and photo-protective properties (99). Natural polyphenols, such as ellagitannins, lignans, isoflavones, and flavanones, which are poorly absorbed or not absorbed at all. Interaction with gut microbiota leads to the biochemical transformations of the native phytochemicals into more bioavailable metabolites. These gut microbiota transformations are grouped into three major catabolic processes: hydrolysis (O-deglycosylations and ester hydrolysis), cleavage (C-ring cleavage; delactonization, demethylation), and reductions (dehydroxylation and double bond reduction) (100). Lactobacillus, Bifidobacterium, Bacteroides, Enterococcus, Enterobacter, and Firmicutes have been demonstrated to participate in hydrolysis (101–103). Cleavage reactions are catalyzed by Clostridium, Coriobacteriaceae, Eubacterium and Eggerthella strains (101, 104). Gordonibacter urolithinfaciens and Lactonifactor longoviformis catalyze different reduction reactions (105–107).
Enterolactone and enterodiol, which are therapeutically relevant polyphenols, are formed as the secondary gut bacterial metabolites of lignans. Urinary levels of enterolactone are associated with lower risk of developing T2D in women in the United States (108). Pre-diagnostic enterolactone concentrations are inversely associated with all-cause and diabetes-specific mortality (109). Enterolactone was found to increase glucose uptake in an AMPK-dependent manner in L6 myotubes and to improve glucose tolerance in db/db mice (110). Many studies have reported that enterolactone may have some benefit for T2D. However, there are also some opposite conclusions. One study did not find a significant association between urine enterolactone levels and T2D risk in Chinese adults (111). Moreover, enterolactone was found to enhanced the hepatic insulin resistance via increased sphingolipid concentrations in the palmitate-rich condition of HepG2 cells (112). Thus, the relationship between enterolactone and T2D should be further studied.
Urolithin A (UA), which is a main gut microbiota-derived metabolite of pomegranate ellagitannins, plays a direct role in improving systemic insulin sensitivity (113). In addition, UA can prevent high-fat-diet-induced insulin resistance and glucose intolerance in mice (114). Further, UA can effectively improve β-cell dysfunction, possibly by regulating autophagy and the AKT/mTOR signaling pathway in the pancreas of diabetic mice (115). Moreover, UA has shown a protective effect in some acute kidney injury animal models via modulation of inflammation, oxidative stress, autophagy, and apoptosis (52, 116–118), which are also some of the main mechanisms of DKD. Thus, UA may be capable of attenuating DKD. Urolithin C (UC), which is another microbiota ellagitannin metabolite, is a glucose-dependent activator of insulin secretion via the facilitation of the opening of L-type Ca2+ channels in β-cells (119). UC can also influence β-cell function by affecting the activation of intracellular signaling proteins, specifically ERK1/2 (120). Urolithin also has been reported have some certain benefits for diabetes complications. Urolithins (includes urolithin A, B, C, D and urolithin B-3-O-glucuronide) may exert positive effects in modulating the pro-inflammatory mediators and growth factors by rat cardiac myocytes and fibroblasts exposed to high glucose concentrations (121). In streptozotocin-induced diabetic rat, both urolithin A and B administration may be able to prevent the initial inflammatory response of myocardial tissue to hyperglycemia and the negative impact of the altered diabetic milieu on cardiac performance (122).
Taken together, polyphenols might have differential microbial metabolites that may improve T2D and its complications. Thus, it is crucial to understand the bacterial pathways involved in the metabolism of polyphenols and their specific roles in T2D and DKD.
BCAAs, including leucine, isoleucine, and valine, are among the nine essential amino acids synthesized by the gut microbiota. The food sources most enriched in BCAAs are meat, fish, dairy products, and eggs (123).
BCAAs are known as biomarkers for insulin resistance and predictors of diabetes development (124). In a recent prospective cohort study, BCAAs showed a strong association with early risk of T2D (125). The serum metabolomes of insulin-resistant and T2D individuals are characterized by increased BCAAs levels (126). Higher intake of total dietary BCAAs, leucine and valine in particular, may increase the incidence of insulin resistance by more than 60% in adults and play an important role in the development of diabetes (127). In contrast, short-term reduction of dietary BCAAs may acutely decrease meal-induced insulin secretion, and improve postprandial insulin sensitivity (128). Furthermore, in both animal and cell models, excess BCAAs have been found to result in liver insulin resistance (129, 130). These results suggest that BCAAs may not only be biomarkers, but also causal agents of insulin resistance and T2D. Evidence of a causal role of BCAAs in human diabetes has also been suggested in some genetic studies. In a mendelian randomization analysis, researchers used genome-wide association studies coupled with large-scale metabolomic measurements to investigate the aetiologic relationship between BCAA metabolism and T2D. They suggested that BCAA-raising polymorphisms were associated with a higher risk of T2D (131). In addition, genetic evidence suggests that genetically elevated insulin resistance is associated with higher concentrations of all BCAAs, supporting the idea that BCAA metabolism lies on a causal pathway from adiposity and insulin resistance to T2D (132). Another study showed that higher BCAA levels do not have a causal effect on insulin resistance while increased insulin resistance drives higher fasting levels of circulating BCAAs (133).
In T2D patients with stages 1 or 2 CKD, high serum BCAA levels are independently associated with a decline in the estimated glomerular filtration rate (134). BCAAs can also directly influence renal function. In one animal model, 5/6 nephrectomized rats receiving a BCAA diet showed a decrease in the estimated glomerular filtration rate and an increase in smooth muscle actin and collagen mRNA expression levels, suggesting renal dysfunction, greater inflammation, and fibrosis in the kidney (135).
However, several studies have suggested that BCAAs may be an effective means of preventing and treating DM and DKD. Moderate intake of BCAA-rich protein was found to improve glucose homeostasis in mice fed a high-fat diet (136). In streptozotocin-induced diabetic rats, treatment with a low dose of BCAAs was found to recover islet function (137). Moreover, one study found that BCAAs countered oxidative stress in the kidney of diabetic rats and alleviated diabetic kidney injury (138), while another study showed that BCAAs protected renal mesangial cells from high-glucose-induced stress (139). More studies are needed to clarify the relationships between BCAAs and T2D. Furthermore, the factors resulting in the differences among these studies needed to be identified.
Many bacterial species are capable of regulating biosynthesis, transport, and metabolism of BCAAs (140). Moreover, transplantation of the microbiota from obese humans to germ-free mice causes a significant increase in circulating BCAAs (141). Additional studies are needed to quantify the microbiota-derived BCAAs in the circulating pool of these metabolites.
Imidazole propionate, which is a microbially produced amino acid-derived metabolite, is present at higher concentrations in people with T2D. A study demonstrated that imidazole propionate directly impaired glucose tolerance and insulin signaling through mTORC1, meaning that imidazole propionate may contribute to the pathogenesis of T2D (142).
4-Cresol is a product of the colonic fermentation of tyrosine and phenylalanine, and it has been reported to be related to T2D. Serum concentrations of 4-cresol are inversely correlated with T2D. The chronic administration of nontoxic doses of 4-cresol was found to reduce adiposity and glucose intolerance in an animal model, and 4-cresol was found to stimulate insulin secretion and β-cell proliferation in vivo and in vitro (143, 144). 4-Cresol may be a regulator of T2D endophenotypes and have potential therapeutic applications.
The metabolism of dietary and host-derived sulfur-containing compounds to hydrogen sulfide (H2S) by the gut microbiota has many prominent connections to host health and disease (145). H2S potentially has both beneficial and toxic effects (146). The plasma H2S levels of Japanese patients with T2D were found to be reduced and significantly associated with their hemoglobin A1c levels (147). H2S also has been suggested to be linked with GLP-1, which has become an important therapeutic target in the treatment of T2D and obesity. By supplementing with a prebiotic chondroitin sulfate, mice were found to enhanced GLP-1 and insulin secretion, improved oral glucose tolerance, and reduced food consumption, suggesting that H2S plays a stimulatory role in GLP-1 secretion (148). However, chronic administration of H2S-releasing agents was found to increase serum glucose, decrease glucose tolerance, and decrease insulin secretion in rats with T2D (149). In the future, studies are needed to clarify the effects of different doses of H2S in T2D. As for DKD, H2S has been reported to have a protective effect and may be used as a novel therapeutic agent.
