 Valentina Gasco
Valentina Gasco Valeria Cambria
Valeria Cambria Fabio Bioletto
Fabio Bioletto Ezio Ghigo
Ezio Ghigo Silvia Grottoli
Silvia Grottoli- Division of Endocrinology, Diabetes and Metabolism, University of Turin, Turin, Italy
Traumatic brain injury (TBI)-related hypopituitarism has been recognized as a clinical entity for more than a century, with the first case being reported in 1918. However, during the 20th century hypopituitarism was considered only a rare sequela of TBI. Since 2000 several studies strongly suggest that TBI-mediated pituitary hormones deficiency may be more frequent than previously thought. Growth hormone deficiency (GHD) is the most common abnormality, followed by hypogonadism, hypothyroidism, hypocortisolism, and diabetes insipidus. The pathophysiological mechanisms underlying pituitary damage in TBI patients include a primary injury that may lead to the direct trauma of the hypothalamus or pituitary gland; on the other hand, secondary injuries are mainly related to an interplay of a complex and ongoing cascade of specific molecular/biochemical events. The available data describe the importance of GHD after TBI and its influence in promoting neurocognitive and behavioral deficits. The poor outcomes that are seen with long standing GHD in post TBI patients could be improved by GH treatment, but to date literature data on the possible beneficial effects of GH replacement therapy in post-TBI GHD patients are currently scarce and fragmented. More studies are needed to further characterize this clinical syndrome with the purpose of establishing appropriate standards of care. The purpose of this review is to summarize the current state of knowledge about post-traumatic GH deficiency.
Introduction
Traumatic brain injury (TBI) is one of the leading causes of disability and mortality affecting many people each year and resulting in a serious burden of devastating health consequences (1–3). The most common mechanism for TBI are falls, especially in older adults and very young children; motor vehicle accidents are instead the most frequent cause of TBI among young adults (1). TBI may lead to permanent or transient pituitary insufficiency (4, 5). The clinical picture presents a very large spectrum determined by the kind, number and severity of hormonal deficiency and could go from mild and non-specific complaints to life-threatening conditions. The reported prevalence of hypopituitarism is quite variable among the available studies (6–9). The pathophysiologic mechanisms underlying pituitary damage in TBI patients include a primary injury that may lead to direct trauma to the hypothalamus or pituitary gland, or to compressive effect from surrounding structures; secondary injuries, on the other hand, are mainly related to an interplay of a complex and ongoing cascade of specific molecular/biochemical events. The diagnosis of pituitary dysfunction is very challenging both due to the common occurrence of TBI, the subtle character of clinical manifestations, the variable course of the disease, as well as the lack of proper diagnostic algorithms. Growth hormone deficiency (GHD) is the most common abnormality, followed by hypogonadism, hypothyroidism, hypocortisolism, and diabetes insipidus (8, 9). The purpose of this review is to summarize the current state of knowledge about post-traumatic hypopituitarism, and especially about post-traumatic GHD. However, well-designed studies are needed to further investigate the pathophysiology, epidemiology, and timing of pituitary dysfunction after a TBI with the purpose of establishing appropriate standards of care.
Epidemiology of TBI and TBI-Related Hypopituitarism
Epidemiology of TBI
TBI is one of the leading causes of disability and mortality in western countries. It can be estimated that in the United States, every year there are around 2.5 million visits at the emergency department, 280,000 hospitalizations and 50,000 deaths related to TBI (1).
The highest rates of TBI are observed in older adults (≥75 years; 2232 per 100,000 population), followed by very young children (0 to 4 years; 1591 per 100,000) and young adults (15 to 24 years; 1081 per 100,000). There is a sex prevalence, with males showing a higher risk than females (959 per 100,000 vs 811 per 100,000). Falls are the most common mechanism for TBI, especially in older adults and very young children; motor vehicle accidents are instead the most frequent cause of TBI among young adults (1).
After the acute phase, TBI-survivors are often forced to deal with relevant and persistent long-term sequelae, with significant neurological and functional impairment. The prevalence of TBI-related long-term disability in the United States is estimated to affect between 3 and 6 million patients, i.e., 1% to 2% of the population (2, 3).
Apart from the individual sequelae, TBI clearly determines significant economic implications for society, related both to direct expenses for medical care and to indirect costs caused by injury-related work loss and disability (10). The former can be as high as 80,000 US dollars per person in the first year after trauma (11). The latter is more difficult to estimate, but it is likely to account for more than 80% of the total economic burden of TBI (10).
Epidemiology of TBI-Related Hypopituitarism
TBI-related hypopituitarism has been recognized as a clinical entity for more than a century, with the first case being reported in 1918 (12). However, during the 20th century it was considered only a rare sequela of TBI.
Most likely, hypopituitarism was under-recognized for such a long time for its generally subtle and nonspecific clinical features that also share a significant overlap with many of the somatic, psychiatric and neurological symptoms directly related to TBI. As a consequence, only patients with the frankest clinical pictures were probably identified as having a TBI-related hypopituitarism, leaving unrecognised the vast majority of TBI patients with some degree of pituitary deficits. This underestimation may probably have affected patient life expectancy and quality, as it happens to every patient living with unrecognised and untreated hypopituitarism (13).
The awareness of the critical relevance of hypopituitarism in TBI patients radically changed during the last two decades. The first two cornerstone studies that solidly proved that TBI-related hypopituitarism was a far more common sequela of head trauma than previously thought have been published in 2000 and 2001 (4, 5). Since then, several other studies of the endocrine function in patients after TBI have been published (6, 7, 14–40).
The reported prevalence of hypopituitarism is quite variable among the available studies, ranging from 1% (6) to 76% (7). However, this relevant heterogeneity should not surprise because the existing studies widely differ in many aspects, such as study design, age of patients, severity of trauma, time point of endocrinological evaluation and testing protocols for the diagnosis of the deficiency of the various pituitary axes.
Given these premises, an overall summary of the available evidence can be found in two major meta-analyses (8, 9). Pooled data show that the proportion of patients with some kind of pituitary disfunction can be estimated to be approximately 27.5-32.0% (8, 9). In most individuals only a single pituitary axis is affected (19.8-25.3%) (8, 9), while involvement of multiple pituitary axes is far less frequent (6.7-7.7%) (8, 9).
Furthermore, it has been consistently proved that not all pituitary axes are equally susceptible to TBI-mediated damage. The most sensitive ones appear to be GH and FSH/LH (12.4-22.1% and 10.2-12.5%, respectively) (8, 9); on the other hand, deficits of ACTH and TSH axes appear to be significantly less frequent (8.2-9.9% and 4.1-6.2%, respectively) (8, 9).
Pathophysiology of TBI-Related Hypopituitarism
Overview on the Pathophysiology of TBI
TBI is a heterogeneous disease. There are many ways to categorize the patients, both in terms of clinical severity and pathophysiological mechanism of injury.
Clinical severity is usually assessed by specific severity scores; the most commonly used is the Glasgow Coma Scale (GCS), which evaluates three neurological domains (eye opening, best verbal response, best motor response) and classifies TBI as mild (GCS 13-15), moderate (GCS 9-12) or severe (GCS ≤ 8) (41).
The pathophysiology of TBI is usually summarized into two separate categories: primary and secondary brain injury (42).
Primary brain injury occurs at the time of trauma, as a consequence of external mechanical forces transferred to intracranial content. The pathologic sequelae of primary brain injury include shearing of white matter tracts (also known as diffuse axonal injury), focal cerebral contusion/hemorrhages, and focal extra-axial hematomas/hemorrhages (i.e. epidural hematomas, subdural hematomas, subarachnoid hemorrhage and intraventricular hemorrhage) (42, 43).
Following this primary injury, extensive and lasting damage is sustained through a complex and ongoing cascade of events referred to as secondary brain injury. Pathogenesis is driven by complex, interacting mechanisms that include, among others, neurotransmitter-mediated excitotoxicity, secondary ischemia (from vasospasm or other secondary vascular injuries, such as focal microvascular occlusion), and inflammatory responses. As a final consequence, these mechanisms of injury lead to neuronal cell death, cerebral edema and increased intracranial pressure, which can further exacerbate brain damage (42, 44, 45).
Pathophysiology of TBI-Related Hypopituitarism
From a general point of view, the pathophysiological mechanisms underlying pituitary damage in TBI-patients are broadly similar to those described for TBI itself.
Primary injury may lead to direct trauma of the hypothalamus or pituitary gland, or to compressive effect from surrounding structures (42, 43); moreover, especially in case of skull base fracture, primary injury may determine pituitary stalk transection (46) (Figure 1).
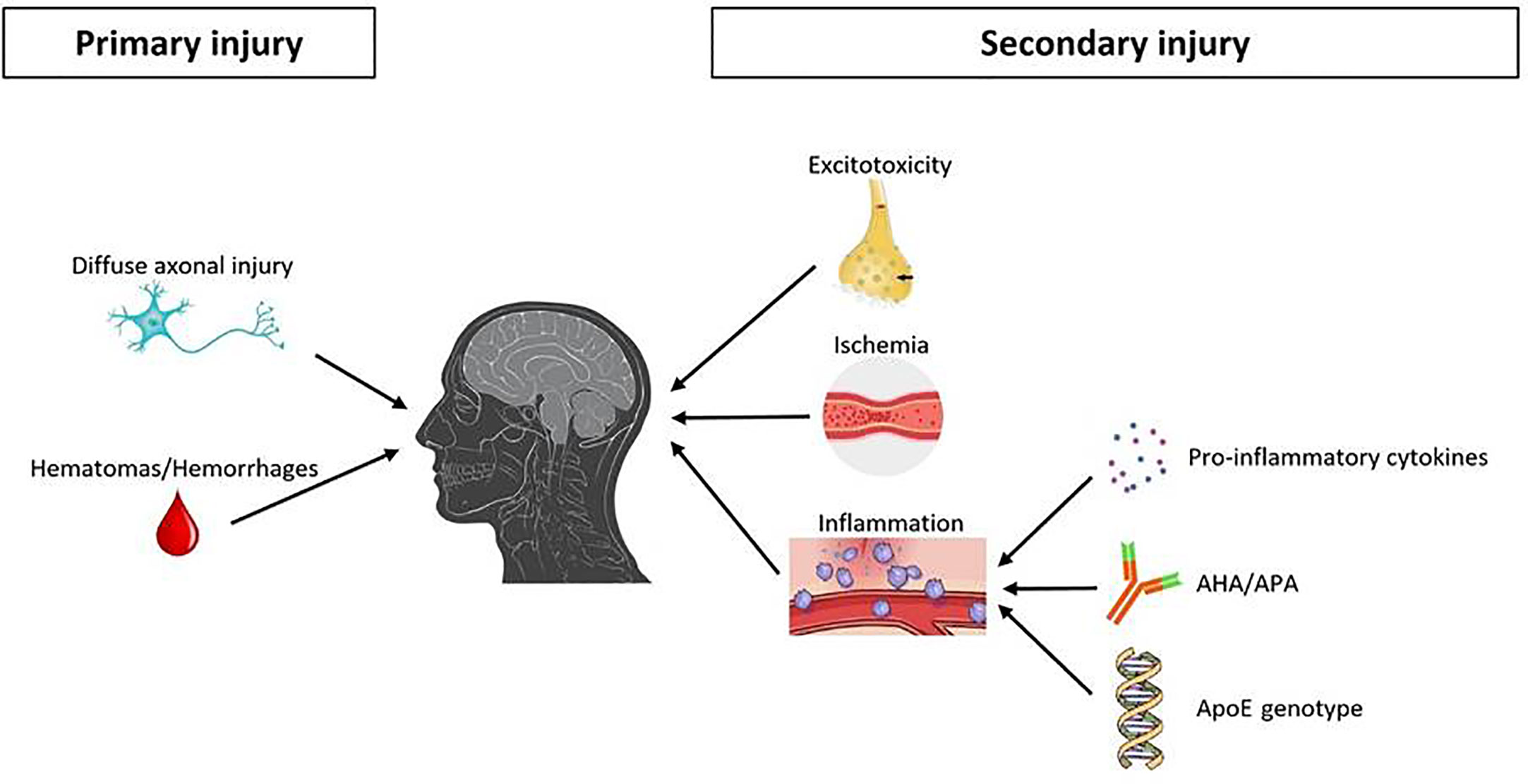
Figure 1 Main pathophysiological mechanisms underlying pituitary damage after TBI. TBI, traumatic brain injury; AHA, anti-hypothalamus antibodies; APA, anti-pituitary antibodies.
On the other hand, secondary injuries are mainly related to an interplay of a complex and ongoing cascade of specific molecular/biochemical events (42, 44, 45) (Figure 1).
As already discussed, one of the three major mechanisms for secondary brain injury after head trauma is represented by excitotoxicity. Excitotoxicity is caused by the abnormal levels of excitatory neurotransmitters (mostly glutamate) that are uncontrollably released in patients with TBI. In fact, at high concentrations, these neurotransmitters act as excitotoxins, operating on ion channels and thus altering cell wall permeability with an unregulated electrolyte shift between intra- and extracellular spaces (47).
Another mechanism for secondary brain damage after TBI is represented by ischemia. Overall, the same pathophysiological events affecting brain are likely to underlie the pituitary-specific ischemic insult as well. However, some distinctive points related to the peculiar vascularization of the hypothalamic-pituitary area are still worth to be discussed. As known, the anterior pituitary receives its blood supply from the hypothalamic-hypophyseal portal circulation (48–50), which likely poses the gland to a greater risk of ischemic harm (Figure 2). In particular, long hypophyseal portal vessels substantially represent the only source of vascularization of the lateral portion and of pars tuberalis (mostly populated by GH, PRL and FSH/LH secreting cells) (52) (Figure 3). Instead, the antero-medial portion and the central wedge (mostly populated by TSH and ACTH secreting cells) (52) (Figure 3) receive a mixed supply by both long and short hypophyseal portal vessels (48–50) (Figure 2). Therefore, the ischemic susceptibility hypothesis may be one of the most plausible explanation for the differential frequency of pituitary axes involvement after TBI. In fact, the most vulnerable axes (GH and FSH/LH) are those whose blood supply only relies on long hypophyseal portal vessels, that are by themselves more prone to vascular damage; instead, the most resilient ones (ACTH and TSH) are those whose blood supply is guaranteed both by long and short hypophyseal portal vessels (48–50, 52) (Figures 2, 3).
