 Nicolas Chevalier
Nicolas Chevalier Charlotte Hinault1,2
Charlotte Hinault1,2 Rachel Paul-Bellon
Rachel Paul-Bellon Patrick Fenichel
Patrick Fenichel
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol. , 26 January 2021
Sec. Molecular and Structural Endocrinology
Volume 11 - 2020 | https://doi.org/10.3389/fendo.2020.600404
This article is part of the Research Topic GPER and Human Pathologies View all 11 articles
The G protein-coupled estrogen receptor (GPER), also known as GPR30, is a widely conserved 7-transmembrane-domain protein which has been identified as a novel 17β-estradiol-binding protein that is structurally distinct from the classic oestrogen receptors (ERα and ERβ). There are still conflicting data regarding the exact role and the natural ligand of GPER/GPR30 in reproductive tracts as both male and female knock-out mice are fertile and have no abnormalities of reproductive organs. Testicular germ cell cancers (TGCCs) are the most common malignancy in young males and the most frequent cause of death from solid tumors in this age group. Clinical and experimental studies suggested that estrogens participate in the physiological and pathological control of male germ cell proliferation. In human seminoma cell line, while 17β-estradiol (E2) inhibits in vitro cell proliferation through an ERβ-dependent mechanism, an impermeable E2 conjugate (E2 coupled to BSA), in vitro cell proliferation is stimulated by activating ERK1/2 and protein kinase A through a membrane GPCR that we further identified as GPER/GPR30. The same effect was observed with low but environmentally relevant doses of BPA, an estrogenic endocrine disrupting compound. Furthermore, GPER/GPR30 is specifically overexpressed in seminomas but not in non-seminomas and this overexpression is correlated with an ERβ-downregulation. This GPER/GPR30 overexpression could be linked to some genetic variations, as single nucleotide polymorphisms, which was also reported in other hormone-dependent cancers. We will review here the implication of GPER/GPR30 in TGCCs pathophysiology and the arguments to consider GPER/GPR30 as a potential therapeutic target in humans.
Although relatively rare, testicular germ cell cancers (TGCC) are the most frequent solid cancer in young people (1, 2). Seminomas represent the most frequent histological form, occurring alone or associated with non-seminoma forms in 50-75% of cases (1, 2). Incidence rates of TGCC have been increasing worldwide for several decades (3, 4).
Risk factors for TGCC are described in Table 1 and are mainly genetic. Indeed, incidence of TGCC is significantly increased in brothers and sons of TGCC patients (5, 7). Consistent with many epidemiological studies, gene variants that might predispose an individual to TGCC were identified by genome-wide association studies (GWAS) (8, 9). These variants included common variations on 12q22 in the KITLG gene, but also on PDE11A, BAK1, SPRY, DMRT1, DAZL, and PRDM14 [reviewed in (10)]. Other classical risk factors are cryptorchidism (or undescended testis), inguinal hernia, and all sexual differentiation disorders (6, 11) (Table 1).
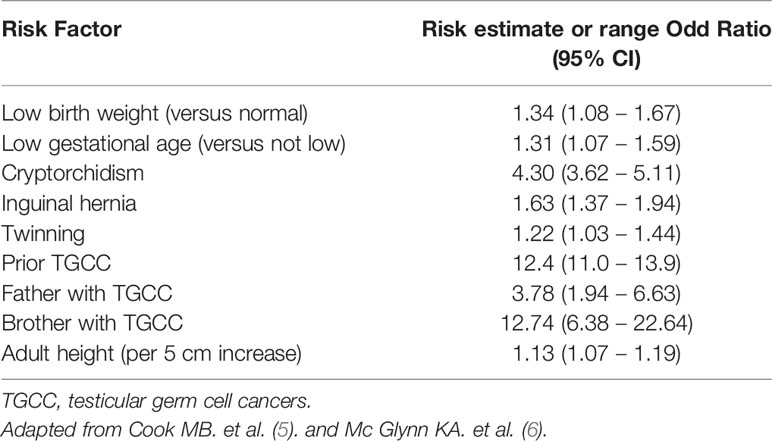
Table 1 Usual risk factors of testicular germ cell cancers.
TGCC are considered to derive from a precursor lesion named “carcinoma in situ of the testis” or “germ cell neoplasia in situ” (GCNIS) (12). This lesion is present before birth, arising from the fetal germ cells (i.e. the gonocytes), and is reactivated after puberty under physiological hormonal stimulation (13). Epidemiological and clinical data have suggested that the increase of TGCC incidence could be related to environmental factors such as fetal exposure to endocrine disruptors (EDCs) with anti-androgenic and/or estrogenic effects (14, 15). However, this hypothesis supposes that TGCCs are estrogen-dependent tumors. In this review, we analyze the implication of classical and non-classical (GPER/GPR30) estrogen receptors in normal and malignant germ cells and the regulation of cell proliferation by xeno-estrogens and discuss how GPER/GPR30 could be considered as a potential therapeutic target in humans.
Several studies have reported abnormalities of male genital tracts in animals that were accidently exposed to endocrine disruptors, such as hypospadias and cryptorchidism in alligators (16) or panthers (17), especially in the case of exposition to the organochloride dichlorodiphenyltrichloroethane (DDT) or its metabolites (DDE, DDD), which exhibit estrogenic properties. However, there is actually no animal model of TGCC, except for transgenic mice with targeted overexpression of GDNF in spermatogonia (18).
In humans, early fetal exposure to diethylstilbestrol (DES), a synthetic estrogen used during the 1960’s, was responsible for an increased incidence rate of cryptorchidism and hypofertility by impairment of sperm quality in sons and in grandsons (19, 20). Such an exposure was also suggested to be responsible for the occurrence of TGCC in the offspring of two meta-analysis (21, 22). In past studies, the association between occupational exposure and risk to develop TGCC (23–25) was well-documented and offered suggestive or strong arguments. However, more recent epidemiological case-control studies reported conflicting data for fetal exposure to p,p′-DDT (estrogenic compound) or to p,p′-DDE (a stable metabolite of DDT with antiandrogenic properties) (26–31).
Testicular concentrations of 17β-estradiol (E2) are 10 to 100 times higher than those measured in blood (32). E2 is produced after testosterone conversion by aromatase in all mammalian testes, including humans (33). Estrogens are essential for spermatogenesis control but the type of estrogen receptors involved and the molecular mechanisms by which estrogens may precisely act during spermatogenesis still remain incompletely understood (34).
Expression of classical and non-classical estrogen receptors expression in mammalian testes is well-established. It exhibits some species specificity and some controversial results, especially in humans [reviewed in (35)]. Indeed, in humans, the classical nuclear estrogen receptor ERβ has been clearly identified in most germ cells, including fetal gonocytes (36), neonatal, prepubertal (37), and adult spermatogonia (38), while ERα is not expressed in human gonocytes (36) or neonatal or prepubertal spermatogonia (37). However, data concerning the expression of ERα by male germ cells are inconsistent, as some authors reported an expression in elongated spermatids and mature spermatozoa (39) and others did not find any expression of ERα at all (38, 40). In fact, these inconsistent observations could be due to the existence of a truncated isoform of ERα lacking exon 1, called ERα46, which has been identified in human adult spermatozoa (41). This isoform could participate in non-genomic membrane signaling. Indeed, one reported case of a man with an inactivating mutation of ERα gene was associated with a normal sperm count but with completely abnormal motility (42).
GPR30 is a widely conserved orphan GPCR, which has been renamed as G protein-coupled estrogen receptor (GPER) (HUGO & MGI Databases). It is a seven-transmembrane domain protein, identified for the first time in a triple-negative breast cancer cell line, that can bind E2 and other estrogenic compounds independently of the classic estrogen receptors (ERα and ERβ). The precise subcellular localization of GPER/GPR30 is still a matter of debate as it has been detected at the plasma membrane but also in the endoplasmic reticulum and Golgi apparatus (43).
