 Katsumi Iizuka
Katsumi Iizuka Ken Takao1
Ken Takao1 Daisuke Yabe
Daisuke Yabe
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 03 December 2020
Sec. Diabetes: Molecular Mechanisms
Volume 11 - 2020 | https://doi.org/10.3389/fendo.2020.587189
Carbohydrate response element-binding protein (ChREBP) plays an important role in the development of type 2 diabetes, dyslipidemia, and non-alcoholic fatty liver disease, as well as tumorigenesis. ChREBP is highly expressed in lipogenic organs, such as liver, intestine, and adipose tissue, in which it regulates the production of acetyl CoA from glucose by inducing Pklr and Acyl expression. It has recently been demonstrated that ChREBP plays a role in the conversion of gut microbiota-derived acetate to acetyl CoA by activating its target gene, Acss2, in the liver. ChREBP regulates fatty acid synthesis, elongation, and desaturation by inducing Acc1 and Fasn, elongation of long-chain fatty acids family member 6 (encoded by Elovl6), and Scd1 expression, respectively. ChREBP also regulates the formation of very low-density lipoprotein by inducing the expression of Mtp. Furthermore, it plays a crucial role in peripheral lipid metabolism by inducing Fgf21 expression, as well as that of Angptl3 and Angptl8, which are known to reduce peripheral lipoprotein lipase activity. In addition, ChREBP is involved in the production of palmitic-acid-5-hydroxystearic-acid, which increases insulin sensitivity in adipose tissue. Curiously, ChREBP is indirectly involved in fatty acid β-oxidation and subsequent ketogenesis. Thus, ChREBP regulates whole-body lipid metabolism by controlling the transcription of lipogenic enzymes and liver-derived cytokines.
Excess carbohydrate intake causes hepatic triglyceride accumulation through the activation of carbohydrate response element binding protein (ChREBP) and de novo lipogenesis. Dietary carbohydrate is metabolized to acetyl CoA, which is a key intermediate in lipid metabolism (1). Acetyl CoA is produced by the oxidation of pyruvate, the end product of glycolysis, through the action of pyruvate dehydrogenase (PDH) and fatty acid β-oxidation, for use as a substrate in triglyceride and cholesterol synthesis, as well as ketogenesis and protein acetylation (1). In the fed state, the resulting high nucleocytosolic acetyl CoA is readily utilized for lipid synthesis and histone acetylation (1, 2). In contrast, in the fasted state or under more extreme conditions, acetyl CoA is preferentially directed to the mitochondria to permit greater synthesis of adenosine triphosphate (ATP) and ketone bodies (1, 2). Acetyl CoA is produced in the liver from glucose and fructose, which are rapidly converted to glyceraldehyde-3 phosphate (GAP) by glycolysis and fructolysis (Figure 1). GAP is then converted to pyruvate by several glycolytic enzymes, including liver-type pyruvate kinase (encoded by Pklr), which is activated by ChREBP and converts phosphoenolpyruvate to pyruvate (1). Pyruvate is then converted to acetyl CoA by the PDH complex in the mitochondria. When acetyl CoA enters the tricarboxylic acid cycle, there is an increase in the production of citrate, which is exported from the mitochondria and converted to acetyl CoA by ATP citrate lyase (encoded by Acly) (1, 2). Cytosolic acetyl CoA is converted into long-chain fatty acyl CoA by lipogenic enzymes such as acetyl CoA carboxylase 1 (encoded by Acc1) and fatty acid synthase (encoded by Fasn).
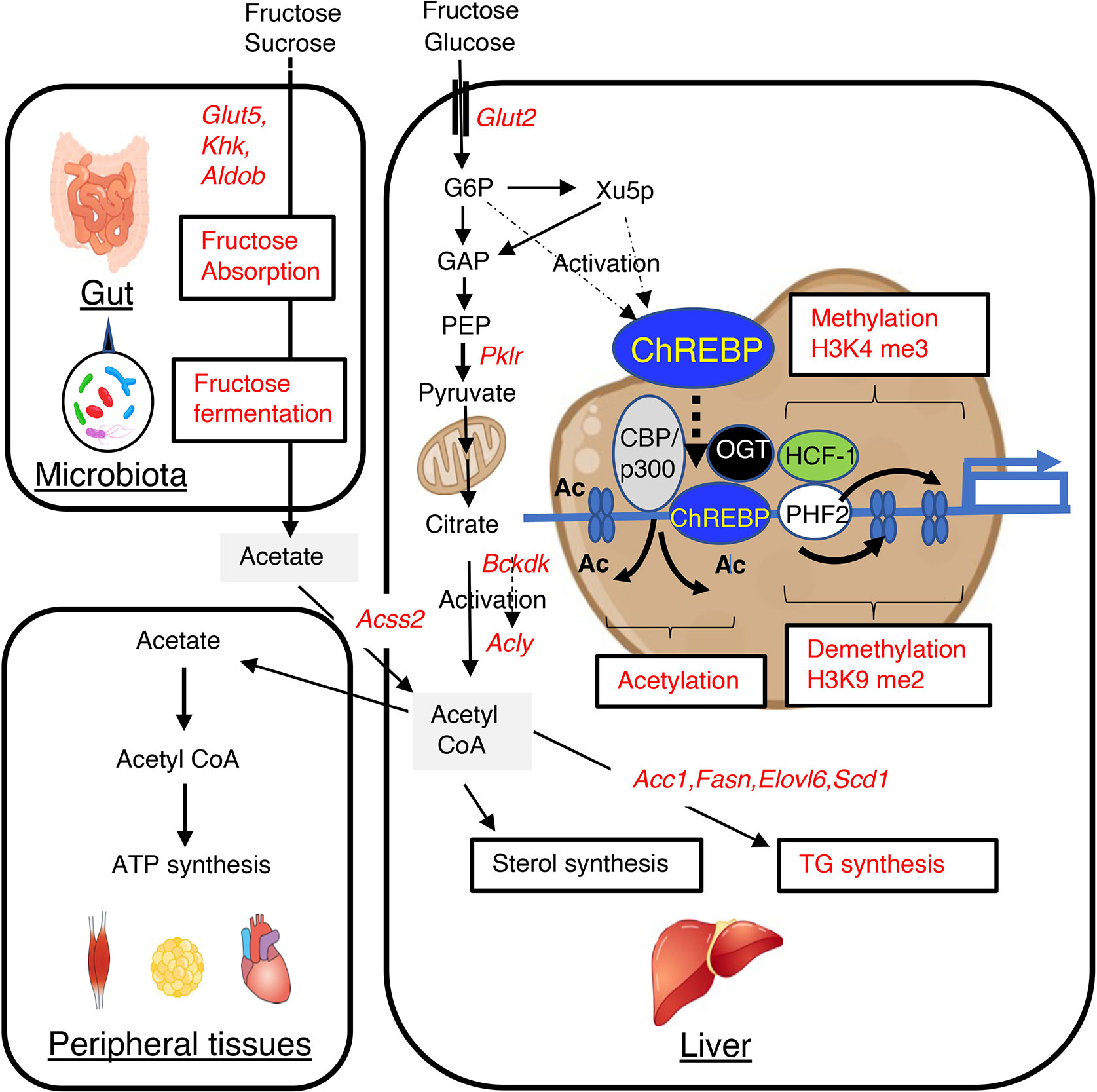
Figure 1 High-fructose/high-sucrose diet-feeding causes an increase in de novo lipogenesis through the regulation of lipogenic gene expression and gut microbiota-derived acetate utilization. Fructose is normally absorbed and converted to glucose and lactate. Glucose, lactate, and a small amount of fructose enter the portal vein. In the liver, glucose and fructose activate ChREBP transcriptional activity through increases in the concentrations of glucose and fructose-derived metabolites, such as xylulose-5-phosphate and glucose-6-phosphate, and this results in greater expression of lipogenic genes, such as Acc1, Fasn, Elovl6, and Scd1. This higher expression causes the metabolism of glucose and fructose to generate acetyl CoA and fatty acyl CoA in the liver. Fructose that is not absorbed in the small intestine is absorbed in the colon and enters the portal vein. In the liver, acetate is converted to acetyl CoA by acyl-coenzyme A synthetase short-chain family member 2 (encoded by Acss2), and this is used for fatty acyl CoA synthesis, sterol synthesis, and histone acetylation. ChREBP transcription activity is also regulated by acetyl CoA and uridine diphosphate-N-acetylglucosamine, through acetylation and O-GlcNAcylation, respectively. These substances are involved in epigenic regulation, such as histone acetylation and histone methylation. GLUT5, glucose transporter 5; KHK, ketohexokinase; ALDOB, aldolase B; GLUT2, glucose transporter 2; PKLR, liver-type pyruvate kinase; ACSS2, acyl-coenzyme A synthetase short-chain family member 2; G6P, glucose 6-phosphate; Xu5P, xylulose 5-phosphate; ACLY, ATP citrate lyase; ACC1, acetyl CoA carboxylase; FASN, fatty acid synthase; ELOVL6, fatty acid elongase 6; SCD1, stearyl CoA desaturase; BCKDK, branched-chain ketoacid dehydrogenase kinase; CBP, CREB binding protein; OGT, O-linked N-acetylglucosamine (GlcNAc) transferase; HCF-1, host cell factor-1; PHF2, plant homeodomain finger 2; H3K4me3, trimethylated H3K4; H3K9me2, dimethylated H3K9.
