 Song Peng1
Song Peng1 Jun Zhang1
Jun Zhang1 Xintao Tan1
Xintao Tan1 Yiqiang Huang1
Yiqiang Huang1 Jing Xu1
Jing Xu1 Natalie Silk2
Natalie Silk2 Dianzheng Zhang2
Dianzheng Zhang2 Qiuli Liu1*
Qiuli Liu1* Jun Jiang1*
Jun Jiang1*- 1Department of Urology, Daping Hospital, Army Medical University, Chongqing, China
- 2Department of Bio-Medical Sciences, Philadelphia College of Osteopathic Medicine, Philadelphia, PA, United States
Pheochromocytomas and paragangliomas (PPGLs) are rare neuroendocrine tumors originating from chromaffin cells in the adrenal medulla (PCCs) or extra-adrenal sympathetic or parasympathetic paraganglia (PGLs). About 40% of PPGLs result from germline mutations and therefore they are highly inheritable. Although dysfunction of any one of a panel of more than 20 genes can lead to PPGLs, mutations in genes involved in the VHL/HIF axis including PHD, VHL, HIF-2A (EPAS1), and SDHx are more frequently found in PPGLs. Multiple lines of evidence indicate that pseudohypoxia plays a crucial role in the tumorigenesis of PPGLs, and therefore PPGLs are also known as metabolic diseases. However, the interplay between VHL/HIF-mediated pseudohypoxia and metabolic disorder in PPGLs cells is not well-defined. In this review, we will first discuss the VHL/HIF axis and genetic alterations in this axis. Then, we will dissect the underlying mechanisms in VHL/HIF axis-driven PPGL pathogenesis, with special attention paid to the interplay between the VHL/HIF axis and cancer cell metabolism. Finally, we will summarize the currently available compounds/drugs targeting this axis which could be potentially used as PPGLs treatment, as well as their underlying pharmacological mechanisms. The overall goal of this review is to better understand the role of VHL/HIF axis in PPGLs development, to establish more accurate tools in PPGLs diagnosis, and to pave the road toward efficacious therapeutics against metastatic PPGLs.
Introduction
Pheochromocytomas (PCCs) are catecholamine-secreting tumors that originated from the chromaffin cells in the adrenal medulla. Paragangliomas (PGLs) are neural crest-derived neuroendocrine neoplasms originating from extra-adrenal sympathetic or parasympathetic ganglia (1). Both PCCs and PGLs are collectively known as PPGLs. PPGLs are rare tumors with the incidence rate between 0.2 and 0.8 per 100,000 (2–4) with great clinical manifestations (5). Due to elevated levels of catecholamines in the circulation, the common clinical presentations of PPGLs include episodes of headache, sweating, palpitation, and hypertension. In addition, about 10% of PCCs are metastatic (6) and 40% of PGLs are considered as metastatic disease (7, 8).
Etiologically, about 70%–80% of PPGLs are caused by genetic abnormalities which affect different signaling pathways (9). Approximately, 40% of PPGLs result from germline mutations, and therefore they are highly inheritable (10). Although dysfunction of any of these related susceptible gene products can lead to PPGLs, mutations in the genes encoding the VHL/HIF axis such as VHL, HIF, and PHD are more commonly found in PPGLs (11). Moreover, multiple lines of evidence suggest that pseudohypoxia plays a crucial role in the tumorigenesis of PPGLs. In this review, we will discuss the genetic alterations affecting the VHL/HIF axis and dissect the underlying molecular mechanisms in pseudohypoxia signaling and PPGLs. We will also summarize the currently available compounds or drugs targeting VHL/HIF axis, their specific targets, and pharmacological mechanisms.
The VHL/HIF Axis
The Von Hippel-Lindau (VHL) gene located on 3p25.5 encodes an ancient tumor suppressor, pVHL. Although pVHL functions in both physiology and pathology, as a component of an E3 ubiquitin-ligase complex, pVHL plays a determinant role in the degradation of hypoxia-induced factors (HIFs) including HIF-1α, HIF-2α, and HIF-3α. The roles of HIF-1α and HIF-2α in sensing and facilitating cellular adaptation to hypoxic conditions as well as their underlying molecular mechanisms are well-established (12). However, much less is known about HIF-3α. Functionally, HIF-1α and HIF-2α heterodimerize with HIF-β by HLH domain, which is also known as ARNT, to transcriptionally regulate a wide spectrum of HIF target genes. Both HIF-α and HIF-β belong to the basic helix-loop-helix-Per-ARNT-Sim (bHLH-PAS) family. They contain a basic DNA binding domain, a conserved NH2-terminal domain (N-TAD), and two specialized transactivation domains located in their variable COOH-terminal domains (C-TAD) (13) (Figure 1). The asparagine residue (N803) in the C-TAD of HIF-α can be hydroxylated by factor-inhibiting HIF (FIH) to interrupt its interaction with CREB-binding protein (CBP)/p300, an essential coactivator of HIF (14–16). The N-TAD also contains an oxygen-dependent domain (ODDD), in which a few prolyl residues (Pro-402 and Pro-564 in HIF-1α; Pro-405 and Pro-531 in HIF-2α) are selectively hydroxylated under normoxic condition and hydroxylated HIFs are subsequently degraded (17–20). The enzymes responsible for HIF-α hydroxylation belong to the egg-laying-defective nine (EGLN) family known as PHD1, PHD2, and PHD3 because they all contain a prolyl-4-hydroxylase domain. These enzymes are dioxygenases and use both molecular oxygen and Fe2+ as their co-substrates to catalyze HIF-α hydroxylation.
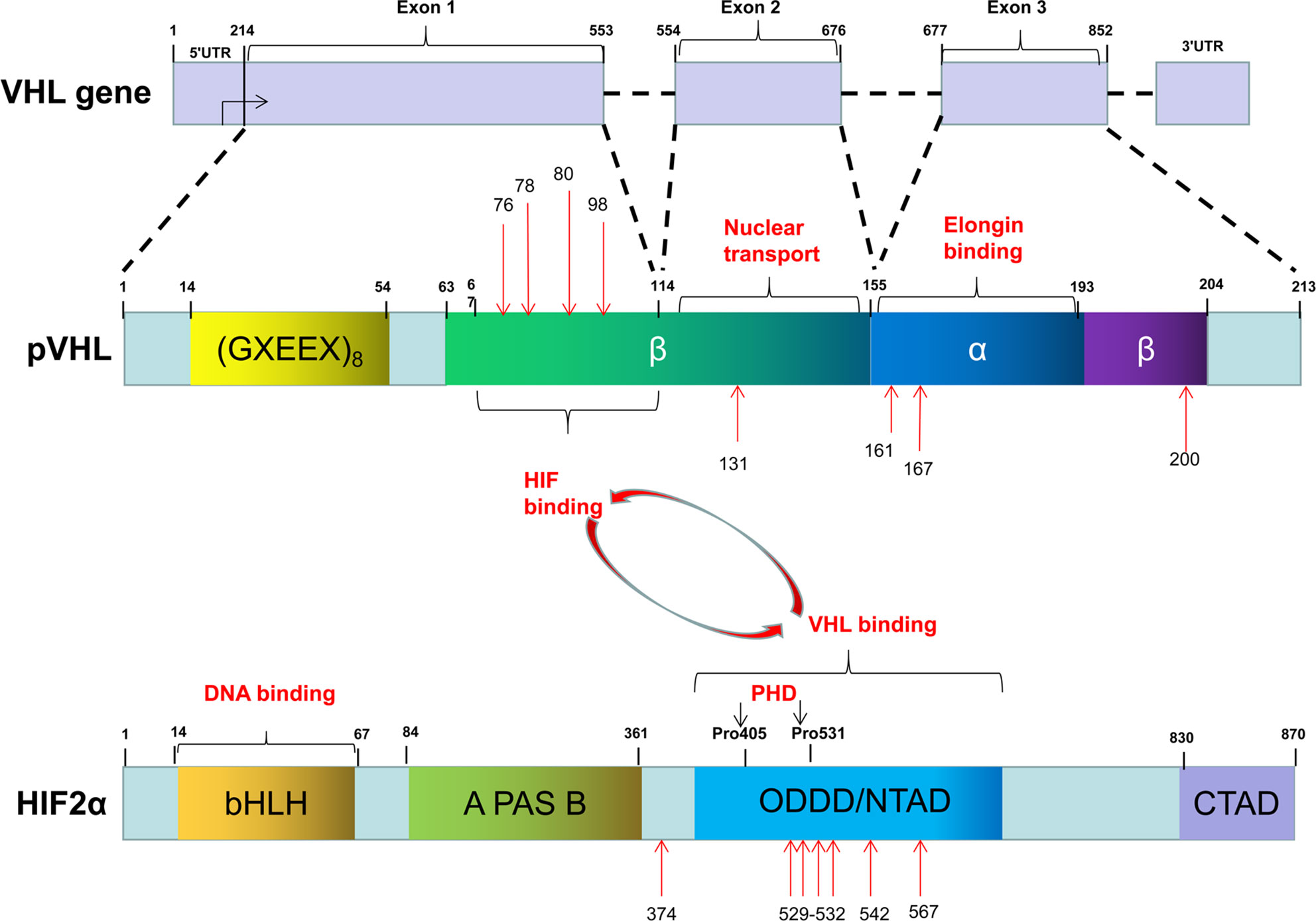
Figure 1 The common mutation sites of VHL and EPAS1 genes in PPGLs.
The VHL/HIF axis responses to reduced oxygen concentration or hypoxia. Although HIF-1α and HIF-2α have about 48% sequence similarity, they regulate two different groups of target genes with limited overlap mainly due to their dissimilar transactivation domains (21, 22). In addition, HIF-1α is widely expressed, while HIF-2α is only expressed in certain cell types (23, 24). For example, the genes involved in glucose metabolism are mainly regulated by HIF-1α. HIF-2α plays a more important role in the adjustment to high altitudes and the regulation of EPO expression (25, 26). As mentioned above that compared to HIF-1α and HIF-2α, much less is known about HIF-3α. Since it lacks the transactivation domain (27), HIF-3α likely does not transcriptionally regulate its target genes. Overall, the levels and functions of both HIF-1α and HIF-2α are oxygen-concentration dependent. Specific proline residues of HIF-1α and HIF-2α are hydroxylated by PHD under normoxic conditions. With the involvement of the molecules such as elongin B, elongin C, cul2, the hydroxylated HIFs are recognized by the pVHL (28–30), subsequently ubiquitinated and ultimately degraded (31, 32). Under hypoxic conditions, the non-hydroxylated HIFs are dissociated from pVHL, accumulated in the cells, and subsequently upregulate their target genes transcriptionally. However, failure in the degradation of HIFs due to either deletion or mutation of either VHL, HIFs, or PHDs can lead to dysregulation of HIFs-regulated genes in a variety of diseases including PPGLs (Figure 2).