GPER/GPR30 has been identified in numerous rodents and human estrogen targets normal or malignant tissues where it can mediate rapid E2-induced non genomic signaling events (43). GPER/GPR30 can activate cell proliferation through several signaling pathways involving MAP kinases, ERK1/2, and PI3K pathways (44, 45) but also microRNA regulation (46–48), EGFR transactivation (49, 50), HIF induced pathway (51, 52), IGF-R pathway (53, 54), NF-kB pathway (55, 56), and crosstalk with other receptors (classical or truncated estrogen receptors, or other steroids receptors) (57–59). Within those pathways, the activation of ERK1/2 is undoubtedly the most consistent pathway across cell types and is usually considered as a key factor in cancer prognosis.
Analyzing normal human testes from a fertile man, we previously reported that GPER/GPR30 was expressed by both somatic (Sertoli and Leydig cells) and germ cells (60). Amazingly, Rago et al. (61). reported a negative staining in adult germ cells, probably due to the use of abnormal granulomatous testes. As expression of GPER/GPR30 in human fetal gonocytes has not yet been studied; it could be possible that only immature germ cells and gonocytes express GPER/GPR30, explaining these inconsistent data [reviewed in (62)].
Estrogen receptor expression is a well-recognized prognosis factor of estrogen-dependent cancers, especially in the case of breast cancer (63–65). Several teams have suggested that TGCCs could be estrogen-dependent cancers as they express both ERβ and GPER/GPR30 (66–70). We previously reported in a large cohort of TGCCs that GPER/GPR30 was overexpressed only in seminoma but not in non-seminoma tumors (60) and promoted seminoma cell proliferation (71). Pais et al. (72) reported that expression of ERβ was decreased in seminoma but remained high in teratomas. In the same way, Boscia et al. (69) showed that ERβ was downregulated in seminomas and reported a negative association between the expression of ERβ and GPER/GPR30 protein. This inverse receptor expression pattern could reflect a switch in estrogen responsiveness from a suppressive (66) to a promoting profile (60, 67), as it has also been observed in other estrogen-dependent cancers and was correlated to a poorer prognosis (63–65).
Genetic factors could of course explain this specific profile of expression. Variants of ERβ were explored but studies reported inconsistent data. Ferlin et al. (73) reported a weak but not significant association between one variant for ERβ and an increase risk of TGCC in Italian men, while Brokken et al. (74) described exactly the opposite in a cohort of 367 Nordic patients with TGCC and two other variants of the ERβ. In our large cohort of 169 TGCCs, we were able to describe that seminomas were characterized by a loss of homozygous ancestral genotype concerning two polymorphisms located in the promoter region of GPER/GPR30 (75). We assumed that this genotype could explain a part of GPER/GPR30 overexpression in seminomas. This expression profile could also be determined by epigenetic modulation of ERβ and GPER/GPR30 genes (low expression of ERβ due to an hypermethylation of its promoter and high expression of GPER/GPR30 gene due to an hypomethylation of its promoter). Indeed, fetal exposure to EDCs is supposed to induce such epigenetic modulation as reported, for example, by Zama et al. (76) who reported that fetal and neonatal exposure to the endocrine disruptor methoxychlor was responsible for a down regulation of ovarian ERβ gene expression.
Anway et al. (77) were the first to observe and to report several epigenetic modifications in rodent DNA male germ cells after gestational exposure to vinclozolin (antiandrogenic compound) or methoxychlor (estrogenic compound). These data have been recently confirmed by Dumasia et al. (78) for xenoestrogens signaling through ERβ. Since this first publication of Anway et al. (77) DNA methylation (hyper- and hypo-) (79, 80), onco-miRNAs expression (miR 371-373) (81, 82), or chromatin modifications have been reported in TGCC (83). However, even if experimental data in rodents suggested that these epigenetic modifications might be induced by fetal exposure to EDCs, it remains to be proven that such epigenetic modifications exist in humans and can be induced by fetal exposure to EDCs.
JKT-1 cell line is derived from a human testicular seminoma (84), which expressed functional aromatase (66) and is able to convert testosterone into E2 and as well as ERβ, but not ERα. At physiological concentrations (10-7 to 10-9 M), we previously reported that E2 was able to inhibit in vitro JKT-1 cell proliferation involving an ERβ pathway (66). We conjugated E2 to bovine serum albumin (E2-BSA) for the purpose that E2 cannot cross the plasma membrane and then cannot link to its canonical receptor ERβ. In this condition, E2-BSA at the same concentrations (10-7 to 10-9 M) stimulated in vitro JKT-1 cell proliferation by activating the ERK1/2 and PKA pathways. E2-BSA is responsible for a rapid (15 min) phosphorylation of CREB. This effect was not inhibited by ICI-182,780, an antagonist of ERβ, but by Pertussis toxin, suggestive of the involvement of a membrane G-protein-coupled receptor (GPCR). Similar results were obtained with bisphenol A (BPA) at low and very low (nM to pM) concentrations (85), the levels already found in male cord blood and in more than 95% of the worldwide population (86, 87).
Among EDCs, BPA is especially a matter of concern as populations exhibit worldwide with detectable blood and/or urine levels of BPA (86), and so it is used as a monomer to manufacture a wide range of objects containing polycarbonate plastic and resins. BPA is considered an estrogenic EDC and is recognized as a substance of very high concern (SVHC) by the European Chemicals Agency (ECHA) because several experimentations and data reported that it is involved in developmental, reproductive, and malignant diseases by mimicking the natural hormone E2 and by interfering with endogenous pathways at selective periods, especially during fetal life (88). However, BPA exhibits a weak affinity for the classical ERs, which is 1,000–2,000 times lower than E2. Thus, it has been suggested that BPA could act through other receptors than classical ERs, for example GPER/GPR30, PPARγ gamma, or ERRγ gamma (88).
In our JKT-1 seminoma cells model, we were able to identify the GPCR involved in the promoting action of E2-BSA and BPA as GPER/GPR30 (71). Indeed, the BPA-induced promotive effect was mimicked by G1 alone, a specific agonist of GPER/GPR30, while it was totally inhibited by G15, a partial antagonist of GPER/GPR30, as well as a selective anti-GPER/GPR30 siRNA (Figure 1A) (60, 71). This GPER/GPR30-mediated signaling of BPA was also reported in other hormone-dependent tumors. For example, Pupo et al. (90) reported that BPA could increase the proliferation of SKBr3 breast cancer cells, which lack the classical ERs, through a GPER/GPR30-EGFR/ERK transduction pathway.