Carbohydrate response element-binding protein (ChREBP) plays a pivotal role in the pathogenesis of metabolic diseases and tumors (3–6). ChREBP was initially identified as a glucose-activated transcription factor that binds to the carbohydrate response element (ChoRE) in the promoter of Pklr (6). ChoREs are also found in the promoters of lipogenic genes, including Acc1 and Fasn (7, 8), and ChREBP, together with its heterodimerization partner Max-like factor X, controls de novo lipogenesis in the liver by upregulating the expression of lipogenic genes (9–12). Studies that used DNA microarray, chromosome immunoprecipitation-sequencing (ChIP-seq) and Chrebp knockout mice have confirmed a critical role for ChREBP in hepatic glucose and lipid metabolism (13–16). Interestingly, ChREBP also modulates lipolysis in adipose tissue by directly regulating the expression of Fgf21 and Angptl genes (14, 17–23). Furthermore, it regulates the hepatic and intestinal metabolism of fructose, which is tightly linked to lipogenesis and the pathogenesis of the metabolic syndrome (24–28). In addition, ChREBP plays a critical role in the conversion of gut microbiota-derived acetate, the production of which is increased by excess dietary fructose intake, to acetyl coenzyme A (CoA) by activating its target, Acss2, in the liver, which contributes to hepatic triglyceride accumulation (29). ChREBP also modulates hepatic β-oxidation and ketogenesis (30–32). This review focuses on recent advances in knowledge of the ChREBP-mediated regulation of lipid metabolism in the liver, adipose tissue, and gut.
As described in the Introduction, ACLY is an important mediator of the supply of acetyl CoA from glycolysis and fatty acid oxidation. Because the ACLY-mediated conversion of citrate to acetyl CoA is critical for the synthesis of triglycerides and sterols, efforts have been made to develop ACLY inhibitors for the treatment of hypertriglyceridemia and hypercholesterolemia (2, 3). Acly is expressed mainly in the liver and white adipose tissue and its expression is upregulated at the transcriptional level by glucose-activated ChREBP and the insulin-induced transcription factor sterol response element-binding protein-1c (SREBP-1c) (13, 14, 33, 34). The enzymatic activity of ACLY is post-translationally regulated by the phosphatidylinositol 3-kinase (PI3K)/Akt pathway and acetylation (35, 36). In addition, recent studies have demonstrated that ACLY is activated by branched-chain ketoacid dehydrogenase kinase (BDK) and mitochondrial protein phosphatase 1K (PPM1K), which are respectively positively and negatively regulated by ChREBP (37–39) (Figure 1). Furthermore, the branched-chain α-ketoacid dehydrogenase complex (BCKDH) is an enzyme that catalyzes the commitment step of branched-chain amino acid (BCAA) catabolism and is negatively and positively regulated by BDK and PPM1K, respectively. Thus, ChREBP regulates the use of BCAAs in triglyceride synthesis by regulating BDK/PPM1K (39). Therefore, ChREBP might contribute to the development of BCAA-induced insulin resistance (39).
Recent studies have revealed that gut microbiota-derived acetate also represents an important source of acetyl CoA for hepatic lipogenesis (29). Dietary fructose is absorbed and metabolized to glucose in the intestine, whereas unabsorbed fructose is fermented by the gut microbiota to produce acetate (26, 27, 29, 40–42). Glucose induces the expression of both ChREBP-β and SREBP-1c, whereas fructose only induces the expression of SREBP-1c in the liver (28). Our group and others have recently reported that ChREBP regulates intestinal fructose absorption by inducing the expression of Glut5, Khk, and Aldob in the intestine and that Chrebp knockout mice consuming a sucrose-based diet show an irritable bowel syndrome-like phenotype, which develops because of fructose malabsorption and an increase in the numbers of acetate-producing bacteria in the intestine (26, 27). These findings suggest that the inhibition of intestinal ChREBP increases microbial acetate production by increasing the entry of unabsorbed fructose into the colon. Gut microbiota-derived acetate is absorbed and reaches the liver via the portal vein. Acetate is then converted to acetyl CoA by acyl-CoA synthetase short-chain family member, which is encoded by Acss2, and used as a substrate for lipogenesis (43, 44). Acss2 is highly expressed in the kidney and liver and is present in both the cytosol and nucleus (45, 46). Importantly, depletion of the gut microbiota using antibiotics suppresses acetate production in the colon, and subsequent acetyl CoA synthesis and de novo lipogenesis in the liver; and the knock-down of Acss2 mRNA using siRNA also reduces the hepatic production of acetyl CoA (29). In addition, it has been shown that SREBP-1c, a critical regulator of hepatic lipogenesis that is transcriptionally activated by insulin, induces Acss2 mRNA expression (47–49). ChREBP also induces Acss2 expression (Figure 1) (14, 29, 33). Thus, inhibition of hepatic ChREBP is likely to prevent fructose-induced triglyceride accumulation by suppressing lipogenic gene expression and hepatic acetyl CoA production from gut microbial acetate, while the inhibition of intestinal ChREBP may increase acetate production in the intestine (27).
Acetyl CoA is converted to fatty acids through fatty acid synthesis, elongation, and desaturation; and the resulting fatty acids are then esterified with glycerol before being packaged into very-low-density lipoprotein (VLDL) particles to be delivered to the periphery (50). These processes are transcriptionally regulated by ChREBP and SREBP-1c (51) (Table 1). In fatty acid synthesis, both ChREBP and SREBP-1c induce Acc, which encodes an enzyme that is required for the conversion of acetyl CoA to malonyl CoA, and Fasn, which encodes an enzyme that is required for the production of palmitoyl CoA from malonyl CoA and acetyl CoA (9, 10, 13, 34, 51). In fatty acid elongation and desaturation, ChREBP induces the expression of the stearoyl CoA desaturase-1 gene (Scd-1) and Elovl6, both of which are required for the synthesis of monounsaturated fatty acids (MUFAs), while SREBP-1c induces Elovl2, fatty acid desaturase 1, and fatty acid desaturase 2, which are required for the synthesis of polyunsaturated fatty acids (PUFAs), as well as Scd-1 and Elovl6 (Figure 2) (52–56). Consistent with this, hepatic Chrebp overexpression reduces the concentrations of saturated fatty acids (SFAs), such as palmitic acid (C16:0) and stearic acid (C18:0); and increases that of the MUFA oleic acid (C18:1 n-9) (32, 57). Furthermore, hepatic SREBP-1c overexpression reduces the concentration of stearic acid (C18:0) and increases that of oleic acid (C18:1 n-9) (58). Importantly, PUFAs suppress the transcriptional activities of both ChREBP and SREBP-1c (59, 60). Moreover, PUFAs also reduce the cleavage of SREBP-1c at the post-transcriptional level (61). Thus, both ChREBP and SREBP-1c regulate de novo lipogenesis, elongation, and desaturation.