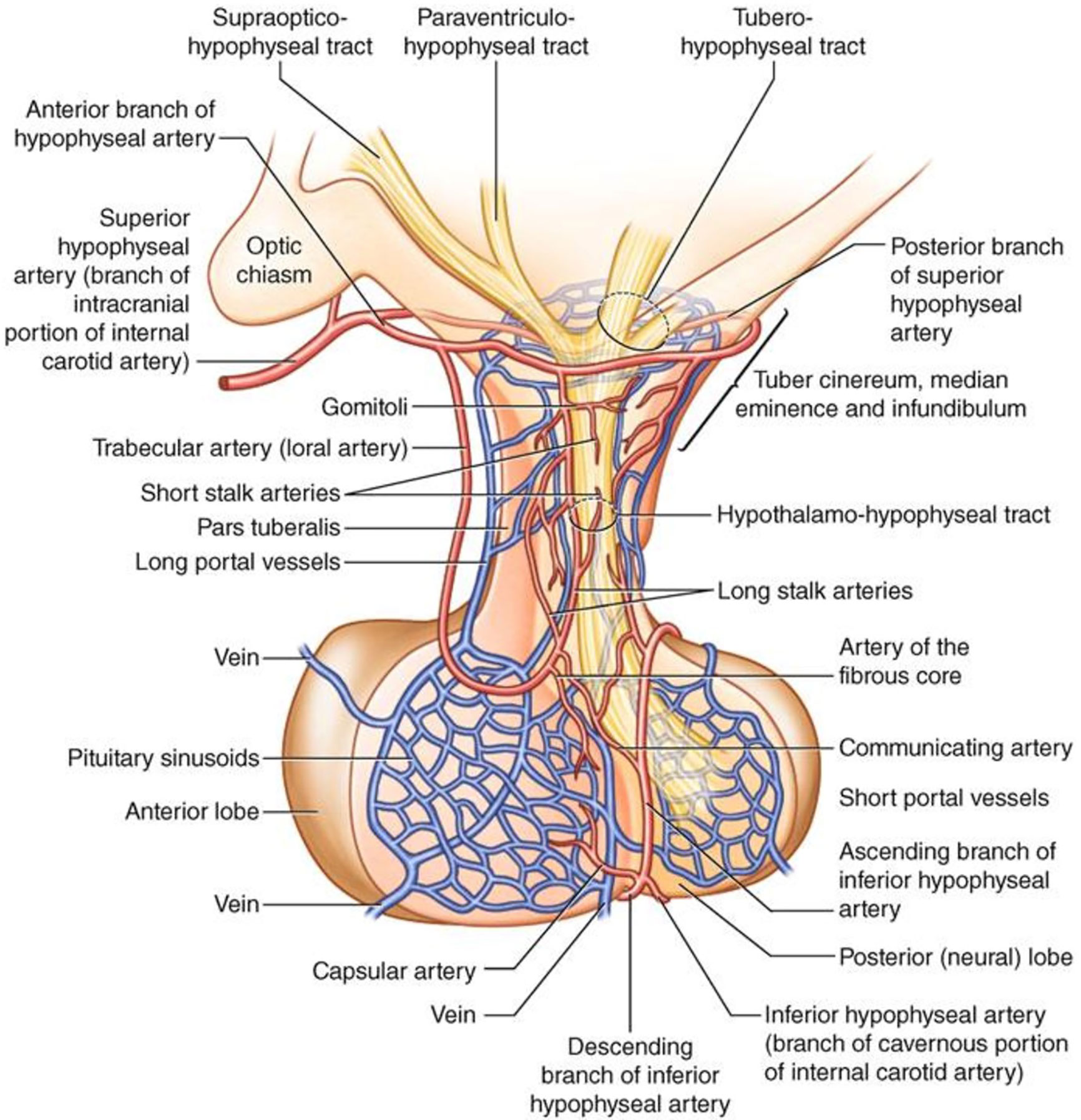
Figure 2 Anatomy and vascularization of the hypothalamus and pituitary gland [reproduced with permission from (51)].

Figure 3 Distribution of anterior pituitary cell subtypes.
The third and last major mechanism implied in the pathophysiology of secondary pituitary injury after trauma is inflammation. Part of the inflammation mechanisms affecting the pituitary gland after TBI are probably shared with the general inflammation mechanisms known to affect the brain parenchyma as a whole, with an uncontrolled and self-sustaining release of pro-inflammatory cytokines such as interleukin 1 (IL-1) and tumor necrosis factor α (TNF-α) (53).
In addition, a pituitary-specific inflammatory mechanism may be related to autoimmunity and, more specifically, to the presence of anti-hypothalamus antibodies (AHA) and/or anti-pituitary antibodies (APA). The positivity to these antibodies was demonstrated to be more frequent in patients with previous TBI (ranging from 44% to 60% depending on the study) than in matched healthy controls (0%) (54, 55). A similar result, even if less pronounced, was found in a cohort of subjects who underwent chronic repetitive head trauma due to amateur boxing activity, in which a higher positivity of AHA (21%) and APA (23%) was found compared to matched controls (0%) (56). Moreover, among patients with previous TBI, various authors have demonstrated a statistically significant correlation between the presence of AHA and/or APA positivity and that of post-traumatic hypopituitarism, with an odds-ratio (OR) ranging from 2.2 to 8.5, depending on the study (54–56). Looking at the whole body of these results, it is therefore reasonable to hypothesize that autoimmunity may play a role in the pathogenesis of TBI-induced hypopituitarism. The genesis of AHA and/or APA production is likely related to hypothalamus and/or pituitary exposure and release of antigens that would have otherwise remained unexposed. Despite the aforementioned association, the available evidence is not clearly sufficient to establish whether AHA/APA positivity may actually play an active pathophysiological role in the propagation/perpetuation of TBI-related pituitary damage, or may instead represent only an epiphenomenon of hypothalamic/pituitary antigen exposure due to necrotic post-TBI alterations of these areas.
Finally, another potential pituitary-specific inflammatory modulation mechanism may be related to individual intrinsic genetic susceptibility. In the general context of traumatic brain injury, ApoE polymorphisms have been widely shown to be associated with various clinical outcomes after TBI, such as the onset of seizures (57), coma duration (58), and subsequent neurobehavioral recovery (59). In fact, ApoE is a key protein in enhancing lipid transport and metabolism within the nervous system and has a role in neuronal repair and maintenance. In the specific context of post-traumatic hypopituitarism, a study from Tanriverdi et al. confirmed the possible role of ApoE polymorphism on neuroendocrinological outcomes; in particular, this study demonstrated a likely protective role of ApoE3/E3 genotype against the development of post-traumatic pituitary disfunction, with an estimated OR of 0.29 (60).
Evidence From Histopathological Studies
Most histopathological studies in patients with TBI-related hypopituitarism have been published several decades ago, as autoptic case series. These studies showed that the most frequent pathological alterations of pituitary gland after TBI were anterior lobe ischemic necrosis, posterior lobe haemorrhage and pituitary stalk haemorrhage, each occurring in approximately 14-42% of patients (61–64). A normal pituitary could be found in a variable proportion of 14-74% of patients, depending on the study (61–64).
More recently, another study by Salehi et al. (65) provided a further insight into histopathological changes of the pituitary gland after TBI. The overall results were not in contrast with previous case series, but a more accurate analysis relating pathological findings to the timing of death after trauma has to be discussed. In fact, the authors showed that no abnormalities of the pituitary gland could be found in patients who died immediately after trauma, both at macroscopic and at microscopic level. On the other hand, several degrees of pituitary haemorrhage and/or ischemic necrosis could be found in 43% of patients who survived between 3 h and 7 days after trauma. Moreover, the extent of histopathological alterations in those patients was significantly related to the time elapsed between injury and death (< 10% of adenohypophysis involved in patients who died within 1 day, > 50% in patients who died between 1 and 7 days) (65).
In conclusion, the whole body of histopathological evidences supports the hypothesis that post-TBI neuroendocrine damage could be mostly related to pituitary vascular vulnerability. However, despite the interest of these results, it must be pointed out that they are based on autoptic studies looking at a very specific and selected subset of TBI patients, i.e. those dying in the acute phase after a severe TBI. This undoubtedly represents an important selection bias, as this category of patients accounts for just a small minority of all patients suffering from TBI. Moreover, this is a category of TBI patients in which correlation with hormonal outcomes is neither feasible nor relevant. Precise pituitary histopathological correlations in long-term TBI survivors are thus lacking, but further research in this field are clearly conditioned by the constitutional limit to conduct this kind of evaluation in vivo.
Evidence From Imaging Studies
Several studies have been published about the possible identification of microstructural abnormalities of the pituitary gland by imaging techniques in patients with TBI-related hypopituitarism. Also in this subset of patients the imaging modality of choice for the evaluation of the pituitary gland was mostly MRI.
No single features were found to predict with high accuracy the presence or the absence of hypopituitarism in patients experiencing TBI. Therefore, in clinical practice the role of imaging in the prediction of post-traumatic hypopituitarism is limited. However, the available evidence is still of significant interest, as it provides information on the possible pathophysiological mechanisms through which post-traumatic hypopituitarism develops.
In this regard, the role of imaging is surely less accurate than that of pathological studies, considering that the deduced evidences about the underlying pathophysiological mechanisms can be only indirect. On the other hand, the clear advantage is that these evidences may be obtained for all patients with TBI, regardless of trauma severity and mortality.
In the acute phase, Maiya et al. (66) showed a significant enlargement of pituitary gland dimensions after TBI compared to healthy controls. In particular, this enlargement was evident not only in those patients with focal changes of the pituitary gland (haemorrhagic foci, heterogeneous signal intensities, macroscopical swelling) but also in those without specific focal pituitary abnormality. This is consistent with a pathophysiological hypothesis of an underlying pituitary edematous change, which is likely to be predominant in the first days after TBI. However, in this study no data about the subsequent pituitary functional outcomes were available, neither in the short nor in the long term. Therefore, it is not possible – based on these data – to explore the potential correlation of pituitary dimension in the acute phase and the subsequent development of pituitary deficits.
In the subacute phase, Zheng et al. (67) showed a reduction in apparent diffusion coefficient (ADC) in diffusion-weighted imaging (DWI) of pituitary gland in patients with previous TBI compared to healthy controls. DWI is based on the capacity of MRI to demonstrate the random, brownian diffusion movement of water. It is widely recognized as one of the best imaging modalities of cerebral ischemia, in which failure of the energy-dependent Na-K-ATPase determines a translocation of water from the interstitial to the intracellular space, and therefore a reduction of its diffusion capacity (68). This diffusion restriction determines a coherent change in signal intensity on DWI and on the derived ADC map, in which ischemic areas appear characteristically hypointense (69, 70). The results obtained by Zheng et al. thus confirm the probable role of ischemia in pituitary microstructural change in the first few weeks after TBI. Interestingly, even if ADC reduction with respect to healthy controls was found to be significant in the whole cohort of TBI patients, this alteration resulted to be more prominent in those which subsequently developed pituitary deficits at the hormonal follow-up; this is a further proof of the plausible relation between the severity of microstructural ischemic abnormalities in the short-term and the final functional outcome in the long-term.
In the chronic phase, Schneider et al. (71) showed that, after TBI, pituitary abnormalities at imaging were more frequent in patients with some degree of hypopituitarism (80%) than in those without pituitary hormone deficits (29%). The most common finding, in particular, was pituitary volume reduction, up to the degree of empty sella in some patients. The pathophysiological process underlying these changes is plausibly related to the long-term sequelae of pituitary tissue necrotization and subsequent fibrosis, which likely contribute to irreversible tissue loss.
The evidence of a pituitary volume reduction in the chronic phase finds further confirmation in a study by Tanriverdi et al. (72), in which the authors examined pituitary function and pituitary volume in a cohort of amateur boxers. As it is known, boxers are exposed during their career to mild chronic repetitive head traumas, which may determine pathophysiological consequences similar to classical TBI in the long term. In this study, 18% of boxers had some degree of pituitary dysfunction, and the presence of one or more hormonal deficits was significantly correlated with a lower pituitary volume at MRI.
TBI-Related Hypopituitarism: Clinical Presentation
Post-traumatic hypopituitarism (PTHP) can present with variable and non-specific clinical features, which may overlap with those resulting from the injury. The consequence is a delay in diagnosis, with a higher morbidity and mortality (39).
The clinical picture presents a very large spectrum determined by the kind, number and severity of hormonal deficiencies and could go from mild and non-specific complaints to life-threatening conditions (73).
Traditionally, an acute and a chronic phase can be distinguished, but pituitary dysfunction during the acute phase does not necessarily lead to long-term hypopituitarism. In a systematic review, the analysis of prospective studies showed that some of the early abnormalities are transient with complete recovery, while hypopituitarism can evolve over time and become detectable only later (8).
The first 2 weeks after trauma are considered the acute stage.
The most common hormonal changes in this phase are represented by gonadotropin and GH deficiency, but in the clinical presentation they are not the most evident.
In fact, the most fearsome endocrinological complication of the acute stage is adrenal insufficiency. Hyponatremia, hypoglycemia, hypotension, fatigue, mental confusion are some of its typical features. Patients affected by hypocortisolism require a higher dose of vasopressors and have a higher mortality rate; therefore hormone replacement therapy is crucial (74, 75).
Impaired vasopressin secretion (Syndrome of Inappropriate Antidiuretic Hormone Hypersecretion - SIADH or Central Diabetes Insipidus - CDI) could also be life-threatening, contributing to the hydro-electrolytic imbalance of the acute phase (76).
