 Miguel Hernandez-Quiles
Miguel Hernandez-Quiles Marjoleine F. Broekema
Marjoleine F. Broekema Eric Kalkhoven
Eric Kalkhoven
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 26 February 2021
Sec. Molecular and Structural Endocrinology
Volume 12 - 2021 | https://doi.org/10.3389/fendo.2021.624112
This article is part of the Research TopicNuclear Receptors and Coregulators in Metabolism and Immunity View all 14 articles
The proliferator-activated receptor γ (PPARγ), a member of the nuclear receptor superfamily, is one of the most extensively studied ligand-inducible transcription factors. Since its identification in the early 1990s, PPARγ is best known for its critical role in adipocyte differentiation, maintenance, and function. Emerging evidence indicates that PPARγ is also important for the maturation and function of various immune system-related cell types, such as monocytes/macrophages, dendritic cells, and lymphocytes. Furthermore, PPARγ controls cell proliferation in various other tissues and organs, including colon, breast, prostate, and bladder, and dysregulation of PPARγ signaling is linked to tumor development in these organs. Recent studies have shed new light on PPARγ (dys)function in these three biological settings, showing unified and diverse mechanisms of action. Classical transactivation—where PPARγ activates genes upon binding to PPAR response elements as a heterodimer with RXRα—is important in all three settings, as underscored by natural loss-of-function mutations in FPLD3 and loss- and gain-of-function mutations in tumors. Transrepression—where PPARγ alters gene expression independent of DNA binding—is particularly relevant in immune cells. Interestingly, gene translocations resulting in fusion of PPARγ with other gene products, which are unique to specific carcinomas, present a third mode of action, as they potentially alter PPARγ’s target gene profile. Improved understanding of the molecular mechanism underlying PPARγ activity in the complex regulatory networks in metabolism, cancer, and inflammation may help to define novel potential therapeutic strategies for prevention and treatment of obesity, diabetes, or cancer.
Since its discovery in the early 1990s by Tontonoz et al (1)., the nuclear receptor PPARγ, encoded by the PPARG gene on chromosome 3p25.2 in humans (Figure 1A) (2), has been recognized as the master regulator of adipose tissue biology. The human PPARG gene, encompassing 9 exons, generates four PPARG splice variants (PPARG1-4) encoding for two protein isoforms via differential promoter usage and alternative splicing (Figure 1B) (3). The mRNAs PPARG1, PPARG3, and PPARG4 all give rise to the PPARγ1 isoform. PPARγ1 is a 477 amino acid protein that is broadly expressed with relative high levels in the adipose tissue, liver, colon, heart, various epithelial cell types, and skeletal muscle. In addition, PPARγ1 is expressed in numerous cells of the immune system, including monocytes/macrophages, dendritic cells, and T lymphocytes. The PPARG2 mRNA transcript translates into the PPARγ2 isoform. PPARγ2, containing an additional 28 amino acids in its NH2-terminus, is almost exclusively expressed in adipose tissue. This isoform is also expressed in urothelial cells (4, 5), which are highly specialized transitional epithelial cells that line the organs of the urinary system, including the bladder, and in regulatory T cells (Tregs) and other T cell populations, albeit that total PPARγ expression is low in non-Tregs (6). Recently, a third and fourth PPARγ protein isoform, denoted as PPARγ1Δ5, and PPARγ2Δ5, respectively, have been reported (Figure 1B) (7). PPARγ2Δ5 is endogenously expressed in adipose tissue and lacks the entire ligand binding domain (LBD) due to physiological exon 5 skipping (7). The endogenous expression PPARγΔ5 positively correlates with body mass index (BMI) in overweight or obese and type 2 diabetic patients. The naturally occurring PPARγΔ5 isoforms impair the adipogenic potential of adipocyte precursor cells by dominant-negative inhibition of PPARγ, which possibly contributes to adipose tissue dysfunction in obesity (7).
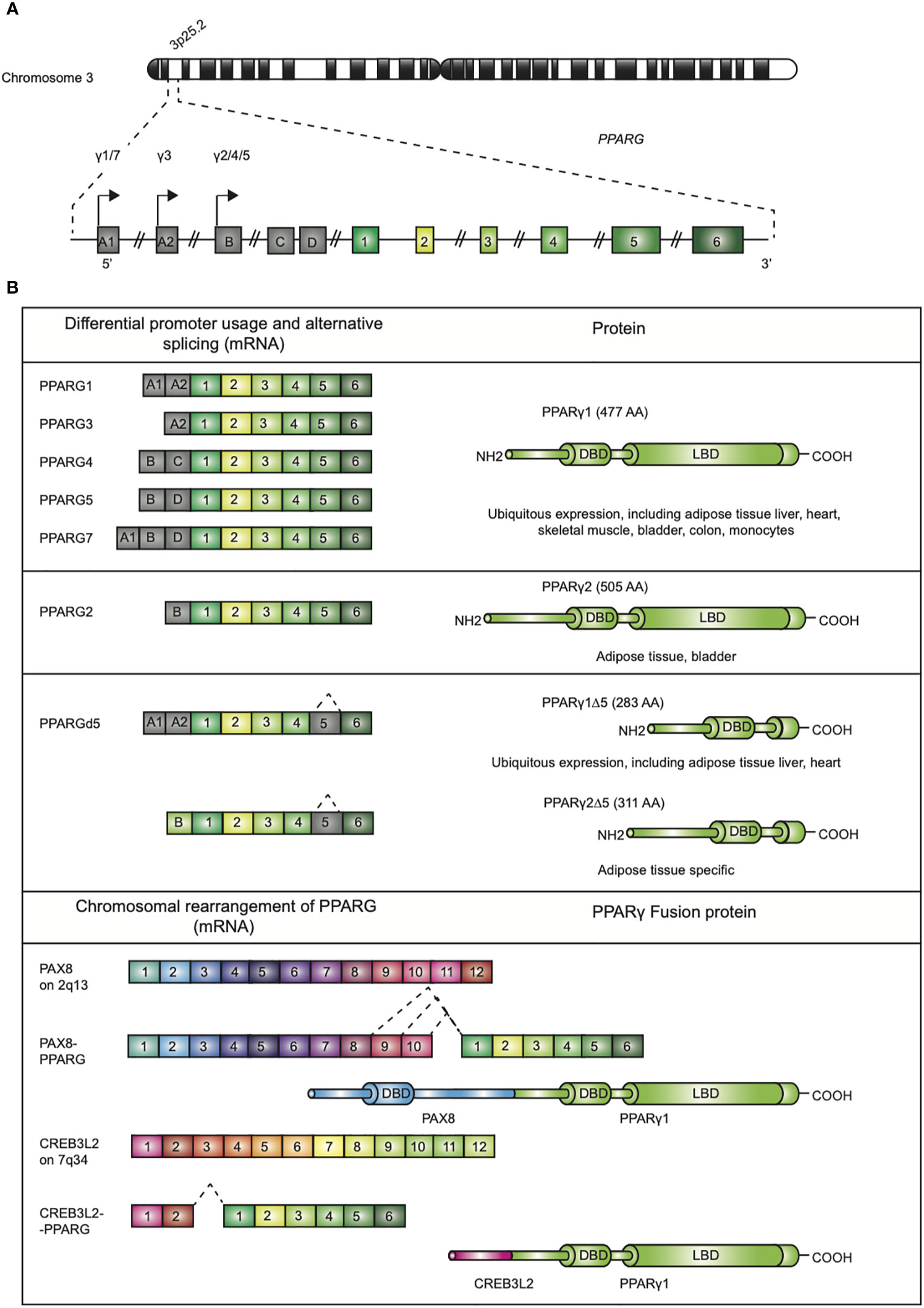
Figure 1 Genomic map of the PPARG gene on chromosome 3p25 and structure of PPARγ isoforms. (A) The gene PPARG is situated on chromosome 3p25. The gene encompassed 9 exons (exon A1-2, exon B-D, and exons 1-6). (B) Alternative promoter and mRNA splicing give rise to several PPARγ mRNA and protein isoforms. The mRNAs PPARG1, -3, and -4 translate into PPARγ1 (477 amino acids; AA). mRNA PPARG2 gives rise to PPARγ2 (505 AA). A third and fourth PPARγ protein isoform, denoted as PPARγ1Δ5 and PPARγ2Δ5, have been reported. These isoforms lack the ligand binding domain (LBD), which is due to alternative splicing. Chromosomal rearrangement of PPARγ leading to PAX8/PPARγ and CREB3L2/PPARγ fusion proteins, contains functional DBDs of both proteins, have been described in carcinogenesis.
PPARγ is a representative member of the nuclear receptor (NR) superfamily. To date, 48 NRs have been identified in human. NRs regulate various critical aspects in development, physiology, reproduction, and homeostasis. NRs are multi-domain ligand-inducible transcription factors that share a structural homology to a varying extent (8). Alike other NRs, PPARγ contains an autonomous transactivation domain 1 (AF-1) in the unstructured N-terminus (Figure 2). The AF-1 domain is implicated in the constitutive ligand-independent activation of PPARγ target genes. Juxtaposed to the AF-1 domains is the DNA binding domain (DBD) that contains two zinc fingers required for DNA binding. The DBD connected to the ligand binding domain (LBD) via a flexible hinge region. In the case of PPARγ, this hinge region physically interacts with the DNA (9). The ligand binding domain (LBD) is situated in the C-terminus. The LBD is a complicated structure that is arranged in a conserved three-layered α-helical sandwich containing 12 α-helices and 4 β-strand elements (8). The LBD overlaps with the ligand-dependent transactivation domain 2 (AF-2). The LBD is a key domain for transactivation of PPARγ target genes as it is implicated in ligand binding, heterodimerization with binding partner retinoid X receptor alpha (RXRα), and interactions with transcriptional co-regulators.
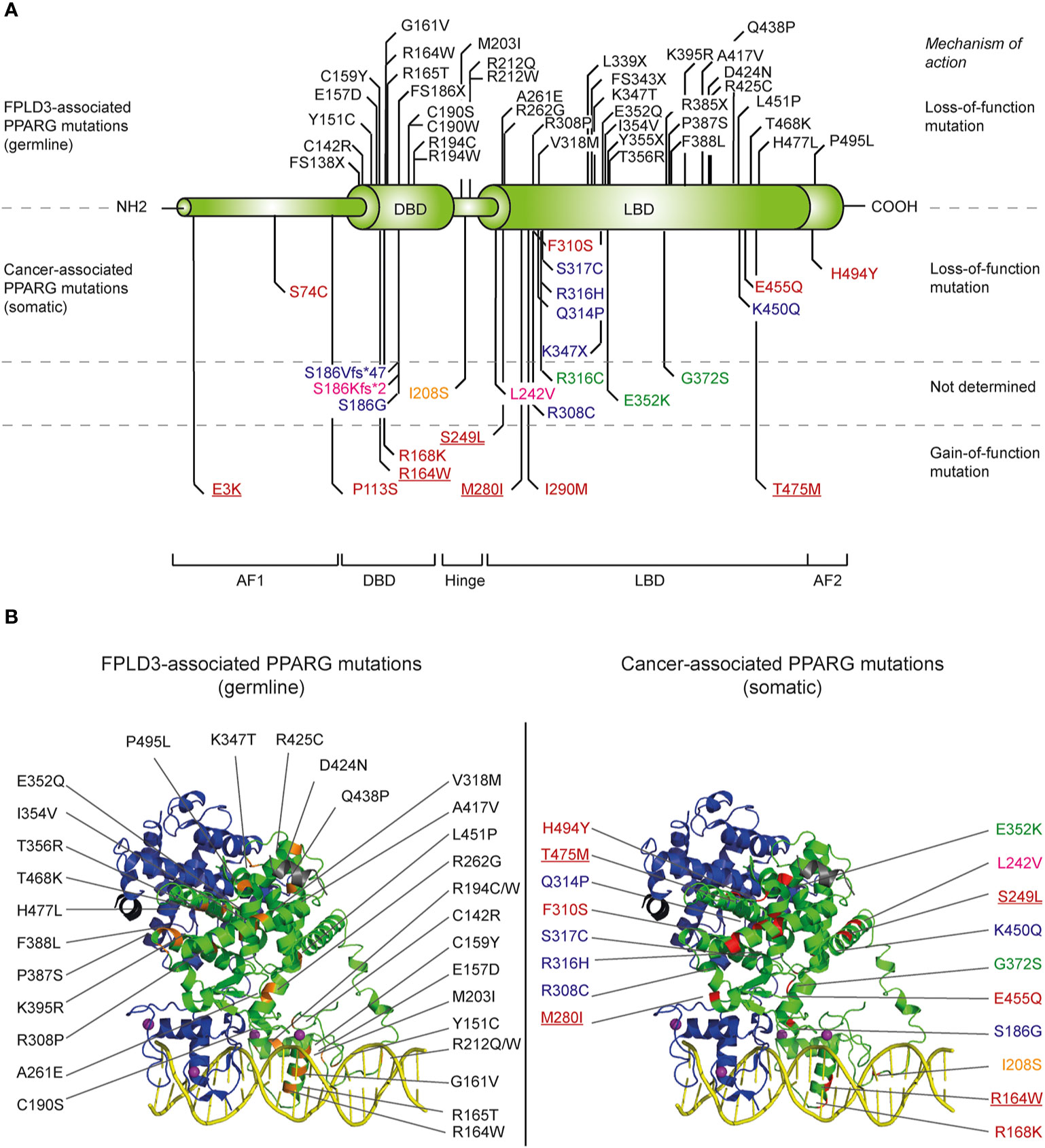
Figure 2 Overview of identified natural PPARG mutations implicated in FPLD3 and cancer. (A) Schematic representation of the distinct domains of PPARγ. Mutations indicated above the PPARγ structure are mutations are germline loss-of-function mutation, implicated in FPLD3. Mutations depicted below the PPARγ structure are somatic loss-of-function or gain-of-function mutations identified in different cancer types. Mutations have been identified in tissue form digestive tract (colon, stomach, oesophagus, and pancreas; indicated in blue), melanoma (green), breast cancer (pink), prostate cancer (yellow), and bladder cancer (red). Some bladder cancer-associated PPARγ mutations (underscored in figure) have also been identified in other types of cancer, including lung cancer (E3K), kidney cancer (R164W), endometrium cancer (S249L), melanoma (M280I), and diffuse glioma (T465M), respectively. (B) FPLD3 (orange, left panel) and cancer associated mutations (red, right panel) indicated in 3D representation, based on the crystal structure of PPARγ (green)-RXRα (blue) on DNA (yellow) with Rosiglitazone, 9-cis retinoic acid and NCOA2 peptide (grey) (PDB entry 3DZY).
