 Kan Chen
Kan Chen Haoyu Wang
Haoyu Wang- Liaoning Provincial Key Laboratory of Endocrine Diseases, Department of Endocrinology and Metabolism, Institute of Endocrinology, The First Hospital, China Medical University, Shenyang, China
Introduction: Kallmann syndrome (KS) is idiopathic hypogonadotropic hypogonadism with olfactory loss or decline. Waardenburg syndrome type II (WS2) is a clinically and genetically heterogeneous disease, characterized by congenital sensorineural deafness and abnormal pigmentation of the iris, hair, and skin. Recently, mutations in the well-known WS pathogenic gene SOX10 have been found in some KS patients with deafness, but whether SOX10 is a co-pathogenic gene of KS and WS remains uncertain. Here, we report a rare case of KS and WS2 co-occurrence due to SOX10 mutations.
Methods: Detailed histories were collected through questionnaires and physical examination. Blood samples of the patient and his family members were collected after obtaining informed consents. Suspected mutations were amplified and verified by Sanger sequencing after the next generation sequencing of related genes. The raw sequence data were compared to the known gene sequence data in publicly available sequence data bases using Burrows-Wheeler Aligner software (BWA, 0.7.12-r1039).
Results: A 28-year-old male patient sought treatment for hypogonadism and the absence of secondary sexual characteristics. In addition, he showed signs of obesity, hyposmia, sensorineural hearing loss, and blue iris. Magnetic resonance imaging (MRI) of the olfactory bulb showed small bilateral olfactory bulbs and tracts and diaphragma cerebri. MRI of the pituitary gland revealed a flat pituitary gland in the sella. Laboratory examination demonstrated hypogonadotropic hypogonadism, pituitary hypothyroidism, subclinical hypothyroidism, and the presence of insulin resistance with normal blood glucose levels. Sequencing of the SOX10 gene showed a 20 bp insertion in between coding bases 1,179 and 1,180 (c.1179_1180insACTATGGCTCAGCCTTCCCC). This results in a frame-shifting mutation of the 394th amino acid serine in exon4 with the resulting the amino acid sequence of the protein predicted to be TMAQPSP PSPAPSLTTL TISPQDPIMA TRARPLASTR PSPIWGPRSG PSTRPSLTPA PQGPSPTAPH TGSSQYIRHC PGPKGGPVAT TPRPAPAPSL CALFLAHLRP GGGSGGG*.
Conclusion: SOX10 plays an important role in some critical stages of neural crest cell development and SOX10 mutation may be a common pathogenic factor for both KS and WS. Therefore, SOX10 mutation analysis should be considered for KS patients with combined WS clinical manifestations, especially deafness.
Introduction
Kallmann syndrome (KS) is a clinically and genetically heterogeneous disease with multiple genetic patterns of inheritance including autosomal dominant (65%), autosomal recessive (25%), and X-linked recessive (10%). The etiology and pathogenesis of KS is a disturbance in the common neural migration pathways of gonadotropin-releasing hormone (GnRH) neurons and olfactory neurons early in embryonic development, and the main clinical manifestations are hypogonadism and anosmia (1). At present, more than 20 pathogenic genes have been found to be associated with KS, of which six are relatively common (KAL1, FGFR1, PROKR2, PROK2, CHD7, FGF8) (2–6). However, only about 30% of the etiology and pathogenesis can be explained by the reported genetic mutations currently and about 70% remains unknown. Although numerous KS cases have been reported in the literature, co-occurrence of KS and Waardenburg syndrome type II (WS2) has only been reported twice (7, 8).
KS is often associated with other developmental deficiencies, such as hearing impairment that is found in about 5% of KS patients. In 2013, Pingault et al. (9) found that approximately 38% of patients with KS and deafness have SOX10 mutations. It is well known that SOX10 mutations are one of the causative gene mutations associated with WS, found in about 15% of WS2 patients and 45% of WS4 patients. The characteristic clinical manifestations of WS2 are congenital sensorineural deafness and abnormal pigmentation of the iris, hair, and skin. WS4 is a combination of clinical manifestations of WS2 and Hirschsprung disease (10, 11). The clinical manifestations of WS3 and WS1 are similar and their striking features are accompanied by abnormal development of the facial or upper limb musculoskeletal structures. However, the genetic link between KS and WS has not been established, and the clinical manifestations caused by SOX10 mutations need to be fully elaborated.
In this case study, we identified a novel compound heterozygous mutation in the SOX10 gene (c.1179_1180insACTATGGCTCAGCCTTCCCC, p.Ser394fs) in a Chinese patient with both KS and WS2.