Hippuric acid is a gut microbial mammalian co-metabolite of benzoic acid; it is subsequently conjugated with glycine in the mitochondria, and then excreted in the urine (150). Several metabolomics studies have shown that hippuric acid levels in patients with impaired glucose tolerance and diabetes are lower than those in healthy people (151). Hippuric acid has also been proposed as a potential urinary biomarker for fruit, vegetable, and polyphenol consumption. Consumption of bilberries and flavonoids, which can increase hippuric acid levels, is associated with a favorable risk factor profile for T2D and better glucose and insulin metabolism (152, 153). Further, hippuric acid has been reported to be decreased in both human diabetic renal pathology studies (154) and animal DKD models (155), and it has been suggested as an additional indicators of DKD. However, there is no evidence to excluded the direct effect of hippuric acid on T2D or DKD. Further studies are needed to infer causality between hippuric acid and T2D or DKD.
In general, none of the widely used current treatments specifically address the underlying molecular processes responsible for DKD. Several interventional strategies have involved multifactorial approaches, including blood pressure and glucose lowering (156).
RAAS inhibitors have been the treatment of choice for DKD, following the publication of clinical trial results demonstrating benefits of angiotensin converting enzyme inhibitors and angiotensin receptor blockers for decreasing albuminuria in patients with DKD (157). Metformin, the most frequently administered medication to decrease blood glucose, has recently been suggested to enrich SCFA-producing microbiota, such as such as Blautia, Bacteroides, Butyricoccus, Bifidobacterium, Prevotella, Megasphaera, Butyrivibrio (158). SGLT2 inhibitors, GLP-1 receptor agonists, and dipeptidyl peptidase 4 (DPP4) inhibitors are three new classes of glucoselowering agents for patients with DKD (159–161). SGLT2 inhibition has been reported to promote elevations in the levels of the SCFAs acetate and butyric acid in cecal contents of hypertensive mice (162). Vildagliptin, a DPP4 inhibitor, has been reported to enrich SCFA-producing bacteria (163). Moreover, mice on HFD with DPP-4 inhibitor PKF-275-055 treatment showed enriched butyrate-producing Rumminococcus and of the acetogen Dorea (164).
Some drugs for the treatment of DKD can increase the level of SCFA by enriching SCFA-producing microbiota. Whether the mechanism of these drugs in the treatment of DKD is mediated by gut microbial metabolites remains unknown. The relationship between these drugs and gut microbial metabolites still needs more research.
There is growing evidence of the roles of gut microbial metabolites as biomarkers of the pathophysiological features or as pathogenic agents of DKD. In this review, we highlighted aspects relating to the involvement of microbiota metabolites in the pathogenesis of DKD (Figure 4). In the future, we should continue to look for microbial metabolites that can be used to diagnose and treat DKD. However, there are still many challenges to be overcome on the path toward using microbial metabolites for therapies.
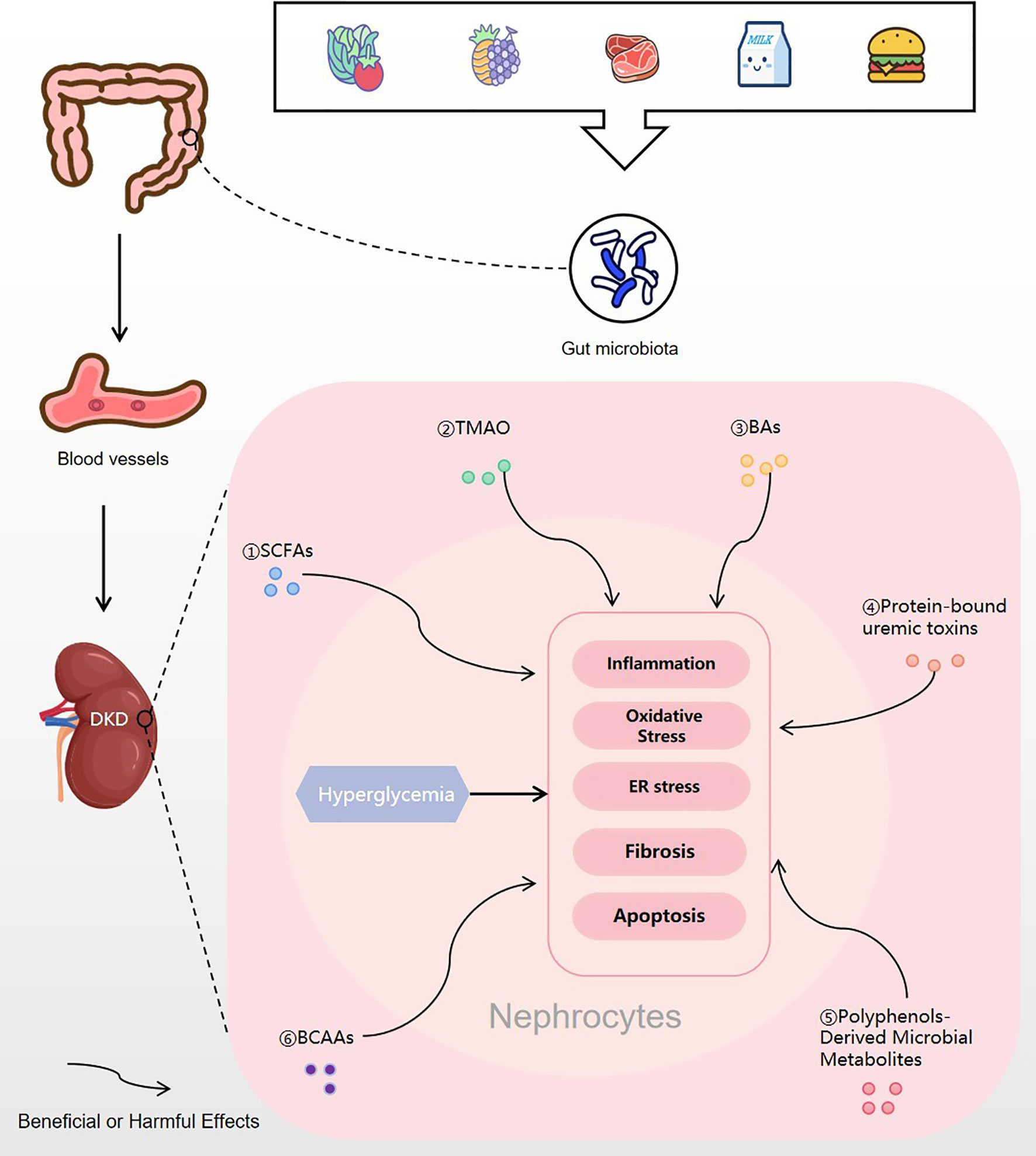
Figure 4 Gut microbial metabolites regulating DKD. Food is digested and absorbed by the gastrointestinal tract, and various gut microbial metabolites are produced under the action of the gut microbiota. Then, the microbial metabolites are absorbed into the blood vessels and finally enter the kidney. In nephrocytes, gut microbial metabolites of SCFAs, TMAO, BAs, protein-bound uremic toxins, polyphenols-derived microbial metabolites and BCAAs may trigger or inhibit inflammation, oxidative stress, ER stress, fibrosis or apoptosis, which will improve or excacerbate the progress of DKD. DKD, diabetic kidney disease; SCFA, short-chain fatty acid; TMAO, trimethylamine N-oxide; BA, bile acids; BCAA, branched-chain amino acid; ER stress, endoplasmic reticulum stress.
First, most microbial metabolites act as signaling molecules by binding to receptors and triggering downstream signaling cascades. Therefore, we should identify the targets of these microbial metabolites in humans. Some metabolites, such as SCFAs and BAs, interact with G-protein-coupled receptors (GPCRs) associated with diverse functions. Many studies have shown that large-scale functional screening for GPCRs can identify microbial metabolites that exert various physiological functions by activating GPCRs. There are many undiscovered receptors similar to GPCRs, and efforts should be made to identify these receptors and generate drugs targeting these receptors in host tissues.
Second, most studies to date have only clarified the correlations between microbial metabolites and T2D, and lack of research on causality. Large prospective cohort studies are needed to determine if microbial metabolites are altered prior to or after disease onset. The result of some studies showing causal effects in rodents should be confirmed in humans. If the results on causality can be confirmed in humans, then further research on the human intestinal microbiota may lead to the development of novel diagnostic and therapeutic tools.