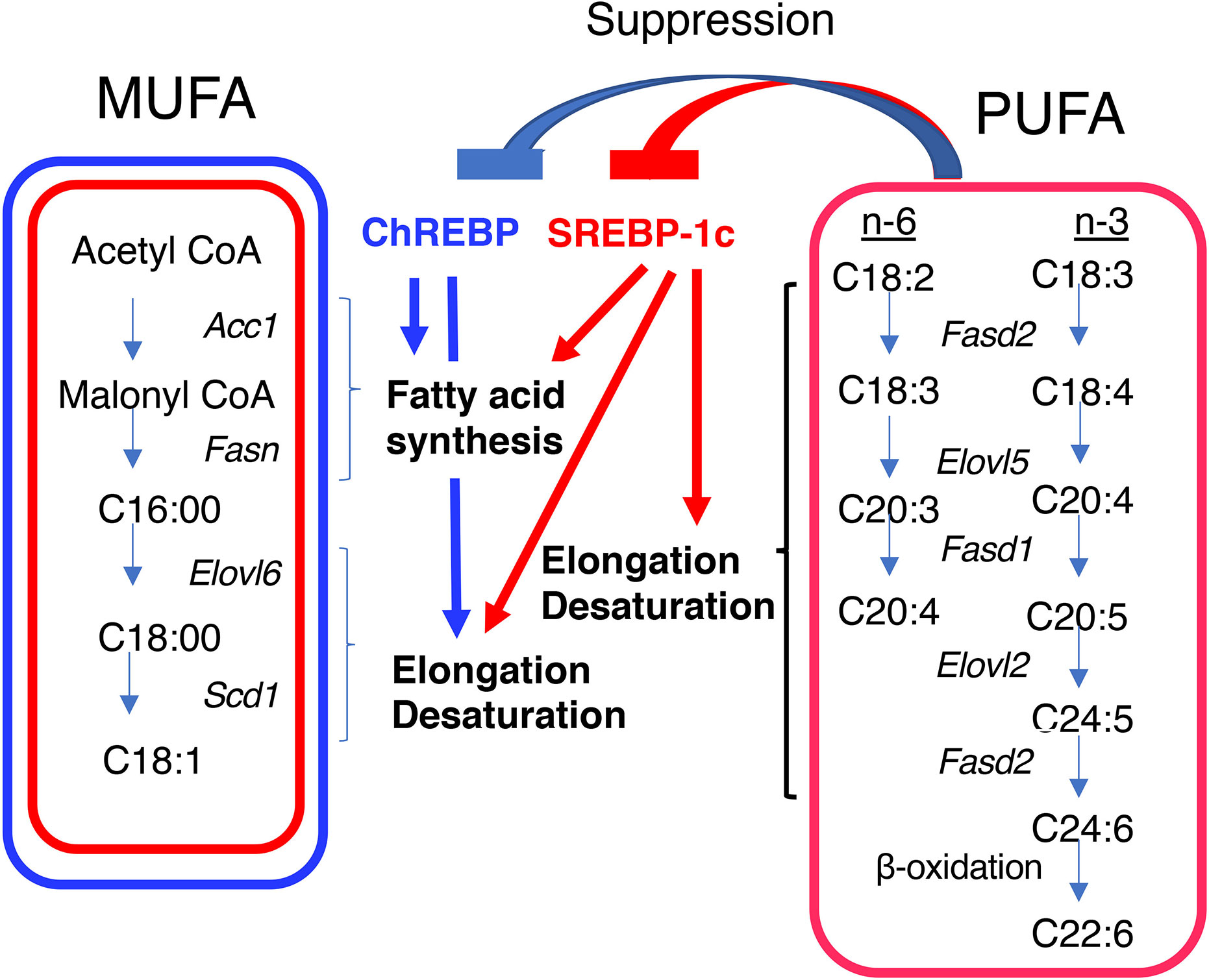
Figure 2 Differential regulation of monounsaturated fatty acid (MUFA) and polyunsaturated fatty acid (PUFA) synthesis by ChREBP and SREBP-1c. Both ChREBP and SREBP-1c regulate MUFA synthesis by regulating the expression of genes involved in fatty acid synthesis (e.g., Acc1 and Fasn), elongation (e.g., Elovl6), and desaturation (e.g., Scd1); whereas SREBP-1c, but not ChREBP, regulates PUFA synthesis by regulating the expression of genes involved in elongation (e.g., Elovl2) and desaturation [e.g., fatty acid desaturase 1 (Fasd1) and fatty acid desaturase 2 (Fasd2)]. PUFAs suppress the transcriptional activities of ChREBP and SREBP-1c. ACC1, acetyl CoA carboxylase; FASN, fatty acid synthase; ELOVL6, fatty acid elongase 6; SCD1, stearyl CoA desaturase; ELOVL5, fatty acid elongase 5; FASD1, fatty acid desaturase 1; FASD2, fatty acid desaturase 2.
The transcriptional activity of ChREBP is activated by glucose-derived metabolites such as glucose-6-phosphate, xylulose-5-phophate, and fructose-2,6-bisphosphate via regulation of its nucleocytoplasmic translocation (62–65). Moreover, ChREBP transcription activity is also regulated by acetyl CoA and uridine diphosphate-N-acetylglucosamine, through acetylation and O-GlcNAcylation, respectively (66–73). These substances are involved in epigenic regulation, such as histone acetylation and histone methylation (Figure 1).
During weaning, Fasn mRNA expression is increased by greater binding of ChREBP and SREBP-1c to Fasn promoter and greater histone H3 and H4 acetylation (70). Moreover, ChREBP induces Fasn mRNA expression and triglyceride synthesis by facilitating H3 and H4 acetylation, but these effects are prevented by the inhibition of histone acetylation using garcinol, a histone acetyltransferase (HAT) inhibitor (71). The role of the histone deacetylase activity of CREB-binding protein (CBP)/p300 in the mechanism of ChREBP-induced histone acetylation is well known (72). In human hepatocytes, the binding of farnesoid X receptor (FXR) to the ChREBP- Hepatocyte Nuclear Factor 4α (HNF4α) complex triggers the release of ChREBP from CBP/p300, leading to the recruitment of the histone deacetylase, silencing mediator of retinoic acid and thyroid hormone receptor (SMRT), to the Lpk promoter, where it acts as a co-repressor of ChREBP transcriptional activity (73). Interestingly, CBP/p300 HAT acetylates ChREBP, which promotes transactivation by this molecule (72). Therefore, CBP/p300 regulates ChREBP transcriptional activity through the acetylation of substrates such as histones and ChREBP (Figure 1).
Glucose activates the hexosamine pathway and thereby O-linked N-acetylglucosamine transferase (OGT)-mediated ChREBP O-GlcNacylation, which enhances ChREBP DNA-binding and protein stability by reducing ubiquitin-mediated degradation (66–68). Recently, host cell factor-1 (HCF-1) has been identified as a ChREBP-interacting protein (74). Glucose stimulates HCF-1 O-GlcNAcylation and cleavage, and interacts with ChREBP, thereby augmenting ChREBP O-GlcNAcylation and the recruitment of OGT to ChREBP (74). HCF-1 also augments the trimethylation of H3K4, which promotes the recruitment of the histone demethylase, PHD Finger Protein 2 (PHF2), resulting in greater transcriptional activity (74). PHF2 is also known to be a coactivator of ChREBP, and the ChREBP-PHF2 interaction causes the expression of Scd1 and MUFA synthesis through H3K9me2 demethylation (75). H3K4 methylation and H3K9me2 contribute to transcriptional activation and repression, respectively. Thus, ChREBP and its co-factors, such as HCF-1, PHF2, and OGT, also regulate lipogenic gene expression through epigenetic modifications (Figure 1).