Hypothyroidism can be also reported, due to the adaptive response after trauma and the use of steroids. It contributes to the clinical picture of marked asthenia, lethargy and confusion, not always easy to discriminate in the acute stage.
GHD is considered a common alteration of the acute phase, reflecting the adaptive response after the traumatic event.
Hyperprolactinemia could be reported as a consequence of pituitary stalk compression or as a physiological reaction to stress. Together with hypogonadism, hyperprolactinemia may lead to the menstrual alteration and the sexual dysfunction of the post-TBI period.
Some of these abnormalities are transient: recovery from hypoadrenalism is described in 50% of patients, from CDI in up to 90% (77). Hypothyroidism and SIADH often resolve in 3-12 months (40).
The chronic phase starts at 3 months after TBI. The clinical features could be very variable and not specific, depending on the different axes involved. Lethargy or insomnia, fatigue, reduced attention, difficulty concentrating, memory impairment, anxiety, depression, irritability and diminished libido are often described (78).
Also in this phase GHD and hypogonadism are the most frequent anomalies, hypocortisolism and central hypothyroidism are relatively rare and CDI could persist in a small percentage of patients (79).
These patients experience metabolic abnormalities, neurocognitive impairment, and a decreased quality of life (34, 80).
Metabolic Alterations
In patients with PTHP, altered function of hypothalamic nuclei and pituitary disfunction itself determine an adverse metabolic profile. Glycemic disorders, dyslipidemia, weight gain with abdominal fat distribution, changes in body composition and reduced bone mineral density determine the higher morbidity and mortality of these patients (78).
Different studies demonstrated that PTHP patients have higher BMI, increased LDL cholesterol and total cholesterol (19), altered glucose levels and insulin resistence (34).
Decreased thyroid function reduces the basal metabolic rate and hypogonadism affects bone and muscle mass, beyond the effect on libido and reproduction. It is however GHD that plays the major role, affecting glycemic and lipidic profile, increasing BMI and waist circumference, reducing bone mineral density and leading also to anemia (78).
GHD can affect also the rehabilitation: patients with post-TBI GHD seem to have lower aerobic capacity, a measure of physical resistance, which may delay or inhibit the recovery process (81).
Neurocognitive Alterations
Recently, more attention was paid to neuropsychiatric symptoms which are due both to PTHP and brain injury itself. Cognitive and affective impairment may be severe, prejudicing patients’ social skills. In fact, attention, memory, executive functions and language can be involved.
Symptoms of PTHP can overlap with cognitive, sleep, mood, and anxiety disorders due to Post-Traumatic Stress Disorder (PTSD). Differential diagnosis can be challenging, but essential for the therapeutic implications.
Hypothyroidism is associated with neurocognitive function: low speed of information processing and deficits in short-term memory are the most described (73).
Hypogonadism is also associated with cognitive dysfunction: patients with lower testosterone levels after TBI seem to have an increased risk for Alzheimer’s disease (82).
Also hypoadrenalism results in mood disorders, decreased memory, and frank psychosis in the chronic phase, in addition to the classical picture of fatigue, weakness, and impaired response to stress (78).
Although every hypothalamic-pituitary axis could be involved in cognitive functions, the role of GH and the effects of its deficiency are the most known and frequently observed (83).
In fact, somatotropic axis has a role in microtubular regeneration, dendritic growth and regrowth, regulation of the use of glucose in the brain and, probably, an action on hippocampal area. So, GHD may lead to impaired neuronal, somatic, and dendritic growth, affecting memory and cognitive function too (78, 84).
Several studies reported that patients with post-traumatic GHD have more severe cognitive impairment, in particular deficit in attention and memory, as well as emotional problems, than those with normal GH values (33, 85, 86).
Patients with post-TBI GHD have a higher risk to achieve poor cognition outcomes than those with an intact somatotropic axis after trauma (87). Park et al. showed that patients with GHD after TBI have decreased cerebral glucose metabolism in specific cortical areas involved in intellectual function, executive function, and working memory (88).
In contrast to all these studies, Pavlovic et al. did not find significant differences between patients with or without GHD after brain injury (89).
Quality of Life (QoL)
Another important feature is QoL: patients with post-TBI GHD are more likely to be depressed and with a poorer quality of life than those with GHD due to other causes. In particular, domains of physical health, energy and fatigue, emotional well-being, pain, and general health seem to be affected (85).
Also perceived poor QoL would negatively impact on rehabilitation after TBI.
TBI-Related Hypopituitarism: Diagnosis
PTHP, and in particular GHD, are often underdiagnosed: patients with post-TBI GHD seem to be diagnosed on average two and a half years later after the primary onset of disease when compared to those with Non-Functioning Pituitary Adenoma (NFPA) (87).
PTHP diagnosis is not different from hypopituitarism due to other causes. However, the time and type of hormonal assessment in TBI patients is controversial.
Who to Test
Despite the severity of trauma itself, hypopituitarism can develop in patients post-TBI, but testing all of them is not feasible because of the great amount of human and economic resources needed. A rational approach is to evaluate life expectancy, avoiding to test patients with poor prognosis who cannot benefit of hormonal replacement therapy. Conversely, patients with mild TBI could develop hypopituitarism, but not all of them achieve medical attention (90) and testing is not considered cost-effective (87).
Epidemiology could be useful to establish who to screen: in fact, PTHP frequency is better established in patients with moderate or severe TBI based on GCS score. Furthermore, other risk factors include age, intracranial hemorrhage, focal cortical contusion, seizures and skull base fracture.
In patients symptomatic for acute hypopituitarism (i.e. electrolyte unbalance and/or acute adrenal insufficiency) an endocrinological evaluation is mandatory.
Patients with mild TBI who need hospitalization (more than 24 h), a neurosurgical intervention, monitoring in an Intensive Care Unit, or those who present anatomical changes on CT scan, are considered complicated and screening is also recommended (87). Conversely, patients with a mild uncomplicated TBI should be screened for hypopituitarism only if the clinical suspect is present (91).
Patients who required hospitalization for at least 24 h, those with radiological abnormalities on CT scan, and those who presents signs and symptoms of PTHP should be screened at three months and one year post-TBI. It is possible to perform hormonal screening even further, if symptoms persist (78).
Biochemical Evaluation: Time and Kind
Pituitary function evaluation could be challenging in the acute phase post-TBI. In fact, in this stage patients have hormonal changes as part of the stress response and acute adaptive response to injury. Hormonal levels are also affected by medications and surgery. Pituitary function during this time could be variable (78).
While some of the alterations are natural consequences of the trauma itself, others are life-threatening and require an immediate hormonal replacement therapy. So, basal hormonal evaluation should be performed in any patient with TBI who has been hospitalized and presents signs and symptoms suggesting adrenal insufficiency (92). For the diagnosis, a morning serum cortisol should be measured. Usually, a level beyond 15 µg/dL indicates a proper function, values between 3 and 15 µg/dL require further investigation using a stimulation test, while a concentration lower than 3 µg/dL allows a diagnosis of hypoadrenalism (93). In the post-TBI context, these cut-points are not considered applicable. In fact, plasma cortisol concentrations rise in parallel to the severity of acute illness, so Hannon et al. proposed that critically sick patients should have plasma cortisol levels >300 nmol/L (=10.875 µg/dL), suggesting the need of hormonal replacement therapy for lower values (74). Also the evaluation of cortisol reserve with a dynamic test seems to be not reliable in TBI patients, at least in the acute phase. In fact, Endocrine Society Clinical Practice Guidelines suggest performing an ACTH stimulation test (Synacthen test) when required for secondary adrenal deficiency diagnosis (93), preferring the low-dose (1 µg) version to reduce false negative results due to a supraphysiological stimulus (94, 95). However, in the setting of the acute phase of TBI a Synacthen test is inappropriate because of the acute nature of ACTH deficiency, that would not have time to lead to secondary adrenal insufficiency. Therefore, patients with TBI would have a normal response also to the low-dose ACTH stimulation test (74). Even an extremely accurate test as Insulin Tolerance Test (ITT) is not feasible for safety concerns in patients with TBI in the acute phase.
First post-acute phase evaluation should be scheduled 3-6 months after TBI. Thyroid and gonadal axes are frequently involved, so they should be evaluated. TSH and fT4 are dosed to confirm or exclude hypothyroidism, while gonadic evaluation is different between males and females. In men with suspected hypogonadism LH, FSH, testosterone and PRL levels should be dosed. In women of reproductive age with menstrual irregularities is recommended to measure PRL, LH, FSH and estradiol (E2), while in postmenopausal women gonadotropins reduction may be sufficient for the diagnosis (78, 83, 93). The adrenal axis should be re-evaluated measuring morning serum cortisol and, eventually, using an appropriate stimulation test.
To assess for CDI in patients with polyuria, serum and urine osmolality should be dosed simultaneously: urine osmolality/plasma osmolality ratio should be ≥2, after excluding glycosuria (78).
Somatotropic axis evaluation is recommended at least six months after TBI because of the possible spontaneous recovery in the post-acute phase (87). Some Authors proposed to postpone the evaluation until 1 year after TBI in adults, while children may require an earlier assessment (83). The diagnosis of GHD could not be based only on IGF-I levels; even if in patients with low IGF-I levels and multihormonal deficiency GHD diagnosis does not require further tests (96), in all other conditions different dynamic tests are available to confirm the suspected somatotropic deficiency with a high accuracy (96, 97). The choice of a test over another is usually based on patient’s characteristics, availability of secretagogues and center experience (87). ITT is considered the gold standard and it evaluates both hypothalamic and pituitary integrity, but it can be used only after excluding contraindications (i.e. seizure, cardiac disease) (8, 90). Up to 22% of patients who had TBI develop seizure disorders, so ITT is often considered not safe in this setting (98).
The administration of GH Releasing Hormone (GHRH) plus Arginine (Arg) or GHRH plus GH Releasing Peptide 6 (GHRP-6) provides a strong stimulus to GH secretion and are considered safe, so they could be used as a dynamic test for GHD evaluation. However, they are not useful for GHD of hypothalamic origin and both GHRH and GHRP-6 are unavailable in many Countries (96). Glucagon Stimulation Test (GST) is not usually the first choice for GHD diagnosis. In fact, it could be labored, requiring a monitoring of at least 3 h, and it could lead to delayed hypoglycaemia (96). The diagnostic accuracy could be lower in patients with glucose intolerance (97).
For all GHRH+Arg, GHRH+GHRP-6 and GST different BMI-related cut-off points are available (99–101). Also for ITT new BMI-related cut-points have been recently proposed, allowing to avoid false positive results due to obesity (102). However, they need further validation.
Lately, Macimorelin test has been proposed for the diagnosis of adult GHD (97), but data about its use in post-TBI patients are lacking until now.
An overview of the main stimulation tests for GHD diagnosis is provided in Table 1.
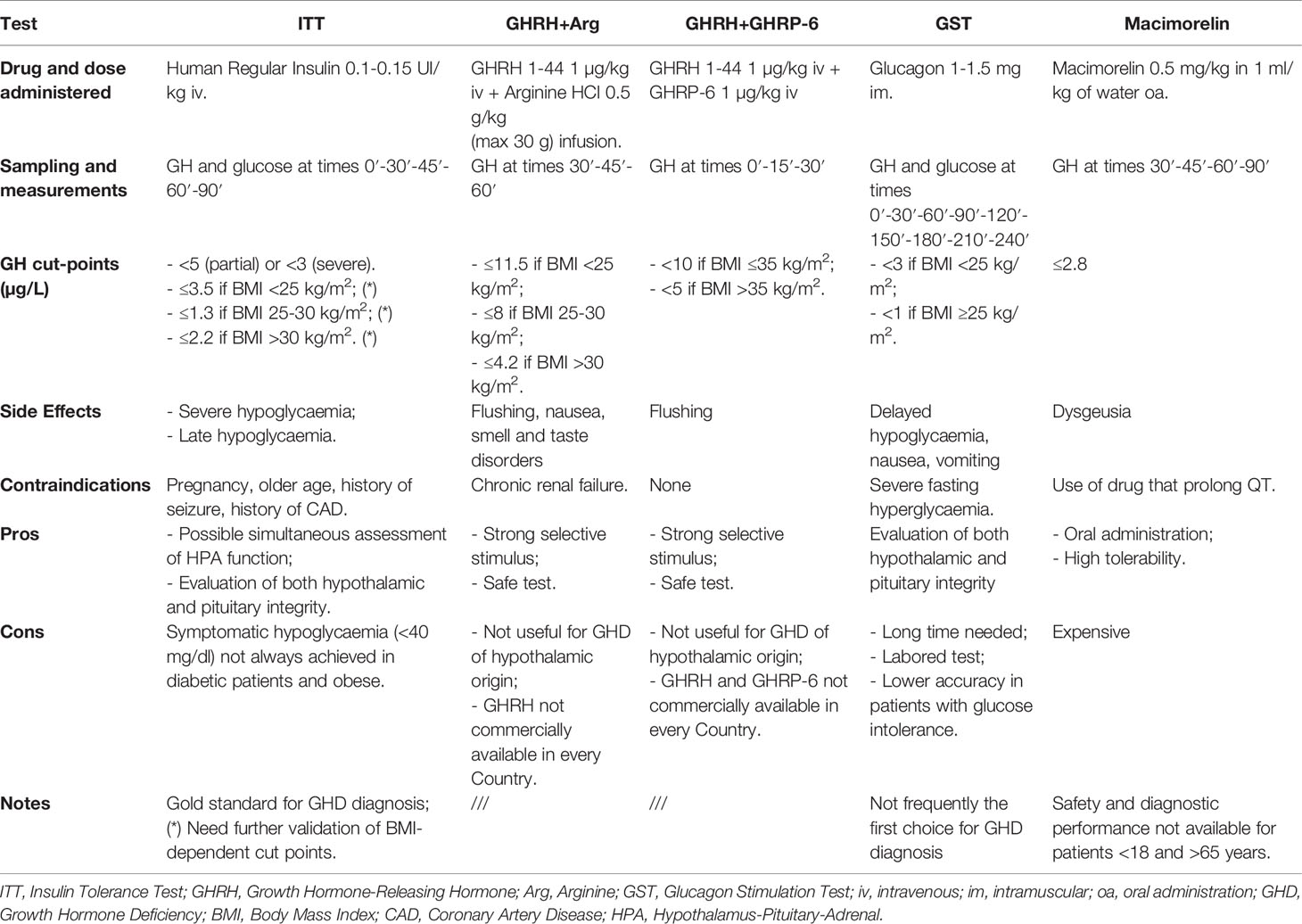
Table 1 Main characteristics of GH stimulation tests.