Finally, the concentrations of intestinal metabolites might be highly context-dependent. If the metabolite is beneficial to human health, it needs to be supplemented once its level falls below the physiological level. The supplementation strategy needs to consider the route and frequency of administration, individual differences in pharmacokinetics, and side effects for doses that exceed physiological concentrations. If the metabolite contributes to the pathophysiology of the disease, then the production of the metabolite needs to be suppressed. Inhibiting related enzymes that produce metabolites is a promising method. Developing inhibitors targeting bacterial enzymes is another therapeutic strategy to prevent the action of harmful microbial metabolites, as in the case of TMA lyase producing TMA, a precursor for TMAO. Metabolites do not act in isolation, and thus the combined signals mediated by different metabolites need to be investigated.
We believe that numerous challenges must be overcome on the path toward using microbial metabolites for therapies. Despite these difficulties, the metabolic pathways involved in the production and signaling of microbiome-related metabolites are huge untapped opportunities to regulate disease susceptibility.
QF, NL, and BZ performed the literature search. QF drafted the manuscript. XZ, FG, and XH provided critical intellectual contributions. DO directed the research and made the critical revision. All authors contributed to the article and approved the submitted version.
This work was supported by the National Development of Key Novel Drugs for Special Projects of China [grant numbers 2017ZX09304014], the Hunan Key Laboratory for Bioanalysis of Complex Matrix Samples [grant number 2017TP1037], the Hunan Province Science and Technology Innovation Project, Significance of TMAO, a metabolite of intestinal flora, in adverse cardiovascular reactions caused by antibiotics [grant number 2018SK52008], the Key R&D Programs of Hunan Province [grant number 2019SK2241], and the International Scientific and Technological Innovation Cooperation Base for Bioanalysis of Complex Matrix Samples in Hunan Province [grant number 2019CB1014].
Author DO was employed by company Changsha Duxact Biotech Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res Clin Pract (2019) 157:107843. doi: 10.1016/j.diabres.2019.107843
2. Al-Hasani K, Khurana I, Farhat T, Eid A, El-Osta A. Epigenetics of diabetic nephropathy: from biology to therapeutics. J EMJ (2020) 5(1):48–57.
3. Raval N, Kumawat A, Kalyane D, Kalia K, Tekade R. Understanding molecular upsets in diabetic nephropathy to identify novel targets and treatment opportunities. Drug Discov Today (2020) 25(5):862–78. doi: 10.1016/j.drudis.2020.01.008
4. Vasanth Rao V A/L B, Tan S, Candasamy M, Bhattamisra S. Diabetic nephropathy: an update on pathogenesis and drug development. Diabetes Metab Syndr (2019) 13(1):754–62. doi: 10.1016/j.dsx.2018.11.054
5. Li J, Albajrami O, Zhuo M, Hawley C, Paik J. Decision algorithm for prescribing SGLT2 Inhibitors and GLP-1 receptor agonists for diabetic kidney disease. Clin J Am Soc Nephrol (2020) 15(11):1678–88. doi: 10.2215/cjn.02690320
6. Kato M, Natarajan R. Epigenetics and epigenomics in diabetic kidney disease and metabolic memory. Nat Rev Nephrol (2019) 15(6):327–45. doi: 10.1038/s41581-019-0135-6
7. Jo J, Oh J, Park C. Microbial community analysis using high-throughput sequencing technology: a beginner’s guide for microbiologists. J Microbiol (2020) 58(3):176–92. doi: 10.1007/s12275-020-9525-5
8. Neurath MF. Host–microbiota interactions in inflammatory bowel disease. Nat Rev Immunol (2020) 17(2):76–7. doi: 10.1038/s41575-019-0248-1
9. Delzenne N, Bindels L. Microbiome metabolomics reveals new drivers of human liver steatosis. Nat Med (2018) 24(7):906–7. doi: 10.1038/s41591-018-0126-3
10. Cani P. Human gut microbiome: hopes, threats and promises. Gut (2018) 67(9):1716–25. doi: 10.1136/gutjnl-2018-316723
11. Cani PD. Microbiota and metabolites in metabolic diseases. Nat Rev Endocrinol (2019) 15(2):69–70. doi: 10.1038/s41574-018-0143-9
12. Garrett W. Immune recognition of microbial metabolites. Nat Rev Immunol (2020) 20(2):91–2. doi: 10.1038/s41577-019-0252-2
13. O’Donnell M, Fox B, Chao P, Schroeder F, Sengupta P. A neurotransmitter produced by gut bacteria modulates host sensory behaviour. Nature (2020) 583(7816):415–20. doi: 10.1038/s41586-020-2395-5
14. Vangipurapu J, Fernandes Silva L, Kuulasmaa T, Smith U, Laakso M. Microbiota-related metabolites and the risk of type 2 diabetes. Diabetes Care (2020) 43(6):1319–25. doi: 10.2337/dc19-2533
15. Kikuchi K, Saigusa D, Kanemitsu Y, Matsumoto Y, Thanai P, Suzuki N, et al. Gut microbiome-derived phenyl sulfate contributes to albuminuria in diabetic kidney disease. Nat Commun (2019) 10(1):1835. doi: 10.1038/s41467-019-09735-4
16. Li Y, Su X, Gao Y, Lv C, Gao Z, Liu Y, et al. The potential role of the gut microbiota in modulating renal function in experimental diabetic nephropathy murine models established in same environment. Biochim Biophys Acta Mol Basis Dis (2020) 1866(6):165764. doi: 10.1016/j.bbadis.2020.165764
17. Levy M, Thaiss CA, Elinav E. Metabolites: messengers between the microbiota and the immune system. Genes Dev (2016) 30(14):1589–97. doi: 10.1101/gad.284091.116
18. Serino M. SCFAs — the thin microbial metabolic line between good and bad. Nat Rev Endocrinol (2019) 15(6):318–9. doi: 10.1038/s41574-019-0205-7
19. Dong W, Jia Y, Liu X, Zhang H, Li T, Huang W, et al. Sodium butyrate activates NRF2 to ameliorate diabetic nephropathy possibly via inhibition of HDAC. J Endocrinol (2017) 232(1):71–83. doi: 10.1530/joe-16-0322
20. Du Y, Tang G, Yuan W. Suppression of HDAC2 by sodium butyrate alleviates apoptosis of kidney cells in db/db mice and HG−induced NRK−52E cells. Int J Mol Med (2020) 45(1):210–22. doi: 10.3892/ijmm.2019.4397
21. Parada Venegas D, De la Fuente M, Landskron G, González M, Quera R, Dijkstra G, et al. Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front Immunol (2019) 10:277. doi: 10.3389/fimmu.2019.00277
22. Khan S, Jena G. Sodium butyrate, a HDAC inhibitor ameliorates eNOS, iNOS and TGF-β1-induced fibrogenesis, apoptosis and DNA damage in the kidney of juvenile diabetic rats. Food Chem Toxicol (2014) 73:127–39. doi: 10.1016/j.fct.2014.08.010
23. Li Y, Chen X, Kwan T, Loh Y, Singer J, Liu Y, et al. Dietary fiber protects against diabetic nephropathy through short-chain fatty acid-mediated activation of G protein-coupled receptors GPR43 and GPR109A. J Am Soc Nephrol (2020) 31(6):1267–81. doi: 10.1681/asn.2019101029
24. Huang J, Pearson JA, Peng J, Hu Y, Sha S, Xing Y, et al. Gut microbial metabolites alter IgA immunity in type 1 diabetes. JCI Insight (2020) 5(10):e135718. doi: 10.1172/jci.insight.135718