It has been suggested that ChREBP may regulate lipoprotein metabolism, because the plasma concentrations of cholesterol, triglyceride, and very low density lipoprotein (VLDL)-triglyceride, as well as the number of VLDL particles, are lower in Chrebp knockout mice (13, 31, 58, 76). Our detailed analysis demonstrated that the mRNA and protein expression of hepatic microsomal triglyceride transfer protein (MTP) is lower in these mice (58, 76). MTP catalyzes the rate-limiting step in the production of apoB-containing VLDL, and therefore plays an important role in VLDL secretion (77). Furthermore, the consumption of a high fat/high sucrose diet is associated with lower VLDL secretion in Chrebp knockout mice than in WT mice, which is consistent with the effect of the genetic manipulation on hepatic MTTP expression (76). Thus, the lower MTP expression, together with the suppression of de novo lipogenesis, in the livers of Chrebp knockout mice may contribute to the lower hepatic VLDL secretion and plasma lipid concentrations (58, 76). Interestingly, hepatic Chrebp overexpression is also associated with lower plasma triglyceride concentrations, but a different mechanism is involved (32). Hepatic Chrebp overexpression is associated with higher hepatic triglyceride content but lower plasma triglyceride concentrations (32). The hepatic Mtp mRNA expression of mice with hepatic Chrebp overexpression is similar to that of control mice (32). Because hepatic Chrebp overexpression reduces the concentrations of VLDL-triglycerides and low-density lipoprotein-triglycerides (32), it has been suggested that lipoprotein lipase (LPL) might be activated by hepatic Chrebp overexpression. Consistent with this, we found that hepatic Chrebp overexpression increased hepatic Fgf21 mRNA expression and the plasma FGF21 concentration (20, 32, 57). We also found that hepatic Chrebp overexpression was associated with lower hepatic mRNA and protein expression of ANGPTL3 (32). Because LPL is activated by FGF21 and suppressed by the ANGPTL3/ANGPTL8 complex, it is conceivable that high FGF21 and low ANGPTL3 might activate peripheral lipoprotein metabolism by increasing LPL activity, and thereby reducing plasma triglyceride concentration (32). Thus, ChREBP controls lipoprotein metabolism through the regulation of de novo lipogenesis, VLDL secretion, and the secretion of hepatokines that influence peripheral lipoprotein metabolism (Figure 3).
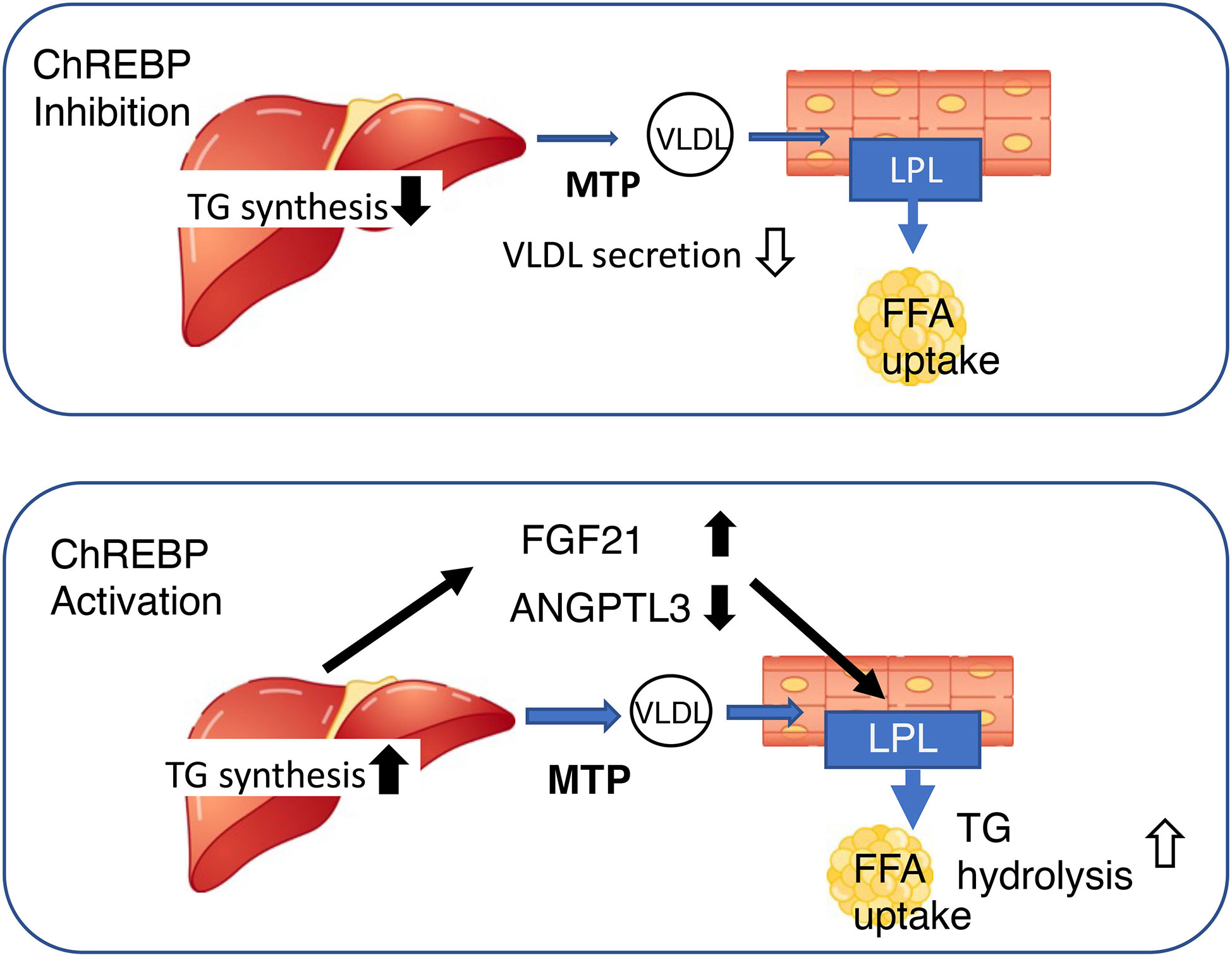
Figure 3 Both the activation and inhibition of ChREBP reduce blood triglyceride concentration, but through differing mechanisms. Upper, ChREBP inhibition reduces de novo lipogenesis and very-low density lipoprotein (VLDL) formation, resulting in lower circulating triglyceride concentration. Lower, ChREBP activation increases hepatic triglyceride synthesis, increases Fgf21 expression, and reduces Angptl3 expression. High FGF21 and low ANGPTL3 concentrations increase lipoprotein lipase activity in adipose tissue, thereby reducing the circulating triglyceride concentration. FGF21, fibroblast growth factor 21; MTTP, microsomal triglyceride transfer protein; LPL, lipoprotein lipase; TG, triglyceride; FFA, free fatty acid; ANGPTL3, angiopoietin-like 3.
Fatty acid esters of hydroxyfatty acids (FAHFAs) are a recently discovered class of endogenous lipids that have anti-diabetic and anti-inflammatory properties (78–83). FAHFAs, and especially palmitic-acid-hydroxy-stearic-acid (PAHSA), have been found to be present in much higher concentrations in mice that overexpress GLUT4 in adipose tissue, which is a major site of FAHFA synthesis (78, 79). PAHSA is hydrolyzed by carboxyl ester lipase, mutations of which are known to cause maturity-onset diabetes of the young type 8 (80). In humans, FAHFAs can be found in serum, breast milk, meconium, and adipose tissue; and the serum PHASA concentration correlates with insulin sensitivity (81). It has been shown that PAHSA activates G-protein coupled receptor 120 (GPR120) to increase insulin-stimulated glucose uptake by adipocytes and glucagon-like-peptide-1 (GLP-1) secretion in the intestine (82). In addition, PAHSA activates G-protein coupled receptor 40 (GPR40) to increase glucose-induced insulin secretion (82) (Figure 4). PAHSA reduces adipose tissue inflammation and potentiates the insulin-induced suppression of hepatic glucose production (78, 79, 83). Whole-body Chrebp knockout mice have lower PAHSA concentrations in their adipose depots (78). Recently, adipose tissue-specific Chrebp knockout mice were also shown to have lower PAHSA concentrations in their serum and adipose tissue (83). Although the relationship between carboxyl ester lipase and ChREBP remain unclear, ChREBP regulates de novo lipogenesis in adipose tissue. Therefore, ChREBP may regulate PAHSA concentration via effects on biosynthetic and/or degradative pathways in adipose tissue. because a reduction in adipose Chrebp expression causes insulin resistance, the administration of PAHSA might be a means of ameliorating this defect, but further investigation is needed to clarify the mechanism involved.