TBI-Related GHD: Management and Outcomes
Rationale for Treatment of Post-Traumatic GHD
GH is expressed not only at pituitary level but also in many other organs and tissues, including the central nervous system (CNS) (103) where it plays an important role in the regulation of cell proliferation and survival (104–107). It is well known that GH plays a role in brain repair, and several preclinical and clinical studies have demonstrated the positive effects of GH treatment on neurogenesis in both animals and humans (108, 109). Moreover, the whole GH system [GH, GH receptor (GHR) and IGF-I] is acutely and strongly upregulated after brain injury (110, 111), and specifically associated with stressed neurons and glia (112–114). However, to date, the role exerted by GH at CNS level and, in particular, its possible contribution to the recovery of neurologic injuries remains poorly understood.
GH exerts its beneficial effects on neural repair through different mechanisms that include regulation of the proliferation, survival, differentiation and migration of both neural progenitors and newly formed neurons.
Both in vitro and in vivo studies support the ability of GH to promote the proliferation of neural precursor. GH treatment promotes proliferation of both human fetal (115) and adult mice neural stem cells (NSCs) (106, 116). It has been demonstrated that peripheral administration of GH is able to induce cell proliferation in the brain of both normal (117) and hypophysectomized (107) adult rats (117); moreover, peripheral administration of GH seems to increase the proliferative response of hippocampal progenitors to kainate-induced injury (118). Despite the main role of GH on the proliferation of neural precursors, it has been suggested that GH may actually have a more prominent effect in the regulation of survival, differentiation, or even migration of newly formed neurons (113). In agreement with the control exerted by GH on cell survival, GH prevents the apoptotic death of both mature neurons (119–121) and primary neurospheres derived from embryonic mouse NSCs (122). On the other hand, an increased apoptotic death of NSCs is observed during the treatment with a GHR antagonist (123). Lastly, GHD has been shown to impair the survival of newborn neurons in the subgranular zone of adult rat dentate gyrus (124), while elevated GH levels within the hippocampus reduce apoptosis (125). All these neuroprotective effects would explain some of the acute consequences of GH therapy. Several studies support a role for GH in enhancing neuronal precursors differentiation (126, 127), while a possible effect of GH on neuronal precursors migration is less clearly established.
Finally, it cannot be ruled out that GH promotes neurogenesis and neurorepair, at least in part, through indirect mechanisms including both the synthesis and the release of IGF-I, epidermal growth factor (EGF) or erythropoietin (EPO) or changes in neurotransmitter turnover.
IGF-I is the main mediator of GH action and it is essential for CNS development (128, 129). IGF-I stimulates the proliferation of neuronal precursors and the survival and differentiation of both neurons and oligodendrocytes (130). As a result, brain growth is increased by IGF-I overexpression and reduced as a consequence of decreased IGF-I levels.
In this context, it must be pointed out that IGF-I has been shown to be a crucial modulator of CNS activity, including higher functions like cognition, and to modulate genes involved in microvascular structure and performance, and synaptic plasticity (131, 132).
On the other side, EGF has been demonstrated to be a powerful mitogen capable of inducing neurogenesis both in basal studies and after experimental injuries (133, 134). Since it has been shown that GH is able to induce the expression of both EGF and its receptor (EGFR) and to mediate EGFR activation (135, 136), it cannot be excluded that the action performed by GH at CNS level is mediated by these interactions.
EPO and its receptor (EPOR) are other factors involved in neurogenesis. Both EPO and EPOR have been identified in numerous areas of the CNS during development, and they are expressed in several neuronal cells like neurons, astrocytes, oligodendrocytes, microglia and cerebral endothelial cells, where EPO activates anti-apoptotic, anti-oxidant and anti-inflammatory signals and stimulates angiogenesis and neurogenesis. It is therefore not surprising that EPO can determine a strong protective effect on neuronal tissue in experimental models of stroke, cerebral hemorrhage, traumatic brain injury, and neuroinflammatory and neurodegenerative diseases (137).
Moreover, it has been shown that the blockade of EPOR in the CNS leads to impairments in neural cell proliferation and survival during embryonic development, and in post-stroke neurogenesis in adult brain, further confirming EPO’s role in neurogenesis (138, 139). Finally, both in vivo and in vitro studies show that EPO is able to increase oligodendrogenesis and remyelination after stroke (140). Since GH induces EPO release from kidneys (141), it can be assumed that GH promotes neurogenesis and neurorepair, at least in part, through indirect mechanisms including EPO mediated effects.
Peripherally, GH is an anabolic hormone that promotes growth in skeletal and soft tissues (142) through the expression of GHR at many levels including liver, muscle, bone, and adipose tissue (143). GH plays an important role in metabolism stimulating lipolysis, reducing hepatic triglyceride secretion, activating the nitric oxide system (and so reducing vascular tone), increasing cardiac performance and exercise capacity, and promoting longitudinal skeletal growth (142). In patients with traditional causes of hypopituitarism such as pituitary tumors, GHD is associated with changes in body composition, worrisome metabolic dysfunction, reduced bone density, a significant decrease in QoL, increased cardiovascular risk, and impaired cardiac function (33, 144). GH replacement therapy has been shown to be partially beneficial in adults with GHD (145). However, the evidence of the benefit from GH replacement therapy in post-traumatic GHD is scant: more robust data are available about improving cognition and QoL, but not about all the other parameters.
Literature data on the possible beneficial effects of recombinant human GH (rhGH) therapy in patients with GHD post-TBI are currently scarce and fragmented. The studies available so far are few (14, 146–153), carried out on small series (147–150, 152–154), extremely variable with respect to the type of patients considered (severity of TBI, age and sex of the subjects, presence or absence of other associated hormonal deficits, time elapsed since TBI), the type of control group (subjects with GHD from another cause, subjects with GHD from TBI, subjects with previous TBI but without GHD), the way in which the condition of GHD is defined (different stimulation tests, some of which not even recognized for GHD diagnosis by the international guidelines), the rhGH dose which in some cases appears supraphysiological (148, 150), the outcomes considered and the tools used for their detection. Furthermore, the studies in general have a short duration (3-12 months) and the drop-out is often high (153). All these factors make the results of every single study difficult to compare and the results of every individual experience on a broader case series are scarcely generalizable.
An overview of the main studies investigating GH replacement therapy in adult post-TBI GHD patients is provided in Table 2.
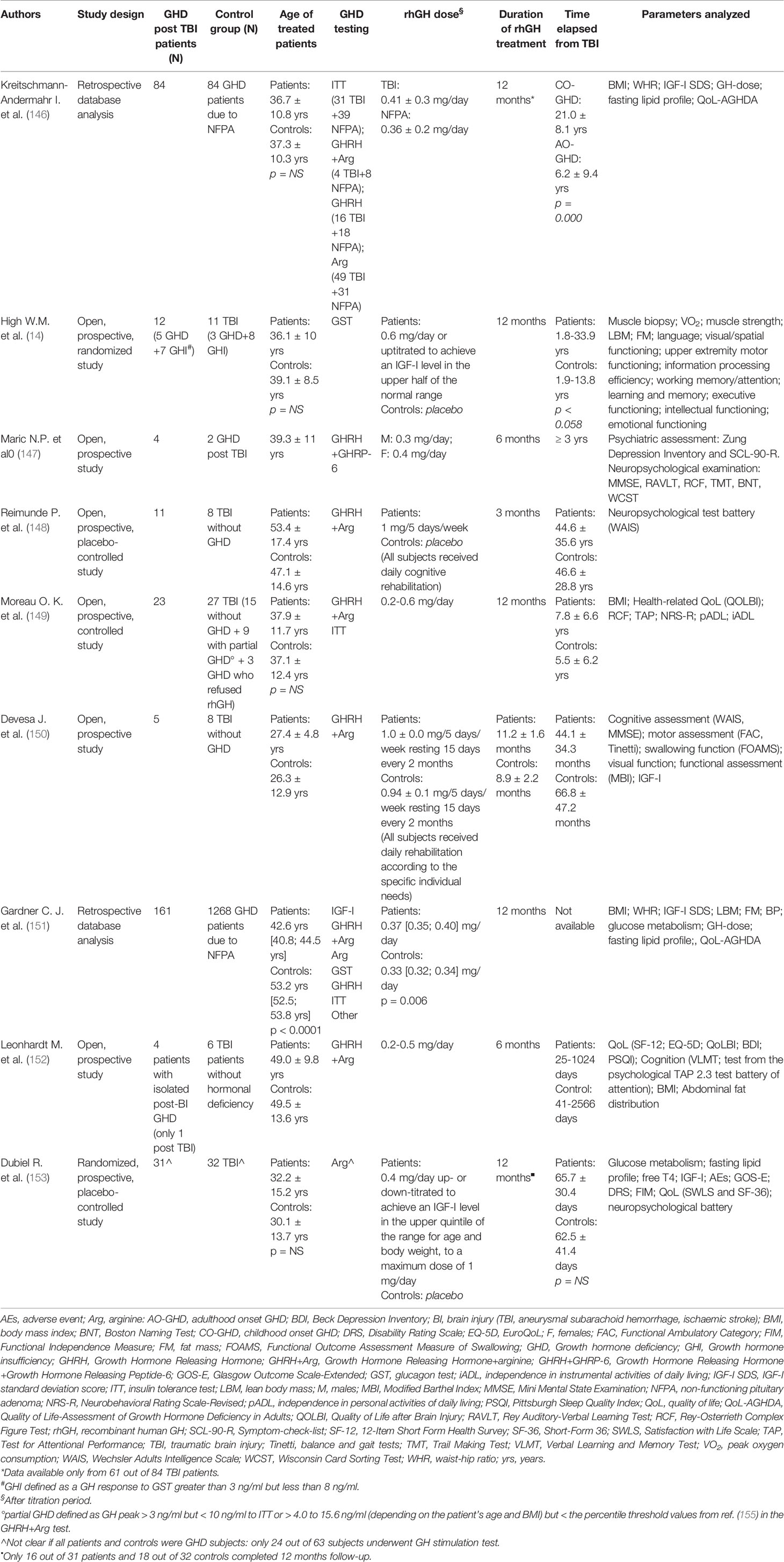
Table 2 Studies investigating GH replacement therapy in adult post-TBI GHD patients.
Evidence for Treatment of Post-Traumatic GHD: Cognition
Several studies reported that patients with post-traumatic GHD have a severe cognitive impairment mainly characterized by deficit in attention and memory, as well as emotional problems, and poor verbal learning (7, 33, 85, 86). GHD also seems to be associated with poor mental health outcomes. The cognition and quality of life problems experienced by patients with GHD may be explained by the reduced expression of GH activity in specific CNS areas involved in memory, learning, and emotions, like the hippocampus and the limbic system (156–158). Park et al. showed that patients with GHD after TBI have decreased cerebral glucose metabolism in specific cortical areas involved in intellectual function, executive function, and working memory (88).
It has been demonstrated that in TBI patients the GH response to stimulation test was negatively correlated to paranoid ideation and somatization (33). GHD patients have a moderate to large impairment in each of the cognitive domains assessed when compared to the matched controls (159) and patients with post-TBI GHD have a higher risk to achieve poor cognition outcomes than those with an intact somatotropic axis after trauma (87). In contrast to all these studies, Pavlovic et al. did not find significant differences between patients with or without GHD after brain injury (89). It is well known that GH replacement therapy in patients with GHD from non-traumatic causes is of benefit (96, 160). A meta-analysis conducted on patients with GHD from any cause showed a moderate improvement in cognitive performance during treatment with rhGH; the improvement concerned mainly memory and attention domains (159).
A beneficial effect of GH replacement therapy on cognition has also been reported in post-TBI GHD patients (14, 148, 150). However, till now, there is not sufficient evidence to support GH treatment with the aim of improving cognition in post-TBI GHD patients.
Recovery during a thorough rehabilitation program after TBI may be positively influenced by normal GH secretion as suggested by Bondanelli et al. who showed that GH peak during GHRH + ARG test was an independent predictor of positive outcomes suggesting that GH replacement therapy may be considered in post-traumatic GHD patients (161). The improvement in cognitive function is more significant and appears earlier than that in motor function; this observation supports the usefulness of GH replacement therapy in the rehabilitation process of these kind of disabilities (159, 162–164). In general, the improvement during GH therapy appears to be more marked in patients with worse cognitive impairment at baseline (149) and it has also been shown that discontinuation of GH therapy is accompanied by a worsening of cognitive performance (147).
Although to date the exact mechanism underlying these effects is not well understood, it cannot be excluded that it could be based on GH stimulating effect on neurogenesis in CNS areas related to recent memory like the hippocampal dentate gyrus. However, stimulation of neurogenesis does not appear to be the mechanism by which GH therapy improves other cognitive processes such as attention and concentration. In this case it has been hypothesized that the effect of GH replacement therapy is mediated by the action of the hormone on some neurotransmitter pathways and this might explain the early responses observed after starting a treatment with rhGH (116, 118, 148, 150).
