 Mengqi Chang
Mengqi Chang Chengxian Yang†
Chengxian Yang† Xinjie Bao
Xinjie Bao Renzhi Wang
Renzhi Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 26 January 2021
Sec. Pituitary Endocrinology
Volume 11 - 2020 | https://doi.org/10.3389/fendo.2020.596554
Pituitary adenomas (PAs) can be classified as non-secreting adenomas, somatotroph adenomas, corticotroph adenomas, lactotroph adenomas, and thyrotroph adenomas. Substantial advances have been made in our knowledge of the pathobiology of PAs. To obtain a comprehensive understanding of the molecular biological characteristics of different types of PAs, we reviewed the important advances that have been made involving genetic and epigenetic variation, comprising genetic mutations, chromosome number variations, DNA methylation, microRNA regulation, and transcription factor regulation. Classical tumor predisposition syndromes include multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4) syndromes, Carney complex, and X-LAG syndromes. PAs have also been described in association with succinate dehydrogenase-related familial PA, neurofibromatosis type 1, and von Hippel–Lindau, DICER1, and Lynch syndromes. Patients with aryl hydrocarbon receptor-interacting protein (AIP) mutations often present with pituitary gigantism, either in familial or sporadic adenomas. In contrast, guanine nucleotide-binding protein G(s) subunit alpha (GNAS) and G protein-coupled receptor 101 (GPR101) mutations can lead to excess growth hormone. Moreover, the deubiquitinase gene USP8, USP48, and BRAF mutations are associated with adrenocorticotropic hormone production. In this review, we describe the genetic and epigenetic landscape of PAs and summarize novel insights into the regulation of pituitary tumorigenesis.
Pituitary adenomas (PAs) are the second most common brain tumors, accounting for approximately 15% of all primary brain tumors (1). PAs can be classified based on the types of hormones that they excessively secrete into the blood. The clinical presentations caused by hormone overproduction in PAs are closely related to the pituitary cell types, as follows: corticotropin-secreting corticotroph adenomas result in Cushing’s disease, growth hormone (GH)-secreting somatotroph adenomas result in acromegaly, prolactin-secreting lactotroph adenomas result in hyperprolactinemia, and thyrotropin-secreting thyrotroph adenomas result in hyperthyroidism (2). Non-secreting adenomas, such as null cell adenomas, silent gonadotroph adenomas, silent corticotroph adenomas, and silent somatotroph adenomas, lead to hypogonadism and often manifest as incidental sellar masses (2). Of the different PA types, only lactotroph adenomas are treated with dopamine agonists as a first-line option. Because of a lack of effective drugs, other PA subtypes are generally treated with transsphenoidal surgery as the first-line therapy (3). Furthermore, because many PAs are invasive and unresectable—or in some cases of Cushing’s disease, because the tumors themselves are too small to be detected or completely removed during surgery—drugs and stereotactic radiosurgery are needed to achieve tumor control or biochemical remission. However, despite current treatments, the 10-year recurrence rate remains as high as 7–12% (3, 4). It is therefore important to obtain a comprehensive understanding of the molecular biological characteristics of different types of PAs, such as gene mutations, DNA methylation, microRNA (miRNA) regulation, and regulation at other levels, to allow the targeted treatment of individuals, thus achieving better prognoses. Herein, we summarize the known variation in the different types of PAs and review the potential molecular targets for future clinical application.
Familial PAs can be divided into two types: isolated and syndromic (5). These familial PAs and their molecular mechanisms are described herein.
MEN1 syndrome is classically characterized by the combined occurrence of parathyroid adenomas, PAs (in approximately 30–40% of cases), and neuroendocrine tumors (6). PAs that develop in MEN1 syndrome include lactotroph adenomas (42–62%), silent PAs (15–42%), somatotroph adenomas (6.5–9%), and corticotroph adenomas (3–4%). In addition, somatic mutations in MEN1 can also be found in sporadic PAs (7). The MEN1 gene is located on chromosome 11q13.1 and encodes a ubiquitously expressed transcription cofactor of cyclins; MEN1 also participates in G1/S checkpoint regulation (8, 9). Approximately 10% of all MEN1-related PA cases can be attributed to de novo mutations, which are sometimes identified as a mosaicism in the proband (10, 11).
Some patients with MEN1 syndrome harbor no MEN1 mutation. Instead, cyclin-dependent kinase inhibitor 1B (CDKN1B) mutations have been detected in these patients. This syndrome is known as MEN4 syndrome (12). Patients with MEN4 syndrome are prone to developing somatotroph adenomas, and can also develop other types of Pas (13). CDKN1B mutations are rarely found in sporadic pituitary tumors (14, 15). CDKN1B encodes a cyclin-dependent kinase inhibitor that regulates the cell cycle and mitosis from the G1 to the S phase (13). CDKN1B-knockout mice develop various types of tumors, including PAs, and this tumorigenesis is associated with accelerated pituitary cell proliferation (16, 17). CDKN1B mutations likely lead to PAs by influencing cell cycle regulation.
Carney complex is characterized by endocrine and non-endocrine tumors with spotty skin pigmentation, as well as by cardiac and cutaneous myxomas (18). More than two-thirds of patients present asymptomatic elevations of insulin-like growth factor 1 (IGF-1), GH, and prolactin caused by pituitary hyperplasia, and 10% of patients present with adenomas and symptomatic acromegaly (19). In some cases, Carney complex is caused by an inactivating mutation of the PRKAR1A gene, which encodes the type 1-alpha regulatory subunit of protein kinase A (20). In addition, a gain-of-function mutation has been described in the gene encoding the catalytic subunit of protein kinase A, PRKACB (21).
X-LAG syndrome is a newly defined syndrome in patients with pituitary gigantism or PA who carry microduplications on chromosome Xq26.3 (22, 23). X-LAG syndrome is generally recognized as an aggressive disease because it is difficult to control the excess GH. Most patients require multiple interventions (both surgical and medical), and subtotal or total hypophysectomy is sometimes necessary. In contrast, radiation therapy is not usually helpful. X-LAG syndrome is probably caused by G protein-coupled receptor 101 (GPR101) overexpression because the GPR101 gene is located on chromosome Xq26.3. GPR101 is coupled to the stimulatory G protein complex and activates adenylate cyclase, increasing cyclic adenosine monophosphate (cAMP) production. In addition, GPR101 amplification has also been identified as a germline or mosaic mutation (22).
This “3PAs” syndrome, which combines PA with pheochromocytoma/paraganglioma (PPGL), is sometimes associated with mutations in PPGL-predisposing genes, such as the genes encoding SDHx (24, 25). Such mutations occur in SDHA-D and SDHA2F, among others (24–28). SDH is a multimeric enzyme that binds to the inner membranes of mitochondria. It has a dual role: it serves both as a critical step of the tricarboxylic acid or Krebs cycle, and as a member of the respiratory chain that transfers electrons directly to the ubiquinone pool (25, 27, 29). Pituitary hyperplasia has been reported to develop in a Sdhb-knockout mouse model (25).
Rarely, optic pathway gliomas cause high GH levels in NF1, while true PAs are extremely rare. Empty sella syndrome and hypopituitarism may also occur in the context of NF1. Lifelong endocrine follow-up is recommended for all NF1 patients (13). A recent case report described a patient with a heterozygous guanine nucleotide-binding protein G(s) subunit alpha (GNAS) R201C mutation in a somatotroph adenoma. This was the first reported rare MEN1-like case of genetically diagnosed NF1 complicated with acromegaly caused by somatotroph adenoma (14).
VHL syndrome is a heritable multisystem cancer syndrome that is caused by germline mutations of the VHL tumor suppressor gene. The incidence of this disorder is as high as 1 in 36,000 live births (15). Patients with VHL syndrome are at risk of developing various benign and malignant tumors of the central nervous system [i.e., pituitary stalk hemangioblastomas (15)], kidneys, adrenal glands, pancreas, and reproductive adnexal organs (16). It has been reported that propranolol can decrease the viability of VHL-related hemangioblastomas and renal cell carcinomas in vitro, likely by modulating vascular endothelial growth factor expression and inducing apoptosis (17). However, propranolol treatment for this disease is limited to early clinical trials.
DICER1 syndrome is caused by heterozygous germline mutations in the DICER1 gene (30). Several cases have been reported of rare infantile-onset pituitary blastoma that were mainly caused by germline mutations in DICER1 (31). Recently, corticotroph adenomas have also been identified in this tumor syndrome (31). DICER1 encodes a cytoplasmic endoribonuclease that processes hairpin precursor miRNAs into short, functional miRNAs that downregulate targeted mRNAs, thereby modulating cellular protein production (32). In addition, specific somatic mutations in the DICER1 RNase III catalytic domain have been identified in several DICER1-associated tumor types (31, 33).
Lynch syndrome is a cancer-predisposing syndrome caused by germline mutations in genes involved in DNA mismatch repair (34). Germline mutations in MLH1 (35) and MSH2 (34) in the mismatch repair pathway, have been identified in Lynch syndrome patients with aggressive corticotropin-secreting adenomas, although these are isolated case reports. Missense mutations have also been detected in four mismatch repair genes (MSH5, MSH6, MLH1, and MLH3) in non-secreting adenomas (3).
The incidence of somatotroph adenomas is approximately 10 cases per 1 million individuals (2, 36, 37). Somatotroph adenomas are GH-secreting somatotropic tumors that exhibit excessive secretion of GH and IGF-1, causing acromegaly and abnormal growth of bones, tissues, and organs in patients. Currently, treatment methods are limited to surgery, radiotherapy, somatostatin receptor (SSTR) ligands, and GH receptor antagonists. Each of these treatments has specific side effects, and the efficacy varies greatly among different patients; thus, it is hard to directly target and inhibit the continuous secretion of GH in postoperative patients (2). It is necessary to further understand the molecular mechanisms of somatotroph adenomas to elucidate new therapeutic targets.
Somatotroph adenomas have greater genomic disruption than corticotroph adenomas or inactive tumors with no clinical evidence of hormone secretion (38). Gene mutations in PAs can be divided into heritable germline variations, mosaic mutations, and non-heritable somatic mutations (29). The former two are often familial and associated with syndromes, while the latter are sporadic (29). Mutations in aryl hydrocarbon receptor-interacting protein (AIP), GNAS, and cadherin-related 23 (CDH23), which are all involved in cAMP-associated pathways, are key for somatotroph tumorigenesis (Figure 1, Table 1).