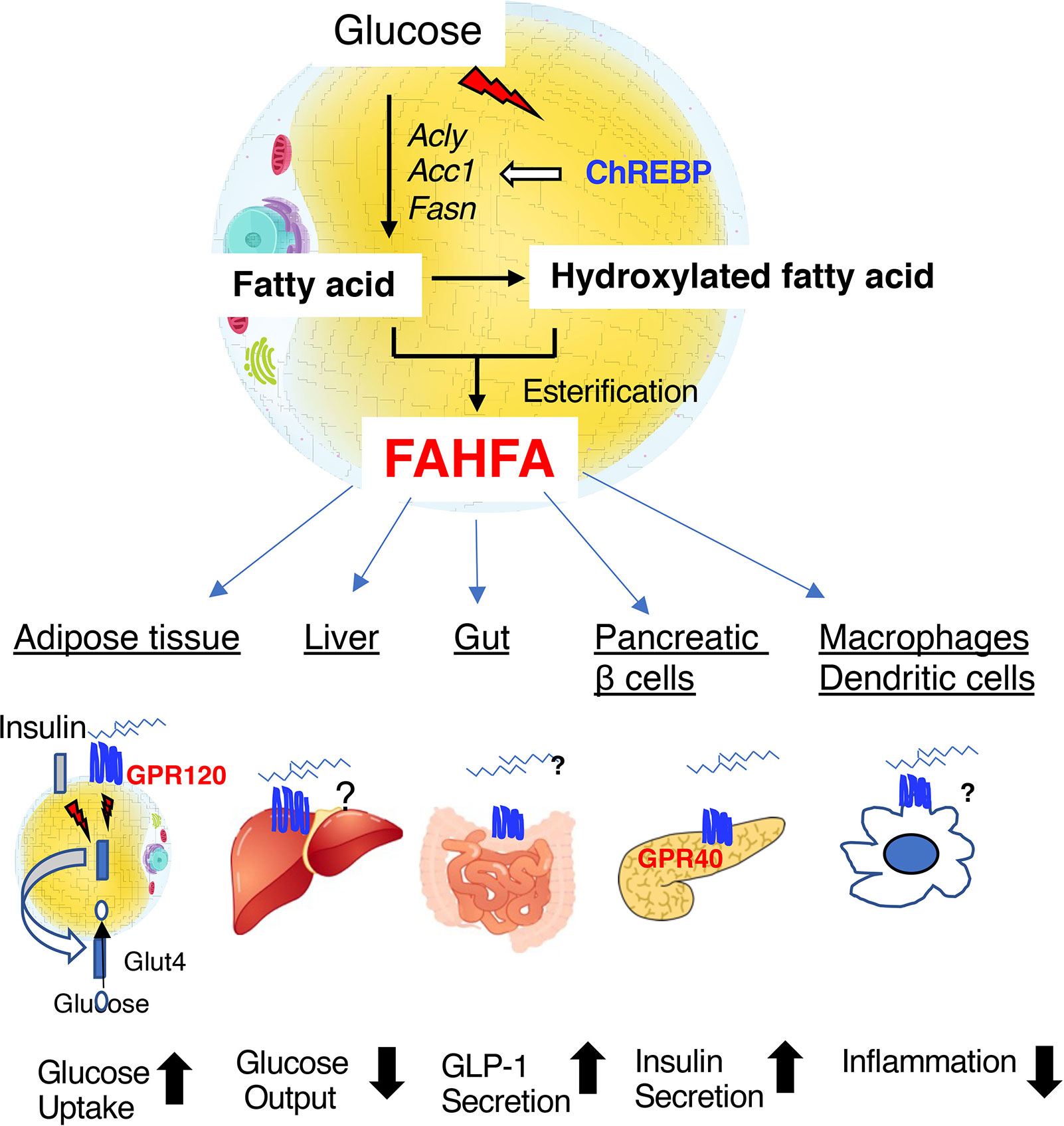
Figure 4 Fatty acid esters of hydroxyfatty acid (FAHFAs) are novel bioactive lipids that are produced in adipose tissue under the regulation of ChREBP. FAHFAs affect glucose uptake in adipose tissue, hepatic glucose output, GLP-1 secretion in the intestine, insulin secretion by pancreatic β-cells, and inflammation mediated by macrophages and dendritic cells by activating GPR120 (adipose tissue), GPR40 (pancreatic β-cells), and other unknown mediators. Abbreviations: GLP-1, glucagon-like peptide 1; Acly, ATP citrate lyase; Acc1, acetyl CoA carboxylase 1; Fasn, fatty acid synthase.
β-oxidation and ketogenesis are upregulated in the fasting state to produce ketone bodies that are used as an energy source in peripheral tissues, including brain, heart, and muscle. Through β-oxidation, fatty acyl CoA is converted to acetyl CoA; and the resulting acetyl CoA is then converted to 3-hydroxybutyric acid (OHBA) or acetoacetyl CoA through ketogenesis. Although ChREBP is activated by feeding, it is also involved in β-oxidation and ketogenesis in the fasting state (30, 32, 76), which was suggested by the observation that hepatic Chrebp overexpression reduces plasma OHBA concentration and increases free fatty acid concentrations (32). Hepatic Chrebp overexpression causes an increase in the expression of Acc2, which catalyzes the conversion of acetyl CoA to malonyl CoA. Malonyl CoA reduces the entry of acyl CoA into mitochondria by inhibiting carnitine palmitoyl transferase-1 (84). Hepatic Chrebp overexpression also reduces the expression of acyl CoA oxidase (58). Therefore, it has been suggested that ChREBP activation suppresses β-oxidation and ketogenesis. However, Chrebp knockout mice have paradoxically low plasma OHBA concentrations, compared to their FFA concentrations, which suggests that ChREBP inhibition also suppresses β-oxidation and ketogenesis (30, 31, 58, 76). Recently, it was reported that peroxisome proliferator-activated receptor-α (PPARα) is required for the ChREBP-mediated transcriptional activation of the Fgf21 gene (85). Although ChREBP and PPARα mRNA expression are reciprocally regulated in other tissues such brown adipose tissues (86), these results suggest that ChREBP may act in concert with PPARα to regulate gene transcription, even in the fasting state, to fine-tune β-oxidation and ketogenesis.
The last two decades of research have revealed critical roles of ChREBP in lipid metabolism. ChREBP regulates hepatic lipogenesis via acetyl-CoA produced by the conventional pathway (glycolysis and fructolysis) and a novel pathway (conversion of gut microbiota-derived acetate). It also regulates lipoprotein metabolism in the liver and PAHSA production in adipose tissue. Furthermore, ChREBP regulates β-oxidation and ketogenesis in concert with PPARα. However, despite remarkable progress in understanding of the roles of ChREBP in these processes, a number of questions remain regarding its relationship with lipid metabolism. First, to what extent does gut microbiota-derived acetate induce hepatic triglyceride accumulation? Although ChREBP regulates Acss2 mRNA, does acetate itself affect ChREBP transcriptional activity? Second, the role of ChREBP in cholesterol metabolism remains to be determined. Chrebp knockout mice have lower plasma concentrations of cholesterol, despite normal cholesterol synthesis in their livers, and the low plasma cholesterol concentrations may affect physiological processes, such as steroidogenesis in the adrenal gland. Third, the physiological and pathophysiological roles of ChREBP in the fasting state require more in-depth investigation. Because ChREBP is normally activated by glucose, it is conceivable that it regulates post-prandial lipid metabolism. However, it remains unclear why the ability of ChREBP to regulate β-oxidation and ketogenesis evolved. Finally, it remains unclear whether ChREBP suppression modifies the microbiota and lipid metabolism in humans. As discussed above, Chrebp knockout mice have an altered gut microbiota due to impaired fructose absorption from the gut (25). Consistent with this, it has been demonstrated that metformin treatment modifies the gut microbiota (87). AMP-activated protein kinase (AMPK) is known to be a major target of metformin, and it also regulates the activity of several transcription factors, such as SREBP-1c, PGC-1, FOXO1, and CREB. In addition, both AMPK and metformin reduce the transcriptional activity of ChREBP. Therefore, the effects of metformin on the microbiota might be mediated partly through ChREBP (88–90). Genome-wide scanning has also identified a human ChREBP homologue, MLXIPL, genetic variants of which are associated with plasma triglyceride concentration (91). Thus, it would be interesting to determine the effects of certain dietary habits (e.g., high fructose consumption) on the gut microbiota of individuals with MLXIPL variants (92). Further investigation is necessary to clarify the various physiological and pathophysiological roles of ChREBP in lipid metabolism.
All the authors (KI, KT, and DY) have made a substantial, direct intellectual contribution to the work, and approved it for publication.
This work was supported by grants from Japan Society for the Promotion of Sciences (JSPS) [KAKENHI Grant 17K00850 and 20K11645 (to K. I.) and 17K09825 (to D.Y.)].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thanks Professor Kosaku Uyeda (University of Texas) for their guidance and interest. The authors also thank the former and present members of the Department of Diabetes and Endocrinology, Gifu University Graduate School of Medicine, especially J. Takeda, Y. Horikawa, Y. Liu, W. Wu, and T. Kato, for helpful discussions. The authors also thank H. Tsuchida for technical assistance and M. Yato, Y. Ogiso, and M. Nozu for their secretarial assistance. Finally, we thank Mark Cleasby, PhD, from Edanz Group (https://en-author-services.edanzgroup.com/ac) for editing a draft of this manuscript.