PPARγ exerts its gene regulatory potential via transactivation and transrepression (Figure 3). Transactivation involves a mechanism by which PPARγ binds as a heterodimer complex with RXRα to PPAR response elements (PPREs) (10). PPREs consist of a hexameric repeat (AGGTCA) spaced by one or two nucleotides (referred to as DR1 and DR2 elements) (11), which are situated in promoter and enhancer regions of PPARγ target genes (12). Noteworthy, enhancers may not only loop to the nearest promoters, but can also increase transcription of their target genes via looping to promoters at greater genomic distances.
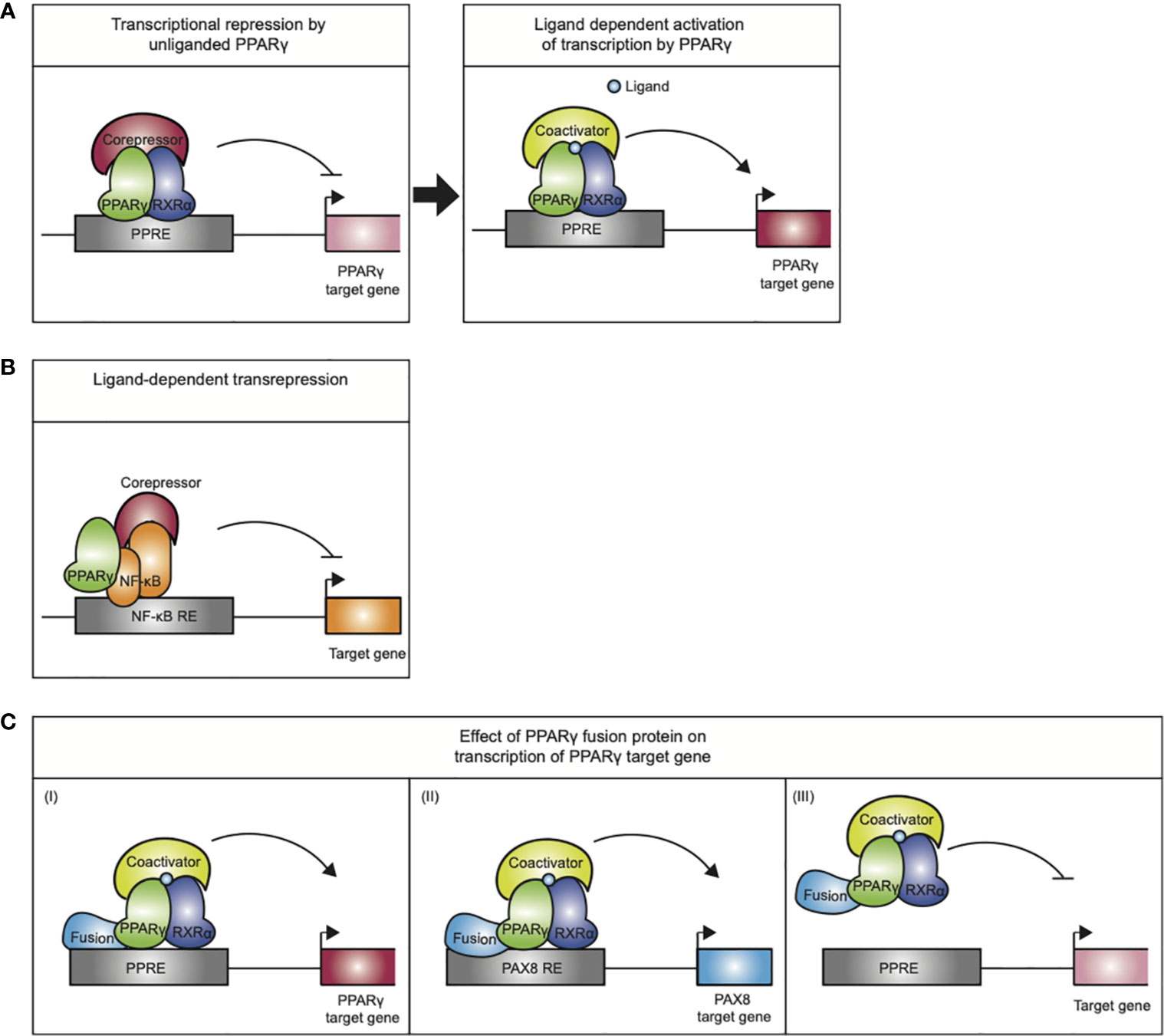
Figure 3 Mechanisms of action exerted by the PPARγ/RXRα heterodimer. (A) Transcriptional repression by unliganded PPARγ. Upon ligand binding the PPARγ/RXRα heterodimer undergoes a conformational change that promotes corepressor release and recruitment of coactivators, initiating transcription. (B) Ligand-dependent transrepression by antagonizing the NF-κB (and AP-1, not indicated) pro-inflammatory signaling pathways. This effect does not require DNA binding by the PPARγ. (C) The mode of action performed by PPARγ-fusion proteins in carcinogenesis is not completely understood. (I) altered expression of PPARγ target genes, (II) altered expression of PAX8 target genes (III) PPARγ fusion protein may act as a negative inhibitor of tumor suppression by inhibiting PPARγ target gene expression.
In the last decade, genome-wide binding profiles of PPARγ have been mapped in different cell types, including adipocytes and macrophages (13–17). These binding profiles have not only indicated that PPARγ binds to thousands of sites in the genome, of which many binding sites are located far from proximal promoters, but also that the PPARγ binding is highly context-dependent as binding sites differ between cell types and even between adipocytes from different anatomical locations (13–17). The context-dependency of PPARγ binding is at least in part mediated by cooperative binding to the chromatin with other adipogenic transcription factors, such as C/EBPα, followed by cooperative recruitment of coactivators (15).
Transcriptional control of the target genes by PPARγ furthermore depends on multiprotein coregulatory complexes that are recruited to the PPREs (18). In basal conditions, i.e., in absence of ligand, PPARγ/RXRα favors stable interactions with corepressor complexes, containing NCoR or SMRT, which recruit chromatin-modifying enzymes such as histone deacetylases that make the chromatin inaccessible to binding of transcription factors or resistant to their actions and thereby actively repress transcription (Figure 3A). Upon ligand binding, the PPARγ/RXRα heterodimer undergoes a conformational change that promotes corepressor release and recruitment of coactivators, like SRC1 and CBP. Coactivators enhance PPARγ transactivation by facilitating acetylation of the histone tails, making the chromatin less restrictive, and assembly of general transcriptional machinery. Next to the “classical” transactivation mechanism described above, PPARγ can also negatively regulate gene expression by a mechanism referred to as ligand-dependent transrepression (Figure 3B). This mechanism involves antagonizing the NF-κB and AP-1 pro-inflammatory signaling pathways, and has been mostly described in immune cells (19–23). In this case, PPARγ does not bind to DNA itself, and several studies indicate that PPARγ transrepresses genes as a monomer, i.e., independent of RXRα (23). While various mechanisms have been postulated for transrepression by different NRs (24–26), the most detailed mechanism proposed for PPARγ involves inhibition of co-repressor degradation. Pascual et al. (27) showed that clearance of NCoR/SMRT-HDAC3 complexes by proteosomal degradation from various AP1- and NFkB-regulated promoters (e.g., IL-8, Mmp12, and iNOS) upon activation is prevented in the presence of liganded, monomeric PPARγ.
Interestingly, the transrepression mechanism described above involves a specific post-translational modification, SUMOylation of lysine 365. In fact, to adequately processes external signals and adapt to relevant gene expression programs PPARγ activity is regulated by several, probably interconnected, post-translational modifications, including phosphorylation, acetylation, and the aforementioned SUMOylation [reviewed in (28)]. Depending on cellular context and the kinases involved, phosphorylation of PPARγ S112 can either impair or increase PPARγ activity (29). Phosphorylation of PPARγ S273 by Cdk5 does not affect its adipogenic capacity, but affects many PPARγ target genes that have been shown to be dysregulated in obesity (30). In addition, acetylation of K268 and K293 correlates with the phosphorylation status of S273 and favors lipid storage and cell proliferation (31). Selective adipocyte deletion of the deacetylase Sirt1 that deacetylates PPARγ K268 and K293 leads to dephosphorylation of S273 and improve metabolic functions (32).
Alike other NRs, PPARγ governs nutrient- and hormone-mediated responses. Despite intensive efforts, it is not clear whether PPARγ is in vivo activated by a specific, high-affinity, and endogenous ligand. PPARγ LBD crystal structures reveal a large ligand binding pocket (LBP), which not only allows for promiscuous binding of ligands with lower affinity, but also allows ligands to occupy the canonical LBP in different conformations (33). Indeed, the activity of PPARγ can be modulated by a variety of natural compounds, including polyunsaturated fatty acids (34), eicosanoids (35, 36), and oxidized lipid components (discussed below) (37), suggesting that PPARγ functions as a general lipid or nutrient sensor (34). However, the physiological relevance of these compounds is not exactly clear. Endogenous ligands not only bind with low affinity for PPARγ, also the physiological concentrations in mammalian cells are often insufficient to function as a physiological ligand (38). Alternatively, the physiological activation of PPARγ could be the resultant of combined effects of multiple ligands that simultaneously bind with different affinities to distinct subregions in the LBP (39), thereby inducing different PPARγ conformations with potential different biological outcomes (39).
PPARγ is the cognate receptor for thiazolidinediones (TZDs), a class of anti-hyperglycaemic drugs, including rosiglitazone and pioglitazone (40). TZDs stimulate adipogenesis (40) and cause a metabolically beneficial shift in lipid repartitioning from storage in visceral to subcutaneous adipose tissue depots as well as from ectopic storage in non-AT organs (e.g., liver muscle) to AT (41–43). TZDs and endogenous ligands have overlapping binding sites in the LBP, which potentially allows for binding competition to the same site. TZDs occupy the canonical LBP of PPARγ and by interacting with residues in helices 3, 5, 6, and 7 and the β-sheet, stabilizes the dynamics of helix 12 and the AF2 surface (44, 45).
Whereas TZDs are commonly referred to as full classical PPARγ agonists, TZDs have a separate biochemical activity: inhibition of the Cdk5-mediated phosphorylation of PPARγ at serine residue 273 (30). Phosphorylation of PPARγ S273 requires a physical interaction between CDK5 and PPARγ (46). The transcriptional corepressor NCoR is an adaptor protein for the physical interaction between CDK5 and PPARγ. Upon rosiglitazone the interaction between NCoR and PPARγ is reduced, which leads to i) derepression of PPARγ and activation of the PPARγ transcriptional program and ii) attenuation of the psychical interaction between CDK5 and PPARγ and subsequent reduced phosphorylation of S273 (46). Interestingly, MRL24 that displays poor agonistic activity but robust anti-diabetic activity in mice (47), was also very effective in inhibiting the Cdk5-mediated phosphorylation (30). This suggests that new classes of antidiabetic drugs that i) bind with high affinity to PPARγ, ii) specifically target the Cdk5-mediated phosphorylation of S273, and iii) completely lack the classical transcriptional agonism, hold promise for treatment of T2DM. The PPARγ ligand SR1664 was essentially displayed no transcriptional activity and was very effective in blocking the Cdk5-mediated phosphorylation (48). In obese mice, SR1664 displayed strong antidiabetic effects without adverse effects (48). However, unfavorable pharmacokinetic properties of SR1664 preclude its administration in human (48). Therefore, SR1664 should rather be considered as a proof-of-principle.
In addition to binding in the canonical LBP, a recent structure-function study shows that some PPARγ ligands denoted as noncanonical agonist ligands (NALs), like the aforementioned compound MRL24, and SR1664, can also bind to an alternate site of PPARγ (49). TZDs, including rosiglitazone and pioglitazone display less prominent alternate site functional effects (49). The alternate binding of PPARγ ligands can occur when the canonical LBP is occupied by the covalent antagonists or endogenous ligands. Although the exact mechanisms are not clear, alternate site binding stabilizes the AF2 surface, most likely indirectly via stabilization of helix 3. Furthermore, coregulator-binding assays indicate that alternate site binding has an impact on coregulator interactions, transactivation, and target gene expression (49). The identification of the alternate binding site has three important implications. Firstly, compounds that block phosphorylation of S273 with little transactivation might be complicated by alternate site binding if this site in vivo contributes to classical PPARγ agonism. Secondly, it needs to be defined whether some of the supposed PPARγ-independent effects of TZDs could in fact be mediated by the alternate site binding. Lastly, allosteric modulators that target the alternate site might be particularly relevant for obese individuals in which the probability that canonical LBP is occupied by oxidized fatty acids due to increased bioavailability of endogenous ligands is increased (49).
