 Valentina Cirello1
Valentina Cirello1 Carla Colombo1,2*
Carla Colombo1,2* Olga Karapanou3
Olga Karapanou3 Gabriele Pogliaghi1
Gabriele Pogliaghi1 Luca Persani1,4
Luca Persani1,4 Laura Fugazzola1,2
Laura Fugazzola1,2- 1Department of Endocrine and Metabolic Diseases, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milan, Italy
- 2Department of Pathophysiology and Transplantation, Università degli Studi di Milano, Milan, Italy
- 3Department of Endocrinology, 401 Military Hospital, Athens, Greece
- 4Department of Medical Biotechnology and Translational Medicine, University of Milan, Milan, Italy
Several low penetration susceptibility risk loci or genes have been proposed in recent years with a possible causative role for familial non-medullary thyroid cancer (FNMTC), though the results are still not conclusive or reliable. Among all the candidates, here fully reviewed, a new extremely rare germline variant c.3607A>G (p.Y1203H) of the DUOX2 gene, has been recently reported to co-segregate with the affected members of one non-syndromic FNMTC family. We aimed to validate this finding in our series of 33 unrelated FNMTC Italian families, previously found to be negative for two susceptibility germline variants in the HABP2 and MAP2K5 genes. Unfortunately, the DUOX2 p.Y1203H variant was not found in either the 74 affected or the 12 not affected family members of our series. We obtained interesting data by comparing the clinico-pathological data of the affected members of our kindreds with a large consecutive series of sporadic cases, followed at our site. We found that familial tumors had a statistically significant more aggressive presentation at diagnosis, though not resulting in a worst outcome. In conclusion, we report genetic and clinical data in a large series of FNMTC kindreds. Our families are negative for variants reported as likely causative, namely those lying in the HABP2, MAP2K5 and DUOX2 genes. The extensive review of the current knowledge on the genetic risk factors for non-syndromic FNMTCs underlies how the management of these tumors remains mainly clinical. Despite the more aggressive presentation of familial cases, an appropriate treatment leads to an outcome similar to that observed for sporadic cases.
Introduction
The term familial non-medullary thyroid cancers (FNMTCs) is used to indicate thyroid tumors, arising from follicular cells, which are observed in two or more first-degree relatives in the absence of predisposing environmental factors. FNMTCs account for 3–9% of all thyroid cancer (TC) cases, and show an autosomal dominant pattern of inheritance with incomplete penetrance and variable expressivity (1). FNMTC is further sub-classified in: syndromic FNMTCs (5% of all FNMTCs) in which TC occurs as a minor component (familial adenomatous polyposis, Gardner’s syndrome, Cowden’s syndrome, Werner’s syndrome, McCune-Albright syndrome, Carney complex type 1, DICER1 syndrome) and non-syndromic FNMTCs (95% of all FNMTCs), in which TC is the predominant feature. Among FNMTCs, papillary thyroid cancer (PTC) is the commonest histological subtype (2, 3). Some studies reported that non-syndromic familial PTCs (FPTCs) are more aggressive than sporadic PTCs, in terms of higher rate of multifocal and bilateral tumors, extrathyroidal extension, lymph node metastasis, younger age of onset, and recurrence rate (4–8). Moreover, the phenomenon of “anticipation” with early and more severe manifestations of the disease has been observed in the second generation (9). On the contrary, other studies did not found clinical differences between non-syndromic FNMTCs and sporadic cases (10, 11). FNMTCs are indistinguishable from sporadic NMTC forms by either morphological examination or immunohistochemical staining, implying that FNMTCs cannot be diagnosed until at least one of the patient’s first-degree relatives is diagnosed with a thyroid cancer. Genetic alterations underlying syndromic forms have been at least in part defined (APC in familial adenomatous polyposis and Gardner’s syndrome; PTEN in Cowden’s syndrome; PRKAR1A in Carney Complex type 1; WNR in Werner’s syndrome, DICER1 in DICER1 syndrome; GNAS in McCune Albright syndrome) (1, 3). On the other hand, the genetic susceptibility for non-syndromic FNMTCs is still not well defined. To date, only few susceptibility risk chromosomal loci [MNG1 (14q32), TCO (19p13.2), fPTC/PRN (1q21), NMTC1 (2q21), FTEN (8p23.1–p22), 6q22, 4q32, and 8q24] (1) and susceptibility genes (TTF-1/NKX.2, SRGAP1, FOXE1, SRRM2, HABP2, MAP2K5) (12–17) have been identified by means of linkage analysis, genome-wide association study (GWAS), whole exome (WES) and next-generation sequencing (Table 1). In addition, an imbalance of the telomere–telomerase complex has been reported in the peripheral blood of familial NMTC patients (9). Nevertheless, the reliability of these genetic alterations is controversial and still under investigation. Very recently, Bann and colleagues identified a novel germline variant, c.3607T>A (p.Y1203H) in Dual Oxidase-2 (DUOX2) gene, segregating as an autosomal dominant trait in all affected individuals from a non-syndromic FNMTC family. This variant is extremely rare in the general population (prevalence of 1 out of 138,000 individuals reported in the dbSNP database), consistent with its role as a causative mutation in a rare condition such as FNMTC (34). DUOX2 gene (MIM*606759) maps on chromosome 15 and encodes a protein comprising an extracellular N-terminal peroxidase-like domain, seven transmembrane domains, two cytosolic Calcium-binding sites (EF hand motifs), and an intracellular C-terminal oxidase domain. The maturation factors DUOXA1 and DUOXA2 are essential components for the enzymatic activity of DUOX2, because they form heterodimeric complexes with DUOX2 and allow the translocation of the dimer from endoplasmic reticulum to the membrane (35). The DUOX2 p.Y1203H variant affects an amino acid highly conserved across mammalian, localized in the fifth transmembrane domain of DUOX2 protein. Due to its location, it is unlikely that the variant could impair the enzymatic active site of the protein or the interaction with DUOX2A.