1. Shi L, Tu BP. Acetyl-CoA and the regulation of metabolism: mechanisms and consequences. Curr Opin Cell Biol (2015) 33:125–31. doi: 10.1016/j.ceb.2015.02.003
2. Feng X, Zhang L, Xu S, Shen AZ. ATP-citrate lyase (ACLY) in lipid metabolism and atherosclerosis: An updated review. Prog Lipid Res (2020) 77:101006. doi: 10.1016/j.plipres.2019.101006
3. Uyeda K, Repa JJ. Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab (2006) 4:107–10. doi: 10.1016/j.cmet.2006.06.008
4. Iizuka K. The transcription factor carbohydrate-response element-binding protein (ChREBP): A possible link between metabolic disease and cancer. Biochim Biophys Acta Mol Basis Dis (2017) 1863:474–85. doi: 10.1016/j.bbadis.2016.11.029
5. Ortega-Prieto P, Postic C. Carbohydrate Sensing Through the Transcription Factor ChREBP. Front Genet (2019) 10:472. doi: 10.3389/fgene.2019.00472
6. Yamashita H, Takenoshita M, Sakurai M, Bruick RK, Henzel WJ, Shillinglaw W, et al. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc Natl Acad Sci U S A (2001) 98:9116–21. doi: 10.1073/pnas.161284298
7. Towle HC, Kaytor EN, Shih HM. Regulation of the expression of lipogenic enzyme genes by carbohydrate. Annu Rev Nutr (1997) 17:405–33. doi: 10.1146/annurev.nutr.17.1.405
8. Girard J, Ferré P, Foufelle F. Mechanisms by which carbohydrates regulate expression of genes for glycolytic and lipogenic enzymes. Annu Rev Nutr (1997) 17:325–52. doi: 10.1146/annurev.nutr.17.1.325
9. Ishii S, Iizuka K, Miller BC, Uyeda K. Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription. Proc Natl Acad Sci U S A (2004) 101:15597–602. doi: 10.1073/pnas.0405238101
10. Stoeckman AK, Ma L, Towle HC. Mlx is the functional heteromeric partner of the carbohydrate response element-binding protein in glucose regulation of lipogenic enzyme genes. J Biol Chem (2004) 279:15662–9. doi: 10.1074/jbc.M311301200
11. Cairo S, Merla G, Urbinati F, Ballabio A, Reymond A. WBSCR14, a gene mapping to the Williams–Beuren syndrome deleted region, is a new member of the Mlx transcription factor network. Hum Mol Genet (2001) 10:617–27. doi: 10.1093/hmg/10.6.617
12. Iizuka K, Takeda J, Horikawa Y. Hepatic overexpression of dominant negative Mlx improves metabolic profile in diabetes-prone C57BL/6J mice. Biochem Biophys Res Commun (2009) 379:499–504. doi: 10.1016/j.bbrc.2008.12.100
13. Iizuka K, Bruick RK, Liang G, Horton JD, Uyeda K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci U S A (2004) 101:7281–6. doi: 10.1073/pnas.0401516101
14. Ma L, Robinson LN, Towle HC. ChREBP*Mlx is the principal mediator of glucose-induced gene expression in the liver. J Biol Chem (2006) 281:28721–30. doi: 10.1074/jbc.M601576200
15. Poungvarin N, Chang B, Imamura M, Chen J, Moolsuwan K, Sae-Lee C, et al. Genome-Wide Analysis of ChREBP Binding Sites on Male Mouse Liver and White Adipose Chromatin. Endocrinology (2015) 156:1982–94. doi: 10.1210/en.2014-1666
16. Jeong YS, Kim D, Lee YS, Kim HJ, Han JY, Im SS, et al. Integrated expression profiling and genome-wide analysis of ChREBP targets reveals the dual role for ChREBP in glucose-regulated gene expression. PloS One (2011) 6:e22544. doi: 10.1016/j.msard.2012.01.001
17. Schlein C, Talukdar S, Heine M, Fischer AW, Krott LM, Nilsson SK, et al. FGF21 Lowers Plasma Triglycerides by Accelerating Lipoprotein Catabolism in White and Brown Adipose Tissues. Cell Metab (2016) 23:441–53. doi: 10.1016/j.cmet.2016.01.006
18. Kersten S. Angiopoietin-like 3 in lipoprotein metabolism. Nat Rev Endocrinol (2017) 13:731–9. doi: 10.1038/nrendo.2017.119
19. Zhang R. The ANGPTL3-4-8 model, a molecular mechanism for triglyceride trafficking. Open Biol (2016) 6:150272. doi: 10.1098/rsob.150272
20. Iizuka K, Takeda J, Horikawa Y. Glucose induces FGF21 mRNA expression through ChREBP activation in rat hepatocytes. FEBS Lett (2009) 583:2882–6. doi: 10.1016/j.febslet.2009.07.053
21. Fisher FM, Kim M, Doridot L, Cunniff JC, Parker TS, Levine DM, et al. A critical role for ChREBP-mediated FGF21 secretion in hepatic fructose metabolism. Mol Metab (2016) 6:14–21. doi: 10.1016/j.molmet.2016.11.008
22. Lundsgaard AM, Fritzen AM, Sjøberg KA, Myrmel LS, Madsen L, Wojtaszewski JFP, et al. Circulating FGF21 in humans is potently induced by short term overfeeding of carbohydrates. Mol Metab (2016) 6:22–9. doi: 10.1016/j.molmet.2016.11.001
23. Fu Z, Berhane F, Fite A, Seyoum B, Abou-Samra AB, Zhang R. Elevated circulating lipasin/betatrophin in human type 2 diabetes and obesity. Sci Rep (2014) 4:5013. doi: 10.1038/srep05013
24. Bantle JP. Dietary fructose and metabolic syndrome and diabetes. J Nutr (2009) 139:1263S–8S. doi: 10.3945/jn.108.098020
25. Iizuka K. The Role of Carbohydrate Response Element Binding Protein in Intestinal and Hepatic Fructose Metabolism. Nutrients (2017) 9:181. doi: 10.3390/nu9020181
26. Kim M, Astapova II, Flier SN, Hannou SA, Doridot L, Sargsyan A, et al. Intestinal, but not hepatic, ChREBP is required for fructose tolerance. JCI Insight (2017) 2:e96703. doi: 10.1172/jci.insight.96703
27. Kato T, Iizuka K, Takao K, Horikawa Y, Kitamura T, Takeda J. ChREBP-Knockout Mice Show Sucrose Intolerance and Fructose Malabsorption. Nutrients (2018) 10:340. doi: 10.3390/nu10030340
28. Softic S, Gupta MK, Wang GX, Fujisaka S, O’Neill BT, Rao TN, et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J Clin Invest (2017) 127:4059–74. doi: 10.1172/JCI94585
29. Zhao S, Jang C, Liu J, Uehara K, Gilbert M, Izzo L, et al. Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature (2020) 579:586–91. doi: 10.1038/s41586-020-2101-7
30. Burgess SC, Iizuka K, Jeoung NH, Harris RA, Kashiwaya Y, Veech RL, et al. Carbohydrate-response element-binding protein deletion alters substrate utilization producing an energy-deficient liver. J Biol Chem (2008) 283:1670–8. doi: 10.1074/jbc.M706540200
31. Iizuka K, Miller B, Uyeda K. Deficiency of carbohydrate-activated transcription factor ChREBP prevents obesity and improves plasma glucose control in leptin-deficient (ob/ob) mice. Am J Physiol Endocrinol Metab (2006) 291:E358–64. doi: 10.1152/ajpendo.00027.2006
32. Iizuka K, Takao K, Kato T, Horikawa Y, Takeda J. ChREBP Reciprocally Regulates Liver and Plasma Triacylglycerol Levels in Different Manners. Nutrients (2018) 10:1699. doi: 10.3390/nu10111699
33. Linden AG, Li S, Choi HY, Fang F, Fukasawa M, Uyeda K, et al. Interplay between ChREBP and SREBP-1c coordinates postprandial glycolysis and lipogenesis in livers of mice. J Lipid Res (2018) 59:475–87. doi: 10.1194/jlr.M081836
34. Liang G, Yang J, Horton JD, Hammer RE, Goldstein JL, Brown MS. Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J Biol Chem (2002) 277:9520–8. doi: 10.1074/jbc.M111421200
