 Livia López–Noriega
Livia López–Noriega Guy A. Rutter
Guy A. Rutter
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 08 February 2021
Sec. Diabetes: Molecular Mechanisms
Volume 11 - 2020 | https://doi.org/10.3389/fendo.2020.610213
This article is part of the Research TopicUnderstanding the Gene Expression of Beta Cell Dysfunction in DiabetesView all 9 articles
Numerous studies have sought to decipher the genetic and other mechanisms contributing to β-cell loss and dysfunction in diabetes mellitus. However, we have yet to fully understand the etiology of the disease or to develop satisfactory treatments. Since the majority of diabetes susceptibility loci are mapped to non-coding regions within the genome, understanding the functions of non-coding RNAs in β-cell biology might provide crucial insights into the pathogenesis of type 1 (T1D) and type 2 (T2D) diabetes. During the past decade, numerous studies have indicated that long non-coding RNAs play important roles in the maintenance of β-cell mass and function. Indeed, lncRNAs have been shown to be involved in controlling β-cell proliferation during development and/or β-cell compensation in response to hyperglycaemia. LncRNAs such as TUG-1 and MEG3 play a role in both β-cell apoptosis and function, while others sensitize β-cells to apoptosis in response to stress signals. In addition, several long non-coding RNAs have been shown to regulate the expression of β-cell-enriched transcription factors in cis or in trans. In this review, we provide an overview of the roles of lncRNAs in maintaining β-function and mass, and discuss their relevance in the development of diabetes.
Diabetes Mellitus (DM) is a metabolic disease characterized by chronic hyperglycaemia, which ultimately results from pancreatic β-cell failure (1). Type 1 diabetes (T1D) involves immune cell-mediated destruction of β-cells (2), as well as more minor changes in beta cell function (3), with many patients having residual beta cell mass (4). The etiology of type 2 diabetes (T2D) remains less clear, but deficiencies in insulin secretion are a hallmark of the disease (5). T2D usually ensues when β-cells are unable to overcome insulin resistance and so maintain physiological glucose levels (6). However, a subset of patients displays defective insulin secretion despite near-normal insulin sensitivity (7) and monogenic forms of the diabetes (maturity onset diabetes of the young; MODY) involve mutations in genes which impact β-cell function (8) with unaltered insulin action.
A change in β-cell “identity” (9, 10) is a likely possibility in all forms of diabetes, though more importantly in T2D than in T1D. Numerous studies have tried to decipher the genetic and molecular mechanisms contributing to β-cell identity loss (11). Fully functional β-cells require the expression of a subset of specific transcription factors (e.g., Pdx1, Mafa, and Pax6), as well as genes involved in glucose sensing and metabolism (e.g., Glut2/Slc2a2, Glucokinase, Gck) and insulin production (insulin, Ins) (10, 11). Another important feature of these cells is the repression of the so-called “disallowed” genes (e.g., Ldha, Hsd11b1). Expression of this group of genes, whose transcripts are present at very low levels in β-cells compared to other tissues, may otherwise interfere with insulin secretion (12, 13).
Altered intercellular coupling between β-cells is implicated in both T2D (14, 15) and T1D (16). In both diseases, the drivers for the above changes in β-cells, and their interconnections, are likely to be both genetic and environmental, but in neither case are they fully characterized.
Genome wide-association studies (GWAS) have revealed that the majority of diabetes (both T1D and T2D) susceptibility loci map outside protein-coding regions, consistent with a role for regulatory regions [i.e., enhancers or promoters which control the expression of protein-coding genes (17)] but also suggesting an important role for non-coding RNAs (ncRNAs) in maintaining a functional β-cell mass (18–23). NcRNAs are a heterogenous group characterized by the lack of protein coding potential, and which include both linear and circular transcripts. They can be classified according to their size into short non-coding RNAs, which are <200 nucleotides (nts) (e.g., miRNAs 19-21 nts, snRNA ~150 nts, snoRNAs 60–140 nts, piRNA 26–31 nts) and long non-coding RNAs (>200 ntds) (24, 25). Long non-coding RNAs (lncRNAs) are further classified, according to their position relative to protein coding genes, into: i) intergenic, located between two protein coding genes, usually in enhancer regions; ii) bidirectional, transcribed in the opposite direction of a protein coding gene and within 1kb of its promoter region; iii) natural antisense transcripts, RNAs overlapping partially or totally with a protein coding gene and transcribed from its opposite strand; iv) sense intronic RNAs, transcribed from intronic regions of protein coding genes in the same direction (26–28) (Figure 1).
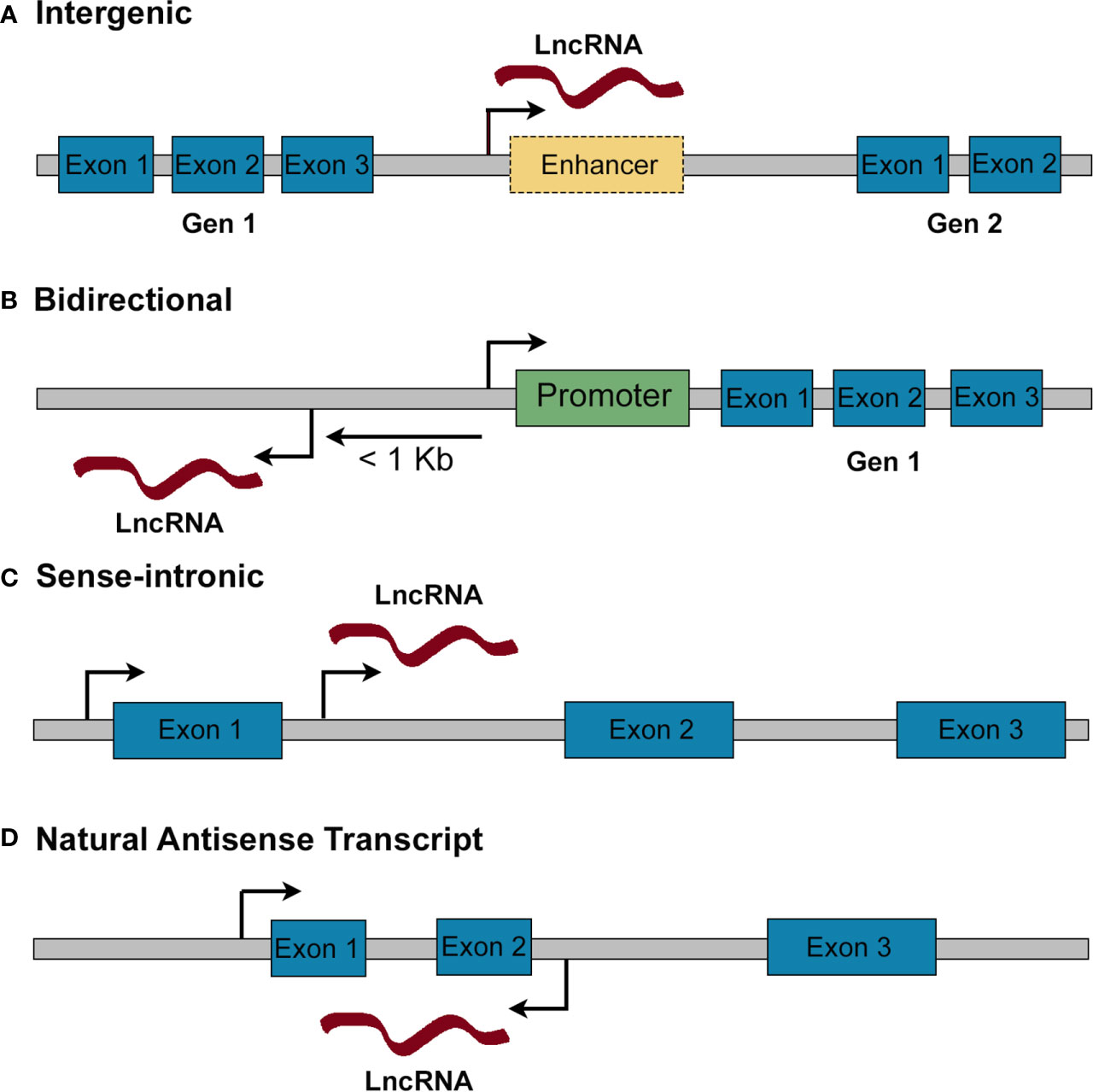
Figure 1 Classification of lncRNAs according to their genomic position. (A) Intergenic lncRNAs are found in gene deserts between two protein coding genes. They may be subclassified into long intergenic RNAs and enhancer RNAs (when they are transcribed from an enhancer). (B) Bidirectional lncRNAs are mapped within 1 kb from the promoter of a protein coding gene and are transcribed in the opposite direction. (C) Sense-intronic lncRNAs are transcribed from an intron of a protein coding gene in the same direction. (D) Natural antisense transcripts are transcribed from the opposite strand of a protein coding gene, overlapping partially or totally with its exons/introns.
LncRNAs share common features with mRNAs as they are usually transcribed by RNA polymerase II, display chromatin marks typical of active transcription and undergo post-transcriptional modifications such as 5’capping, splicing, and polyadenylation (29). However, lncRNAs are normally expressed at lower levels, contain fewer exons and are less evolutionarily conserved between species than protein-coding genes. Moreover, lncRNAs are expressed in a highly cell-type specific manner, making them well placed to be involved in cell lineage specification (29, 30). Moran et al. identified more than 1,100 lncRNAs expressed in human islets with 55% of intergenic lncRNAs and 40% of antisense transcripts being islet-specific (31). These authors also found that the expression of several of the islet-specific lncRNAs was modulated during pancreatic development, indicating that they are involved in the differentiation process of pancreatic endocrine cells. Moreover, several studies have reported differential expression of lncRNAs in islets from T1D (32) and T2D mouse models (33–35) as well as in patients with T2D (31, 36). In addition, differentially expressed lncRNAs have also been found in peripheral blood mononuclear cells from T1D patients, being proposed as biomarkers for early diagnosis (37). Therefore, it is tempting to speculate that lncRNAs are crucial players in the development of both T1D and T2D and could be used as novel biomarkers or targets for future therapies. In this review, we provide an overview of the roles of lncRNAs in maintaining β-function and mass and discuss their relevance in DM development.