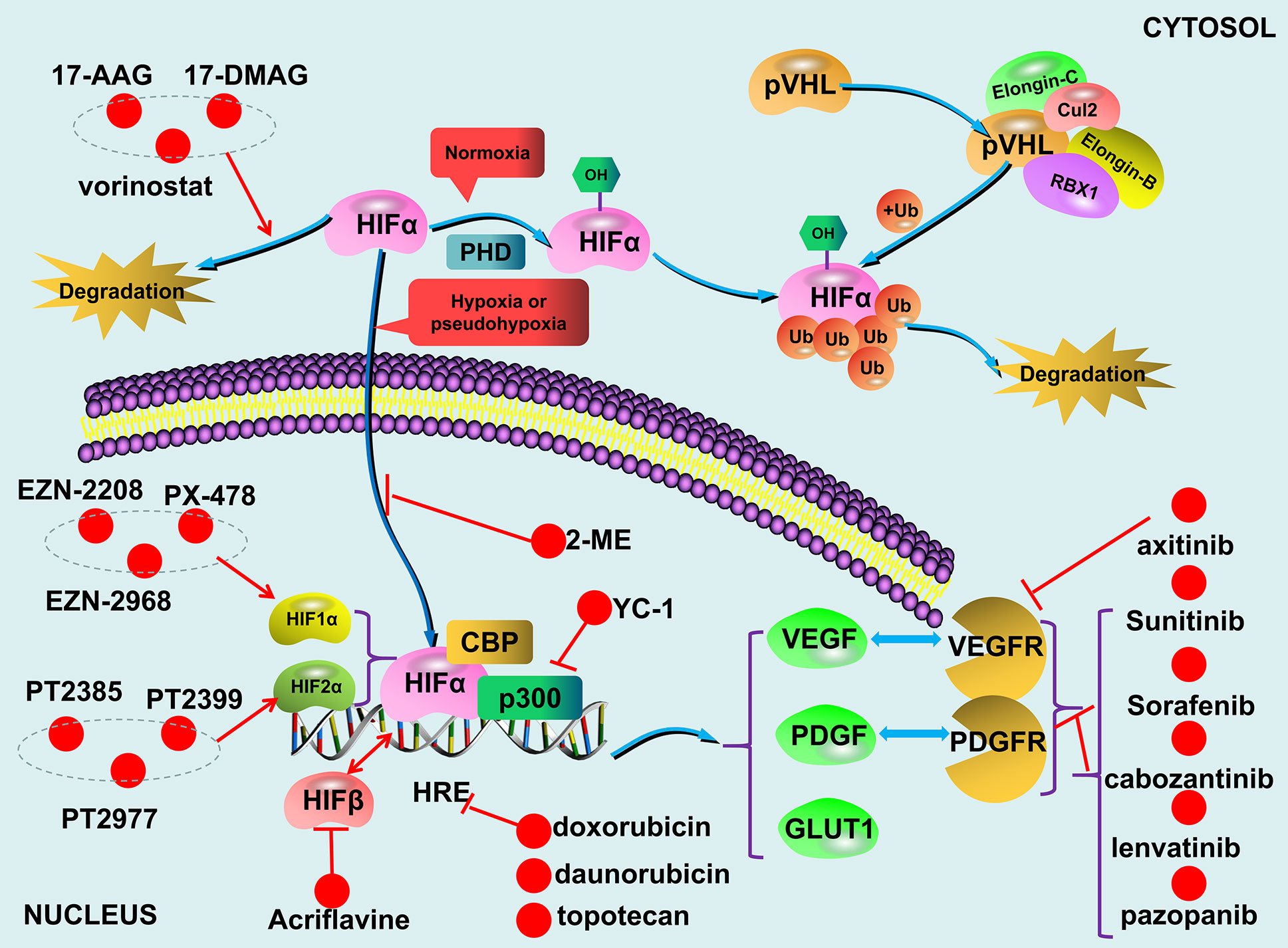
Figure 2 The VHL/HIF axis and compounds targeting the axis.
Dysregulation of the VHL/HIF Axis and PPGLs
As mentioned above that mutation in either the three genes encoding pVHL, HIFs and PHDs can lead to abnormal accumulation of HIFs. Minor alteration of this axis usually causes erythrocytosis; whereas major dysregulation of the axis is associated with tumorigenesis (33). Although a wide spectrum of tumors including hemangioblastomas, renal cell carcinoma (RCC), pancreatic neuroendocrine tumor, and PPGLs can result from dysregulation of the VHL/HIF axis (34–37), this review will only focus on the relationship between aberrations of these genes and PPGLs.
VHL Mutations
After the VHL mutations were first described in an ophthalmic disease (34), multiple studies subsequently confirmed that VHL mutations can cause a variety of diseases including cancers (35–37). To honor the contributions of the German ophthalmologist Eugen von Hippel and the Swedish pathologist Arvid Lindau, the gene responsible for these diseases is, therefore, named as VHL. Of note, VHL disease caused by heterozygous germline mutations is autosomal dominant and almost completely penetrant (97%) (38). VHL diseases are generally classified into two types, type 1 (without PCCs) and type 2 (with PCCs). The type 2 disease is manifested as RCCs, PCCs, central nervous system, retinal hemangioblastomas, pancreatic neuroendocrine tumors and pancreatic and renal cysts and can be further divided into three subtypes (34), PCCs with all types of VHL disease manifestations without RCC (Type 2A), PCC with all types of VHL disease including RCC (Type 2B), and isolated PCCs (Type 2C).
To date, more than 1,000 mutations in VHL gene have been identified. These mutations can be categorized as missense mutation (52%), frameshift mutation (13%), nonsense mutation (11%), in-frame deletion/insertion mutation (6%), large/complete deletion mutation (11%), and splicing mutation (7%) (39). The common germline mutations in VHL are delPhe76, Asn78Ser, Argl61Stop, Arg167Gln, Argl67Trp, and Leu178Pro (40) (Figure 1). Recently, we reported four missense mutations in five Chinese unrelated families c.239G>T (p.Ser80Ile), c.232A>T (p.Asn78Tyr), c.500G>A (p.Arg167Gln), c.293A>G (p.Try98Cys), and all four mutations predispose the patients to VHL disease (41). Notably, type 2 VHL disease mainly resulted from missense mutations (85%–92%) (40, 42), especially mutations in codons 167 and 238, are mainly associated with PPGLs (43, 44). In contrast, homozygous germline mutations are rare or barely cause tumors. Sonny et al. found a c.598C>T (p.Arg200Trp) homozygous missense germline mutation of VHL caused Chuvash polycythemia (45). In addition, somatic VHL mutations were found in majority (50%–70%) of clear-cell RCC cases (38).
It has been reported that different mutations in VHL lead to diverse clinical symptoms (41, 46–49), and sometimes even the same mutation can lead to different phenotypes (50–53). Since pVHL has multiple functional domains, one of the potential explanations for this phenomenon is that a specific mutation causes particular dysfunction. It appears that missense mutations are more likely linked with type 2 disease and truncating mutations are responsible for type 1 disease (54). However, Liu et al. further stratified the missense mutations as HIF-α binding site missense mutations (HM) group and non-HIF-α binding site missense mutations (nHM) group, and found that the missense mutations in HM group had similar risks of most tumors with truncating mutations with the exception that the HM group had a lower risk of RCC. Moreover, compared to nHM, missense mutations in HM had a higher risk of pancreatic cyst or tumor and a lower risk of PCCs (55). Secondly, some functions of pVHL are O2-independent (56, 57) or unrelated to HIF regulation, these functions may also be involved in PPGLs pathogenesis. Michael et al. found that RCCs with deficient pVHL exhibited deficiency in fibronectin matrix assembly (58). Intriguingly, Clifford et al. reported that mutations associated with type 2C phenotype could even promote, rather than inhibit, HIF-α ubiquitylation and degradation (39). These findings altogether supported the notion that disturbing the functions of pVHL contributes to the development of PPGLs. Additionally, based on Knudson’s Two-Hit model (59), it is understandable that the diverse phenotypes of VHL diseases could be the result of two different “hits”.
The VHL/HIF axis also can be affected by dysregulated epigenetic modifications such as gene silencing by methylation of the CpG islands in the promoter of related genes. Indeed, promoter hypermethylation occurs in about 3%–42% of clear-cell RCC (60). Adam Andreasson found that the promoter methylation of the VHL gene is not only elevated in PPGLs compared with normal tissue (57% vs. 27%) but also significantly higher in malignancies than that in tumors (63% vs. 55%) (61). However, the precise molecular mechanisms in the pathogenesis of PPGLs related to loss-of-function of pVHL are still largely unknown and therefore need further investigation.
HIF-A Mutations
As mentioned above that HIF-α family composed three members, HIF-1α, HIF-2α, and HIF-3α. But little is known about HIF-3α. Compared with HIF-2A, HIF-1A has relatively few mutations, ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) only collects 30 records. Morris et al. reported a somatic mutation (p.Val116Glu) and a germline missense mutation (p.Ala475Ser) of HIF-1A in a clear-cell RCC with VHL inactivation. Of note, the germline mutation (p.Ala475Ser) was likely to be a benign variant (62). Furthermore, Gladek et al. found that HIF-1A Single-Nucleotide Polymorphisms (SNPs) are association with the phenotypes of many tumors (63). In PPGLs patients, only copy number aberration (TCGA-QT-A5XP, https://portal.gdc.cancer.gov/), not HIF-1A mutation, have been found. On the other hand, both germline and somatic mutation in HIF-2A have been identified in patients with polycythemia and/or PPGLs. However, it appears that germline mutations of HIF-2A including p.Met535Val, p.Gly537Arg, p.Gly537Trp only leads to polycythemias, not tumors (64, 65). A gain-of-function germline mutation in HIF-2A alone is not sufficient for tumorigenesis presumably that simultaneous loss-of-function in some tumor suppressors is needed. In fact, we recently reported that germline mutations in HIF-2A (c.1609G>A, p.Gly537Arg) are responsible for polycythemia formation and additional somatic VHL mutations are needed for the development of clear-cell RCC (66). Similarly, a germline mutation in HIF-2A exon 9 (c.1121T>A, p.F374Y) leads to polycythemia and predisposes the patients for PPGLs development (67). In addition, somatic mutations in HIF-2A appear to be more frequent genetic events in PPGLs (68). For example, Zhang et al. reported two gain-of-function somatic mutations (c.1588G>A, p.Ala530Thr and c.1589C>T, p.Ala530Val) in exon 12 of HIF-2A resulting in paraganglioma and polycythemia, respectively. Further analyses suggest that mutations in the vicinity of the hydroxylation site Pro-531 affect the catalytic activity of PHD and then lead to the interrupted interaction between HIF-2α and pVHL (69). Moreover, Karel Pacak et al. reported two somatic mutations of HIF-2A (c.1595A>G p.Y532C and c.1586T>C p.L529P) in patients with either congenital polycythemia, multiple recurrent PPGLs, or somatostatinoma (70). We recently found that a gain-of-function mutation of HIF-2A (c.1589C>T) leads to PPGLs with polycythemia simultaneously (26) and a mutation in HIF-2A immediately distal to its DNA binding domain (p.Ser71Tyr) has been identified in sporadic PPGLs (71) (Figure 1). Germline or somatic mutations of HIF-2A can be mosaic. Buffet et al. reported two cases of HIF-2A-related Polycythemia-Paraganglioma Syndrome resulted from mosaicism mutations. They found that these patients could present with young age and multiplicity; and also the mutations could be transmitted to the offspring (72). In addition, HIF-2A mosaic mutation might be involved in high secretion of catecholamines and cyanotic congenital heart disease (73).
Mutation in PHD and Other Related Factors
Heterozygous germline mutations in PHD2 gene were first reported in familial erythrocytosis (74, 75). Later, Ladroue et al. reported a heterozygous loss-of-function mutation of PHD2 (c.1121A>G, p.His374Arg) with the development of both erythrocytosis and recurrent paraganglioma. Functional analysis indicates that His374 is important in the binding of cofactor Fe2+, and mutation of this residue is expected to impair the catalytic function of PHDs (76). Yang et al. reported heterozygous germline mutations in PHD1 (c.188T>A, p.Ser61Arg and c.682G>T, p.Ala228Ser) in patients with polycythemia and PPGLs, respectively. Further research found that the half-lives of both PHD1 and PHD2 are reduced with these PHD1 mutants (77). These findings collectively demonstrated that mutant PHDs are indeed associated with susceptibility to PPGLs. However, compared to VHL and HIF-A, mutations in PHDs are relatively rare in patients with PPGLs (78). Additionally, mutations of enzymes in the TCA cycle can affect VHL/HIF axis indirectly. For example, elevated levels of HIFs can be caused by the mutations in SDHx, FH, MDH, and IDH with subsequent accumulation of specific metabolites and reactive oxygen species (31, 79–89). In addition, multiple lines of evidence indicated that mutations in cluster 2 (Kinase Signaling Cluster) genes, including NF1, RET, TMEM127, ERK, MAX, and H-RAS could affect the VHL/HIF axis indirectly (90–94), although these mutations were initially thought to drive PPGLs through the oxygen-independent kinase signaling pathway, such as mTOR axis.