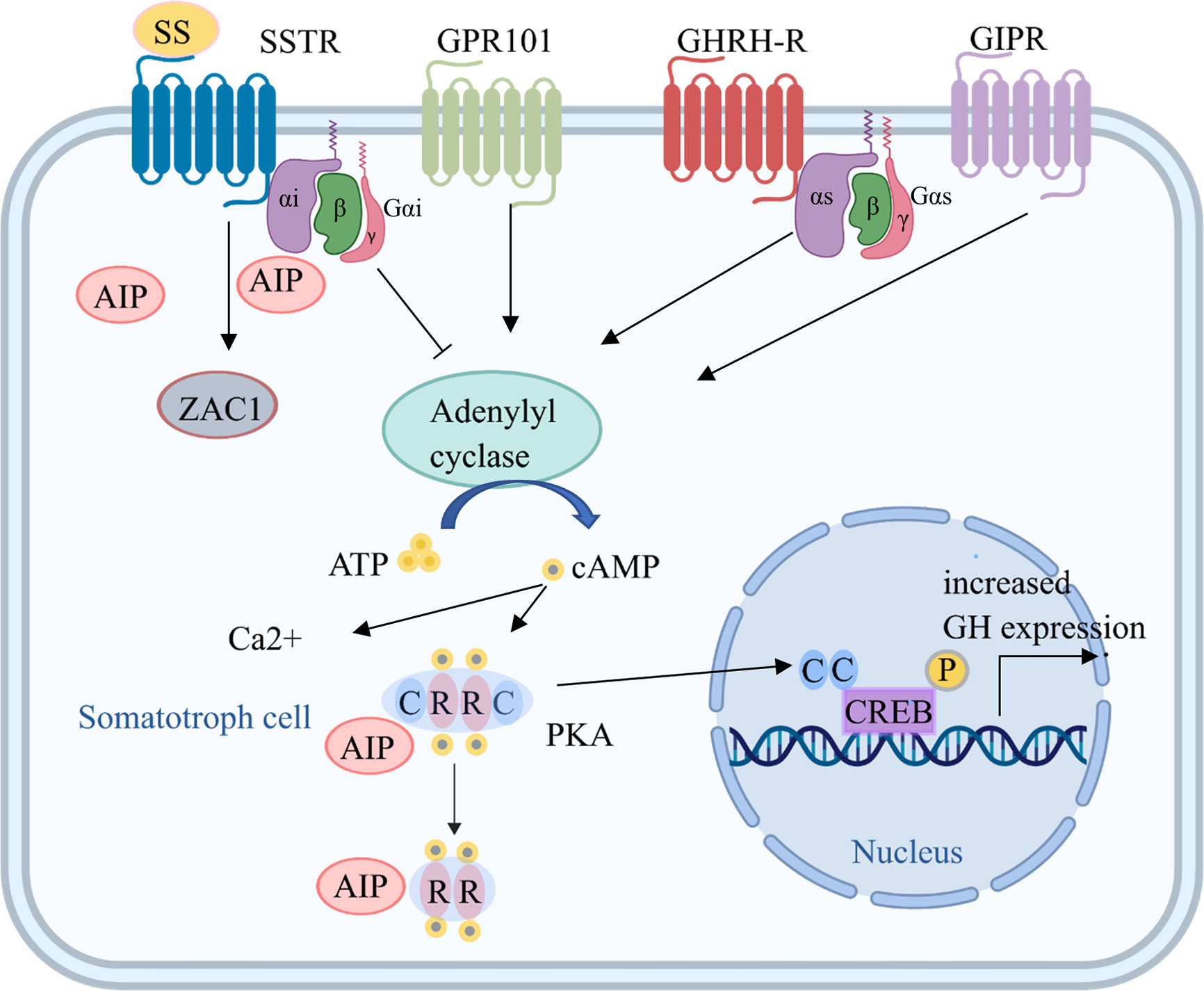
Figure 1 Tumorigenic mechanisms in somatotroph cells. Several mechanisms increase cAMP production, which is key for somatotroph tumorigenesis. Hormones bind to receptors, including GHRH-R, SSTR, GPR101, and GIPR, on the somatotroph cell membrane and increase the activation of adenylyl cyclase through Gsα. The consequent increase in cAMP production leads to the dissociation of the regulatory subunits of PKA from the catalytic subunits, which then translocate to phosphorylate CREB in the nucleus and other targets, leading to increased GH expression and cell proliferation. Gsα activation induced by GNAS mutations also leads to upregulation of the cAMP pathway. In addition, ectopic expression of GIPR may lead to an activated cAMP pathway, and GPR101 is a Gsα-coupled constitutively active receptor that leads to increased cAMP signaling. AIP, aryl hydrocarbon receptor-interacting protein; ATP, adenosine triphosphate; C, catalytic subunit; cAMP, cyclic adenosine monophosphate; CREB, cAMP response element; GHRH, growth hormone-releasing hormone; GHRH-R, GHRH receptor; GIPR, gastric inhibitory polypeptide receptor; GPR101, G protein-coupled receptor 101; Gsα, G protein stimulatory alpha subunit; GTP, guanine triphosphate; PKA, protein kinase A; R, regulatory subunit; SSTR, somatostatin receptor; ZAC1, zinc finger protein PLAGL1.
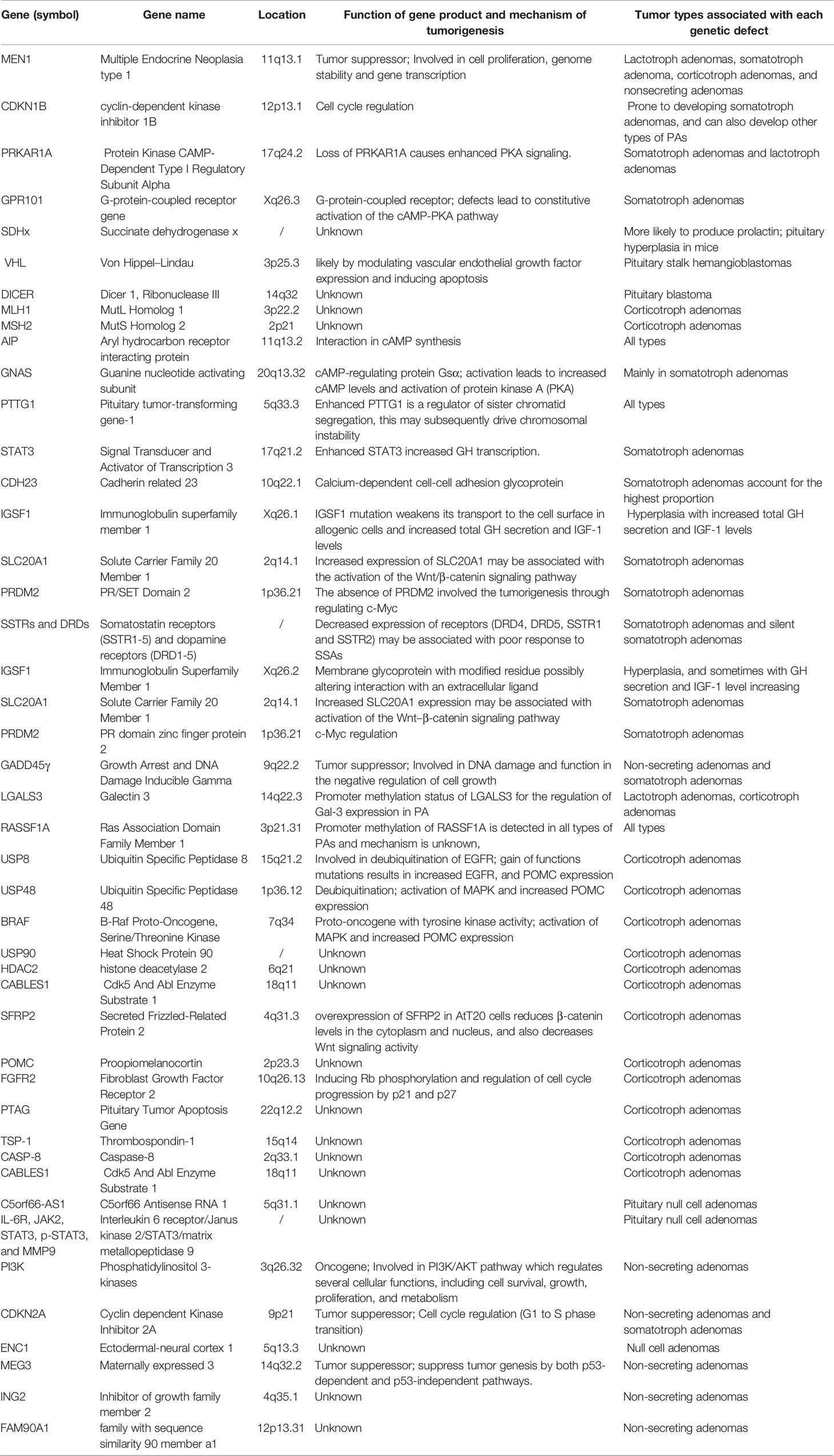
Table 1 Genes affected in different pituitary tumors via genetic mutation or DNA methylation.
Familial isolated pituitary adenoma (FIPA) is characterized by the familial occurrence of PAs in the absence of other clinical features (39). Germline mutations in the AIP gene are detected in approximately 20% of FIPA families and 50% of familial acromegaly families (40–43). AIP is located on human chromosome 11q13.2, and acts as a tumor suppressor in PAs (44). Mutations in AIP have been identified as causing a predisposition for PAs of variable penetrance in 20% of FIPA families (41). AIP mutations are usually associated with somatotropinomas, but prolactinomas, non-functioning PAs (NF-PAs), Cushing’s disease, and other infrequent clinical adenoma types can also occur (40–43). AIP mutations are common in male pediatric acrogigantism patients, and tend to cause large and invasive tumors; densely granulated subtypes rarely occur, and patients with AIP mutations are often resistant to somatostatin analogue (SSA) treatment (42, 43, 45, 46). Families with AIP mutations show incomplete penetrance, of approximately 15–30% (40, 42, 47). Genetic screening can identify carrier family members, and clinical screening has been reported to result in the earlier recognition of clinically relevant disease in approximately 20% of patients (22/187) (43).
Mechanically, some of the mutations lead to truncation of the AIP protein and loss of the C-terminal sequence, which affects protein interactions and leads to disrupted AIP function. A number of mechanisms may explain the resistance of AIP-mutated patients to SSAs. First, one mechanism may involve the reduced expression of the inhibitory G protein subtype, Gαi-2, which mediates the inhibitory effects of SSAs (44). Second, AIP has been reported to interact with both phosphodiesterase (PDE) and guanine nucleotide-binding proteins (G proteins); PDE4 expression is lower in AIP-mutated PAs, and interactions between PDE4 and AIP are disrupted by such mutations (44, 48, 49). Consistent with these findings, both PDE isoforms are reportedly overexpressed in GH cells from sporadic AIP mutation-negative GH-secreting adenomas (49). Third, AIP interacts with the protein kinase A (PKA) complex. AIP mutations affect the PKA pathway, thus affecting cell proliferation and development and the inflammatory response (42, 49). Fourth, another mechanism of SSA resistance may be related to the SSTR2–zinc-finger protein 1 PLAGL1 (ZAC1) pathway (50). AIP is upregulated by SSAs, and AIP can in turn upregulate ZAC1 mRNA expression (51, 52). Disordered cAMP regulation is important for the resistance of AIP mutations to SSA treatment (42, 49).
GNAS encodes the stimulatory α subunit of the G protein complex, which plays an important role in transmembrane signal transduction (53). Its mutation rate is the highest of somatic mutations in somatotroph adenomas (up to 40%). GNAS-mutated tumors are often smaller and less invasive, respond better to SSAs, and are usually densely granulated somatotroph adenomas (52, 54, 55). In addition, GNAS-mutated tumors have relatively high expression of dopamine receptor (DRD) 2, which suggests a good response to dopamine agonists (38). Somatic mutations in GNAS can result in sporadic somatotroph adenomas, while mosaic mutations for codon 201 likely result in McCune–Albright syndrome. This syndrome is characterized by polyostotic fibrous dysplasia, skin hyperpigmentation, and autonomous endocrine hyperfunction (56).
GNAS mutations can also lead to disruption of the cAMP signaling pathway. Especially, mutations in codon 201 or 227 result in the inhibition of G proteins and the activation of adenylyl cyclase, promote cAMP synthesis in cells, and drive tumorigenesis (57). Recently, a GNAS mutation (p.Arg201Cys) has been detected as a recurrent somatic event, and this mutation is shared with chromosome losses (58, 59).
Recent studies have indicated that increased cAMP in tumorigenesis can probably induce DNA damage, leading to somatic CNVs and genome instability (60). Of these, arm-level CNVs are the most common abnormalities in somatotroph adenomas. Specifically, whole chromosome losses (chromosomes 1, 6, 13, 14, 15, 16, 18, and 22) and gains (chromosomes 3, 5, 7, 10, 19, 20, and X) have been observed (58, 59, 61). Interestingly, GNAS mutation-positive adenomas have relatively low CNV levels, whereas GNAS mutation-negative adenomas have a high degree of genomic disruption (58, 59, 61). These CNVs likely affect the Ca2+ and ATP pathways, which are involved in PA tumorigenesis (58, 59, 61). Thus, the CNVs in GNAS mutation-negative somatotroph adenomas provide an alternative tumorigenic pathway, which is linked to genomic instability (58, 59, 61).
Pituitary tumor-transforming gene 1 (PTTG1) is overexpressed in various pituitary tumors, and its expression is higher in more aggressive tumors (62–64). Somatotroph adenomas with recurrent aneuploidy have relatively high expression of PTTG1; as a regulator of sister chromatid segregation, this may subsequently drive chromosomal instability (64–66).
STAT3 is a member of the STAT family, and participates in cellular responses to cytokines and growth factors (67). Its expression is enhanced in somatotroph adenomas, leading to GH hypersecretion, which in turn promotes STAT3 expression (68). In primary human somatotroph adenoma-derived cell cultures, the specific inhibitor S3I-201 can inhibit STAT3 expression, thus decreasing GH transcription and reducing GH secretion (68).
CDH23 is involved in Wnt pathway regulation. The CDH23 c.4136G>T (p.Arg1379Leu) mutation leads to an amino acid substitution in the calcium-binding motif of the extracellular cadherin domain, which is predicted to disrupt cell–cell adhesion. The incidence rate of CDH23 mutations is 33% in FIPA patients and 12% in sporadic PA patients. Of the CDH23-mutated PAs, somatotroph adenomas account for the highest proportion (25.9%) (69). PAs with functional CDH23 variants are smaller and less aggressive compared with non-mutated PAs (69, 70). In addition, variants in this gene are associated with Usher syndrome (70). However, CDH23 variations have been reported in only one study, and functional validation studies are needed.