LncRNAs can be located in the nucleus or the cytoplasm and regulate the expression of protein-coding genes both transcriptionally and post-transcriptionally (Figure 2) (38). In the nucleus, long non-coding RNAs have been found to modulate transcription through their interaction with chromatin remodeling proteins, recruiting, or sequestering transcription factors to their target sites and interacting with enhancer regions. Moreover, several nuclear lncRNAs can also affect alternative splicing of protein-coding genes or regulate protein stability in the nucleus (39, 40). In the cytoplasm, lncRNAs may regulate translation, control mRNA stability, act as sponges for miRNAs or affect protein-protein interactions (41). Although, classically, lncRNAs were thought to be enriched in the nucleus, a growing body of evidence suggests that they may be just as abundant in the cytoplasm (42–44). Indeed, and in contrast to previous publications (42, 45), Heesch et al. (46). reported an enrichment of lncRNAs in the cytoplasm and ribosomal fractions compared to the nucleus. Interestingly, several of the well-established nuclear lncRNAs have also been found to play important roles in the cytoplasm, suggesting that many lncRNAs are, in fact, bi-compartmental (40, 47–49). A good example is provided by HOTAIR, first described as a lncRNA involved in the transcriptional silencing of the HOXD cluster by the modulation of histone methylation patterns (50, 51). Nevertheless, a later study (47) reported the presence of HOTAIR in the cytoplasm, where it binds to two E3 ubiquitin ligases (Mex3b and Dzip3), acting as a scaffold to promote protein ubiquitination. Similarly, MALAT1 was first described as a nuclear lncRNA involved in alternative splicing (52). However, numerous studies in different cell types have reported a function of MALAT1 in the cytoplasm, where it acts as a sponge for miRNAs (49). In β-cells, MALAT1 has been reported to modulate active histone marks of the Pdx1 promoter in MIN6 cells and mouse islets (53) and to promote the stability of polypyrimidine tract binding protein 1 (Ptbp1) protein in the nucleus. Additionally, MALAT1 is implicated in the deleterious effects of cigarette smoke on the β-cell, acting in the cytoplasm through the inhibition of miRNA-17 (54) (Figure 2). In summary, although subcellular localization has been regarded as a main determinant of the functions of a given lncRNA, a substantial body of recent evidence suggests that several lncRNAs have multiple actions in different cell compartments even within the same cell lineage.
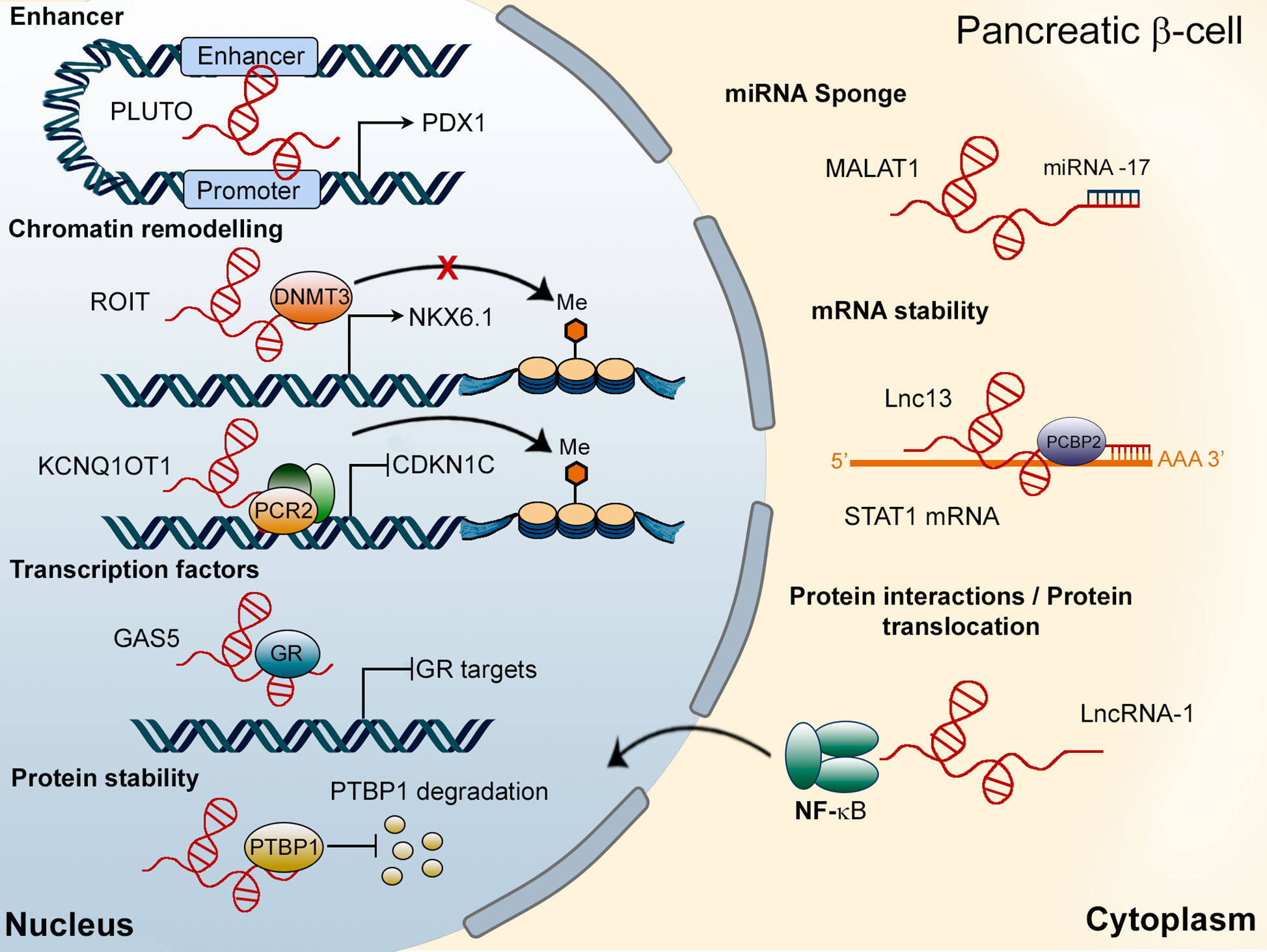
Figure 2 Mechanisms of action of lncRNAs in pancreatic β-cells. In the nucleus, lncRNAs, such as PLUTO, regulate the transcription of target genes (PDX1) by modulating enhancer/promoter interactions. Other LncRNAs, including ROIT and KCNQ1OT1, interact with chromatin remodeling proteins. The lncRNA GAS5 has been shown to act as a decoy for the glucocorticoid receptor (GR), inhibiting the expression of GR target genes. Finally, MALAT1 has been found to be located in the nucleus (regulating PTBP1 protein stability) as well as in the cytoplasm of β-cells, where it interacts with miRNA-17. Cytosolic lncRNAs can also modulate mRNA stability or affect protein-protein interactions or their subcellular localization.
Regeneration and replacement of β-cells have been proposed as promising strategies for the treatment of insulin-requiring and insulin-dependent diabetes (55, 56). An increase in β-cell proliferation is well documented in animal models of T1D (57, 58) and T2D (59) as well as during pregnancy in rodents (60), while it remains controversial whether β-cell proliferation is significant in the human pancreas (61). However, several studies have provided evidence of β-cell proliferation in patients with recent onset T1D (62, 63). In addition, β-cell mass increases in humans during obesity and pregnancy, although immunohistochemical studies suggest that neogenesis rather than proliferation may be the main source of new β-cells in humans (64–66). The lack of unequivocal and sensitive markers to measure proliferation may complicate the task of assessing the real rate of human β-cell proliferation in this context (67). As opposed to earlier studies, Hanley et al. (68) observed increased proliferation rates using cell nuclear antigen (PCNA) staining in obese non-diabetic individuals when compared with lean non-diabetic controls. The authors also found decreased β-cell proliferation in islets from T2D donors, while a more recent study (69) indicated increased numbers of PCNA-positive β-cells in specimens from individuals with T2D when compared to nondiabetic subjects. Overall, it can be concluded that mitogenic signalling pathways are difficult to activate in human β-cells, but we still do not fully understand the mechanisms involved in β-cell proliferation and β-cell mass compensation in humans (70). Of note, recent findings point to inhibition of the protein kinase DYRK1A as a possible target for the activation of β-cell growth (71), though no evidence exists at present of a role for lncRNAs in the control of this enzyme.
In order to understand the molecular mechanisms driving β-cell mass compensation during insulin resistance or in pre-diabetes, Sisino et al., combined a NCodelncRNA Microarray with analyses from published RNA-seq data to identify lncRNAs that were differentially expressed in islets from female mice at day 14.5 of gestation, corresponding with the peak in β-cell mass expansion. The authors identified six lncRNAs that were differentially expressed in pancreatic islets during gestation. Interestingly, one of them, Gm16308, was strongly induced by prolactin treatment and was required for β-cell proliferation induced by the hormone. Moreover, overexpression of Gm16308 alone was sufficient to induce β-proliferation (72), suggesting that it may be a good target to increase β-cell mass, although the impact of Gm16308 in human β-cell proliferation needs to be validated. The lncRNA DANCR (Differentiation Antagonizing Non-Protein Coding RNA) may also be involved in the compensation of β-cell mass during gestation. DANCR has been shown to act as a sponge for miR-33a-5p, which is upregulated in blood samples from patients with gestational diabetes mellitus (GDM). Overexpressing miR-33a-5p in INS-1 cells inhibited β-cell proliferation, insulin content and secretion at both low and high glucose. Remarkably, forced expression of DANCR rescued miR-33a-5p-mediated inhibition effects on cell proliferation and insulin content.