Methods and Result
Case Presentation
The 28-year-old male patient was born to non-consanguineous Chinese parents after a normal pregnancy. He had bilateral sensory deafness and blue irises at the time of birth. His mother noticed his lack of sense of smell in childhood. His mental development was normal. He had no Hirschsprung disease or episodes of constipation. The abnormality of microphallus was first noticed by his family 10 years ago, but he had not been medically examined for it, and no improvement was seen after taking traditional Chinese medicine. Therefore, he was first referred to the First Affiliated Hospital of China Medical University for evaluation of the short penis at the age of 28. Both the patient’s mother and brother were healthy. However, his father had died.
Physical Examination
The patient showed the following features: height, 180 cm; weight, 103 kg; body mass index (BMI), 31.97 kg/m2; body temperature, 36.5°C; blood pressure, 120/76 mmHg; pulse rate, 74/min; respirations, 18/min; blue irises (Figure 1A); lack of facial hair; white forehead; lack of acne; lack of moon face; lack of underarm hair; lack of thyroid swelling; enlarged breasts and mild tenderness; palpable breast nodules; lack of galactorrhea; sparse pubic hair; a stretched penis length of 3 cm (Figure 1B); a small bilateral testicular volume of 6.0 ml (Figure 1C).
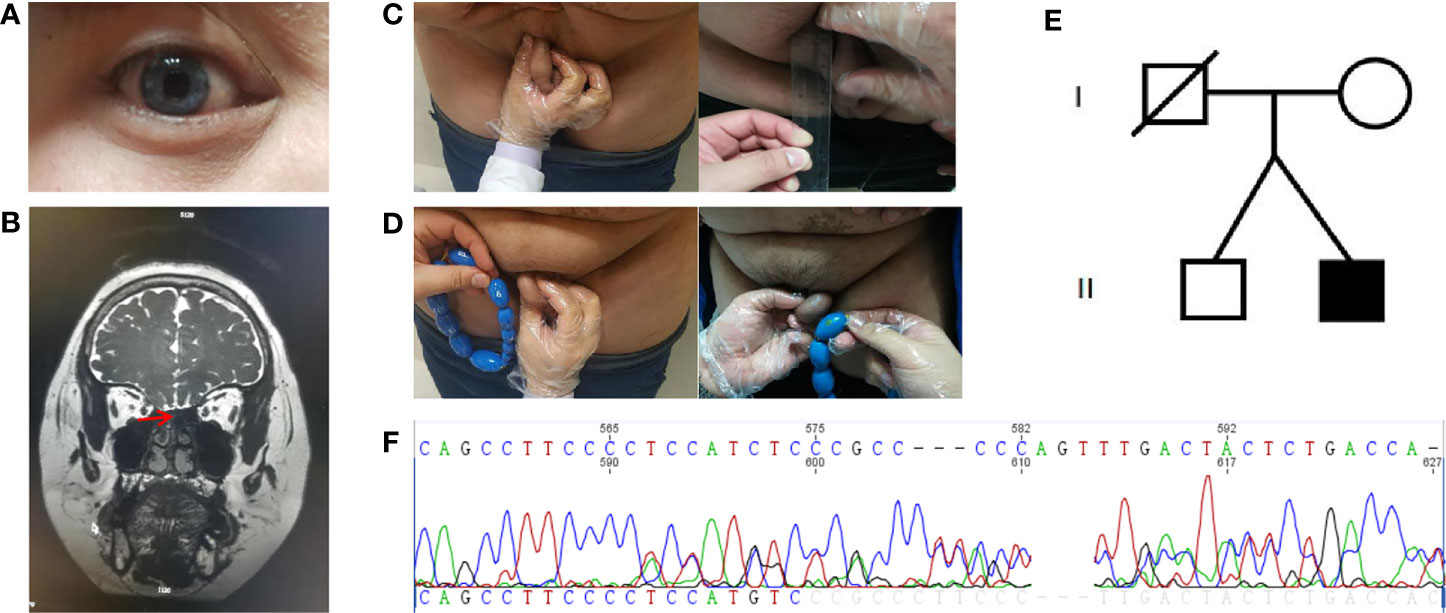
Figure 1 Clinical characteristics and gene sequencing results of the patient and his family members. (A) Depigmentation of the irides. (B) Magnetic resonance imaging of the olfactory bulb. (C) Comparison of stretched penis length before and after treatment. (D) Comparison of testicular volume before and after treatment. (E) The proband’s family pedigree. (F) The de novo mutation in SOX10 gene (c.1179_1180insACTATGGCTCAGCCTTCCCC, p.Ser394fs).