White, beige, and brown adipocytes have been identified in mammals. Although these three type of adipocytes rise from different precursors and differ significantly in their morphology and function, the cells all go through a well-orchestrated differentiation process to become mature and fully functional (50). During the various stages of the adipocyte lifespan, PPARγ is a well-established key player. Recently, a fourth type of adipocyte, denoted as pink adipocytes, has been described in in mammary glands of pregnant mice (51). During pregnancy, lactation, and post-lactation subcutaneous white adipocytes in murine mammary gland undergo a transdifferentiation process ending in milk-producing epithelial glandular cells that contain abundant cytoplasmic lipid droplets to meet the nutritional needs of the pups (51, 52). As the number of studies in pink adipocytes is limited so far, we will focus in this review on the role of PPARγ in white, brown, and beige adipocytes. In these cells, PPARγ exerts its essential functions primarily via “classical” transactivation of target genes.
White adipose tissue (WAT) is the most abundant adipose tissue in the human body (53). Mature white adipocytes are unilocular cells composed of a large lipid droplet occupying ~95% of the cellular volume. Depending on the size of the lipid droplet, the cell size varies from 20 to 200 µM (54). The in vivo regulation of adipocyte development, including the stem cell commitment toward white adipocytes, is poorly understood. Adipocyte-lineage tracing, which so far can only be performed in mice, indicate that white adipocytes can be derived from both Myf5− and Myf5+ precursor cells (55). The Myf5-lineage distribution in adipose tissue is dynamic and can be affected by ageing and diet. The Myf5− and Myf5+ white adipocytes can compensate for each other during development, reflecting adipose tissue plasticity (55). In mice, depot-dependent variations were observed among the degree of plasticity (55). Although it remains to be defined whether this concept also applies to human adipocytes, a heterogeneity in adipocyte origins may explain the heterogeneity in adipose tissue depot function and contribute to adipose tissue patterning variations in the human population (55). After stem cell commitment toward white adipocyte lineage, the expression and activation of PPARγ is both sufficient and crucial to initiate the adipogenic differentiation program and maintain adipocyte phenotype, integrity, and function, based on a large set of different genetic mouse models (56). PPARγ primarily regulates the expression of genes implicated in adipocyte differentiation and adipocyte maintenance. In addition, PPARγ governs the expression genes involved in various processes in lipid and glucose metabolism including lipogenesis (e.g., LPL, ANGTPL4, and CIDEC), fatty acid transport (e.g., FABP4), and gluconeogenesis (e.g., PEPCK, GYK, and AQP7).
The importance of PPARγ for white adipose tissue biology in humans is underscored in patients suffering from familial partial lipodystrophy subtype 3 (FPLD3), a rare autosomal dominant inherited condition caused by loss-of-function mutations in the PPARG gene [reviewed in (28)]. Patients with FPLD3 lack subcutaneous adipose tissue in the extremities and gluteal region combined with lipohypertrophy in the face, neck, and trunk, and suffer from multiple metabolic complications including type 2 diabetes mellitus (T2DM). Since the first report of a germline loss-of-function mutation in PPARG in patients with FPLD3 (57) an increasing number of FPLD3-associated mutations in PPARG has been identified [reviewed in (28)]. The FPLD3-associated PPARγ mutations are mainly situated in either the DBD or LBD (Figure 2). Mutations in the DBD interfere in efficient DNA binding. Mutations affecting the LBD—which are scattered over the whole LBD, based on crystal structures (Figure 2)—often cause multiple molecular defects by impairing heterodimerization with RXRα, ligand- and/or cofactor binding (18).
Taken together, genetic mouse models together with the FPLD3-associated PPARγ mutations indicate that PPARγ plays a key role in white AT differentiation, function, and maintenance. The dominant mode of action in this biological setting appears to be “classical” transactivation: the majority of genes regulated by PPARγ in white adipocytes rely on direct DNA binding, and FPLD3-associated PPARγ mutations do not alter transrepression, although this is not studied frequently (58).
Brown adipose tissue (BAT) emerged approximately 150 million years ago in mammals (59). BAT is unique for endothermic placental mammals and makes it possible to maintain a body temperature that is higher than the ambient temperature by producing heat independently of shivering and locomotor activity. This process is also referred to as non-shivering thermogenesis (59). BAT is richly innervated and vascularized and is composed of brown adipocytes (~40 µM in size) that contain multilocular lipid droplets and a large number of mitochondria (54). BAT derives its brown color from the conspicuous iron-rich mitochondrial mass. BAT uniquely expresses the gene UCP1, which encodes for uncoupling protein 1 (UCP1), located in the inner mitochondrial membrane. When activated, UCP1 mediates non-shivering thermogenesis by uncoupling of the oxidative phosphorylation from ATP synthesis, thereby provoking 1) dissipation of chemical energy in the form of heat and 2) stimulating high levels of fatty acid oxidation (60).
BAT is present in dedicated depots. In rodents, BAT is abundantly present throughout life. In human adults, BAT is located mainly cervical/axillary, perirenal/adrenal, and in the mediastinum along large blood vessels, trachea, and surrounding the intercostal arteries (59). In new-born infants, BAT is also situated between the shoulder blades as a thin kite-shaped layer (60). Although BAT depots regress with increasing age and can become even indistinguishable from WAT, healthy adults retain metabolically active BAT (61–63). For instance, positron emission tomography (PET) and computer tomography (CT) in human indicated that BAT-mediated thermogenesis is activated and increases in size by cold exposure (61–63). This process is also known as BAT recruitment. Depending on the size of the BAT depots, thermogenesis can account for up to approximately 15% of the total daily energy expenditure (64). Therefore, increasing energy expenditure by activation of BAT has been suggested as a therapeutic strategy for treating obesity (65).
Mice studies indicate that PPARγ functions is a master regulator in BAT (66). BAT-specific PPARγ knock out mice showed reduced wet weight of BAT, smaller brown adipocytes, and smaller lipid droplets when compare to wild type animals. However, there was no difference in total body weight or body composition (67). Furthermore, it was also shown that loss of PPARγ inhibited the ability of brown adipocytes to respond to β -adrenergic stimulus in in vitro cultures (67). An increase in non-shivering thermogenesis was observed in mice treated with TZDs (68, 69), and in vitro studies showed that activation of PPARγ in brown adipocytes leads to increase in adipogenesis and increase in lipid metabolism (70). Additional studies pointed at PPARγ as crucial regulator of UCP1 expression and BAT function (71). Specific BAT PPARγ target genes have been described (FABP3 and GYK), and particularly the de-acetylation of K268 and K293 of PPARγ by SIRT1 have been linked to BAT (32). De-acetylation of these residues is required for the recruitment of Prdm16, an essential cofactor in BAT (72). Moreover PGC1a, one of the most well-known regulators of BAT, has also been identified as a cofactor of PPARγ in BAT (73).
Collectively, PPARγ plays a key role in BAT differentiation and function, which most likely relies on “classical” transactivation, although transrepression cannot be excluded given the limited number of studies. BAT-specific molecular mechanisms, which may be different from WAT, could involve for example specific transcriptional cofactors (73), but details remain to be fully elucidated.
Mammals possess a second type of thermogenic adipocytes: beige adipocytes, also denoted as “brite” (brown-like in white) adipocytes (74). Beige adipocytes are inducible thermogenic cells that are sporadically located in white adipose tissue depots (74). Beige adipocytes share many morphological and biochemical features with brown adipocytes (Figure 1) (60). Alike brown adipocytes, beige adipocytes contain multiple small lipid droplets and a large number of mitochondria that express UCP1. Recruitment of beige adipocytes, referred to as “browning” or “beigeing/beiging” of white adipose tissue, is induced in response to environmental conditions, including chronic cold exposure, exercise, long-term treatment with PPARγ agonists or β3-adrenergic receptor agonists, cancer cachexia, and tissue injury (75). It is currently unknown whether beige adipocytes arise through transdifferentiation from pre-existing white adipocytes or by de novo adipogenesis from a precursor cell pool, or both (76).
Although, the exact mechanism by which PPARγ agonists induce browning of white adipocytes is not exactly known, PPARγ agonist require full agonism to activate the browning fat program. The effect is at least in part mediated by PRDM16, a factor that as described above is essential in the development of classical brown fat (77). Therefore, it is likely that in beige adipocytes, alike brown adipocytes, “classical” transactivation by PPARγ is an important mechanism of action.
Even though PPARγ is the master regulator of adipocyte differentiation and function (78), already in one of the first publications showed high PPARγ expression in mouse spleen (79) suggesting a role for PPARγ in immune cells. In fact, PPARγ is expressed in a variety of immune cells and its role and importance have been investigated during the last twenty years (80–82). Although PPARγ expression have been described in several types of immune cells we will focus on monocyte/macrophages and dendritic cells as part of the innate immune system, and T cells of the adaptative immune system.
As described above for adipocytes, PPARγ plays a role in determining the cellular phenotype by regulating differentiation (adipogenesis) and function (e.g., lipid metabolism and secretome) by directly activating the transcription of so-called PPARγ target genes. Similar molecular mechanisms are in place in immune cells, and also here PPARγ can deterimine cellular phenotype: amongst others, PPARγ 1) regulates macrophage differentiation, 2) regulates classical/alternative macrophage activation (“polarization”), 3) controls lipid metabolism in multiple immune cell types, and 4) plays an immune-modulatory role. PPARγ function in immune cells could also be categorized according to its mechanism of action, with the regulation of lipid metabolism and the ability to induce differentiation of immune cells more linked to “classical” transactivation, while the transrepression activity of PPARγ is more important in its immunomodulatory role and both mechanisms are involved in macrophage activation.
PPARγ can directly activate the transcription of target genes in immune cells through direct DNA binding, similar to its activity in adipocytes described above. As mentioned earlier, the genomic locations where PPARγ binds and the target genes partly overlap between, for example, adipocytes and macrophages, but cell-type specific regulation may depend on cooperation with other transcription factors like PU.1 and STAT6 (17, 83).
PPARγ expression is highly induced during monocyte to macrophage differentiation (84–86), and although initial studies using embryonic stem cells suggested that PPARγ is dispensable in this process (87), more recent studies have demonstrated that PPARγ is essential for the differentiation of fetal monocytes into alveolar macrophages (88). In mature macrophages, PPARγ was found to cooperate with PU1 specifically on monocyte-unique target genes (17), reminiscent of the interplay between PPARγ and C/EBPa in adipocytes mentioned earlier. PPARγ is also expressed in several dendritic cell (DC) subtypes and is also highly upregulated in monocyte-derived DC differentiation (89, 90). Although the importance of PPARγ in immune cell differentiation is evident, little is known about the exact function of the receptor in these differentiation processes. Better models are required as well as studying the contribution of PPARγ in a more cell-type specific way.
Next to macrophage differentiation, PPARγ is also an important regulator in macrophage polarization, where PPARγ activation drives the alternative M2 macrophage phenotype (91–93). Alternatively activated macrophages (M2 phenotype) can be induced by IL-4, IL-10, and IL-13 and are characterized by the expression of several genes including Arg1 and Mgl1/CD301a, CD-204 and mannose receptor/CD163, and IL-10 and transforming growth factor beta (TGF-β). Some of these, including Arg1 and Mgl1 (94), are direct PPARγ target genes. Furthermore, PPARγ expression is induced by IL-4/STAT6 signaling as well as IL-13 (95), and STAT6 functions as a “facilitator” of PPARγ signaling, all supporting the idea that PPARγ is crucial for the anti-inflammatory M2 phenotype in macrophages. It was recently found that PPARγ contributes to maintain a chromatin structure that facilitates the binding of STAT6 and polymerase II upon repeated IL-4 treatments. PPARγ recruits the coactivator P300 and RAD21 to the DNA and thus reinforcing a M2-like phenotype in macrophages (96), is worth mention that this function of PPARγ is independent of ligand binding.
Next to macrophage and DC differentiation and macrophage polarization, PPARγ can also directly regulate lipid metabolism in immune cells (37, 87, 92, 97, 98), reminiscent of its role in white and brown adipocytes. In monocytes, macrophages, and dendritic cells, PPARγ directly regulates the expression of genes involve in lipid transport and metabolism such as the class B scavenger receptor CD36 (99), FABP4, LXRA, and PGAR (86). The use of PPARγ ligands in these cells has shown that the expression of these genes is upregulated upon treatment and downregulated when treated with PPARγ antagonists (100). The CD36 protein is also involved in macrophage uptake of oxLDL, but at the same time PPARγ directly activates an LXR-ABCA1 pathway for cholesterol efflux (97). In DCs PPARγ also plays a key role in lipid homeostasis by directly regulating many “known suspects” (101) but it also regulates another aspect of lipid homeostasis and lipid antigen presentation. Activation of PPARγ gives higher expression of CD1d, a molecule involved in the presentation of lipid antigens to T cells, resulting in a DC subtype with increased potential to activate iNKT cells (100, 102, 103). These findings indicate that PPARγ has a functional role in the modulation of the immune response through DCs beyond regulation of more classical lipid metabolism pathways.
Changes in the lipid microenvironment can trigger different DC functions that regulate the immune response (104). PPARγ classical transactivation role bridges the lipid microenvironment and the DC function by activating genes involve in lipid transport, metabolism, and presentation.