As stated earlier, a subgroup of patients with non-Mendelian forms of the disease develop T2D without showing significant insulin resistance suggesting that defects in β-cell mass and/or function are the principal disease drivers in these patients. In any case, relative insulin deficiency is a sine qua non for the emergence of the frank disease even in the presence of insulin resistance. In both instances, inadequate neonatal β-cell expansion may predispose to the appearance of diabetes later in life. Therefore, it is important to identify genetic factors driving β-cell proliferation during neonatal development. H19 is a maternally imprinted intergenic lncRNA located at the Igf2 locus that has recently been shown to drive β-cell mass expansion during development (73). Ding et al. (74) first described differential expression of H19 in islets from the offspring of a mouse model for gestational diabetes (GD), which develop glucose intolerance and impaired glucose stimulated insulin secretion (GSIS) with age. Recently, H19 was found to be required for normal β-cell mass expansion during the neonatal period through the activation of the Akt/PKB pathway. H19 mediated β-cell proliferation was also dependent on the presence of Argonaute 2 (Ago2), an essential component of the RNA-induced silencing complex (RISC), suggesting that H19 may act as a miRNA sponge in β-cells. Supporting this view, removal of binding sites in H19 for the miRNA let-7 reduced the proliferative capacities of the long non-coding RNA (Figure 3). Therefore, and although other mechanisms were not excluded, the authors proposed that H19 may induce β-cell proliferation at least partially through inhibiting let-7, which in turns may activate the PI3K/Akt pathway, as it does in skeletal muscle. Importantly, although H19 expression decreased considerably in adult islets compared to those from neonates, this lncRNA was re-expressed under conditions of high insulin demand. The latter findings suggest that H19 may also be involved in β-cell mass compensation in rodents, although further studies are needed to assess the role of H19 in human β-cells (73).
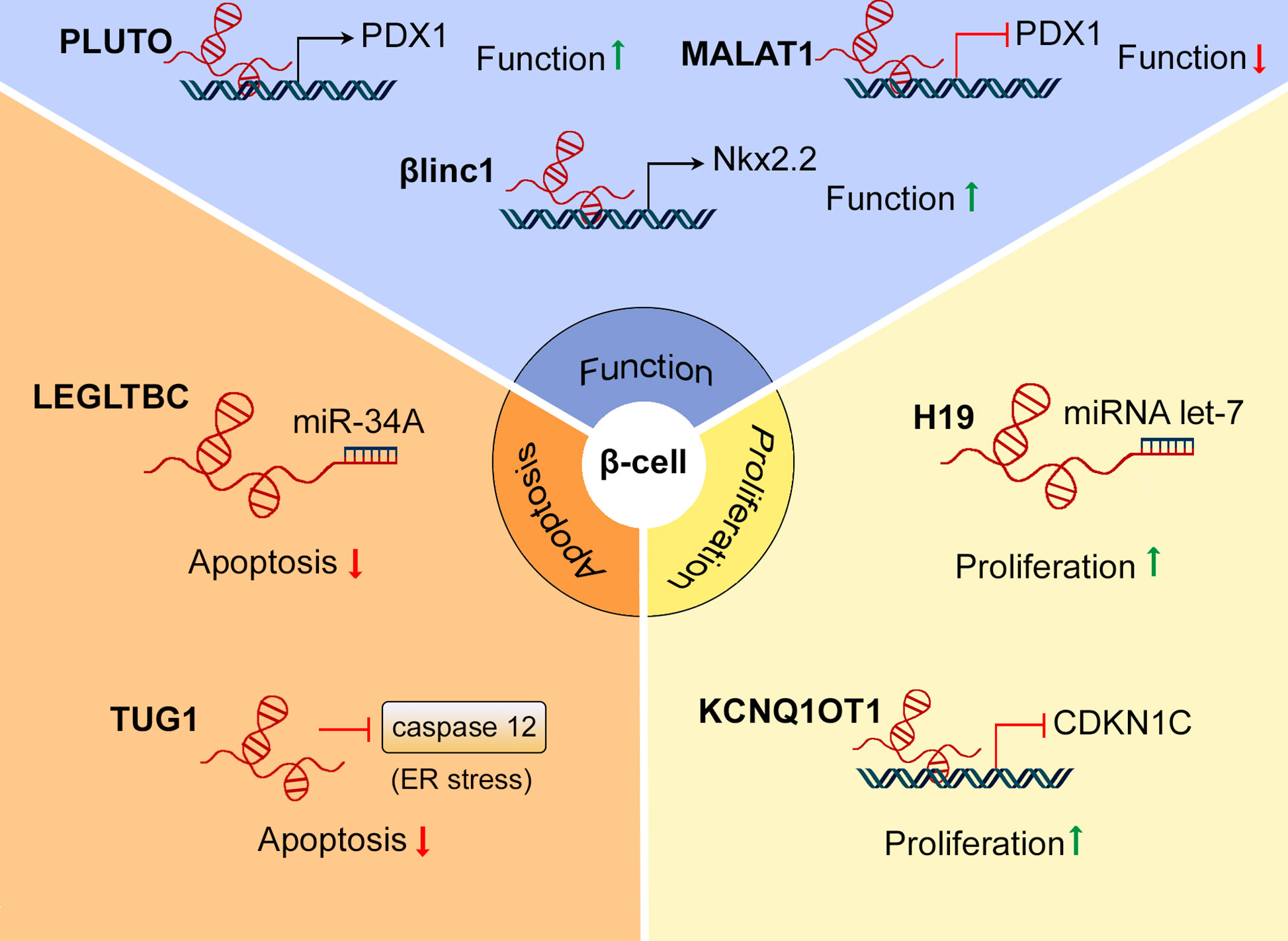
Figure 3 Roles of lncRNAs in β-cell mass and function. Several lncRNAs, including PLUTO, MALAT1, and βlinc1 have been found to regulate β-cell function by modulating the expression of β-cell signature genes. LncRNAs as LEGLTBC and TUG1 protect β-cells from apoptosis in response to different stimuli. H19 and KCNQ1OT1 are involved in β-cell mass expansion during development, promoting β-cell proliferation.
GWAS have revealed that several single nucleotide polymorphisms (SNPs) in the KCNQ1 region, containing the lncRNA KCNQ1OT1, are strongly associated with T2D risk (21, 75). In early development, KCNQ1OT1 is expressed exclusively from the paternal allele and has been linked to regulate the imprinted expression of neighbouring target genes, including the Kv7.1 voltage-gated potassium channel subunit (KCNQ1) and cyclin-dependent kinase inhibitor 1C (CDKN1C), resulting in these genes being expressed exclusively from the maternal allele (Figure 2). However, the imprinting of KCNQ1 is lost during adulthood, whereas CDKN1C expression remains monoallelic throughout life (76). Asahara et al. showed that truncation of the paternal allele of Kcnq1ot1 in mice increased the expression of Cdkn1c in a tissue-specific manner, resulting in decreased β-cell mass at birth and at 24 weeks of age (Figure 3). Moreover, mice harbouring paternally-truncated Kcnq1ot1 displayed glucose intolerance and defects in insulin secretion (77). It is therefore tempting to speculate that decreased expression of KCNQ1OT1, and subsequent upregulation of CDKN1C, mediates diabetes susceptibility at the KCNQ1 locus.
In addition, several SNPs at the CDKN2A/B locus that encodes the lncRNA ANRIL, MTAP, and the cell cycle inhibitors p14, p15, and p16, have been linked to increased T2D risk. The homozygous risk variants of two of these SNPs, rs2383208 and rs10811661, were associated with high expression of ANRIL in samples from young donors (between 10 and 50 years old), while only protective variants showed an age-dependent increase. Moreover, the risk allele of rs564398, a SNP located within exon 2 of ANRIL, was associated with reduced β-cell proliferation rate. Therefore, these results suggest a potential role for ANRIL in the maintenance of human β-cell mass (78).
Numerous studies have shown that there is a significant reduction of β-cell mass in both T1D and T2D (79), although the extent of the “loss” of β-cell mass in T2D – usually reported as the difference in β-cell mass between populations of diabetic and control subjects; prospective measurements of β-cell mass in the same individual over time are not currently feasible (80) – is disputed, ranging from 25% to 50% (81). Although β-cell apoptosis is certainly a major aspect of the pathogenesis of T1D (32), there is a debate regarding the relative contributions of apoptosis, β-cell dedifferentiation and dysfunction, in the development of T2D (82).