Assessment
Scrotal ultrasound showed a reduced size of the testes and the presence of microcalcification in them. His sense of smell was tested formally (Toyota-Takagi olfactometer test) and hyposmia was discovered. Audiologic examination demonstrated a sensorineural hearing loss. Magnetic resonance imaging (MRI) of the olfactory bulb showed small bilateral olfactory bulbs and tracts, and diaphragma cerebri (Figure 1D). Bone mineral density analysis revealed decreased bone density in the hip joint and lumbar vertebrae. MRI of the pituitary gland revealed no enlargement of the sella turcica or collapse of the saddle bottom but a flat pituitary gland in the sella. Ultrasound of breasts and bilateral axillary lymph nodes revealed bilateral male breast development. Thyroid gland and bilateral cervical lymph node ultrasound demonstrated a single hypoechoic nodule in the right thyroid lobe (TI-RADS 3+level). Liver ultrasound showed a fatty liver. No definite abnormality was found on computed tomography of the lungs and adrenal glands. Chromosomal analysis revealed a 46, XY karyotype. Laboratory examination demonstrated hypogonadotropic hypogonadism, pituitary hypothyroidism, subclinical hypothyroidism, and the presence of insulin resistance with normal blood glucose levels (Table 1).
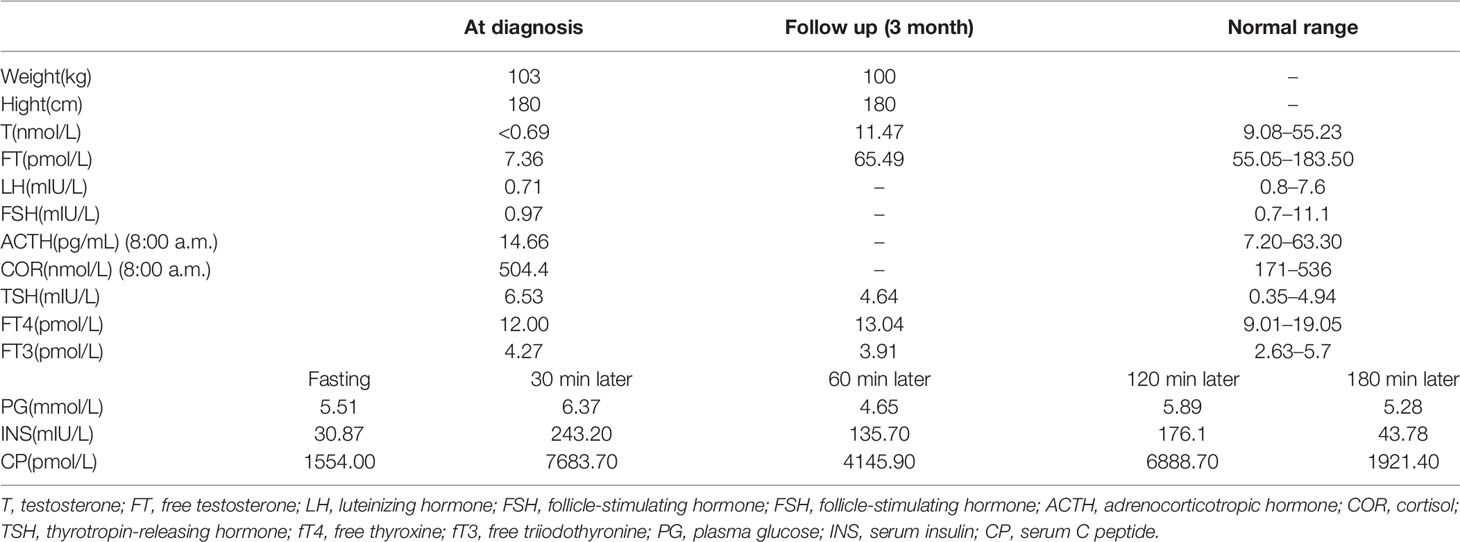
Table 1 Clinical and laboratory data of the patient at diagnosis.
Genetic Testing
Blood samples were collected from the patient and his family members (mother and brother). Genomic DNA was extracted using a blood extraction kit (Tian Jing Biochemical Technology Beijing, Ltd.). Roche NimbleGen SeqCap EZ Choice XL Library was used for exon trapping (including 4,132 genes). Illumina NextSeq500 was used for high-throughput sequencing. Burrows-Wheeler Aligner software (BWA, 0.7.12-r1039) was applied to align the sequencing data to the human genome. ANNOVAR was used to annotate the genetic variants detected. Emphasis was laid on the analysis of the known genes involved in KS and WS pathology. The results revealed that a large fragment of base insertion at the site 1,179–1,180 of SOX10 (c.1179_1180insACTATGGCTCAGCCTTCCCC, p.Ser394fs) resulted in a heterozygous mutation in exon4 (Figure 1E). The frame-shifting mutation of the 394th amino acid serine resulted in a series of code changes downstream. Predicted by MutationTaster (disease-causing, score=1) software, it is related to the disease. After querying in databases such as The Single Nucleotide Polymorphism Database, The Human Gene Mutation Database, 1000 Genomes Project, ClinVar, Exome Sequencing Project v.6500 database, and related literature, it was found that this mutation had not been reported as a new mutation. When the coding region of exon4 of SOX10 was resequenced in the samples of the patient’s mother and brother, the insertion was not detected (Figure 1F).