The Mechanisms in Dysregulated VHL/HIF Axis and PPGLs
Under normal physiological conditions, HIFs are degraded during normoxic condition and HIFs accumulation only occur during hypoxia. The undegraded HIF-α translocates to the nucleus and dimerizes with HIF-β (95). Together with p300/CBP, the HIF-α/HIF-β heterodimer is recruited to the hypoxia-responsive elements (HREs) located on the promoter regions of HIF-regulated targets to transcriptionally upregulate the expression of the genes including vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), and glucose transporter (GLUT) (93, 96–98) (Figure 2). The combined effects of these upregulated gene products result in an increased supply of blood and nutrients to the hypoxic tissues and switch glucose metabolism from aerobic to anaerobic glycolysis. Due to the fast growth of tumor tissues, this process occurs in all solid tumors (99, 100), and dysregulated VHL/HIF axis further exacerbate the development of certain tumors such as PPGLs.
Aerobic glycolysis, also known as Warburg Effect (9, 101, 102), occurs in all solid tumor cells. However, dysregulated VHL/HIF axis plays a more important role in certain cancer types such as clear-cell RCC and PPGLs. Pseudohypoxia, mimicking the hypoxic condition, can affect different cancer processes including tumorigenesis and malignant transformation by promoting epithelial-mesenchymal transition and enhancing stem cell-like property. Of note, metabolic reprogramming can affect each of these processes and the role of VHL/HIF axis in cancer metabolic reprogramming has been well defined. HIF-1 aberrant activation due to either VHL or PHD mutations increases glucose uptake and glycolysis with a concomitant decrease in mitochondrial mass (103). HIF-α, especially HIF-1α, controls a wide spectrum of enzymes including GLUT1, GLUT3, hexokinase 1/2, lactate dehydrogenase-A (LDH-A), and pyruvate dehydrogenase kinase 1 (PDK1) (104–108). Upregulating these enzymes collectively shifts glycolysis from aerobic to anaerobic (109).
PPGLs are also considered as metabolic diseases due to the increased secretion of one or more catecholamines (epinephrine, norepinephrine, and dopamine). Catecholamines play a crucial role in the regulation of multiple metabolic pathways. Patients with PPGLs usually manifest with impaired insulin secretion, increased insulin resistance, elevated lipolysis, and the bone resorption marker C-terminal telopeptide of type I collagen (110). Many studies have revealed that oncometabolite such as succinate, fumarate, and 2-hydroxyglutarate (2HG) are increased in PPGLs (83, 111, 112). Another study found that compared to PPGLs without SDHx mutation, PPGLs with a deficient SDH have 25-fold higher succinate and 80% lower levels of fumarate, cis-aconitate, and isocitrate (113). Mutation in FH and IDH lead to the accumulation of fumarate and (R)-2-hydroxyglutarate, respectively (88, 114). Mechanistically, these oncometabolite modulate the activity of α-ketoglutarate-dependent dioxygenases such as PDH, which are involved in the induction of the pseudohypoxia pathway and activation of HIF axis (10, 31, 115). In addition, PPGLs with a germline mutation in genes encoding enzymes in the TCA cycle belong to Cluster I tumors, characterized by a pseudohypoxia signature (31). Together with the other intermediate metabolites of the TCA cycle, succinate can increase the chance of tumor development and progression through an ill-defined mechanism (83).
Results from more recent researches indicate that HIFs can regulate non-coding RNA (ncRNA) either directly or indirectly. Direct regulation is achieved by the recruiting HIFs to the HREs located on the promoter regions of ncRNAs. Whereas indirect regulation of ncRNA is achieved by epigenetic modification (116). One of the HIFs targets microRNA 210 (miR-210) (117) participates in a variety of biological processes including carcinogenesis, cancer cell proliferation, apoptosis, angiogenesis, and metastasis (118–120). On the other hand, miRNA can also activate HIF via mTOR indirectly. Calsina et al. reported miR-21-3p can regulate TSC2/mTOR axis in metastatic PPGLs and proposed that miR-21-3p can be the predictive markers of metastases (121). In addition, some lncRNA such as H19, MALAT1, HOTAIR, and lncRNA-SARCC play important roles in the activity of VHL/HIF axis (122).
Inhibitors Targeting The VHL/HIF Axis
Since the VHL/HIF axis plays a critical role in the development of PPGLs, targeting this axis could be a promising therapeutic strategy. Multiple reagents targeting the VHL/HIF axis have been explored and some of them have been applied clinically (123–127). Among them, the tyrosine kinase inhibitors (TKIs) are most widely used because TKIs can repress angiogenesis by inhibiting the VEGF pathway (128–130). Some compounds targeting the VHL/HIF axis can inhibit tumor growth in both animal models and clinical trials (Table 1).
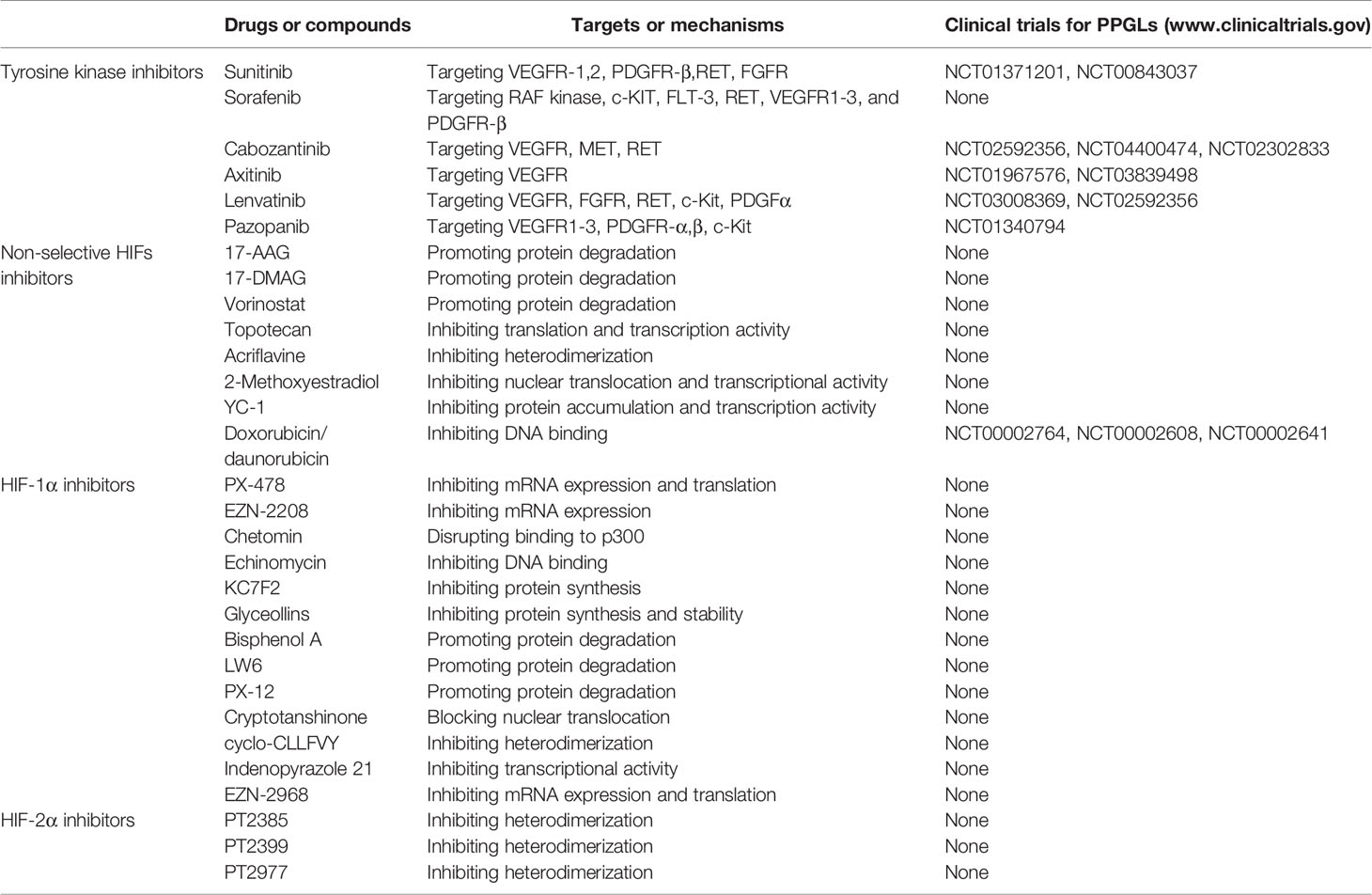
Table 1 The inhibitors targeting the VHL/HIF axis.
Tyrosine Kinase Inhibitors
To date, more than 40 protein kinase inhibitors have been approved by the FDA for cancer treatment (131). Several TKIs including sunitinib, cabozantinib, axitinib, Lenvatinib, and pazopanib are currently being evaluated in phase II clinical trials (www.ClinicalTrials.gov). By repressing the tyrosine kinase receptors, these reagents can inhibit cancer cell growth, metastasis, and the development of therapeutic resistance (132). More recently, several case studies and/or clinical trials in small cohorts suggest that TKIs could be a promising treatment for metastatic PPGLs or the syndrome-associated PPGLs.
Sunitinib, an orally administered TKI, can target both VEGFR and PDGFR (133), and therefore, it could potentially serve as a therapeutic reagent for PPGLs treatment. Early in vitro studies showed that sunitinib can repress the growth of PCCs (134), inhibit both synthesis and secretion of catecholamine (135). Several clinical trials have suggested that patients with metastatic PPGLs responded well to sunitinib (136–140). Results from one of our recent studies also suggested that sunitinib could be an optional therapy for patients with VHL disease-associated PCCs (141). Results from the SNIPP trial showed that sunitinib at 50mg daily benefited most patients with progressive PPGLs. Of 23 evaluable cases, the disease control rate (DCR) was 83% and median progression free survival (PFS) was 13.4 months, 3 (13%) patients with germline variants in RET or SDHx achieved a partial response (PR), 16 (70%) patients had stable disease (SD) (142). Currently, a phase II clinical trial (the First International Randomized Study in Malignant Progressive Pheochromocytoma and Paraganglioma, FIRSTMAPP) studying the effect of sunitinib on PPGLs is ongoing. In addition, results from sdhb knockout tumors bearing mice showed that sunitinib treatment can prevent tumor growth and vessel development in the first 2 weeks; thereafter, resistance will develop (143). Another study by using both in vivo and vitro models demonstrated that sunitinib and sorafenib can inhibit the growth of PCCs (144, 145). Previous study reported that a patient with recurrence and metastatic PPGLs responded well to 12 weeks of sorafenib treatment evidenced by regressed metastatic and decreased catecholamine level (146).
In addition, cabozantinib also appears to be a promising TKI for patients with PPGLs, especially for those with bone metastases. A trial (NCT02302833) enrolled 11 PPGLs patients with bone metastases is currently ongoing. Preliminary results identified 4 patients with PR (37%) and 6 patients with SD (55%); all patients with SD had tumor regression (18%–29%). The DCR was 92%, PFS was 16 months. None of the patients had any serious hypertension or cardiovascular events (147). A recent trial (NCT01967576) showed that 36% of patients with metastatic PPGLs achieved a PR when treated with axitinib (148); while only one of seven patients with metastatic PPGLs who received pazopanib showed a PR (149). Finally, recruitment for a phase II clinical trial has just begun to test if lenvatinib can be used as an anti-angiogenic medication for metastatic PPGLs (www.ClinicalTrials.gov) (Figure 2).