The classical role of PPARγ as a gene activator has also been studied in T cells and again relates to lipid metabolism (81, 82). T cells can be subdivided into cytotoxic T cells, T helper, and regulatory T cells (Treg), and the T helper cells can be further classified depending on the phenotype into Th1, Th2, and Th17; less well characterized are Th9 and Th22 subsets. Regardless of the subtype of T cell, activation of PPARγ is linked to an activation of genes related to lipid metabolism (CD36 and FABPs) indicating the importance of PPARγ in this process. Special mention deserves the visceral adipose tissue resident regulatory T cells (VAT Tregs), in which PPARγ has been implicated in its function and development (6). VAT Tregs represents a unique subtype of cells in which the expression of PPARγ positively correlates with the expression of chemokines and chemokines receptors (Ccr2, Cxcl3, and Cxcr6) that regulates leukocyte migration and infiltration, lipid metabolism genes, and IL10. Interestingly, the PPARγ1 and PPARγ2 isoforms induce the same genes upon activation in VAT Tregs (mainly related to lipid metabolism) but differ in the genes that they downregulate (6), the latter happening most likely through the mechanism of transrepression.
The role of PPARγ as an immune-modulator, and in particular a repressor of inflammation, has been studied in most detail in macrophages and T cells (19–22, 93). Although the transrepression activity of PPARγ is probably not exclusive to immune cells, this immunomodulatory role is a good example of the importance of this specific mechanism of action of PPARγ.
In macrophages it has been shown that activation of PPARγ using TZDs suppresses the production of pro-inflammatory cytokine, such as TNFα, IL-1B, and IL-6 (19, 93) and the expression of other genes involved in inflammation, including iNOS and MMP9, in a dose-dependent manner. As described above, inhibition of the transcription factors NFkB and AP-1 is the most widely studied mechanism, but other mechanisms are also possible (23). Similarly, in DCs PPARγ ligands downregulate chemokines and receptors (IL-12, CD80, CXCL10, RANTES) that recruit Th1 lymphocytes (100, 102). In addition, PPARγ activation in DC may impair the migration of these cells to the lymph nodes, and this might be partially due to inhibition of CCR7 by PPARγ (102, 105).
The role of transrepression by PPARγ in T cells has been the object of intensive discussion during the last two decades (81, 82, 106), as this mechanism of action was implicated in seemingly conflicting biological processes. Initial studies suggested that PPARγ had an inhibitory effect on T cell proliferation (107), and that the underlying mechanism involved transrepression of the IL2 gene: activated PPARγ was shown to bind to nuclear factor of activated T cells (NFAT) and repress its activity and binding to the IL-2 promotor (107, 108). Besides T cell proliferation, PPARy-mediated transrepression was reported as a repressor of excessive Th1 response, by on the one hand inhibiting production of the Th1 cytokine and antigen-specific proliferation and on the other hand controlling Th2 sensitivity to IL-33 (109, 110). In fact, Cunard and colleagues showed that PPARγ binds to the IFNγ promoter and is able to repress its expression when T cell were treated with PPARγ ligands, and that IFNγ expression was enhanced when cells were treated with PPARγ antagonist GW9662 (111). The underlying mechanism was proposed to be inhibition of AP-1 activity, similar to the transrepression mechanism in macrophages. However, while these studies suggest a pro-Th2 role for PPARy mediated transrepression, PPARγ was also reported to be involved in the downregulation of well-known Th2 cytokines like IL-4, IL-5, and IL-13, again through interaction with NFAT (112). Altogether, these studies indicate that the role of PPARγ in the modulation of the Th2 response in T cells remains unclear and further research is needed to fully elucidate its function. Finally, PPARy-mediated repression is important for Th17 differentiation, as lack of PPARγ leads to increased Th17 differentiation while activation of PPARγ was shown to have inhibitory effects (22). PPARγ recruits NCoR and SMRT to the Rorc promoter, thereby inhibiting IL-17a expression, and blocks IL-6 signaling by inhibiting the DNA binding activity of STAT3 (20, 21).
In summary, transrepression by PPARγ—where it counteracts other transcription factors like NFkB, AP-1, NFAT, and STAT3—may be a major molecular mechanism that drives the functional phenotype(s) and secretory output of macrophages, dendritic cells, and T cells. Findings in T cells appear sometimes conflicting, which makes it difficult to assign a clear pro-Th1 or pro-Th2 role to PPARy activation. It also indicates that the use of ligands in these cells might “hide” some of the PPARγ functions and more subtle approaches, such as the use of cels harboring specific PPARγ mutations or selective PPARγ modulators, must be used in order to fully elucidated PPARγ role in immune cells, taking the complex interactions between immune cell population into account.
Cancer is driven by the acquisition of genome instability. The cancer genome landscape contains an enormous diverse repertoire of amplifications, deletions, inversions, translocations, point mutations, loss of heterozygosity, and epigenetic changes that collectively result in tumorigenesis. The role of PPARγ in tumorigenesis is controversial. A large body of evidence suggests that PPARγ functions as a tumor suppressor, as activation of the PPARγ/RXRα signaling pathway in different types of cancer, including colon (113), lung (114, 115), pancreatic (116), prostate (117), and breast (118, 119) cancers, leads to inhibition of cell growth, decreased tumor invasiveness, and reduced production of proinflammatory cytokines. In addition, treatment with TZDs was shown to increase sensitivity to chemotherapy through downregulation of Metallothionein genes (120) and/or endotrophin (121), which may be linked to ligand-mediated prevention of S273 phosphorylation (122).
Furthermore, in lung cancer cells, a tumor suppressive function of PPARγ was contributed metabolic reprogramming (123), an essential biochemical adaptation required for cancer viability that is considered to be a crucial emerging hallmark of cancer (124). In contrast, a protumorigenic role for PPARγ has been suggested in a variety of cancers as well (5, 125, 126). Here, we will discuss several loss-of-function and gain-of-function mechanisms by which PPARγ can be implicated in tumor initiation and progression in several major cancers. In addition, we will address the yet partly undefined role of PPARγ fusion proteins in cancer.
As discussed above, the PPARγ1 isoform is highly expressed in colon epithelial cells. The role of PPARγ in the development of normal colon epithelium and colorectal cancers is not completely understood and seems to be dual. The growth and differentiation of many colorectal cancers can be considerably inhibited upon ligand activation of PPARγ1 (113). This finding suggests that PPARγ functions as a tumor suppressor during colorectal carcinogenesis. In line with this, somatic PPARG mutations have been reported in ~8% of sporadic colorectal cancers (Figure 2). Genetic and epigenetic phenomena due to genetic alterations in other genes, like RAS, can further decrease PPARγ function in colon cancer. Activating mutations in RAS for example can result in hyperactivation of ERK1/2 and JNK pathways and ultimately impair PPARγ activity (28). Whereas all FPLD3-associated PPARG mutations that have been reported to date lead to mutant proteins that show a consistent and profound impairment in the transcriptional activity of PPARγ, the functional effects of colon cancer-associated PPARG mutations vary considerably (127). So far, six unique somatic PPARG mutations in colorectal cancers have been reported (128, 129). A side-by-side analysis of these colon-cancer associated mutants with some FPLD3-associated PPARγ mutants, shows that the colon-cancer associated mutants do not consistently display profound intra- and/or intermolecular defects (127). Moreover, while the abovementioned studies suggest that PPARγ functions as a tumor suppressor during colorectal carcinogenesis, it should be noted that other studies suggest that PPARγ activation increases the risk of developing colorectal cancer. Ligand-activation of PPARγ in min mice, an animal model for familial adenomatous polyposis due to mutations in the APC gene, results in a considerably greater number of polyps in the colon (125). Follow-up studies are clearly needed to reconcile these apparently conflicting findings and assign a clear role to PPARy in colon cancer.
In basal bladder tumors, four non-recurrent loss-of-function PPARγ mutations (S74C, F310S, E455Q, and H494Y, Figure 2) have been identified (130). All four PPARγ mutants display significantly reduced transcriptional activities. Biochemical and biophysical analysis of amino acid residues F310 and H494, situated in helix 3 and 12, respectively, indicated that both residues are essential for proper stabilization of helix 12. F310S and H494Y favor an inactive conformation, impairing both a proper release of corepressors and recruitment of coactivators (130). Basal tumors rely on EGFR signaling for growth (131). Interestingly, in basal cell lines the overexpression of wildtype but not H494Y, downregulates EGFR signaling.
Although the cancer-related PPARγ mutants—which are mainly scattered throughout the LBD (Figure 2)—may display variable and more subtle, i.e., context-dependent, intra- and/or intermolecular defects than the FPLD3-associated PPARγ, the cancer-related PPARγ mutants (Figure 2) are impaired in their ability to exert “classical” transactivation.
In addition to its well-established role as master regulator in adipocyte biology, PPARγ has also been shown to be involved in the terminal differentiation of urothelium (4), a layer of specialized epithelial cells lining the lower urinary tract. However, little is known about its function in the bladder and in the pathogenesis of bladder cancer. In 12–17% of the muscle-invasive bladder carcinomas (MIBC) and in 10% of the non-muscle-invasive bladder carcinomas, PPARγ focal amplifications leading to PPARγ overexpression have been reported, suggesting a role for PPARγ in the initiation and maintenance of bladder cancer. MIBC are biologically heterogeneous and can further be grouped into basal and luminal subtypes (132). PPARγ has a protumorigenic role in luminal MIBCS, as the loss of PPARγ expression impairs the bladder cancer cell viability (133). These luminal tumors maintain molecular urothelial differentiation, even in the loss of morphological differentiation (133). This molecular differentiation depends on PPARγ (133).
In approximately 5% of the MIBCs and the luminal subgroup of MIBCs hotspot mutations of RXRα (S427F/Y) has been identified. These RXRα mutations rely on the introduction of an aromatic amino acid residue that enhances the ligand-independent activation of PPARγ (134). Tumors harboring RXR S427F/Y display enhanced expression of genes implicated in adipogenesis and lipid metabolism, including ACOX1, ACSL1, ACSL5, and FABP4 (135). In addition, the RXRα hotspot mutations stimulate the proliferation of urothelial organoids, render bladder tumor cell growth PPARγ-dependent, and favor tumor evasion by the immune system.
Recently, seven recurrent driver gain-of-function PPARγ mutations have been identified in luminal bladder tumors (E3K, S249L, M280I, K164W, and T475M) (5). The mutations occur throughout the protein, affecting the N-terminus, DNA-binding domain, and ligand-binding domain (Figure 2). One recurrent mutation (E3K) was specific to the PPARγ isoform as it was situated in the N-terminal end. Functional analysis indicates that five mutations promote the transcriptional activity of PPARγ, which renders PPARγ-dependence to the cells. The three recurrent LBD-mutations promote, in absence of PPARγ ligands, the adoption of the active conformation of PPARγ by stabilizing helix 12 and induce recruitment of co-activators. Interestingly, four of the seven recurrent PPARγ mutations have also been identified in other types of cancer, including lung cancer, kidney cancer, cutaneous melanoma, and diffuse glioma (Figure 2) (5). Furthermore, other recurrent mutations that have not been identified in bladder cancer, have been identified in other types of cancer, including melanoma and prostate cancer (Figure 2) (5). Surprisingly, one of these recurrent PPARγ mutations, which are yet functionally uncharacterized, results in the same amino acid changes as FPLD3-associated loss-of-function PPARγ mutations (e.g., R164W and E352Q/K). This may indicate that a potential loss-of-function or gain-of-function effect is context dependent.
Although, not all recently identified gain-of-function PPARγ mutants have extensively been characterized and even affect different domains in the protein, at least some of the mutants have implications for “classical” transactivation of PPARγ target genes in bladder cancer.
Besides the loss- and gain-of-function mechanisms described above, a third way in which PPARy may be involved in carcinogenesis is represented by PPARG gene fusions observed in follicular thyroid carcinomas (FTCs). The t(2;3)(q13;p25) chromosomal translocation results in a PAX8/PPARG fusion gene that is detected in approximately 35% of FTCs and in a subset of follicular variant of papillary thyroid carcinomas (136). This chromosomal rearrangement is occasionally present in follicular adenomas as well (137). The gene paired-box gene 8 (PAX8) encodes for a member of the paired box (PAX) family of transcription factors and is a critical regulator in physiological thyroid development (138). In addition, PAX8 promotes the thyroid progenitor survival en in the mature thyroid it drives the expression thyroid specific genes, including genes encoding for thyroglobin and thyroid peroxidase (138, 139). The endogenous expression of PPARG in the thyroid is extremely low and it remains to be defined whether PPARγ has a physiological function in the thyroid (140). The translocation t(2;3)(q13;p25) results in a fusion transcript, driven by the PAX8 promoter, wherein most of the coding sequence of PAX8 is fused in-frame to the entire coding sequence of PPARγ1 (141). The PAX8-PPARγ fusion protein (PPFP) contains functional DBDs of both the PAX8 and PPARγ (142). In vitro and in vivo evidence indicates that the PAX8-PPARγ fusion protein can function as an oncoprotein i) by acting as a negative inhibitor of tumor suppressor PPARγ or as ii) a novel transcriptional factor with proto-oncogene activity. Nevertheless, the expression of PAX8-PPARG in FTCs does not affect prognosis (143).