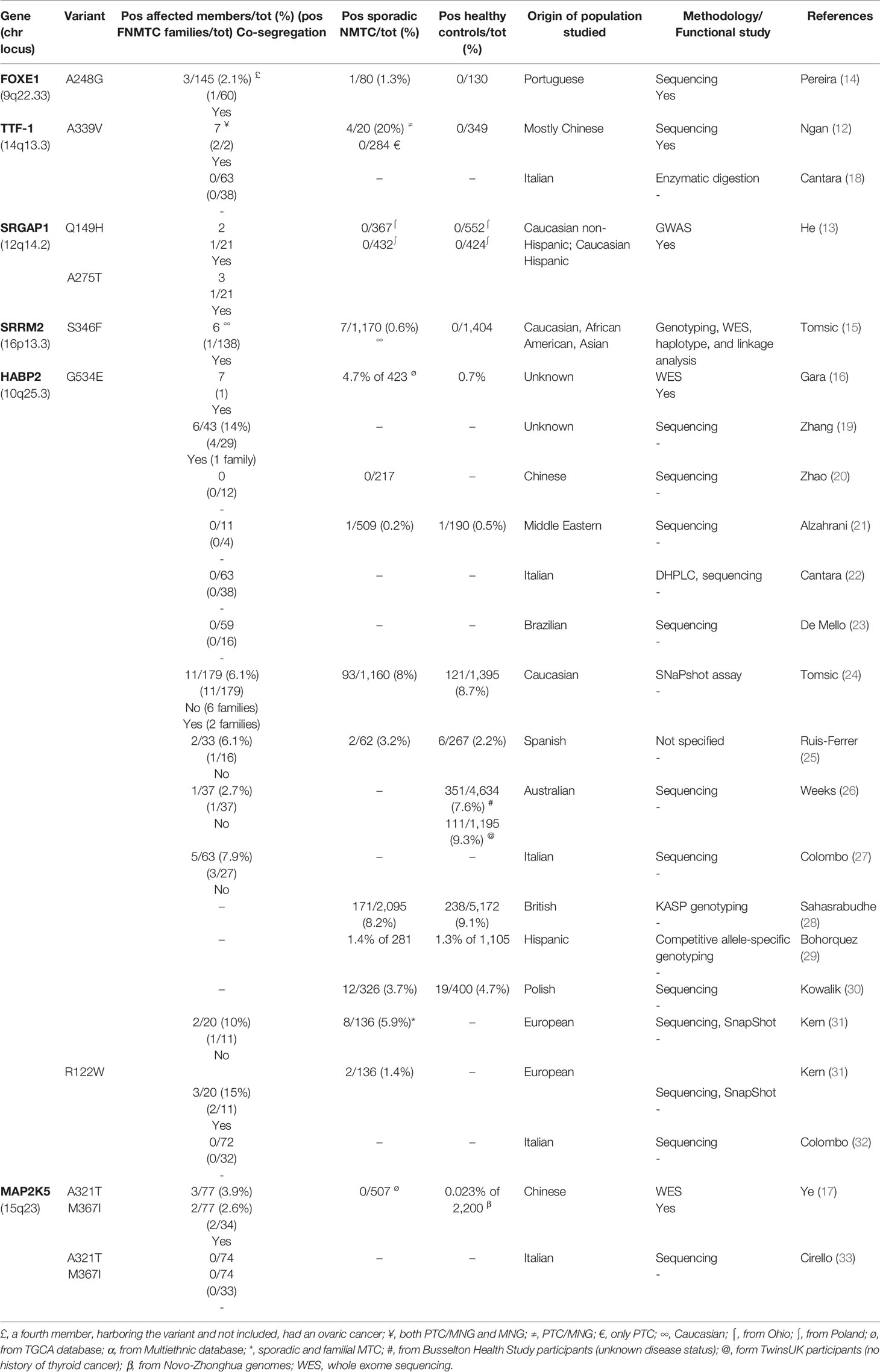
Table 1 Susceptibility genes and variants identified in non-syndromic familial non-medullary thyroid cancer (FNMTC).
In the present study, we aimed to validate the hypothesized role of this DUOX2 p.Y1203H germline variant in our series of FPTC Italian families. We also carried out a review of literature in order to summarize the current knowledge on the genetic risk factors for FNMTCs. Finally, the clinical features and the outcome of our familial cases were compared with those of our consecutive series of sporadic PTC cases.