Although the promising therapeutic effects of TKIs on PPGLs have been widely reported, the toxicity of TKIs should also be mentioned. The side effects of TKIs include fatigue, nausea, thrombocytopenia, hypertension, myocardial infarction, and restrictive cardiomyopathy and so on. O’Kane et al. reported that due to severe adverse events, several patients needed to reduce the dose of sunitinib, and even 20% patients discontinued trial participation (142). A phase III clinical trial compared the safety of pazopanib and sunitinib in metastatic RCC, the results showed that patients treated with sunitinib had a higher incidence of fatigue, the hand-foot syndrome and thrombocytopenia than patients treated with pazopanib. Although the rate of cardiovascular adverse events of pazopanib were similar to that of sunitinib, the abnormal liver tests leading to discontinuation in pazopanib-treatment patients should be noted (139). Furthermore, the tolerance of axitinib was similar to that of other VEGFR inhibitors. Rini et al. reported that axtinib more frequently causes hypertension than sorafenib (40% vs. 29%) (NCT00678392) (140). Similarly, Van Geel et al. reported that the incidence of hypertension in axtinib-treatment patients was higher than that in pazopanib-treatment patients (150). Burotto Pichun et al. reported that even 80% axtinib-treatment patients developed severe hypertension (148). Recently, a phase III randomized ATLAS trial assessed the safety of axitinib versus placebo, axitinib-treated patients had more grade 3/4 adverse events and discontinuations (151). Taken together, the safety of TKIs needs to be further evaluated in the future.
HIFs Inhibitors
Transcription factors including HIFs have been historically considered undruggable. This is one of the reasons that research in the pharmaceutical field has been mainly focusing on HIF’s downstream pathways, such as VEGF. However, based on the structure of HIF-2α (152), two compounds PT2385 and PT2399 targeting HIF-2α were successfully identified (145, 153). Subsequent in vitro and in vivo studies showed that these compounds can inhibit the growth of clear-cell RCC (154). A phase I trial found that for patients with progressive clear-cell RCC the complete response, partial response, and stabilized disease to PT2385 were 2%, 12%, and 52%, respectively (155). It has been proposed that HIF-2α inhibitors possess a great potential for the treatment of advanced PPGLs (156). These initial results could also spearhead a multitude of preclinical and clinical studies assessing the efficiency of the compounds in other tumor types. In fact, PT2385 has entered its phase II clinical trial (NCT03108066) evaluating its efficacy in patients with advanced cancers carrying a VHL germline mutation. Recently, second-generation allosteric inhibitor of HIF-2α PT2977 (MK-6482) was identified. Compared to PT2385, PT2977 have increased potency and improved pharmacokinetic profile (157). The result of phase I/II trial of PT2977 in 55 patients with advanced RCCs revealed that 24% patients experienced a confirmed PR and 54% had SD, with a clinical benefit rate of 78%. Moreover, a PT2977 monotherapy Phase III trial in patients with previously treated advanced RCC is planned (158). Notably, previous studies reported that HIF-2α was overexpressed in VHL and in SDH-related PPGLs compared to HIF-1α (159, 160). Therefore, inhibitors targeting HIF-2α appear to be more promising than inhibitors targeting HIF-1α.
Other Compounds Targeting VHL/HIF Axis
Theoretically, any compounds capable of inhibiting the VHL/HIF axis can potentially become therapeutic reagents for the treatment of metastatic PPGLs. For example, the HSP90 inhibitors, 17-N-allylamino-17-demethoxy geldanamycin (17-AAG) and 17-dimethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG) (161–163), and histone deacetylase inhibitor, vorinostat (164, 165), are capable of inducing HIF-α degradation. Topotecan can downregulate HIF-α by inhibiting topoisomerase I (TOP-I) (166–168). Of note, topotecan has already been used as a therapeutic reagent for the treatment of metastatic ovarian carcinoma, recurrent small cell lung cancer, and recurrent cervical cancer (169–171). Acriflavine can inhibit dimerization between HIF-α and HIF-β and subsequently repress the expression of HIFs target genes (172). 2-Methoxyestradiol (2-ME), an active metabolite of 17β-estradiol, can inhibit the synthesis, nuclear translocation, and transcriptional activity of HIF-α (173, 174). In addition, an antiplatelet aggregation agent YC-1 can not only suppress HIFs transcriptional activity by inhibiting p300 recruitment but also promote HIF-α degradation by enhancing FIH binding (175). Finally, two anthracyclines, doxorubicin and daunorubicin, have been demonstrated to inhibit the expression of HIFs targets efficiently by interrupting HIF-α recruitment (176).
There are also compounds inhibiting HIF-1α synthesis. For example, PX-478 is capable of downregulating both the mRNA and protein levels of HIF-1α (177–179). EZN-2208 (PEG-SN38) can downregulate the expression of HIF-1α in lymphocytic leukemia (180). By hybridizing with HIF-1α mRNA, EZN-2968, a 3rd generation antisense oligonucleotide, can specifically inhibit HIF-1α translation (181, 182). Chetomin is capable of repressing xenograft growth in vivo by disrupting HIF-1α and p300 interaction (183). Finally, there is a myriad of compounds including echinomycin, CAY10585, KC7F2, glyceollins, bisphenol A, LW6, PX-12, cryptotanshinone (CPT), cyclo-CLLFVY, and indenopyrazoles 21 that have all been validated as selective inhibitors of HIF-1α with different molecular mechanisms (184–196).
Conclusion
The VHL/HIF axis plays an important role in oxygen homeostasis and cellular metabolism in both physiology and pathology. Dysregulation of this axis due to either germline mutations, somatic mutations, and epigenetic dysregulation can be involved in tumorigenesis and progression of different cancer types including PPGLs. Mechanistically, by reprogramming metabolic pathways the abnormally activated HIFs drive cancer cells toward aerobic glycolysis. Based on the underlying molecular mechanisms of VHL/HIF axis in PPGLs development, a wide spectrum of drugs specifically targeting this axis have been and will continue to be developed as PPGL therapeutics. With a better understanding of the relationship between VHL/HIF axis and PPGLs, more accurate diagnosis and prognosis of PPGLs, as well as efficacious therapeutics against PPGLs, are expected in the near future.
Author Contributions
SP, QL, JZ, XT, JX, and YH contributed to the writing of the manuscript. NS, JJ, and DZ provided consultation and contributed to the revising of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (81972398, JJ) and University Research Project of Army Medical University (218XLC3073, JZ).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Drs. Farhadul Islam, Ichiro Abe, Alfred King-yin Lam, and Suja Pillai for inviting this review submission.
References
1. Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet (2005) 366(9486):665–75. doi: 10.1016/S0140-6736(05)67139-5
2. Stenstrom G, Svardsudd K. Pheochromocytoma in Sweden 1958-1981. An analysis of the National Cancer Registry Data. Acta Med Scand (1986) 220(3):225–32. doi: 10.1111/j.0954-6820.1986.tb02755.x
3. Ariton M, Juan CS, AvRuskin TW. Pheochromocytoma: clinical observations from a Brooklyn tertiary hospital. Endocr Pract (2000) 6(3):249–52. doi: 10.4158/EP.6.3.249
4. Beard CM, Sheps SG, Kurland LT, Carney JA, Lie JT. Occurrence of pheochromocytoma in Rochester, Minnesota, 1950 through 1979. Mayo Clin Proc (1983) 58(12):802–4.
5. Eisenhofer G, Pacak K, Huynh TT, Qin N, Bratslavsky G, Linehan WM, et al. Catecholamine metabolomic and secretory phenotypes in phaeochromocytoma. Endocr Relat Cancer (2011) 18(1):97–111. doi: 10.1677/ERC-10-0211
6. Nomura K, Kimura H, Shimizu S, Kodama H, Okamoto T, Obara T, et al. Survival of patients with metastatic malignant pheochromocytoma and efficacy of combined cyclophosphamide, vincristine, and dacarbazine chemotherapy. J Clin Endocrinol Metab (2009) 94(8):2850–6. doi: 10.1210/jc.2008-2697
7. Welander J, Soderkvist P, Gimm O. Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Endocr Relat Cancer (2011) 18(6):R253–76. doi: 10.1530/ERC-11-0170
8. Ayala-Ramirez M, Feng L, Johnson MM, Ejaz S, Habra MA, Rich T, et al. Clinical risk factors for malignancy and overall survival in patients with pheochromocytomas and sympathetic paragangliomas: primary tumor size and primary tumor location as prognostic indicators. J Clin Endocrinol Metab (2011) 96(3):717–25. doi: 10.1210/jc.2010-1946
9. Taieb D, Pacak K. Genetic Determinants of Pheochromocytoma and Paraganglioma Imaging Phenotypes. J Nucl Med (2020) 61(5):643–5. doi: 10.2967/jnumed.120.245613
10. Dahia PL. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat Rev Cancer (2014) 14(2):108–19. doi: 10.1038/nrc3648
11. Fishbein L, Leshchiner I, Walter V, Danilova L, Robertson AG, Johnson AR, et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell (2017) 31(2):181–93. doi: 10.1016/j.ccell.2017.01.001
12. Samanta D, Prabhakar NR, Semenza GL. Systems biology of oxygen homeostasis. Wiley Interdiscip Rev Syst Biol Med (2017) 9(4). doi: 10.1002/wsbm.1382
13. Wu D, Rastinejad F. Structural characterization of mammalian bHLH-PAS transcription factors. Curr Opin Struct Biol (2017) 43:1–9. doi: 10.1016/j.sbi.2016.09.011