A second chromosomal translocation, t(3;7)(p25;q34) resulting in a CREB3L2/PPARG fusion gene, is a low incidence fusion mutation that is found in <3% of the FTCs (144). The gene cAMP Responsive Element Binding Protein 3 Like 2 (CREB1L2) encodes for a member of the bZIP transcription factor family. The CREB3L2/PPARγ fusion protein consists of amino acids 1 to 106 of wildtype CREB3L2, a new glutamic acid at position 107 juxtaposed to the all 477 amino acids of wildtype PPARγ1 (144). The CREB3L2/PPARγ fusion protein stimulates cell growth of transduced primary thyroid cells by inducing proliferation (144). The fusion protein seems to be unresponsive to thiazolidinediones. In addition, CREB3L2/PPARγ interferes in the CRE-related transcription as overexpression of CREB3L2/PPARγ inhibits the transcription of native cAMP-responsive genes in normal thyroid cells (144). The impaired ability to stimulate transcription is consistent with the loss of CREB3L2 bZIP domain, implicated in dimerization and DNA binding, in the CREB3L2/PPARγ fusion protein (144). The oncogenic activities of the CREB3L2/PPARγ fusion protein are most likely (at least in part) due to 1) disruption one functional CREB3L2 allele and 2) inhibition of cAMP responsive genes by interfering in CREB3L2 DNA-binding (144).
Taken together, the PPARγ fusion proteins display a third mode of PPARγ action, as they potentially alter the target gene profile of both parent proteins in the chimeric protein (Figure 3C) and will target multiple signaling pathways implicated in cancer.
Since the identification of the PPARG gene in the early 1990s the role of PPARγ in cancer has extensively been studied in many different human cancer cells and animal models. However, the biological significance of PPARγ in cancer development and progression is far from completely understood and for some cancers appears to be even inconsistent and contradicting. At best, the overall conclusion from these studies is that the context, e.g., specific tumor type, tumor stage, and tumor microenvironment, determines the exact role and function of PPARγ in human cancer. Therefore, cell-culture studies are limited in representing the complex gene-gene and gene-environment molecular interactions that are implicated in cancer onset and progression.
For many years, PPARγ was referred to mainly as the master regulator of adipocyte function, and although its expression in the immune system was already described in early research, its actual role in these cells only became apparent later (Figure 4). Nowadays, the immunomodulatory role of PPARγ in several immune cells is well-established as described in this review. While PPARy clearly functions in gene transactivation in both adipocytes and immune cells, gene repression by PPARy has been predominantly investigated in immune cells. PPARγ has also emerged as a factor involved in cancer onset and progression of several cancer types in recent years. Also, in this case, transactivation mechanisms are clearly relevant, underscored by both loss-of-function and gain-of-function mutations. It should be noted however that no single unifying role for PPARy in human cancer emerges, and that transrepression has not always been studied specifically. Finally, gene fusions with other gene products (PAX8, CREB3L2) as reported in specific carcinoma presents a third way in which PPARy regulates gene expression, resulting in either altered target gene sets and/or loss of activation.
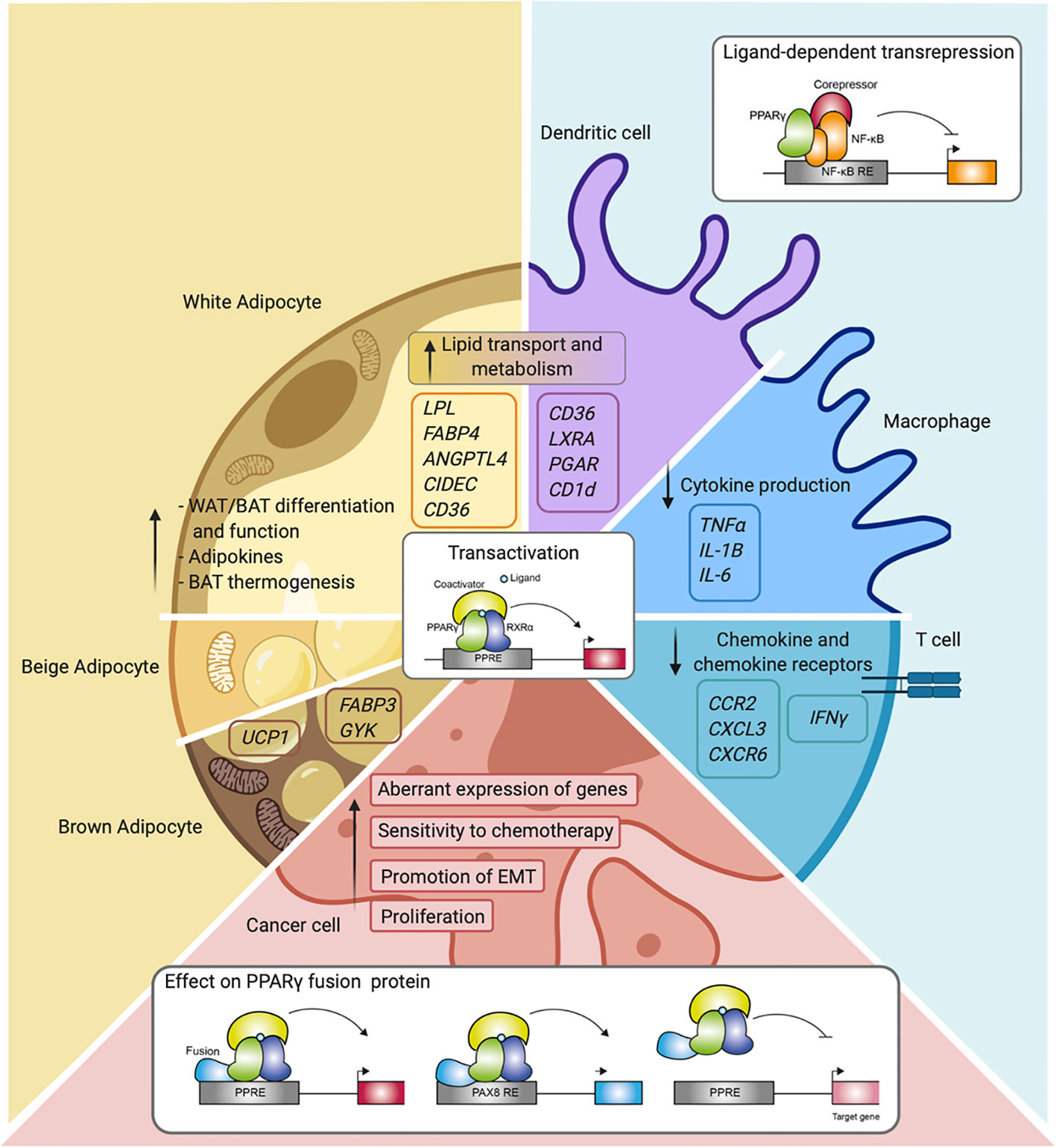
Figure 4 Overview of PPARγ function and mechanisms in the different cell types. Schematic representation of PPARγ in adipocytes (white, beige, and brown adipocytes), immune cells (macrophages, dendritic cells, and T cells), and cancer cells. Indicated are different cellular processes and mechanisms of action in which PPARγ is involved.
It is well known that PPARy is the molecular target for TZDs, these drugs have been widely used for the treatment of hyperglycemia and T2DM. TZDs stimulate the expression of genes implicated in lipid uptake and storage (145) and consequently the levels of ectopically stored and circulating lipids are decreased. In addition, TZDs also increase the expression of adiponectin, which contributes to enhance insulin sensitivity of the liver, and improves hepatic steatosis (145). Given its central role in adipocyte biology and energy homeostasis, there is a clear rationale behind therapeutically targeting PPARy and improving insulin sensitivity. However, the use of TZDs is curtailed due to serious side-effects [review in (146)]. Although some side-effects, such as troglitazone-associated hepatotoxicity and rosiglitazone-associated myocardial infarction have been solved (147), others are still present. These common side-effects include weight gain, fluid retention, and osteoporosis. These unwanted side-effects are due to the ubiquitous expression of PPARy1 in combination with the full agonism characteristics of TZDs. As indicated earlier, new generations of ligands, referred to as noncanonical agonist ligands (NALs) and selective PPARy modulators (SPPARMs), hold promise in that respect. In fact, very recently, it has been shown how selective modulators of PPARy can improve liver histology without affecting body weight in biopsy-confirmed mouse model of nonalcoholic steatohepatitis (NASH) (148).
Similar to being a potential drug target in metabolism, PPARy could represent a therapeutic target for a variety of cancers because of its ability to be selectively activated through its LBD. As indicated above, various parameters including tumor type and genetic background must be taken into account, as PPARy displays oncogenic and tumor suppressor roles. Nonetheless, targeting PPARγ in the cancer context can be effective. In pancreatic ductal adenocarcinoma for example, the fourth most frequent cause in cancer-related deaths, PPARγ ligands have shown promising results in vitro and in vivo increasing apoptosis and reducing tumor growth, respectively (149, 150).
While we have described above that PPARy is expressed in multiple cancer cell types, and PPARy ligands can affect cancer cell function and behavior (e.g., proliferation and sensitivity to chemotherapy), some of the anti-cancer effects may actually occur indirectly through adipocytes surrounding the tumor or distal adipose tissue. PPARγ plays a crucial role in AT, and as it has been shown before, AT influences cancer initiation and progression through several mechanisms (151). It is estimated that obesity contributes to up to 20% of cancer-related deaths. Obesity is associated with increased risk of cancer development (i.e., colorectal, post-menopausal breast, and kidney among others) but the association with poor prognosis is even stronger for some of these cancer types. Obese AT is characterized by a chronic low-grade inflammation that leads to dysfunctional adipocytes, metabolic dysregulation, and secretion of pro-inflammatory cytokines are some of the factors that have been correlated with increased risk of cancer death. A clear example of this is the adipokine endotrophin (152), a cleavage product of the collagen VIα3 chain. Endotrophin has been shown to promote tumor growth by enhancing the ability of breast cancer cells to undergo epithelial to mesenchymal transition (EMT) in mice and humans (153). Interestingly, TZDs have been shown to decrease levels of endotrophin in obese patients (154).
A second exciting option to consider when considering the use of PPARγ ligands in cancer treatment is the role of the receptor in epithelial to mesenchymal transition (EMT). Epithelial cells that undergo EMT in the primary tumor acquire crucial features that increase their invasiveness, migratory phenotype, and resistance to apoptosis that are essential for the development of metastasis (155). Transdifferentiation of breast cancer epithelial cells undergoing EMT into post-mitotic adipocytes cells using TZDs and MEK inhibitors have been shown to be a promising therapeutic approach to repress primary tumor invasion and metastasis formation (156). The ability of PPARγ to drive or inhibit EMT might be subjected to the specific cell type from which the tumor arises however, as for example different studies in lung cancer cells have shown PPARγ ligands to inhibit and promote EMT (157). More research is needed to study the implication of PPARγ in EMT to fully determine its role and if it can be a real cancer treatment option.
PPARγ plays a pivotal role in the crossroad between obesity, immunity, and cancer. Understanding the common and unique molecular mechanism underlying the function of PPARγ in these situations will allow the development of new therapies. In order to do so, some challenges have to be overcome; achieving a selective modulation of PPARγ and a cell-specific delivery of these modulators are two of them. In order to maximize the beneficial effects of targeting PPARγ, the key might be that PPARγ has to be targeted in one specific cell type, and not indiscriminately throughout the whole body. The use of nanoparticles coupled to biological ligands that binds to specific membrane receptors for drug delivery is a technique that is been study for cancer treatment and it could have a bright future in the nuclear receptor field if its proven successful. Given the different and complex roles of PPARy in metabolism, immunity, and cancer, which rely on overlapping and diverse mechsmisms of action, cell-specific delivery of PPARγ ligands, especially noncanonical agonist ligands (NALs) and selective PPARy modulators (SPPARMs), represent a promising field of study for future research.
MH-Q, MB, and EK drafted, edited, and revised the manuscript. All authors contributed to the article and approved the submitted version.
MH-Q was funded by the European Training Networks Project (H2020-MSCA-ITN-308 2016 721532).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM. mPPAR gamma 2: tissue-specific regulator of an adipocyte enhancer. Genes Dev (1994) 8(10):1224–34. doi: 10.1101/gad.8.10.1224
2. Greene ME, Blumberg B, McBride OW, Yi HF, Kronquist K, Kwan K, et al. Isolation of the human peroxisome proliferator activated receptor gamma cDNA: expression in hematopoietic cells and chromosomal mapping. Gene Expr (1995) 4(4–5):281–99.