Interestingly, several lncRNAs that have been shown to impact β-cell function also play an important role in β-cell apoptosis. Silencing the lncRNA Taurine-upregulated gene 1 (TUG1) was sufficient to induce β-cell apoptosis in MIN6 cells and pancreatic islets. This involved upregulated expression of the effector caspases 3 and 7 as well as caspase 12, which is associated with ER-stress-mediated apoptosis (Figure 3). In addition, TUG1 knockdown also reduced Ins1 and Ins2 mRNA levels and impaired GSIS in MIN6 cells. Importantly, wild-type (wt) mice injected with an siRNA targeting TUG1 showed reduced plasma insulin levels and increased fasting glycaemia (83). Similarly, the lncRNA Maternally expressed gene 3 (Meg3) has been shown to regulate insulin secretion as well as β-cell apoptosis (84). Meg3 expression is down-regulated in pancreatic islets from T1D and T2D mouse models (NOD and hyperglycaemic db/db mice, respectively) (85) as well as in pancreatic islets from T2D donors (86). In addition, a SNP located within an intron of Meg3 is strongly associated with T1D risk (87). Meg3 down-regulation reduces the expression of ins2, Pdx1 and Mafa, impairing GSIS both in vitro and in vivo (85). Moreover, this manoeuvre upregulated the pro-apoptotic proteins Bax and caspase-3, increasing β-cell apoptosis. Furthermore, wild-type mice injected with an siRNA targeting Meg3 display reduced β-cell mass (84). Nevertheless, despite the association studies linking Meg3 expression with T1D and T2D in humans, there are no current studies assessing the impact of Meg3 in human β-cells.
In contrast, Morlette et al. (35) identified two different lncRNAs that were differentially expressed in mice fed with a high fat diet (HFD), as well as in db/db mice, and which had an impact in β-cell apoptosis without affecting β-cell function. βlinc2, a lncRNA mapped in the mouse to chromosome 14, was up-regulated in pancreatic islets from T2D models and its expression was positively correlated with weight, glycaemia and hyperinsulinemia. The lncRNA βlinc3 was downregulated in pancreatic islets from T2D mouse models and its expression was negatively correlated with weight and glycaemia. Remarkably, a human ortholog for βlinc3 was also found to be downregulated in islets from T2D patients, while no orthologs were found for βlinc2 in humans. Overexpression of βlinc2, or downregulation of βlinc3, had no effects on pro-insulin mRNA levels, total insulin content or GSIS. However, overexpression of βlinc2 or knockdown of βlinc3 increased β-cell apoptosis in Min6 cells (35).
A number of lncRNAs have also been shown to be involved in β-cell apoptosis mediated by different stress stimuli, such as glucolipotoxicity or inflammation. Kong et al. (88) identified ten different lncRNAs that were modulated in the rat β-cell line INS-1 after treatment with a combination of high glucose and palmitate. One of the lncRNAs identified, named LEGLTBC (low expression in glucolipotoxicity-treated beta cells), was shown to reduce oxidative stress and glucolipotoxicity-mediated β-cell apoptosis, acting as a sponge for miR-34a, which targets SIRT1 (Figure 3). Correspondingly, silencing of SIRT1 diminished the protective effects of LEGLTBC overexpression (89). In contrast, increased expression of the lncRNA PVT1 after streptozotozin (STZ) treatment, partially mediates the increase in oxidative stress and apoptosis induced by the drug, while knocking down PVT1 reduces STZ-mediated β-cell apoptosis (90). In another study (32), the expression of several lncRNAs was found to be up-regulated in MIN6 cells and mouse islets after 24 h of cytokine treatment. Importantly, the expression of three of these lncRNAs was also modulated during the progression of insulitis in non-obese diabetic (NOD) mice, suggesting a role for these lncRNAs in inflammatory processes. Overexpression of these lncRNAs in MIN6 cells did not affect insulin content or secretion, but sensitized β-cells to apoptosis. Interestingly, the overexpression of one of the lncRNAs [referred as lncRNA-1 in the study (32)] promoted the translocation of NF-κB to the nucleus to a similar extent as treatment with interleukin 1 β (IL-1β) and interferon-γ (IFN-γ), indicating a role for this lncRNA in cytokine-mediated apoptosis (32). More recently, a lncRNA associated with T1D, Lnc13, was shown to promote STAT1 mRNA stability (91), the latter considered a master modulator of inflammation-mediated β-cell apoptosis (92). Several studies have identified a number of SNPs located on chromosome 12q13, surrounding the ERBB3 gene and its antisense lncRNA NONHSAG011351, that are associated with T1D risk. NONHSAG011351 may regulate ERBB3 expression, which is downregulated in human islets after exposure to pro-inflammatory cytokines. Furthermore, knocking down Erbb3 in INS1-cells reduced cytokine-induced apoptosis, indicating a role for ERBB3 and possibly NONHSAG011351 in modulating β-cell survival in response to inflammation (93).
For a number of years, it was believed that β-cell apoptosis played a major role in the pathogenesis of T2D (6, 66). However, later studies (82) indicated that the apoptosis level detected in pancreata from T2D donors did not correlate with the decline in β-cell mass and function observed. During the last decade, loss of β-cell identity has increasingly been recognized as a major contributor to compromised insulin secretion in T2D (10, 82). Moreover, several studies have also reported that a decrease in β-cell function precedes β-cell mass decline in T1D (94, 95). Indeed, defects in β-cell secretory responses to glucose have been reported in T1D patients five years before the diagnosis of the disease (96). As noted in the Introduction, maintenance of fully differentiated and functional β-cells requires the expression of key β-cell transcription factors, including Pdx1 (97), MafA (98), Nkx6.1, Nkx2.2, and Pax6 (99), alongside with the inhibition of the so-called “disallowed” or “forbidden” genes, such as Ldha and Hsd11b1 (12, 13). Changes in the expression of any or all of the above may therefore contribute to impaired β-cell identity in T2D (13, 15). Importantly, several of these critical genes appear to be the target of lncRNA action.
Many of the lncRNAs identified in pancreatic islets are mapped to the proximity of transcription factors required for β-cell function and maturation. A number of these lncRNAs have been shown to regulate these transcription factors in cis (36). The lncRNA PLUTO, transcribed antisense to Pdx1, modulates the expression of the latter transcription factor by promoting the interaction of an enhancer region within its promoter (Figure 2). Consistent with a downregulation of Pdx1, silencing of PLUTO in human-derived EndoC-βH3 cells impaired GSIS (Figure 3) (36). Another islet specific lncRNA, known as βlinc1 in mouse and HI-LNC15 in humans, has been shown to regulate the expression of Nkx2.2 in cis. The lncRNA βlinc1/HI-LNC15 is mapped in an intergenic region between Nkx2-2 and Pax1 on chromosome 2 in mice and chromosome 20 in humans. Although the specific mechanism(s) of action of βlinc1/HI-LNC15 remains to be elucidated, silencing of this lncRNA in MIN6 and EndoC-βH1 cells resulted in the down-regulation of Nkx2-2 together with a large number of Nkx2-2 target genes (Figure 3). Moreover, mice null for βlinc1 are glucose intolerant and display defects in pancreatic endocrine cell lineage specification, showing a similar phenotype to Nkx2-2 knockout (KO) mice (100). However, a functional role for HI-LNC15 in human β-cells still remains to be confirmed.
Two different lncRNAs, Paupar and Pax6os1/PAX6-AS1, are transcribed antisense from the Pax6 locus (101, 102). PAX6 is another transcription factor required for β-cell differentiation in the embryo and β-cell function during adulthood (103, 104). Paupar is expressed predominantly in α-cells within the islets, where it acts in cis to regulate the alternative splicing of Pax6. Paupar-silencing decreases the expression of the Pax6 isoform Pax6 5a, resulting in decreased expression of several α-cell signature genes and defective glucagon secretion (101). In contrast, knockdown of Pax6os1/PAX6-AS1, which is more abundant in β than α cells, up-regulates insulin (Ins1) expression in MIN6 and human EndoC-βH1 cells and enhances GSIS in human islets (102). Interestingly, Paupar is down-regulated in islets from db/db mice (101), while Pax6os1/PAX6-AS1 expression is increased in islets from mice fed a HFD and in human T2D donors versus non-diabetic controls (102).
A number of lncRNAs have also been shown to regulate the expression of β-cell signature transcription factors in trans (53, 105). As indicated above, MALAT1 downregulates Pdx1 expression by inhibiting H3 histone acetylation in its promoter region (Figure 3). Ding et al. (53) showed that IL-1β treatment down-regulated Pdx1 and impaired GSIS in MIN6 cells, while knocking down MALAT1 restored both Pdx1 levels and insulin secretion in response to glucose. Interestingly, MALAT1 expression was induced by IL-1β treatment and during the progression of hyperglycaemia in the non-obese diabetic (NOD) T1D mouse model, indicating that this lncRNA might play a role in the decline of β-cell function during the progression of T1D. In the context of T2D, however, silencing MALAT1 has been suggested to impair β-cell function, reducing GSIS and increasing β-cell apoptosis. Xiong et al (40). reported a decrease in MALAT1 expression in db/db mice as well as MIN6 cells treated with palmitate. In this study, MALAT1 was found to inhibit Polypyrimidine tract binding protein (Ptbp) protein degradation in the nucleus, which, according to the authors, mediated the increased insulin expression and reduced β-cell apoptosis observed after exendin-4 treatment. In contrast, Sun et al. (54) reported that increased levels of MALAT1 contributed to β-cell dysfunction caused by cigarette smoke. These authors found that MALAT1 acts as a sponge for mir-17, which inhibits TXNIP expression, the latter serving as an important regulator of β-cell survival and function (106). Thus, increased levels of MALAT1 were found to decrease mir-17 expression, increase TXNIP mRNA levels and decrease the expression of MAFA. Silencing MALAT1 in MIN6 cells down-regulated TXNIP, recovered mir-17 and MAFA expression, increased insulin content and enhanced GSIS (54). Nevertheless, the role of MALAT1 in β-cell function under different physiological circumstances and in human β-cells is not fully understood and should be clarified before proposing this lncRNA as a possible target to treat diabetes.