25. Zhao L, Zhang F, Ding X, Wu G, Lam Y, Wang X, et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science (2018) 359(6380):1151–6. doi: 10.1126/science.aao5774
26. Huang W, Guo H-L, Deng X, Zhu T-T, Xiong J-F, Xu Y-H, et al. Short-chain fatty acids inhibit oxidative stress and inflammation in mesangial cells induced by high glucose and lipopolysaccharide. Exp Clin Endocrinol Diabetes (2017) 125(2):98–105. doi: 10.1055/s-0042-121493
27. Huang W, Man Y, Gao C, Zhou L, Gu J, Xu H, et al. Short-chain fatty acids ameliorate diabetic nephropathy via GPR43-mediated inhibition of oxidative stress and NF-κB signaling. Oxid Med Cell Longev (2020) 2020:4074832. doi: 10.1155/2020/4074832
28. Dong W, Jia Y, Liu X, Zhang H, Li T, Huang W, et al. Sodium butyrate activates NRF2 to ameliorate diabetic nephropathy possibly via inhibition of HDAC. J Endocrinol (2017) 232(1):71–83. doi: 10.1530/joe-16-0322
29. Gu J, Huang W, Zhang W, Zhao T, Gao C, Gan W, et al. Sodium butyrate alleviates high-glucose-induced renal glomerular endothelial cells damage via inhibiting pyroptosis. Int Immunopharmacol (2019) 75:105832. doi: 10.1016/j.intimp.2019.105832
30. Du Y, Yang Y, Tang G, Jia J, Zhu N, Yuan W. Butyrate alleviates diabetic kidney disease by mediating the miR-7a-5p/P311/TGF-β1 pathway. FASEB J (2020) 34(8):10462–75. doi: 10.1096/fj.202000431R
31. Xu Y, Gao C, Guo H, Zhang W, Huang W, Tang S, et al. Sodium butyrate supplementation ameliorates diabetic inflammation in db/db mice. J Endocrinol (2018) 238(3):231–44. doi: 10.1530/joe-18-0137
32. Huang W, Xu Y, Xu Y, Zhou L, Gao C. Short-chain fatty acids prevent diabetic nephropathy in vivo and in vitro. Am Diabetes Assoc (2018) 67(Supplement 1):92–OR. doi: 10.2337/db18-92-OR
33. Lu C-C, Hu Z-B, Wang R, Hong Z-H, Lu J, Chen P-P, et al. Gut microbiota dysbiosis-induced activation of the intrarenal renin–angiotensin system is involved in kidney injuries in rat diabetic nephropathy. Acta Pharmacol Sin (2020) 10(10):1398. doi: 10.1038/s41401-019-0326-5
34. Hu ZB, Lu J, Chen PP, Lu CC, Zhang JX, Li XQ, et al. Dysbiosis of intestinal microbiota mediates tubulointerstitial injury in diabetic nephropathy via the disruption of cholesterol homeostasis. Theranostics (2020) 10(6):2803–16. doi: 10.7150/thno.40571
35. Zeisel S, Warrier M. Trimethylamine N-Oxide, the microbiome, and heart and kidney disease. Annu Rev Nutr (2017) 37:157–81. doi: 10.1146/annurev-nutr-071816-064732
36. Falony G, Vieira-Silva S, Raes J. Microbiology meets big data: the case of gut microbiota-derived trimethylamine. Annu Rev Microbiol (2015) 69:305–21. doi: 10.1146/annurev-micro-091014-104422
37. Janeiro MH, Ramírez MJ, Milagro FI, Martínez JA, Solas M. Implication of Trimethylamine N-Oxide (TMAO) in disease: potential biomarker or new therapeutic target. Nutrients (2018) 10(10):57–63. doi: 10.3390/nu10101398
38. Zhu W, Gregory JC, Org E, Buffa JA, Gupta N, Wang Z, et al. Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell (2016) 165(1):111–24. doi: 10.1016/j.cell.2016.02.011
39. Tang WH, Wang Z, Shrestha K, Borowski AG, Wu Y, Troughton RW, et al. Intestinal microbiota-dependent phosphatidylcholine metabolites, diastolic dysfunction, and adverse clinical outcomes in chronic systolic heart failure. J Card Fail (2015) 21(2):91–6. doi: 10.1016/j.cardfail.2014.11.006
40. Roncal C, Martínez-Aguilar E, Orbe J, Ravassa S, Fernandez-Montero A, De Pipaon GS, et al. Trimethylamine (Tma) and Trimethylamine-N-Oxide (Tmao) as predictors of cardiovascular mortality in peripheral artery disease. atherosclerosis (2019) 287:e233. doi: 10.1016/j.atherosclerosis.2019.06.716
41. Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature (2011) 472(7341):57–63. doi: 10.1038/nature09922
42. Tang WH, Wang Z, Kennedy DJ, Wu Y, Buffa JA, Agatisa-Boyle B, et al. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ Res (2015) 116(3):448–55. doi: 10.1161/CIRCRESAHA.116.305360
43. Chen S, Henderson A, Petriello MC, Romano KA, Gearing M, Miao J, et al. Trimethylamine N-oxide binds and activates PERK to promote metabolic dysfunction. Cell Metab (2019) 74(6):558–65. doi: 10.1016/j.cmet.2019.08.021
44. Vogt N, Romano K, Darst B, Engelman C, Johnson S, Carlsson C, et al. The gut microbiota-derived metabolite trimethylamine N-oxide is elevated in Alzheimer’s disease. Alzheimers Res Ther (2018) 10(1):124. doi: 10.1186/s13195-018-0451-2
45. Yue C, Yang X, Li J, Chen X, Zhao X, Chen Y, et al. Trimethylamine N-oxide prime NLRP3 inflammasome via inhibiting ATG16L1-induced autophagy in colonic epithelial cells. Biochem Biophys Res Commun (2017) 490(2):541–51. doi: 10.1016/j.bbrc.2017.06.075
46. Chen ML, Zhu XH, Ran L, Lang HD, Yi L, Mi MT. Trimethylamine-N-oxide induces vascular inflammation by activating the NLRP3 inflammasome through the SIRT3-SOD2-mtROS signaling pathway. J Am Heart Assoc (2017) 6(9):e002767. doi: 10.1161/JAHA.117.006347
47. Yamagata K, Hashiguchi K, Yamamoto H, Tagami M. Dietary apigenin reduces induction of LOX-1 and NLRP3 expression, leukocyte adhesion and acetylated low-density lipoprotein uptake in human endothelial cells exposed to trimethylamine-N-oxide. J Cardiovasc Pharmacol (2019) 94:203–12. doi: 10.1097/fjc.0000000000000747
48. Zhang X, Li Y, Yang P, Liu X, Lu L, Chen Y, et al. Trimethylamine-N-oxide promotes vascular calcification through activation of NLRP3 (Nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3) inflammasome and NF-kappaB (Nuclear factor kappaB) signals. Arterioscler Thromb Vasc Biol (2020) 363:ATVBAHA119313414. doi: 10.1161/ATVBAHA.119.313414
49. Ma G, Pan B, Chen Y, Guo C, Zhao M, Zheng L, et al. Trimethylamine N-oxide in atherogenesis: impairing endothelial self-repair capacity and enhancing monocyte adhesion. Biosci Rep (2017) 37(2):88–100. doi: 10.1042/bsr20160244
50. Seldin MM, Meng Y, Qi H, Zhu W, Wang Z, Hazen SL, et al. Trimethylamine N-oxide promotes vascular inflammation through signaling of mitogen-activated protein kinase and nuclear factor-kappaB. J Am Heart Assoc (2016) 5(2):3570. doi: 10.1161/JAHA.115.002767
51. Ren D, Liu Y, Zhao Y, Yang X. Hepatotoxicity and endothelial dysfunction induced by high choline diet and the protective effects of phloretin in mice. Food Chem Toxicol (2016) 94:203–12. doi: 10.1016/j.fct.2016.06.004
52. Guada M, Ganugula R, Vadhanam M, Ravi Kumar MNV. Urolithin A mitigates cisplatin-Induced nephrotoxicity by inhibiting renal inflammation and apoptosis in an experimental rat model. J Pharmacol Exp Ther (2017) 363(1):58–65. doi: 10.1124/jpet.117.242420
53. Ke Y, Li D, Zhao M, Liu C, Liu J, Zeng A, et al. Gut flora-dependent metabolite Trimethylamine-N-oxide accelerates endothelial cell senescence and vascular aging through oxidative stress. Free Radic Biol Med (2018) 116:88–100. doi: 10.1016/j.freeradbiomed.2018.01.007