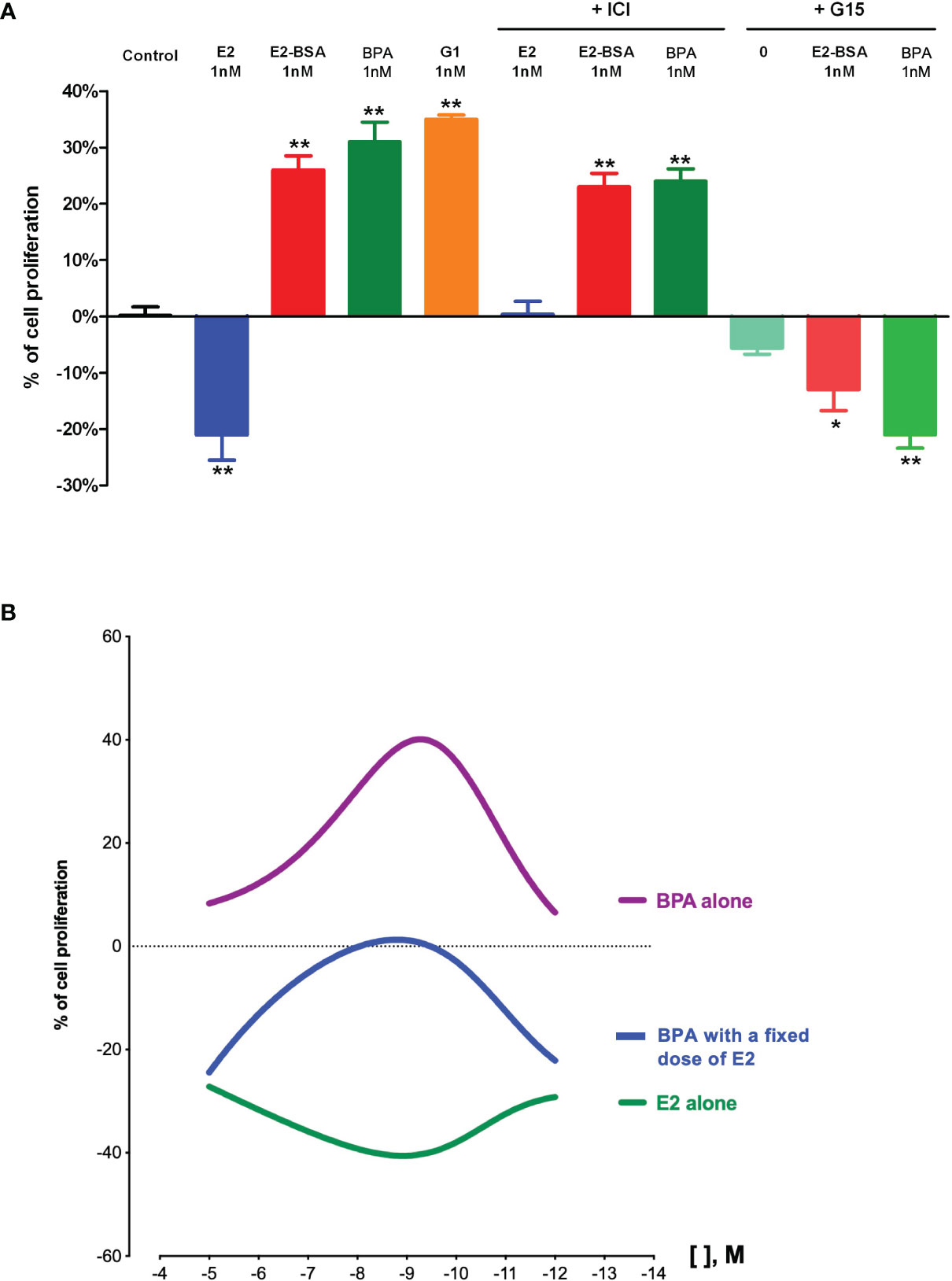
Figure 1 Effects of estrogens and bisphenol A on human testicular seminoma cell (JKT-1) proliferation in vitro. (A) Analysis of JKT-1 cells proliferation in vitro, adapted from Chevalier et al. (71) JKT-1 cells were seeded in six-well plates (0.6 × 106 cells/well). After 48 h, the JKT-1 cells were washed and estrogen starved overnight in phenol red-free DMEM (Dulbeccos’s Modified Eagle Medium) supplemented with 1% charcoal-stripped fetal bovine serum. Serum-deprived JKT-1 cells were then incubated for 24 hours with 17β-estradiol (E2; 1 nM), E2-BSA (1 nM), or bisphenol A (BPA; 1 nM), after a pre-treatment with G15 (1 nM) or ICI-182,780 (1 µM). G1 (1 nM) was used as a positive control. Values shown are expressed in percent change in cell number compared to control (steroid-free medium containing DMSO for bisphenol A or medium containing ethanol for estrogens, G1, and G15) given as the mean ± SE of at least three independent experiments. Cell counting was performed using a Malassez hemocytometer and confirmed using Vi-CELL automate (Beckman Coulter, Fullerton, CA). *p < 0.05; **p < 0.001. (B) Dose-response curves obtained with 17β-estradiol (E2) and bisphenol A (BPA) in JKT-1 cells in vitro, adapted from Fenichel et al. (89). and Bouskine et al. (85). JKT-1 cells were seeded in six-well plates (0.6 × 106 cells/well). After 48 h, the JKT-1 cells were washed and estrogen starved overnight in phenol red-free DMEM (Dulbeccos’s Modified Eagle Medium) supplemented with 1% charcoal-stripped fetal bovine serum. Serum-deprived JKT-1 cells were then incubated for 24 hours with 17β-estradiol (E2) alone or bisphenol A (BPA) alone at variable doses from 10-5 M to 10-12 M obtained by serial dilutions, or with a fixed dose of E2 (10-9 M) and BPA at variable doses (same range, from 10-5 M to 10-12 M). Values shown are expressed in percent change in cell number compared to control (steroid-free medium containing DMSO for bisphenol A or medium containing ethanol for estrogens) given as the mean ± SE of nine independent experiments for each condition. Cell counting was performed using a Malassez hemocytometer and confirmed using Vi-CELL automate (Beckman Coulter, Fullerton, CA). Modeling of dose-response curves were performed using GraphPad Prism version 8.4.3 for Mac OS X, GraphPad Software, San Diego, California USA, www.graphpad.com.
Interestingly, the dose-response curve that we obtained for BPA in our model was non-monotonic and showed an inverted U-shape curve (Figure 1B). Non-monotonic dose response curves (NMDRC) have already been reported and well-documented for natural hormones. NMDRC have also been suggested for EDCs, especially in the case of BPA, but there are few consistent data available in the literature (91). Most authors explained that these atypical dose-response curves resulted from the complex interactions between the ligand (i.e., the natural hormone or an EDC) and a hormone receptor. In our model, it could, for example, be explained by the resultant of the double opposite effect of BPA on ERβ and GPER/GPR30 (60, 85). Indeed, at low doses (nM or pM), BPA acts only through GPER/GPR30 by a promotive effect while it acts also through ERβ at higher dose (mM), which counteracted the promotive GPER/GPR30-mediated effect (66). In order to confirm this hypothesis, we exposed JKT-1 cells to variable doses of BPA together with a fixed dose of E2. The BPA dose-response curve that we obtained kept its inverted U-shape aspect but was down-translated, confirming that BPA can act either through ERβ or GPER/GPR30 depending on the other estrogenic compounds that are present in the cell environment. This parameter is particularly important to consider since in most cases we are exposed to EDC mixtures.
Furthermore, in the same cellular model, the effects of several EDCs on in vitro proliferation were totally different and dependent on the resultant of the two expressed receptors, ERβ and GPER/GPR30. For example, atrazine, another estrogenic pesticide, induced a suppressive effect on seminoma cell proliferation in vitro involving a GPER/GPR30-dependent pathway (92). In the same way, an alkylphenol mix promoted seminoma cell proliferation through a GPER/GPR30-dependent pathway (93). However, in this case, the promoting effect is also mediated through ERα36, which is a truncated form of the canonical ERα66 (without both transcriptional activation domains (AF-1 and AF-2)) and was first described first by Wang et al. (94) in 2005. It seems to participate in non-genomic estrogen signaling concurrently to and/or associated with GPER/GPR30, as demonstrated in breast cancer cell lines (94) and in seminoma-like TCam-2 cell line (95). Thus, the presence of ERα36 in tumors is an important parameter to consider before considering selective antagonists of GPER/GPR30 as a therapeutic target in TGCC or other estrogen-dependent cancers.
The crosstalk among GPER/GPR30 signaling, classical estrogen receptors, and other nuclear receptors involved in testis physiology regulation is also important to consider (96). Through such interactions, GPER/GPR30 could probably modulate the tumor microenvironment and through this mediate TGCC progression and aggressiveness, especially by inducing epithelial-mesenchymal-transition (97, 98), as has been reported in breast cancer (98, 99) and in pancreatic adenocarcinoma (100).