Evidence for Treatment of Post-Traumatic GHD: Metabolic and Cardiovascular Risk Factors
Regardless of pathogenesis, GHD is associated with adverse effects on body composition, alterations in glucose and lipids metabolism, reduced physical and cardiovascular performance (165); all these alterations are thought to be the cause of the incresed cardiovascular mortality observed in hypopituitaric patients (166–168). A systematic review showed a small but significant improvement on lean and fat body mass, low-density lipoprotein-(LDL) and total cholesterol, and diastolic blood pressure in GHD adults during GH replacemente therapy (169). In contrast, plasma glucose and insulin levels were increased during GH therapy (169). However, improvement in hard outcomes, such as reductions in cardiovascular events and mortality, has yet to be demonstrated (96, 170).
Several studies argue metabolic disorders in patients with post-traumatic GHD. In patients with post-TBI hypopituitarism, mainly GHD, high LDL and total cholesterol levels, waist circumference, and total fat mass have been showed (19). Treatment of these patients has shown variable results. In one observational study, weight or waist to hip ratio did not change in post-traumatic GHD patients treated with rhGH for a year (146), while in another study there was an improvement in blood pressure, total and LDL cholesterol after 1-year treatment in patients with GHD post TBI (151). A case study of two patients with GHD secondary to sport-related TBI showed some improvement in lipid profile and body composition after a 6-month treatment with rhGH (171).
Patients with TBI have been found to have a reduced aerobic capacity, a well-established measure of physical endurance and fatigue resistance, which may further delay or hinder the rehabilitative process (81). In particular, patients with TBI and a normal GH axis show suboptimal aerobic capacity and those with GHD performed even worse (81). Although GH treatment seems to improve skeletal muscle mass in patients with GHD from non-traumatic causes, data available are not so convincing in post-traumatic GHD patients and the improvement, if any, seems to concern only male patients with TBI and GHD (172). A case study of one patient showed an improvement in muscle force production, body composition, and aerobic capacity after treatment with rhGH for 12 months (173).
Evidence for Treatment of Post-Traumatic GHD: Bone Health
A higher risk of osteopenia, osteoporosis, and vertebral fractures is observed in hypopituitaric patients adequately replaced with glucocorticoids and thyroid hormones (174) suggesting a fundamental role of both GH and gonadotropins secretion disturbances in the pathogenesis of the reduced bone health observed in these patients. Apparently, in GHD patients there is no difference related to gender and age. GH replacement therapy increases bone mineral density (BMD) (175, 176) and seems to mitigate the increased fracture risk observed in GHD patients (174), but specific data for skeletal outcomes in TBI induced GHD are lacking.
Evidence for Treatment of Post-Traumatic GHD: QoL
Regardless of the cause, GH is characterized by a compromised QoL (177, 178). Patients with post-traumatic GHD are more likely to be depressed and with a poorer QoL than those with GHD due to other causes (85, 160). Interestingly, patients with post-TBI GHD, when compared to those with GHD secondary to NFPA, seemed to have a less severe GHD at biochemical level, but worse QoL scores (151). In particular, domains of physical health, energy and fatigue, emotional well-being, pain, and general health seem to be affected (85). This perceived poor QoL would negatively impact on rehabilitation after TBI. In general, the treatment of GHD of any cause seems to improve QoL as measured by the QoL-AGHDA (Quality of Life Assessment of Growth Hormone Deficiency in Adults) score and other evaluation tools (179). The QoL improvement is higher in post-traumatic GHD patients than in patients with GHD due to NFPA, especially in the domains of socialization, self-confidence, and tenseness (151). The observed improvement was maintained for a long time, up to eight years, and always based on continuation of treatment.
Conclusions
TBI is one of the leading causes of disability and mortality affecting many people each year and resulting in a serious burden of devastating health consequences. TBI may lead to transient or permanent pituitary insufficiency. After the initial primary injury, secondary mechanisms that involve an interplay of ischemia, inflammation, and cytotoxicity seem to result in hypopituitarism that causes adverse changes in body composition, worrisome metabolic dysfunction, decreased bone density, and a significant reduced QoL. GHD, the most common pituitary hormone deficiency after TBI, is associated with adverse sequelae, which may impair recovery and rehabilitation. The poor outcomes that are seen with long standing GHD in post TBI patients could be improved by treatment with rhGH, but literature data on the possible beneficial effects of GH replacement therapy in post-TBI GHD patients are currently scarce and fragmented. More studies are needed to further characterize the post-TBI GHD syndrome with the purpose of establishing appropriate standards of care.
Author Contributions
VG, VC, and FB performed a literature search, wrote the first draft, and designed tables and figures. EG and SG supervised the work and revised the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare the absence of any commercial or financial relationship that could be construed as a potential conflict of interest.
References
1. Taylor C, Bell J, Breiding M, Xu L. Morbidity and Mortality Weekly Report Traumatic Brain Injury-Related Emergency Department Visits, Hospitalizations, and Deaths-United States, 2007 and 2013. MMWR Surveill Summ (2017) 66(9):1–16. doi: 10.15585/mmwr.ss6609a1
2. Thurman D, Alverson C, Dunn K, Guerrero J, Sniezek J. Traumatic Brain Injury in the UnitedStates: A Public Health Perspective. J Head Trauma Rehabil (1999) 14(6):602–15. doi: 10.1097/00001199-199912000-00009
3. Zaloshnja E, Miller T, Langlois JA, Selassie AW. Prevalence of Long-Term Disability From Traumatic Brain Injury in the Civilian Population of the United States, 2005. J Head Trauma Rehabil (2008) 23(6):394–400. doi: 10.1097/01.HTR.0000341435.52004.ac
4. Kelly DF, Gaw Gonzalo IT, Berman N, Swerdloff R, Wang C. Hypopituitarism following traumatic brain injury and aneurysmal subarachnoid hemorrhage: a preliminary report. J Neurosurg (2000) 93(5):743–52. doi: 10.3171/jns.2000.93.5.0743
5. Lieberman SA, Oberoi AL, Gilkison CR, Masel BE, Urban RJ. Prevalence of Neuroendocrine Dysfunction in Patients Recovering from Traumatic Brain Injury. J Clin Endocrinol Metab (2001) 86(6):2752–6. doi: 10.1210/jcem.86.6.7592
6. Van Der Eerden AW, Twickler MTB, Sweep FCGJ, Beems T, Hendricks HT, Hermus ARMM, et al. Should anterior pituitary function be tested during follow-up of all patients presenting at the emergency department because of traumatic brain injury? Eur J Endocrinol (2010) 162(1):19–28. doi: 10.1530/EJE-09-0436
7. Moreau OK, Yollin E, Merlen E, Daveluy W, Rousseaux M. Lasting pituitary hormone deficiency after traumatic brain injury. J Neurotrauma (2012) 29(1):81–9. doi: 10.1089/neu.2011.2048
8. Schneider HJ, Kreitschmann-Andermahr I, Ghigo E, Stalla GK, Agha A. Hypothalamopituitary dysfunction following traumatic brain injury and aneurysmal subarachnoid hemorrhage: A systematic review. J Am Med Assoc (2007) 298(12):1429–38. doi: 10.1001/jama.298.12.1429
9. Emelifeonwu J, Flower H, Loan J, McGivern K, Andrews P. Prevalence of Anterior Pituitary Dysfunction Twelve Months or More following Traumatic Brain Injury in Adults: A Systematic Review and Meta-Analysis. J Neurotrauma (2020) 37(2):217–26. doi: 10.1089/neu.2018.6349
10. Max W, MacKenzie E, Rice D. Head injuries: costs and consequences. J Head Trauma Rehabil (1991) 6(2):76–91. doi: 10.1097/00001199-199106000-00010
11. Thompson HJ, Weir S, Rivara FP, Wang J, Sullivan SD, Salkever D, et al. Utilization and costs of health care after geriatric traumatic brain injury. J Neurotrauma (2012) 29(10):1864–71. doi: 10.1089/neu.2011.2284
13. Schneider H, Aimaretti G, Kreitschmann-Andermahr I, Stalla G, Ghigo E. Hypopituitarism. Lancet (2007) 369(9571):1461–70. doi: 10.1016/S0140-6736(07)60673-4
14. High WM, Briones-Galang M, Clark JA, Gilkison C, Mossberg KA, Zgaljardic DJ, et al. Effect of growth hormone replacement therapy on cognition after traumatic brain injury. J Neurotrauma (2010) 27(9):1565–75. doi: 10.1089/neu.2009.1253
15. Agha A, Rogers B, Sherlock M, O’Kelly P, Tormey W, Phillips J, et al. Anterior pituitary dysfunction in survivors of traumatic brain injury. J Clin Endocrinol Metab (2004) 89(10):4929–36. doi: 10.1210/jc.2004-0511
16. Cuesta M, Hannon MJ, Crowley RK, Behan LA, Tormey W, Rawluk D, et al. Symptoms of gonadal dysfunction are more predictive of hypopituitarism than nonspecific symptoms in screening for pituitary dysfunction following moderate or severe traumatic brain injury. Clin Endocrinol (Oxf) (2016) 84(1):92–8. doi: 10.1111/cen.12874
17. Jeong JH, Kim YZ, Cho YW, Kim JS. Negative effect of hypopituitarism following brain trauma in patients with diffuse axonal injury: Clinical article. J Neurosurg (2010) 113(3):532–8. doi: 10.3171/2009.10.JNS091152
18. Klose M, Juul A, Poulsgaard L, Kosteljanetz M, Brennum J, Feldt-Rasmussen U. Prevalence and predictive factors of post-traumatic hypopituitarism. Clin Endocrinol (Oxf) (2007) 67(2):193–201. doi: 10.1111/j.1365-2265.2007.02860.x
19. Klose MC, Watt T, Brennum J, Feldt-Rasmussen U. Posttraumatic hypopituitarism is associated with an unfavorable body composition and lipid profile, and decreased quality of life 12 months after injury. J Clin Endocrinol Metab (2007) 92(10):3861–8. doi: 10.1210/jc.2007-0901
20. Nourollahi S, Wille J, Weiß V, Wedekind C, Lippert-Grüner M. Quality-of-life in patients with post-traumatic hypopituitarism. Brain Inj (2014) 28(11):1425–9. doi: 10.3109/02699052.2014.917201
21. Schneider HJ, Schneider M, Saller B, Petersenn S, Uhr M, Husemann B, et al. Prevalence of anterior pituitary insufficiency 3 and 12 months after traumatic brain injury. Eur J Endocrinol (2006) 154(2):259–65. doi: 10.1530/eje.1.02071
22. Silva PPB, Bhatnagar S, Herman SD, Zafonte R, Klibanski A, Miller KK, et al. Predictors of Hypopituitarism in Patients with Traumatic Brain Injury. J Neurotrauma (2015) 32(22):1789–95. doi: 10.1089/neu.2015.3998
23. Ulfarsson T, Arnar Gudnason G, Rosén T, Blomstrand C, Stibrant Sunnerhagen K, Lundgren-Nilsson Å, et al. Pituitary function and functional outcome in adults after severe traumatic brain injury: The long-term perspective. J Neurotrauma (2013) 30(4):271–80. doi: 10.1089/neu.2012.2494
24. Zgaljardic DJ, Guttikonda S, Grady JJ, Gilkison CR, Mossberg KA, High WM, et al. Serum IGF-1 concentrations in a sample of patients with traumatic brain injury as a diagnostic marker of growth hormone secretory response to glucagon stimulation testing. Clin Endocrinol (Oxf) (2011) 74(3):365–9. doi: 10.1111/j.1365-2265.2010.03935.x
25. Aimaretti G, Ambrosio MR, Di Somma C, Gasperi M, Cannavò S, Scaroni C, et al. Residual pituitary function after brain injury-induced hypopituitarism: A prospective 12-month study. J Clin Endocrinol Metab (2005) 90(11):6085–92. doi: 10.1210/jc.2005-0504
26. Kleindienst A, Brabant G, Bock C, Maser-Gluth C, Buchfelder M. Neuroendocrine function following traumatic brain injury and subsequent intensive care treatment: A prospective longitudinal evaluation. J Neurotrauma (2009) 26(9):1435–46. doi: 10.1089/neu.2008.0601
27. Aimaretti G, Ambrosio MR, Di Somma C, Gasperi M, Cannavò S, Scaroni C, et al. Hypopituitarism induced by traumatic brain injury in the transition phase. J Endocrinol Invest (2005) 28(2):984–9. doi: 10.1007/BF03345336
28. Berg C, Oeffner A, Schumm-Draeger PM, Badorrek F, Brabant G, Gerbert B, et al. Prevalence of anterior pituitary dysfunction in patients following traumatic brain injury in a german multi-centre screening program. Exp Clin Endocrinol Diabetes (2010) 118(2):139–44. doi: 10.1055/s-0029-1225611
29. Bondanelli M, De Marinis L, Ambrosio MR, Monesi M, Valle D, Zatelli MC, et al. Occurrence of pituitary dysfunction following traumatic brain injury. J Neurotrauma (2004) 21(6):685–96. doi: 10.1089/0897715041269713
30. Herrmann BL, Rehder J, Kahlke S, Wiedemayer H, Doerfler A, Ischebeck W, et al. Hypopituitarism following severe traumatic brain injury. Exp Clin Endocrinol Diabetes (2006) 114(6):316–21. doi: 10.1055/s-2006-924254
31. Hari Kumar KVS, Swamy M, Khan M. Prevalence of hypothalamo pituitary dysfunction in patients of traumatic brain injury. Indian J Endocrinol Metab (2016) 20(6):772–8. doi: 10.4103/2230-8210.192917
32. Nemes O, Kovacs N, Czeiter E, Kenyeres P, Tarjanyi Z, Bajnok L, et al. Predictors of post-traumatic pituitary failure during long-term follow-up. Hormones (2015) 14(3):383–91. doi: 10.14310/horm.2002.1564
33. Popovic V, Pekic S, Pavlovic D, Maric N, Jasovic-Gasic M, Djurovic B, et al. Hypopituitarism as a consequence of traumatic brain injury (TBI) and its possible relation with cognitive disabilities and mental distress. J Endocrinol Invest (2004) 27(11):1048–54. doi: 10.1007/BF03345308
34. Prodam F, Gasco V, Caputo M, Zavattaro M, Pagano L, Marzullo P, et al. Metabolic alterations in patients who develop traumatic brain injury (TBI)-induced hypopituitarism. Growth Horm IGF Res (2013) 23(4):109–13. doi: 10.1016/j.ghir.2013.04.001
35. Wachter D, Gündling K, Oertel MF, Stracke H, Böker DK. Pituitary insufficiency after traumatic brain injury. J Clin Neurosci (2009) 16(2):202–8. doi: 10.1016/j.jocn.2008.01.009
36. Klose M, Stochholm K, Janukonyté J, Christensen LL, Frystyk J, Andersen M, et al. Prevalence of posttraumatic growth hormone deficiency is highly dependent on the diagnostic set-up: Results from the danish national study on posttraumatic hypopituitarism. J Clin Endocrinol Metab (2014) 99(1):101–10. doi: 10.1210/jc.2013-2397
37. Kokshoorn NE, Smit JWA, Nieuwlaat WA, Tiemensma J, Bisschop PH, Groote Veldman R, et al. Low prevalence of hypopituitarism after traumatic brain injury: A multicenter study. Eur J Endocrinol (2011) 165(2):225–31. doi: 10.1530/EJE-11-0365
38. Krahulik DD, Zapletalova J, Frysak Z, Vaverka M. Dysfunction of hypothalamic-hypophysial axis after traumatic brain injury in adults: Clinical article. J Neurosurg (2010) 113(3):581–4. doi: 10.3171/2009.10.JNS09930
39. Park KD, Kim DY, Lee JK, Nam HS, Park YG. Anterior pituitary dysfunction in moderate-to-severe chronic traumatic brain injury patients and the influence on functional outcome. Brain Inj (2010) 24(11):1330–5. doi: 10.3109/02699052.2010.506863
40. Tanriverdi F, Senyurek H, Unluhizarci K, Selcuklu A, Casanueva FF, Kelestimur F. High risk of hypopituitarism after traumatic brain injury: A prospective investigation of anterior pituitary function in the acute phase and 12 months after trauma. J Clin Endocrinol Metab (2006) 91(6):2105–11. doi: 10.1210/jc.2005-2476
41. Teasdale B, Jennett G. Assessment of coma and impaired consciousness. A practical scale. Lancet (1974) 2(7872):81–4. doi: 10.1016/S0140-6736(74)91639-0
42. Kaur P, Sharma S. Recent Advances in Pathophysiology of Traumatic Brain Injury. Curr Neuropharmacol (2018) 16(8):1224–38. doi: 10.2174/1570159X15666170613083606
43. Grandhi R, Bonfield C, Newman W, Okonkwo D. Surgical management of traumatic brain injury: a review of guidelines, pathophysiology, neurophysiology, outcomes, and controversies. J Neurosurg Sci (2014) 58(4):249–59.