Patients and Methods
Patients
The presence of DUOX2 p.Y1203H germline variant was evaluated in a series of 33 unrelated Italian FNMTC kindreds, followed-up in our tertiary center for thyroid cancer care during the period 2003–2019. All affected members had a papillary thyroid cancer (FPTC). By definition, all families had a PTC in first-degree relatives and, in particular, 12/33 (36%) families had a parent-child relationship, 19/33 (58%) had a sibling relationship and 2/33 (6%) families had an uncle-nephew relationship. The number of affected members was two in 27 families, three in 4 families, and four in 2 families. Families came from different regions of Italy, North, South, and islands. Tumors were classified and staged according to the thyroid malignancy World Health Organization classification and the 8th edition of TNM staging (36). Patients were diagnosed and treated according to the recent guidelines for the management of thyroid cancer (37, 38). To identify remission or persistent/recurrent disease, ATA guidelines and Italian consensus recommendations were followed (37, 38). Clinico-pathological features at diagnosis and the final outcome, after a mean follow-up of 93 months, were available for all FPTC probands and for additional 10 affected members. None of kindreds analyzed had clinical features consistent with a possible familial cancer syndrome and none of the family members reported had a history of other primary cancers, except one member who was affected with a tongue carcinoma in remission. The clinical data of the FPTC patients were compared with those coming from all the sporadic PTCs (n = 1,186) included in the database of our tertiary Center. An Institutional Review Board approval was obtained for the analysis of the clinical and molecular data, and informed consent was given by all screened subjects.
Genotyping
Germline DNA was extracted from the peripheral blood of 86 family members (74 affected with FPTC and 12 unaffected), using standard methods. The exon 28 of the DUOX2 gene, where the genetic variant c.3607A>G (p.Y1203H) lies, was amplified by PCR and sequenced on ABI PRISM 3100 automated genetic analyzer (Applied Biosystems, Waltham MA, USA) both in forward and reverse directions, as previously reported (39). A subset of 14 thyroid tumor samples from our kindreds were analyzed using the custom PTC-MA assay, based on matrix-assisted laser desorption/ionization time-of-flight mass spectrometry, and previously set up for the simultaneous identification of 13 known hotspot mutations BRAFV600E; AKT1E17K; EIF1AX c.338-1G>C; NRASQ61R and NRASQ61K; HRASG13C, HRASQ61K, and HRASQ61R; KRASG12V and KRASG13C; TERT c.-124C>T and TERT c.-146C>T; and PIK3CAE542K and 7 recurrent fusion genes typical of PTC: RET/PTC1 (RET/CCDC6), RET/PTC2 (RET/PRKAR1A), and RET/PTC3 (RET/NCOA4) and TRK (NTRK1/TPM3), TRK-T1 (NTRK-T1/TPR), and TRK-T3 (NTRK1/TFG) (40, 41).
Statistical Analysis
Relations between discrete variables were evaluated by means of t-test or chi2 test, as appropriate. Statistical significance was defined as P <0.05. All statistical analyses were performed using MedCalc Analyses (Version 18.11.3 of the MedCalc Software, B-8400 Ostend, Belgium).
Results
Clinico-Pathological and Molecular Features
Clinical data were available for all probands and for 10 additional affected family members, for a total of 43 patients (38 females and 5 males), with a median age at diagnosis of 44 years (Table 2). In particular, most patients underwent surgery due to a malignant (67%) or suspicious (5%) cytology, while thyroid carcinoma evidence was incidental in 28% of cases. Mean tumor diameter was 13.9 mm, and the prevalence of papillary microcarcinomas was of 37%. Moreover, the prevalence of multifocal carcinomas, unilateral or bilateral, was 56%, and the prevalent variant of papillary histotype was the classical one (81%). Gross extrathyroidal extension, defined by the 8th edition of TNM staging as the presence of macroscopic tumor extending outside the thyroid gland, was recorded in 35% of cases, while metastatic lymph nodes and lung metastases were found in 26 and 7% of cases, respectively.
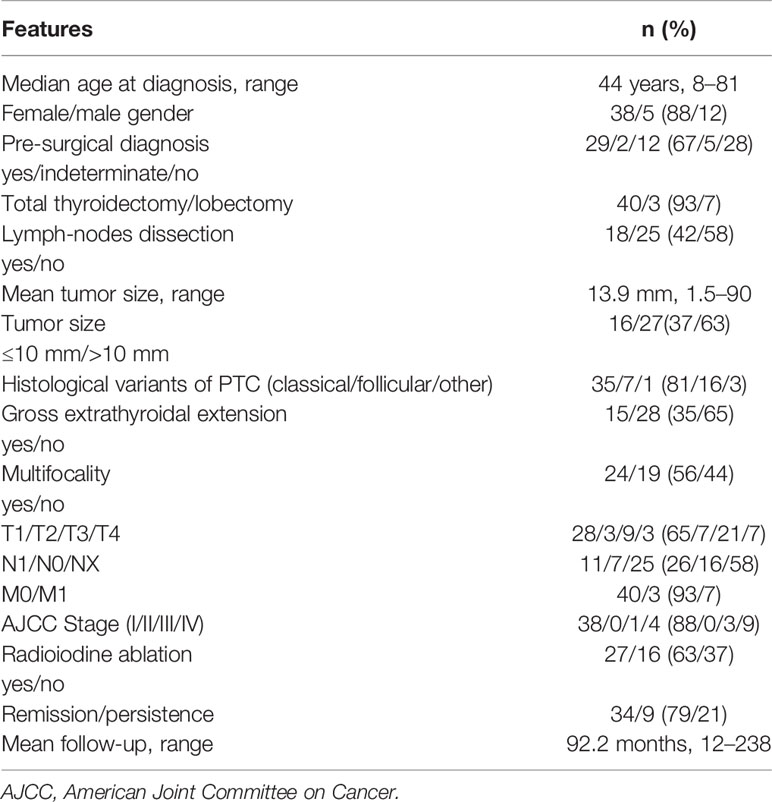
Table 2 Clinico-pathological features at diagnosis of 43 patients with familial papillary thyroid cancer (FPTC).