NKX6.1 is another transcription factor required for maintaining functional and fully mature β-cells and Nkx6.1/NKX6.1 expression is decreased during the development of T2D in both rodents and humans (107). Recently, two lncRNAs, 810019D21Rik (also referred as ROIT) and Gm10451, were shown to regulate Nkx6.1 expression in trans by modulating different epigenetic marks. ROIT, which is transcribed antisense to Espr2 from a locus syntenic block on human chromosome 16 and mouse chromosome 8, promotes the degradation of DNA methyltransferase 3A (DNMT3A), thus affecting Nkx6.1 methylation. Knocking down ROIT, decreased insulin expression and impaired GSIS in MIN6 cells, while ROIT overexpression upregulated insulin and enhanced GSIS in vitro (105). In addition, inhibition of ROIT in wt mice, impaired insulin synthesis and glucose tolerance. Remarkably, ROIT expression was down-regulated in islets from multiple mouse models of T2D (HFD-fed, db/db and ob/ob mice) as well as in the serum of patients with T2D (105). These data provide compelling evidence of a role for ROIT in the development of the disease. The lncRNA Gm10451 seems to regulate Nkx6.1 expression during development by targeting miR-338-3p as a competitive endogenous RNA (cerna). This microRNA regulates the expression of the histone H3K4 methyltransferase complex PTIP (Pax Transcription activation domain Interacting Protein), which is required for appropriate expression of Nkx6.1, along with other mature β-cell markers such as Insulin and MafA (108).
Reduced levels of the lncRNA GAS5 (growth arrest-specific transcript 5) in human serum have been correlated with T2D (109). Moreover, GAS5 expression has been found to be downregulated in islets from db/db mice (110). Decreased expression of this lncRNA in MIN6 cells reduces insulin synthesis and secretion, pointing to a role for GAS5 in β-cell dysfunction during T2D (110). Interestingly, glucocorticoid treatment impairs insulin secretion and reduces the expression of GAS5 in EndoC-βH1 cells cultured at high glucose concentrations. Introducing the active segment of GAS5, called the hormone response element mimic (HREM), rescued GSIS in glucocorticoid-treated cells, suggesting that GAS5 could modulate the glucocorticoid-glucocorticoid receptor (GC-GR) pathway (33). In Hela cells, GAS5 has been shown to act as a decoy for the glucocorticoid receptor that acts as a ligand-dependent transcription factor since it binds to the DNA binding domain of the protein (Figure 2) (111). As opposed to the previous study, however, the latter authors found that GAS5 expression was up-regulated in the islets of T2D donors and in a rat model of T2D. Since GAS5 down-regulation robustly impaired β-cell function, the authors hypothesized that GAS5 induction might be part of a β-cell compensatory mechanism to hyperglycaemia (33). Similarly, the lncRNA p3134 may also play a role in β-cell compensation in diabetes. Indeed, p3134 expression is increased in the blood of T2D patients, while overexpressing this lncRNA has been shown to enhance GSIS in MIN6 cells and restore β-cell function db/db mice (112). In contrast, knocking down the lncRNA linc-p21, whose levels are also increased in the serum of T2D versus normoglycemic patients, enhances GSIS and β-cell proliferation, while overexpressing it has opposite effects (113).
We argue here that understanding the functions of non-coding RNAs in β-cell biology may provide crucial insights into the pathogenesis of T1D and T2D. Over the past decade, numerous studies have indicated that lncRNAs play important roles in the maintenance of β-cell mass and function. Importantly, several long non-coding RNAs that are differentially expressed during diabetes progression have also been shown to regulate β-cell proliferation, apoptosis, or to modulate the expression of genes required for a fully functional and mature β-cell phenotype (36, 84, 102). A summary of these roles is provided in Figure 3.
The highly cell- or tissue-specific expression profile of lncRNAs make them exceptional therapeutic targets, as they could – in theory – be specifically targeted in a given cell-type, limiting off-target effects (114). Furthermore, sequence-based nucleic acid therapeutics (such as antisense oligonucleotides) are evolving rapidly, and several oligonucleotide-based drugs have already been approved by the U.S. Food and Drug Administration (FDA) (115, 116). Clinical trials of novel treatments targeting lncRNAs are already being conducted in the context of other diseases, such as cancer. For instance, BC-819, which is a DNA plasmid that drives the expression of the diphtheria toxin gene under the regulation of the H19 promoter, has been tested in several trials as a treatment for cancers overexpressing H19, including pancreatic cancer (117). However, a better understanding of the roles of specific lncRNAs in β-cell biology, and their mechanisms of action, is needed before they can be considered as therapeutic targets. This point is illustrated by the conflicting results obtained for MALAT1 downregulation in β-cells under different pathophysiological conditions (40, 53, 54). In addition, some lncRNAs may play redundant functions in β-cells. For example, the lncRNA A830019P07Rik is differentially expressed in pancreatic islets during different stages of development and its expression is reduced in pancreatic islets from both db/db and ob/ob mice. However, mice null for this lncRNA do not display any changes in glucose tolerance or insulin secretion (34). Likewise, knocking down the β-cell specific lncRNA G3R1 does not affect glucose metabolism or islet architecture in mice (118). Furthermore, many functional studies of lncRNAs have only been performed in rodent cell lines up to now, although a few have been undertaken in human-derived β-cells, such as EndoC-βH1 (e.g., PLUTO and PAX6-AS1). It will be important, therefore, to validate these findings in human systems.
In conclusion, although several lncRNAs have been shown to play important roles in β-cell function and survival, further detailed investigations of their mechanism of action are needed before they can be considered bona fide therapeutic targets to treat T1D or T2D.
LL and GR jointly conceived and wrote the manuscript. LL provided the original draft of the article and prepared the figures. All authors contributed to the article and approved the submitted version.
GAR was supported by a Wellcome Trust Investigator Award (212625/Z/18/Z), MRC Programme grants (MR/R022259/1, MR/J0003042/1, MR/L020149/1) and by Diabetes UK (BDA/11/0004210, BDA/15/0005275, BDA 16/0005485) project grants. This project has received funding from the European Union’s Horizon 2020 research and innovation programme via the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement No 115881 (RHAPSODY). This Joint Undertaking receives support from the European Union’s Horizon 2020 research and innovation programme and EFPIA.
GR has received research grant funding from Sun Pharmaceuticals Inc and Les Laboratoires Servier, and is a consultant for Sun Pharmaceuticals. These funders had no involvement with the present study.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
FDA, U.S. Food and Drug Administration; GD, Gestational diabetes; GSIS, Glucose stimulated insulin secretion; GWAS, Genome wide-association studies; HFD, High fat diet; KO, knockout; LncRNAs, Long non-coding RNAs; miRNA, micro-RNA; MODY, maturity onset diabetes of the young; NcRNAS, non-coding RNAs; NOD, non-obese diabetic; Nts, nucleotides; SNP, single nucleotide polymorphism; STZ, streptozotocin; T1D, Type 1 diabetes; T2D, Type 2 diabetes; WT, wild-type.