54. Singh GB, Zhang Y, Boini KM, Koka S. High mobility group box 1 mediates TMAO-induced endothelial dysfunction. Int J Mol Sci (2019) 20(14):158–71. doi: 10.3390/ijms20143570
55. Sun G, Yin Z, Liu N, Bian X, Yu R, Su X, et al. Gut microbial metabolite TMAO contributes to renal dysfunction in a mouse model of diet-induced obesity. Biochem Biophys Res Commun (2017) 493(2):964–70. doi: 10.1016/j.bbrc.2017.09.108
56. Li T, Gua C, Wu B, Chen Y. Increased circulating trimethylamine N-oxide contributes to endothelial dysfunction in a rat model of chronic kidney disease. Biochem Biophys Res Commun (2018) 495(2):2071–7. doi: 10.1016/j.bbrc.2017.12.069
57. Gupta N, Buffa JA, Roberts AB, Sangwan N, Skye SM, Li L, et al. Targeted inhibition of gut microbial trimethylamine N-oxide production reduces renal tubulointerstitial fibrosis and functional impairment in a murine model of chronic kidney disease. Am Heart Assoc (2020) 40(5):1239–55. doi: 10.1161/ATVBAHA.120.314139
58. Winston J, Theriot C. Diversification of host bile acids by members of the gut microbiota. Gut Microbes (2020) 11(2):158–71. doi: 10.1080/19490976.2019.1674124
59. Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res (2006) 47(2):241–59. doi: 10.1194/jlr.R500013-JLR200
60. Ridlon JM, Harris SC, Bhowmik S, Kang DJ, Hylemon PB. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes (2016) 7(1):22–39. doi: 10.1080/19490976.2015.1127483
61. Begley M, Gahan CG, Hill C. The interaction between bacteria and bile. FEMS Microbiol Rev (2005) 29(4):625–51. doi: 10.1016/j.femsre.2004.09.003
62. Chiang J, Ferrell J. Bile acids as metabolic regulators and nutrient sensors. Annu Rev Nutr (2019) 39:175–200. doi: 10.1146/annurev-nutr-082018-124344
63. Wang X, Wang D, Luo Y, Myakala K, Dobrinskikh E, Rosenberg A, et al. FXR/TGR5 dual agonist prevents progression of nephropathy in diabetes and obesity. J Am Soc Nephrol (2018) 29(1):118–37. doi: 10.1681/asn.2017020222
64. Jiang T, Wang X, Scherzer P, Wilson P, Tallman J, Takahashi H, et al. Farnesoid X receptor modulates renal lipid metabolism, fibrosis, and diabetic nephropathy. Diabetes (2007) 56(10):2485–93. doi: 10.2337/db06-1642
65. Wang XX, Wang D, Luo Y, Myakala K, Dobrinskikh E, Rosenberg AZ, et al. FXR/TGR5 dual agonist prevents progression of nephropathy in diabetes and obesity. J Am Soc Nephrol (2018) 29(1):118–37. doi: 10.1681/ASN.2017020222
66. Marquardt A, Al-Dabet MDM, Ghosh S, Kohli S, Manoharan J, ElWakiel A, et al. Farnesoid X receptor agonism protects against diabetic tubulopathy: potential add-on therapy for diabetic nephropathy. J Am Soc Nephrol (2017) 28(11):3182–9. doi: 10.1681/ASN.2016101123
67. Han SY, Song HK, Cha JJ, Han JY, Kang YS, Cha DR. Farnesoid X receptor (FXR) agonist ameliorates systemic insulin resistance, dysregulation of lipid metabolism, and alterations of various organs in a type 2 diabetic kidney animal model. Acta Diabetol (2021) 54(3):657–70. doi: 10.1007/s00592-020-01652-z
68. Wang X, Edelstein M, Gafter U, Qiu L, Luo Y, Dobrinskikh E, et al. G protein-coupled bile acid receptor TGR5 activation inhibits kidney disease in obesity and diabetes. J Am Soc Nephrol (2016) 27(5):1362–78. doi: 10.1681/asn.2014121271
69. Xiao H, Sun X, Liu R, Chen Z, Lin Z, Yang Y, et al. Gentiopicroside activates the bile acid receptor Gpbar1 (TGR5) to repress NF-kappaB pathway and ameliorate diabetic nephropathy. Pharmacol Res (2020) 151:104559. doi: 10.1016/j.phrs.2019.104559
70. Yang Z, Xiong F, Wang Y, Gong W, Huang J, Chen C, et al. TGR5 activation suppressed S1P/S1P2 signaling and resisted high glucose-induced fibrosis in glomerular mesangial cells. Pharmacol Res (2016) 111:226–36. doi: 10.1016/j.phrs.2016.05.035
71. Xiong F, Li X, Yang Z, Wang Y, Gong W, Huang J, et al. TGR5 suppresses high glucose-induced upregulation of fibronectin and transforming growth factor-β1 in rat glomerular mesangial cells by inhibiting RhoA/ROCK signaling. Endocrine (2016) 54(3):657–70. doi: 10.1007/s12020-016-1032-4
72. Sun L, Xie C, Wang G, Wu Y, Wu Q, Wang X, et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat Med (2018) 24(12):1919–29. doi: 10.1038/s41591-018-0222-4
73. Sonne D. Mechanisms in endocrinology : FXR signalling - a novel target in metabolic diseases. Eur J Endocrinol (2021) 8(1):109–19. doi: 10.1530/eje-20-1410
74. Niewczas M, Sirich T, Mathew A, Skupien J, Mohney R, Warram J, et al. Uremic solutes and risk of end-stage renal disease in type 2 diabetes: metabolomic study. Kidney Int (2014) 85(5):1214–24. doi: 10.1038/ki.2013.497
75. Ottiger M, Nickler M, Steuer C, Bernasconi L, Huber A, Christ-Crain M, et al. Gut, microbiota-dependent trimethylamine-N-oxide is associated with long-term all-cause mortality in patients with exacerbated chronic obstructive pulmonary disease. Nutrition (2018) 45:135–41.e1. doi: 10.1016/j.nut.2017.07.001
76. Keszthelyi D, Troost FJ, Masclee AAM. Understanding the role of tryptophan and serotonin metabolism in gastrointestinal function. Neurogastroenterol Motil (2009) 21(12):1239–49. doi: 10.1111/j.1365-2982.2009.01370.x
77. van der Kloet F, Tempels F, Ismail N, van der Heijden R, Kasper P, Rojas-Cherto M, et al. Discovery of early-stage biomarkers for diabetic kidney disease using ms-based metabolomics (FinnDiane study). Metabolomics (2012) 8(1):109–19. doi: 10.1007/s11306-011-0291-6
78. Atoh K, Itoh H, Haneda M. Serum indoxyl sulfate levels in patients with diabetic nephropathy: relation to renal function. Diabetes Res Clin Pract (2009) 83(2):220–6. doi: 10.1016/j.diabres.2008.09.053
79. Chiu C, Lu L, Yu T, Hung W, Chung F, Tsai I, et al. Increased levels of total P-Cresylsulphate and indoxyl sulphate are associated with coronary artery disease in patients with diabetic nephropathy. Rev Diabetes Stud (2010) 7(4):275–84. doi: 10.1900/rds.2010.7.275
80. Men L, Pi Z, Zhou Y, Liu Y, Wei M, Song F, et al. Metabolomics insights into diabetes nephropathy and protective effects of Radix Scutellariae on rats using ultra-high performance liquid chromatography coupled with quadrupole time-of-flight mass spectrometry. RSC Adv (2017) 7(27):16494–504. doi: 10.1039/C6RA28595C
81. Gooding J, Cao L, Whitaker C, Mwiza J, Fernander M, Ahmed F, et al. Meprin β metalloproteases associated with differential metabolite profiles in the plasma and urine of mice with type 1 diabetes and diabetic nephropathy. BMC Nephrol (2019) 20(1):141. doi: 10.1186/s12882-019-1313-2
82. Zhao T, Zhang H, Zhao T, Zhang X, Lu J, Yin T, et al. Intrarenal metabolomics reveals the association of local organic toxins with the progression of diabetic kidney disease. J Pharm BioMed Anal (2012) 60:32–43. doi: 10.1016/j.jpba.2011.11.010
83. Ji C, Luo Y, Zou C, Huang L, Tian R, Lu Z. Effect of astragaloside IV on indoxyl sulfate-induced kidney injury in mice via attenuation of oxidative stress. BMC Pharmacol Toxicol (2018) 19(1):53. doi: 10.1186/s40360-018-0241-2