3. Fajas L, Auboeuf D, Raspé E, Schoonjans K, Lefebvre A-M, Saladin R, et al. The Organization, Promoter Analysis, and Expression of the Human PPARγ Gene. J Biol Chem (1997) 272(30):18779–89. doi: 10.1074/jbc.272.30.18779
4. Varley CL, Stahlschmidt J, Lee W-C, Holder J, Diggle C, Selby PJ, et al. Role of PPAR γ and EGFR signalling in the urothelial terminal differentiation programme. J Cell Sci (2004) 117(10):2029–36. doi: 10.1242/jcs.01042
5. Rochel N, Krucker C, Coutos-Thévenot L, Osz J, Zhang R, Guyon E, et al. Recurrent activating mutations of PPARγ associated with luminal bladder tumors. Nat Commun (2019) 10(1):253. doi: 10.1038/s41467-018-08157-y
6. Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, et al. PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature (2012) 486(7404):549–53. doi: 10.1038/nature11132
7. Aprile M, Cataldi S, Ambrosio MR, D’Esposito V, Lim K, Dietrich A, et al. PPARγΔ5, a Naturally Occurring Dominant-Negative Splice Isoform, Impairs PPARγ Function and Adipocyte Differentiation. Cell Rep [Internet] (2018) 25(6):1577–1592.e6. doi: 10.1016/j.celrep.2018.10.035
8. Rastinejad F, Huang P, Chandra V, Khorasanizadeh S. Understanding nuclear receptor form and function using structural biology. J Mol Endocrinol (2013) 51(3):T1–21. doi: 10.1530/JME-13-0173
9. Chandra V, Huang P, Hamuro Y, Raghuram S, Wang Y, Burris TP, et al. Structure of the intact PPAR-γ–RXR-α nuclear receptor complex on DNA. Nature (2008) 456:350. doi: 10.1038/nature07413
10. Tontonoz P, Graves RA, Budavari AI, Erdjument-Bromage H, Lui M, Hu E, et al. Adipocyte-specific transcription factor ARF6 is a heterodimeric complex of two nuclear hormone receptors, PPARγ and RXRa. Nucleic Acids Res (1994) 22(25):5628–34. doi: 10.1093/nar/22.25.5628
11. Dreyer C, Krey G, Keller H, Givel F, Helftenbein G, Wahli W. Control of the peroxisomal β-oxidation pathway by a novel family of nuclear hormone receptors. Cell (1992) 68(5):879–87. doi: 10.1016/0092-8674(92)90031-7
12. Lefterova MI, Haakonsson AK, Lazar MA, Mandrup S. PPARγ and the global map of adipogenesis and beyond. Trends Endocrinol Metab (2014) 25(6):293–302. doi: 10.1016/j.tem.2014.04.001
13. Nielsen R, Grøntved L, Stunnenberg HG, Mandrup S. Peroxisome proliferator-activated receptor subtype- and cell-type-specific activation of genomic target genes upon adenoviral transgene delivery. Mol Cell Biol (2006) 26(15):5698–714. doi: 10.1128/MCB.02266-05
14. Nielsen R, Pedersen TAÅ, Hagenbeek D, Moulos P, Siersbaek R, Megens E, et al. Genome-wide profiling of PPARγ:RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis. Genes Dev (2008) 22(21):2953–67. doi: 10.1101/gad.501108
15. Madsen MS, Siersbæk R, Boergesen M, Nielsen R, Mandrup S. Peroxisome Proliferator-Activated Receptor γ and C/EBPα Synergistically Activate Key Metabolic Adipocyte Genes by Assisted Loading. Mol Cell Biol (2014) 34(6):939–54. doi: 10.1128/MCB.01344-13
16. Siersbaek MS, Loft A, Aagaard MM, Nielsen R, Schmidt SF, Petrovic N, et al. Genome-Wide Profiling of Peroxisome Proliferator-Activated Receptor γ in Primary Epididymal, Inguinal, and Brown Adipocytes Reveals Depot-Selective Binding Correlated with Gene Expression. Mol Cell Biol (2012) 32(17):3452–63. doi: 10.1128/MCB.00526-12
17. Lefterova MI, Steger DJ, Zhuo D, Qatanani M, Mullican SE, Tuteja G, et al. Cell-Specific Determinants of Peroxisome Proliferator-Activated Receptor γ Function in Adipocytes and Macrophages. Mol Cell Biol (2010) 30(9):2078–89. doi: 10.1128/MCB.01651-09
18. Jeninga EH, Gurnell M, Kalkhoven E. Functional implications of genetic variation in human PPARγ. Trends Endocrinol Metab (2009) 20(8):380–7. doi: 10.1016/j.tem.2009.04.005
19. Welch JS, Ricote M, Akiyama TE, Gonzalez FJ, Glass CK. PPARγ and PPARδ negatively regulate specific subsets of lipopolysaccharide and IFN-γ target genes in macrophages. Proc Natl Acad Sci U S A (2003) 100(11):6712–17. doi: 10.1073/pnas.1031789100
20. Li B, Reynolds JM, Stout RD, Bernlohr DA, Suttles J. Regulation of Th17 Differentiation by Epidermal Fatty Acid-Binding Protein. J Immunol (2009) 182(12):7625–33. doi: 10.4049/jimmunol.0804192
21. Wang LH, Yang XY, Zhang X, Huang J, Hou J, Li J, et al. Transcriptional Inactivation of STAT3 by PPARγ Suppresses IL-6-Responsive Multiple Myeloma Cells. Immunity (2004) 20(2):205–18. doi: 10.1016/S1074-7613(04)00030-5
22. Klotz L, Burgdorf S, Dani I, Saijo K, Flossdorf J, Hucke S, et al. The nuclear receptor PPARγ selectively inhibits Th17 differentiation in a T cell-intrinsic fashion and suppresses CNS autoimmunity. J Exp Med (2009) 206(10):2079–89. doi: 10.1084/jem.20082771
23. Ricote M, Glass CK. PPARs and molecular mechanisms of transrepression. Biochim Biophys Acta - Mol Cell Biol Lipids (2007) 1771(8):926–35. doi: 10.1016/j.bbalip.2007.02.013
24. Chinenov Y, Gupte R, Rogatsky I. Nuclear receptors in inflammation control: repression by GR and beyond. Mol Cell Endocrinol (2013) 380(1–2):55–64. doi: 10.1016/j.mce.2013.04.006
25. Pascual G, Glass CK. Nuclear receptors versus inflammation: mechanisms of transrepression. Trends Endocrinol Metab [Internet] (2006) 17(8):321–7. doi: 10.1016/j.tem.2006.08.005
26. Hollman DAA, Milona A, van Erpecum KJ, van Mil SWC. Anti-inflammatory and metabolic actions of FXR: Insights into molecular mechanisms. Biochim Biophys Acta - Mol Cell Biol Lipids [Internet] (2012) 1821(11):1443–52. doi: 10.1016/j.bbalip.2012.07.004
27. Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature (2005) 437(7059):759–63. doi: 10.1038/nature03988
28. Broekema MF, Savage DB, Monajemi H, Kalkhoven E. Gene-gene and gene-environment interactions in lipodystrophy: Lessons learned from natural PPARγ mutants. Biochim Biophys Acta - Mol Cell Biol Lipids (2019) 1864(5):715–32. doi: 10.1016/j.bbalip.2019.02.002
29. van Beekum O, Fleskens V, Kalkhoven E. Posttranslational modifications of PPAR-gamma: fine-tuning the metabolic master regulator. Obesity (Silver Spring) (2009) 17(2):213–9. doi: 10.1038/oby.2008.473
30. Choi JH, Banks AS, Estall JL, Kajimura S, Bostrom P, Laznik D, et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature (2010) 466(7305):451–6. doi: 10.1038/nature09291
31. Qiang L, Wang L, Kon N, Zhao W, Lee S, Zhang Y, et al. Brown Remodeling of White Adipose Tissue by SirT1-Dependent Deacetylation of Pparγ. Cell (2012) 150(3):620–32. doi: 10.1016/j.cell.2012.06.027
32. Mayoral R, Osborn O, McNelis J, Johnson AM, Oh DY, Izquierdo CL, et al. Adipocyte SIRT1 knockout promotes PPARγ activity, adipogenesis and insulin sensitivity in chronic-HFD and obesity. Mol Metab [Internet] (2015) 4(5):378–91. doi: 10.1016/j.molmet.2015.02.007
33. Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, Kurokawa R, et al. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature (1998) 395(6698):137–43. doi: 10.1038/25931
34. Kliewer SA, Sundseth SS, Jones SA, Brown PJ, Wisely GB, Koble CS, et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors α and γ. Proc Natl Acad Sci (1997) 94(9):4318–23. doi: 10.1073/pnas.94.9.4318
35. Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-Deoxy-Δ12,14-Prostaglandin J2 is a ligand for the adipocyte determination factor PPARγ. Cell (1995) 83(5):803–12. doi: 10.1016/0092-8674(95)90193-0
36. Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor γ and promotes adipocyte differentiation. Cell (1995) 83(5):813–9. doi: 10.1016/0092-8674(95)90194-9
37. Nagy L, Tontonoz P, Alvarez JGA, Chen H, Evans RM. Oxidized LDL Regulates Macrophage Gene Expression through Ligand Activation of PPARγ. Cell (1998) 93(2):229–40. doi: 10.1016/S0092-8674(00)81574-3
38. Bell-Parikh LC, Ide T, Lawson JA, McNamara P, Reilly M, FitzGerald GA. Biosynthesis of 15-deoxy-Δ12,14-PGJ2 and the ligation of PPARγ. J Clin Invest (2003) 112(6):945–55. doi: 10.1172/JCI200318012
39. Balint BL, Nagy L. Selective Modulators of PPAR Activity as New Therapeutic Tools in Metabolic Diseases. Endocr Metab Immune Disord - Drug Targets (2006) 6:33–43. doi: 10.2174/187153006776056620
40. Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An Antidiabetic Thiazolidinedione Is a High Affinity Ligand for Peroxisome Proliferator-activated Receptor γ (PPARγ). J Biol Chem [Internet] (1995) 270(22):12953–6. doi: 10.1074/jbc.270.22.12953
41. Kelly IE, Han TS, Walsh K, Lean ME. Effects of a thiazolidinedione compound on body fat and fat distribution of patients with type 2 diabetes. Diabetes Care (1999) 22(2):288–93. doi: 10.2337/diacare.22.2.288
42. Nakamura T, Funahashi T, Yamashita S, Nishida M, Nishida Y, Takahashi M, et al. Thiazolidinedione derivative improves fat distribution and multiple risk factors in subjects with visceral fat accumulation—double-blind placebo-controlled trial. Diabetes Res Clin Pract (2001) 54(3):181–90. doi: 10.1016/S0168-8227(01)00319-9
43. Akazawa S, Sun F, Ito M, Kawasaki E, Eguchi K. Efficacy of troglitazone on body fat distribution in type 2 diabetes. Diabetes Care (2000) 23(8):1067–71. doi: 10.2337/diacare.23.8.1067
44. Kroker AJ, Bruning JB. Review of the Structural and Dynamic Mechanisms of PPARγ Partial Agonism. PPAR Res (2015) 2015:816856. doi: 10.1155/2015/816856
45. Kojetin DJ, Burris TP. Small Molecule Modulation of Nuclear Receptor Conformational Dynamics: Implications for Function and Drug Discovery. Mol Pharmacol (2013) 83(1):1–8. doi: 10.1124/mol.112.079285
46. Li P, Fan W, Xu J, Lu M, Yamamoto H, Auwerx J, et al. Adipocyte NCoR Knockout Decreases PPARγ Phosphorylation and Enhances PPARγ Activity and Insulin Sensitivity. Cell (2011) 147(4):815–26. doi: 10.1016/j.cell.2011.09.050
47. Acton JJ, Black RM, Jones AB, Moller DE, Colwell L, Doebber TW, et al. Benzoyl 2-methyl indoles as selective PPARγ modulators. Bioorg Med Chem Lett (2005) 15(2):357–62. doi: 10.1016/j.bmcl.2004.10.068
48. Choi JH, Banks AS, Kamenecka TM, Busby SA, Chalmers MJ, Kumar N, et al. Antidiabetic actions of a non-agonist PPARγ ligand blocking Cdk5-mediated phosphorylation. Nature (2011) 477:477. doi: 10.1038/nature10383
49. Hughes TS, Giri PK, de Vera IMS, Marciano DP, Kuruvilla DS, Shin Y, et al. An alternate binding site for PPARγ ligands. Nat Commun (2014) 5:3571. doi: 10.1038/ncomms4571
50. Rosen ED, Spiegelman BM. What we talk about when we talk about fat. Cell (2014) 156(1–2):20–44. doi: 10.1016/j.cell.2013.12.012
51. Morroni M, Giordano A, Zingaretti MC, Boiani R, De Matteis R, Kahn BB, et al. Reversible transdifferentiation of secretory epithelial cells into adipocytes in the mammary gland. Proc Natl Acad Sci U S A (2004) 101(48):16801–6. doi: 10.1073/pnas.0407647101
52. Cinti S. Pink Adipocytes. Trends Endocrinol Metab (2018) 29(9):651–66. doi: 10.1016/j.tem.2018.05.007
53. Hausman DB, DiGirolamo M, Bartness TJ, Hausman GJ, Martin RJ. The biology of white adipocyte proliferation. Obes Rev (2001) 2(4):239–54. doi: 10.1046/j.1467-789X.2001.00042.x
54. Cedikova M, Kripnerová M, Dvorakova J, Pitule P, Grundmanova M, Babuska V, et al. Mitochondria in White, Brown, and Beige Adipocytes. Stem Cells Int (2016) 2016:6067349. doi: 10.1155/2016/6067349
55. Sanchez-Gurmaches J, Guertin DA. Adipocytes arise from multiple lineages that are heterogeneously and dynamically distributed. Nat Commun (2014) 5:4099. doi: 10.1038/ncomms5099
56. Rosen ED, Hsu C-H, Wang X, Sakai S, Freeman MW, Gonzalez FJ, et al. C/EBPα induces adipogenesis through PPARγ: a unified pathway. Genes Dev (2002) 16(1):22–6. doi: 10.1101/gad.948702
57. Barroso I, Gurnell M, Crowley VEF, Agostini M, Schwabe JW, Soos MA, et al. Dominant negative mutations in human PPAR gamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature (1999) 402(December):880–3. doi: 10.1038/47254
58. Jeninga EH, van Beekum O, van Dijk ADJ, Hamers N, Hendriks-Stegeman BI, Bonvin AMJJ, et al. Impaired peroxisome proliferator-activated receptor gamma function through mutation of a conserved salt bridge (R425C) in familial partial lipodystrophy. Mol Endocrinol [Internet] (2007) 21(5):1049–65. doi: 10.1210/me.2006-0485
59. Enerbäck S. Human Brown Adipose Tissue. Cell Metab (2010) 11(4):248–52. doi: 10.1016/j.cmet.2010.03.008
60. Enerback S. Brown adipose tissue in humans. Int J Obes (2010) 34:S43–6. doi: 10.1038/ijo.2010.183
61. van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JMAFL, Kemerink GJ, Bouvy ND, et al. Cold-Activated Brown Adipose Tissue in Healthy Men. N Engl J Med (2009) 360(15):1500–8. doi: 10.1056/NEJMoa0808718
62. Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, et al. Functional Brown Adipose Tissue in Healthy Adults. N Engl J Med (2009) 360(15):1518–25. doi: 10.1056/NEJMoa0808949
63. Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, et al. Identification and Importance of Brown Adipose Tissue in Adult Humans. N Engl J Med (2009) 360(15):1509–17. doi: 10.1056/NEJMoa0810780
64. van Marken Lichtenbelt WD, Schrauwen P. Implications of nonshivering thermogenesis for energy balance regulation in humans. Am J Physiol Integr Comp Physiol (2011) 301(2):R285–96. doi: 10.1152/ajpregu.00652.2010
65. Li Z, de Jonge WJ, Wang Y, Rensen PCN, Kooijman S. Electrical Neurostimulation Promotes Brown Adipose Tissue Thermogenesis. Front Endocrinol (Lausanne) (2020) 11:567545. doi: 10.3389/fendo.2020.567545
66. Nedergaard J, Petrovic N, Lindgren EM, Jacobsson A, Cannon B. PPARγ in the control of brown adipocyte differentiation. Biochim Biophys Acta - Mol Basis Dis (2005) 1740(2):293–304. doi: 10.1016/j.bbadis.2005.02.003
67. Lasar D, Rosenwald M, Kiehlmann E, Balaz M, Tall B, Opitz L, et al. Peroxisome Proliferator Activated Receptor Gamma Controls Mature Brown Adipocyte Inducibility through Glycerol Kinase. Cell Rep (2018) 22(3):760–73. doi: 10.1016/j.celrep.2017.12.067
68. Thurlby PL, Wilson S, Arch JRS. Ciglitazone is not itself thermogenic but increases the potential for thermogenesis in lean mice. Biosci Rep (1987) 7(7):573–7. doi: 10.1007/BF01119774
69. Mercer SW, Trayhurn P. Effects of ciglitazone on insulin resistance and thermogenic responsiveness to acute cold in brown adipose tissue of genetically obese (ob/ob) mice. FEBS Lett (1986) 195(1–2):12–6. doi: 10.1016/0014-5793(86)80120-X
70. Tai TAC, Jennermann C, Brown KK, Oliver BB, MacGinnitie MA, Wilkison WO, et al. Activation of the nuclear receptor peroxisome proliferator-activated receptor γ promotes brown adipocyte differentiation. J Biol Chem (1996) 271(47):29909–14. doi: 10.1074/jbc.271.47.29909
71. Rosen ED, Walkey CJ, Puigserver P, Spiegelman BM. Transcriptional regulation of adipogenesis. Genes Dev (2000) 14(11):1293–307. doi: 10.1101/gad.14.11.1293
72. Seale P, Kajimura S, Yang W, Chin S, Rohas LM, Uldry M, et al. Transcriptional Control of Brown Fat Determination by PRDM16. Cell Metab (2007) 6(1):38–54. doi: 10.1016/j.cmet.2007.06.001
73. Koppen A, Kalkhoven E. Brown vs white adipocytes: The PPARγ coregulator story. FEBS Lett (2010) 584(15):3250–9. doi: 10.1016/j.febslet.2010.06.035
74. Wu J, Boström P, Sparks LM, Ye L, Choi JH, Giang A-H, et al. Beige Adipocytes Are a Distinct Type of Thermogenic Fat Cell in Mouse and Human. Cell (2012) 150(2):366–76. doi: 10.1016/j.cell.2012.05.016
75. Ikeda K, Maretich P, Kajimura S. The Common and Distinct Features of Brown and Beige Adipocytes. Trends Endocrinol Metab (2018) 29(3):191–200. doi: 10.1016/j.tem.2018.01.001
76. Giralt M, Villarroya F. White, Brown, Beige/Brite: Different Adipose Cells for Different Functions? Endocrinology (2013) 154(9):2992–3000. doi: 10.1210/en.2013-1403
77. Ohno H, Shinoda K, Spiegelman BM, Kajimura S. PPARγ agonists induce a white-to-brown fat conversion through stabilization of PRDM16 protein. Cell Metab [Internet] (2012) 15(3):395–404. doi: 10.1016/j.cmet.2012.01.019
78. Tontonoz P, Spiegelman BM. Fat and beyond: The diverse biology of PPARγ. Annu Rev Biochem (2008) 77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829
79. Kliewer SA, Forman BM, Blumberg B, Ong ES, Borgmeyer U, Mangelsdorf DJ, et al. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc Natl Acad Sci U S A (1994) 91(15):7355–9. doi: 10.1073/pnas.91.15.7355
80. Széles L, Töröcsik D, Nagy L. PPARγ in immunity and inflammation: cell types and diseases. Biochim Biophys Acta - Mol Cell Biol Lipids (2007) 1771(8):1014–30. doi: 10.1016/j.bbalip.2007.02.005
81. Choi JM, Bothwell ALM. The nuclear receptor PPARs as important regulators of T-cell functions and autoimmune diseases. Molecules Cells (2012) 33(3):217–22. doi: 10.1007/s10059-012-2297-y
82. Le Menn G, Neels JG. Regulation of immune cell function by PPARs and the connection with metabolic and neurodegenerative diseases. Int J Mol Sci (2018) 19(6):1575. doi: 10.3390/ijms19061575
83. Szanto A, Balint BL, Nagy ZS, Barta E, Dezso B, Pap A, et al. STAT6 transcription factor is a facilitator of the nuclear receptor PPARγ-regulated gene expression in macrophages and dendritic cells. Immunity (2010) 33(5):699–712. doi: 10.1016/j.immuni.2010.11.009
84. Ricote M, Huang J, Fajas L, Li A, Welch J, Najib J, et al. Expression of the peroxisome proliferator-activated receptor γ (PPARγ) in human atherosclerosis and regulation in macrophages by colony stimulating factors and oxidized low density lipoprotein. Proc Natl Acad Sci U S A (1998) 95(13):7614–9. doi: 10.1073/pnas.95.13.7614
85. Moore KJ, Rosen ED, Fitzgerald ML, Randow F, Andersson LP, Altshuler D, et al. The role of PPAR-γ in macrophage differentiation and cholesterol uptake. Nat Med (2001) 7(1):41–7. doi: 10.1038/83328
86. Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM. PPAR gamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. PPARg promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell (1998) 93(2):241–52. doi: 10.1016/s0092-8674(00)81575-5
87. Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM. PPAR-γ dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med (2001) 7(1):48–52. doi: 10.1038/83336
88. Schneider C, Nobs SP, Kurrer M, Rehrauer H, Thiele C, Kopf M. Induction of the nuclear receptor PPAR-γ 3 by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat Immunol (2014) 15(11):1026–37. doi: 10.1038/ni.3005
89. Gosset P, Charbonnier AS, Delerive P, Fontaine J, Staels B, Pestel J, et al. Peroxisome proliferator-activated receptor γ activators affect the maturation of human monocyte-derived dendritic cells. Eur J Immunol (2001) 31(10):2857–65. doi: 10.1002/1521-4141(2001010)31:10<2857::AID-IMMU2857>3.0.CO;2-X
90. Le Naour F, Hohenkirk L, Grolleau A, Misek DE, Lescure P, Geiger JD, et al. Profiling Changes in Gene Expression during Differentiation and Maturation of Monocyte-derived Dendritic Cells Using Both Oligonucleotide Microarrays and Proteomics. J Biol Chem (2001) 276(21):17920–31. doi: 10.1074/jbc.M100156200
91. Bouhlel MA, Derudas B, Rigamonti E, Dièvart R, Brozek J, Haulon S, et al. PPARγ Activation Primes Human Monocytes into Alternative M2 Macrophages with Anti-inflammatory Properties. Cell Metab (2007) 6(2):137–43. doi: 10.1016/j.cmet.2007.06.010
92. Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature (2007) 447(7148):1116–20. doi: 10.1038/nature05894
93. Jiang C, Ting AT, Seed B. PPAR-γ agonists inhibit production of monocyte inflammatory cytokines. Nature (1998) 391(6662):82–6. doi: 10.1038/34184
94. Gallardo-Soler A, Gómez-Nieto C, Campo ML, Marathe C, Tontonoz P, Castrillo A, et al. Arginase I induction by modified lipoproteins in macrophages: A peroxisome proliferator-activated receptor-γ/δ-mediated effect that links lipid metabolism and immunity. Mol Endocrinol (2008) 22(6):1394–402. doi: 10.1210/me.2007-0525
95. Huang JT, Welch JS, Ricote M, Binder CJ, Willson TM, Kelly C, et al. Interleukin-4-dependent production of PPAR-γ ligands in macrophages by 12/15-lipoxygenase. Nature (1999) 400(6742):378–82. doi: 10.1038/22572
96. Daniel B, Nagy G, Czimmerer Z, Horvath A, Hammers DW, Cuaranta-Monroy I, et al. The Nuclear Receptor PPARγ Controls Progressive Macrophage Polarization as a Ligand-Insensitive Epigenomic Ratchet of Transcriptional Memory. Immunity (2018) 49(4):615–26.e6. doi: 10.1016/j.immuni.2018.09.005
97. Chawla A, Boisvert WA, Lee CH, Laffitte BA, Barak Y, Joseph SB, et al. A PPARγ-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell (2001) 7(1):161–71. doi: 10.1016/S1097-2765(01)00164-2
98. Chinetti G, Lestavel S, Bocher V, Remaley AT, Neve B, Torra IP, et al. PPAR-α and PPAR-γ activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat Med (2001) 7(1):53–8. doi: 10.1038/83348
99. Albert ML, Pearce SFA, Francisco LM, Sauter B, Roy P, Silverstein RL, et al. Immature dendritic cells phagocytose apoptotic cells via αvβ5 and CD36, and cross-present antigens to cytotoxic T lymphocytes. J Exp Med (1998) 188(7):1359–68. doi: 10.1084/jem.188.7.1359
100. Szatmari I, Gogolak P, Im JS, Dezso B, Rajnavolgyi E, Nagy L. Activation of PPARγ specifies a dendritic cell subtype capable of enhanced induction of iNKT cell expansion. Immunity (2004) 21(1):95–106. doi: 10.1016/j.immuni.2004.06.003
101. Szatmari I, Töröcsik D, Agostini M, Nagy T, Gurnell M, Barta E, et al. PPARγ regulates the function of human dendritic cells primarily by altering lipid metabolism. Blood (2007) 110(9):3271–80. doi: 10.1182/blood-2007-06-096222
102. Nencioni A, Grünebach F, Zobywlaski A, Denzlinger C, Brugger W, Brossart P. Dendritic Cell Immunogenicity Is Regulated by Peroxisome Proliferator-Activated Receptor γ. J Immunol (2002) 169(3):1228–35. doi: 10.4049/jimmunol.169.3.1228
103. Szatmari I, Pap A, Rühl R, Ma JX, Illarionov PA, Besra GS, et al. PPARγ controls CD1d expression by turning on retinoic acid synthesis in developing human dendritic cells. J Exp Med (2006) 203(10):2351–62. doi: 10.1083/JCB1751OIA1
104. Cuaranta-Monroy I, Kiss M, Simandi Z, Nagy L. Genomewide effects of peroxisome proliferator-activated receptor gamma in macrophages and dendritic cells - Revealing complexity through systems biology. Eur J Clin Invest (2015) 45(9):964–75. doi: 10.1111/eci.12491
105. Angeli V, Hammad H, Staels B, Capron M, Lambrecht BN, Trottein F. Peroxisome Proliferator-Activated Receptor γ Inhibits the Migration of Dendritic Cells: Consequences for the Immune Response. J Immunol (2003) 170(10):5295–301. doi: 10.4049/jimmunol.170.10.5295
106. Park BV, Pan F. The role of nuclear receptors in regulation of Th17/Treg biology and its implications for diseases. Cell Mol Immunol (2015) 12(5):533–42. doi: 10.1038/cmi.2015.021
107. Yang XY, Wang LH, Chen T, Hodge DR, Resau JH, DaSilva L, et al. Activation of human T lymphocytes is inhibited by peroxisome proliferator-activated receptor γ (PPARγ) agonists. PPARγ co-association with transcription factor NFAT. J Biol Chem (2000) 275(7):4541–4. doi: 10.1074/jbc.275.7.4541
108. Clark RB, Bishop-Bailey D, Estrada-Hernandez T, Hla T, Puddington L, Padula SJ. The Nuclear Receptor PPARγ and Immunoregulation: PPARγ Mediates Inhibition of Helper T Cell Responses. J Immunol (2000) 164(3):1364–71. doi: 10.4049/jimmunol.164.3.1364
109. Hontecillas R, Bassaganya-Riera J. Peroxisome Proliferator-Activated Receptor γ Is Required for Regulatory CD4 + T Cell-Mediated Protection against Colitis. J Immunol (2007) 178(5):2940–9. doi: 10.4049/jimmunol.178.5.2940
110. Nobs SP, Natali S, Pohlmeier L, Okreglicka K, Schneider C, Kurrer M, et al. PPARγ in dendritic cells and T cells drives pathogenic type-2 effector responses in lung inflammation. J Exp Med (2017) 214(10):3015–35. doi: 10.1084/jem.20162069
111. Cunard R, Eto Y, Muljadi JT, Glass CK, Kelly CJ, Ricote M. Repression of IFN-γ Expression by Peroxisome Proliferator-Activated Receptor γ. J Immunol (2004) 172(12):7530–6. doi: 10.4049/jimmunol.172.12.7530
112. Chung SW, Kang BY, Kim TS. Inhibition of Interleukin-4 Production in CD4+ T Cells by Peroxisome Proliferator-Activated Receptor-γ (PPAR-γ) Ligands: Involvement of Physical Association between PPAR-γ and the Nuclear Factor of Activated T Cells Transcription Factor. Mol Pharmacol (2003) 64(5):1169–79. doi: 10.1124/mol.64.5.1169
113. Sarraf P, Mueller E, Jones D, King FJ, DeAngelo DJ, Partridge JB, et al. Differentiation and reversal of malignant changes in colon cancer through PPARγ. Nat Med (1998) 4(9):1046–52. doi: 10.1038/2030
114. Tsubouchi Y, Sano H, Kawahito Y, Mukai S, Yamada R, Kohno M, et al. Inhibition of human lung cancer cell growth by the peroxisome proliferator-activated receptor-gamma agonists through induction of apoptosis. Biochem Biophys Res Commun (2000) 270(2):400—405. doi: 10.1006/bbrc.2000.2436
115. Bren-Mattison Y, Van Putten V, Chan D, Winn R, Geraci MW, Nemenoff RA. Peroxisome proliferator-activated receptor-gamma (PPAR(gamma)) inhibits tumorigenesis by reversing the undifferentiated phenotype of metastatic non-small-cell lung cancer cells (NSCLC). Oncogene (2005) 24(8):1412—1422. doi: 10.1038/sj.onc.1208333
116. Motomura W, Okumura T, Takahashi N, Obara T, Kohgo Y. Activation of peroxisome proliferator-activated receptor gamma by troglitazone inhibits cell growth through the increase of p27KiP1 in human. Pancreatic carcinoma cells. Cancer Res (2000) 60(19):5558–64.