Accumulating evidence supports the role of GPER/GPR30 in cancer progression and metastasis in estrogen-dependent cancers (especially in breast cancer), even though GPER/GPR30 signaling can differently affect the development of cancer depending on the type of tissue, but also in the same tissue depending on the type of ligand (92). A better comprehension of the molecular pathways involved in TGCC development, in particular the role of GPER/GPR30 in tumor progression, points out new tools like agonists or antagonists of GPER/GPR30, which could be used going forward by clinicians to target cancer cells and improve the patient’s chance of survival (68, 101).
Three pharmacological GPER/GPR30-ligands were routinely available to study GPER/GPR30 functions. The first one, G-1, was identified by Bologa in 2006 and is a specific agonist of GPER/GPR30, while G-15 and G-36, identified respectively in 2009 and 2011 by Dennis, are GPER/GPR30 antagonists. However, G-15 exhibits a partial cross-reactivity with ERα explaining why G-36 is mainly used in the study of GPER/GPR30 (102). Other pharmacological ligands were synthetized (GPER/GPR30-L1 and GPER/GPR30-L2) (102, 103) but they exhibit variable affinities for GPER/GPR30 and potential cross-reactivity with classical ERs, explaining why they cannot be considered as therapeutic tools at this time (104). These small molecules were used especially in vitro, as we did with seminoma cells; in our model, G-1 was able to mimic the proliferative effect of BPA while G-15 neutralized this effect and reduced cell proliferation in the presence of BPA (71). Thus, G-15 may be a helpful adjuvant in the treatment of TGCC. Nevertheless, to date, no studies have reported the use of GPER/GPR30 antagonists in this way.
However, agonists and antagonists of GPER/GPR30 were tested in the treatment of other tumors. For example, as we observed in vitro in seminoma cells, G-15 was also able to decrease the in vitro proliferation of non-small cell lung cancer (105) while G-1 was reported to induce malignant cell proliferation, invasion, and migration in primary cultured lung cancer cells (106) and in ER-negative breast cancer cells (107, 108) involving SIRT1 (108). At the opposite end, G-1 was able to decrease in vivo the tumor volume of pancreatic ductal adenocarcinoma in mice (109) and of adrenocortical carcinoma in a xenograft model (110, 111).
Interestingly, G-1 was also able to reduce the side effects of chemotherapy, as, for example, the cardiac toxicity of doxorubicin is usually used as an adjuvant therapy in breast cancer (112). This beneficial effect is related to the well-documented GPER/GPR30 actions on the vascular system, involving in this specific case the Nox1 pathway, which could constitute new therapeutic tools (113, 114).
Actually, only one clinical study is registered in Clinical Trials involving a GPER/GPR30 agonist. The NCT04130516 is a phase 1, first-in-human, open-label, multicenter study (up to six study sites in the United States) designed to characterize the safety, tolerability, and antitumor effects of LNS8801 administered orally in patients with advanced cancer (solid tumor or lymphoma). The recruitment is still on-going, and the estimated primary completion date is the end of 2021.
Finally, even though GPER/GPR30 modulation represents a potential novel strategy in cancer therapy, there remains a lack of solid clinical evidence supporting the specificity of GPER/GPR30 antagonists, especially in TGCC.
When compared with normal tissues, GPER/GPR30 is highly expressed in breast cancer and its high expression at the plasma membrane is strongly correlated with a poor prognosis, especially in triple negative tumors (115). This overexpression of GPER/GPR30 was also related to tamoxifen resistance (116, 117). Thus, GPER/GPR30 could be considered as a potential therapeutic target in such estrogen-dependent cancers.
Since its discovery in breast cancer, the role of GPER/GPR30 in estrogen-dependent malignancies has been of great interest. TGCC, the most common solid cancer in young men, expresses classical estrogen receptors (ERβ) but also GPER/GPR30. While E2 is responsible for a suppressive effect through an ERβ-dependent pathway, EDCs like BPA could induce in vitro seminoma cell proliferation by binding to GPER/GPR30. Furthermore, GPER/GPR30 is overexpressed in seminoma, probably due to genetic and/or epigenetic modulations that could be induced by fetal exposure to some EDCs. As proposed by Skakkebaek (4), an estrogenic environment might impair normal differentiation and proliferation of normal fetal, perinatal, and peripubertal germ stem cells, and then predispose an individual to TGCC, meaning it may be considered as an estrogen-dependent cancer. In our model, we have showed that G-15, a partial antagonist of GPER/GPR30, was able to reduce in vitro the BPA-induced cell proliferation (71) and may constitute a potential adjuvant in the treatment of TGCC. However, there remains a lack of solid clinical evidence to consider its clinical use. Direct regulation of GPER/GPR30 expression by siRNA silencing and/or nanotechnology could offer, at last, another tool to target GPER/GPR30 in cancer therapy.
NC and PF designed the study and contributed to the discussions and manuscript. NC and RP-B researched and interpreted data. SC and CH contributed to discussions and manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Société Française d’Endocrinologie (SFE), the Institut National de la Santé et de la Recherche (INSERM), and the Fondation pour la Recherche Médicale (FRM) (to NC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors greatly acknowledge Eric R. Prossnitz, Ph.D. (University of New Mexico, Albuquerque, USA) for kindly providing the selective GPER/GPR30 antagonist G15.
1. Bosl GJ, Motzer RJ. Testicular germ-cell cancer. New Engl J Med (1997) 337(4):242–53. doi: 10.1056/NEJM199707243370406
2. Bray F, Richiardi L, Ekbom A, Pukkala E, Cuninkova M, Moller H. Trends in testicular cancer incidence and mortality in 22 European countries: continuing increases in incidence and declines in mortality. Int J Cancer (2006) 118(12):3099–111. doi: 10.1002/ijc.21747
3. Le Cornet C, Lortet-Tieulent J, Forman D, Beranger R, Flechon A, Fervers B, et al. Testicular cancer incidence to rise by 25% by 2025 in Europe? Model-based predictions in 40 countries using population-based registry data. Eur J Cancer (2014) 50(4):831–9. doi: 10.1016/j.ejca.2013.11.035
4. Skakkebaek NE. A Brief Review of the Link between Environment and Male Reproductive Health: Lessons from Studies of Testicular Germ Cell Cancer. Hormone Res Paediatrics (2016) 86(4):240–6. doi: 10.1159/000443400
5. Cook MB, Akre O, Forman D, Madigan MP, Richiardi L, McGlynn KA. A systematic review and meta-analysis of perinatal variables in relation to the risk of testicular cancer–experiences of the son. Int J Epidemiol (2010) 39(6):1605–18. doi: 10.1093/ije/dyq120
6. McGlynn KA, Trabert B. Adolescent and adult risk factors for testicular cancer. Nat Rev Urol (2012) 9(6):339–49. doi: 10.1038/nrurol.2012.61
7. Hemminki K, Chen B. Familial risks in testicular cancer as aetiological clues. Int J Androl (2006) 29(1):205–10. doi: 10.1111/j.1365-2605.2005.00599.x
8. Rapley EA, Turnbull C, Al Olama AA, Dermitzakis ET, Linger R, Huddart RA, et al. A genome-wide association study of testicular germ cell tumor. Nat Genet (2009) 41(7):807–10. doi: 10.1038/ng.394
9. Ferlin A, Pengo M, Pizzol D, Carraro U, Frigo AC, Foresta C. Variants in KITLG predispose to testicular germ cell cancer independently from spermatogenic function. Endocrine-Related Cancer (2012) 19(1):101–8. doi: 10.1530/ERC-11-0340
10. Xing JS, Bai ZM. Is testicular dysgenesis syndrome a genetic, endocrine, or environmental disease, or an unexplained reproductive disorder? Life Sci (2018) 194:120–9. doi: 10.1016/j.lfs.2017.11.039
11. Cook MB, Akre O, Forman D, Madigan MP, Richiardi L, McGlynn KA. A systematic review and meta-analysis of perinatal variables in relation to the risk of testicular cancer–experiences of the mother. Int J Epidemiol (2009) 38(6):1532–42. doi: 10.1093/ije/dyp287
12. Skakkebaek NE. Possible carcinoma-in-situ of the testis. Lancet (1972) 2(7776):516–7. doi: 10.1016/s0140-6736(72)91909-5
13. Sonne SB, Almstrup K, Dalgaard M, Juncker AS, Edsgard D, Ruban L, et al. Analysis of gene expression profiles of microdissected cell populations indicates that testicular carcinoma in situ is an arrested gonocyte. Cancer Res (2009) 69(12):5241–50. doi: 10.1158/0008-5472.CAN-08-4554
14. Olesen IA, Sonne SB, Hoei-Hansen CE, Rajpert-De Meyts E, Skakkebaek NE. Environment, testicular dysgenesis and carcinoma in situ testis. Best Pract Res Clin Endocrinol Metab (2007) 21(3):462–78. doi: 10.1016/j.beem.2007.04.002
15. Lymperi S, Giwercman A. Endocrine disruptors and testicular function. Metabol: Clin Exp (2018) 86:79–90. doi: 10.1016/j.metabol.2018.03.022
16. Guillette LJ Jr., Gross TS, Masson GR, Matter JM, Percival HF, Woodward AR. Developmental abnormalities of the gonad and abnormal sex hormone concentrations in juvenile alligators from contaminated and control lakes in Florida. Environ Health Perspect (1994) 102(8):680–8. doi: 10.1289/ehp.94102680
17. Facemire CF, Gross TS, Guillette LJ Jr. Reproductive impairment in the Florida panther: nature or nurture? Environ Health Perspect (1995) 103(Suppl 4):79–86. doi: 10.1289/ehp.103-1519283
18. Meng X, de Rooij DG, Westerdahl K, Saarma M, Sariola H. Promotion of seminomatous tumors by targeted overexpression of glial cell line-derived neurotrophic factor in mouse testis. Cancer Res (2001) 61(8):3267–71.