84. Tanaka S, Watanabe H, Nakano T, Imafuku T, Kato H, Tokumaru K, et al. Indoxyl sulfate contributes to adipose tissue inflammation through the activation of NADPH oxidase. Toxins (Basel) (2020) 12(8):523–35. doi: 10.3390/toxins12080502
85. Ellis R, Small D, Ng K, Vesey D, Vitetta L, Francis R, et al. Indoxyl sulfate induces apoptosis and hypertrophy in human kidney proximal tubular cells. Toxicol Pathol (2018) 46(4):449–59. doi: 10.1177/0192623318768171
86. Park J, Choi H, Bae E, Ma S, Kim S. Paricalcitol attenuates indoxyl sulfate-induced apoptosis through the inhibition of MAPK, Akt, and NF-kB activation in HK-2 cells. Korean J Intern Med (2019) 34(1):146–55. doi: 10.3904/kjim.2016.298
87. Gryp T, Vanholder R, Vaneechoutte M, Glorieux G. p-Cresyl sulfate. Toxins (2017) 9(2):52. doi: 10.3390/toxins9020052
88. Russell WR, Duncan SH, Scobbie L, Duncan G, Cantlay L, Calder AG, et al. Major phenylpropanoid-derived metabolites in the human gut can arise from microbial fermentation of protein. Mol Nutr Food Res (2013) 57(3):523–35. doi: 10.1002/mnfr.201200594
89. Mutsaers H, Caetano-Pinto P, Seegers A, Dankers A, van den Broek P, Wetzels J, et al. Proximal tubular efflux transporters involved in renal excretion of p-cresyl sulfate and p-cresyl glucuronide: Implications for chronic kidney disease pathophysiology. Toxicol In Vitro (2015) 29(7):1868–77. doi: 10.1016/j.tiv.2015.07.020
90. Park J, Choi H, Kim D, Kim C, Bae E, Ma S, et al. Alpha-lipoic acid attenuates p-cresyl sulfate-induced renal tubular injury through suppression of apoptosis and autophagy in human proximal tubular epithelial cells. BioMed Pharmacother (2019) 112:108679. doi: 10.1016/j.biopha.2019.108679
91. Poveda J, Sanchez-Niño M, Glorieux G, Sanz A, Egido J, Vanholder R, et al. p-cresyl sulphate has pro-inflammatory and cytotoxic actions on human proximal tubular epithelial cells. Nephrol Dial Transplant (2014) 29(1):56–64. doi: 10.1093/ndt/gft367
92. Watanabe H, Miyamoto Y, Honda D, Tanaka H, Wu Q, Endo M, et al. p-Cresyl sulfate causes renal tubular cell damage by inducing oxidative stress by activation of NADPH oxidase. Kidney Int (2013) 83(4):582–92. doi: 10.1038/ki.2012.448
93. Smith EA, Macfarlane GT. Enumeration of human colonic bacteria producing phenolic and indolic compounds: effects of pH, carbohydrate availability and retention time on dissimilatory aromatic amino acid metabolism. J Appl Bacteriol (1996) 81(3):288–302. doi: 10.1111/j.1365-2672.1996.tb04331.x
94. Walker AW, Duncan SH, Louis P, Flint HJ. Phylogeny, culturing, and metagenomics of the human gut microbiota. Trends Microbiol (2014) 22(5):267–74. doi: 10.1016/j.tim.2014.03.001
95. Mikkaichi T, Suzuki T, Onogawa T, Tanemoto M, Mizutamari H, Okada M, et al. Isolation and characterization of a digoxin transporter and its rat homologue expressed in the kidney. Proc Natl Acad Sci U S A (2004) 101(10):3569–74. doi: 10.1073/pnas.0304987101
96. Konishi K, Nakano S, Tsuda S, Nakagawa A, Kigoshi T, Koya D. AST-120 (Kremezin) initiated in early stage chronic kidney disease stunts the progression of renal dysfunction in type 2 diabetic subjects. Diabetes Res Clin Pract (2008) 81(3):310–5. doi: 10.1016/j.diabres.2008.04.024
97. Ohtake T, Mochida Y, Ishioka K, Moriya H, Hidaka S, Hirata M, et al. Improvement of microcirculatory impairment in patients with predialysis chronic kidney disease after AST-120 administration. Renal Replacement Ther (2020) 6(1):1–8. doi: 10.1186/s41100-020-00276-x
98. Toyoda S, Hashimoto R, Tezuka T, Sakuma M, Abe S, Ishikawa T, et al. Antioxidative effect of an oral adsorbent, AST-120, and long-term outcomes in chronic kidney disease patients with cardiovascular disease. Hypertens Res (2020) 43(10):1128–31. doi: 10.1038/s41440-020-0465-6
99. Eid HM, Wright ML, Anil Kumar NV, Qawasmeh A, Hassan STS, Mocan A, et al. Significance of microbiota in obesity and metabolic diseases and the modulatory potential by medicinal plant and food ingredients. Front Pharmacol (2017) 8:387. doi: 10.3389/fphar.2017.00387
100. Espín J, González-Sarrías A, Tomás-Barberán F. The gut microbiota: a key factor in the therapeutic effects of (poly)phenols. Biochem Pharmacol (2017) 139:82–93. doi: 10.1016/j.bcp.2017.04.033
101. Braune A, Blaut M. Bacterial species involved in the conversion of dietary flavonoids in the human gut. Gut Microbes (2016) 7(3):216–34. doi: 10.1080/19490976.2016.1158395
102. Martinez-Zapata MJ, Vernooij RW, Uriona Tuma SM, Stein AT, Moreno RM, Vargas E, et al. Phlebotonics for venous insufficiency. Cochrane Database Syst Rev (2016) 4(4):Cd003229. doi: 10.1002/14651858.CD003229.pub3
103. Quartieri A, García-Villalba R, Amaretti A, Raimondi S, Leonardi A, Rossi M, et al. Detection of novel metabolites of flaxseed lignans in vitro and in vivo. Mol Nutr Food Res (2016) 60(7):1590–601. doi: 10.1002/mnfr.201500773
104. Tomas-Barberan F, García-Villalba R, Quartieri A, Raimondi S, Amaretti A, Leonardi A, et al. In vitro transformation of chlorogenic acid by human gut microbiota. Mol Nutr Food Res (2014) 58(5):1122–31. doi: 10.1002/mnfr.201300441
105. Selma MV, Beltrán D, García-Villalba R, Espín JC, Tomás-Barberán FA. Description of urolithin production capacity from ellagic acid of two human intestinal Gordonibacter species. Food Funct (2014) 5(8):1779–84. doi: 10.1039/c4fo00092g
106. Tomás-Barberán FA, García-Villalba R, González-Sarrías A, Selma MV, Espín JC. Ellagic acid metabolism by human gut microbiota: consistent observation of three urolithin phenotypes in intervention trials, independent of food source, age, and health status. J Agric Food Chem (2014) 62(28):6535–8. doi: 10.1021/jf5024615
107. Clavel T, Lippman R, Gavini F, Doré J, Blaut M. Clostridium saccharogumia sp. nov. and Lactonifactor longoviformis gen. nov., sp. nov., two novel human faecal bacteria involved in the conversion of the dietary phytoestrogen secoisolariciresinol diglucoside. Syst Appl Microbiol (2007) 30(1):16–26. doi: 10.1016/j.syapm.2006.02.003
108. Sun Q, Wedick NM, Pan A, Townsend MK, Cassidy A, Franke AA, et al. Gut microbiota metabolites of dietary lignans and risk of type 2 diabetes: a prospective investigation in two cohorts of U.S. women. Diabetes Care (2014) 37(5):1287–95. doi: 10.2337/dc13-2513
109. Eriksen AK, Kyrø C, Nørskov NP, Frederiksen K, Bach Knudsen K-E, Overvad K, et al. Pre-diagnostic plasma enterolactone concentrations are associated with lower mortality among individuals with type 2 diabetes: a case-cohort study in the Danish Diet, Cancer and Health cohort. Diabetologia (2019) 62(6):959–69. doi: 10.1007/s00125-019-4854-9
110. Zhou F, Furuhashi K, Son MJ, Toyozaki M, Yoshizawa F, Miura Y, et al. Antidiabetic effect of enterolactone in cultured muscle cells and in type 2 diabetic model db/db mice. Cytotechnology (2017) 69(3):493–502. doi: 10.1007/s10616-016-9965-2