44. Jassam YN, Izzy S, Whalen M, McGavern DB, El Khoury J. Neuroimmunology of Traumatic Brain Injury: Time for a Paradigm Shift. Neuron (2017) 95(6):1246–65. doi: 10.1016/j.neuron.2017.07.010
45. Corps KN, Roth TL, McGavern DB. Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol (2015) 72(3):355–62. doi: 10.1001/jamaneurol.2014.3558
46. Daniel P, Prichard M, Schurr P. Extent of the infarct in the anterior lobe of the human pituitary gland after stalk section. Lancet (1958) 1(7030):1101–3. doi: 10.1016/S0140-6736(58)91852-X
47. Bullock R, Zauner A, Woodward J, Myseros J, Choi S, Ward J, et al. Factors affecting excitatory amino acid release following severe human head injury. J Neurosurg (1998) 89(4):507–18. doi: 10.3171/foc.1998.5.2.1
48. Xuereb G, Prichard M, Daniel P. The arterial supply and venous drainage of the human hypophysis cerebri. Q J Exp Physiol Cognit Med Sci (1954) 39(3):199–217. doi: 10.1113/expphysiol.1954.sp001072
49. Xuereb BG P, Prichard MM L, Daniel PM. The hypophyseal portal system of vessels in man. Q J Exp Physiol Cognit Med Sci (1954) 39(3):219–30. doi: 10.1113/expphysiol.1954.sp001073
50. Gorczyca W, Hardy J. Arterial supply of the human anterior pituitary gland. Neurosurgery (1987) 20(3):369–78. doi: 10.1227/00006123-198703000-00003
51. Famini P, Melmed S. Pituitary Apoplexy. In: Loriaux L, editor. Endocrine Emergencies. Contemporary Endocrinology, vol. 74 . Totowa, NJ: Humana Press (2014). p. 175–211.
52. Horvath E, Kovacs K. Fine Structural Cytology of the Adenohypophysis in Rat and Man. J Electron Microsc Tech (1988) 8:401–32. doi: 10.1002/jemt.1060080410
53. Kasturi BS, Stein DG. Traumatic Brain Injury Causes Long-Term Reduction in Serum Growth Hormone and Persistent Astrocytosis in the Cortico-Hypothalamo-Pituitary Axis of Adult Male Rats. J Neurotrauma (2009) 26(8):1315–24. doi: 10.1089/neu.2008-0751
54. Tanriverdi F, De Bellis A, Bizzarro A, Sinisi AA, Bellastella G, Pane E, et al. Antipituitary antibodies after traumatic brain injury: Is head trauma-induced pituitary dysfunction associated with autoimmunity? Eur J Endocrinol (2008) 159(1):7–13. doi: 10.1530/EJE-08-0050
55. Tanriverdi F, De Bellis A, Ulutabanca H, Bizzarro A, Sinisi AA, Bellastella G, et al. A five year prospective investigation of anterior pituitary function after traumatic brain injury: Is hypopituitarism long-term after head trauma associated with autoimmunity? J Neurotrauma (2013) 30(16):1426–33. doi: 10.1089/neu.2012.2752
56. Tanriverdi F, De Bellis A, Battaglia M, Bellastella G, Bizzarro A, Sinisi AA, et al. Investigation of antihypothalamus and antipituitary antibodies in amateur boxers: Is chronic repetitive head trauma-induced pituitary dysfunction associated with autoimmunity? Eur J Endocrinol (2010) 162(5):861–7. doi: 10.1530/EJE-09-1024
57. Diaz-Arrastia R, Gong Y, Fair S, Scott KD, Garcia MC, Carlile MC, et al. Increased Risk of Late Posttraumatic Seizures Associated With Inheritance of APOE 4 Allele. Arch Neurol (2003) 60(6):818–22. doi: 10.1001/archneur.60.6.818
58. Sorbi S, Nacmias B, Piacentini S, Repice A, Latorraca S, Forleo P, et al. ApoE as a prognostic factor for post-traumatic coma. Nat Med (1995) 1(9):852. doi: 10.1038/nm0995-852
59. Crawford FC, Vanderploeg RD, Freeman MJ, Singh S, Waisman M, Michaels L, et al. APOE genotype influences acquisition and recall following traumatic brain injury. Neurology (2002) 58(7):1115–8. doi: 10.1212/WNL.58.7.1115
60. Tanriverdi F, Taheri S, Ulutabanca H, Caglayan AO, Ozkul Y, Dundar M, et al. Apolipoprotein E3/E3 genotype decreases the risk of pituitary dysfunction after traumatic brain injury due to various causes: Preliminary data. J Neurotrauma (2008) 25(9):1071–7. doi: 10.1089/neu.2007.0456
61. Ceballos R. Pituitary changes in head trauma (analysis of 102 consecutive cases of head injury). Ala J Med Sci (1966) 3(2):185–98.
62. Kornblum R, Fisher RS. Pituitary lesions in craniocerebral injuries. Arch Pathol (1969) 88(3):242–8.
63. Crompton M. Hypothalamic lesions following closed head injury. Brain (1971) 94(1):165–72. doi: 10.1093/brain/94.1.165
64. Harper C, Doyle D, Hume Adams J, Graham D. Analysis of abnormalities in pituitary gland in non-missile head injury: study of 100 consecutive cases. J Clin Pathol (1986) 39(7):769–73. doi: 10.1136/jcp.39.7.769).
65. Salehi F, Kovacs K, Scheithauer BW, Pfeifer EA, Cusimano M. Histologic study of the human pituitary gland in acute traumatic brain injury. Brain Inj (2007) 21(6):651–6. doi: 10.1080/02699050701426956
66. Maiya B, Newcombe V, Nortje J, Bradley P, Bernard F, Chatfield D, et al. Magnetic resonance imaging changes in the pituitary gland following acute traumatic brain injury. Intensive Care Med (2008) 34(3):468–75. doi: 10.1007/s00134-007-0902-x
67. Zheng P, He B, Guo Y, Zeng J, Tong W. Decreased apparent diffusion coefficient in the pituitary and correlation with hypopituitarism in patients with traumatic brain injury. J Neurosurg (2015) 123(1):75–80. doi: 10.3171/2014.12.JNS132308
68. Beauchamp NJ, Barker PB, Wang PY, Vanzijl PCM. Imaging of Acute Cerebral Ischemia. Radiology (1999) 212(2):307–24. doi: 10.1148/radiology.212.2.r99au16307
69. Warach S, Gaa J, Siewert B, Wielopolski P, Edelman RR. Acute Human Stroke Studied by Whole Brain Echo Planar Dfision-weighted Magnetic Resonance Imaging. Ann Neurol (1995) 37(2):231–41. doi: 10.1002/ana.410370214
70. Kuhl CK, Textor J, Gieseke J, Von Falkenhausen M, Cernert S, Urbach H, et al. Acute and subacute ischemic stroke at high-field-strength (3.0-T) diffusion-weighted MR imaging: Intraindividual comparative study. Radiology (2005) 234(2):509–16. doi: 10.1148/radiol.2342031323
71. Schneider H, Samann P, Schneider M, Croce C, Corneli G, Sievers C, et al. Pituitary imaging abnormalities in patients with and without hypopituitarism after traumatic brain injury. J Endocrinol Investig (2007) 30(4):RC9–12. doi: 10.1007/BF03346291
72. Tanriverdi F, Unluhizarci K, Kocyigit I, Tuna IS, Karaca Z, Durak AC, et al. Brief Communication: Pituitary Volume and Function in Competing and Retired Male Boxers. Ann Intern Med (2008) 148(11):827–31. doi: 10.7326/0003-4819-148-11-200806030-00005
73. Caputo M, Mele C, Prodam F, Marzullo P, Aimaretti G. Clinical picture and the treatment of TBI-induced hypopituitarism. Pituitary (2019) 22(3):261–9. doi: 10.1007/s11102-019-00956-w
74. Hannon MJ, Crowley RK, Behan LA, O’Sullivan EP, O’Brien MMC, Sherlock M, et al. Acute glucocorticoid deficiency and diabetes insipidus are common after acute traumatic brain injury and predict mortality. J Clin Endocrinol Metab (2013) 98(8):3229–37. doi: 10.1210/jc.2013-1555
75. Bensalah M, Donaldson M, Aribi Y, Iabassen M, Cherfi L, Nebbal M, et al. Cortisol evaluation during the acute phase of traumatic brain injury—A prospective study. Clin Endocrinol (Oxf) (2018) 88(5):627–36. doi: 10.1111/cen.13562
76. Agha A, Thornton E, O’Kelly P, Tormey W, Phillips J, Thompson CJ. Posterior pituitary dysfunction after traumatic brain injury. J Clin Endocrinol Metab (2004) 89(12):5987–92. doi: 10.1210/jc.2004-1058
77. Agha A, Phillips J, O’Kelly P, Tormey W, Thompson CJ. The natural history of post-traumatic hypopituitarism: Implications for assessment and treatment. Am J Med (2005) 118(12):1416.e1–1416.e7. doi: 10.1016/j.amjmed.2005.02.042
78. Gray S, Bilski T, Dieudonne B, Saeed S. Hypopituitarism After Traumatic Brain Injury. Cureus (2019) 11(3):e4163. doi: 10.7759/cureus.4163
79. Glynn N, Agha A. Which patient requires neuroendocrine assessment following traumatic brain injury, when and how? Clin Endocrinol (Oxf) (2013) 78(1):17–20. doi: 10.1111/cen.12010
80. Prodam F, Caputo M, Belcastro S, Garbaccio V, Zavattaro M, Samà M, et al. Quality of life, mood disturbances and psychological parameters in adult patients with GH deficiency. Panminerva Med (2012) 54(4):323–31.