35. Berwick DC, Hers I, Heesom KJ, Moule SK, Tavare JM. The identification of ATP-citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. J Biol Chem (2002) 277:33895–900. doi: 10.1074/jbc.M204681200
36. Lin R, Tao R, Gao X, Li T, Zhou X, Guan KL, et al. Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth. Mol Cell (2013) 51:506–18. doi: 10.1016/j.molcel.2013.07.002
37. Brosnan JT, Brosnan ME. Branched-chain amino acids: enzyme and substrate regulation. J Nutr (2006) 136:207S–11S. doi: 10.1093/jn/136.1.207S
38. Biswas D, Duffley L, Pulinilkunnil T. Role of Branched-Chain Amino Acid-Catabolizing Enzymes in Intertissue Signaling, Metabolic Remodeling, and Energy Homeostasis. FASEB J (2019) 33:8711–31. doi: 10.1096/fj.201802842RR
39. White PJ, McGarrah RW, Grimsrud PA, Tso SC, Yang WH, Haldeman JM, et al. The BCKDH Kinase and Phosphatase Integrate BCAA and Lipid Metabolism via Regulation of ATP-Citrate Lyase. Cell Metab (2018) 27:1281–93.e7. doi: 10.1016/j.cmet.2018.04.015
40. Jang C, Hui S, Lu W, Cowan AJ, Morscher RJ, Lee G, et al. The Small Intestine Converts Dietary Fructose into Glucose and Organic Acids. Cell Metab (2018) 27:351–61.e3. doi: 10.1016/j.cmet.2017.12.016
41. Ferraris RP, Choe JY, Patel CR. Intestinal Absorption of Fructose. Annu Rev Nutr (2018) 38:41–67. doi: 10.1146/annurev-nutr-082117-051707
42. Douard V, Ferraris RP. The role of fructose transporters in diseases linked to excessive fructose intake. J Physiol (2013) 591:401–14. doi: 10.1113/jphysiol.2011.215731
43. Jones ME, Lipmann F, Hilz H, Lynen F. On the enzymatic mechanism of coenzyme A acetylation with adenosine triphosphate and acetate. J Am Chem Soc (1953) 75:3285–6. doi: 10.1021/ja01109a517
44. Berg P. Acyl adenylates: An enzymatic mechanism of acetate activation. J Biol Chem (1956) 222:991–1013.
45. Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK, et al. Acetate dependence of tumors. Cell (2014) 159:1591–602. doi: 10.1016/j.cell.2014.11.020
46. Fujino T, Kondo J, Ishikawa M, Morikawa K, Yamamoto TT. Acetyl-CoA Synthetase 2, a Mitochondrial Matrix Enzyme Involved in the Oxidation of Acetate. J Biol Chem (2001) 276:11420–6. doi: 10.1074/jbc.M008782200
47. Sone H, Shimano H, Sakakura Y, Inoue N, Amemiya-Kudo M, Yahagi N, et al. Acetyl-coenzyme A Synthetase Is a Lipogenic Enzyme Controlled by SREBP-1 and Energy Status. Am J Physiol Endocrinol Metab (2002) 282:E222–30. doi: 10.1152/ajpendo.00189.2001
48. Ikeda Y, Yamamoto J, Okamura M, Fujino T, Takahashi S, Takeuchi K, et al. Transcriptional Regulation of the Murine acetyl-CoA Synthetase 1 Gene Through Multiple Clustered Binding Sites for Sterol Regulatory Element-Binding Proteins and a Single Neighboring Site for Sp1. J Biol Chem (2001) 276:34259–69. doi: 10.1074/jbc.M103848200
49. Luong A, Hannah VC, Brown MS, Goldstein JL. Molecular Characterization of Human acetyl-CoA Synthetase, an Enzyme Regulated by Sterol Regulatory Element-Binding Proteins. J Biol Chem (2000) 275:26458–66. doi: 10.1074/jbc.M004160200
50. Alves-Bezerra M, Cohen DE. Triglyceride Metabolism in the Liver. Compr Physiol (2017) 8:1–8. doi: 10.1002/cphy.c170012
51. Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest (2004) 114:147–52. doi: 10.1172/JCI22422
52. Czumaj A, Śledziński T. Biological Role of Unsaturated Fatty Acid Desaturases in Health and Disease. Nutrients (2020) 12:356. doi: 10.3390/nu12020356
53. Nakamura MT, Nara TY. Structure, function, and dietary regulation of delta6, delta5, and delta9 desaturases. Annu Rev Nutr (2004) 24:345–76. doi: 10.1146/annurev.nutr.24.121803.063211
54. Wang Y, Botolin D, Christian B, Busik J, Xu J, Jump DB. Tissue-specific, nutritional, and developmental regulation of rat fatty acid elongases. J Lipid Res (2005) 46:706–15. doi: 10.1194/jlr.M400335-JLR200
55. Jump DB, Botolin D, Wang Y, Xu J, Christian B, Demeure O. Fatty acid regulation of hepatic gene transcription. J Nutr (2005) 135:2503–06. doi: 10.1093/jn/135.11.2503
56. Wang Y, Botolin D, Xu J, Christian B, Mitchell E, Jayaprakasam B, et al. Regulation of hepatic fatty acid elongase and desaturase expression in diabetes and obesity. J Lipid Res (2006) 47:2028–41. doi: 10.1194/jlr.M600177-JLR200
57. Benhamed F, Denechaud PD, Lemoine M, Robichon C, Moldes M, Bertrand-Michel J, et al. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J Clin Invest (2012) 122:2176–94. doi: 10.1172/JCI41636
58. Niwa H, Iizuka K, Kato T, Wu W, Tsuchida H, Takao K, et al. ChREBP Rather Than SHP Regulates Hepatic VLDL Secretion. Nutrients (2018) 10:321. doi: 10.3390/nu10030321
59. Yahagi N, Shimano H, Hasty AH, Amemiya-Kudo M, Okazaki H, Tamura Y, et al. A crucial role of sterol regulatory element-binding protein-1 in the regulation of lipogenic gene expression by polyunsaturated fatty acids. J Biol Chem (1999) 274:35840–4. doi: 10.1074/jbc.274.50.35840
60. Dentin R, Benhamed F, Pégorier JP, Foufelle F, Viollet B, Vaulont S, et al. Polyunsaturated fatty acids suppress glycolytic and lipogenic genes through the inhibition of ChREBP nuclear protein translocation. J Clin Invest (2005) 115:2843–54. doi: 10.1172/JCI25256
61. Takeuchi Y, Yahagi N, Izumida Y, Nishi M, Kubota M, Teraoka Y, et al. Polyunsaturated fatty acids selectively suppress sterol regulatory element-binding protein-1 through proteolytic processing and autoloop regulatory circuit. J Biol Chem (2010) 285:11681–91. doi: 10.1074/jbc.M109.096107
62. Kabashima T, Kawaguchi T, Wadzinski BE, Uyeda K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc Natl Acad Sci U S A (2003) 100:5107–12. doi: 10.1073/pnas.0730817100
63. Dentin R, Tomas-Cobos L, Foufelle F, Leopold J, Girard J, Postic C, et al. Glucose 6-phosphate, rather than xylulose 5-phosphate, is required for the activation of ChREBP in response to glucose in the liver. J Hepatol (2012) 56:199–209. doi: 10.1016/j.jhep.2011.07.019
64. Iizuka K, Wu W, Horikawa Y, Takeda J. Role of glucose-6-phosphate and xylulose-5-phosphate in the regulation of glucose-stimulated gene expression in the pancreatic β cell line, INS-1E. Endocr J (2013) 60:473–82. doi: 10.1507/endocrj.EJ12-0413
65. Arden C, Tudhope SJ, Petrie JL, Al-Oanzi ZH, Cullen KS, Lange AJ, et al. Fructose 2,6-bisphosphate is essential for glucose-regulated gene transcription of glucose-6-phosphatase and other ChREBP target genes in hepatocytes. Biochem J (2012) 443:111–23. doi: 10.1042/BJ20111280
66. Guinez C, Filhoulaud G, Rayah-Benhamed F, Marmier S, Dubuquoy C, Dentin R, et al. O-GlcNAcylation increases ChREBP protein content and transcriptional activity in the liver. Diabetes (2011) 60:1399–413. doi: 10.2337/db10-0452