111. Talaei M, Lee BL, Ong CN, van Dam RM, Yuan JM, Koh WP, et al. Urine phyto-oestrogen metabolites are not significantly associated with risk of type 2 diabetes: the Singapore Chinese health study. Br J Nutr (2016) 115(9):1607–15. doi: 10.1017/S0007114516000581
112. Charytoniuk T, Iłowska N, Berk K, Drygalski K, Chabowski A, Konstantynowicz-Nowicka K. The effect of enterolactone on sphingolipid pathway and hepatic insulin resistance development in HepG2 cells. Life Sci (2019) 217:1–7. doi: 10.1016/j.lfs.2018.11.044
113. Toney A, Fan R, Xian Y, Chaidez V, Ramer-Tait A, Chung S. Urolithin A, a gut metabolite, improves insulin sensitivity through augmentation of mitochondrial function and biogenesis. Obes (Silver Spring) (2019) 27(4):612–20. doi: 10.1002/oby.22404
114. Xia B, Shi XC, Xie BC, Zhu MQ, Chen Y, Chu XY, et al. Urolithin A exerts antiobesity effects through enhancing adipose tissue thermogenesis in mice. PLoS Biol (2020) 18(3):e3000688. doi: 10.1371/journal.pbio.3000688
115. Tuohetaerbaike B, Zhang Y, Tian Y, Zhang N, Kang J, Mao X, et al. Pancreas protective effects of Urolithin A on type 2 diabetic mice induced by high fat and streptozotocin via regulating autophagy and AKT/mTOR signaling pathway. J Ethnopharmacol (2020) 250:112479. doi: 10.1016/j.jep.2019.112479
116. Jing T, Liao J, Shen K, Chen X, Xu Z, Tian W, et al. Protective effect of urolithin a on cisplatin-induced nephrotoxicity in mice via modulation of inflammation and oxidative stress. Food Chem Toxicol (2019) 129:108–14. doi: 10.1016/j.fct.2019.04.031
117. Chen P, Lei J, Chen F, Zhou B. Ameliorative effect of urolithin A on d-gal-induced liver and kidney damage in aging mice via its antioxidative, anti-inflammatory and antiapoptotic properties. RSC Adv (2020) 10(14):8027–38. doi: 10.1039/D0RA00774A
118. Wang Y, Huang H, Jin Y, Shen K, Chen X, Xu Z, et al. Role of TFEB in autophagic modulation of ischemia reperfusion injury in mice kidney and protection by urolithin A. Food Chem Toxicol (2019) 131:110591. doi: 10.1016/j.fct.2019.110591
119. Bayle M, Neasta J, Dall’Asta M, Gautheron G, Virsolvy A, Quignard J, et al. The ellagitannin metabolite urolithin C is a glucose-dependent regulator of insulin secretion through activation of L-type calcium channels. Br J Pharmacol (2019) 176(20):4065–78. doi: 10.1111/bph.14821
120. Toubal S, Oiry C, Bayle M, Cros G, Neasta J. Urolithin C increases glucose-induced ERK activation which contributes to insulin secretion. Fundam Clin Pharmacol (2020) 34(5):571–80. doi: 10.1111/fcp.12551
121. Sala R, Mena P, Savi M, Brighenti F, Crozier A, Miragoli M, et al. Urolithins at physiological concentrations affect the levels of pro-inflammatory cytokines and growth factor in cultured cardiac cells in hyperglucidic conditions. J Funct Foods (2015) 15:97–105. doi: 10.1016/j.jff.2015.03.019
122. Savi M, Bocchi L, Mena P, Dall’Asta M, Crozier A, Brighenti F, et al. In vivo administration of urolithin A and B prevents the occurrence of cardiac dysfunction in streptozotocin-induced diabetic rats. Cardiovasc Diabetol (2017) 16(1):80. doi: 10.1186/s12933-017-0561-3
123. White PJ, Newgard CB. Branched-chain amino acids in disease. Science (2019) 363(6427):582–3. doi: 10.1126/science.aav0558
124. Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, et al. Metabolite profiles and the risk of developing diabetes. Nat Med (2011) 17(4):448–53. doi: 10.1038/nm.2307
125. Ahola-Olli AV, Mustelin L, Kalimeri M, Kettunen J, Jokelainen J, Auvinen J, et al. Circulating metabolites and the risk of type 2 diabetes: a prospective study of 11,896 young adults from four Finnish cohorts. Diabetologia (2019) 62(12):2298–309. doi: 10.1007/s00125-019-05001-w
126. Pedersen HK, Gudmundsdottir V, Nielsen HB, Hyotylainen T, Nielsen T, Jensen BAH, et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature (2016) 535(7612):376–81. doi: 10.1038/nature18646
127. Asghari G, Farhadnejad H, Teymoori F, Mirmiran P, Tohidi M, Azizi F. High dietary intake of branched-chain amino acids is associated with an increased risk of insulin resistance in adults. J Diabetes (2018) 10(5):357–64. doi: 10.1111/1753-0407.12639
128. Karusheva Y, Koessler T, Strassburger K, Markgraf D, Mastrototaro L, Jelenik T, et al. Short-term dietary reduction of branched-chain amino acids reduces meal-induced insulin secretion and modifies microbiome composition in type 2 diabetes: a randomized controlled crossover trial. Am J Clin Nutr (2019) 110(5):1098–107. doi: 10.1093/ajcn/nqz191
129. Maguire M, Surugihalli C, Hughes A, Sunny N. Impact of branched chain amino acids on mitochondrial metabolism and insulin signaling in mouse hepatocytes. Curr Dev Nutr (2019) 3(Supplement_1):873–8. doi: 10.1093/cdn/nzz044.P08-111-19
130. Zhao H, Zhang F, Sun D, Wang X, Zhang X, Zhang J, et al. Branched-chain amino acids exacerbate obesity-related hepatic glucose and lipid metabolic disorders via attenuating Akt2 signaling. Diabetes (2020) 69(6):1164–77. doi: 10.2337/db19-0920
131. Lotta LA, Scott RA, Sharp SJ, Burgess S, Luan J, Tillin T, et al. Genetic predisposition to an impaired metabolism of the branched-chain amino acids and risk of type 2 diabetes: a mendelian randomisation analysis. PLoS Med (2016) 13(11):e1002179–e. doi: 10.1371/journal.pmed.1002179
132. Wang Q, Holmes M, Davey Smith G, Ala-Korpela M. Genetic support for a causal role of insulin resistance on circulating branched-chain amino acids and inflammation. Diabetes Care (2017) 40(12):1779–86. doi: 10.2337/dc17-1642
133. Mahendran Y, Jonsson A, Have CT, Allin KH, Witte DR, Jørgensen ME, et al. Genetic evidence of a causal effect of insulin resistance on branched-chain amino acid levels. Diabetologia (2017) 60(5):873–8. doi: 10.1007/s00125-017-4222-6
134. Lim L-L, Lau ES, Lee HM, Tam CH, Lim CKP, Luk A, et al. 533-P: Association of serum branched-chain amino acids with kidney function decline in type 2 diabetes: The Hong Kong diabetes register. Diabetes (2019) 68(Supplement 1):533–P. doi: 10.2337/db19-533-P
135. Pillai S, Herzog B, Seebeck P, Pellegrini G, Roth E, Verrey F. Differential impact of dietary branched chain and aromatic amino acids on chronic kidney disease progression in rats. Front Physiol (2019) 10:1460. doi: 10.3389/fphys.2019.01460
136. Roquetto A, Moura C, de Almeida Santos-Junior V, Oliveira P, Machado K, Carvalho G, et al. Moderate intake of BCAA-rich protein improves glucose homeostasis in high-fat-fed mice. J Nutr Biochem (2020) 80:108332. doi: 10.1016/j.jnutbio.2019.108332
137. Lu M, Zhang X, Zheng D, Jiang X, Chen Q. Branched-chain amino acids supplementation protects streptozotocin-induced insulin secretion and the correlated mechanism. Biofactors (2015) 41(2):127–33. doi: 10.1002/biof.1188