IGSF1 is a membrane glycoprotein. IGSF1 mutations can weaken its transport to the cell surface in allogenic cells, resulting in a novel X-related syndrome. This syndrome is characterized by central hypothyroidism, macro-orchidism, and prolactin deficiency (71–73). It can also be associated with acromegaloid facial features, increased head circumference, and increased total GH secretion and IGF-1 levels. However, considering that patients present with hyperplasia rather than adenomas, these symptoms may be secondary to the failure of regulatory and feedback mechanisms.
SLC20A1 levels are positively associated with tumor size, invasive behavior, and recurrence in somatotroph adenomas. In addition, increased SLC20A1 expression may be associated with activation of the Wnt–β−catenin signaling pathway (74).
PRDM2, a tumor suppressor, plays an important role in cancer and obesity, including PAs. The absence of PRDM2 is likely to be involved in the tumorigenesis of somatotroph adenomas by regulating c-Myc (75). However, this discovery remains to be validated by more investigation groups.
SSTR (SSTR1–5) and DRD (DRD1–5) subtypes play critical roles in the regulation of hormone secretion (76). Decreased expression of some of these receptors (DRD4, DRD5, SSTR1, and SSTR2) may be associated with a poor response to SSAs (77). In particular, SSTR2 expression might be a good predictor of a patient’s response to SSAs (78).
DNA methylation is the most frequently studied epigenetic phenomenon, in which alterations of CpG dinucleotides block the transcriptional mechanism and silence gene expression (79). Approximately 80% of CpG dinucleotides are methylated in the human genome throughout the lifespan, and nearly 70% of CpG islands are methylated at any time, suggesting a widespread regulatory scope of DNA methylation (80). Here, we summarize the methylated genes in somatotroph adenomas (Table 1).
Growth arrest and DNA damage-inducible gene (GADD45γ) is a negative regulator of cell growth that is involved in DNA damage repair. In one study, most PAs (22/33, 67%) did not have GADD45γ expression, and 57.6% (19/33) of PAs were detected with GADD45γ methylation; there was significantly associated between GADD45γ methylated tumors and tumors in which GADD45γ transcript was not expressed (18 of 22; 82%; P = 0.002) (81). The silencing of GADD45γ is likely to confer a selective growth advantage during PA tumorigenesis (82).
The importance of the promoter methylation status of LGALS3 (the gene encoding galectin-3) for the regulation of galectin-3 expression in PA was confirmed by Ruebel et al. Ikaros, a factor with transcriptional functions and chromatin-remodeling properties that determine the fate of hypothalamic neuroendocrine and pituitary cell populations during development, may also contribute to the expression of galectin-3 (83).
The human Ras-association domain family 1A (RASSF1A) gene has been reported as frequently (38%, 20/52) hypermethylated in its promoter region in all types of PAs. RASSF1A promoter methylation is relatively low in gonadotroph cell adenomas, higher in the most aggressive adenomas, and potentially correlated with Ki-67 expression. Reduced expression of RASSF1A has been identified in 18 of 20 (90%) adenomas with hypermethylation of RASSF1A (84).
miRNA binding to the 3’ and 5’ untranslated regions and coding sequences of target RNA is a form of post-transcriptional modification that results in differential gene expression. miRNAs play an important role in various pathways in tumors.
Palumbo et al (85). reported that miRNAs that target HMGA, including miR-15, miR-16, miR-26a, let-7a, miR-196a2, and other miRNAs, are downregulated in PAs. These HMGA-targeting miRNAs have also been demonstrated to inhibit the proliferation of a somatotroph adenoma cell line (GH3) and promote pituitary tumorigenesis. Furthermore, D’Angelo et al. reported that downregulated miRNAs, including miR-326, miR-570, and miR-432, target HMGA1 and HMGA2; while miR-34b and miR-548c-3p target HMGA2; and miR-603 and miR-326 target E2F transcription factor 1 (E2F1). In addition, some miRNAs are downregulated in somatotroph adenomas, including miR-34b, miR-326, miR-432, miR-548c-3p, miR-570, and miR-603 (86). The long non-coding RNA (lncRNA) ribosomal protein SA pseudogene 52 (RPSAP52) is overexpressed in PAs. It promotes cell proliferation in a competing endogenous RNA (ceRNA)-dependent manner by competitively binding to miR-15a, miR-15b, and miR-16, and by upregulating the expression of HMGA1 and HMGA2 (87, 88).
Downregulated and upregulated miRNAs sometimes work together. For example, the downregulation of miR-26b expression together with the upregulation of miR-128 suppresses colony formation ability and invasiveness, and regulates the activity of the PTEN–AKT pathway in somatotroph adenomas (85).
Multiple miRNAs are associated with the increased expression of PTTG1. First, miR-338-3p is upregulated in invasive somatotroph adenomas, and probably mediates the increased expression of PTTG1 (89). Second, miR-423-5p, which targets PTTG1, shows decreased expression in somatotroph adenomas, and inhibits the expression of PTTG1 at both the mRNA and protein levels (90). Third, overexpression of miR-524-5p downregulates the expression of PTTG1-binding factor, which interacts with PTTG1 to mediate downstream effects, and significantly attenuates proliferation, migration, and invasion in vitro (91).
Corticotropin-secreting adenomas account for 15% of PAs, with an incidence of 1.6 cases per 1 million individuals. These adenomas are typically small, and the excessive secretion of corticotropin leads to adrenal hypercortisolemia (92). Although 75% of patients achieve remission after surgical treatment, recurrence occurs in approximately 10% of these patients (93). Current pituitary-targeted drugs, including cabergoline and pasireotide, can improve the clinical features of excessive hormone secretion, but 60–75% of patients are insensitive to these drugs, which cannot control the symptoms long after drug treatment (94). Therefore, an understanding of the molecular characteristics of these tumors is necessary to identify additional drug targets. Here, we summarize the abnormal alterations in corticotropin-secreting adenomas that may serve as potential therapeutic targets (Figure 2, Table 1).
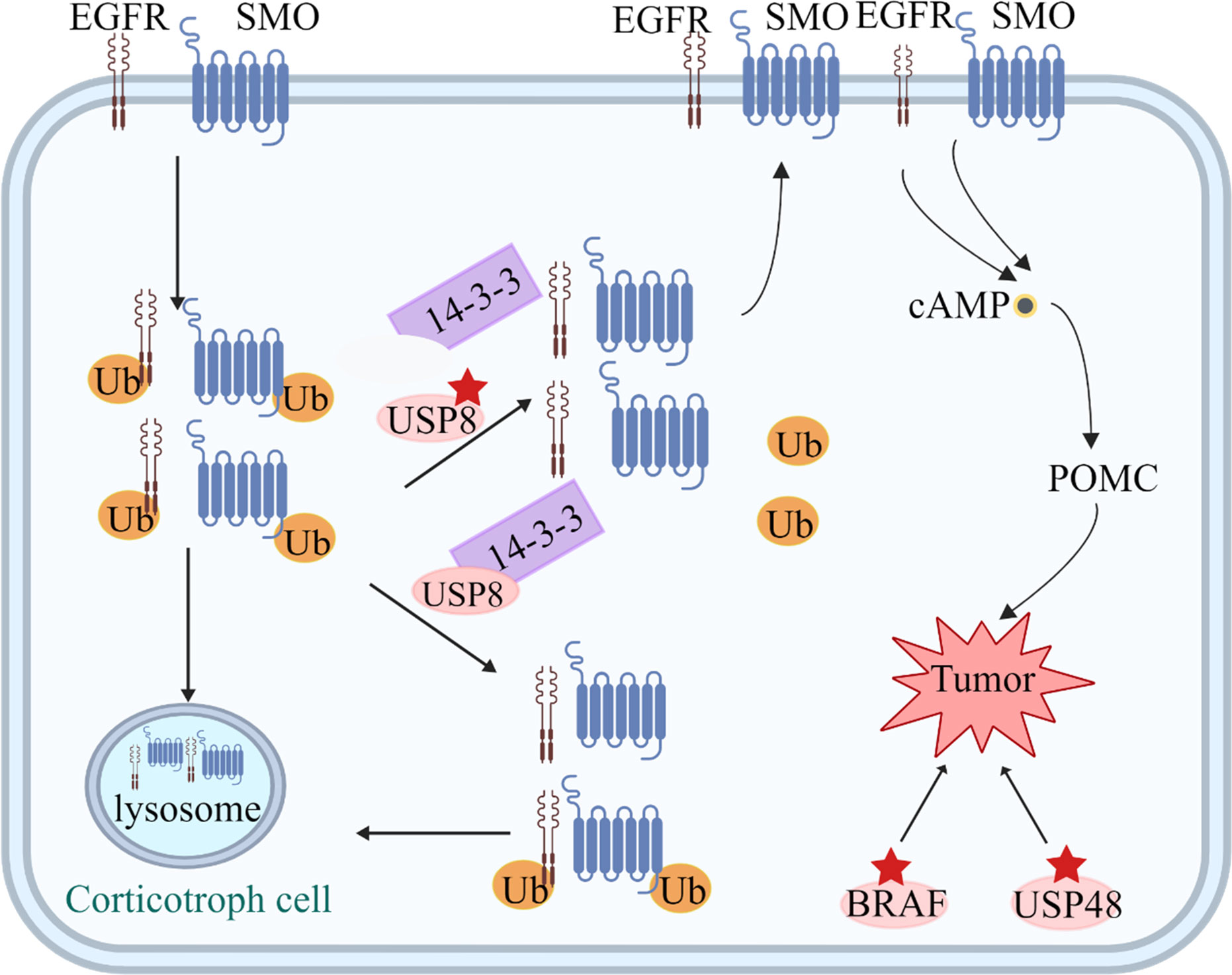
Figure 2 Tumorigenic mechanisms in corticotroph cells. USP8 removes ubiquitin tags from targets, such as EGFR and Smoothened (SMO), preventing them from undergoing proteasomal degradation and allowing recycling back to the cell surface. Increased EGFR and SMO activity leads to increased cAMP and POMC levels. Mutated USP8 cannot bind 14-3-3 protein and undergoes cleavage, which increase enzymatic activity, leading to increased deubiquitination of EGFR and SMO and higher expression of the two proteins on the cell membrane. USP48 mutations and gain-of-function mutations of BRAF probably play a similar role to USP8.
USP8 encodes a deubiquitinase enzyme that protects epidermal growth factor receptor (EGFR) from degradation. Up to 62.4% of corticotropin-secreting adenomas were found to have USP8 mutations that block 14-3-3 protein binding, leading to increased activity of USP8 (95–97). Gain-of-function mutations in USP8 increase the deubiquitination of EGFR, which inhibits its degradation, leading to the activation of EGFR signaling. This mechanism likely leads to the synthesis and secretion of adrenocorticotropic hormone (ACTH) and promotes tumorigenesis (95–98). In USP8-mutated corticotropin-secreting adenoma samples, the cell cycle inhibitor p27, heat shock protein 90 (HSP90), and phosphorylated cAMP-response element binding protein (pCREB) were significantly reduced, suggesting that these proteins are direct or indirect clients of USP8 and could therefore be potential targets for treatment (99).
USP8 mutations have also been identified in silent corticotroph adenomas (100). Transcriptomic profiles show significant differences between functioning and silent corticotroph adenomas. However, USP8 mutations have pleiotropic effects in both functioning and silent corticotroph adenomas, and affect the expression levels of many genes that are involved in a range of different pathways (100).
Germline USP8 mutations, which are commonly found as somatic mutations in corticotropin-secreting adenomas, have also recently been described in a child with dysmorphic features, developmental delay, and a corticotroph adenoma (101). In addition, an overall prevalence of USP8 mutations of 32% has been reported in corticotropin-secreting adenomas. USP8-mutated tumors are more common in females, and are associated with earlier onset (96, 102), a smaller size (95), and increased ACTH production (95, 102). Patients with USP8-mutated tumors are more likely to go into initial remission after surgery, but may also be more likely to show recurrence later in the clinical course (102–104). In contrast, USP8-mutation-negative tumors are more likely to show sphenoid invasion with an increased epithelial–mesenchymal transition signature (38). Lapatinib, an EGFR inhibitor, decreases proliferation in vitro and reduces tumor weight in vivo (105). In addition, SSTR5 expression is higher in USP8-mutated tumors (38, 106), potentially allowing mutation status to be used as a predictor of response to pasireotide (a second-generation SSA with a greater affinity for SSTR5) (107).
With the recognition of the importance of USP8 mutations, another two mutated genes in the mitogen-activated protein kinase (MAPK) pathway, USP48 and BRAF, have also been detected in corticotropin-secreting adenomas (108). Missense mutations in USP48 include M415I/V substitutions, while V600E is an activation mutation of BRAF. Among tumors without USP8 mutations, 23% of corticotropin-secreting adenomas have USP48 mutations and 16% have BRAF mutations (108). Both mutations enhance the promoter activity and transcription of the ACTH precursor, the proopiomelanocortin (POMC) gene, and are potential therapeutic targets for the excess secretion of ACTH in corticotropin-secreting adenomas. Furthermore, USP48 variants are associated with smaller tumors and a better response to corticotropin-releasing hormone (CRH) stimulation (109). Therefore, variations in EGFR regulation, the MAPK pathway, and POMC-related genes play a certain role in corticotropin-secreting adenomas.