1. DeFronzo RA. Insulin resistance, lipotoxicity, type 2 diabetes and atherosclerosis: the missing links. The Claude Bernard Lecture 2009. Diabetologia (2010) 53(7):1270–87. doi: 10.1007/s00125-010-1684-1
2. Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. Lancet (2014) 383(9911):69–82. doi: 10.1016/S0140-6736(13)60591-7
3. Rui J, Deng S, Arazi A, Perdigoto AL, Liu Z, Herold KC. β Cells that Resist Immunological Attack Develop during Progression of Autoimmune Diabetes in NOD Mice. Cell Metab (2017) 25(3):727–38. doi: 10.1016/j.cmet.2017.01.005
4. Oram RA, McDonald TJ, Shields BM, Hudson MM, Shepherd MH, Hammersley S, et al. Most people with long-duration type 1 diabetes in a large population-based study are insulin microsecretors. Diabetes Care (2015) 38(2):323–8. doi: 10.2337/dc14-0871
5. Esser N, Legrand-Poels S, Piette J, Scheen AJ, Paquot N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res Clin Pract (2014) 105(2):141–50. doi: 10.1016/j.diabres.2014.04.006
6. Weir GC, Bonner-Weir S. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes (2004) 53 Suppl 3:S16–21. doi: 10.2337/diabetes.53.suppl_3.S16
7. Ahlqvist E, Storm P, Käräjämäki A, Martinell M, Dorkhan M, Carlsson A, et al. Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol (2018) 6(5):361–9. doi: 10.1016/S2213-8587(18)30051-2
8. Hattersley AT, Patel KA. Precision diabetes: learning from monogenic diabetes. Diabetologia (2017) 60(5):769–77. doi: 10.1007/s00125-017-4226-2
9. Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell (2012) 150(6):1223–34. doi: 10.1016/j.cell.2012.07.029
10. Rutter GA, Pullen TJ, Hodson DJ, Martinez-Sanchez A. Pancreatic β-cell identity, glucose sensing and the control of insulin secretion. Biochem J (2015) 466(2):203–18. doi: 10.1042/BJ20141384
11. Del Guerra S, Lupi R, Marselli L, Masini M, Bugliani M, Sbrana S, et al. Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes (2005) 54(3):727–35. doi: 10.2337/diabetes.54.3.727
12. Pullen TJ, Khan AM, Barton G, Butcher SA, Sun G, Rutter GA. Identification of genes selectively disallowed in the pancreatic islet. Islets (2010) 2(2):89–95. doi: 10.4161/isl.2.2.11025
13. Pullen TJ, Huising MO, Rutter GA. Analysis of Purified Pancreatic Islet Beta and Alpha Cell Transcriptomes Reveals 11β-Hydroxysteroid Dehydrogenase (Hsd11b1) as a Novel Disallowed Gene. Front Genet (2017) 8:41. doi: 10.3389/fgene.2017.00041
14. Rutter GA, Hodson DJ. Beta cell connectivity in pancreatic islets: a type 2 diabetes target? Cell Mol Life Sci (2015) 72(3):453–67. doi: 10.1007/s00018-014-1755-4
15. Rutter GA, Georgiadou E, Martinez-Sanchez A, Pullen TJ. Metabolic and functional specialisations of the pancreatic beta cell: gene disallowance, mitochondrial metabolism and intercellular connectivity. Diabetologia (2020) 63(10):1990–8. doi: 10.1007/s00125-020-05205-5
16. Benninger RKP, Dorrell C, Hodson DJ, Rutter GA. The Impact of Pancreatic Beta Cell Heterogeneity on Type 1 Diabetes Pathogenesis. Curr Diabetes Rep (2018) 18(11):112. doi: 10.1007/s11892-018-1085-2
17. Miguel-Escalada I, Bonàs-Guarch S, Cebola I, Ponsa-Cobas J, Mendieta-Esteban J, Atla G, et al. Human pancreatic islet three-dimensional chromatin architecture provides insights into the genetics of type 2 diabetes. Nat Genet (2019) 51(7):1137–48. doi: 10.1038/s41588-019-0457-0
18. Billings LK, Florez JC. The genetics of type 2 diabetes: what have we learned from GWAS? Ann N Y Acad Sci (2010) 1212:59–77. doi: 10.1111/j.1749-6632.2010.05838.x
19. Fadista J, Vikman P, Laakso EO, Mollet IG, Esguerra JL, Taneera J, et al. Global genomic and transcriptomic analysis of human pancreatic islets reveals novel genes influencing glucose metabolism. Proc Natl Acad Sci U S A (2014) 111(38):13924–9. doi: 10.1073/pnas.1402665111
20. Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet (2007) 39(7):857–64. doi: 10.1038/ng2068
21. Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet (2010) 42(7):579–89. doi: 10.1038/ng.609
22. Aashiq M, Simranjeet K, Flemming P. Long non-coding RNAs as novel players in β cell function and type 1 diabetes. Hum Genomics (2017) 11:17. doi: 10.1186/s40246-017-0113-7
23. Sathishkumar C, Prabu P, Mohan V, Balasubramanyam M. Linking a role of lncRNAs (long non-coding RNAs) with insulin resistance, accelerated senescence, and inflammation in patients with type 2 diabetes. Hum Genomics (2018) 12(1):41. doi: 10.1186/s40246-018-0173-3
24. Wong WKM, Sørensen AE, Joglekar MV, Hardikar AA, Dalgaard LT. Non-Coding RNA in Pancreas and β-Cell Development. Noncod RNA (2018) 4(4):41. doi: 10.3390/ncrna4040041
25. Singer RA, Arnes L, Sussel L. Noncoding RNAs in β cell biology. Curr Opin Endocrinol Diabetes Obes (2015) 22(2):77–85. doi: 10.1097/MED.0000000000000141
26. Esguerra JL, Eliasson L. Functional implications of long non-coding RNAs in the pancreatic islets of Langerhans. Front Genet (2014) 5:209. doi: 10.3389/fgene.2014.00209
27. Lanzafame M, Bianco G, Terracciano LM, Ng CKY, Piscuoglio S. The Role of Long Non-Coding RNAs in Hepatocarcinogenesis. Int J Mol Sci (2018) 19(3).:682 doi: 10.3390/ijms19030682
28. Salviano-Silva A, Lobo-Alves SC, Almeida RC, Malheiros D, Petzl-Erler ML. Besides Pathology: Long Non-Coding RNA in Cell and Tissue Homeostasis. Noncod RNA (2018) 4(1):3. doi: 10.3390/ncrna4010003
29. Krchnáková Z, Thakur PK, Krausová M, Bieberstein N, Haberman N, Müller-McNicoll M, et al. Splicing of long non-coding RNAs primarily depends on polypyrimidine tract and 5’ splice-site sequences due to weak interactions with SR proteins. Nucleic Acids Res (2019) 47(2):911–28. doi: 10.1093/nar/gky1147
30. Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res (2012) 22(9):1775–89. doi: 10.1101/gr.132159.111
31. Morán I, Akerman I, van de Bunt M, Xie R, Benazra M, Nammo T, et al. Human β cell transcriptome analysis uncovers lncRNAs that are tissue-specific, dynamically regulated, and abnormally expressed in type 2 diabetes. Cell Metab (2012) 16(4):435–48. doi: 10.1016/j.cmet.2012.08.010
32. Motterle A, Gattesco S, Caille D, Meda P, Regazzi R. Involvement of long non-coding RNAs in beta cell failure at the onset of type 1 diabetes in NOD mice. Diabetologia (2015) 58(8):1827–35. doi: 10.1007/s00125-015-3641-5
33. Esguerra JLS, Ofori JK, Nagao M, Shuto Y, Karagiannopoulos A, Fadista J, et al. Glucocorticoid induces human beta cell dysfunction by involving riborepressor GAS5 LincRNA. Mol Metab (2020) 32:160–7. doi: 10.1016/j.molmet.2019.12.012
34. Guay C, Abdulkarim B, Tan JY, Dubuis G, Rütti S, Laybutt DR, et al. Loss-of-function of the long non-coding RNA A830019P07Rik in mice does not affect insulin expression and secretion. Sci Rep (2020) 10(1):6413. doi: 10.1038/s41598-020-65550-8
35. Motterle A, Gattesco S, Peyot ML, Esguerra JLS, Gomez-Ruiz A, Laybutt DR, et al. Identification of islet-enriched long non-coding RNAs contributing to β-cell failure in type 2 diabetes. Mol Metab (2017) 6(11):1407–18. doi: 10.1016/j.molmet.2017.08.005
36. Akerman I, Tu Z, Beucher A, Rolando DMY, Sauty-Colace C, Benazra M, et al. Human Pancreatic β Cell lncRNAs Control Cell-Specific Regulatory Networks. Cell Metab (2017) 25(2):400–11. doi: 10.1016/j.cmet.2016.11.016
37. Geng G, Zhang Z, Cheng L. Identification of a Multi-Long Noncoding RNA Signature for the Diagnosis of Type 1 Diabetes Mellitus. Front Bioeng Biotechnol (2020) 8:553. doi: 10.3389/fbioe.2020.00553
38. Chen LL. Linking Long Noncoding RNA Localization and Function. Trends Biochem Sci (2016) 41(9):761–72. doi: 10.1016/j.tibs.2016.07.003
39. Yao RW, Wang Y, Chen LL. Cellular functions of long noncoding RNAs. Nat Cell Biol (2019) 21(5):542–51. doi: 10.1038/s41556-019-0311-8
40. Xiong L, Gong Y, Wu L, Li J, He W, Zhu X, et al. LncRNA-Malat1 is Involved in Lipotoxicity-Induced ß-cell Dysfunction and the Therapeutic Effect of Exendin-4 via Ptbp1. Endocrinology (2020) 161(7):bqaa065. doi: 10.1210/endocr/bqaa065
41. Noh JH, Kim KM, McClusky WG, Abdelmohsen K, Gorospe M. Cytoplasmic functions of long noncoding RNAs. Wiley Interdiscip Rev RNA (2018) 9(3):e1471. doi: 10.1002/wrna.1471
42. Cabili MN, Dunagin MC, McClanahan PD, Biaesch A, Padovan-Merhar O, Regev A, et al. Localization and abundance analysis of human lncRNAs at single-cell and single-molecule resolution. Genome Biol (2015) 16:20. doi: 10.1186/s13059-015-0586-4
43. Rashid F, Shah A, Shan G. Long Non-coding RNAs in the Cytoplasm. Genomics Proteomics Bioinf (2016) 14(2):73–80. doi: 10.1016/j.gpb.2016.03.005