Therapeutic Intervention and Follow-Up
For hypogonadism, 2,000 U human chorionic gonadotropin (hCG) was intramuscularly injected twice a week, and testosterone and free testosterone levels were re-examined periodically at outpatient visits. Additionally, 75 U of human menopausal gonadotropin (hMG) was added to the patient’s medications 3 months later. For subclinical hypothyroidism, regular monitoring of thyroid function was carried out. Subsequently, the underarm and pubic hair of the patient showed significant growth. The length of the penis increased to 5 cm (Figure 1B), testicular volume increased to 8 ml (Figure 1C), and his testosterone levels increased as well and reached the normal range (Table 1). Thyroid function also returned to normal (Table 1).
Discussion
Herein, we report a case of a man with KS and WS2, who carried a novel insertion of a 20 bp fragment into the coding sequence of the exon4 of the SOX10 gene. Our patient exhibited hypogonadotropic hypogonadism and anosmia, which are characteristics of KS. Interestingly, he also manifested characteristics of WS2 such as bilateral sensorineural deafness and abnormal pigmentation (blue irises, white forehead hair). To date, 18 patients with KS have been identified to have the SOX10 mutation, of which 15 patients had unilateral or bilateral sensorineural deafness, accounting for 83% of the KS patients with SOX10 mutation. In addition, five of the 18 patients demonstrated abnormal pigmentation and other typical clinical manifestations of WS. However, Hirschsprung disease was not described in any of the 18 patients (Table 2). Our report and previous findings indicate the importance of SOX10 haploinsufficiency as one of the genetic causes of KS with WS-characteristic clinical features. It is well known that SOX10 is an important pathogenic gene associated with WS2 and WS4, and KS with deafness is similar to WS with anosmia in many clinical manifestations, such as perceptive nervous deafness, abnormal pigmentation, and hyposmia. Therefore, it can be speculated that there is a certain association between the two diseases.
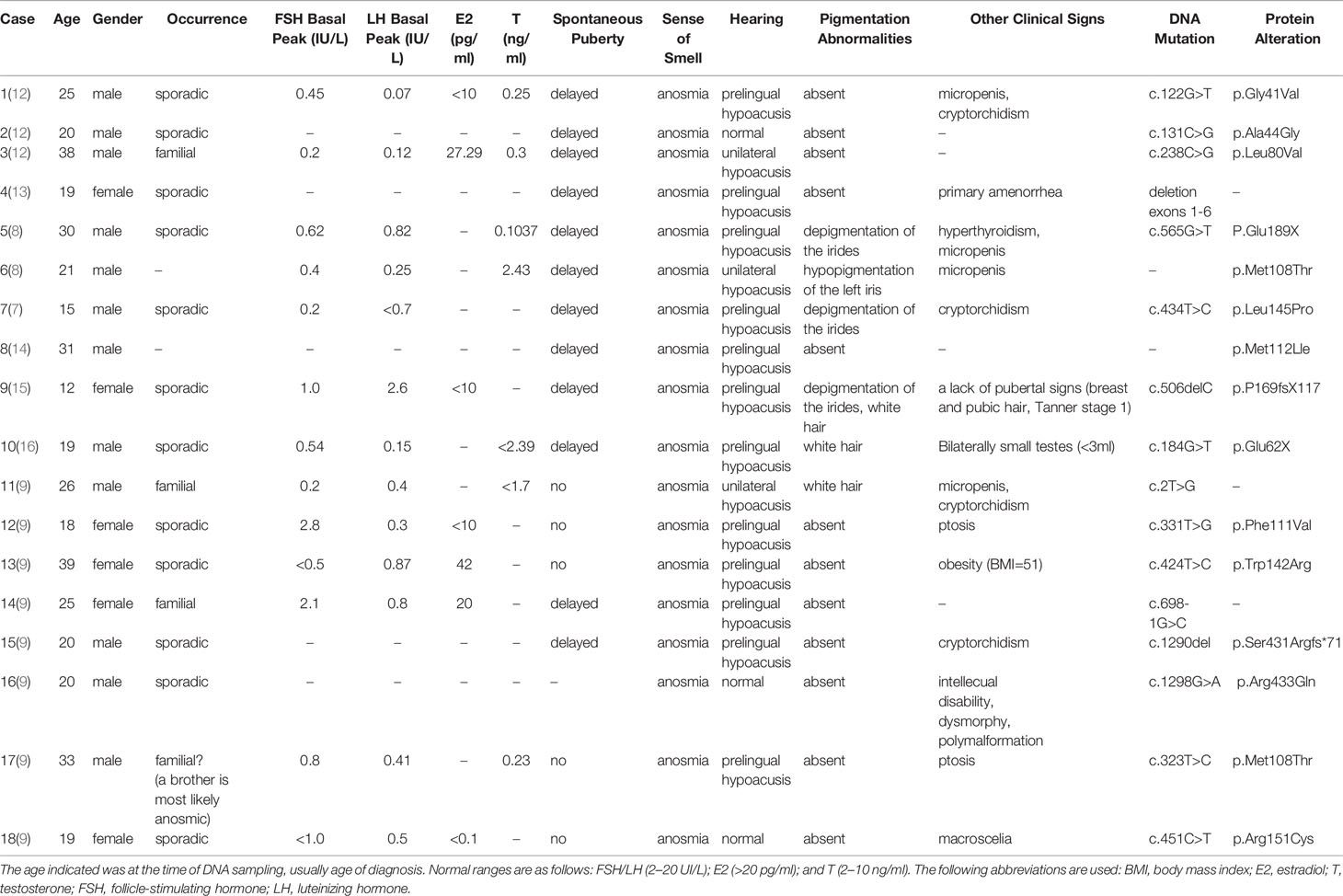
Table 2 Phenotypes of the KS Individuals Carrying SOX10 Mutations.
The SOX10 gene is located on chromosome 22q 13.1 and includes five exons, of which exons 3, 4, and 5 encode proteins. The SOX10 protein is a member of the 20 SOX gene family and contains 466 amino acids with a relative molecular mass of about 51kDa. The main functions are attributed to the highly conserved high mobility group (HMG) and the C-terminal transactivation domain (TAD) (17). The HMG box is composed of 80 amino acid residues (bits 102–181) and the stability of the whole structure is maintained by a hydrophobic inner core. It is able to specifically recognize and bind to the promoter DNA of the target gene, enabling the protein to act as a transcription factor (TF). When SOX10 