Corticotroph adenomas overexpress HSP90 compared with the normal pituitary gland. N- and C-terminal HSP90 inhibitors act at different stages of the HSP90 catalytic cycle to regulate corticotropic cell proliferation and glucocorticoid (Gc) receptor (GR) transcriptional activity. The C-terminal HSP90 inhibitor silibinin has been reported to have anti-tumorigenic effects, partially decrease hormonal secretions, and alleviate the symptoms of Cushing’s disease in a mouse model (110).
The negative feedback regulation of POMC by GRs is a critical feature of the hypothalamo–pituitary–adrenal axis. Loss of Brg1 or HDAC2 should therefore produce Gc resistance, and we have previously shown that approximately 50% of Gc-resistant human and dog corticotropin-secreting adenomas, which are the hallmark of Cushing’s disease, have deficient nuclear expression of either of these proteins. In addition to providing a molecular basis for Gc resistance, Brg1 and HDAC2 deficiencies may also contribute to the tumorigenic process (111).
CABLES1 is a cell cycle regulator that participates in the adrenal–pituitary negative feedback loop. Four heterozygous germline missense variants have been identified in CABLES1 in four sporadic patients from a cohort of 182 patients with corticotropin-secreting adenomas, with functional evidence. The four variants affected residues within or close to the predicted cyclin-dependent kinase-3 (CDK3)-binding region of the CABLES1 protein and impaired its ability to block cell growth in a mouse corticotropinoma cell line (AtT20/D16v-F2). However, further studies are needed to assess the prevalence of CABLES1 mutations in patients with other types of PAs, and to elucidate the pituitary-specific functions of this gene (111).
The RNA and protein expression of SFRP2 is decreased in corticotroph adenomas compared with normal pituitary glands (112). In addition, the overexpression of SFRP2 in AtT20 cells reduces β−catenin levels in the cytoplasm and nucleus, and also decreases Wnt signaling activity. SFRP2 may therefore act as a tumor suppressor in Cushing’s disease by regulating Wnt signaling pathway activity. Clinically, there is an association between lower SFRP2 expression and aggressive adenoma characteristics, including a larger size and invasiveness (112).
Although some corticotropin-secreting adenomas have been found to have genetic mutations, the pathogenesis of non-mutated adenomas remains unknown. DNA methylation, a complementary mechanism for gene mutations, also plays an important role in PAs.
Hypomethylation of the promoter of POMC, which encodes the precursor of ACTH, leads to the occurrence of corticotropin-secreting adenomas. A comparison of the methylation profiles of ACTH-PAs and NF-PAs showed that the overexpression of POMC likely accounts for promoter hypomethylation (3). As a pituitary hormone, ACTH leads to increased serum cortisol levels in patients with Cushing’s disease, which is associated with the occurrence of corticotropin-secreting adenomas.
FGF2 is a potent growth factor that regulates stem cell maintenance and neurogenesis during embryonic development and in response to challenges such as stress or injury in the adult brain (113–117). FGFR2 encodes a growth factor receptor, and was found to be methylated by a 5’ promoter in mouse AtT20 cells, leading to significantly downregulated expression (80).
In a model pituitary tumor cell line (AtT20), enforced expression of PTAG is associated with significantly increased sensitivity to the apoptotic effects induced by bromocriptine challenge (118).
The secreted angioinhibitory factor TSP-1 is an adhesive glycoprotein that mediates cell-to-cell and cell-to-matrix interactions, and is associated with platelet aggregation, angiogenesis, and tumorigenesis. Overexpression of TSP-1 in a murine AtT20 pituitary corticotroph tumor cell line leads to increased ACTH secretion (119).
The methylation levels of ESR1 and CASP8 in corticotropin-secreting adenomas are also increased compared with those of silent corticotroph adenomas. CASP8 plays an important role in apoptosis. Furthermore, CASP8 has been suggested to affect the functional behavior of corticotroph adenomas, but the specific mechanisms remain unknown. The methylation of POMC, FGFR2, RASSF1A, LGALS3, and apoptosis-related factors plays a potential role in the development of corticotropin-secreting adenomas.
The tumor suppressors miR-15a, miR-16, and miR-132 are downregulated in corticotroph adenomas. These miRNAs inhibit the proliferation, invasion, and migration of pituitary tumor cells by targeting sex-determining region Y-box 5 (SOX5) (120). In addition, the following miRNAs are also reportedly downregulated in corticotroph adenomas: miR-145 (2.0 fold), miR-21 (2.4 fold), miR-141 (2.6 fold), let-7a (3.3 fold), miR-150 (3.8 fold), and miR-143 (6.4 fold), suggesting that they may play a role in the tumorigenesis of corticotropin-secreting adenomas. miR-26a is overexpressed in corticotroph adenomas, and one of its direct targets is protein kinase Cδ (PRKCD) (85). PRKCD silencing is associated with increased EGFR expression, indicating PRKCD as a possible molecular target for the treatment of corticotroph adenomas (121, 122).
Non-secreting adenomas account for 20–35% of PAs originating from different hormone-secreting cells. Many of these are gonadotroph adenomas, and some show somatotroph, corticotroph, thyrotroph, or lactotroph differentiation. In older studies, the diagnosis of non-functional adenomas was mainly based on the normal results of hormone secretion rather than the results of immunohistochemical staining. However, with recently updated pituitary tumor classifications, the diagnosis of non-functional adenomas is made according to detailed pituitary cell types. Because excessive hormone levels are not found in the blood with non-secreting adenomas, they may not be detected for years in patients, and are often diagnosed incidentally (92). Although mostly benign, some invasive pituitary tumors require adjuvant radiation after surgery, often resulting in pituitary failure and a risk of recurrence. In an effort to aid in the development of additional treatment methods, we herein summarize the molecular signatures of non-secreting adenomas (Table 1).
C5orf66-AS1 encodes a lncRNA that is differentially expressed between pituitary null cell adenoma tissues and normal pituitary tissues, as well as between invasive and non-invasive tumors (123). Co-expression analysis in RNA sequencing data revealed that PAQR7 [a membrane progesterone receptor that may mediate a reduction in gonadotropin-releasing hormone in the progesterone negative feedback action in a progesterone receptor (A/B)-independent way (124)] was the gene that was most correlated with C5orf66-AS1, and several predicted trans-acting target genes, including SCGB3A1 (encoding secretoglobin family 3A member 1), were also highly correlated with C5orf66-AS1 (123). These results indicate that C5orf66-AS1 suppresses the development and invasion of pituitary null cell adenomas (123).
Integrative proteomics and transcriptomics have revealed the activation of IL6R, JAK2, and STAT3, and the overexpression of IL-6R, JAK2, STAT3, p-STAT3, and MMP9, in invasive pituitary null cell adenomas (125). Therefore, activation of the IL-6R–JAK2–STAT3–MMP9 signaling pathway is correlated with the invasiveness of pituitary null cell adenomas.
There is a negative correlation between SSTR2 and tumor size in silent somatotroph adenomas. Additionally, levels of DRD2 expression are reportedly similar between silent and functioning somatotroph adenomas, suggesting a possible basis for the treatment of these tumors with SSAs and dopamine (126).
PI3K is an important regulator of cell growth, transformation, adhesion, apoptosis, survival, and movement. The catalytic subunit encoding the PIK3CA gene is located on chromosome 3q26.3 and is often found to be deficient in cancer (127). Somatic mutations have been detected in 8 out of 91 (9%) of invasive pituitary tumors versus 0 out of 262 (0%) non-invasive tumors (128). In addition to mutations of PI3K, mutations of MEN1, AIP (129), and CDH23 (69) have also been detected in non-secreting adenomas. In this section, we summarize the gene mutations in cell cycle regulators, phosphokinase, the cAMP signaling pathway, and cadherin that are associated with the occurrence of non-secreting adenomas.
The p16 protein, encoded by the tumor suppressor gene CDKN2A, is located on chromosome 9p21. CDKN2A has been found to exhibit deletions, point mutations, or methylation inactivation in a variety of tumors. Methylation of the CpG island of CDKN2A was detected in 32/46 (70%) non-secreting adenomas, in contrast to 2/21 (9.5%) somatotroph adenomas and 0/15 histologically normal postmortem pituitaries (130). Methylation of CDKN2A corresponds to a loss of p16 expression on immunohistochemical analysis (130). When p16 is silenced, retinoblastoma protein (encoded by RB1) becomes phosphorylated, which enables cell cycle progression via the activation of E2F transcription factors (80). These observations indicate that the CDKN2A-related pathway is a potential target for non-secreting adenoma cell proliferation.
MEG3 is a maternally expressed imprinted lncRNA that is transcribed from multiple transcriptional variants with different splicing patterns, all of which are lncRNAs. Several studies have shown that MEG3-encoded lncRNA is a tumor suppressor that interacts with p53 and regulates expression of the p53 gene (TP53) (79, 131). In NF-PAs, MEG3 was found to be hypermethylated in both the 5’ region of the first exon and approximately 1.6–2.1 kb upstream of the first exon, leading to the silencing of gene expression (79). Hypermethylation in the regulated area of MEG3 is an important mechanism that likely leads to the loss of MEG3 expression in clinical NF-PAs.
Methylated ENC1 has been found in tumor samples, and decreased levels were detected compared with normal pituitary glands (132). Notably, ENC1 expression levels are reportedly lower in invasive null cell adenomas than in non-invasive adenomas (133, 134).
ING2 is a member of the growth inhibitor family, and participates in regulating the activity of histone acetyltransferase and histone deacetylase complexes, and plays a role in DNA repair and apoptosis. Methylation sequencing of NF-PAs revealed the presence of ING2 DNA methylation in NF-PAs, and the methylation and expression levels were correlated with tumor recurrence. Furthermore, clinical data have indicated that ING2 is an independent prognostic marker of NF-PAs (135).
FAM90A1 is a primate-specific gene that is associated with ING2. FAM90A1 is also hypermethylated in NF-PAs, and the methylation level is significantly correlated with patient prognosis (135). FAM90A1 methylation likely plays a role in NF-PA tumorigenesis and is a potential marker of PA.
Similar to somatotroph adenomas, NF-PAs also exhibit hypermethylation in the CpG island of the GADD45γ gene. In addition, the methylation of CDKN2A, MEG3, and RASSF1A has also been detected in NF-PAs. Most methylated genes are involved in the cell cycle and DNA damage repair. However, novel abnormally methylated genes, such as ING2 and FAM90A1, have also been detected, and their specific regulatory mechanisms remain unknown.
Lactotroph adenomas are the most common secretory tumors, accounting for 60% of all PAs. Microprolactinomas are usually stable and slow growing, and continued growth after diagnosis occurs in less than 15% of cases (2). Lactotroph adenomas are ideally managed with dopamine agonists (136). Dopamine agonists can reduce prolactin levels and shrink tumors; however, they have side effects, and 15% of patients are not sensitive to these drugs (2). Additional targets are therefore needed to improve lactotroph adenoma treatment. Here, we summarize the molecular variations that occur in lactotroph adenomas.
HMGA proteins comprise a family of transcriptional regulating factors that are highly expressed during embryogenesis and play an important role in tumorigenesis in various tissues, including the pituitary gland. HMGA2 has been shown to be rearranged and amplified in lactotroph adenomas, and transgenic mice with Hmga2 overexpression develop PAs with prolactin and GH secretion (137). The primary mechanism by which HMGA2 mutations lead to PAs involves an increase in E2F1 activity (138). It has also been reported that MIA is one of the most commonly downregulated genes in lactotroph adenomas, and this may serve as a downstream mechanism of HMGA2 (137, 139). The HMGA2 protein can bind to the DNA elements of pituitary-specific positive transcription factor 1 (PIT-1) and cause a PIT-1 reaction, thus positively regulating the production of the PIT-1 promoter. It is therefore speculated that HMGA2 leads to the proliferation of PIT-1-expressing cells or the abnormal proliferation of GH- and prolactin-secreting embryonic cells (137, 140). In addition, the downregulation of miRNAs may be related to the accumulation of HMGA2, which might lead to the proliferation of pituitary cells. In one study, the expression of miR-let-7 was reported to be decreased in 23 of 55 (42%) lactotroph adenomas, and was negatively correlated with the expression of HMGA2 (141). The overexpression of HMGA2 protein is also positively correlated with pituitary tumor phenotype, and overexpression of Hmga2 in transgenic mice results in the development of PA (142). Therefore, HMGA2 is a specific oncogene for pituitary transformation. However, according to the current evidence that has been collected on genes in the HMGA family, only HMGA2 has a direct causal role in human pituitary tumorigenesis because it is amplified or rearranged in human lactotroph adenomas. It has been reported that HMGA1 and HMGA2 nuclear expression levels are significantly higher in invasive adenomas than in non-invasive adenomas (143, 144).