14. Yamashita K, Discher DJ, Hu J, Bishopric NH, Webster KA. Molecular regulation of the endothelin-1 gene by hypoxia. Contributions of hypoxia-inducible factor-1, activator protein-1, GATA-2, AND p300/CBP. J Biol Chem (2001) 276(16):12645–53. doi: 10.1074/jbc.M011344200
15. Dayan F, Roux D, Brahimi-Horn MC, Pouyssegur J, Mazure NM. The oxygen sensor factor-inhibiting hypoxia-inducible factor-1 controls expression of distinct genes through the bifunctional transcriptional character of hypoxia-inducible factor-1alpha. Cancer Res (2006) 66(7):3688–98. doi: 10.1158/0008-5472.CAN-05-4564
16. Kaelin WG Jr., Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell (2008) 30(4):393–402. doi: 10.1016/j.molcel.2008.04.009
17. Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science (2001) 294(5545):1337–40. doi: 10.1126/science.1066373
18. Favier J, Buffet A, Gimenez-Roqueplo AP. HIF2A mutations in paraganglioma with polycythemia. N Engl J Med (2012) 367(22):2161. doi: 10.1056/NEJMc1211953
19. Ivan M, Haberberger T, Gervasi DC, Michelson KS, Gunzler V, Kondo K, et al. Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proc Natl Acad Sci USA (2002) 99(21):13459–64. doi: 10.1073/pnas.192342099
20. Baek JH, Mahon PC, Oh J, Kelly B, Krishnamachary B, Pearson M, et al. OS-9 interacts with hypoxia-inducible factor 1alpha and prolyl hydroxylases to promote oxygen-dependent degradation of HIF-1alpha. Mol Cell (2005) 17(4):503–12. doi: 10.1016/j.molcel.2005.01.011
21. Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer (2011) 12(1):9–22. doi: 10.1038/nrc3183
22. Al Tameemi W, Dale TP, Al-Jumaily RMK, Forsyth NR. Hypoxia-Modified Cancer Cell Metabolism. Front Cell Dev Biol (2019) 7:4. doi: 10.3389/fcell.2019.00004
23. Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer (2008) 8(12):967–75. doi: 10.1038/nrc2540
24. Triner D, Shah YM. Hypoxia-inducible factors: a central link between inflammation and cancer. J Clin Invest (2016) 126(10):3689–98. doi: 10.1172/JCI84430
25. Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol (2003) 23(24):9361–74. doi: 10.1128/mcb.23.24.9361-9374.2003
26. Liu Q, Wang Y, Tong D, Liu G, Yuan W, Zhang J, et al. A Somatic HIF2alpha Mutation-Induced Multiple and Recurrent Pheochromocytoma/Paraganglioma with Polycythemia: Clinical Study with Literature Review. Endocr Pathol (2017) 28(1):75–82. doi: 10.1007/s12022-017-9469-4
27. Heikkila M, Pasanen A, Kivirikko KI, Myllyharju J. Roles of the human hypoxia-inducible factor (HIF)-3alpha variants in the hypoxia response. Cell Mol Life Sci (2011) 68(23):3885–901. doi: 10.1007/s00018-011-0679-5
28. Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science (2001) 292(5516):468–72. doi: 10.1126/science.1059796
29. Loboda A, Jozkowicz A, Dulak J. HIF-1 and HIF-2 transcription factors–similar but not identical. Mol Cells (2010) 29(5):435–42. doi: 10.1007/s10059-010-0067-2
30. Semenza GL. Oxygen sensing, homeostasis, and disease. N Engl J Med (2011) 365(6):537–47. doi: 10.1056/NEJMra1011165
31. Jochmanova I, Zhuang Z, Pacak K. Pheochromocytoma: Gasping for Air. Horm Cancer (2015) 6(5-6):191–205. doi: 10.1007/s12672-015-0231-4
32. Kaelin WG Jr. The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer (2008) 8(11):865–73. doi: 10.1038/nrc2502
33. Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature (2006) 441(7092):437–43. doi: 10.1038/nature04871
34. Gossage L, Eisen T, Maher ER. VHL, the story of a tumour suppressor gene. Nat Rev Cancer (2015) 15(1):55–64. doi: 10.1038/nrc3844
35. Richard S, Graff J, Lindau J, Resche F. Von Hippel-Lindau disease. Lancet (2004) 363(9416):1231–4. doi: 10.1016/S0140-6736(04)15957-6
36. Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, et al. von Hippel-Lindau disease. Lancet (2003) 361(9374):2059–67. doi: 10.1016/S0140-6736(03)13643-4
37. Chittiboina P, Lonser RR. Von Hippel-Lindau disease. Handb Clin Neurol (2015) 132:139–56. doi: 10.1016/B978-0-444-62702-5.00010-X
38. Richards FM. Molecular pathology of von HippelLindau disease and the VHL tumour suppressor gene. Expert Rev Mol Med (2001) 2001:1–27. doi: 10.1017/S1462399401002654
39. Clifford SC, Cockman ME, Smallwood AC, Mole DR, Woodward ER, Maxwell PH, et al. Contrasting effects on HIF-1alpha regulation by disease-causing pVHL mutations correlate with patterns of tumourigenesis in von Hippel-Lindau disease. Hum Mol Genet (2001) 10(10):1029–38. doi: 10.1093/hmg/10.10.1029
40. Zbar B, Kishida T, Chen F, Schmidt L, Maher ER, Richards FM, et al. Germline mutations in the Von Hippel-Lindau disease (VHL) gene in families from North America, Europe, and Japan. Hum Mutat (1996) 8(4):348–57. doi: 10.1002/(SICI)1098-1004(1996)8:4<348::AID-HUMU8>3.0.CO;2-3
41. Liu Q, Yuan G, Tong D, Liu G, Yi Y, Zhang J, et al. Novel genotype-phenotype correlations in five Chinese families with Von Hippel-Lindau disease. Endocr Connect (2018) 7(7):870–8. doi: 10.1530/EC-18-0167
42. Nordstrom-O’Brien M, van der Luijt RB, van Rooijen E, van den Ouweland AM, Majoor-Krakauer DF, Lolkema MP, et al. Genetic analysis of von Hippel-Lindau disease. Hum Mutat (2010) 31(5):521–37. doi: 10.1002/humu.21219
43. Beroud C, Joly D, Gallou C, Staroz F, Orfanelli MT, Junien C. Software and database for the analysis of mutations in the VHL gene. Nucleic Acids Res (1998) 26(1):256–8. doi: 10.1093/nar/26.1.256
44. Garcia A, Matias-Guiu X, Cabezas R, Chico A, Prat J, Baiget M, et al. Molecular diagnosis of von Hippel-Lindau disease in a kindred with a predominance of familial phaeochromocytoma. Clin Endocrinol (Oxf) (1997) 46(3):359–63. doi: 10.1046/j.1365-2265.1997.00149.x
45. Ang SO, Chen H, Hirota K, Gordeuk VR, Jelinek J, Guan Y, et al. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat Genet (2002) 32(4):614–21. doi: 10.1038/ng1019
46. Gallou C, Chauveau D, Richard S, Joly D, Giraud S, Olschwang S, et al. Genotype-phenotype correlation in von Hippel-Lindau families with renal lesions. Hum Mutat (2004) 24(3):215–24. doi: 10.1002/humu.20082
47. Weirich G, Klein B, Wohl T, Engelhardt D, Brauch H. VHL2C phenotype in a German von Hippel-Lindau family with concurrent VHL germline mutations P81S and L188V. J Clin Endocrinol Metab (2002) 87(11):5241–6. doi: 10.1210/jc.2002-020651
48. Fukino K, Teramoto A, Adachi K, Takahashi H, Emi M. A family with hydrocephalus as a complication of cerebellar hemangioblastoma: identification of Pro157Leu mutation in the VHL gene. J Hum Genet (2000) 45(1):47–51. doi: 10.1007/s100380050009
49. Hong B, Ma K, Zhou J, Zhang J, Wang J, Liu S, et al. Frequent Mutations of VHL Gene and the Clinical Phenotypes in the Largest Chinese Cohort With Von Hippel-Lindau Disease. Front Genet (2019) 10:867. doi: 10.3389/fgene.2019.00867
50. Martin RL, Walpole I, Goldblatt J. Identification of two sporadically derived mutations in the Von Hippel-Lindau gene. Hum Mutat (1996) 7(2):185. doi: 10.1002/(SICI)1098-1004(1996)7:2<185::AID-HUMU22>3.0.CO;2-Y
51. Crossey PA, Foster K, Richards FM, Phipps ME, Latif F, Tory K, et al. Molecular genetic investigations of the mechanism of tumourigenesis in von Hippel-Lindau disease: analysis of allele loss in VHL tumours. Hum Genet (1994) 93(1):53–8. doi: 10.1007/BF00218913
52. Wang H, Shepard MJ, Zhang C, Dong L, Walker D, Guedez L, et al. Deletion of the von Hippel-Lindau Gene in Hemangioblasts Causes Hemangioblastoma-like Lesions in Murine Retina. Cancer Res (2018) 78(5):1266–74. doi: 10.1158/0008-5472.CAN-17-1718
53. Lee JS, Lee JH, Lee KE, Kim JH, Hong JM, Ra EK, et al. Genotype-phenotype analysis of von Hippel-Lindau syndrome in Korean families: HIF-alpha binding site missense mutations elevate age-specific risk for CNS hemangioblastoma. BMC Med Genet (2016) 17(1):48. doi: 10.1186/s12881-016-0306-2
54. Ong KR, Woodward ER, Killick P, Lim C, Macdonald F, Maher ER. Genotype-phenotype correlations in von Hippel-Lindau disease. Hum Mutat (2007) 28(2):143–9. doi: 10.1002/humu.20385
55. Liu SJ, Wang JY, Peng SH, Li T, Ning XH, Hong BA, et al. Genotype and phenotype correlation in von Hippel-Lindau disease based on alteration of the HIF-alpha binding site in VHL protein. Genet Med (2018) 20(10):1266–73. doi: 10.1038/gim.2017.261
56. Bishop T, Lau KW, Epstein AC, Kim SK, Jiang M, O’Rourke D, et al. Genetic analysis of pathways regulated by the von Hippel-Lindau tumor suppressor in Caenorhabditis elegans. PloS Biol (2004) 2(10):e289. doi: 10.1371/journal.pbio.0020289
57. Bommi-Reddy A, Almeciga I, Sawyer J, Geisen C, Li W, Harlow E, et al. Kinase requirements in human cells: III. Altered kinase requirements in VHL-/- cancer cells detected in a pilot synthetic lethal screen. Proc Natl Acad Sci USA (2008) 105(43):16484–9. doi: 10.1073/pnas.0806574105
58. Ohh M, Yauch RL, Lonergan KM, Whaley JM, Stemmer-Rachamimov AO, Louis DN, et al. The von Hippel-Lindau tumor suppressor protein is required for proper assembly of an extracellular fibronectin matrix. Mol Cell (1998) 1(7):959–68. doi: 10.1016/s1097-2765(00)80096-9
59. Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U.S.A. (1971) 68(4):820–3. doi: 10.1073/pnas.68.4.820
60. Joosten SC, Smits KM, Aarts MJ, Melotte V, Koch A, Tjan-Heijnen VC, et al. Epigenetics in renal cell cancer: mechanisms and clinical applications. Nat Rev Urol (2018) 15(7):430–51. doi: 10.1038/s41585-018-0023-z
61. Andreasson A, Kiss NB, Caramuta S, Sulaiman L, Svahn F, Backdahl M, et al. The VHL gene is epigenetically inactivated in pheochromocytomas and abdominal paragangliomas. Epigenetics (2013) 8(12):1347–54. doi: 10.4161/epi.26686
62. Morris MR, Hughes DJ, Tian YM, Ricketts CJ, Lau KW, Gentle D, et al. Mutation analysis of hypoxia-inducible factors HIF1A and HIF2A in renal cell carcinoma. Anticancer Res (2009) 29(11):4337–43.