81. Mossberg KA, Masel BE, Gilkison CR, Urban RJ. Aerobic capacity and growth hormone deficiency after traumatic brain injury. J Clin Endocrinol Metab (2008) 93(7):2581–7. doi: 10.1210/jc.2008-0368
82. Jellinger KA, Paulus W, Wrocklage C, Litvan I. Traumatic brain injury as a risk factor for Alzheimer disease. Comparison of two retrospective autopsy cohorts with evaluation of ApoE genotype. BMC Neurol (2001) 1:3. doi: 10.1186/1471-2377-1-3
83. Gilis-Januszewska A, Kluczyński Ł, Hubalewska-Dydejczyk A. Traumatic brain injuries induced pituitary dysfunction: A call for algorithms. Endocr Connect (2020) 9(5):R112–23. doi: 10.1530/EC-20-0117
84. Cheng CM, Mervis RF, Niu SL, Salem N, Witters LA, Tseng V, et al. Insulin-like growth factor 1 is essential for normal dendritic growth. J Neurosci Res (2003) 73(1):1–9. doi: 10.1002/jnr.10634
85. Kelly DF, McArthur DL, Levin H, Swimmer S, Dusick JR, Cohan P, et al. Neurobehavioral and quality of life changes associated with growth hormone insufficiency after complicated mild, moderate, or severe traumatic brain injury. J Neurotrauma (2006) 23(6):928–42. doi: 10.1089/neu.2006.23.928
86. León-Carrión J, Leal-Cerro A, Cabezas FM, Atutxa AM, Gomez SG, Cordero JMF, et al. Cognitive deterioration due to GH deficiency in patients with traumatic brain injury: A preliminary report. Brain Inj (2007) 21(8):871–5. doi: 10.1080/02699050701484849
87. Kgosidialwa O, Hakami O, Zia-Ul-Hussnain HM, Agha A. Growth hormone deficiency following traumatic brain injury. Int J Mol Sci (2019) 20(13):3323. doi: 10.3390/ijms20133323
88. Park KD, Lim OK, Yoo CJ, Kim YW, Lee S, Park Y, et al. Voxel-based statistical analysis of brain metabolism in patients with growth hormone deficiency after traumatic brain injury. Brain Inj (2016) 30(4):407–13. doi: 10.3109/02699052.2015.1127997
89. Pavlovic D, Pekic S, Stojanovic M, Zivkovic V, Djurovic B, Jovanovic V, et al. Chronic cognitive sequelae after traumatic brain injury are not related to growth hormone deficiency in adults. Eur J Neurol (2010) 17(5):696–702. doi: 10.1111/j.1468-1331.2009.02910.x
90. Gasco V, Prodam F, Pagano L, Grottoli S, Belcastro S, Marzullo P, et al. Hypopituitarism following brain injury: When does it occur and how best to test? Pituitary (2012) 15(1):20–4. doi: 10.1007/s11102-010-0235-6
91. Glynn N, Agha A. The frequency and the diagnosis of pituitary dysfunction after traumatic brain injury. Pituitary (2019) 22(3):249–60. doi: 10.1007/s11102-019-00938-y
92. Ghigo E, Masel B, Aimaretti G, Léon-Carrión J, Casanueva FF, Dominguez-Morales MR, et al. Consensus guidelines on screening for hypopituitarism following traumatic brain injury. Brain Inj (2005) 19(9):711–24. doi: 10.1080/02699050400025315
93. Fleseriu M, Hashim IA, Karavitaki N, Melmed S, Murad MH, Salvatori R, et al. Hormonal replacement in hypopituitarism in adults: An endocrine society clinical practice guideline. J Clin Endocrinol Metab (2016) 101(11):3888–921. doi: 10.1210/jc.2016-2118
94. Thaler LM, Blevins LS. The Low Dose (1-μg) Adrenocorticotropin Stimulation Test in the Evaluation of Patients with Suspected Central Adrenal Insufficiency. J Clin Endocrinol Metab (1998) 83(8):2726–9. doi: 10.1210/jc.83.8.2726
95. Ceccato F, Scaroni C. Central adrenal insufficiency: Open issues regarding diagnosis and glucocorticoid treatment. Clin Chem Lab Med (2018) 57(8):1125–35. doi: 10.1515/cclm-2018-0824
96. Molitch ME, Clemmons DR, Malozowski S, Merriam GR, Vance ML. Evaluation and treatment of adult growth hormone deficiency: An endocrine society clinical practice guideline. J Clin Endocrinol Metab (2011) 96(6):1587–609. doi: 10.1210/jc.2011-0179
97. Yuen KCJ, Biller BMK, Radovick S, Carmichael JD, Jasim S, Pantalone KM, et al. American Association of Clinical endocrinologists and American College of Endocrinology guidelines for management of growth hormone deficiency in adults and patients transitioning from pediatric to adult care. Endocr Pract (2019) 25(11):1191–232. doi: 10.4158/GL-2019-0405
98. Vespa PM, Nuwer MR, Nenov V, Ronne-Engstrom E, Hovda DA, Bergsneider M, et al. Increased incidence and impact of nonconvulsive and convulsive seizures after traumatic brain injury as detected by continuous electroencephalographic monitoring. J Neurosurg (1999) 91(5):750–60. doi: 10.3171/jns.1999.91.5.0750
99. Corneli G, Di Somma C, Baldelli R, Rovere S, Gasco V, Croce CG, et al. The cut-off limits of the GH response to GH-releasing hormone-arginine test related to body mass index. Eur J Endocrinol (2005) 153(2):257–64. doi: 10.1530/eje.1.01967
100. Gómez JM, Espadero RM, Escobar-Jiménez F, Hawkins F, Picó A, Herrera-Pombo JL, et al. Growth hormone release after glucagon as a reliable test of growth hormone assessment in adults. Clin Endocrinol (Oxf) (2002) 56(3):329–34. doi: 10.1046/j.1365-2265.2002.01472.x
101. Kelestimur F, Popovic V, Leal A, Van Dam PS, Torres E, Perez Mendez LF, et al. Effect of obesity and morbid obesity on the growth hormone (GH) secretion elicited by the combined GHRH + GHRP-6 test. Clin Endocrinol (Oxf) (2006) 64(6):667–71. doi: 10.1111/j.1365-2265.2006.02525.x
102. Gasco V, Ferrero A, Bisceglia A, Prencipe N, Cambria V, Bioletto F, et al. The cut-off limits of GH response to insulin tolerance test related to body mass index for the diagnosis of adult GH deficiency. Neuroendocrinology (2020). doi: 10.1159/000508103
103. Devesa J, Devesa P, Reimunde P. Growth hormone revisited. Med Clin (2010) 135(14):665–70. doi: 10.1016/j.medcli.2009.10.017
104. Costoya JA, Finidori J, Moutoussamy S, Señaris R, Devesa J, Arce VM. Activation of growth hormone receptor delivers an antiapoptotic signal: Evidence for a role of Akt in this pathway. Endocrinology (1999) 140(12):5937–43. doi: 10.1210/endo.140.12.7209
105. Sanders EJ, Baudet ML, Parker E, Harvey S. Signaling mechanisms mediating local GH action in the neural retina of the chick embryo. Gen Comp Endocrinol (2009) 163(1–2):63–9. doi: 10.1016/j.ygcen.2009.01.005
106. McLenachan S, Lum MG, Waters MJ, Turnley AM. Growth hormone promotes proliferation of adult neurosphere cultures. Growth Horm IGF Res (2009) 19(3):212–8. doi: 10.1016/j.ghir.2008.09.003
107. Åberg ND, Johansson I, Åberg MAI, Lind J, Johansson UE, Cooper-Kuhn CM, et al. Peripheral administration of GH induces cell proliferation in the brain of adult hypophysectomized rats. J Endocrinol (2009) 201(1):141–50. doi: 10.1677/JOE-08-0495
108. Scheepens A, Sirimanne E, Beilharz E, Breier BH, Waters MJ, Gluckman PD, et al. Alterations in the neural growth hormone axis following hypoxic-ischemic brain injury. Mol Brain Res (1999) 68(1–2):88–100. doi: 10.1016/s0169-328x(99)00051-0
109. Scheepens A, Sirimanne ES, Breier BH, Clark RG, Gluckman PD, Williams CE. Growth hormone as a neuronal rescue factor during recovery from CNS injury. Neuroscience (2001) 104(3):677–87. doi: 10.1016/s0306-4522(01)00109-9
110. Madathil SK, Evans HN, Saatman KE. Temporal and regional changes in IGF-1/IGF-1R signaling in the mouse brain after traumatic brain injury. J Neurotrauma (2010) 27(1):95–107. doi: 10.1089/neu.2009.1002
111. Walter HJ, Berry M, Hill DJ, Logan A. Spatial and temporal changes in the insulin-like growth factor (IGF) axis indicate autocrine/paracrine actions of IGF-I within wounds of the rat brain. Endocrinology (1997) 138(7):3024–34. doi: 10.1210/endo.138.7.5284
112. Scheepens A, Williams CE, Breier BH, Guan J, Gluckman PD. A role for the somatotropic axis in neural development, injury and disease. Journal of Pediatric Endocrinology and Metabolism. Freund Publishing House Ltd (2000) 13 Suppl 6:1483–91. doi: 10.1515/jpem-2000-s623
113. Christophidis LJ, Gorba T, Gustavsson M, Williams CE, Werther GA, Russo VC, et al. Growth hormone receptor immunoreactivity is increased in the subventricular zone of juvenile rat brain after focal ischemia: A potential role for growth hormone in injury-induced neurogenesis. Growth Horm IGF Res (2009) 19(6):497–506. doi: 10.1016/j.ghir.2009.05.001
114. Li RC, Guo SZ, Raccurt M, Moudilou E, Morel G, Brittian KR, et al. Exogenous growth hormone attenuates cognitive deficits induced by intermittent hypoxia in rats. Neuroscience (2011) 196:237–50. doi: 10.1016/j.neuroscience.2011.08.029
115. Pathipati P, Gorba T, Scheepens A, Goffin V, Sun Y, Fraser M. Growth hormone and prolactin regulate human neural stem cell regenerative activity. Neuroscience (2011) 190:409–27. doi: 10.1016/j.neuroscience.2011.05.029
116. Devesa P, Reimunde P, Gallego R, Devesa J, Arce VM. Growth hormone (GH) treatment may cooperate with locally-produced GH in increasing the proliferative response of hippocampal progenitors to kainate-induced injury. Brain Inj (2011) 25(5):503–10. doi: 10.3109/02699052.2011.559611
117. Åberg ND, Lind J, Isgaard J, Kuhn HG. Peripheral growth hormone induces cell proliferation in the intact adult rat brain. Growth Horm IGF Res (2010) 20(3):264–9. doi: 10.1016/j.ghir.2009.12.003
118. Devesa J, Alonso, Casteleiro, Couto, Castanón, Zas, et al. Effects of recombinant growth hormone (GH) replacement and psychomotor and cognitive stimulation in the neurodevelopment of GH-deficient (GHD) children with cerebral palsy: a pilot study. Ther Clin Risk Manage (2011) 7:199. doi: 10.2147/TCRM.S21403
119. Möderscheim TAE, Christophidis LJ, Williams CE, Scheepens A. Distinct neuronal growth hormone receptor ligand specificity in the rat brain. Brain Res (2007) 1137(1):29–34. doi: 10.1016/j.brainres.2006.12.040
120. Byts N, Samoylenko A, Fasshauer T, Ivanisevic M, Hennighausen L, Ehrenreich H, et al. Essential role for Stat5 in the neurotrophic but not in the neuroprotective effect of erythropoietin. Cell Death Differ (2008) 15(4):783–92. doi: 10.1038/cdd.2008.1
121. Silva C, Zhang K, Tsutsui S, Holden JK, Gill MJ, Power C. Growth Hormone Prevents Human Immunodeficiency Virus-Induced Neuronal p53 Expression. Ann Neurol (2003) 54(5):605–14. doi: 10.1002/ana.10729
122. Van Marle G, Antony JM, Silva C, Sullivan A, Power C. Aberrant cortical neurogenesis in a pediatric neuroAIDS model: Neurotrophic effects of growth hormone. AIDS (2005) 19(16):1781–91. doi: 10.1097/01.aids.0000189854.06194.87