The deletion of the PRLR-encoding gene leads to aggressive pituitary tumors in mice, while homozygous deletion mutants of PRLR in patients with hyperprolactinemia and agalactia do not lead to pituitary tumors (145). In contrast, a gain-of-function mutation was found in 9 of 46 patients with lactotroph adenomas, representing a potential novel mechanism for lactotroph adenoma tumorigenesis. In addition, three other rare variants and two low-frequency variants found in this cohort may represent benign changes (146). However, further investigations are needed to confirm the underlying mechanisms.
Thyrotroph adenomas are the least common type of PAs, accounting for approximately 1% of all adenomas. Thyrotroph adenomas lead to elevated or inappropriately suppressed thyrotropin levels, with normal or elevated thyroid hormone levels. Sapkota et al. confirmed six DNA variants as candidate driver mutations; two of these mutations were identified in genes with an established role in malignant tumorigenesis (SMOX and SYTL3), while four had unknown roles (ZSCAN23, ASTN2, R3HDM2, and CWH43) (147). Similarly, a single nucleotide polymorphism array analysis revealed frequent chromosomal regions of copy number gains, including recurrent gains at the loci harboring four of these six genes. In addition, Ando et al. reported a somatic mutation in the ligand-binding domain of thyroid hormone receptor beta (TRbeta) that causes a His to Tyr substitution at codon 435 of TRbeta1, corresponding to codon 450 of TRbeta2 (148). Unlike other PAs, thyrotroph adenomas are rarely associated with genetic syndromes or common somatic mutations.
Gain or loss of chromosome. A non‐random gain in pituitary tumors has been reported in chromosomes 5, 8, 12, and X (149, 150). Gains of chromosomes 8 and 12 were found in prolactinomas and non-secreting adenomas, whereas a combined loss of chromosomes 5 and 8 was observed in corticotroph and somatotroph adenomas. In addition, recurrent structural rearrangements affecting chromosomes 1, 3, and 12 have also been identified in prolactinomas, which appear to be the only PA subtype with a defined trend of tumor‐specific chromosomal changes (149). In another whole-exome sequencing investigation, 75% of the highly disrupted group were functional adenomas or atypical null cell adenomas, whereas 87% of the less-disrupted group were non-functional adenomas (151). The disrupted samples were characterized by expression changes associated with poor outcomes in other cancers. Furthermore, arm-level losses of chromosomes 1, 2, 11, and 18 were significantly recurrent (151). These data indicate that sporadic PAs have distinct copy-number profiles that are associated with hormonal and histological subtypes and influence gene expression.
Transcription factors. Cell lineages of the pituitary are dependent on the expression of the pituitary transcription factors PIT1, TPIT, and SF1 that, in concert with ERα and GATA2/3, regulate cellular differentiation and hormone secretion (152). Recently, these transcription factors (PIT1 for GH, prolactin, and TSH lineages; SF1 for gonadotroph lineages; and TPIT for ACTH lineages) have been added to the classification (152, 153).
DNA methylation. Frequently methylated genes (i.e., CDKN2A, GADD45y, FGFR2, CASP8, and PTAG) demonstrate methylation in over 50% of PAs; moderately methylated genes (i.e., TSP-1, RASSF1A, RB1, p73, MGMT, and CDH1) demonstrate methylation in 20–50% of PAs; and infrequently methylated genes (i.e., p14, DAPK1, TIMP3, p21, and p27) are methylated in less than 20% of PAs (79, 154). These variable rates of methylation may reflect a variety of factors.
Implications for therapy. In a subset of pituitary tumors that are refractory to routine therapy, both MGMT promoter hypermethylation in aggressive PAs and pituitary carcinomas and low protein expression have been implicated. Hypermethylation of MGMT indicates a better response to treatment with temozolomide (TMZ) in some aggressive PAs (155). In one study, combination treatment with TMZ and pyrimethamine (PYR) produced synergistic antitumor activity both in vivo and in vitro. Moreover, TMZ/PYR treatment induced cell cycle arrest, increased DNA damage, and upregulated the expression of cathepsin B, BAX, cleaved PARP, and phosphorylated histone H2AX; it also elevated caspase 3/7, 8, and 9 activities. Therefore, PYR may enhance the efficacy of TMZ by triggering both cathepsin B- and caspase-dependent apoptotic pathways. A combination of PYR and TMZ may thus provide a novel regimen for invasive PAs that are refractory to standard therapy and TMZ alone (156).
We reviewed older and more recent discoveries of molecular variations in somatotroph adenomas, corticotroph adenomas, lactotroph adenomas, non-secreting adenomas, and thyrotroph adenomas. Summarizing the genetic and epigenetic markers of different types of PAs and elucidating their comprehensive molecular mechanisms will be helpful to identify the physiological pathways of PAs. These findings will provide a better understanding of the occurrence of PAs and lead to the development of novel therapeutic treatments.
CM wrote the manuscript of this paper, and YC helped in modifying the manuscript. BX and WR directed the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Key R&D Program of China (2018YFA0108602, 2018YFA0108600 to XB), China Postdoctoral Science Foundation (BX20190045 to MC), Beijing Municipal Natural Science Foundation (7182134 to XB), CAMS Initiative for Innovative Medicine (CAMSI2M) (2016-I2M-1-017 to XB), CAMS Young Talents Award Project (2018RC320003 to XB), and Beijing Nova Program (Z181100006218003 to XB).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Ostrom QT, Gittleman H, Liao P, Rouse C, Chen Y, Dowling J, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007-2011. Neuro Oncol (2014) 16 Suppl 4(Suppl 4):iv1–63. doi: 10.1093/neuonc/nou223
2. Melmed S. Pituitary-Tumor Endocrinopathies. N Engl J Med (2020) 382(10):937–50. doi: 10.1056/NEJMra1810772
3. Salomon MP, Wang X, Marzese DM, Hsu SC, Nelson N, Zhang X, et al. The Epigenomic Landscape of Pituitary Adenomas Reveals Specific Alterations and Differentiates Among Acromegaly, Cushing’s Disease and Endocrine-Inactive Subtypes. Clin Cancer Res (2018) 24(17):4126–36. doi: 10.1158/1078-0432.ccr-17-2206
4. Reddy R, Cudlip S, Byrne JV, Karavitaki N, Wass JA. Can we ever stop imaging in surgically treated and radiotherapy-naive patients with non-functioning pituitary adenoma? Eur J Endocrinol (2011) 165(5):739–44. doi: 10.1530/eje-11-0566
5. Caimari F, Korbonits M. Novel Genetic Causes of Pituitary Adenomas. Clin Cancer Res (2016) 22(20):5030–42. doi: 10.1158/1078-0432.Ccr-16-0452
6. Maxwell JE, Sherman SK, Howe JR. Translational Diagnostics and Therapeutics in Pancreatic Neuroendocrine Tumors. Clin Cancer Res (2016) 22(20):5022–9. doi: 10.1158/1078-0432.ccr-16-0435
7. Thakker RV. Multiple endocrine neoplasia type 1 (MEN1). Best Pract Res Clin Endocrinol Metab (2010) 24(3):355–70. doi: 10.1016/j.beem.2010.07.003
8. Longuini VC, Lourenço DM Jr., Sekiya T, Meirelles O, Goncalves TD, Coutinho FL, et al. Association between the p27 rs2066827 variant and tumor multiplicity in patients harboring MEN1 germline mutations. Eur J Endocrinol (2014) 171(3):335–42. doi: 10.1530/eje-14-0130
9. Balogh K, Patócs A, Hunyady L, Rácz K. Menin dynamics and functional insight: take your partners. Mol Cell Endocrinol (2010) 326(1-2):80–4. doi: 10.1016/j.mce.2010.04.011
10. Beijers H, Stikkelbroeck NML, Mensenkamp AR, Pfundt R, van der Luijt RB, Timmers H, et al. Germline and somatic mosaicism in a family with multiple endocrine neoplasia type 1 (MEN1) syndrome. Eur J Endocrinol (2019) 180(2):K15–k19. doi: 10.1530/eje-18-0778
11. Coppin L, Ferrière A, Crépin M, Haissaguerre M, Ladsous M, Tabarin A, et al. Diagnosis of mosaic mutations in the MEN1 gene by next generation sequencing. Eur J Endocrinol (2019) 180(2):L1–l3. doi: 10.1530/eje-18-0852
12. Agarwal SK, Mateo CM, Marx SJ. Rare germline mutations in cyclin-dependent kinase inhibitor genes in multiple endocrine neoplasia type 1 and related states. J Clin Endocrinol Metab (2009) 94(5):1826–34. doi: 10.1210/jc.2008-2083
13. Kyritsi EM, Hasiotou M, Kanaka-Gantenbein C. Partial empty sella syndrome, GH deficiency and transient central adrenal insufficiency in a patient with NF1. Endocrine (2020) 69(2):377–85. doi: 10.1007/s12020-020-02351-z
14. Hozumi K, Fukuoka H, Odake Y, Takeuchi T, Uehara T, Sato T, et al. Acromegaly caused by a somatotroph adenoma in patient with neurofibromatosis type 1. Endocr J (2019) 66(10):853–7. doi: 10.1507/endocrj.EJ19-0035
15. Lonser RR, Butman JA, Kiringoda R, Song D, Oldfield EH. Pituitary stalk hemangioblastomas in von Hippel-Lindau disease. J Neurosurg (2009) 110(2):350–3. doi: 10.3171/2008.4.17532
16. Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, et al. von Hippel-Lindau disease. Lancet (2003) 361(9374):2059–67. doi: 10.1016/s0140-6736(03)13643-4
17. Shepard MJ, Bugarini A, Edwards NA, Lu J, Zhang Q, Wu T, et al. Repurposing propranolol as an antitumor agent in von Hippel-Lindau disease. J Neurosurg (2018) 131(4):1–9. doi: 10.3171/2018.5.jns172879
18. Courcoutsakis NA, Tatsi C, Patronas NJ, Lee CC, Prassopoulos PK, Stratakis CA. The complex of myxomas, spotty skin pigmentation and endocrine overactivity (Carney complex): imaging findings with clinical and pathological correlation. Insights Imaging (2013) 4(1):119–33. doi: 10.1007/s13244-012-0208-6
19. Correa R, Salpea P, Stratakis CA. Carney complex: an update. Eur J Endocrinol (2015) 173(4):M85–97. doi: 10.1530/eje-15-0209
20. Boikos SA, Stratakis CA. Carney complex: pathology and molecular genetics. Neuroendocrinology (2006) 83(3-4):189–99. doi: 10.1159/000095527
21. Forlino A, Vetro A, Garavelli L, Ciccone R, London E, Stratakis CA, et al. PRKACB and Carney complex. N Engl J Med (2014) 370(11):1065–7. doi: 10.1056/NEJMc1309730
22. Trivellin G, Daly AF, Faucz FR, Yuan B, Rostomyan L, Larco DO, et al. Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. N Engl J Med (2014) 371(25):2363–74. doi: 10.1056/NEJMoa1408028
23. Iacovazzo D, Caswell R, Bunce B, Jose S, Yuan B, Hernández-Ramírez LC, et al. Germline or somatic GPR101 duplication leads to X-linked acrogigantism: a clinico-pathological and genetic study. Acta Neuropathol Commun (2016) 4(1):56. doi: 10.1186/s40478-016-0328-1