67. Ido-Kitamura Y, Sasaki T, Kobayashi M, Kim HJ, Lee YS, Kikuchi O, et al. Hepatic FoxO1 integrates glucose utilization and lipid synthesis through regulation of Chrebp O-glycosylation. PloS One (2012) 7:e47231. doi: 10.1371/journal.pone.0047231
68. Sakiyama H, Fujiwara N, Noguchi T, Eguchi H, Yoshihara D, Uyeda K, et al. The role of O-linked GlcNAc modification on the glucose response of ChREBP. Biochem Biophys Res Commun (2010) 402:784–9. doi: 10.1016/j.bbrc.2010.10.113
69. Benhamed F, Filhoulaud G, Caron S, Lefebvre P, Staels B, Postic C. O-GlcNAcylation Links ChREBP and FXR to Glucose-Sensing. Front Endocrinol (Lausanne) (2015) 5:230. doi: 10.3389/fendo.2014.00230
70. Morishita S, Mochizuki K, Goda T. Bindings of ChREBP and SREBP1, and histone acetylation around the rat liver fatty acid synthase gene are associated with induction of the gene during the suckling-weaning transition. J Nutr Sci Vitaminol (Tokyo) (2014) 60:94–100. doi: 10.3177/jnsv.60.94
71. Cai C, Yu H, Huang G, Du X, Yu X, Zhou Y, et al. Histone modifications in fatty acid synthase modulated by carbohydrate responsive element binding protein are associated with non−alcoholic fatty liver disease. Int J Mol Med (2018) 42:1215–28. doi: 10.3892/ijmm.2018.3702
72. Cha-Molstad H, Saxena G, Chen J, Shalev A. Glucose-stimulated expression of Txnip is mediated by carbohydrate response element-binding protein, p300, and histone H4 acetylation in pancreatic beta cells. J Biol Chem (2009) 284:16898–905. doi: 10.1074/jbc.M109.010504
73. Caron S, Huaman Samanez C, Dehondt H, Ploton M, Briand O, Lien F, et al. Farnesoid X receptor inhibits the transcriptional activity of carbohydrate response element binding protein in human hepatocytes. Mol Cell Biol (2013) 33:2202–11. doi: 10.1128/MCB.01004-12
74. Lane EA, Choi DW, Garcia-Haro L, Levine ZG, Tedoldi M, Walker S, et al. HCF-1 Regulates De Novo Lipogenesis through a Nutrient-Sensitive Complex with ChREBP. Mol Cell (2019) 75:357–71.e7. doi: 10.1016/j.molcel.2019.05.019
75. Bricambert J, Alves-Guerra MC, Esteves P, Prip-Buus C, Bertrand-Michel J, Guillou H, et al. The histone demethylase Phf2 acts as a molecular checkpoint to prevent NAFLD progression during obesity. Nat Commun (2018) 9:2092. doi: 10.1038/s41467-018-04361-y
76. Wu W, Tsuchida H, Kato T, Niwa H, Horikawa Y, Takeda J, et al. Fat and Carbohydrate in Western Diet Contribute Differently to Hepatic Lipid Accumulation. Biochem Biophys Res Commun (2015) 46:681–6. doi: 10.1016/j.bbrc.2015.04.092
77. Hussain MM, Rava P, Walsh M, Rana M, Iqbal J. Multiple functions of microsomal triglyceride transfer protein. Nutr Metab (Lond) (2012) 9:14. doi: 10.1186/1743-7075-9-14
78. Yore MM, Syed I, Moraes-Vieira PM, Zhang T, Herman MA, Homan EA, et al. Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell (2014) 159:318–32. doi: 10.1016/j.cell.2014.09.035
79. Kahn BB. Adipose Tissue, Inter-Organ Communication, and the Path to Type 2 Diabetes: The 2016 Banting Medal for Scientific Achievement Lecture. Diabetes (2019) 68:3–14. doi: 10.2337/dbi18-0035
80. Kolar MJ, Kamat SS, Parsons WH, Homan EA, Maher T, Peroni OD, et al. Branched Fatty Acid Esters of Hydroxy Fatty Acids Are Preferred Substrates of the MODY8 Protein Carboxyl Ester Lipase. Biochemistry (2016) 55:4636–41. doi: 10.1021/acs.biochem.6b00565
81. Zhou P, Santoro A, Peroni OD, Nelson AT, Saghatelian A, Siegel D, et al. PAHSAs enhance hepatic and systemic insulin sensitivity through direct and indirect mechanisms. J Clin Invest (2019) 129:4138–50. doi: 10.1172/JCI127092
82. Syed I, Lee J, Moraes-Vieira PM, Donaldson CJ, Sontheimer A, Aryal P, et al. Palmitic Acid Hydroxystearic Acids Activate GPR40, Which Is Involved in Their Beneficial Effects on Glucose Homeostasis. Cell Metab (2018) 27:419–27.e4. doi: 10.1016/j.cmet.2018.01.001
83. Vijayakumar A, Aryal P, Wen J, Syed I, Vazirani RP, Moraes-Vieira PM, et al. Absence of Carbohydrate Response Element Binding Protein in Adipocytes Causes Systemic Insulin Resistance and Impairs Glucose Transport. Cell Rep (2017) 21:1021–35. doi: 10.1016/j.celrep.2017.09.091
84. Abu-Elheiga L, Matzuk MM, Abo-Hashema KA, Wakil SJ. Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science (2001) 291:2613–6. doi: 10.1126/science.1056843
85. Iroz A, Montagner A, Benhamed F, Levavasseur F, Polizzi A, Anthony E, et al. A Specific ChREBP and PPARα Cross-Talk Is Required for the Glucose-Mediated FGF21 Response. Cell Rep (2017) 21:403–16. doi: 10.1016/j.celrep.2017.09.065
86. Iizuka K, Wu W, Horikawa Y, Saito M, Takeda J. Feedback looping between ChREBP and PPARα in the regulation of lipid metabolism in brown adipose tissues. Endocr J (2013) 60:1145–53. doi: 10.1507/endocrj.ej13-0079
87. Bryrup T, Thomsen CW, Kern T, Allin KH, Brandslund I, Jørgensen NR, et al. Metformin-induced changes of the gut microbiota in healthy young men: results of a non-blinded, one-armed intervention study. Diabetologia (2019) 62:1024–35. doi: 10.1007/s00125-019-4848-7
88. Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest (2001) 108:1167–74. doi: 10.1172/JCI13505
89. Cantó C, Auwerx J. AMP-activated protein kinase and its downstream transcriptional pathways. Cell Mol Life Sci (2010) 67:3407–23. doi: 10.1007/s00018-010-0454-z
90. Sato S, Jung H, Nakagawa T, Pawlosky R, Takeshima T, Lee WR, et al. Metabolite Regulation of Nuclear Localization of Carbohydrate-response Element-binding Protein (ChREBP): ROLE OF AMP AS AN ALLOSTERIC INHIBITOR. J Biol Chem (2016) 291:10515–27. doi: 10.1074/jbc.M115.708982
91. Kooner JS, Chambers JC, Aguilar-Salinas CA, Hinds DA, Hyde CL, Warnes GR, et al. Genome-wide scan identifies variation in MLXIPL associated with plasma triglycerides. Nat Genet (2008) 40:149–51. doi: 10.1038/ng.2007.61
Keywords: fatty acid synthesis, lipoprotein metabolism, β-oxidation, ketogenesis, carbohydrate response element-binding protein (Chrebp), gut microbiota
Citation: Iizuka K, Takao K and Yabe D (2020) ChREBP-Mediated Regulation of Lipid Metabolism: Involvement of the Gut Microbiota, Liver, and Adipose Tissue. Front. Endocrinol. 11:587189. doi: 10.3389/fendo.2020.587189
Received: 25 July 2020; Accepted: 09 November 2020;
Published: 03 December 2020.
Edited by:
Anca Dana Dobrian, Eastern Virginia Medical School, United StatesReviewed by:
Hitoshi Shimano, University of Tsukuba, JapanCopyright © 2020 Iizuka, Takao and Yabe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katsumi Iizuka, a2lpenVrYUBnaWZ1LXUuYWMuanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.