None of the clinico-pathological parameters analyzed were found to be significantly different among the 27 families with 2 affected members and the 6 families with 3 or more affected members (data not shown).
Clinical data related to the 1186 sporadic PTC followed at our site are reported in Table 3. Interestingly, statistically significant differences were found between FPTC and sporadic PTC cases (Figure 1), with the former presenting with worse prognostic features. In particular, in FPTCs the histological variants other than the classical one were more represented (19 vs 10%, P < 0.0001),
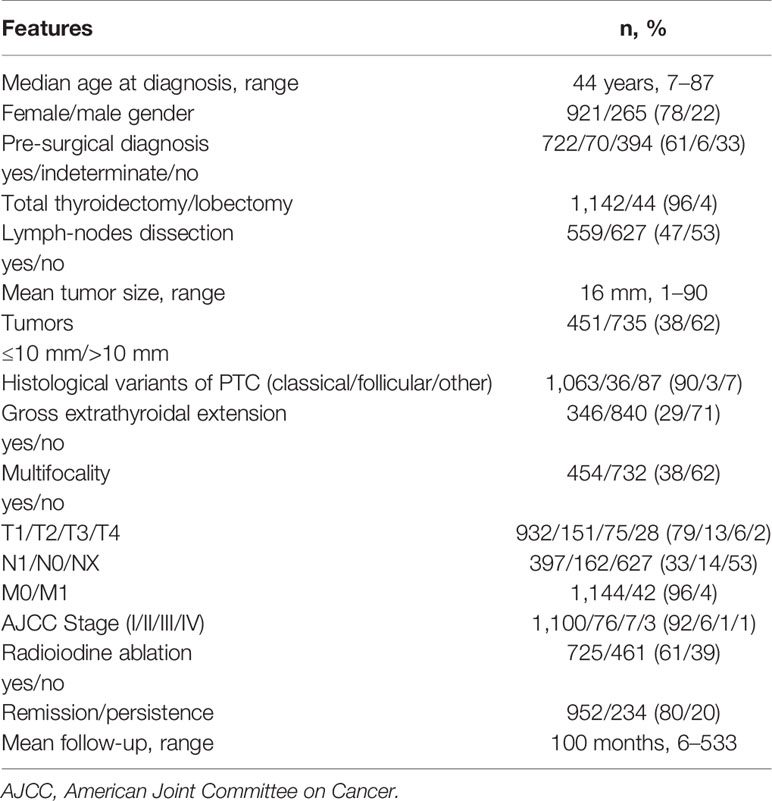
Table 3 Clinico-pathological features of 1,186 sporadic papillary thyroid cancer patients at diagnosis.
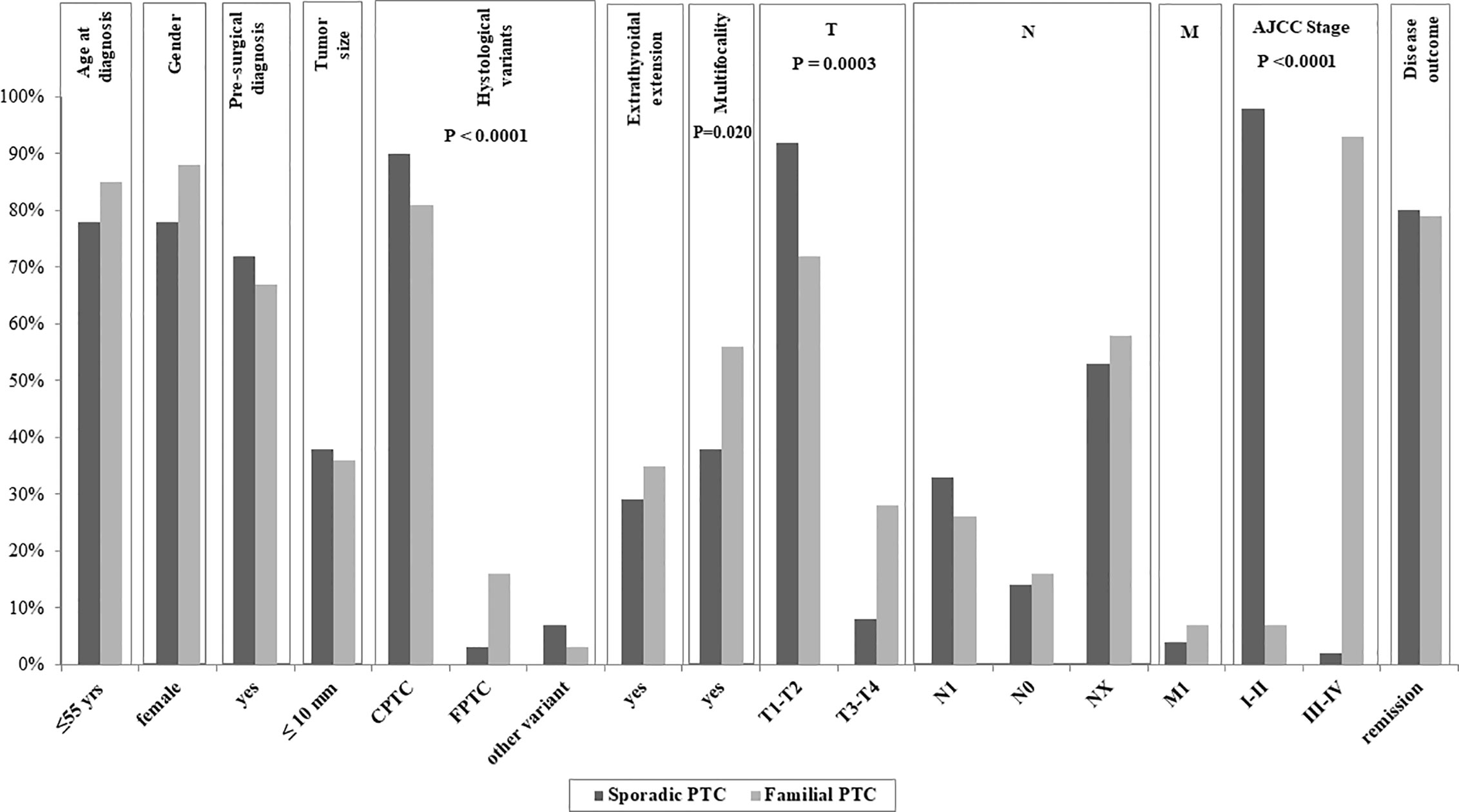
Figure 1 Clinico-pathological data of the affected members of our Familial Papillary Thyroid Cancer (FPTC) kindreds compared with those of the sporadic PTC cases followed at our site. The P value is reported only for the features with significant differences between familial and sporadic cases.