24. Gill AJ, Toon CW, Clarkson A, Sioson L, Chou A, Winship I, et al. Succinate dehydrogenase deficiency is rare in pituitary adenomas. Am J Surg Pathol (2014) 38(4):560–6. doi: 10.1097/pas.0000000000000149
25. Xekouki P, Szarek E, Bullova P, Giubellino A, Quezado M, Mastroyannis SA, et al. Pituitary adenoma with paraganglioma/pheochromocytoma (3PAs) and succinate dehydrogenase defects in humans and mice. J Clin Endocrinol Metab (2015) 100(5):E710–9. doi: 10.1210/jc.2014-4297
26. O’Toole SM, Dénes J, Robledo M, Stratakis CA, Korbonits M. 15 YEARS OF PARAGANGLIOMA: The association of pituitary adenomas and phaeochromocytomas or paragangliomas. Endocr Relat Cancer (2015) 22(4):T105–22. doi: 10.1530/erc-15-0241
27. Dénes J, Swords F, Rattenberry E, Stals K, Owens M, Cranston T, et al. Heterogeneous genetic background of the association of pheochromocytoma/paraganglioma and pituitary adenoma: results from a large patient cohort. J Clin Endocrinol Metab (2015) 100(3):E531–41. doi: 10.1210/jc.2014-3399
28. Xekouki P, Pacak K, Almeida M, Wassif CA, Rustin P, Nesterova M, et al. Succinate dehydrogenase (SDH) D subunit (SDHD) inactivation in a growth-hormone-producing pituitary tumor: a new association for SDH? J Clin Endocrinol Metab (2012) 97(3):E357–66. doi: 10.1210/jc.2011-1179
29. Xekouki P, Stratakis CA. Succinate dehydrogenase (SDHx) mutations in pituitary tumors: could this be a new role for mitochondrial complex II and/or Krebs cycle defects? Endocr Relat Cancer (2012) 19(6):C33–40. doi: 10.1530/erc-12-0118
30. Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet (2010) 11(9):597–610. doi: 10.1038/nrg2843
31. de Kock L, Sabbaghian N, Plourde F, Srivastava A, Weber E, Bouron-Dal Soglio D, et al. Pituitary blastoma: a pathognomonic feature of germ-line DICER1 mutations. Acta Neuropathol (2014) 128(1):111–22. doi: 10.1007/s00401-014-1285-z
32. Choong CS, Priest JR, Foulkes WD. Exploring the endocrine manifestations of DICER1 mutations. Trends Mol Med (2012) 18(9):503–5. doi: 10.1016/j.molmed.2012.07.003
33. Vedanayagam J, Chatila WK, Aksoy BA, Majumdar S, Skanderup AJ, Demir E, et al. Cancer-associated mutations in DICER1 RNase IIIa and IIIb domains exert similar effects on miRNA biogenesis. Nat Commun (2019) 10(1):3682. doi: 10.1038/s41467-019-11610-1
34. Bengtsson D, Joost P, Aravidis C, Askmalm Stenmark M, Backman AS, Melin B, et al. Corticotroph Pituitary Carcinoma in a Patient With Lynch Syndrome (LS) and Pituitary Tumors in a Nationwide LS Cohort. J Clin Endocrinol Metab (2017) 102(11):3928–32. doi: 10.1210/jc.2017-01401
35. Uraki S, Ariyasu H, Doi A, Furuta H, Nishi M, Sugano K, et al. Atypical pituitary adenoma with MEN1 somatic mutation associated with abnormalities of DNA mismatch repair genes; MLH1 germline mutation and MSH6 somatic mutation. Endocr J (2017) 64(9):895–906. doi: 10.1507/endocrj.EJ17-0036
36. Fernandez A, Karavitaki N, Wass JA. Prevalence of pituitary adenomas: a community-based, cross-sectional study in Banbury (Oxfordshire, UK). Clin Endocrinol (Oxf) (2010) 72(3):377–82. doi: 10.1111/j.1365-2265.2009.03667.x
37. Burton T, Le Nestour E, Neary M, Ludlam WH. Incidence and prevalence of acromegaly in a large US health plan database. Pituitary (2016) 19(3):262–7. doi: 10.1007/s11102-015-0701-2
38. Neou M, Villa C, Armignacco R, Jouinot A, Raffin-Sanson ML, Septier A, et al. Pangenomic Classification of Pituitary Neuroendocrine Tumors. Cancer Cell (2020) 37(1):123–4.e5. doi: 10.1016/j.ccell.2019.11.002
39. Pepe S, Korbonits M, Iacovazzo D. Germline and mosaic mutations causing pituitary tumours: genetic and molecular aspects. J Endocrinol (2019) 240(2):R21–r45. doi: 10.1530/joe-18-0446
40. Hernández-Ramírez LC, Gabrovska P, Dénes J, Stals K, Trivellin G, Tilley D, et al. Landscape of Familial Isolated and Young-Onset Pituitary Adenomas: Prospective Diagnosis in AIP Mutation Carriers. J Clin Endocrinol Metab (2015) 100(9):E1242–54. doi: 10.1210/jc.2015-1869
41. Beckers A, Aaltonen LA, Daly AF, Karhu A. Familial isolated pituitary adenomas (FIPA) and the pituitary adenoma predisposition due to mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene. Endocr Rev (2013) 34(2):239–77. doi: 10.1210/er.2012-1013
42. Daly AF, Tichomirowa MA, Petrossians P, Heliövaara E, Jaffrain-Rea ML, Barlier A, et al. Clinical characteristics and therapeutic responses in patients with germ-line AIP mutations and pituitary adenomas: an international collaborative study. J Clin Endocrinol Metab (2010) 95(11):E373–83. doi: 10.1210/jc.2009-2556
43. Marques P, Caimari F, Hernández-Ramírez LC, Collier D, Iacovazzo D, Ronaldson A, et al. Significant Benefits of AIP Testing and Clinical Screening in Familial Isolated and Young-onset Pituitary Tumors. J Clin Endocrinol Metab (2020) 105(6):e2247–60. doi: 10.1210/clinem/dgaa040
44. Tuominen I, Heliövaara E, Raitila A, Rautiainen MR, Mehine M, Katainen R, et al. AIP inactivation leads to pituitary tumorigenesis through defective Gαi-cAMP signaling. Oncogene (2015) 34(9):1174–84. doi: 10.1038/onc.2014.50
45. Raitila A, Lehtonen HJ, Arola J, Heliövaara E, Ahlsten M, Georgitsi M, et al. Mice with inactivation of aryl hydrocarbon receptor-interacting protein (Aip) display complete penetrance of pituitary adenomas with aberrant ARNT expression. Am J Pathol (2010) 177(4):1969–76. doi: 10.2353/ajpath.2010.100138
46. Williams F, Hunter S, Bradley L, Chahal HS, Storr HL, Akker SA, et al. Clinical experience in the screening and management of a large kindred with familial isolated pituitary adenoma due to an aryl hydrocarbon receptor interacting protein (AIP) mutation. J Clin Endocrinol Metab (2014) 99(4):1122–31. doi: 10.1210/jc.2013-2868
47. Srirangam Nadhamuni V, Korbonits M. Novel Insights into Pituitary Tumorigenesis: Genetic and Epigenetic Mechanisms. Endocr Rev (2020) 41(6):821–6. doi: 10.1210/endrev/bnaa006
48. Schernthaner-Reiter MH, Trivellin G, Stratakis CA. Interaction of AIP with protein kinase A (cAMP-dependent protein kinase). Hum Mol Genet (2018) 27(15):2604–13. doi: 10.1093/hmg/ddy166
49. Bizzi MF, Pinheiro SVB, Bolger GB, Schweizer J, Giannetti AV, Dang MN, et al. Reduced protein expression of the phosphodiesterases PDE4A4 and PDE4A8 in AIP mutation positive somatotroph adenomas. Mol Cell Endocrinol (2018) 476:103–9. doi: 10.1016/j.mce.2018.04.014
50. Theodoropoulou M, Zhang J, Laupheimer S, Paez-Pereda M, Erneux C, Florio T, et al. Octreotide, a somatostatin analogue, mediates its antiproliferative action in pituitary tumor cells by altering phosphatidylinositol 3-kinase signaling and inducing Zac1 expression. Cancer Res (2006) 66(3):1576–82. doi: 10.1158/0008-5472.Can-05-1189
51. Chahal HS, Trivellin G, Leontiou CA, Alband N, Fowkes RC, Tahir A, et al. Somatostatin analogs modulate AIP in somatotroph adenomas: the role of the ZAC1 pathway. J Clin Endocrinol Metab (2012) 97(8):E1411–20. doi: 10.1210/jc.2012-1111
52. Gadelha MR, Kasuki L, Korbonits M. Novel pathway for somatostatin analogs in patients with acromegaly. Trends Endocrinol Metab (2013) 24(5):238–46. doi: 10.1016/j.tem.2012.11.007
53. Hayward BE, Barlier A, Korbonits M, Grossman AB, Jacquet P, Enjalbert A, et al. Imprinting of the G(s)alpha gene GNAS1 in the pathogenesis of acromegaly. J Clin Invest (2001) 107(6):R31–6. doi: 10.1172/jci11887
54. Buchfelder M, Fahlbusch R, Merz T, Symowski H, Adams EF. Clinical correlates in acromegalic patients with pituitary tumors expressing GSP oncogenes. Pituitary (1999) 1(3-4):181–5. doi: 10.1023/a:1009905131334
55. Zhou Y, Zhang X, Klibanski A. Genetic and epigenetic mutations of tumor suppressive genes in sporadic pituitary adenoma. Mol Cell Endocrinol (2014) 386(1-2):16–33. doi: 10.1016/j.mce.2013.09.006
56. Salpea P, Stratakis CA. Carney complex and McCune Albright syndrome: an overview of clinical manifestations and human molecular genetics. Mol Cell Endocrinol (2014) 386(1-2):85–91. doi: 10.1016/j.mce.2013.08.022
57. Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med (1991) 325(24):1688–95. doi: 10.1056/nejm199112123252403
58. Välimäki N, Demir H, Pitkänen E, Kaasinen E, Karppinen A, Kivipelto L, et al. Whole-Genome Sequencing of Growth Hormone (GH)-Secreting Pituitary Adenomas. J Clin Endocrinol Metab (2015) 100(10):3918–27. doi: 10.1210/jc.2015-3129
59. Välimäki N, Schalin-Jäntti C, Karppinen A, Paetau A, Kivipelto L, Aaltonen LA, et al. Genetic and Epigenetic Characterization of Growth Hormone-Secreting Pituitary Tumors. Mol Cancer Res (2019) 17(12):2432–43. doi: 10.1158/1541-7786.mcr-19-0434
60. Ben-Shlomo A, Deng N, Ding E, Yamamoto M, Mamelak A, Chesnokova V, et al. DNA damage and growth hormone hypersecretion in pituitary somatotroph adenomas. J Clin Invest (2020) 130(11):5738–55. doi: 10.1172/jci138540
61. Hage M, Viengchareun S, Brunet E, Villa C, Pineau D, Bouligand J, et al. Genomic Alterations and Complex Subclonal Architecture in Sporadic GH-Secreting Pituitary Adenomas. J Clin Endocrinol Metab (2018) 103(5):1929–39. doi: 10.1210/jc.2017-02287
62. Vlotides G, Eigler T, Melmed S. Pituitary tumor-transforming gene: physiology and implications for tumorigenesis. Endocr Rev (2007) 28(2):165–86. doi: 10.1210/er.2006-0042
63. McCabe CJ, Khaira JS, Boelaert K, Heaney AP, Tannahill LA, Hussain S, et al. Expression of pituitary tumour transforming gene (PTTG) and fibroblast growth factor-2 (FGF-2) in human pituitary adenomas: relationships to clinical tumour behaviour. Clin Endocrinol (Oxf) (2003) 58(2):141–50. doi: 10.1046/j.1365-2265.2003.01598.x
64. Zhang X, Horwitz GA, Heaney AP, Nakashima M, Prezant TR, Bronstein MD, et al. Pituitary tumor transforming gene (PTTG) expression in pituitary adenomas. J Clin Endocrinol Metab (1999) 84(2):761–7. doi: 10.1210/jcem.84.2.5432
65. Larkin S, Reddy R, Karavitaki N, Cudlip S, Wass J, Ansorge O. Granulation pattern, but not GSP or GHR mutation, is associated with clinical characteristics in somatostatin-naive patients with somatotroph adenomas. Eur J Endocrinol (2013) 168(4):491–9. doi: 10.1530/eje-12-0864
66. Pei L, Melmed S. Isolation and characterization of a pituitary tumor-transforming gene (PTTG). Mol Endocrinol (1997) 11(4):433–41. doi: 10.1210/mend.11.4.9911
67. Darnell JE Jr., Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science (1994) 264(5164):1415–21. doi: 10.1126/science.8197455
68. Zhou C, Jiao Y, Wang R, Ren SG, Wawrowsky K, Melmed S. STAT3 upregulation in pituitary somatotroph adenomas induces growth hormone hypersecretion. J Clin Invest (2015) 125(4):1692–702. doi: 10.1172/jci78173