63. Gladek I, Ferdin J, Horvat S, Calin GA, Kunej T. HIF1A gene polymorphisms and human diseases: Graphical review of 97 association studies. Genes Chromosomes Cancer (2017) 56(6):439–52. doi: 10.1002/gcc.22449
64. Percy MJ, Beer PA, Campbell G, Dekker AW, Green AR, Oscier D, et al. Novel exon 12 mutations in the HIF2A gene associated with erythrocytosis. Blood (2008) 111(11):5400–2. doi: 10.1182/blood-2008-02-137703
65. Percy MJ, Furlow PW, Lucas GS, Li X, Lappin TR, McMullin MF, et al. A gain-of-function mutation in the HIF2A gene in familial erythrocytosis. N Engl J Med (2008) 358(2):162–8. doi: 10.1056/NEJMoa073123
66. Liu Q, Tong D, Liu G, Yi Y, Zhang D, Zhang J, et al. HIF2A germline-mutation-induced polycythemia in a patient with VHL-associated renal-cell carcinoma. Cancer Biol Ther (2017) 18(12):944–7. doi: 10.1080/15384047.2017.1394553
67. Lorenzo FR, Yang C, Ng Tang Fui M, Vankayalapati H, Zhuang Z, Huynh T, et al. A novel EPAS1/HIF2A germline mutation in a congenital polycythemia with paraganglioma. J Mol Med (Berl) (2013) 91(4):507–12. doi: 10.1007/s00109-012-0967-z
68. Comino-Mendez I, de Cubas AA, Bernal C, Alvarez-Escola C, Sanchez-Malo C, Ramirez-Tortosa CL, et al. Tumoral EPAS1 (HIF2A) mutations explain sporadic pheochromocytoma and paraganglioma in the absence of erythrocytosis. Hum Mol Genet (2013) 22(11):2169–76. doi: 10.1093/hmg/ddt069
69. Zhuang Z, Yang C, Lorenzo F, Merino M, Fojo T, Kebebew E, et al. Somatic HIF2A gain-of-function mutations in paraganglioma with polycythemia. N Engl J Med (2012) 367(10):922–30. doi: 10.1056/NEJMoa1205119
70. Pacak K, Jochmanova I, Prodanov T, Yang C, Merino MJ, Fojo T, et al. New syndrome of paraganglioma and somatostatinoma associated with polycythemia. J Clin Oncol (2013) 31(13):1690–8. doi: 10.1200/JCO.2012.47.1912
71. Toledo RA, Qin Y, Srikantan S, Morales NP, Li Q, Deng Y, et al. In vivo and in vitro oncogenic effects of HIF2A mutations in pheochromocytomas and paragangliomas. Endocr Relat Cancer (2013) 20(3):349–59. doi: 10.1530/ERC-13-0101
72. Buffet A, Smati S, Mansuy L, Menara M, Lebras M, Heymann MF, et al. Mosaicism in HIF2A-related polycythemia-paraganglioma syndrome. J Clin Endocrinol Metab (2014) 99(2):E369–73. doi: 10.1210/jc.2013-2600
73. Vaidya A, Flores SK, Cheng ZM, Nicolas M, Deng Y, Opotowsky AR, et al. EPAS1 Mutations and Paragangliomas in Cyanotic Congenital Heart Disease. N Engl J Med (2018) 378(13):1259–61. doi: 10.1056/NEJMc1716652
74. Minamishima YA, Moslehi J, Bardeesy N, Cullen D, Bronson RT, Kaelin WG Jr. Somatic inactivation of the PHD2 prolyl hydroxylase causes polycythemia and congestive heart failure. Blood (2008) 111(6):3236–44. doi: 10.1182/blood-2007-10-117812
75. Percy MJ, Furlow PW, Beer PA, Lappin TR, McMullin MF, Lee FS. A novel erythrocytosis-associated PHD2 mutation suggests the location of a HIF binding groove. Blood (2007) 110(6):2193–6. doi: 10.1182/blood-2007-04-084434
76. Ladroue C, Carcenac R, Leporrier M, Gad S, Le Hello C, Galateau-Salle F, et al. PHD2 mutation and congenital erythrocytosis with paraganglioma. N Engl J Med (2008) 359(25):2685–92. doi: 10.1056/NEJMoa0806277
77. Yang C, Zhuang Z, Fliedner SM, Shankavaram U, Sun MG, Bullova P, et al. Germ-line PHD1 and PHD2 mutations detected in patients with pheochromocytoma/paraganglioma-polycythemia. J Mol Med (Berl) (2015) 93(1):93–104. doi: 10.1007/s00109-014-1205-7
78. Lee S, Nakamura E, Yang H, Wei W, Linggi MS, Sajan MP, et al. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: developmental culling and cancer. Cancer Cell (2005) 8(2):155–67. doi: 10.1016/j.ccr.2005.06.015
79. Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science (2000) 287(5454):848–51. doi: 10.1126/science.287.5454.848
80. Niemann S, Muller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet (2000) 26(3):268–70. doi: 10.1038/81551
81. Korpershoek E, Favier J, Gaal J, Burnichon N, van Gessel B, Oudijk L, et al. SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J Clin Endocrinol Metab (2011) 96(9):E1472–6. doi: 10.1210/jc.2011-1043
82. Burnichon N, Briere JJ, Libe R, Vescovo L, Riviere J, Tissier F, et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet (2010) 19(15):3011–20. doi: 10.1093/hmg/ddq206
83. Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell (2005) 7(1):77–85. doi: 10.1016/j.ccr.2004.11.022
84. Philip B, Ito K, Moreno-Sanchez R, Ralph SJ. HIF expression and the role of hypoxic microenvironments within primary tumours as protective sites driving cancer stem cell renewal and metastatic progression. Carcinogenesis (2013) 34(8):1699–707. doi: 10.1093/carcin/bgt209
85. Pan Y, Mansfield KD, Bertozzi CC, Rudenko V, Chan DA, Giaccia AJ, et al. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol Cell Biol (2007) 27(3):912–25. doi: 10.1128/MCB.01223-06
86. Wang L, Lam G, Thummel CS. Med24 and Mdh2 are required for Drosophila larval salivary gland cell death. Dev Dyn (2010) 239(3):954–64. doi: 10.1002/dvdy.22213
87. Calsina B, Curras-Freixes M, Buffet A, Pons T, Contreras L, Leton R, et al. Role of MDH2 pathogenic variant in pheochromocytoma and paraganglioma patients. Genet Med (2018) 20(12):1652–62. doi: 10.1038/s41436-018-0068-7
88. Castro-Vega LJ, Buffet A, De Cubas AA, Cascon A, Menara M, Khalifa E, et al. Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum Mol Genet (2014) 23(9):2440–6. doi: 10.1093/hmg/ddt639
89. Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science (2009) 324(5924):261–5. doi: 10.1126/science.1170944
90. Baba M, Hirai S, Yamada-Okabe H, Hamada K, Tabuchi H, Kobayashi K, et al. Loss of von Hippel-Lindau protein causes cell density dependent deregulation of CyclinD1 expression through hypoxia-inducible factor. Oncogene (2003) 22(18):2728–38. doi: 10.1038/sj.onc.1206373
91. Hudson CC, Liu M, Chiang GG, Otterness DM, Loomis DC, Kaper F, et al. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol (2002) 22(20):7004–14. doi: 10.1128/mcb.22.20.7004-7014.2002
92. Mayerhofer M, Valent P, Sperr WR, Griffin JD, Sillaber C. BCR/ABL induces expression of vascular endothelial growth factor and its transcriptional activator, hypoxia inducible factor-1alpha, through a pathway involving phosphoinositide 3-kinase and the mammalian target of rapamycin. Blood (2002) 100(10):3767–75. doi: 10.1182/blood-2002-01-0109
93. Jochmanova I, Yang C, Zhuang Z, Pacak K. Hypoxia-inducible factor signaling in pheochromocytoma: turning the rudder in the right direction. J Natl Cancer Inst (2013) 105(17):1270–83. doi: 10.1093/jnci/djt201
94. Vicha A, Musil Z, Pacak K. Genetics of pheochromocytoma and paraganglioma syndromes: new advances and future treatment options. Curr Opin Endocrinol Diabetes Obes (2013) 20(3):186–91. doi: 10.1097/MED.0b013e32835fcc45
95. Semenza GL. A compendium of proteins that interact with HIF-1alpha. Exp Cell Res (2017) 356(2):128–35. doi: 10.1016/j.yexcr.2017.03.041
96. Ajith TA. Current insights and future perspectives of hypoxia-inducible factor-targeted therapy in cancer. J Basic Clin Physiol Pharmacol (2018) 30(1):11–8. doi: 10.1515/jbcpp-2017-0167
97. Shen C, Kaelin WG Jr. The VHL/HIF axis in clear cell renal carcinoma. Semin Cancer Biol (2013) 23(1):18–25. doi: 10.1016/j.semcancer.2012.06.001
98. Jang M, Kim SS, Lee J. Cancer cell metabolism: implications for therapeutic targets. Exp Mol Med (2013) 45:e45. doi: 10.1038/emm.2013.85
99. Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, et al. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res (1999) 59(22):5830–5.
100. Maxwell PH, Dachs GU, Gleadle JM, Nicholls LG, Harris AL, Stratford IJ, et al. Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc Natl Acad Sci USA (1997) 94(15):8104–9. doi: 10.1073/pnas.94.15.8104
101. Liberti MV, Locasale JW. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci (2016) 41(3):211–8. doi: 10.1016/j.tibs.2015.12.001
102. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324(5930):1029–33. doi: 10.1126/science.1160809
103. Semenza GL. Regulation of cancer cell metabolism by hypoxia-inducible factor 1. Semin Cancer Biol (2009) 19(1):12–6. doi: 10.1016/j.semcancer.2008.11.009
104. Maxwell PH, Pugh CW, Ratcliffe PJ. Activation of the HIF pathway in cancer. Curr Opin Genet Dev (2001) 11(3):293–9. doi: 10.1016/s0959-437x(00)00193-3
105. Masoud GN, Li W. HIF-1alpha pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sin B (2015) 5(5):378–89. doi: 10.1016/j.apsb.2015.05.007
106. Gordan JD, Thompson CB, Simon MC. HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell (2007) 12(2):108–13. doi: 10.1016/j.ccr.2007.07.006
107. Icard P, Lincet H. A global view of the biochemical pathways involved in the regulation of the metabolism of cancer cells. Biochim Biophys Acta (2012) 1826(2):423–33. doi: 10.1016/j.bbcan.2012.07.001
108. Kruspig B, Zhivotovsky B, Gogvadze V. Mitochondrial substrates in cancer: drivers or passengers? Mitochondrion (2014) 19 Pt A:8–19. doi: 10.1016/j.mito.2014.08.007
109. Weidemann A, Johnson RS. Biology of HIF-1alpha. Cell Death Differ (2008) 15(4):621–7. doi: 10.1038/cdd.2008.12
110. Erlic Z, Beuschlein F. Metabolic Alterations in Patients with Pheochromocytoma. Exp Clin Endocrinol Diabetes (2019) 127(2-03):129–36. doi: 10.1055/a-0649-0960
111. Richter S, Gieldon L, Pang Y, Peitzsch M, Huynh T, Leton R, et al. Metabolome-guided genomics to identify pathogenic variants in isocitrate dehydrogenase, fumarate hydratase, and succinate dehydrogenase genes in pheochromocytoma and paraganglioma. Genet Med (2019) 21(3):705–17. doi: 10.1038/s41436-018-0106-5
112. Gottlieb E, Tomlinson IP. Mitochondrial tumour suppressors: a genetic and biochemical update. Nat Rev Cancer (2005) 5(11):857–66. doi: 10.1038/nrc1737
113. Richter S, Peitzsch M, Rapizzi E, Lenders JW, Qin N, de Cubas AA, et al. Krebs cycle metabolite profiling for identification and stratification of pheochromocytomas/paragangliomas due to succinate dehydrogenase deficiency. J Clin Endocrinol Metab (2014) 99(10):3903–11. doi: 10.1210/jc.2014-2151
114. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature (2009) 462(7274):739–44. doi: 10.1038/nature08617
115. Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev (2012) 26(12):1326–38. doi: 10.1101/gad.191056.112
116. Peng X, Gao H, Xu R, Wang H, Mei J, Liu C. The interplay between HIF-1alpha and noncoding RNAs in cancer. J Exp Clin Cancer Res (2020) 39(1):27. doi: 10.1186/s13046-020-1535-y
117. Huang X, Zuo J. Emerging roles of miR-210 and other non-coding RNAs in the hypoxic response. Acta Biochim Biophys Sin (Shanghai) (2014) 46(3):220–32. doi: 10.1093/abbs/gmt141
118. Li L, Huang K, You Y, Fu X, Hu L, Song L, et al. Hypoxia-induced miR-210 in epithelial ovarian cancer enhances cancer cell viability via promoting proliferation and inhibiting apoptosis. Int J Oncol (2014) 44(6):2111–20. doi: 10.3892/ijo.2014.2368
119. Ying Q, Liang L, Guo W, Zha R, Tian Q, Huang S, et al. Hypoxia-inducible microRNA-210 augments the metastatic potential of tumor cells by targeting vacuole membrane protein 1 in hepatocellular carcinoma. Hepatology (2011) 54(6):2064–75. doi: 10.1002/hep.24614
120. Yang W, Sun T, Cao J, Liu F, Tian Y, Zhu W. Downregulation of miR-210 expression inhibits proliferation, induces apoptosis and enhances radiosensitivity in hypoxic human hepatoma cells in vitro. Exp Cell Res (2012) 318(8):944–54. doi: 10.1016/j.yexcr.2012.02.010
121. Calsina B, Castro-Vega LJ, Torres-Perez R, Inglada-Perez L, Curras-Freixes M, Roldan-Romero JM, et al. Integrative multi-omics analysis identifies a prognostic miRNA signature and a targetable miR-21-3p/TSC2/mTOR axis in metastatic pheochromocytoma/paraganglioma. Theranostics (2019) 9(17):4946–58. doi: 10.7150/thno.35458
122. Flippot R, Beinse G, Boileve A, Vibert J, Malouf GG. Long non-coding RNAs in genitourinary malignancies: a whole new world. Nat Rev Urol (2019) 16(8):484–504. doi: 10.1038/s41585-019-0195-1
123. Kim HC, Lee JS, Kim SH, So HS, Woo CY, Lee JL. Sunitinib treatment for metastatic renal cell carcinoma in patients with von hippel-lindau disease. Cancer Res Treat (2013) 45(4):349–53. doi: 10.4143/crt.2013.45.4.349
124. Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene (2010) 29(5):625–34. doi: 10.1038/onc.2009.441
125. Melillo G. Hypoxia-inducible factor 1 inhibitors. Methods Enzymol (2007) 435:385–402. doi: 10.1016/S0076-6879(07)35020-9
126. Melillo G. Targeting hypoxia cell signaling for cancer therapy. Cancer Metastasis Rev (2007) 26(2):341–52. doi: 10.1007/s10555-007-9059-x
127. Xia Y, Choi HK, Lee K. Recent advances in hypoxia-inducible factor (HIF)-1 inhibitors. Eur J Med Chem (2012) 49:24–40. doi: 10.1016/j.ejmech.2012.01.033
128. Wang Y, Li Z, Zhang H, Jin H, Sun L, Dong H, et al. HIF-1alpha and HIF-2alpha correlate with migration and invasion in gastric cancer. Cancer Biol Ther (2010) 10(4):376–82. doi: 10.4161/cbt.10.4.12441
129. Joshi S, Singh AR, Zulcic M. Durden DL. A macrophage-dominant PI3K isoform controls hypoxia-induced HIF1alpha and HIF2alpha stability and tumor growth, angiogenesis, and metastasis. Mol Cancer Res (2014) 12(10):1520–31. doi: 10.1158/1541-7786.MCR-13-0682
130. Philips GK, Atkins MB. New agents and new targets for renal cell carcinoma. Am Soc Clin Oncol Educ Book (2014) e222–7. doi: 10.14694/EdBook_AM.2014.34.e222
131. Roskoski R Jr. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol Res (2019) 144:19–50. doi: 10.1016/j.phrs.2019.03.006
132. McCormack PL. Pazopanib: a review of its use in the management of advanced renal cell carcinoma. Drugs (2014) 74(10):1111–25. doi: 10.1007/s40265-014-0243-3
133. Faivre S, Demetri G, Sargent W, Raymond E. Molecular basis for sunitinib efficacy and future clinical development. Nat Rev Drug Discovery (2007) 6(9):734–45. doi: 10.1038/nrd2380
134. Saito Y, Tanaka Y, Aita Y, Ishii KA, Ikeda T, Isobe K, et al. Sunitinib induces apoptosis in pheochromocytoma tumor cells by inhibiting VEGFR2/Akt/mTOR/S6K1 pathways through modulation of Bcl-2 and BAD. Am J Physiol Endocrinol Metab (2012) 302(6):E615–25. doi: 10.1152/ajpendo.00035.2011
135. Aita Y, Ishii KA, Saito Y, Ikeda T, Kawakami Y, Shimano H, et al. Sunitinib inhibits catecholamine synthesis and secretion in pheochromocytoma tumor cells by blocking VEGF receptor 2 via PLC-gamma-related pathways. Am J Physiol Endocrinol Metab (2012) 303(8):E1006–14. doi: 10.1152/ajpendo.00156.2012
136. Joshua AM, Ezzat S, Asa SL, Evans A, Broom R, Freeman M, et al. Rationale and evidence for sunitinib in the treatment of malignant paraganglioma/pheochromocytoma. J Clin Endocrinol Metab (2009) 94(1):5–9. doi: 10.1210/jc.2008-1836
137. Jimenez C, Cabanillas ME, Santarpia L, Jonasch E, Kyle KL, Lano EA, et al. Use of the tyrosine kinase inhibitor sunitinib in a patient with von Hippel-Lindau disease: targeting angiogenic factors in pheochromocytoma and other von Hippel-Lindau disease-related tumors. J Clin Endocrinol Metab (2009) 94(2):386–91. doi: 10.1210/jc.2008-1972
138. Ayala-Ramirez M, Chougnet CN, Habra MA, Palmer JL, Leboulleux S, Cabanillas ME, et al. Treatment with sunitinib for patients with progressive metastatic pheochromocytomas and sympathetic paragangliomas. J Clin Endocrinol Metab (2012) 97(11):4040–50. doi: 10.1210/jc.2012-2356
139. Motzer RJ, Hutson TE, Cella D, Reeves J, Hawkins R, Guo J, et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N Engl J Med (2013) 369(8):722–31. doi: 10.1056/NEJMoa1303989
140. Rini BI, Escudier B, Tomczak P, Kaprin A, Szczylik C, Hutson TE, et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet (2011) 378(9807):1931–9. doi: 10.1016/S0140-6736(11)61613-9
141. Yuan G, Liu Q, Tong D, Liu G, Yi Y, Zhang J, et al. A retrospective case study of sunitinib treatment in three patients with Von Hippel-Lindau disease. Cancer Biol Ther (2018) 19(9):766–72. doi: 10.1080/15384047.2018.1470732
142. O’Kane GM, Ezzat S, Joshua AM, Bourdeau I, Leibowitz-Amit R, Olney HJ, et al. A phase 2 trial of sunitinib in patients with progressive paraganglioma or pheochromocytoma: the SNIPP trial. Br J Cancer (2019) 120(12):1113–9. doi: 10.1038/s41416-019-0474-x
143. Facchin C, Perez-Liva M, Garofalakis A, Viel T, Certain A, Balvay D, et al. Concurrent imaging of vascularization and metabolism in a mouse model of paraganglioma under anti-angiogenic treatment. Theranostics (2020) 10(8):3518–32. doi: 10.7150/thno.40687
144. Denorme M, Yon L, Roux C, Gonzalez BJ, Baudin E, Anouar Y, et al. Both sunitinib and sorafenib are effective treatments for pheochromocytoma in a xenograft model. Cancer Lett (2014) 352(2):236–44. doi: 10.1016/j.canlet.2014.07.005
145. Wallace EM, Rizzi JP, Han G, Wehn PM, Cao Z, Du X, et al. A Small-Molecule Antagonist of HIF2alpha Is Efficacious in Preclinical Models of Renal Cell Carcinoma. Cancer Res (2016) 76(18):5491–500. doi: 10.1158/0008-5472.CAN-16-0473
146. Gunaldi M, Kara IO, Duman BB, Afsar CU, Ergin M, Avci A. A new approach to the treatment of metastatic paraganglioma: sorafenib. Cancer Res Treat (2014) 46(4):411–4. doi: 10.4143/crt.2013.093
147. Jimenez C, Fazeli S, Roman-Gonzalez A. Antiangiogenic therapies for pheochromocytoma and paraganglioma. Endocr Relat Cancer (2020) 27(7):R239–R54. doi: 10.1530/ERC-20-0043
148. Pichun MEB, Edgerly M, Velarde M, Bates SE, Daerr R, Adams K, et al. Phase II clinical trial of axitinib in metastatic pheochromocytomas and paraganlgiomas (P/PG): Preliminary results. J Clin Oncol (2015) 33:457–. doi: 10.1200/jco.2015.33.7_suppl.457
149. Jasim S, Suman VJ, Jimenez C, Harris P, Sideras K, Burton JK, et al. Phase II trial of pazopanib in advanced/progressive malignant pheochromocytoma and paraganglioma. Endocrine (2017) 57(2):220–5. doi: 10.1007/s12020-017-1359-5
150. van Geel RM, Beijnen JH, Schellens JH. Concise drug review: pazopanib and axitinib. Oncologist (2012) 17(8):1081–9. doi: 10.1634/theoncologist.2012-0055
151. Gross-Goupil M, Kwon TG, Eto M, Ye D, Miyake H, Seo SI, et al. Axitinib versus placebo as an adjuvant treatment of renal cell carcinoma: results from the phase III, randomized ATLAS trial. Ann Oncol (2018) 29(12):2371–8. doi: 10.1093/annonc/mdy454
152. Erbel PJ, Card PB, Karakuzu O, Bruick RK, Gardner KH. Structural basis for PAS domain heterodimerization in the basic helix–loop–helix-PAS transcription factor hypoxia-inducible factor. Proc Natl Acad Sci USA (2003) 100(26):15504–9. doi: 10.1073/pnas.2533374100
153. Cho H, Du X, Rizzi JP, Liberzon E, Chakraborty AA, Gao W, et al. On-target efficacy of a HIF-2alpha antagonist in preclinical kidney cancer models. Nature (7627) 2016) 539:107–11. doi: 10.1038/nature19795
154. Chen W, Hill H, Christie A, Kim MS, Holloman E, Pavia-Jimenez A, et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature (2016) 539(7627):112–7. doi: 10.1038/nature19796
155. Courtney KD, Infante JR, Lam ET, Figlin RA, Rini BI, Brugarolas J, et al. Phase I Dose-Escalation Trial of PT2385, a First-in-Class Hypoxia-Inducible Factor-2alpha Antagonist in Patients With Previously Treated Advanced Clear Cell Renal Cell Carcinoma. J Clin Oncol (2018) 36(9):867–74. doi: 10.1200/JCO.2017.74.2627
156. Toledo RA. New HIF2alpha inhibitors: potential implications as therapeutics for advanced pheochromocytomas and paragangliomas. Endocr Relat Cancer (2017) 24(9):C9–C19. doi: 10.1530/ERC-16-0479
157. Xu R, Wang K, Rizzi JP, Huang H, Grina JA, Schlachter ST, et al. 3-[(1S,2S,3R)-2,3-Difluoro-1-hydroxy-7-methylsulfonylindan-4-yl]oxy-5-fluorobenzo nitrile (PT2977), a Hypoxia-Inducible Factor 2alpha (HIF-2alpha) Inhibitor for the Treatment of Clear Cell Renal Cell Carcinoma. J Med Chem (2019) 62(15):6876–93. doi: 10.1021/acs.jmedchem.9b00719
158. Choueiri TK, Plimack ER, Bauer TM, Merchan JR, Papadopoulos KP, McDermott DF, et al. (2019). A First-in-Human Phase 1/2 Trial of the Oral HIF-2α Inhibitor PT2977 in Patients with Advanced RCC, in: Presented at the 14th European International Kidney Cancer Symposium, Dubrovnik, Croatia, March 29–30. doi: 10.1093/annonc/mdz249.010