protein binds to the target DNA, the DNA gap binds to the HMG box, resulting in a conformational change of the DNA, and the TAD architecture activates the transcription of the gene (18). Originally defined as a glial TF, SOX10 TF was found to be essential for the development of neural crest cells (NCC), such as olfactory ensheathing cells, melanocytes, intestinal ganglion cells, and many autonomic and sensory ganglion cells (19–21).
In 2013, Pingault et al. (9) found a mutation in the SOX10 gene by performing a genetic test on a WS patient with an atrophied olfactory bulb whose clinical manifestations included sensorineural deafness and anosmia. It is speculated that SOX10 mutation may lead to KS because KS patients with deafness have similar clinical manifestations to WS patients with anosmia. Pingault et al. (9) tested 17 KS patients who had been excluded from having known pathogenic gene mutations (e.g., KAL1, FGFR1, FGF8, PROKR, PROK2, WDR11, HS6ST1, CHD7, SEMA3A, etc.) for SOX10 gene mutations and found seven SOX10 mutations, six of which were pathogenic. The team then created a SOX10 knockout mouse model and found that they exhibited a massive loss of olfactory ensheathing cells that hindered the migration signals of olfactory and GnRH neurons, causing characteristic clinical manifestations of KS such as anosmia and hypogonadism. Based on the above studies, a massive loss of olfactory ensheathing cells is considered as the main pathogenesis of KS patients with SOX10 mutations.
The development and differentiation of NCC is a complex process regulated by multiple TFs and signaling pathways. SOX10 plays an important role in this process and can regulate the development of NCC, especially melanocytes, either individually or synergistically with other TFs (22). In the pathogenesis of deafness in WS patients, studies (23, 24) have found that the potassium pump function of melanocytes in microphthalmia-associated transcription factor (MITF) mutant mouse models is abnormal and cannot maintain endolymphatic potential. Subsequently, no action potential was generated leading to secondary hair cell apoptosis. Moreover, SOX10 can act on MITF and play its regulatory role in monomeric form or dimer form with PAX3 (25, 26). As research continues, it has been reported that semicircular canal (SC) malformation is the most common malformation in WS patients with inner ear malformations. However, the phenotype of the inner ear malformation has not been linked to the genetic characteristics (27). In 2013, Elmaleh et al. (28) found bilateral SC malformations with vestibular and cochlear malformations along with SOX10 mutations by genetic testing in 15 WS patients with inner ear malformations. Another study (29) has found that SOX10 protein is highly expressed during the early development of the inner ear and is consistently expressed throughout the development of the inner ear.