19. Palmer JR, Herbst AL, Noller KL, Boggs DA, Troisi R, Titus-Ernstoff L, et al. Urogenital abnormalities in men exposed to diethylstilbestrol in utero: a cohort study. Environ Health Global Access Sci Source (2009) 8:37. doi: 10.1186/1476-069X-8-37
20. Fenichel P, Brucker-Davis F, Chevalier N. The history of Distilbene(R) (Diethylstilbestrol) told to grandchildren–the transgenerational effect. Annal D’endocrinol (2015) 76(3):253–9. doi: 10.1016/j.ando.2015.03.008
21. Strohsnitter WC, Noller KL, Hoover RN, Robboy SJ, Palmer JR, Titus-Ernstoff L, et al. Cancer risk in men exposed in utero to diethylstilbestrol. J Natl Cancer Institute (2001) 93(7):545–51. doi: 10.1093/jnci/93.7.545
22. Martin OV, Shialis T, Lester JN, Scrimshaw MD, Boobis AR, Voulvoulis N. Testicular dysgenesis syndrome and the estrogen hypothesis: a quantitative meta-analysis. Environ Health Perspect (2008) 116(2):149–57. doi: 10.1289/ehp.10545
23. Ryder SJ, Crawford PI, Pethybridge RJ. Is testicular cancer an occupational disease? A case-control study of Royal Naval personnel. J R Naval Med Service (1997) 83(3):130–46.
24. Hardell L, Malmqvist N, Ohlson CG, Westberg H, Eriksson M. Testicular cancer and occupational exposure to polyvinyl chloride plastics: a case-control study. Int J Cancer (2004) 109(3):425–9. doi: 10.1002/ijc.11709
25. Knight JA, Marrett LD, Weir HK. Occupation and risk of germ cell testicular cancer by histologic type in Ontario. J Occup Environ Med (1996) 38(9):884–90. doi: 10.1097/00043764-199609000-00010
26. Giannandrea F, Paoli D, Figa-Talamanca I, Lombardo F, Lenzi A, Gandini L. Effect of endogenous and exogenous hormones on testicular cancer: the epidemiological evidence. Int J Dev Biol (2013) 57(2-4):255–63. doi: 10.1387/ijdb.130015fg
27. Hardell L, van Bavel B, Lindstrom G, Carlberg M, Dreifaldt AC, Wijkstrom H, et al. Increased concentrations of polychlorinated biphenyls, hexachlorobenzene, and chlordanes in mothers of men with testicular cancer. Environ Health Perspect (2003) 111(7):930–4. doi: 10.1289/ehp.5816
28. Purdue MP, Engel LS, Langseth H, Needham LL, Andersen A, Barr DB, et al. Prediagnostic serum concentrations of organochlorine compounds and risk of testicular germ cell tumors. Environ Health Perspect (2009) 117(10):1514–9. doi: 10.1289/ehp.0800359
29. McGlynn KA, Quraishi SM, Graubard BI, Weber JP, Rubertone MV, Erickson RL. Persistent organochlorine pesticides and risk of testicular germ cell tumors. J Natl Cancer Institute (2008) 100(9):663–71. doi: 10.1093/jnci/djn101
30. Bonde JP, Flachs EM, Rimborg S, Glazer CH, Giwercman A, Ramlau-Hansen CH, et al. The epidemiologic evidence linking prenatal and postnatal exposure to endocrine disrupting chemicals with male reproductive disorders: a systematic review and meta-analysis. Hum Reprod Update (2016) 23(1):104–25. doi: 10.1093/humupd/dmw036
31. Cohn BA, Cirillo PM, Christianson RE. Prenatal DDT exposure and testicular cancer: a nested case-control study. Arch Environ Occup Health (2010) 65(3):127–34. doi: 10.1080/19338241003730887
32. Carreau S, Bourguiba S, Lambard S, Galeraud-Denis I, Genissel C, Levallet J. Reproductive system: aromatase and estrogens. Mol Cell Endocrinol (2002) 193(1-2):137–43. doi: 10.1016/S0303-7207(02)00107-7
33. Hess RA. Estrogen in the adult male reproductive tract: a review. Reprod Biol Endocrinol RB&E (2003) 1:52. doi: 10.1186/1477-7827-1-52
34. Carreau S, Bouraima-Lelong H, Delalande C. Role of estrogens in spermatogenesis. Front Biosci (2012) 4:1–11. doi: 10.2741/e356
35. Carreau S, Hess RA. Oestrogens and spermatogenesis. Philos Trans R Soc London Ser B Biol Sci (1546) 2010) 365:1517–35. doi: 10.1098/rstb.2009.0235
36. Boukari K, Ciampi ML, Guiochon-Mantel A, Young J, Lombes M, Meduri G. Human fetal testis: source of estrogen and target of estrogen action. Hum Reprod (2007) 22(7):1885–92. doi: 10.1093/humrep/dem091
37. Berensztein EB, Baquedano MS, Gonzalez CR, Saraco NI, Rodriguez J, Ponzio R, et al. Expression of aromatase, estrogen receptor alpha and beta, androgen receptor, and cytochrome P-450scc in the human early prepubertal testis. Pediatr Res (2006) 60(6):740–4. doi: 10.1203/01.pdr.0000246072.04663.bb
38. Makinen S, Makela S, Weihua Z, Warner M, Rosenlund B, Salmi S, et al. Localization of oestrogen receptors alpha and beta in human testis. Mol Hum Reprod (2001) 7(6):497–503. doi: 10.1093/molehr/7.6.497
39. Cavaco JE, Laurentino SS, Barros A, Sousa M, Socorro S. Estrogen receptors alpha and beta in human testis: both isoforms are expressed. Syst Biol Reprod Med (2009) 55(4):137–44. doi: 10.3109/19396360902855733
40. Saunders PT, Millar MR, Macpherson S, Irvine DS, Groome NP, Evans LR, et al. ERbeta1 and the ERbeta2 splice variant (ERbetacx/beta2) are expressed in distinct cell populations in the adult human testis. J Clin Endocrinol Metab (2002) 87(6):2706–15. doi: 10.1210/jcem.87.6.8619
41. Lambard S, Galeraud-Denis I, Saunders PT, Carreau S. Human immature germ cells and ejaculated spermatozoa contain aromatase and oestrogen receptors. J Mol Endocrinol (2004) 32(1):279–89. doi: 10.1677/jme.0.0320279
42. Smith EP, Boyd J, Frank GR, Takahashi H, Cohen RM, Specker B, et al. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. New Engl J Med (1994) 331(16):1056–61. doi: 10.1056/NEJM199410203311604
43. Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science (2005) 307(5715):1625–30. doi: 10.1126/science.1106943
44. Jacenik D, Cygankiewicz AI, Krajewska WM. The G protein-coupled estrogen receptor as a modulator of neoplastic transformation. Mol Cell Endocrinol (2016) 429:10–8. doi: 10.1016/j.mce.2016.04.011
45. Xu S, Yu S, Dong D, Lee LTO. G Protein-Coupled Estrogen Receptor: A Potential Therapeutic Target in Cancer. Front Endocrinol (2019) 10:725:725. doi: 10.3389/fendo.2019.00725
46. Tao S, He H, Chen Q, Yue W. GPER mediated estradiol reduces miR-148a to promote HLA-G expression in breast cancer. Biochem Biophys Res Commun (2014) 451(1):74–8. doi: 10.1016/j.bbrc.2014.07.073
47. Teng Y, Radde BN, Litchfield LM, Ivanova MM, Prough RA, Clark BJ, et al. Dehydroepiandrosterone Activation of G-protein-coupled Estrogen Receptor Rapidly Stimulates MicroRNA-21 Transcription in Human Hepatocellular Carcinoma Cells. J Biol Chem (2015) 290(25):15799–811. doi: 10.1074/jbc.M115.641167