159. Favier J, Briere JJ, Burnichon N, Riviere J, Vescovo L, Benit P, et al. The Warburg effect is genetically determined in inherited pheochromocytomas. PloS One (2009) 4(9):e7094. doi: 10.1371/journal.pone.0007094
160. Morin A, Goncalves J, Moog S, Castro-Vega LJ, Job S, Buffet A, et al. TET-Mediated Hypermethylation Primes SDH-Deficient Cells for HIF2alpha-Driven Mesenchymal Transition. Cell Rep (2020) 30(13):4551–66.e7. doi: 10.1016/j.celrep.2020.03.022
161. Newman B, Liu Y, Lee HF, Sun D, Wang Y. HSP90 inhibitor 17-AAG selectively eradicates lymphoma stem cells. Cancer Res (2012) 72(17):4551–61. doi: 10.1158/0008-5472.CAN-11-3600
162. Bohonowych JE, Peng S, Gopal U, Hance MW, Wing SB, Argraves KM, et al. Comparative analysis of novel and conventional Hsp90 inhibitors on HIF activity and angiogenic potential in clear cell renal cell carcinoma: implications for clinical evaluation. BMC Cancer (2011) 11:520. doi: 10.1186/1471-2407-11-520
163. Ibrahim NO, Hahn T, Franke C, Stiehl DP, Wirthner R, Wenger RH, et al. Induction of the hypoxia-inducible factor system by low levels of heat shock protein 90 inhibitors. Cancer Res (2005) 65(23):11094–100. doi: 10.1158/0008-5472.CAN-05-1877
164. Wigerup C, Pahlman S, Bexell D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol Ther (2016) 164:152–69. doi: 10.1016/j.pharmthera.2016.04.009
165. Hutt DM, Roth DM, Vignaud H, Cullin C, Bouchecareilh M. The histone deacetylase inhibitor, Vorinostat, represses hypoxia inducible factor 1 alpha expression through translational inhibition. PloS One (2014) 9(8):e106224. doi: 10.1371/journal.pone.0106224
166. Puppo M, Battaglia F, Ottaviano C, Delfino S, Ribatti D, Varesio L, et al. Topotecan inhibits vascular endothelial growth factor production and angiogenic activity induced by hypoxia in human neuroblastoma by targeting hypoxia-inducible factor-1alpha and -2alpha. Mol Cancer Ther (2008) 7(7):1974–84. doi: 10.1158/1535-7163.MCT-07-2059
167. Rapisarda A, Uranchimeg B, Sordet O, Pommier Y, Shoemaker RH, Melillo G. Topoisomerase I-mediated inhibition of hypoxia-inducible factor 1: mechanism and therapeutic implications. Cancer Res (2004) 64(4):1475–82. doi: 10.1158/0008-5472.can-03-3139
168. Bertozzi D, Marinello J, Manzo SG, Fornari F, Gramantieri L, Capranico G, et al. camptothecin, modulates HIF-1alpha activity by changing miR expression patterns in human cancer cells. Mol Cancer Ther (2014) 13(1):239–48. doi: 10.1158/1535-7163.MCT-13-0729
169. Lorusso D, Pietragalla A, Mainenti S, Masciullo V, Di Vagno G, Scambia G. Review role of topotecan in gynaecological cancers: current indications and perspectives. Crit Rev Oncol Hematol (2010) 74(3):163–74. doi: 10.1016/j.critrevonc.2009.08.001
170. Horita N, Yamamoto M, Sato T, Tsukahara T, Nagakura H, Tashiro K, et al. Topotecan for Relapsed Small-cell Lung Cancer: Systematic Review and Meta-Analysis of 1347 Patients. Sci Rep (2015) 5:15437. doi: 10.1038/srep15437
171. Musa F, Blank S, Muggia F. A pharmacokinetic evaluation of topotecan as a cervical cancer therapy. Expert Opin Drug Metab Toxicol (2013) 9(2):215–24. doi: 10.1517/17425255.2013.758249
172. Lee K, Zhang H, Qian DZ, Rey S, Liu JO, Semenza GL. Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc Natl Acad Sci USA (2009) 106(42):17910–5. doi: 10.1073/pnas.0909353106
173. Ma L, Li G, Zhu H, Dong X, Zhao D, Jiang X, et al. 2-Methoxyestradiol synergizes with sorafenib to suppress hepatocellular carcinoma by simultaneously dysregulating hypoxia-inducible factor-1 and -2. Cancer Lett (2014) 355(1):96–105. doi: 10.1016/j.canlet.2014.09.011
174. Zhou X, Liu C, Lu J, Zhu L, Li M. 2-Methoxyestradiol inhibits hypoxia-induced scleroderma fibroblast collagen synthesis by phosphatidylinositol 3-kinase/Akt/mTOR signalling. Rheumatol (Oxford) (2018) 57(9):1675–84. doi: 10.1093/rheumatology/key166
175. Li SH, Shin DH, Chun YS, Lee MK, Kim MS, Park JW. A novel mode of action of YC-1 in HIF inhibition: stimulation of FIH-dependent p300 dissociation from HIF-1{alpha}. Mol Cancer Ther (2008) 7(12):3729–38. doi: 10.1158/1535-7163.MCT-08-0074
176. Lee K, Qian DZ, Rey S, Wei H, Liu JO, Semenza GL. Anthracycline chemotherapy inhibits HIF-1 transcriptional activity and tumor-induced mobilization of circulating angiogenic cells. Proc Natl Acad Sci USA (2009) 106(7):2353–8. doi: 10.1073/pnas.0812801106
177. Pan X, Lv Y. Effects and Mechanism of Action of PX-478 in Oxygen-Induced Retinopathy in Mice. Ophthalmic Res (2020) 63(2):182–93. doi: 10.1159/000504023
178. Lee K, Kim HM. A novel approach to cancer therapy using PX-478 as a HIF-1alpha inhibitor. Arch Pharm Res (2011) 34(10):1583–5. doi: 10.1007/s12272-011-1021-3
179. Sun K, Halberg N, Khan M, Magalang UJ, Scherer PE. Selective inhibition of hypoxia-inducible factor 1alpha ameliorates adipose tissue dysfunction. Mol Cell Biol (2013) 33(5):904–17. doi: 10.1128/MCB.00951-12
180. Coltella N, Valsecchi R, Ponente M, Ponzoni M, Bernardi R. Synergistic Leukemia Eradication by Combined Treatment with Retinoic Acid and HIF Inhibition by EZN-2208 (PEG-SN38) in Preclinical Models of PML-RARalpha and PLZF-RARalpha-Driven Leukemia. Clin Cancer Res (2015) 21(16):3685–94. doi: 10.1158/1078-0432.CCR-14-3022
181. Jeong W, Rapisarda A, Park SR, Kinders RJ, Chen A, Melillo G, et al. Pilot trial of EZN-2968, an antisense oligonucleotide inhibitor of hypoxia-inducible factor-1 alpha (HIF-1alpha), in patients with refractory solid tumors. Cancer Chemother Pharmacol (2014) 73(2):343–8. doi: 10.1007/s00280-013-2362-z
182. Greenberger LM, Horak ID, Filpula D, Sapra P, Westergaard M, Frydenlund HF, et al. A RNA antagonist of hypoxia-inducible factor-1alpha, EZN-2968, inhibits tumor cell growth. Mol Cancer Ther (2008) 7(11):3598–608. doi: 10.1158/1535-7163.MCT-08-0510
183. Kung AL, Zabludoff SD, France DS, Freedman SJ, Tanner EA, Vieira A, et al. Small molecule blockade of transcriptional coactivation of the hypoxia-inducible factor pathway. Cancer Cell (2004) 6(1):33–43. doi: 10.1016/j.ccr.2004.06.009
184. Kong D, Park EJ, Stephen AG, Calvani M, Cardellina JH, Monks A, et al. Echinomycin, a small-molecule inhibitor of hypoxia-inducible factor-1 DNA-binding activity. Cancer Res (2005) 65(19):9047–55. doi: 10.1158/0008-5472.CAN-05-1235
185. Hu N, Jiang D, Huang E, Liu X, Li R, Liang X, et al. BMP9-regulated angiogenic signaling plays an important role in the osteogenic differentiation of mesenchymal progenitor cells. J Cell Sci (2013) 126(Pt 2):532–41. doi: 10.1242/jcs.114231
186. Narita T, Yin S, Gelin CF, Moreno CS, Yepes M, Nicolaou KC, et al. Identification of a novel small molecule HIF-1alpha translation inhibitor. Clin Cancer Res (2009) 15(19):6128–36. doi: 10.1158/1078-0432.CCR-08-3180
187. Lee SH, Jee JG, Bae JS, Liu KH, Lee YM. A group of novel HIF-1alpha inhibitors, glyceollins, blocks HIF-1alpha synthesis and decreases its stability via inhibition of the PI3K/AKT/mTOR pathway and Hsp90 binding. J Cell Physiol (2015) 230(4):853–62. doi: 10.1002/jcp.24813
188. Pham TH, Lecomte S, Efstathiou T, Ferriere F, Pakdel F. An Update on the Effects of Glyceollins on Human Health: Possible Anticancer Effects and Underlying Mechanisms. Nutrients (2019) 11(1):79. doi: 10.3390/nu11010079
189. Kubo T, Maezawa N, Osada M, Katsumura S, Funae Y, Imaoka S, et al. an environmental endocrine-disrupting chemical, inhibits hypoxic response via degradation of hypoxia-inducible factor 1alpha (HIF-1alpha): structural requirement of bisphenol A for degradation of HIF-1alpha. Biochem Biophys Res Commun (2004) 318(4):1006–11. doi: 10.1016/j.bbrc.2004.04.125
190. Fu B, Xue J, Li Z, Shi X, Jiang BH, Fang J. Chrysin inhibits expression of hypoxia-inducible factor-1alpha through reducing hypoxia-inducible factor-1alpha stability and inhibiting its protein synthesis. Mol Cancer Ther (2007) 6(1):220–6. doi: 10.1158/1535-7163.MCT-06-0526
191. Lee K, Kang JE, Park SK, Jin Y, Chung KS, Kim HM, et al. LW6, a novel HIF-1 inhibitor, promotes proteasomal degradation of HIF-1alpha via upregulation of VHL in a colon cancer cell line. Biochem Pharmacol (2010) 80(7):982–9. doi: 10.1016/j.bcp.2010.06.018
192. Kim YH, Coon A, Baker AF, Powis G. Antitumor agent PX-12 inhibits HIF-1alpha protein levels through an Nrf2/PMF-1-mediated increase in spermidine/spermine acetyl transferase. Cancer Chemother Pharmacol (2011) 68(2):405–13. doi: 10.1007/s00280-010-1500-0
193. Jordan BF, Runquist M, Raghunand N, Gillies RJ, Tate WR, Powis G, et al. The thioredoxin-1 inhibitor 1-methylpropyl 2-imidazolyl disulfide (PX-12) decreases vascular permeability in tumor xenografts monitored by dynamic contrast enhanced magnetic resonance imaging. Clin Cancer Res (2005) 11(2 Pt 1):529–36.
194. Zhang L, Chen C, Duanmu J, Wu Y, Tao J, Yang A, et al. Cryptotanshinone inhibits the growth and invasion of colon cancer by suppressing inflammation and tumor angiogenesis through modulating MMP/TIMP system, PI3K/Akt/mTOR signaling and HIF-1alpha nuclear translocation. Int Immunopharmacol (2018) 65:429–37. doi: 10.1016/j.intimp.2018.10.035
195. Miranda E, Nordgren IK, Male AL, Lawrence CE, Hoakwie F, Cuda F, et al. A cyclic peptide inhibitor of HIF-1 heterodimerization that inhibits hypoxia signaling in cancer cells. J Am Chem Soc (2013) 135(28):10418–25. doi: 10.1021/ja402993u
Keywords: pheochromocytomas, paragangliomas, VHL, HIF, metabolism, inhibitor
Citation: Peng S, Zhang J, Tan X, Huang Y, Xu J, Silk N, Zhang D, Liu Q and Jiang J (2020) The VHL/HIF Axis in the Development and Treatment of Pheochromocytoma/Paraganglioma. Front. Endocrinol. 11:586857. doi: 10.3389/fendo.2020.586857
Received: 24 July 2020; Accepted: 23 October 2020;
Published: 24 November 2020.
Edited by:
Alfred King-yin Lam, Griffith University, AustraliaReviewed by:
Judith Favier, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceKan Gong, Peking University First Hospital, China
Copyright © 2020 Peng, Zhang, Tan, Huang, Xu, Silk, Zhang, Liu and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Jiang, amlhbmdqdW5fNjRAMTYzLmNvbQ==; Qiuli Liu, bGl1cWl1bGk5MDA4MjdAMTYzLmNvbQ==