69. Zhang Q, Peng C, Song J, Zhang Y, Chen J, Song Z, et al. Germline Mutations in CDH23, Encoding Cadherin-Related 23, Are Associated with Both Familial and Sporadic Pituitary Adenomas. Am J Hum Genet (2017) 100(5):817–23. doi: 10.1016/j.ajhg.2017.03.011
70. Bolz H, von Brederlow B, Ramírez A, Bryda EC, Kutsche K, Nothwang HG, et al. Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D. Nat Genet (2001) 27(1):108–12. doi: 10.1038/83667
71. Sun Y, Bak B, Schoenmakers N, van Trotsenburg AS, Oostdijk W, Voshol P, et al. Loss-of-function mutations in IGSF1 cause an X-linked syndrome of central hypothyroidism and testicular enlargement. Nat Genet (2012) 44(12):1375–81. doi: 10.1038/ng.2453
72. Kiyokawa H, Kineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, et al. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1). Cell (1996) 85(5):721–32. doi: 10.1016/s0092-8674(00)81238-6
73. Faucz FR, Horvath AD, Azevedo MF, Levy I, Bak B, Wang Y, et al. Is IGSF1 involved in human pituitary tumor formation? Endocr Relat Cancer (2015) 22(1):47–54. doi: 10.1530/erc-14-0465
74. Li J, Dong W, Li Z, Wang H, Gao H, Zhang Y. Impact of SLC20A1 on the Wnt/β−catenin signaling pathway in somatotroph adenomas. Mol Med Rep (2019) 20(4):3276–84. doi: 10.3892/mmr.2019.10555
75. Wei D, Yiyuan C, Qian L, Jianhua L, Yazhuo Z, Hua G. The absence of PRDM2 involved the tumorigenesis of somatotroph adenomas through regulating c-Myc. Gene (2020) 737:144456. doi: 10.1016/j.gene.2020.144456
76. Ben-Shlomo A, Liu NA, Melmed S. Somatostatin and dopamine receptor regulation of pituitary somatotroph adenomas. Pituitary (2017) 20(1):93–9. doi: 10.1007/s11102-016-0778-2
77. Venegas-Moreno E, Vazquez-Borrego MC, Dios E, Gros-Herguido N, Flores-Martinez A, Rivero-Cortés E, et al. Association between dopamine and somatostatin receptor expression and pharmacological response to somatostatin analogues in acromegaly. J Cell Mol Med (2018) 22(3):1640–9. doi: 10.1111/jcmm.13440
78. Franck SE, Gatto F, van der Lely AJ, Janssen J, Dallenga AHG, Nagtegaal AP, et al. Somatostatin Receptor Expression in GH-Secreting Pituitary Adenomas Treated with Long-Acting Somatostatin Analogues in Combination with Pegvisomant. Neuroendocrinology (2017) 105(1):44–53. doi: 10.1159/000448429
79. Pease M, Ling C, Mack WJ, Wang K, Zada G. The role of epigenetic modification in tumorigenesis and progression of pituitary adenomas: a systematic review of the literature. PLoS One (2013) 8(12):e82619. doi: 10.1371/journal.pone.0082619
80. Zhu X, Lee K, Asa SL, Ezzat S. Epigenetic silencing through DNA and histone methylation of fibroblast growth factor receptor 2 in neoplastic pituitary cells. Am J Pathol (2007) 170(5):1618–28. doi: 10.2353/ajpath.2007.061111
81. Bahar A, Bicknell JE, Simpson DJ, Clayton RN, Farrell WE. Loss of expression of the growth inhibitory gene GADD45gamma, in human pituitary adenomas, is associated with CpG island methylation. Oncogene (2004) 23(4):936–44. doi: 10.1038/sj.onc.1207193
82. Zhang X, Sun H, Danila DC, Johnson SR, Zhou Y, Swearingen B, et al. Loss of expression of GADD45 gamma, a growth inhibitory gene, in human pituitary adenomas: implications for tumorigenesis. J Clin Endocrinol Metab (2002) 87(3):1262–7. doi: 10.1210/jcem.87.3.8315
83. Righi A, Jin L, Zhang S, Stilling G, Scheithauer BW, Kovacs K, et al. Identification and consequences of galectin-3 expression in pituitary tumors. Mol Cell Endocrinol (2010) 326(1-2):8–14. doi: 10.1016/j.mce.2010.04.026
84. Qian ZR, Sano T, Yoshimoto K, Yamada S, Ishizuka A, Mizusawa N, et al. Inactivation of RASSF1A tumor suppressor gene by aberrant promoter hypermethylation in human pituitary adenomas. Lab Invest (2005) 85(4):464–73. doi: 10.1038/labinvest.3700248
85. Palumbo T, Faucz FR, Azevedo M, Xekouki P, Iliopoulos D, Stratakis CA. Functional screen analysis reveals miR-26b and miR-128 as central regulators of pituitary somatomammotrophic tumor growth through activation of the PTEN-AKT pathway. Oncogene (2013) 32(13):1651–9. doi: 10.1038/onc.2012.190
86. D’Angelo D, Palmieri D, Mussnich P, Roche M, Wierinckx A, Raverot G, et al. Altered microRNA expression profile in human pituitary GH adenomas: down-regulation of miRNA targeting HMGA1, HMGA2, and E2F1. J Clin Endocrinol Metab (2012) 97(7):E1128–38. doi: 10.1210/jc.2011-3482
87. D’Angelo D, Mussnich P, Sepe R, Raia M, Del Vecchio L, Cappabianca P, et al. RPSAP52 lncRNA is overexpressed in pituitary tumors and promotes cell proliferation by acting as miRNA sponge for HMGA proteins. J Mol Med (Berl) (2019) 97(7):1019–32. doi: 10.1007/s00109-019-01789-7
88. D’Angelo D, Arra C, Fusco A. RPSAP52 lncRNA Inhibits p21Waf1/CIP Expression by Interacting With the RNA Binding Protein HuR. Oncol Res (2020) 28(2):191–201. doi: 10.3727/096504019x15761465603129
89. Lee YJ, Cho JM, Moon JH, Ku CR, Kim J, Kim SH, et al. Increased miR-338-3p expression correlates with invasiveness of GH-producing pituitary adenomas. Endocrine (2017) 58(1):184–9. doi: 10.1007/s12020-017-1390-6
90. Zhao S, Li J, Feng J, Li Z, Liu Q, Lv P, et al. Identification of Serum miRNA-423-5p Expression Signature in Somatotroph Adenomas. Int J Endocrinol (2019) 2019:8516858. doi: 10.1155/2019/8516858
91. Zhen W, Qiu D, Zhiyong C, Xin W, Mengyao J, Dimin Z, et al. MicroRNA-524-5p Functions as a Tumor Suppressor in a Human Pituitary Tumor-Derived Cell Line. Horm Metab Res (2017) 49(7):550–7. doi: 10.1055/s-0043-106437
92. Ragnarsson O, Olsson DS, Chantzichristos D, Papakokkinou E, Dahlqvist P, Segerstedt E, et al. The incidence of Cushing’s disease: a nationwide Swedish study. Pituitary (2019) 22(2):179–86. doi: 10.1007/s11102-019-00951-1
93. Nieman LK, Biller BM, Findling JW, Murad MH, Newell-Price J, Savage MO, et al. Treatment of Cushing’s Syndrome: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab (2015) 100(8):2807–31. doi: 10.1210/jc.2015-1818
94. Feelders RA, Newell-Price J, Pivonello R, Nieman LK, Hofland LJ, Lacroix A. Advances in the medical treatment of Cushing’s syndrome. Lancet Diabetes Endocrinol (2019) 7(4):300–12. doi: 10.1016/s2213-8587(18)30155-4
95. Ma ZY, Song ZJ, Chen JH, Wang YF, Li SQ, Zhou LF, et al. Recurrent gain-of-function USP8 mutations in Cushing’s disease. Cell Res (2015) 25(3):306–17. doi: 10.1038/cr.2015.20
96. Perez-Rivas LG, Theodoropoulou M, Ferraù F, Nusser C, Kawaguchi K, Stratakis CA, et al. The Gene of the Ubiquitin-Specific Protease 8 Is Frequently Mutated in Adenomas Causing Cushing’s Disease. J Clin Endocrinol Metab (2015) 100(7):E997–1004. doi: 10.1210/jc.2015-1453
97. Reincke M, Sbiera S, Hayakawa A, Theodoropoulou M, Osswald A, Beuschlein F, et al. Mutations in the deubiquitinase gene USP8 cause Cushing’s disease. Nat Genet (2015) 47(1):31–8. doi: 10.1038/ng.3166
98. Jian FF, Li YF, Chen YF, Jiang H, Chen X, Zheng LL, et al. Inhibition of Ubiquitin-specific Peptidase 8 Suppresses Adrenocorticotropic Hormone Production and Tumorous Corticotroph Cell Growth in AtT20 Cells. Chin Med J (Engl) (2016) 129(17):2102–8. doi: 10.4103/0366-6999.189047
99. Weigand I, Knobloch L, Flitsch J, Saeger W, Monoranu CM, Höfner K, et al. Impact of USP8 Gene Mutations on Protein Deregulation in Cushing Disease. J Clin Endocrinol Metab (2019) 104(7):2535–46. doi: 10.1210/jc.2018-02564
100. Bujko M, Kober P, Boresowicz J, Rusetska N, Paziewska A, Dąbrowska M, et al. USP8 mutations in corticotroph adenomas determine a distinct gene expression profile irrespective of functional tumour status. Eur J Endocrinol (2019) 181(6):615–27. doi: 10.1530/eje-19-0194
101. Cohen M, Persky R, Stegemann R, Hernández-Ramírez LC, Zeltser D, Lodish MB, et al. Germline USP8 Mutation Associated With Pediatric Cushing Disease and Other Clinical Features: A New Syndrome. J Clin Endocrinol Metab (2019) 104(10):4676–82. doi: 10.1210/jc.2019-00697
102. Albani A, Pérez-Rivas LG, Dimopoulou C, Zopp S, Colón-Bolea P, Roeber S, et al. The USP8 mutational status may predict long-term remission in patients with Cushing’s disease. Clin Endocrinol (Oxf) (2018) 89(4):454–8. doi: 10.1111/cen.13802
103. Faucz FR, Tirosh A, Tatsi C, Berthon A, Hernández-Ramírez LC, Settas N, et al. Somatic USP8 Gene Mutations Are a Common Cause of Pediatric Cushing Disease. J Clin Endocrinol Metab (2017) 102(8):2836–43. doi: 10.1210/jc.2017-00161
104. Wanichi IQ, de Paula Mariani BM, Frassetto FP, Siqueira SAC, de Castro Musolino NR, Cunha-Neto MBC, et al. Cushing’s disease due to somatic USP8 mutations: a systematic review and meta-analysis. Pituitary (2019) 22(4):435–42. doi: 10.1007/s11102-019-00973-9
105. Asari Y, Kageyama K, Sugiyama A, Kogawa H, Niioka K, Daimon M. Lapatinib decreases the ACTH production and proliferation of corticotroph tumor cells. Endocr J (2019) 66(6):515–22. doi: 10.1507/endocrj.EJ18-0491
106. Hayashi K, Inoshita N, Kawaguchi K, Ibrahim Ardisasmita A, Suzuki H, Fukuhara N, et al. The USP8 mutational status may predict drug susceptibility in corticotroph adenomas of Cushing’s disease. Eur J Endocrinol (2016) 174(2):213–26. doi: 10.1530/eje-15-0689
107. Bruns C, Lewis I, Briner U, Meno-Tetang G, Weckbecker G. SOM230: a novel somatostatin peptidomimetic with broad somatotropin release inhibiting factor (SRIF) receptor binding and a unique antisecretory profile. Eur J Endocrinol (2002) 146(5):707–16. doi: 10.1530/eje.0.1460707
108. Chen J, Jian X, Deng S, Ma Z, Shou X, Shen Y, et al. Identification of recurrent USP48 and BRAF mutations in Cushing’s disease. Nat Commun (2018) 9(1):3171. doi: 10.1038/s41467-018-05275-5
109. Sbiera S, Perez-Rivas LG, Taranets L, Weigand I, Flitsch J, Graf E, et al. Driver mutations in USP8 wild-type Cushing’s disease. Neuro Oncol (2019) 21(10):1273–83. doi: 10.1093/neuonc/noz109
110. Riebold M, Kozany C, Freiburger L, Sattler M, Buchfelder M, Hausch F, et al. A C-terminal HSP90 inhibitor restores glucocorticoid sensitivity and relieves a mouse allograft model of Cushing disease. Nat Med (2015) 21(3):276–80. doi: 10.1038/nm.3776
111. Bilodeau S, Vallette-Kasic S, Gauthier Y, Figarella-Branger D, Brue T, Berthelet F, et al. Role of Brg1 and HDAC2 in GR trans-repression of the pituitary POMC gene and misexpression in Cushing disease. Genes Dev (2006) 20(20):2871–86. doi: 10.1101/gad.1444606
112. Ren J, Jian F, Jiang H, Sun Y, Pan S, Gu C, et al. Decreased expression of SFRP2 promotes development of the pituitary corticotroph adenoma by upregulating Wnt signaling. Int J Oncol (2018) 52(6):1934–46. doi: 10.3892/ijo.2018.4355