It has the function of maintaining the survival and differentiation of cochlear precursor cells, suggesting that there may be other pathways regulating the development of the inner ear in addition to the MITF pathway and in the mechanism of deafness caused by SOX10 mutation. This pathway explains why WS patients with SOX10 mutations are more prone to inner ear malformations, but relevant pathway has not been reported yet. In addition, SOX10 can also cooperate with MITF to activate dopachrome tautomerase (DTC) which acts in the biosynthesis of pigments. Therefore, SOX10 mutation may cause abnormal pigmentation such as blue irises and white forehead hair (30, 31). In the nervous system, SOX10 can directly regulate c-Ret which plays an important role in the development of the enteric nervous system (32). However, our patient did not suffer from neurological diseases (NIDs) such as Hirschsprung disease, which reflected the genetic and phenotypic heterogeneity. It has been proposed that this may be associated with SOX10 haploinsufficiency (7). Consequently, the pathogenesis of WS is associated with the MITF pathway which is regulated by SOX10 along with other pathways that can regulate inner ear development, pigmentation, and nervous system.
A study found that the peripheral gonadal tissue of KS patients can still retain certain functions, and early hormone replacement therapy can increase testicular volume in some male patients and even produce sperm to gain fertility (33). In 2016, Maione et al. reported that a KS patient with SOX10 mutation received testosterone injections in 1996 (age 21), 1999 (age 24), and 2000 (age 25). After treatment, the function of the hypothalamic-pituitary-gonadal axis returned to normal autonomously, and serum testosterone, gonadotropin, testicular volume, and sperm volume returned to normal as well. There was no recurrence for 20 years, but the olfactory bulb and SC remained hypoplastic (12). This was the first reported case till date of a KS patient with the SOX10 mutation who recovered after treatment. Hence, it is essential to ensure the early and timely diagnosis and treatment of KS patients.
In conclusion, we identified a novel 20 bp insertion into the coding sequence of the exon4 of the SOX10 gene causing a frameshift and denaturation of the SOX10 protein of a patient with KS and WS2, suggesting that SOX10 plays an important role in some critical stages of the development of neural crest cells. This supports previous data that SOX10 mutations may be a common pathogenic factor for the development of both KS and WS. Therefore, SOX10 mutation analysis should be considered for KS patients with combined WS clinical manifestations, especially deafness.
Ethics Statement
The hospital ethics committee of China Medical University approved the study, and the patient and his family members provided written informed consent for publication of their clinical details and clinical images.
Author Contributions
KC and YL designed the study. YL collected the data. KC drafted manuscript. YL and HW interpreted the datat and revised the manuscript. KC, HW, and YL approved the final version of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (grant no. 81300645).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank our patient and his family members who agreed to participate in this study.
References
1. Stamou M II, Georgopoulos NA. Kallmann Syndrome: Phenotype and Genotype of Hypogonadotropic Hypogonadism. Metabolism (2018) 86:124–34. doi: 10.1016/j.metabol.2017.10.012
2. Nair S, Jadhav S, Lila A, Jagtap V, Bukan A, Pandit R, et al. Spectrum of Phenotype and Genotype of Congenital Isolated Hypogonadotropic Hypogonadism in Asian Indians. Clin Endocrinol (Oxf) (2016) 85(1):100–9. doi: 10.1111/cen.13009