multifocality was significantly more frequent (56 vs 38%, P = 0.02), T3–T4 tumors were more prevalent (28 vs 8%, P = 0.0003), as well as the AJCC III-IV stage at diagnosis (12 vs 2%, P < 0.0001). Other clinico-pathological features did not differ between the two groups, namely age at diagnosis, gender, tumor size, pre-surgical diagnosis, gross extrathyroidal extension, local and distant metastases, and disease outcome (Figure 1).
Moreover, the prevalence of patients undergoing radioiodine therapy was similar between two PTC groups (63% in FPTC vs 61% in sporadic PTC, P = 0.826), as well as the mean follow-up (92.2 vs 100 months, respectively, P = 0.587).
Among the 33 families studied, 74 patients with FPTC and 12 healthy subjects were submitted to genetic analyses. The molecular analysis of DUOX2 exon 28 did not show the presence of the p.Y1203H germline variant in any affected or healthy family member.
Interestingly, the genetic characterization of the 14 FPTCs, for whom the tumor tissue was available, revealed a BRAFV600E mutation in 4/14 (28.6%) cases, a RET fusion in 2/14 (14.3%) (1 with ret/PTC1 and 1 ret/PTC3), a TERT c.-124C>T mutation in 2/14 (14.3%), while 1/14 (7.1%) tumor harbored both NRASQ61K and TERT c.-146C>T mutations, and 1/14 both BRAFV600E and TERT c.-124C>T genetic variations. Finally, 4/14 (28.6%) cases were wild type for the mutations and fusions tested. Of note, 9/10 patients harboring somatic mutations owned to families with two affected members, while only 1 patient belonged to a family with three affected members.
Discussion
The genetic susceptibility for non-syndromic FNMTCs is still not well defined, with few susceptibility risk chromosomal loci (1) and susceptibility genes (12–17, 31) identified whose reliability is controversial and still under investigation.
In the present study, our large series of Italian unrelated FNMTC kindreds, in which we previously excluded a role for HABP2 and MAP2K5 germline variants (27, 32, 33), has been found not to harbor the DUOX2 p.Y1203H variant, recently described in a non-syndromic FNMTC family (34). Mutations in the DUOX2 gene, whose protein function is to generate hydrogen peroxidase required for thyroid hormone synthesis in the follicular cells, are involved in the development of congenital hypothyroidism, and are typically found in the N-terminal extra cellular peroxidase-like domain and in the EF hand domains, where they compromise the enzymatic activity of the protein. On the contrary, only few mutations have been described in transmembrane domains (35). The p.Y1203H variant has a transmembrane localization and the derived mutant protein could be mislocalized, thus resulting in the dysregulation of H2O2 metabolism. Thus, since hydrogen peroxidase could be involved in tumorigenesis through oxidative DNA stress, cell signaling and proliferation, regulation of gene expression, and cell senescence and apoptosis (42), the variant was considered of potential interest. Moreover, functional studies demonstrated that the mutated DUOX2 protein is functionally active and capable of producing H2O2, with an increase production rate of reactive oxygen species (ROS) at plasma membrane level than the wild type (34). Unfortunately, the possible causative role suggested for this variant (34), has not been confirmed in our series of 33 unrelated kindreds.
Among other candidate genes, some involved in thyroid morphogenesis (FOXE1 and TTF-1/NKX.2), or coding for a protein implicated in the regulation of the small G-protein CDC42 (SRGAP1), or involved in the RNA splicing machinery (SRRM2) have been proposed. Still, for the HABP2 gene recently indicated as tumor suppressor, and for the MAP2K5 gene, encoding a protein kinase that belongs to the MAP kinase family, a possible pathogenic role has been suggested. Unfortunately, none of the variants identified in these genes has not been further validated, mainly due to their absence in series of different ethnicity (18, 20–23, 32, 33), or due to the lack of co-segregation with affected members (24–27, 31), or due to their presence even in sporadic PTCs or in healthy controls (15, 24, 26, 28–31). These shortcomings could be the consequence of poor characterization of the kindreds, to ethnic differences, or to the inclusion of families with two affected members. Indeed, the presence of only two affected members in a family does not necessarily imply the hereditary origin of a TC. It has been calculated that in the presence of two affected members, the probability for a sporadic origin is 47% and for a familial origin is 53%, rising to 99.9% in the case of three affected members (43). The possible misclassification as hereditary PTCs can be applied to our series too, since the majority of our kindreds includes just two affected members. The finding of a high prevalence of somatic mutations (mainly BRAFV600E, followed by mutations in TERT or ret/PTC rearrangements, and some double mutations in TERT and in NRAS or BRAF), mainly in families with only 2 affected members, could favor the sporadic origin of the PTCs present in some families, rather than their familial origin. Nevertheless, it is worth to note that we found a somatic mutation BRAFV600E also in the proband from a FPTC family composed by three affected members. In accordance with other Authors (2, 14, 34, 44), we can speculate that some of our FNMTC patients could have inherited a defective gene, not yet identified, which predispose the genome to the acquisition of somatic mutations in genes typically involved in thyroid tumorigenesis.