117. Kubota T, Koshizuka K, Williamson EA, Asou H, Said JW, Holden S, et al. Ligand for peroxisome proliferator-activated receptor gamma (troglitazone) has potent antitumor effect against human prostate cancer both in vitro and in vivo. Cancer Res (1998) 58(15):3344–52.
118. Bonofiglio D, Cione E, Qi H, Pingitore A, Perri M, Catalano S, et al. Combined low doses of PPARgamma and RXR ligands trigger an intrinsic apoptotic pathway in human breast cancer cells. Am J Pathol (2009) 175(3):1270–80. doi: 10.2353/ajpath.2009.081078
119. Mueller E, Sarraf P, Tontonoz P, Evans RM, Martin KJ, Zhang M, et al. Terminal Differentiation of Human Breast Cancer through PPARγ. Mol Cell (1998) 1(3):465–70. doi: 10.1016/S1097-2765(00)80047-7
120. Girnun GD, Naseri E, Vafai SB, Qu L, Szwaya JD, Bronson R, et al. Synergy between PPARγ Ligands and Platinum-Based Drugs in Cancer. Cancer Cell (2007) 11(5):395–406. doi: 10.1016/j.ccr.2007.02.025
121. Park J, Morley TS, Scherer PE. Inhibition of endotrophin, a cleavage product of collagen VI, confers cisplatin sensitivity to tumours. EMBO Mol Med (2013) 5(6):935–48. doi: 10.1002/emmm.201202006
122. Khandekar MJ, Banks AS, Laznik-Bogoslavski D, White JP, Choi JH, Kazak L, et al. Noncanonical agonist PPARγ ligands modulate the response to DNA damage and sensitize cancer cells to cytotoxic chemotherapy. Proc Natl Acad Sci U S A (2018) 115(3):561–6. doi: 10.1073/pnas.1717776115
123. Phan ANH, Vo VTA, Hua TNM, Kim MK, Jo SY, Choi JW, et al. PPARγ sumoylation-mediated lipid accumulation in lung cancer. Oncotarget (2017) 8(47):82491–505. doi: 10.18632/oncotarget.19700
124. Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
125. Saez E, Tontonoz P, Nelson MC, Alvarez JGA, Ming UT, SM B, et al. Activators of the nuclear receptor PPARγ enhance colon polyp formation. Nat Med (1998) 4(9):1058–61. doi: 10.1038/2042
126. Lefebvre AM, Chen I, Desreumaux P, Najib J, Fruchart JC, Geboes K, et al. Activation of the peroxisome proliferator-activated receptor gamma promotes the development of colon tumors in C57BL/6J-APCMin/+ mice. Nat Med (1998) 4(9):1053—1057. doi: 10.1038/2036
127. Broekema MF, Massink MPG, Donato C, de Ligt J, Schaarschmidt J, Borgman A, et al. Natural helix 9 mutants of PPARγ differently affect its transcriptional activity. Mol Metab (2019) 20:115–27. doi: 10.1016/j.molmet.2018.12.005
128. Sarraf P, Mueller E, Smith WM, Wright HM, Kum JB, Aaltonen LA, et al. Loss-of-function mutations in PPAR gamma associated with human colon cancer. Mol Cell (1999) 3:799–804. doi: 10.1016/S1097-2765(01)80012-5
129. Gupta RA, Sarraf P, Mueller E, Brockman JA, Prusakiewicz JJ, Eng C, et al. Peroxisome proliferator-activated receptor γ-mediated differentiation: A mutation in colon cancer cells reveals divergent and cell type-specific mechanisms. J Biol Chem (2003) 278(25):22669–77. doi: 10.1074/jbc.M300637200
130. Coutos-Thévenot L, Beji S, Neyret-Kahn H, Pippo Q, Fontugne J, Osz J, et al. PPARγ is a tumor suppressor in basal bladder tumors offering new potential therapeutic opportunities. bioRxiv (2019), 868190. doi: 10.1101/868190
131. Rebouissou S, Bernard-Pierrot I, de Reyniès A, Lepage M-L, Krucker C, Chapeaublanc E, et al. EGFR as a potential therapeutic target for a subset of muscle-invasive bladder cancers presenting a basal-like phenotype. Sci Transl Med (2014) 6(244):244ra91–244ra91. doi: 10.1126/scitranslmed.3008970
132. Choi W, Porten S, Kim S, Willis D, Plimack ER, Hoffman-Censits J, et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell (2014) 25(2):152–65. doi: 10.1016/j.ccr.2014.01.009
133. Biton A, Bernard-Pierrot I, Lou Y, Krucker C, Chapeaublanc E, Rubio-Pérez C, et al. Independent Component Analysis Uncovers the Landscape of the Bladder Tumor Transcriptome and Reveals Insights into Luminal and Basal Subtypes. Cell Rep (2014) 9(4):1235–45. doi: 10.1016/j.celrep.2014.10.035
134. Halstead AM, Kapadia CD, Bolzenius J, Chu CE, Schriefer A, Wartman LD, et al. Bladder-cancer-associated mutations in RXRA activate peroxisome proliferator-activated receptors to drive urothelial proliferation. Levine RL, editor. Elife (2017) 6:e30862. doi: 10.7554/eLife.30862
135. Weinstein JN, Akbani R, Broom BM, Wang W, Verhaak RGW, McConkey D, et al. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature (2014) 507(7492):315–22. doi: 10.1038/nature12965
136. Kroll TG, Sarraf P, Pecciarini L, Chen C-J, Mueller E, Spiegelman BM, et al. PAX8-PPARgamma1 fusion oncogene in human thyroid carcinoma. Science (2000) 289(5483):1357–60. doi: 10.1126/science.289.5483.1357
137. Cheung L, Messina M, Gill A, Clarkson A, Learoyd D, Delbridge L, et al. Detection of the PAX8-PPARγ Fusion Oncogene in Both Follicular Thyroid Carcinomas and Adenomas. J Clin Endocrinol Metab (2003) 88(1):354–7. doi: 10.1210/jc.2002-021020
138. Pasca di Magliano M, Di Lauro R, Zannini M. Pax8 has a key role in thyroid cell differentiation. Proc Natl Acad Sci [Internet] (2000) 97(24):13144–9. doi: 10.1073/pnas.240336397
139. Macchia PE, Lapi P, Krude H, Pirro MT, Missero C, Chiovato L, et al. PAX8 mutations associated with congenital hypothyroidism caused by thyroid dysgenesis. Nat Genet (1998) 19(1):83–6. doi: 10.1038/ng0598-83
140. Karger S, Berger K, Eszlinger M, Tannapfel A, Dralle H, Paschke R, et al. Evaluation of Peroxisome Proliferator-Activated Receptor-γ Expression in Benign and Malignant Thyroid Pathologies. Thyroid (2005) 15(9):997–1003. doi: 10.1089/thy.2005.15.997
141. Raman P, Koenig RJ. Pax-8–PPAR-γ fusion protein in thyroid carcinoma. Nat Rev Endocrinol (2014) 10(10):616–23. doi: 10.1038/nrendo.2014.115
142. Zhang Y, Yu J, Grachtchouk V, Qin T, Lumeng C, Sartor M, et al. Genomic binding of PAX8-PPARG fusion protein regulates cancer-related pathways and alters the immune landscape of thyroid cancer. Oncotarget (2017) 5(0):5761–73. doi: 10.18632/oncotarget.14050
143. Boos LA, Dettmer M, Schmitt A, Rudolph T, Steinert H, Moch H, et al. Diagnostic and prognostic implications of the PAX8–PPARγ translocation in thyroid carcinomas—a TMA-based study of 226 cases. Histopathology (2013) 63(2):234–41. doi: 10.1111/his.12150
144. Lui W-O, Zeng L, Rehrmann V, Deshpande S, Tretiakova M, Kaplan EL, et al. CREB3L2-PPARγ Fusion Mutation Identifies a Thyroid Signaling Pathway Regulated by Intramembrane Proteolysis. Cancer Res (2008) 68(17):7156–64. doi: 10.1158/0008-5472.CAN-08-1085
145. Yki-Järvinen H. Thiazolidinediones. N Engl J Med [Internet] (2004) 351(11):1106–18. doi: 10.1056/NEJMra041001
146. Soccio RE, Chen ER, Lazar MA. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab [Internet] (2014) 20(4):573–91. doi: 10.1016/j.cmet.2014.08.005
147. FDA Drug Safety Communication. FDA eliminates the Risk Evaluation and Mitigation Strategy (REMS) for rosiglitazone-containing diabetes medicines [Internet]. Available at: https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-fda-eliminates-risk-evaluation-and-mitigation-strategy-rems, [cited 2019 Jun 10].
148. Perakakis N, Joshi A, Peradze N, Stefanakis K, Li G, Feigh M, et al. The Selective Peroxisome Proliferator-Activated Receptor Gamma Modulator CHS-131 Improves Liver Histopathology and Metabolism in a Mouse Model of Obesity and Nonalcoholic Steatohepatitis. Hepatol Commun (2020) 4(9):1302–15. doi: 10.1002/hep4.1558
149. Ninomiya I, Yamazaki K, Oyama K, Hayashi H, Tajima H, Kitagawa H, et al. Pioglitazone inhibits the proliferation and metastasis of human pancreatic cancer cells. Oncol Lett (2014) 8(6):2709–14. doi: 10.3892/ol.2014.2553
150. Ceni E, Mello T, Tarocchi M, Crabb DW, Caldini A, Invernizzi P, et al. Antidiabetic thiazolidinediones induce ductal differentiation but not apoptosis in pancreatic cancer cells. World J Gastroenterol (2005) 11(8):1122–30. doi: 10.3748/wjg.v11.i8.1122
151. Quail DF, Dannenberg AJ. The obese adipose tissue microenvironment in cancer development and progression. Nat Rev Endocrinol (2019) 15(3):139–54. doi: 10.1038/s41574-018-0126-x
152. Bu D, Crewe C, Kusminski CM, Gordillo R, Ghaben AL, Kim M, et al. Human endotrophin as a driver of malignant tumor growth. JCI Insight (2019) 5(9):e125094. doi: 10.1172/jci.insight.125094
153. Park J, Scherer PE. Adipocyte-derived endotrophin promotes malignant tumor progression. J Clin Invest (2012) 122(11):4243–56. doi: 10.1172/JCI63930
154. Sun K, Park J, Kim M, Scherer PE. Endotrophin, a multifaceted player in metabolic dysregulation and cancer progression, is a predictive biomarker for the response to PPARγ agonist treatment. Diabetologia (2017) 60(1):24–29. doi: 10.1007/s00125-016-4130-1
155. Dongre A, Weinberg RA. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol (2019) 20(2):69–84. doi: 10.1038/s41580-018-0080-4
156. Ishay-Ronen D, Diepenbruck M, Kalathur RKR, Sugiyama N, Tiede S, Ivanek R, et al. Gain Fat—Lose Metastasis: Converting Invasive Breast Cancer Cells into Adipocytes Inhibits Cancer Metastasis. Cancer Cell (2019) 35(1):17–32.e6. doi: 10.1016/j.ccell.2018.12.002
Keywords: PPARy, adipocyte, immune cell, cancer cell, mechanism
Citation: Hernandez-Quiles M, Broekema MF and Kalkhoven E (2021) PPARgamma in Metabolism, Immunity, and Cancer: Unified and Diverse Mechanisms of Action. Front. Endocrinol. 12:624112. doi: 10.3389/fendo.2021.624112
Received: 30 October 2020; Accepted: 08 January 2021;
Published: 26 February 2021.
Edited by:
Nicolas Venteclef, Sorbonne Universités, FranceReviewed by:
Fraydoon Rastinejad, University of Oxford, United KingdomCopyright © 2021 Hernandez-Quiles, Broekema and Kalkhoven. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric Kalkhoven, ZS5rYWxraG92ZW5AdW1jdXRyZWNodC5ubA==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.