113. Woodbury ME, Ikezu T. Fibroblast growth factor-2 signaling in neurogenesis and neurodegeneration. J Neuroimmune Pharmacol (2014) 9(2):92–101. doi: 10.1007/s11481-013-9501-5
114. Vaccarino FM, Grigorenko EL, Smith KM, Stevens HE. Regulation of cerebral cortical size and neuron number by fibroblast growth factors: implications for autism. J Autism Dev Disord (2009) 39(3):511–20. doi: 10.1007/s10803-008-0653-8
115. Vaccarino FM, Schwartz ML, Raballo R, Nilsen J, Rhee J, Zhou M, et al. Changes in cerebral cortex size are governed by fibroblast growth factor during embryogenesis. Nat Neurosci (1999) 2(3):246–53. doi: 10.1038/6350
116. Korada S, Zheng W, Basilico C, Schwartz ML, Vaccarino FM. Fibroblast growth factor 2 is necessary for the growth of glutamate projection neurons in the anterior neocortex. J Neurosci (2002) 22(3):863–75. doi: 10.1523/jneurosci.22-03-00863.2002
117. Zheng W, Nowakowski RS, Vaccarino FM. Fibroblast growth factor 2 is required for maintaining the neural stem cell pool in the mouse brain subventricular zone. Dev Neurosci (2004) 26(2-4):181–96. doi: 10.1159/000082136
118. Farrell WE. A novel apoptosis gene identified in the pituitary gland. Neuroendocrinology (2006) 84(4):217–21. doi: 10.1159/000097486
119. Ren J, Gu C, Yang Y, Xue J, Sun Y, Jian F, et al. TSP-1 is downregulated and inversely correlates with miR-449c expression in Cushing’s disease. J Cell Mol Med (2019) 23(6):4097–110. doi: 10.1111/jcmm.14297
120. Renjie W, Haiqian L. MiR-132, miR-15a and miR-16 synergistically inhibit pituitary tumor cell proliferation, invasion and migration by targeting Sox5. Cancer Lett (2015) 356(2 Pt B):568–78. doi: 10.1016/j.canlet.2014.10.003
121. Gentilin E, Tagliati F, Filieri C, Molè D, Minoia M, Rosaria Ambrosio M, et al. miR-26a plays an important role in cell cycle regulation in ACTH-secreting pituitary adenomas by modulating protein kinase Cδ. Endocrinology (2013) 154(5):1690–700. doi: 10.1210/en.2012-2070
122. Gentilin E, Di Pasquale C, Gagliano T, Tagliati F, Benfini K, Ambrosio MR, et al. Protein Kinase C Delta restrains growth in ACTH-secreting pituitary adenoma cells. Mol Cell Endocrinol (2016) 419:252–8. doi: 10.1016/j.mce.2015.10.025
123. Yu G, Li C, Xie W, Wang Z, Gao H, Cao L, et al. Long non-coding RNA C5orf66-AS1 is downregulated in pituitary null cell adenomas and is associated with their invasiveness. Oncol Rep (2017) 38(2):1140–8. doi: 10.3892/or.2017.5739
124. Sleiter N, Pang Y, Park C, Horton TH, Dong J, Thomas P, et al. Progesterone receptor A (PRA) and PRB-independent effects of progesterone on gonadotropin-releasing hormone release. Endocrinology (2009) 150(8):3833–44. doi: 10.1210/en.2008-0774
125. Feng J, Yu SY, Li CZ, Li ZY, Zhang YZ. Integrative proteomics and transcriptomics revealed that activation of the IL-6R/JAK2/STAT3/MMP9 signaling pathway is correlated with invasion of pituitary null cell adenomas. Mol Cell Endocrinol (2016) 436:195–203. doi: 10.1016/j.mce.2016.07.025
126. Martínez-López S, García-Martínez A, Torregrosa-Quesada ME, López-Muñoz B, Cámara R, Fajardo C, et al. Is Somatostatin Receptor and Dopamine Receptor profiling useful in the management of silent somatotroph tumors? J Endocrinol Invest (2020) 43(6):859–63. doi: 10.1007/s40618-019-01166-8
127. Karakas B, Bachman KE, Park BH. Mutation of the PIK3CA oncogene in human cancers. Br J Cancer (2006) 94(4):455–9. doi: 10.1038/sj.bjc.6602970
128. Lin Y, Jiang X, Shen Y, Li M, Ma H, Xing M, et al. Frequent mutations and amplifications of the PIK3CA gene in pituitary tumors. Endocr Relat Cancer (2009) 16(1):301–10. doi: 10.1677/erc-08-0167
129. Cazabat L, Bouligand J, Salenave S, Bernier M, Gaillard S, Parker F, et al. Germline AIP mutations in apparently sporadic pituitary adenomas: prevalence in a prospective single-center cohort of 443 patients. J Clin Endocrinol Metab (2012) 97(4):E663–70. doi: 10.1210/jc.2011-2291
130. Seemann N, Kuhn D, Wrocklage C, Keyvani K, Hackl W, Buchfelder M, et al. CDKN2A/p16 inactivation is related to pituitary adenoma type and size. J Pathol (2001) 193(4):491–7. doi: 10.1002/path.833
131. Zhou Y, Zhong Y, Wang Y, Zhang X, Batista DL, Gejman R, et al. Activation of p53 by MEG3 non-coding RNA. J Biol Chem (2007) 282(34):24731–42. doi: 10.1074/jbc.M702029200
132. Hernandez MC, Andres-Barquin PJ, Holt I, Israel MA. Cloning of human ENC-1 and evaluation of its expression and regulation in nervous system tumors. Exp Cell Res (1998) 242(2):470–7. doi: 10.1006/excr.1998.4109
133. Feng J, Hong L, Wu Y, Li C, Wan H, Li G, et al. Identification of a subtype-specific ENC1 gene related to invasiveness in human pituitary null cell adenoma and oncocytomas. J Neurooncol (2014) 119(2):307–15. doi: 10.1007/s11060-014-1479-1
134. Holm R. Null cell adenomas and oncocytomas of the pituitary gland. Pathol Res Pract (1995) 191(4):348–52. doi: 10.1016/s0344-0338(11)80888-5
135. Cheng S, Li C, Xie W, Miao Y, Guo J, Wang J, et al. Integrated analysis of DNA methylation and mRNA expression profiles to identify key genes involved in the regrowth of clinically non-functioning pituitary adenoma. Aging (Albany NY) (2020) 12(3):2408–27. doi: 10.18632/aging.102751
136. Melmed S, Casanueva FF, Hoffman AR, Kleinberg DL, Montori VM, Schlechte JA, et al. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab (2011) 96(2):273–88. doi: 10.1210/jc.2010-1692
137. Fedele M, Palmieri D, Fusco A. HMGA2: A pituitary tumour subtype-specific oncogene? Mol Cell Endocrinol (2010) 326(1-2):19–24. doi: 10.1016/j.mce.2010.03.019
138. Fedele M, Visone R, De Martino I, Troncone G, Palmieri D, Battista S, et al. HMGA2 induces pituitary tumorigenesis by enhancing E2F1 activity. Cancer Cell (2006) 9(6):459–71. doi: 10.1016/j.ccr.2006.04.024
139. Blesch A, Bosserhoff AK, Apfel R, Behl C, Hessdoerfer B, Schmitt A, et al. Cloning of a novel malignant melanoma-derived growth-regulatory protein, MIA. Cancer Res (1994) 54(21):5695–701.
140. Palmieri D, Valentino T, De Martino I, Esposito F, Cappabianca P, Wierinckx A, et al. PIT1 upregulation by HMGA proteins has a role in pituitary tumorigenesis. Endocr Relat Cancer (2012) 19(2):123–35. doi: 10.1530/erc-11-0135
141. Qian ZR, Asa SL, Siomi H, Siomi MC, Yoshimoto K, Yamada S, et al. Overexpression of HMGA2 relates to reduction of the let-7 and its relationship to clinicopathological features in pituitary adenomas. Mod Pathol (2009) 22(3):431–41. doi: 10.1038/modpathol.2008.202
142. Fedele M, Pentimalli F, Baldassarre G, Battista S, Klein-Szanto AJ, Kenyon L, et al. Transgenic mice overexpressing the wild-type form of the HMGA1 gene develop mixed growth hormone/prolactin cell pituitary adenomas and natural killer cell lymphomas. Oncogene (2005) 24(21):3427–35. doi: 10.1038/sj.onc.1208501
143. Wang EL, Qian ZR, Rahman MM, Yoshimoto K, Yamada S, Kudo E, et al. Increased expression of HMGA1 correlates with tumour invasiveness and proliferation in human pituitary adenomas. Histopathology (2010) 56(4):501–9. doi: 10.1111/j.1365-2559.2010.03495.x
144. Portovedo S, Gaido N, de Almeida Nunes B, Nascimento AG, Rocha A, Magalhães M, et al. Differential Expression of HMGA1 and HMGA2 in pituitary neuroendocrine tumors. Mol Cell Endocrinol (2019) 490:80–7. doi: 10.1016/j.mce.2019.04.010
145. Kobayashi T, Usui H, Tanaka H, Shozu M. Variant Prolactin Receptor in Agalactia and Hyperprolactinemia. N Engl J Med (2018) 379(23):2230–6. doi: 10.1056/NEJMoa1805171
146. Gorvin CM, Newey PJ, Rogers A, Stokes V, Neville MJ, Lines KE, et al. Association of prolactin receptor (PRLR) variants with prolactinomas. Hum Mol Genet (2019) 28(6):1023–37. doi: 10.1093/hmg/ddy396
147. Sapkota S, Horiguchi K, Tosaka M, Yamada S, Yamada M. Whole-Exome Sequencing Study of Thyrotropin-Secreting Pituitary Adenomas. J Clin Endocrinol Metab (2017) 102(2):566–75. doi: 10.1210/jc.2016-2261
148. Ando S, Sarlis NJ, Oldfield EH, Yen PM. Somatic mutation of TRbeta can cause a defect in negative regulation of TSH in a TSH-secreting pituitary tumor. J Clin Endocrinol Metab (2001) 86(11):5572–6. doi: 10.1210/jcem.86.11.7984
149. Finelli P, Giardino D, Rizzi N, Buiatiotis S, Virduci T, Franzin A, et al. Non-random trisomies of chromosomes 5, 8 and 12 in the prolactinoma sub-type of pituitary adenomas: conventional cytogenetics and interphase FISH study. Int J Cancer (2000) 86(3):344–50. doi: 10.1002/(sici)1097-0215(20000501)86:3<344::aid-ijc7>3.0.co;2-8
150. Bettio D, Rizzi N, Giardino D, Persani L, Pecori-Giraldi F, Losa M, et al. Cytogenetic study of pituitary adenomas. Cancer Genet Cytogenet (1997) 98(2):131–6. doi: 10.1016/s0165-4608(96)00426-8
151. Bi WL, Horowitz P, Greenwald NF, Abedalthagafi M, Agarwalla PK, Gibson WJ, et al. Landscape of Genomic Alterations in Pituitary Adenomas. Clin Cancer Res (2017) 23(7):1841–51. doi: 10.1158/1078-0432.ccr-16-0790
152. Lopes MBS. The 2017 World Health Organization classification of tumors of the pituitary gland: a summary. Acta Neuropathol (2017) 134(4):521–35. doi: 10.1007/s00401-017-1769-8
153. Mete O, Asa SL. Structure, Function, and Morphology in the Classification of Pituitary Neuroendocrine Tumors: the Importance of Routine Analysis of Pituitary Transcription Factors. Endocr Pathol (2020) 31(4):330–6. doi: 10.1007/s12022-020-09646-x
154. Yoshino A, Katayama Y, Ogino A, Watanabe T, Yachi K, Ohta T, et al. Promoter hypermethylation profile of cell cycle regulator genes in pituitary adenomas. J Neurooncol (2007) 83(2):153–62. doi: 10.1007/s11060-006-9316-9
155. Salehi F, Scheithauer BW, Kros JM, Lau Q, Fealey M, Erickson D, et al. MGMT promoter methylation and immunoexpression in aggressive pituitary adenomas and carcinomas. J Neurooncol (2011) 104(3):647–57. doi: 10.1007/s11060-011-0532-6
Keywords: pituitary adenomas, molecular markers, acromegaly, Cushing’s disease, non-secreting adenomas
Citation: Chang M, Yang C, Bao X and Wang R (2021) Genetic and Epigenetic Causes of Pituitary Adenomas. Front. Endocrinol. 11:596554. doi: 10.3389/fendo.2020.596554
Received: 19 August 2020; Accepted: 23 November 2020;
Published: 26 January 2021.
Edited by:
Hidenori Fukuoka, Kobe University, JapanCopyright © 2021 Chang, Yang, Bao and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinjie Bao, YmFveGluamllMUBwdW1jaC5jbg==; Renzhi Wang, d2FuZ3J6QDEyNi5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.