We obtained interesting data by comparing the clinico-pathological data of the affected members of our kindreds with a large consecutive series of sporadic PTCs followed at our site. We found that familial cases were more frequently multifocal, with a higher frequency of T3-T4 tumors and a higher AJCC stage, though the outcome did not differ with respect to sporadic cases. The higher aggressiveness of familial with respect to sporadic forms of PTC is debated, though most studies reported more advanced features at diagnosis in FPTCs (4–8, 43). Nevertheless, consistent with our findings, no differences in the outcome are reported in the majority of published series (2, 4, 8, 11, 43).
In conclusion, we report genetic and clinical data in a large series of FPTC kindreds. Our families are negative for variants previously reported as likely causative, namely those lying in the HABP2, MAP2K5, and DUOX2 genes. To confirm a possible pathogenic role for these and other susceptibility genes, careful replication studies are certainly needed. Moreover, alternative mechanisms of inheritance, such as epigenetic factors, likely warrant further investigation.
The extensive review of the current knowledge on the genetic risk factors for non-syndromic FNMTCs, here reported, underlies how the management of non-syndromic FNMTCs remains mainly clinical. FPTCs have a more aggressive clinical presentation, though, when recognized and treated appropriately, do not associate with a worst outcome with respect to sporadic cases.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethical Committee of the Istituto Auxologico Italiano IRCCS. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
VC: she was involved in the design of the study, in the molecular analysis of the DUOX2 gene, in the interpretation of the results and in the first compilation of the manuscript. CC: she was involved in the evaluation of clinical and histological characteristics of all patients and in the molecular characterization of tumor tissues available. OK: she was involved in the molecular analysis of the DUOX2 gene. GP: he was involved in the molecular analysis of the DUOX2 gene. LP: he revised the design of the study and the final version of the manuscript. LF: she designed and supervised all the experiments of the study and wrote the paper. All authors contributed to the article and approved the submitted version.
Funding
This work was partially supported by the Ministero dell’Istruzione, dell’Università e della Ricerca (PRIN 2017YTWKWH).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Peiling Yang S, Ngeow J. Familial non-medullary thyroid cancer: unraveling the genetic maze. Endocr Relat Cancer (2016) 23:R577–95. doi: 10.1530/ERC-16-0067
2. Moses W, Weng J, Kebebew E. Prevalence, clinicopathologic features, and somatic genetic mutation profile in familial versus sporadic nonmedullary thyroid cancer. Thyroid (2011) 21:367–71. doi: 10.1089/thy.2010.0256
3. Guilmette J, Nosé V. Hereditary and familial thyroid tumours. Histopathology (2018) 72:70–81. doi: 10.1111/his.13373
4. Uchino S, Noguchi S, Kawamoto H, Yamashita H, Watanabe S, Yamashita H, et al. Familial nonmedullary thyroid carcinoma characterized by multifocality and a high recurrence rate in a large study population. World J Surg (2002) 26:897–902. doi: 10.1007/s00268-002-6615-y
5. Lee YM, Yoon JH, Yi O, Sung TY, Chung KW, Kim WB, et al. Familial history of non-medullary thyroid cancer is an independent prognostic factor for tumor recurrence in younger patients with conventional papillary thyroid carcinoma. J Surg Oncol (2014) 109:168–73. doi: 10.1002/jso.23447
6. Tavarelli M, Russo M, Terranova R, Scollo C, Spadaro A, Sapuppo G, et al. Familial non-medullary thyroid cancer represents an independent risk factor for increased cancer aggressiveness: a retrospective analysis of 74 families. Front Endocrinol (Lausanne) (2015) 6:117. doi: 10.3389/fendo.2015.00117
7. Jiwang L, Zhendong L, Shuchun L, Bo B, Yanguo L. Clinicopathologic characteristics of familial versus sporadic papillary thyroid carcinoma. Acta Otorhinolaryngol Ital (2015) 35:234–42.