48. Vivacqua A, De Marco P, Santolla MF, Cirillo F, Pellegrino M, Panno ML, et al. Estrogenic gper signaling regulates mir144 expression in cancer cells and cancer-associated fibroblasts (cafs). Oncotarget (2015) 6(18):16573–87. doi: 10.18632/oncotarget.4117
49. Filardo EJ, Quinn JA, Bland KI, Frackelton AR Jr. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol (2000) 14(10):1649–60. doi: 10.1210/mend.14.10.0532
50. Filardo EJ. Epidermal growth factor receptor (EGFR) transactivation by estrogen via the G-protein-coupled receptor, GPR30: a novel signaling pathway with potential significance for breast cancer. J Steroid Biochem Mol Biol (2002) 80(2):231–8. doi: 10.1016/s0960-0760(01)00190-x
51. Recchia AG, De Francesco EM, Vivacqua A, Sisci D, Panno ML, Ando S, et al. The G protein-coupled receptor 30 is up-regulated by hypoxia-inducible factor-1alpha (HIF-1alpha) in breast cancer cells and cardiomyocytes. J Biol Chem (2011) 286(12):10773–82. doi: 10.1074/jbc.M110.172247
52. De Francesco EM, Lappano R, Santolla MF, Marsico S, Caruso A, Maggiolini M. HIF-1alpha/GPER signaling mediates the expression of VEGF induced by hypoxia in breast cancer associated fibroblasts (CAFs). Breast Cancer Res (2013) 15(4):R64. doi: 10.1186/bcr3458
53. De Marco P, Bartella V, Vivacqua A, Lappano R, Santolla MF, Morcavallo A, et al. Insulin-like growth factor-I regulates GPER expression and function in cancer cells. Oncogene (2013) 32(6):678–88. doi: 10.1038/onc.2012.97
54. Avino S, De Marco P, Cirillo F, Santolla MF, De Francesco EM, Perri MG, et al. Stimulatory actions of IGF-I are mediated by IGF-IR cross-talk with GPER and DDR1 in mesothelioma and lung cancer cells. Oncotarget (2016) 7(33):52710–28. doi: 10.18632/oncotarget.10348
55. Okamoto M, Mizukami Y. GPER negatively regulates TNFalpha-induced IL-6 production in human breast cancer cells via NF-kappaB pathway. Endocr J (2016) 63(5):485–93. doi: 10.1507/endocrj.EJ15-0571
56. Zhu P, Liao LY, Zhao TT, Mo XM, Chen GG, Liu ZM. GPER/ERK&AKT/NF-kappaB pathway is involved in cadmium-induced proliferation, invasion and migration of GPER-positive thyroid cancer cells. Mol Cell Endocrinol (2017) 442:68–80. doi: 10.1016/j.mce.2016.12.007
57. Feldman RD, Ding Q, Hussain Y, Limbird LE, Pickering JG, Gros R. Aldosterone mediates metastatic spread of renal cancer via the G protein-coupled estrogen receptor (GPER). FASEB J (2016) 30(6):2086–96. doi: 10.1096/fj.15-275552
58. Rigiracciolo DC, Scarpelli A, Lappano R, Pisano A, Santolla MF, Avino S, et al. GPER is involved in the stimulatory effects of aldosterone in breast cancer cells and breast tumor-derived endothelial cells. Oncotarget (2016) 7(1):94–111. doi: 10.18632/oncotarget.6475
59. Parker BM, Wertz SL, Pollard CM, Desimine VL, Maning J, McCrink KA, et al. Novel Insights into the Crosstalk between Mineralocorticoid Receptor and G Protein-Coupled Receptors in Heart Adverse Remodeling and Disease. Int J Mol Sci (2018) 19(12):3764. doi: 10.3390/ijms19123764
60. Chevalier N, Vega A, Bouskine A, Siddeek B, Michiels JF, Chevallier D, et al. GPR30, the non-classical membrane G protein related estrogen receptor, is overexpressed in human seminoma and promotes seminoma cell proliferation. PloS One (2012) 7(4):e34672. doi: 10.1371/journal.pone.0034672
61. Rago V, Giordano F, Brunelli E, Zito D, Aquila S, Carpino A. Identification of G protein-coupled estrogen receptor in human and pig spermatozoa. J Anat (2014) 224(6):732–6. doi: 10.1111/joa.12183
62. Fenichel P, Chevalier N. Is Testicular Germ Cell Cancer Estrogen Dependent? The Role of Endocrine Disrupting Chemicals. Endocrinology (2019) 160(12):2981–9. doi: 10.1210/en.2019-00486
63. Filardo EJ, Graeber CT, Quinn JA, Resnick MB, Giri D, DeLellis RA, et al. Distribution of GPR30, a seven membrane-spanning estrogen receptor, in primary breast cancer and its association with clinicopathologic determinants of tumor progression. Clin Cancer Res (2006) 12(21):6359–66. doi: 10.1158/1078-0432.CCR-06-0860
64. Smith HO, Arias-Pulido H, Kuo DY, Howard T, Qualls CR, Lee SJ, et al. GPR30 predicts poor survival for ovarian cancer. Gynecol Oncol (2009) 114(3):465–71. doi: 10.1016/j.ygyno.2009.05.015
65. Prossnitz ER, Arterburn JB, Smith HO, Oprea TI, Sklar LA, Hathaway HJ. Estrogen signaling through the transmembrane G protein-coupled receptor GPR30. Annu Rev Physiol (2008) 70:165–90. doi: 10.1146/annurev.physiol.70.113006.100518
66. Roger C, Lambard S, Bouskine A, Mograbi B, Chevallier D, Nebout M, et al. Estrogen-induced growth inhibition of human seminoma cells expressing estrogen receptor beta and aromatase. J Mol Endocrinol (2005) 35(1):191–9. doi: 10.1677/jme.1.01704
67. Bouskine A, Nebout M, Mograbi B, Brucker-Davis F, Roger C, Fenichel P. Estrogens promote human testicular germ cell cancer through a membrane-mediated activation of extracellular regulated kinase and protein kinase A. Endocrinology (2008) 149(2):565–73. doi: 10.1210/en.2007-1318
68. Franco R, Boscia F, Gigantino V, Marra L, Esposito F, Ferrara D, et al. GPR30 is overexpressed in post-puberal testicular germ cell tumors. Cancer Biol Ther (2011) 11(6):609–13. doi: 10.4161/cbt.11.6.14672
69. Boscia F, Passaro C, Gigantino V, Perdona S, Franco R, Portella G, et al. High levels of GPR30 protein in human testicular carcinoma in situ and seminomas correlate with low levels of estrogen receptor-beta and indicate a switch in estrogen responsiveness. J Cell Physiol (2015) 230(6):1290–7. doi: 10.1002/jcp.24864
70. Rago V, Romeo F, Giordano F, Maggiolini M, Carpino A. Identification of the estrogen receptor GPER in neoplastic and non-neoplastic human testes. Reprod Biol Endocrinol RB&E (2011) 9:135. doi: 10.1186/1477-7827-9-135
71. Chevalier N, Bouskine A, Fenichel P. Bisphenol A promotes testicular seminoma cell proliferation through GPER/GPR30. Int J Cancer (2012) 130(1):241–2. doi: 10.1002/ijc.25972
72. Pais V, Leav I, Lau KM, Jiang Z, Ho SM. Estrogen receptor-beta expression in human testicular germ cell tumors. Clin Cancer Res (2003) 9(12):4475–82.