138. Mi N, Zhang X, Ding Y, Li G, Wang W, Xian H, et al. Branched-chain amino acids attenuate early kidney injury in diabetic rats. Biochem Biophys Res Commun (2015) 466(2):240–6. doi: 10.1016/j.bbrc.2015.09.017
139. Zhang X, Liu D, He Y, Lou K, Zheng D, Han W. Branched chain amino acids protects rat mesangial cells from high glucose by modulating TGF-β1 and BMP-7. Diabetes Metab Syndr Obes (2019) 12:2433–40. doi: 10.2147/DMSO.S221642
140. Zeng S-L, Li S-Z, Xiao P-T, Cai Y-Y, Chu C, Chen B-Z, et al. Citrus polymethoxyflavones attenuate metabolic syndrome by regulating gut microbiome and amino acid metabolism. Sci Adv (2020) 6(1):eaax6208. doi: 10.1126/sciadv.aax6208
141. Ridaura V, Faith J, Rey F, Cheng J, Duncan A, Kau A, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science (2013) 341(6150):1241214. doi: 10.1126/science.1241214
142. Koh A, Molinaro A, Stahlman M, Khan MT, Schmidt C, Manneras-Holm L, et al. Microbially produced imidazole propionate impairs insulin signaling through mTORC1. Cell (2018) 175(4):947–61.e17. doi: 10.1016/j.cell.2018.09.055
143. Brial F, Alzaid F, Sonomura K, Meneyrol K, Le Lay A, Péan N, et al. The microbial metabolite 4-cresol improves glucose homeostasis and enhances β-cell function. bioRxiv (2018) 56:444893. doi: 10.1016/j.celrep.2020.01.066
144. Brial F, Alzaid F, Sonomura K, Kamatani Y, Meneyrol K, Le Lay A, et al. The natural metabolite 4-cresol improves glucose homeostasis and enhances β-cell function. Cell Rep (2020) 30(7):2306–20.e5. doi: 10.1016/j.celrep.2020.01.066
145. Peck S, Denger K, Burrichter A, Irwin S, Balskus E, Schleheck D. Bilophila wadsworthiaA glycyl radical enzyme enables hydrogen sulfide production by the human intestinal bacterium. Proc Natl Acad Sci U S A (2019) 116(8):3171–6. doi: 10.1073/pnas.1815661116
146. Kalantar-Zadeh K, Berean KJ, Burgell RE, Muir JG, Gibson PR. Intestinal gases: influence on gut disorders and the role of dietary manipulations. Nat Rev Gastroenterol Hepatol (2019) 16(12):733–47. doi: 10.1038/s41575-019-0193-z
147. Suzuki K, Sagara M, Aoki C, Tanaka S, Aso Y. Clinical implication of plasma hydrogen sulfide levels in Japanese patients with type 2 diabetes. Intern Med (2017) 56(1):17–21. doi: 10.2169/internalmedicine.56.7403
148. Pichette J, Fynn-Sackey N, Gagnon J. Hydrogen sulfide and sulfate prebiotic stimulates the secretion of GLP-1 and improves glycemia in male mice. Endocrinology (2017) 158(10):3416–25. doi: 10.1210/en.2017-00391
149. Gheibi S, Jeddi S, Kashfi K, Ghasemi A. Effects of hydrogen sulfide on carbohydrate metabolism in obese type 2 diabetic rats. Molecules (2019) 24(1):1700019. doi: 10.3390/molecules24010190
150. Sun H, Wu Z, Cao L, Zhu M, Liu T, Guo L, et al. Hydrogen sulfide: recent progression and perspectives for the treatment of diabetic nephropathy. Molecules (2019) 24(15):1159–72. doi: 10.3390/molecules24152857
151. Calvani R, Miccheli A, Capuani G, Tomassini Miccheli A, Puccetti C, Delfini M, et al. Gut microbiome-derived metabolites characterize a peculiar obese urinary metabotype. Int J Obes (Lond) (2010) 34(6):1095–8. doi: 10.1038/ijo.2010.44
152. Zhao X, Fritsche J, Wang J, Chen J, Rittig K, Schmitt-Kopplin P, et al. Metabonomic fingerprints of fasting plasma and spot urine reveal human pre-diabetic metabolic traits. Metabolomics (2010) 6(3):362–74. doi: 10.1007/s11306-010-0203-1
153. de Mello V, Lankinen M, Lindström J, Puupponen-Pimiä R, Laaksonen D, Pihlajamäki J, et al. Fasting serum hippuric acid is elevated after bilberry (Vaccinium myrtillus) consumption and associates with improvement of fasting glucose levels and insulin secretion in persons at high risk of developing type 2 diabetes. Mol Nutr Food Res (2017) 61(9):77–87. doi: 10.1002/mnfr.201700019
154. Penczynski K, Herder C, Krupp D, Rienks J, Egert S, Wudy S, et al. Flavonoid intake from fruit and vegetables during adolescence is prospectively associated with a favourable risk factor profile for type 2 diabetes in early adulthood. Eur J Nutr (2019) 58(3):1159–72. doi: 10.1007/s00394-018-1631-3
155. Sharma K, Karl B, Mathew A, Gangoiti J, Wassel C, Saito R, et al. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. J Am Soc Nephrol (2013) 24(11):1901–12. doi: 10.1681/asn.2013020126
156. Gross JL, de Azevedo MJ, Silveiro SP, Canani LH, Caramori ML, Zelmanovitz T. Diabetic nephropathy: diagnosis, prevention, and treatment. Diabetes Care (2005) 28(1):164–76. doi: 10.2337/diacare.28.1.164
157. Roscioni SS, Heerspink HJL, de Zeeuw D. The effect of RAAS blockade on the progression of diabetic nephropathy. Nat Rev Nephrol (2014) 10(2):77–87. doi: 10.1038/nrneph.2013.251
158. Wu H, Esteve E, Tremaroli V, Khan MT, Caesar R, Mannerås-Holm L, et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat Med (2017) 23(7):850–8. doi: 10.1038/nm.4345
159. Hocher B, Tsuprykov O. Diabetic nephropathy: renoprotective effects of GLP1R agonists and SGLT2 inhibitors. Nat Rev Nephrol (2017) 13(12):728–30. doi: 10.1038/nrneph.2017.140
160. Zou H, Zhou B, Xu G. SGLT2 inhibitors: a novel choice for the combination therapy in diabetic kidney disease. Cardiovasc Diabetol (2017) 16(1):65. doi: 10.1186/s12933-017-0547-1
161. Górriz JL, Soler MJ, Navarro-González JF, García-Carro C, Puchades MJ, D’Marco L, et al. GLP-1 receptor agonists and diabetic kidney disease: a call of attention to nephrologists. J Clin Med (2020) 9(4). doi: 10.3390/jcm9040947
162. Herat LY, Ward NC, Magno AL, Rakoczy EP, Kiuchi MG, Schlaich MP, et al. Sodium glucose co-transporter 2 inhibition reduces succinate levels in diabetic mice. World J Gastroenterol (2020) 26(23):3225–35. doi: 10.3748/wjg.v26.i23.3225
163. Zhang Q, Xiao X, Li M, Yu M, Ping F, Zheng J, et al. Vildagliptin increases butyrate-producing bacteria in the gut of diabetic rats. PLoS One (2017) 12(10):e0184735. doi: 10.1371/journal.pone.0184735
164. Du F, Hinke SA, Cavanaugh C, Polidori D, Wallace N, Kirchner T, et al. Potent sodium/glucose cotransporter sglt1/2 dual inhibition improves glycemic control without marked gastrointestinal adaptation or colonic microbiota changes in rodents. J Pharmacol Exp Ther (2018) 365(3):676–87. doi: 10.1124/jpet.118.248575
Keywords: gut microbiota, microbial metabolites, diabetic kidney disease, diabetes, short-chain fatty acids, Trimethylamine-N-oxide
Citation: Fang Q, Liu N, Zheng B, Guo F, Zeng X, Huang X and Ouyang D (2021) Roles of Gut Microbial Metabolites in Diabetic Kidney Disease. Front. Endocrinol. 12:636175. doi: 10.3389/fendo.2021.636175
Received: 15 December 2020; Accepted: 29 March 2021;
Published: 20 May 2021.
Edited by:
Mora Murri, University of Málaga, SpainReviewed by:
Neil G. Docherty, University College Dublin, IrelandCopyright © 2021 Fang, Liu, Zheng, Guo, Zeng, Huang and Ouyang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dongsheng Ouyang, ODAxOTQwQGNzdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.