8. Sezer H, Demirkol MO, Yazici D, Kapran Y, Alagöl MF. The clinicopathologic characteristics of familial and sporadic papillary thyroid carcinoma in Turkish patients. Turk J Med Sci (2020) 250:360–8. doi: 10.3906/sag-1907-94
9. Capezzone M, Marchisotta S, Cantara S, Busonero G, Brilli L, Pazaitou-Panayiotou K, et al. Familial non-medullary thyroid carcinoma displays the features of clinical anticipation suggestive of a distinct biological entity. Endocr Relat Cancer (2008) 15:1075–81. doi: 10.1677/ERC-08-0080
10. Loh KC. Familial nonmedullary thyroid carcinoma: a meta-review of case series. Thyroid (1997) 7:107–13. doi: 10.1089/thy.1997.7.107
11. Pinto AE, Silva GL, Henrique R, Menezes FD, Teixeira MR, Leite V, et al. Familial vs sporadic papillary thyroid carcinoma: a matched-case comparative study showing similar clinical/prognostic behaviour. Eur J Endocrinol (2013) 170:321–7. doi: 10.1530/EJE-13-0865
12. Ngan ES, Lang BH, Liu T, Shum CK, So MT, Lau DK, et al. A germline mutation (A339V) in thyroid transcription factor-1 (TITF-1/NKX2.1) in patients with multinodular goiter and papillary thyroid carcinoma. J Natl Cancer Inst (2009) 101:162–75. doi: 10.1093/jnci/djn471
13. He H, Bronisz A, Liyanarachchi S, Nagy R, Li W, Huang Y, et al. SRGAP1 is a candidate gene for papillary thyroid carcinoma susceptibility. J Clin Endocrinol Metab (2013) 98:E973–80. doi: 10.1210/jc.2012-3823
14. Pereira JS, da Silva JG, Tomaz RA, Pinto AE, Bugalho MJ, Leite V, et al. Identification of a novel germline foxe1 variant in patients with familial non-medullary thyroid carcinoma (FNMTC). Endocrine (2015) 49:204–14. doi: 10.1007/s12020-014-0470-0
15. Tomsic J, He H, Akagi K, Liyanarachchi S, Pan Q, Bertani B, et al. A germline mutation in SRRM2, a splicing factor gene, is implicated in papillary thyroid carcinoma predisposition. Sci Rep (2015) 5:10566. doi: 10.1038/srep10566
16. Gara SK, Jia L, Merino MJ, Agarwal SK, Zhang L, Cam M, et al. Germline HABP2 mutation causing familial nonmedullary thyroid cancer. N Engl J Med (2015) 373:448–55. doi: 10.1056/NEJMoa1502449
17. Ye F, Gao H, Xiao L, Zuo Z, Liu Y, Zhao Q, et al. Whole exome and target sequencing identifies MAP2K5 as novel susceptibility gene for familial non-medullary thyroid carcinoma. Int. J Cancer (2019) 144:1321–30. doi: 10.1002/ijc.318252
18. Cantara S, Capuano S, Formichi C, Pisu M, Capezzone M, Pacini F. Lack of germline A339V mutation in thyroid transcription factor-1 (TITF-1/NKX2.1) gene in familial papillary thyroid cancer. Thyroid Res (2010) 3:4. doi: 10.1186/1756-6614-3-4
19. Zhang T, Xing M. HABP2 G534E mutation in familial nonmedullary thyroid cancer. J Natl Cancer Inst (2016) 108:djv415. doi: 10.1093/jnci/djv415
20. Zhao X, Li X, Zhang X. HABP2 mutation and nonmedullary thyroid cancer. N Engl J Med (2015) 373:2084. doi: 10.1056/NEJMc1511631
21. Alzahrani AS, Murugan AK, Qasem E, Al-Hindi H. HABP2 gene mutations do not cause familial or sporadic non-medullary thyroid cancer in a highly inbred middle eastern population. Thyroid (2016) 26:667–71. doi: 10.1089/thy.2015.0537
22. Cantara S, Marzocchi C, Castagna MG, Pacini F. HABP2 G534E variation in familial non-medullary thyroid cancer: an Italian series. J Endocrinol Invest (2017) 40:557–60. doi: 10.1007/s40618-016-0583-9
23. de Mello LEB, Araujo AN, Alves CX, de Paiva FJP, Brandão-Neto J, Cerutti JM. The G534E variant in HABP2 is not associated with increased risk of familial nonmedullary thyroid cancer in Brazilian Kindreds. Clin Endocrinol (Oxf) (2017) 87:113–4. doi: 10.1111/cen.13352
24. Tomsic J, Fultz R, Liyanarachchi S, He H, Senter L, de la Chapelle A. HABP2 G534E variant in papillary thyroid carcinoma. PLoS One (2016) 11:e0146315. doi: 10.1371/journal.pone.0146315
25. Ruiz-Ferrer M, Fernández RM, Navarro E, Antiñolo G, Borrego S. G534E variant in HABP2 and nonmedullary thyroid cancer. Thyroid (2016) 26:987–8. doi: 10.1089/thy.2016.0193
26. Weeks AL, Wilson SG, Ward L, Goldblatt J, Hui J, Walsh JP. HABP2 germline variants are uncommon in familial nonmedullary thyroid cancer. BMC Med Genet (2016) 17:60. doi: 10.1186/s12881-016-0323-1
27. Colombo C, Muzza M, Proverbio MC, Ercoli G, Perrino M, Cirello V, et al. Segregation and expression analyses of hyaluronan-binding protein 2 (HABP2): insights from a large series of familial non-medullary thyroid cancers and literature review. Clin Endocrinol (Oxf) (2017) 86:837–44. doi: 10.1111/cen.13316
28. Sahasrabudhe R, Stultz J, Williamson J, Lott P, Estrada A, Bohorquez M, et al. The HABP2 G534E variant is an unlikely cause of familial non-medullary thyroid cancer. J Clin Endocrinol Metab (2016) 10:1098–103. doi: 10.1210/jc.2015-3928