44. Carlevaro-Fita J, Johnson R. Global Positioning System: Understanding Long Noncoding RNAs through Subcellular Localization. Mol Cell (2019) 73(5):869–83. doi: 10.1016/j.molcel.2019.02.008
45. Bergmann JH, Spector DL. Long non-coding RNAs: modulators of nuclear structure and function. Curr Opin Cell Biol (2014) 26:10–8. doi: 10.1016/j.ceb.2013.08.005
46. van Heesch S, van Iterson M, Jacobi J, Boymans S, Essers PB, de Bruijn E, et al. Extensive localization of long noncoding RNAs to the cytosol and mono- and polyribosomal complexes. Genome Biol (2014) 15(1):R6. doi: 10.1186/gb-2014-15-1-r6
47. Yoon JH, Abdelmohsen K, Kim J, Yang X, Martindale JL, Tominaga-Yamanaka K, et al. Scaffold function of long non-coding RNA HOTAIR in protein ubiquitination. Nat Commun (2013) 4:2939. doi: 10.1038/ncomms3939
48. Miao H, Wang L, Zhan H, Dai J, Chang Y, Wu F, et al. A long noncoding RNA distributed in both nucleus and cytoplasm operates in the PYCARD-regulated apoptosis by coordinating the epigenetic and translational regulation. PLoS Genet (2019) 15(5):e1008144. doi: 10.1371/journal.pgen.1008144
49. Amodio N, Raimondi L, Juli G, Stamato MA, Caracciolo D, Tagliaferri P, et al. MALAT1: a druggable long non-coding RNA for targeted anti-cancer approaches. J Hematol Oncol (2018) 11(1):63. doi: 10.1186/s13045-018-0606-4
50. Kaneko S, Li G, Son J, Xu CF, Margueron R, Neubert TA, et al. Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes Dev (2010) 24(23):2615–20. doi: 10.1101/gad.1983810
51. Biswas A, Shettar A, Mukherjee G, Kondaiah P, Desai KV. JMJD6 induces HOTAIR, an oncogenic lincRNA, by physically interacting with its proximal promoter. Biochem J (2018) 475(1):355–71. doi: 10.1042/BCJ20170664
52. Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell (2010) 39(6):925–38. doi: 10.1016/j.molcel.2010.08.011
53. Ding H, Wang F, Shi X, Ma H, Du Y, Hou L, et al. LncRNA MALAT1 induces the dysfunction of β cells via reducing the histone acetylation of the PDX-1 promoter in type 1 diabetes. Exp Mol Pathol (2020) 114:104432. doi: 10.1016/j.yexmp.2020.104432
54. Sun Q, Xu H, Xue J, Yang Q, Chen C, Yang P, et al. MALAT1 via microRNA-17 regulation of insulin transcription is involved in the dysfunction of pancreatic β-cells induced by cigarette smoke extract. J Cell Physiol (2018) 233(11):8862–73. doi: 10.1002/jcp.26800
55. Bouwens L. Beta cell regeneration. Curr Diabetes Rev (2006) 2(1):3–9. doi: 10.2174/157339906775473644
56. Karaca M, Magnan C, Kargar C. Functional pancreatic beta-cell mass: involvement in type 2 diabetes and therapeutic intervention. Diabetes Metab (2009) 35(2):77–84. doi: 10.1016/j.diabet.2008.09.007
57. Wang RN, Bouwens L, Klöppel G. Beta-cell proliferation in normal and streptozotocin-treated newborn rats: site, dynamics and capacity. Diabetologia (1994) 37(11):1088–96. doi: 10.1007/s001250050221
58. López-Noriega L, Cobo-Vuilleumier N, Narbona-Pérez Á, Araujo-Garrido JL, Lorenzo PI, Mellado-Gil JM, et al. Levothyroxine enhances glucose clearance and blunts the onset of experimental type 1 diabetes mellitus in mice. Br J Pharmacol (2017) 174(21):3795–810. doi: 10.1111/bph.13975
59. Salpeter SJ, Klein AM, Huangfu D, Grimsby J, Dor Y. Glucose and aging control the quiescence period that follows pancreatic beta cell replication. Development (2010) 137(19):3205–13. doi: 10.1242/dev.054304
60. Kim H, Toyofuku Y, Lynn FC, Chak E, Uchida T, Mizukami H, et al. Serotonin regulates pancreatic beta cell mass during pregnancy. Nat Med (2010) 16(7):804–8. doi: 10.1038/nm.2173
61. Gianani R. Beta cell regeneration in human pancreas. Semin Immunopathol (2011) 33(1):23–7. doi: 10.1007/s00281-010-0235-7
62. Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Evidence of increased islet cell proliferation in patients with recent-onset type 1 diabetes. Diabetologia (2010) 53(9):2020–8. doi: 10.1007/s00125-010-1817-6
63. Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Immunohistochemical analysis of the relationship between islet cell proliferation and the production of the enteroviral capsid protein, VP1, in the islets of patients with recent-onset type 1 diabetes. Diabetologia (2011) 54(9):2417–20. doi: 10.1007/s00125-011-2192-7
64. Dirice E, De Jesus DF, Kahraman S, Basile G, Ng RWS, El Ouaamari A, et al. Human duct cells contribute to β cell compensation in insulin resistance. JCI Insight (2019) 4(8):e99576. doi: 10.1172/jci.insight.99576
65. Guney MA, Lorberbaum DS, Sussel L. Pancreatic β cell regeneration: To β or not to β. Curr Opin Physiol (2020) 14:13–20. doi: 10.1016/j.cophys.2019.10.019
66. Butler AE, Cao-Minh L, Galasso R, Corradin A, Rizza A, Corradin RA, et al. Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia (2010) 53:2167–76. doi: 10.1007/s00125-010-1809-6
67. Linnemann AK, Baan M, Davis DB. Pancreatic β-cell proliferation in obesity. Adv Nutr (2014) 5(3):278–88. doi: 10.3945/an.113.005488
68. Hanley S, Austin E, Assouline-Thomas B, Kapeluto J, Blaichman J, Moosavi M, et al. Beta-cell mass dynamics and islet cell plasticity in human type 2 diabetes. Endocrinology (2010) 151:1462–72. doi: 10.1210/en.2009-1277
69. Folli F, Okada T, Perego C, Gunton J, Liew CW, Akiyama M, et al. Altered insulin receptor signalling and β-cell cycle dynamics in type 2 diabetes mellitus. PLoS One (2011) 6(11):e28050. doi: 10.1371/journal.pone.0028050
70. Wang P, Fiaschi-Taesch NM, Vasavada RC, Scott DK, García-Ocaña A, Stewart AF. Diabetes mellitus–advances and challenges in human β-cell proliferation. Nat Rev Endocrinol (2015) 11(4):201–12. doi: 10.1038/nrendo.2015.9
71. Wang P, Karakose E, Liu H, Swartz E, Ackeifi C, Zlatanic V, et al. Combined Inhibition of DYRK1A, SMAD, and Trithorax Pathways Synergizes to Induce Robust Replication in Adult Human Beta Cells. Cell Metab (2019) 29(3):638–52.e5. doi: 10.1016/j.cmet.2018.12.005
72. Sisino G, Zhou AX, Dahr N, Sabirsh A, Soundarapandian MM, Perera R, et al. Long noncoding RNAs are dynamically regulated during β-cell mass expansion in mouse pregnancy and control β-cell proliferation in vitro. PLoS One (2017) 12(8):e0182371. doi: 10.1371/journal.pone.0182371
73. Sanchez-Parra C, Jacovetti C, Dumortier O, Lee K, Peyot ML, Guay C, et al. Contribution of the Long Noncoding RNA H19 to β-Cell Mass Expansion in Neonatal and Adult Rodents. Diabetes (2018) 67(11):2254–67. doi: 10.2337/db18-0201
74. Ding GL, Wang FF, Shu J, Tian S, Jiang Y, Zhang D, et al. Transgenerational glucose intolerance with Igf2/H19 epigenetic alterations in mouse islet induced by intrauterine hyperglycemia. Diabetes (2012) 61(5):1133–42. doi: 10.2337/db11-1314
75. Ohshige T, Iwata M, Omori S, Tanaka Y, Hirose H, Kaku K, et al. Association of new loci identified in European genome-wide association studies with susceptibility to type 2 diabetes in the Japanese. PLoS One (2011) 6(10):e26911. doi: 10.1371/journal.pone.0026911
76. Travers ME, Mackay DJ, Dekker Nitert M, Morris AP, Lindgren CM, Berry A, et al. Insights into the molecular mechanism for type 2 diabetes susceptibility at the KCNQ1 locus from temporal changes in imprinting status in human islets. Diabetes (2013) 62(3):987–92. doi: 10.2337/db12-0819
77. Asahara S, Etoh H, Inoue H, Teruyama K, Shibutani Y, Ihara Y, et al. Paternal allelic mutation at the Kcnq1 locus reduces pancreatic β-cell mass by epigenetic modification of Cdkn1c. Proc Natl Acad Sci U S A (2015) 112(27):8332–7. doi: 10.1073/pnas.1422104112
78. Kong Y, Sharma RB, Ly S, Stamateris RE, Jesdale WM, Alonso LC. T2D Genome-Wide Association Study Risk SNPs Impact Locus Gene Expression and Proliferation in Human Islets. Diabetes (2018) 67(5):872–84. doi: 10.2337/db17-1055
79. Bouwens L, Rooman I. Regulation of pancreatic beta-cell mass. Physiol Rev (2005) 85(4):1255–70. doi: 10.1152/physrev.00025.2004
80. Laurent D, Vinet L, Lamprianou S, Daval M, Filhoulaud G, Ktorza A, et al. Pancreatic β-cell imaging in humans: fiction or option? Diabetes Obes Metab (2016) 18(1):6–15. doi: 10.1111/dom.12544
81. Martinez-Sanchez A, Rutter GA, Latreille M. MiRNAs in β-Cell Development, Identity, and Disease. Front Genet (2016) 7:226. doi: 10.3389/fgene.2016.00226
82. Sun T, Han X. Death versus dedifferentiation: The molecular bases of beta cell mass reduction in type 2 diabetes. Semin Cell Dev Biol (2020) 103:76–82. doi: 10.1016/j.semcdb.2019.12.002