123. Devesa P. Effects of growth hormone on the central and peripheral neural repair. PhD Thesis (2011).
124. Lichtenwalner RJ, Forbes ME, Sonntag WE, Riddle DR. Adult-onset deficiency in growth hormone and insulin-like growth factor-I decreases survival of dentate granule neurons: Insights into the regulation of adult hippocampal neurogenesis. J Neurosci Res (2006) 83(2):199–210. doi: 10.1002/jnr.20719
125. Sun LY, Bartke A. Adult neurogenesis in the hippocampus of long-lived mice during aging. J Gerontol Ser A Biol Sci Med Sci (2007) 62(2):117–25. doi: 10.1093/gerona/62.2.117
126. Ajo R, Cacicedo L, Navarro C, Sánchez-Franco F. Growth hormone action on proliferation and differentiation of cerebral cortical cells from fetal rat. Endocrinology (2003) 144(3):1086–97. doi: 10.1210/en.2002-220667
127. Lyuh E, Kim HJ, Kim M, Lee JK, Park KS, Yoo KY, et al. Dose-specific or dose-dependent effect of growth hormone treatment on the proliferation and differentiation of cultured neuronal cells. Growth Horm IGF Res (2007) 17(4):315–22. doi: 10.1016/j.ghir.2007.03.002
128. Russo VC, Gluckman PD, Feldman EL, Werther GA. The insulin-like growth factor system and its pleiotropic functions in brain. Endocr Rev (2005) 26(7):916–43. doi: 10.1210/er.2004-0024
129. Popken GJ, Dechert-Zeger M, Ye P, D’Ercole AJ. Brain development. Adv Exp Med Biol (2005) 567:187–220. doi: 10.1007/0-387-26274-1_8
130. D’Ercole AJ, Ye P. Minireview: Expanding the mind: Insulin-like growth factor I and brain development. Endocrinology (2008) 149(12):5958–62. doi: 10.1210/en.2008-0920
131. Torres-Aleman I. Toward a comprehensive neurobiology of IGF-I. Dev Neurobiol (2010) 70(5):384–96. doi: 10.1002/dneu.20778
132. Yan H, Mitschelen M, Bixler GV, Brucklacher RM, Farley JA, Han S, et al. Circulating IGF1 regulates hippocampal IGF1 levels and brain gene expression during adolescence. J Endocrinol (2011) 211(1):27–37. doi: 10.1530/JOE-11-0200
133. Calzà L, Giuliani A, Fernandez M, Pirondi S, D’Intino G, Aloe L, et al. Neural stem cells and cholinergic neurons: Regulation by immunolesion and treatment with mitogens, retinoic acid, and nerve growth factor. Proc Natl Acad Sci U S A (2003) 100(12):7325–30. doi: 10.1073/pnas.1132092100
134. Türeyen K, Vemuganti R, Bowen KK, Sailor KA, Dempsey RJ. EGF and FGF-2 infusion increases post-ischemic neural progenitor cell proliferation in the adult rat brain. Neurosurgery (2005) 57(6):1254–62. doi: 10.1227/01.neu.0000186040.96929.8a
135. Yamauchi T, Ueki K, Tobe K, Tamemoto H, Sekine N, Wada M, et al. Growth hormone-induced tyrosine phosphorylation of EGF receptor as an essential element leading to MAP kinase activation and gene expression. Endocr J (1998) 45(SUPPL.):S27–31. doi: 10.1507/endocrj.45.suppl_s27
136. Pan SN, Ma HM, Su Z, Zhang CX, Zhu SY, Du ML. Epidermal growth factor receptor signalling mediates growth hormone-induced growth of chondrocytes from sex hormone-inhibited adolescent rats. Clin Exp Pharmacol Physiol (2011) 38(8):534–42. doi: 10.1111/j.1440-1681.2011.05547.x
137. Byts N, Sirén A-L. Erythropoietin: a multimodal neuroprotective agent. Exp Transl Stroke Med (2009) 1(1):4. doi: 10.1186/2040-7378-1-4
138. Chen ZY, Asavaritikrai P, Prchal JT, Noguchi CT. Endogenous erythropoietin signaling is required for normal neural progenitor cell proliferation. J Biol Chem (2007) 282(35):25875–83. doi: 10.1074/jbc.M701988200
139. Tsai PT, Ohab JJ, Kertesz N, Groszer M, Matter C, Gao J, et al. A critical role of erythropoietin receptor in neurogenesis and post-stroke recovery. J Neurosci (2006) 26(4):1269–74. doi: 10.1523/JNEUROSCI.4480-05.2006
140. Zhang L, Chopp M, Zhang RL, Wang L, Zhang J, Wang Y, et al. Erythropoietin amplifies stroke-induced oligodendrogenesis in the rat. PloS One (2010) 5(6):e11016. doi: 10.1371/journal.pone.0011016
141. Sohmiya M, Kato Y. Effect of long-term administration of recombinant human growth hormone (rhGH) on plasma erythropoietin (EPO) and haemoglobin levels in anaemic patients with adult gh deficiency. Clin Endocrinol (Oxf) (2001) 55(6):749–54. doi: 10.1046/j.1365-2265.2001.01417.x
142. Devesa J, Almengló C, Devesa P. Multiple effects of growth hormone in the body: Is it really the hormone for growth? Clin Med Insights Endocrinol Diabetes (2016) 9:47–71. doi: 10.4137/CMED.S38201
143. Lu M, Flanagan JU, Langley RJ, Hay MP, Perry JK. Targeting growth hormone function: strategies and therapeutic applications. Signal Transduct Target Ther (2019) 4(1):3. doi: 10.1038/s41392-019-0036-y
144. Reed ML, Merriam GR, Kargi AY. Adult growth hormone deficiency - benefits, side effects, and risks of growth hormone replacement. Front Endocrinol (Lausanne) (2013) 4:64. doi: 10.3389/fendo.2013.00064
145. Gasco V, Caputo M, Lanfranco F, Ghigo E, Grottoli S. Management of GH treatment in adult GH deficiency. Best Pract Res Clin Endocrinol Metab (2017) 31(1):13–24. doi: 10.1016/j.beem.2017.03.001
146. Kreitschmann-Andermahr I, Poll EM, Reineke A, Gilsbach JM, Brabant G, Buchfelder M, et al. Growth hormone deficient patients after traumatic brain injury - Baseline characteristics and benefits after growth hormone replacement - An analysis of the German KIMS database. Growth Horm IGF Res (2008) 18(6):472–8. doi: 10.1016/j.ghir.2008.08.007
147. Maric NP, Doknic M, Pavlovic D, Pekic S, Stojanovic M, Jasovic-Gasic M, et al. Psychiatric and neuropsychological changes in growth hormone-deficient patients after traumatic brain injury in response to growth hormone therapy. J Endocrinol Invest (2010) 33(11):770–5. doi: 10.1007/BF03350340
148. Reimunde P, Quintana A, Castañón B, Casteleiro N, Vilarnovo Z, Otero A, et al. Effects of growth hormone (GH) replacement and cognitive rehabilitation in patients with cognitive disorders after traumatic brain injury. Brain Inj (2011) 25(1):65–73. doi: 10.3109/02699052.2010.536196
149. Moreau OK, Cortet-Rudelli C, Yollin E, Merlen E, Daveluy W, Rousseaux M. Growth hormone replacement therapy in patients with traumatic brain injury. J Neurotrauma (2013) 30(11):998–1006. doi: 10.1089/neu.2012.2705
150. Devesa J, Reimunde P, Devesa P, Barberá M, Arce V. Growth hormone (GH) and brain trauma. Horm Behav (2013) 63(2):331–44. doi: 10.1016/j.yhbeh.2012.02.022
151. Gardner CJ, Mattsson AF, Daousi C, Korbonits M, Koltowska-Haggstrom M, Cuthbertson DJ. GH deficiency after traumatic brain injury: Improvement in quality of life with GH therapy: Analysis of the KIMS database. Eur J Endocrinol (2015) 172(4):371–81. doi: 10.1530/EJE-14-0654
152. Leonhardt M, Kopczak A, Schäpers B, Limbrock J, Sämann PG, Czisch M, et al. Low Prevalence of Isolated Growth Hormone Deficiency in Patients After Brain Injury: Results From a Phase II Pilot Study. Front Endocrinol (Lausanne) (2018) 9:723. doi: 10.3389/fendo.2018.00723
153. Dubiel R, Callender L, Dunklin C, Harper C, Bennett M, Kreber L, et al. Phase 2 randomized, placebo-controlled clinical trial of recombinant human growth hormone (rhGH) during rehabilitation from traumatic brain injury. Front Endocrinol (Lausanne) (2018) 9:520. doi: 10.3389/fendo.2018.00520
154. Gadelha MR, Bronstein MD, Brue T, Coculescu M, Fleseriu M, Guitelman M, et al. Pasireotide versus continued treatment with octreotide or lanreotide in patients with inadequately controlled acromegaly (PAOLA): A randomised, phase 3 trial. Lancet Diabetes Endocrinol (2014) 2(11):875–84. doi: 10.1016/S2213-8587(14)70169-X
155. Colao A, Di Somma C, Savastano S, Rota F, Savanelli MC, Aimaretti G, et al. A reappraisal of diagnosing GH deficiency in adults: Role of gender, age, waist circumference, and body mass index. J Clin Endocrinol Metab (2009) 94(11):4414–22. doi: 10.1210/jc.2009-1134
156. Zhai Q, Lai Z, Roos P, Nyberg F. Characterization of growth hormone binding sites in rat brain. Acta Pædiatrica (1994) 83:92–5. doi: 10.1111/j.1651-2227.1994.tb13433.x
157. Lai Z, Roos P, Zhai Q, Olsson Y, Fhölenhag K, Larsson C, et al. Age-related reduction of human growth hormone-binding sites in the human brain. Brain Res (1993) 621(2):260–6. doi: 10.1016/0006-8993(93)90114-3
158. Nyberg F. Growth hormone in the brain: Characteristics of specific brain targets for the hormone and their functional significance. Front Neuroendocrinol (2000) 21(4):330–48. doi: 10.1006/frne.2000.0200
159. Falleti MG, Maruff P, Burman P, Harris A. The effects of growth hormone (GH) deficiency and GH replacement on cognitive performance in adults: A meta-analysis of the current literature. Psychoneuroendocrinology (2006) 31(6):681–91. doi: 10.1016/j.psyneuen.2006.01.005
160. Ho KKY. Consensus guidelines for the diagnosis and treatment of adults with GH deficiency II: A statement of the GH Research Society in association with the European Society for Pediatric Endocrinology, Lawson Wilkins Society, European Society of Endocrinology. J Eur J Endocrinol (2007) 157(6):695–700. doi: 10.1530/EJE-07-0631
161. Bondanelli M, Ambrosio MR, Cavazzini L, Bertocchi A, Zatelli MC, Carli A, et al. Anterior pituitary function may predict functional and cognitive outcome in patients with traumatic brain injury undergoing rehabilitation. J Neurotrauma (2007) 24(11):1687–97. doi: 10.1089/neu.2007.0343
162. Maruff P, Falleti M. Cognitive function in growth hormone deficiency and growth hormone replacement. Hormone Res Horm Res (2005) 64(Suppl 3):100–8. doi: 10.1159/000089325
163. Van Dam PS. Somatropin therapy and cognitive function in adults with growth hormone deficiency: A critical review. Treat Endocrinol (2006) 5(3):159–70. doi: 10.2165/00024677-200605030-00004
164. Nieves-Martinez E, Sonntag WE, Wilson A, Donahue A, Molina DP, Brunso-Bechtold J, et al. Early-onset GH deficiency results in spatial memory impairment in mid-life and is prevented by GH supplementation. J Endocrinol (2010) 204(1):31–6. doi: 10.1677/JOE-09-0323
165. Cuneo RC, Salomon F, McGauley GA, Sonksen PH. The growth hormone deficiency syndrome in adults. Clin Endocrinol (Oxf) (1992) 37(5):387–97. doi: 10.1111/j.1365-2265.1992.tb02347.x
166. Rosén T, Bengtsson BÅ. Premature mortality due to cardiovascular disease in hypopituitarism. Lancet (1990) 336(8710):285–8. doi: 10.1016/0140-6736(90)91812-O
167. Svensson J, Bengtsson BÅ, Rosén T, Odén A, Johannsson G. Malignant disease and cardiovascular morbidity in hypopituitary adults with or without growth hormone replacement therapy. J Clin Endocrinol Metab (2004) 89(7):3306–12. doi: 10.1210/jc.2003-031601
168. Bülow B, Hagmar L, Mikoczy Z, Nordström CH, Erfurth EM. Increased cerebrovascular mortality in patients with hypopituitarism. Clin Endocrinol (Oxf) (1997) 46(1):75–81. doi: 10.1046/j.1365-2265.1997.d01-1749.x
169. Maison P, Griffin S, Nicoue-Beglah M, Haddad N, Balkau B, Chanson P. Impact of Growth Hormone (GH) Treatment on Cardiovascular Risk Factors in GH-Deficient Adults: A Metaanalysis of Blinded, Randomized, Placebo-Controlled Trials. J Clin Endocrinol Metab (2004) 89(5):2192–9. doi: 10.1210/jc.2003-030840
170. Giustina A, Barkan A, Chanson P, Grossman A, Hoffman A, Ghigo E, et al. Guidelines for the treatment of growth hormone excess and growth hormone deficiency in adults. J Endocrinol Invest (2008) 31(9):820–38. doi: 10.1007/BF03349263
171. Tanriverdi F, Unluhizarci K, Karaca Z, Casanueva FF, Kelestimur F. Hypopituitarism due to sports related head trauma and the effects of growth hormone replacement in retired amateur boxers. Pituitary (2010) 13(2):111–4. doi: 10.1007/s11102-009-0204-0
172. Mossberg KA, Durham WJ, Zgaljardic DJ, Gilkison CR, Danesi CP, Sheffield-Moore M, et al. Functional Changes after Recombinant Human Growth Hormone Replacement in Patients with Chronic Traumatic Brain Injury and Abnormal Growth Hormone Secretion. J Neurotrauma (2017) 34(4):845–52. doi: 10.1089/neu.2016.4552
173. Bhagia V, Gilkison C, Fitts RH, Zgaljardic DJ, High WM, Masel BE, et al. Effect of recombinant growth hormone replacement in a growth hormone deficient subject recovering from mild traumatic brain injury: A case report. Brain Inj (2010) 24(3):560–7. doi: 10.3109/02699051003601705
174. Wüster C, Abs R, Bengtsson BÅ, Bennmarker H, Feldt-Rasmussen U, Hernberg-Ståhl E, et al. The influence of growth hormone deficiency, growth hormone replacement therapy, and other aspects of hypopituitarism on fracture rate and bone mineral density. J Bone Miner Res (2001) 16(2):398–405. doi: 10.1359/jbmr.2001.16.2.398
175. O’Halloran DJ, Tsatsoulis A, Whitehouse RW, Holmes SJ, Shalet SM. Increased bone density after recombinant human growth hormone (GH) therapy in adults with isolated GH deficiency. J Clin Endocrinol Metab (1993) 76(5):1344–8. doi: 10.1210/jcem.76.5.8496328
176. Götherström G, Bengtsson BÅ, Bossæus I, Johansson G, Svensson J. Ten-year GH replacement increase bone mineral density in hypopituitary patients with adult onset GH deficiency. Eur J Endocrinol (2007) 156(1):55–64. doi: 10.1530/eje.1.02317
177. Wallymahmed ME, Foy P, MacFarlane IA. The quality of life of adults with growth hormone deficiency: Comparison with diabetic patients and control subjects. Clin Endocrinol (Oxf) (1999) 51(3):333–8. doi: 10.1046/j.1365-2265.1999.00802.x
178. Badia X, Lucas A, Sanmarti A, Roset M, Ulied A. One-year follow-up of quality of life in adults with untreated growth hormone deficiency. Clin Endocrinol (Oxf) (1998) 49(6):765–71. doi: 10.1046/j.1365-2265.1998.00634.x
Keywords: traumatic brain injury, hypopituitarism, growth hormone deficiency, pituitary, brain damage
Citation: Gasco V, Cambria V, Bioletto F, Ghigo E and Grottoli S (2021) Traumatic Brain Injury as Frequent Cause of Hypopituitarism and Growth Hormone Deficiency: Epidemiology, Diagnosis, and Treatment. Front. Endocrinol. 12:634415. doi: 10.3389/fendo.2021.634415
Received: 27 November 2020; Accepted: 16 February 2021;
Published: 15 March 2021.
Edited by:
Antonio Mancini, Catholic University of the Sacred Heart, Rome, ItalyReviewed by:
Maria Chiara Zatelli, University of Ferrara, ItalyAndrzej Lewinski, Medical University of Lodz, Poland
Francesco Doglietto, University of Brescia, Italy
Copyright © 2021 Gasco, Cambria, Bioletto, Ghigo and Grottoli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valentina Gasco, dmFsZW50aW5hLmdhc2NvQHVuaXRvLml0