29. Bohórquez ME, Estrada AP, Stultz J, Sahasrabudhe R, Williamson J, Lott P, et al. The HABP2 G534E polymorphism does not increase nonmedullary thyroid cancer risk in Hispanics. Endocr Connect (2016) 5:123–7. doi: 10.1530/EC-16-0017
30. Kowalik A, Gąsior-Perczak D, Gromek M, Siołek M, Walczyk A, Pałyga I, et al. The p.G534E variant of HABP2 is not associated with sporadic papillary thyroid carcinoma in a Polish population. Oncotarget (2017) 8:58304–8. doi: 10.18632/oncotarget.16870
31. Kern B, Coppin L, Romanet P, Crépin M, Szuster I, Renaud F, et al. Multiple HABP2 variants in familial papillary thyroid carcinoma: Contribution of a group of “thyroid-checked” controls. Eur J Med Genet (2017) 60:178–84. doi: 10.1016/j.ejmg.2017.01.001
32. Colombo C, Fugazzola L, Muzza M, Proverbio MC, Cirello V. Letter regarding the article: “Multiple HABP2 variants in familial papillary thyroid carcinoma: Contribution of a group of “thyroid-checked” controls” by Kern et al. Eur J Med Genet (2018) 61:104–5. doi: 10.1016/j.ejmg.2017.07.012
33. Cirello V, Colombo C, Persani L, Fugazzola L. Absence of the MAP2K5 germline variants c.G961A and c.T1100C in a wide series of familial nonmedullary thyroid carcinoma Italian families. Int J Cancer (2019) 145:600. doi: 10.1002/ijc.32244
34. Bann DV, Jin Q, Sheldon KE, Houser KR, Nguyen L, Warrick JI, et al. Genetic variants implicate dual oxidase-2 in familial and sporadic nonmedullary thyroid cancer. Cancer Res (2019) 79:5490–9. doi: 10.1158/0008-5472.CAN-19-0721
35. Muzza M, Fugazzola L. Disorders of H2O2 generation. Best Pract Res Clin Endocrinol Metab (2017) 31:225–40. doi: 10.1016/j.beem.2017.04.006
36. Amin MB, Edge S, Greene F, Byrd DR, Brookland RK, Washington MK, et al. AJCC Cancer Staging Manual. 8th. New York: Springer (2017).
37. Haugen BR, Alexander EK, Bible KC, Doherty GM, Mandel SJ, Nikiforov YE, et al. 2015 American Thyroid Association management guidelines for adult patients with thyroid nodules and differentiated thyroid cancer: The American Thyroid Association guidelines task force on thyroid nodules and differentiated thyroid cancer. Thyroid (2016) 26:1–133. doi: 10.1089/thy.2015.0020
38. Pacini F, Basolo F, Bellantone R, Boni G, Cannizzaro MA, De Palma M, et al. Italian consensus on diagnosis and treatment of differentiated thyroid cancer: joint statements of six Italian societies. J Endocrinol Invest (2018) 41:849–76. doi: 10.1007/s40618-018-0884-2
39. Muzza M, Rabbiosi S, Vigone MC, Zamproni I, Cirello V, Maffini MA, et al. The clinical and molecular characterization of patients with dyshormonogenic congenital hypothyroidism reveals specific diagnostic clues for DUOX2 defects. J Clin Endocrinol Metab (2014) 99:E544–53. doi: 10.1210/jc.2013-3618
40. Pesenti C, Muzza M, Colombo C, Proverbio MC, Farè C, Ferrero S, et al. MassARRAY-based simultaneous detection of hotspot somatic mutations and recurrent fusion genes in papillary thyroid carcinoma: the PTC-MA assay. Endocrine (2018) 61:36–41. doi: 10.1007/s12020-017-1483-2
41. Colombo C, Muzza M, Proverbio MC, Tosi D, Soranna D, Pesenti C, et al. Impact of mutation density and heterogeneity on papillary thyroid cancer clinical features and remission probability. Thyroid (2019) 29:237–51. doi: 10.1089/thy.2018.0339
42. Ameziane-El-Hassani R, Schlumberger M, Dupuy C. NADPH oxidases: new actors in thyroid cancer? Nat Rev Endocrinol (2016) 12:485–94. doi: 10.1038/nrendo.2016.64
43. Triponez F, Wong M, Sturgeon C, Caron N, Ginzinger DG, Segal MR, et al. Does familial non-medullary thyroid cancer adversely affect survival? World J Surg (2006) 30:787–93. doi: 10.1007/s00268-005-0398-x
44. Cavaco BM, Batista PF, Martins C, Banito A, do Rosário F, Limbert E, et al. Familial non-medullary thyroid carcinoma (FNMTC): analysis of fPTC/PRN, NMTC1, MNG1 and TCO susceptibility loci and identification of somatic BRAF and RAS mutations. Endocr Relat Cancer (2008) 15:207–15. doi: 10.1677/ERC-07-0214
Keywords: FNMTC, familial non-medullary thyroid cancer, DUOX2, familial thyroid cancer, outcome, HABP2, MAP2K5
Citation: Cirello V, Colombo C, Karapanou O, Pogliaghi G, Persani L and Fugazzola L (2021) Clinical and Genetic Features of a Large Monocentric Series of Familial Non-Medullary Thyroid Cancers. Front. Endocrinol. 11:589340. doi: 10.3389/fendo.2020.589340
Received: 30 July 2020; Accepted: 23 November 2020;
Published: 07 January 2021.
Edited by:
Efisio Puxeddu, University of Perugia, ItalyReviewed by:
Paolo Piero Limone, Hospital Mauritian Turin, ItalyMarco Centanni, Sapienza University of Rome, Italy
Copyright © 2021 Cirello, Colombo, Karapanou, Pogliaghi, Persani and Fugazzola. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carla Colombo, Y2FybGEuY29sb21ibzFAdW5pbWkuaXQ=