73. Ferlin A, Ganz F, Pengo M, Selice R, Frigo AC, Foresta C. Association of testicular germ cell tumor with polymorphisms in estrogen receptor and steroid metabolism genes. Endocrine-Related Cancer (2010) 17(1):17–25. doi: 10.1677/ERC-09-0176
74. Brokken LJ, Lundberg-Giwercman Y, Rajpert De-Meyts E, Eberhard J, Stahl O, Cohn-Cedermark G, et al. Association of polymorphisms in genes encoding hormone receptors ESR1, ESR2 and LHCGR with the risk and clinical features of testicular germ cell cancer. Mol Cell Endocrinol (2012) 351(2):279–85. doi: 10.1016/j.mce.2011.12.018
75. Chevalier N, Paul-Bellon R, Camparo P, Michiels JF, Chevallier D, Fenichel P. Genetic variants of GPER/GPR30, a novel estrogen-related G protein receptor, are associated with human seminoma. Int J Mol Sci (2014) 15(1):1574–89. doi: 10.3390/ijms15011574
76. Zama AM, Uzumcu M. Fetal and neonatal exposure to the endocrine disruptor methoxychlor causes epigenetic alterations in adult ovarian genes. Endocrinology (2009) 150(10):4681–91. doi: 10.1210/en.2009-0499
77. Anway MD, Cupp AS, Uzumcu M, Skinner MK. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science (2005) 308(5727):1466–9. doi: 10.1126/science.1108190
78. Dumasia K, Kumar A, Deshpande S, Balasinor NH. Estrogen signaling, through estrogen receptor beta, regulates DNA methylation and its machinery in male germ line in adult rats. Epigenetics (2017) 12(6):476–83. doi: 10.1080/15592294.2017.1309489
79. Markulin D, Vojta A, Samarzija I, Gamulin M, Beceheli I, Jukic I, et al. Association Between RASSF1A Promoter Methylation and Testicular Germ Cell Tumor: A Meta-analysis and a Cohort Study. Cancer Genomics Proteomics (2017) 14(5):363–72. doi: 10.21873/cgp.20046
80. Killian JK, Dorssers LC, Trabert B, Gillis AJ, Cook MB, Wang Y, et al. Imprints and DPPA3 are bypassed during pluripotency- and differentiation-coupled methylation reprogramming in testicular germ cell tumors. Genome Res (2016) 26(11):1490–504. doi: 10.1101/gr.201293.115
81. Voorhoeve PM, le Sage C, Schrier M, Gillis AJ, Stoop H, Nagel R, et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell (2006) 124(6):1169–81. doi: 10.1016/j.cell.2006.02.037
82. Lobo J, Gillis AJM, Jeronimo C, Henrique R, Looijenga LHJ. Human Germ Cell Tumors are Developmental Cancers: Impact of Epigenetics on Pathobiology and Clinic. Int J Mol Sci (2019) 20(2):258. doi: 10.3390/ijms20020258
83. Almstrup K, Nielsen JE, Mlynarska O, Jansen MT, Jorgensen A, Skakkebaek NE, et al. Carcinoma in situ testis displays permissive chromatin modifications similar to immature foetal germ cells. Br J Cancer (2010) 103(8):1269–76. doi: 10.1038/sj.bjc.6605880
84. Bouskine A, Vega A, Nebout M, Benahmed M, Fenichel P. Expression of embryonic stem cell markers in cultured JKT-1, a cell line derived from a human seminoma. Int J Androl (2010) 33(1):54–63. doi: 10.1111/j.1365-2605.2009.00950.x
85. Bouskine A, Nebout M, Brucker-Davis F, Benahmed M, Fenichel P. Low doses of bisphenol A promote human seminoma cell proliferation by activating PKA and PKG via a membrane G-protein-coupled estrogen receptor. Environ Health Perspect (2009) 117(7):1053–8. doi: 10.1289/ehp.0800367
86. Chevalier N, Fenichel P. Endocrine disruptors: new players in the pathophysiology of type 2 diabetes? Diabetes Metab (2015) 41(2):107–15. doi: 10.1016/j.diabet.2014.09.005
87. Fenichel P, Dechaux H, Harthe C, Gal J, Ferrari P, Pacini P, et al. Unconjugated bisphenol A cord blood levels in boys with descended or undescended testes. Hum Reprod (2012) 27(4):983–90. doi: 10.1093/humrep/der451
88. Vandenberg LN, Maffini MV, Sonnenschein C, Rubin BS, Soto AM. Bisphenol-A and the great divide: a review of controversies in the field of endocrine disruption. Endocrine Rev (2009) 30(1):75–95. doi: 10.1210/er.2008-0021
89. Fenichel P, Chevalier N, Brucker-Davis F. Bisphenol A: an endocrine and metabolic disruptor. Annal D’endocrinol (2013) 74(3):211–20. doi: 10.1016/j.ando.2013.04.002
90. Pupo M, Pisano A, Lappano R, Santolla MF, De Francesco EM, Abonante S, et al. Bisphenol A induces gene expression changes and proliferative effects through GPER in breast cancer cells and cancer-associated fibroblasts. Environ Health Perspect (2012) 120(8):1177–82. doi: 10.1289/ehp.1104526
91. Vandenberg LN. Non-monotonic dose responses in studies of endocrine disrupting chemicals: bisphenol a as a case study. Dose Response (2014) 12(2):259–76. doi: 10.2203/dose-response.13-020.Vandenberg
92. Chevalier N, Paul-Bellon R, Fenichel P. Comment on “Effects of Atrazine on Estrogen Receptor alpha- and G Protein-Coupled Receptor 30-Mediated Signaling and Proliferation in Cancer Cells and Cancer-Associated Fibroblasts”. Environ Health Perspect (2016) 124(4):A64–5. doi: 10.1289/ehp.1510927
93. Ajj H, Chesnel A, Pinel S, Plenat F, Flament S, Dumond H. An alkylphenol mix promotes seminoma derived cell proliferation through an ERalpha36-mediated mechanism. PloS One (2013) 8(4):e61758. doi: 10.1371/journal.pone.0061758
94. Wang Z, Zhang X, Shen P, Loggie BW, Chang Y, Deuel TF. Identification, cloning, and expression of human estrogen receptor-alpha36, a novel variant of human estrogen receptor-alpha66. Biochem Biophys Res Commun (2005) 336(4):1023–7. doi: 10.1016/j.bbrc.2005.08.226
95. Wallacides A, Chesnel A, Ajj H, Chillet M, Flament S, Dumond H. Estrogens promote proliferation of the seminoma-like TCam-2 cell line through a GPER-dependent ERalpha36 induction. Mol Cell Endocrinol (2012) 350(1):61–71. doi: 10.1016/j.mce.2011.11.021
96. Chimento A, De Luca A, Nocito MC, Avena P, La Padula D, Zavaglia L, et al. Role of GPER-Mediated Signaling in Testicular Functions and Tumorigenesis. Cells (2020) 9(9):2115. doi: 10.3390/cells9092115