83. Yin DD, Zhang EB, You LH, Wang N, Wang LT, Jin FY, et al. Downregulation of lncRNA TUG1 affects apoptosis and insulin secretion in mouse pancreatic β cells. Cell Physiol Biochem (2015) 35(5):1892–904. doi: 10.1159/000373999
84. You L, Wang N, Yin D, Wang L, Jin F, Zhu Y, et al. Downregulation of Long Noncoding RNA Meg3 Affects Insulin Synthesis and Secretion in Mouse Pancreatic Beta Cells. J Cell Physiol (2016) 231(4):852–62. doi: 10.1002/jcp.25175
85. Wang N, Zhu Y, Xie M, Wang L, Jin F, Li Y, et al. Long Noncoding RNA Meg3 Regulates Mafa Expression in Mouse Beta Cells by Inactivating Rad21, Smc3 or Sin3α. Cell Physiol Biochem (2018) 45(5):2031–43. doi: 10.1159/000487983
86. Kameswaran V, Bramswig NC, McKenna LB, Penn M, Schug J, Hand NJ, et al. Epigenetic regulation of the DLK1-MEG3 microRNA cluster in human type 2 diabetic islets. Cell Metab (2014) 19(1):135–45. doi: 10.1016/j.cmet.2013.11.016
87. Wallace C, Smyth DJ, Maisuria-Armer M, Walker NM, Todd JA, Clayton DG. The imprinted DLK1-MEG3 gene region on chromosome 14q32.2 alters susceptibility to type 1 diabetes. Nat Genet (2010) 42(1):68–71. doi: 10.1038/ng.493
88. Kong X, Liu CX, Wang GD, Yang H, Yao XM, Hua Q, et al. LncRNA LEGLTBC Functions as a ceRNA to Antagonize the Effects of miR-34a on the Downregulation of SIRT1 in Glucolipotoxicity-Induced INS-1 Beta Cell Oxidative Stress and Apoptosis. Oxid Med Cell Longev (2019) 2019:4010764. doi: 10.1155/2019/4010764
89. Fu B, Li X, Sun R, Tong X, Ling B, Tian Z, et al. Natural killer cells promote immune tolerance by regulating inflammatory T(H)17 cells at the human maternal-fetal interface. Proc Natl Acad Sci U S A (2013) 110(3):E231–E40. doi: 10.1073/pnas.1206322110
90. Cheng Y, Hu Q, Zhou J. Silencing of lncRNA PVT1 ameliorates streptozotocin-induced pancreatic β cell injury and enhances insulin secretory capacity via regulating miR-181a-5p. Can J Physiol Pharmacol (2020). doi: 10.1139/cjpp-2020-0268
91. Gonzalez-Moro I, Olazagoitia-Garmendia A, Colli ML, Cobo-Vuilleumier N, Postler TS, Marselli L, et al. The T1D-associated lncRNA. Proc Natl Acad Sci U S A (2020) 117(16):9022–31. doi: 10.1073/pnas.1914353117
92. Moore F, Naamane N, Colli ML, Bouckenooghe T, Ortis F, Gurzov EN, et al. STAT1 is a master regulator of pancreatic {beta}-cell apoptosis and islet inflammation. J Biol Chem (2011) 286(2):929–41. doi: 10.1074/jbc.M110.162131
93. Kaur S, Mirza AH, Brorsson CA, Fløyel T, Størling J, Mortensen HB, et al. The genetic and regulatory architecture of ERBB3-type 1 diabetes susceptibility locus. Mol Cell Endocrinol (2016) 419:83–91. doi: 10.1016/j.mce.2015.10.002
94. Tersey SA, Nishiki Y, Templin AT, Cabrera SM, Stull ND, Colvin SC, et al. Islet β-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes (2012) 61(4):818–27. doi: 10.2337/db11-1293
95. Sims EK, Mirmira RG, Evans-Molina C. The role of beta-cell dysfunction in early type 1 diabetes. Curr Opin Endocrinol Diabetes Obes (2020) 27(4):215–24. doi: 10.1097/MED.0000000000000548
96. Evans-Molina C, Sims EK, DiMeglio LA, Ismail HM, Steck AK, Palmer JP, et al. β Cell dysfunction exists more than 5 years before type 1 diabetes diagnosis. JCI Insight (2018) 3(15). doi: 10.1172/jci.insight.120877
97. Neelankal John A, Ram R, Jiang FX. RNA-Seq Analysis of Islets to Characterise the Dedifferentiation in Type 2 Diabetes Model Mice db/db. Endocr Pathol (2018) 29(3):207–21. doi: 10.1007/s12022-018-9523-x
98. Moin ASM, Butler AE. Alterations in Beta Cell Identity in Type 1 and Type 2 Diabetes. Curr Diabetes Rep (2019) 19(9):83. doi: 10.1007/s11892-019-1194-6
99. Thompson P, Bhushan A. β Cells led astray by transcription factors and the company they keep. J Clin Invest (2017) 127(1):94–7. doi: 10.1172/JCI91304
100. Arnes L, Akerman I, Balderes DA, Ferrer J, Sussel L. βlinc1 encodes a long noncoding RNA that regulates islet β-cell formation and function. Genes Dev (2016) 30(5):502–7. doi: 10.1101/gad.273821.115
101. Singer RA, Arnes L, Cui Y, Wang J, Gao Y, Guney MA, et al. The Long Noncoding RNA Paupar Modulates PAX6 Regulatory Activities to Promote Alpha Cell Development and Function. Cell Metab (2019) 30(6):1091–106.e8. doi: 10.1016/j.cmet.2019.09.013
102. Lopez-Noriega L, Callingham R, Martinez-Sánchez A, Pizza G, Haberman N, Cvetesic N, et al. The long non-coding RNA Pax6os1/PAX6-AS1 modulates pancreatic β-cell identity and function. BiorXiv (Prepr) (2020). doi: 10.1101/2020.07.17.209015
103. Mitchell RK, Nguyen-Tu MS, Chabosseau P, Callingham RM, Pullen TJ, Cheung R, et al. The transcription factor Pax6 is required for pancreatic β cell identity, glucose-regulated ATP synthesis, and Ca2+ dynamics in adult mice. J Biol Chem (2017) 292(21):8892–906. doi: 10.1074/jbc.M117.784629
104. Swisa A, Avrahami D, Eden N, Zhang J, Feleke E, Dahan T, et al. PAX6 maintains β cell identity by repressing genes of alternative islet cell types. J Clin Invest (2017) 127(1):230–43. doi: 10.1172/JCI88015
105. Zhang FF, Liu YH, Wang DW, Liu TS, Yang Y, Guo JM, et al. Obesity-induced reduced expression of the lncRNA ROIT impairs insulin transcription by downregulation of Nkx6.1 methylation. Diabetologia (2020) 63(4):811–24. doi: 10.1007/s00125-020-05090-y
106. Thielen L, Shalev A. Diabetes pathogenic mechanisms and potential new therapies based upon a novel target called TXNIP. Curr Opin Endocrinol Diabetes Obes (2018) 25(2):75–80. doi: 10.1097/MED.0000000000000391
107. Taylor BL, Liu FF, Sander M. Nkx6.1 is essential for maintaining the functional state of pancreatic beta cells. Cell Rep (2013) 4(6):1262–75. doi: 10.1016/j.celrep.2013.08.010
108. Huang Y, Xu Y, Lu Y, Zhu S, Guo Y, Sun C, et al. lncRNA Gm10451 regulates PTIP to facilitate iPSCs-derived β-like cell differentiation by targeting miR-338-3p as a ceRNA. Biomaterials (2019) 216:119266. doi: 10.1016/j.biomaterials.2019.119266
109. Carter G, Miladinovic B, Patel AA, Deland L, Mastorides S, Patel NA. Circulating long noncoding RNA GAS5 levels are correlated to prevalence of type 2 diabetes mellitus. BBA Clin (2015) 4:102–7. doi: 10.1016/j.bbacli.2015.09.001
110. Jin F, Wang N, Zhu Y, You L, Wang L, De W, et al. Downregulation of Long Noncoding RNA Gas5 Affects Cell Cycle and Insulin Secretion in Mouse Pancreatic β Cells. Cell Physiol Biochem (2017) 43(5):2062–73. doi: 10.1159/000484191
111. Kino T, Hurt DE, Ichijo T, Nader N, Chrousos GP. Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci Signal (2010) 3(107):ra8. doi: 10.1126/scisignal.2000568
112. Ruan Y, Lin N, Ma Q, Chen R, Zhang Z, Wen W, et al. Circulating LncRNAs Analysis in Patients with Type 2 Diabetes Reveals Novel Genes Influencing Glucose Metabolism and Islet β-Cell Function. Cell Physiol Biochem (2018) 46(1):335–50. doi: 10.1159/000488434
113. Cao Z, Yao F, Lang Y, Feng X. Elevated Circulating LINC-P21 Serves as a Diagnostic Biomarker of Type 2 Diabetes Mellitus and Regulates Pancreatic β-cell Function by Sponging miR-766-3p to Upregulate NR3C2. Exp Clin Endocrinol Diabetes (2020). doi: 10.1055/a-1247-4978
114. Pullen TJ, Rutter GA. Roles of lncRNAs in pancreatic beta cell identity and diabetes susceptibility. Front Genet (2014) 5:193. doi: 10.3389/fgene.2014.00193
115. Roberts TC, Langer R, Wood MJA. Advances in oligonucleotide drug delivery. Nat Rev Drug Discov (2020) 19(10):673–94. doi: 10.1038/s41573-020-0075-7
116. Arun G, Diermeier SD, Spector DL. Therapeutic Targeting of Long Non-Coding RNAs in Cancer. Trends Mol Med (2018) 24(3):257–77. doi: 10.1016/j.molmed.2018.01.001
117. Lavie O, Edelman D, Levy T, Fishman A, Hubert A, Segev Y, et al. A phase 1/2a, dose-escalation, safety, pharmacokinetic, and preliminary efficacy study of intraperitoneal administration of BC-819 (H19-DTA) in subjects with recurrent ovarian/peritoneal cancer. Arch Gynecol Obstet (2017) 295(3):751–61. doi: 10.1007/s00404-017-4293-0
Keywords: lncRNA, beta cell, diabetes, islet, insulin
Citation: López–Noriega L and Rutter GA (2021) Long Non-Coding RNAs as Key Modulators of Pancreatic β-Cell Mass and Function. Front. Endocrinol. 11:610213. doi: 10.3389/fendo.2020.610213
Received: 25 September 2020; Accepted: 21 December 2020;
Published: 08 February 2021.
Edited by:
Anca Dana Dobrian, Eastern Virginia Medical School, United StatesReviewed by:
Jeffery Sivert Tessem, Brigham Young University, United StatesCopyright © 2021 López–Noriega and Rutter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guy A. Rutter, Zy5ydXR0ZXJAaW1wZXJpYWwuYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.