3. Chen J, Yuan K, He M-F, Wang C-L, Chen C, Fang Y-L, et al. Clinical and Genetic Features of Kallmann Syndrome: An Analysis of 5 Cases. Chin J Contemp Pediatr (2018) 20(11):925–9. doi: 10.7499/j.issn.1008-8830.2018.11.009
4. Kim JH, Seo GH, Kim G-H, Huh J, Hwang IlT, Jang J-H, et al. Targeted Gene Panel Sequencing for Molecular Diagnosis of Kallmann Syndrome and Normosmic Idiopathic Hypogonadotropic Hypogonadism. Exp Clin Endocrinol Diabetes (2019) 127(8):538–44. doi: 10.1055/a-0681-6608
5. Wen J, Pan L, Xu X, Wang J, Hu C. Clinical Data and Genetic Mutation in Kallmann Syndrome With CHARGE Syndrome: Case Report and Pedigree Analysis. Med (Baltimore) (2018) 97(27):e11284. doi: 10.1097/MD.0000000000011284
6. Kemal Topaloglu A, Damla Kotan L. Genetics of Hypogonadotropic Hypogonadism. Endocr Dev (2016) 29:36–49. doi: 10.1159/000438841
7. Suzuki E, Izumi Y, Chiba Y, Horikawa R, Matsubara Y, Tanaka M, et al. Loss-of-Function SOX10 Mutation in a Patient With Kallmann Syndrome, Hearing Loss, and Iris Hypopigmentation. Horm Res Paediatr (2015) 84(3):212–6. doi: 10.1159/000436965
8. Guimaraes Lima Amato L, Montenegro LR, Marcondes Lerario A, Augusto Lima Jorge A, Guerra Junior G, Schnoll C, et al. New Genetic Findings in a Large Cohort of Congenital Hypogonadotropic Hypogonadism. Eur J Endocrinol (2019) 181(2):103–19. doi: 10.1530/EJE-18-0764
9. Pingault V, Bodereau V, Baral V, Marcos S, Watanabe Y, Chaoui A, et al. Loss-of-function Mutations in SOX10 Cause Kallmann Syndrome With Deafness. Am J Hum Genet (2013) 92(5):707–24. doi: 10.1016/j.ajhg
10. Pingault V, Ente D, Dastot-Le Moal F, Goossens M, Marlin S, Bondurand N. Review and Update of Mutations Causing Waardenburg Syndrome. Hum Mutat (2010) 31(4):391–406. doi: 10.1002/humu.21211
11. Bondurand N, Dastot-Le Moal F, Stanchina L, Collot N, Baral V, Marlin S, et al. Deletions at the SOX10 Gene Locus Cause Waardenburg Syndrome Types 2 and 4. Am J Hum Genet (2007) 81(6):1169–85. doi: 10.1086/522090
12. Dai W, Wu J, Zhao Y, Jiang F, Zheng R, Chen D-N, et al. Functional Analysis of SOX10 Mutations Identified in Chinese Patients With Kallmann Syndrome. Gene (2019) 702:99–106. doi: 10.1016/j.gene.2019.03.039
13. Depenbusch M, von Eckardstein S, Simoni M, Nieschlag E. Maintenance of Spermatogenesis in Hypogonadotropic Hypogonadal Men With Human Chorionic Gonadotropin Alone. Eur J Endocrinol (2002) 147(5):617–24. doi: 10.1530/eje.0.1470617
14. Shin S-J, Sul Y, Kim JH, Cho JH, Kim G-H, Kim JH, et al. Clinical, Endocrinological, and Molecular Characterization of Kallmann Syndrome and Normosmic Idiopathic Hypogonadotropic Hypogonadism: A Single Center Experience. Ann Pediatr Endocrinol Metab (2015) 20(1):27–33. doi: 10.6065/apem.2015.20.1.27
15. Izumi Y, Musha I, Suzuki E, Iso M, Jinno T, Horikawa R, et al. Hypogonadotropic Hypogonadism in a Female Patient Previously Diagnosed as Having Waardenburg Syndrome Due to a sox10 Mutation. Endocrine (2015) 49(2):553–6. doi: 10.1007/s12020-014-0434-4
16. Vaaralahti K, Tommiska J, Tillmann V, Liivak N, Känsäkoski J, Laitinen E-M, et al. De Novo SOX10 Nonsense Mutation in a Patient With Kallmann Syndrome and Hearing Loss. Pediatr Res (2014) 76(1):115–6. doi: 10.1038/pr.2014.60
17. Harris ML, Baxter LL, Loftus SK, Pavan WJ. Sox Proteins in Melanocyte Development and Melanoma. Pigment Cell Melanoma Res (2010) 23(4):496–513. doi: 10.1111/j.1755-148X.2010.00711.x
18. Kuhlbrodt K, Schmidt C, Sock E, Pingault V, Bondurand N, Goossens M, et al. Functional Analysis of Sox10 Mutations Found in Human Waardenburg-Hirschsprung Patients. J Biol Chem (1998) 273(36):23033–8. doi: 10.1074/jbc.273.36.23033
19. Kuhlbrodt K, Herbarth B, Sock E, Hermans-Borgmeyer I, Wegner M. Sox10, a Novel Transcriptional Modulator in Glial Cells. J Neurosci (1998) 18(1):237–50. doi: 10.1523/JNEUROSCI.18-01-00237.1998