97. Filardo EJ. A role for G-protein coupled estrogen receptor (GPER) in estrogen-induced carcinogenesis: Dysregulated glandular homeostasis, survival and metastasis. J Steroid Biochem Mol Biol (2018) 176:38–48. doi: 10.1016/j.jsbmb.2017.05.005
98. De Francesco EM, Maggiolini M, Musti AM. Crosstalk between Notch, HIF-1alpha and GPER in Breast Cancer EMT. Int J Mol Sci (2018) 19(7):2011. doi: 10.3390/ijms19072011
99. De Francesco EM, Sims AH, Maggiolini M, Sotgia F, Lisanti MP, Clarke RB. GPER mediates the angiocrine actions induced by IGF1 through the HIF-1alpha/VEGF pathway in the breast tumor microenvironment. Breast Cancer Res (2017) 19(1):129. doi: 10.1186/s13058-017-0923-5
100. Rice A, Cortes E, Lachowski D, Oertle P, Matellan C, Thorpe SD, et al. GPER Activation Inhibits Cancer Cell Mechanotransduction and Basement Membrane Invasion via RhoA. Cancers (Basel) (2020) 12(2):289. doi: 10.3390/cancers12020289
101. Chieffi P. An up-date on novel molecular targets in testicular germ cell tumors subtypes. Intractable Rare Dis Res (2019) 8(2):161–4. doi: 10.5582/irdr.2019.01055
102. Prossnitz ER, Arterburn JB. International Union of Basic and Clinical Pharmacology. XCVII. G Protein-Coupled Estrogen Receptor and Its Pharmacologic Modulators. Pharmacol Rev (2015) 67(3):505–40. doi: 10.1124/pr.114.009712
103. Zacarias-Lara OJ, Mendez-Luna D, Martinez-Ruiz G, Garcia-Sanchez JR, Fragoso-Vazquez MJ, Bello M, et al. Synthesis and In Vitro Evaluation of Tetrahydroquinoline Derivatives as Antiproliferative Compounds of Breast Cancer via Targeting the GPER. Anticancer Agents Med Chem (2019) 19(6):760–71. doi: 10.2174/1871520618666181119094144
104. DeLeon C, Wang HH, Gunn J, Wilhelm M, Cole A, Arnett S, et al. A novel GPER antagonist protects against the formation of estrogen-induced cholesterol gallstones in female mice. J Lipid Res (2020) 61(5):767–77. doi: 10.1194/jlr.RA119000592
105. Liu C, Liao Y, Fan S, Fu X, Xiong J, Zhou S, et al. G-Protein-Coupled Estrogen Receptor Antagonist G15 Decreases Estrogen-Induced Development of Non-Small Cell Lung Cancer. Oncol Res (2019) 27(3):283–92. doi: 10.3727/096504017X15035795904677
106. Liu C, Liao Y, Fan S, Tang H, Jiang Z, Zhou B, et al. G protein-coupled estrogen receptor (GPER) mediates NSCLC progression induced by 17beta-estradiol (E2) and selective agonist G1. Med Oncol (2015) 32(4):104. doi: 10.1007/s12032-015-0558-2
107. De Francesco EM, Pellegrino M, Santolla MF, Lappano R, Ricchio E, Abonante S, et al. GPER mediates activation of HIF1alpha/VEGF signaling by estrogens. Cancer Res (2014) 74(15):4053–64. doi: 10.1158/0008-5472.CAN-13-3590
108. Santolla MF, Avino S, Pellegrino M, De Francesco EM, De Marco P, Lappano R, et al. SIRT1 is involved in oncogenic signaling mediated by GPER in breast cancer. Cell Death Dis (2015) 6:e1834. doi: 10.1038/cddis.2015.201
109. Natale CA, Li J, Pitarresi JR, Norgard RJ, Dentchev T, Capell BC, et al. Pharmacologic Activation of the G Protein-Coupled Estrogen Receptor Inhibits Pancreatic Ductal Adenocarcinoma. Cell Mol Gastroenterol Hepatol (2020). 10(4):868–80.e1. doi: 10.1016/j.jcmgh.2020.04.016
110. Chimento A, Sirianni R, Casaburi I, Zolea F, Rizza P, Avena P, et al. GPER agonist G-1 decreases adrenocortical carcinoma (ACC) cell growth in vitro and in vivo. Oncotarget (2015) 6(22):19190–203. doi: 10.18632/oncotarget.4241
111. Casaburi I, Avena P, De Luca A, Sirianni R, Rago V, Chimento A, et al. GPER-independent inhibition of adrenocortical cancer growth by G-1 involves ROS/Egr-1/BAX pathway. Oncotarget (2017) 8(70):115609–19. doi: 10.18632/oncotarget.23314
112. De Francesco EM, Rocca C, Scavello F, Amelio D, Pasqua T, Rigiracciolo DC, et al. Protective Role of GPER Agonist G-1 on Cardiotoxicity Induced by Doxorubicin. J Cell Physiol (2017) 232(7):1640–9. doi: 10.1002/jcp.25585
113. da Silva JS, Sun X, Ahmad S, Wang H, Sudo RT, Varagic J, et al. G-Protein-Coupled Estrogen Receptor Agonist G1 Improves Diastolic Function and Attenuates Cardiac Renin-Angiotensin System Activation in Estrogen-Deficient Hypertensive Rats. J Cardiovasc Pharmacol (2019) 74(5):443–52. doi: 10.1097/FJC.0000000000000721
114. Barton M, Meyer MR, Prossnitz ER. Nox1 downregulators: A new class of therapeutics. Steroids (2019) 152:108494. doi: 10.1016/j.steroids.2019.108494
115. Tutzauer J, Sjostrom M, Bendahl PO, Ryden L, Ferno M, Leeb-Lundberg LMF, et al. Plasma membrane expression of G protein-coupled estrogen receptor (GPER)/G protein-coupled receptor 30 (GPR30) is associated with worse outcome in metachronous contralateral breast cancer. PloS One (2020) 15(4):e0231786. doi: 10.1371/journal.pone.0231786
116. Sjostrom M, Hartman L, Grabau D, Fornander T, Malmstrom P, Nordenskjold B, et al. Lack of G protein-coupled estrogen receptor (GPER) in the plasma membrane is associated with excellent long-term prognosis in breast cancer. Breast Cancer Res Treat (2014) 145(1):61–71. doi: 10.1007/s10549-014-2936-4
Keywords: testicular germ cell cancer, estrogen receptors, GPR30/GPER, endocrine disrupting compounds, fetal exposure, bisphenol A
Citation: Chevalier N, Hinault C, Clavel S, Paul-Bellon R and Fenichel P (2021) GPER and Testicular Germ Cell Cancer. Front. Endocrinol. 11:600404. doi: 10.3389/fendo.2020.600404
Received: 29 August 2020; Accepted: 07 December 2020;
Published: 26 January 2021.
Edited by:
Marilena Kampa, University of Crete, GreeceReviewed by:
Ernestina Marianna De Francesco, University of Catania, ItalyCopyright © 2021 Chevalier, Hinault, Clavel, Paul-Bellon and Fenichel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicolas Chevalier, Y2hldmFsaWVAdW5pY2UuZnI=; Y2hldmFsaWVyLm5AY2h1LW5pY2UuZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.