20. Barraud P, St John JA, Stolt CC, Wegner M, Baker CVH. Olfactory Ensheathing Glia Are Required for Embryonic Olfactory Axon Targeting and the Migration of Gonadotropin-Releasing Hormone Neurons. Biol Open (2013) 2(7):750–9. doi: 10.1242/bio.20135249
21. Hou L, Arnheiter H, Pavan WJ. Interspecies Difference in the Regulation of Melanocyte Development by SOX10 and MITF. (2006) Proc Natl Acad Sci U S A 103: (24):9081–5. doi: 10.1073/pnas.0603114103
22. Chen H, Jiang L, Xie Z, Mei L, He C, Hu Z, et al. Novel Mutations of PAX3, MITF, and SOX10 Genes in Chinese Patients With Type I or Type II Waardenburg Syndrome. Biochem Biophys Res Commun (2010) 397(1):70–4. doi: 10.1016/j.bbrc.2010.05.066
23. Locher H, de Groot JCMJ, van Iperen L, Huisman MA, Frijns JHM, Chuva de Sousa Lopes SM. Development of the Stria Vascularis and Potassium Regulation in the Human Fetal Cochlea: Insights Into Hereditary Sensorineural Hearing Loss. Dev Neurobiol (2015) 75(11):1219–40. doi: 10.1002/dneu.22279
24. Jin Z, Mannström P, Järlebark L, Ulfendahl M. Malformation of Stria Vascularis in the Developing Inner Ear of the German Waltzing Guinea Pig. Cell Tissue Res (2007) 328(2):257–70. doi: 10.1007/s00441-006-0369-z
25. Wang H-H, Chen H-S, Li H-B, Zhang H, Mei L-Y, He C-F, et al. Identification and Functional Analysis of a Novel Mutation in the SOX10 Gene Associated With Waardenburg Syndrome Type IV. Gene (2014) 538(1):36–41. doi: 10.1016/j.gene.2014.01.026
26. Chaoui A, Watanabe Y, Touraine R, Baral V, Goossens M, Pingault V, et al. Identification and Functional Analysis of SOX10 Missense Mutations in Different Subtypes of Waardenburg Syndrome. Hum Mutat (2011) 32(12):1436–49. doi: 10.1002/humu.21583
27. Oysu C, Oysu A, Aslan I, Tinaz M. Temporal Bone Imaging Findings in Waardenburg’s Syndrome. Int J Pediatr Otorhinolaryngol (2001) 58(3):215–21. doi: 10.1016/s0165-5876(01)00443-8
28. Elmaleh-Bergès M, Baumann C, Noël-Pétroff N, Sekkal A, Couloigner V, Devriendt K, et al. Spectrum of Temporal Bone Abnormalities in Patients With Waardenburg Syndrome and SOX10 Mutations. AJNR Am J Neuroradiol (2013) 34(6):1257–63. doi: 10.3174/ajnr.A3367
29. Locher H, Frijns JHM, van Iperen L, de Groot JCMJ, Huisman MA, Chuva de Sousa Lopes SM. Neurosensory Development and Cell Fate Determination in the Human Cochlea. Neural Dev (2013) 8:20. doi: 10.1186/1749-8104-8-20
30. Verastegui C, Bille K, Ortonne JP, Ballotti R. Regulation of the Microphthalmia-Associated Transcription Factor Gene by the Waardenburg Syndrome Type 4 Gene, SOX10. J Biol Chem (2000) 275(40):30757–60. doi: 10.1074/jbc.C000445200
31. Jiao Z, Mollaaghababa R, Pavan WJ, Antonellis A, Green ED, Hornyak TJ. Direct Interaction of Sox10 With the Promoter of Murine Dopachrome Tautomerase (Dct) and Synergistic Activation of Dct Expression With Mitf. Pigment Cell Res (2004) 17(4):352–62. doi: 10.1111/j.1600-0749.2004.00154.x
32. Schriemer D, Sribudiani Y, IJpma A, Natarajan D, MacKenzie KC, Metzger M, et al. Regulators of Gene Expression in Enteric Neural Crest Cells Are Putative Hirschsprung Disease Genes. Dev Biol (2016) 416(1):255–65. doi: 10.1016/j.ydbio.2016.06.004
Keywords: Kallmann syndrome, Waardenburg syndrome type Ⅱ, SOX10 mutations, sensorineural deafness, hypogonadotropic hypogonadism, co-occurrence
Citation: Chen K, Wang H and Lai Y (2021) Kallmann Syndrome Due to Heterozygous Mutation in SOX10 Coexisting With Waardenburg Syndrome Type II: Case Report and Review of Literature. Front. Endocrinol. 11:592831. doi: 10.3389/fendo.2020.592831
Received: 08 August 2020; Accepted: 14 December 2020;
Published: 01 February 2021.
Edited by:
Ishwar Parhar, Monash University Malaysia, MalaysiaReviewed by:
Nils Lambrecht, VA Long Beach Healthcare System, United StatesErik Hrabovszky, Institute of Experimental Medicine (MTA), Hungary
Copyright © 2021 Chen, Wang and Lai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yaxin Lai, bGFpeWF4aW44MTEwMDVAMTI2LmNvbQ==