 Julia Hanchard1,2,3†
Julia Hanchard1,2,3† Coral M. Capó-Vélez1†
Coral M. Capó-Vélez1† Kai Deusch1
Kai Deusch1 Darcy Lidington1,2,3
Darcy Lidington1,2,3 Steffen-Sebastian Bolz1,2,3*
Steffen-Sebastian Bolz1,2,3*- 1Aphaia Pharma AG, Zug, Switzerland
- 2Department of Physiology, University of Toronto, Toronto, ON, Canada
- 3Toronto Centre for Microvascular Medicine at The Ted Rogers Centre for Heart Research Translational Biology and Engineering Program, University of Toronto, Toronto, ON, Canada
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and its clinical manifestation (COVID-19; coronavirus disease 2019) have caused a worldwide health crisis. Disruption of epithelial and endothelial barriers is a key clinical turning point that differentiates patients who are likely to develop severe COVID-19 outcomes: it marks a significant escalation in respiratory symptoms, loss of viral containment and a progression toward multi-organ dysfunction. These barrier mechanisms are independently compromised by known COVID-19 risk factors, including diabetes, obesity and aging: thus, a synergism between these underlying conditions and SARS-CoV-2 mechanisms may explain why these risk factors correlate with more severe outcomes. This review examines the key cellular mechanisms that SARS-CoV-2 and its underlying risk factors utilize to disrupt barrier function. As an outlook, we propose that glucagon-like peptide 1 (GLP-1) may be a therapeutic intervention that can slow COVID-19 progression and improve clinical outcome following SARS-CoV-2 infection. GLP-1 signaling activates barrier-promoting processes that directly oppose the pro-inflammatory mechanisms commandeered by SARS-CoV-2 and its underlying risk factors.
Introduction
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) outbreak, first reported in December 2019 in Wuhan, China (1), rapidly evolved into a global pandemic. Comprehensive lock-down measures in most affected jurisdictions have slowed down the spread of the virus, however, in the face of the massive collateral societal and economic damage, these measures are clearly unsustainable. The core issue at hand is the lack of specific treatments for SARS-CoV-2: several antiviral strategies are undergoing clinical study, but thus far, efficacy is limited (2, 3); vaccines are in development, but these will likely not be ready in time to mitigate the current wave or prevent a second global wave. Thus, it is incumbent that we identify interventions that increase the resilience to SARS-CoV-2 infection, particularly in populations at risk of severe responses. To do so, a comprehensive understanding of the disease pathology and the risk factors that increase susceptibility for severe disease progression is required.
SARS-CoV-2 infection can be symptomless in some individuals, while on the other end of the continuum, severe cases elicit terminal multi-organ failure (4, 5). The majority of cases are mild; ~20% of cases require clinical intervention, with ~5% progressing to critically ill stages where mortality is high (5.8% global case fatality ratio, ranging from 0.1 to 16% by country) (1, 6, 7). Overall susceptibility, the likelihood of developing severe symptoms, and mortality all correlate with several known risk factors, including high BMI (>30) (8), diabetes (9–11), hypertension (8, 10) and age [corrected for comorbidities (8–10)]. In all likelihood, these risk factors compromise immune responses and/or permit systemic viral entry and replication. With regard to the latter, the SARS-CoV-2 virus is capable of compromising two critical barriers: the epithelial-endothelial barrier in lung alveoli and the vascular endothelial barrier in the systemic circulation. Indeed, alveolar-endothelial barrier failure is likely the key turning point differentiating patients who will quickly worsen into severe cases, as it marks a significant escalation in respiratory symptoms, the loss of viral containment and a progression toward multi-organ dysfunction (12–14).
This review will focus on the key cellular mechanisms that SARS-CoV-2 utilizes to disrupt epithelial and endothelial barriers. These barrier mechanisms are independently compromised by known coronavirus disease 2019 (COVID-19) risk factors; a combination effect may explain why these risk factors correlate with more severe outcomes. As an outlook, we propose a therapeutic intervention that may slow COVID-19 progression and improve clinical outcome following SARS-CoV-2 infection. In this regard, glucagon-like peptide 1 (GLP-1) signaling activates barrier-promoting processes that directly oppose the pro-inflammatory mechanisms commandeered by SARS-CoV-2 and its underlying risk factors. Thus, medications that stimulate GLP-1 signaling, e.g., exendin-4, may have unappreciated utility for COVID-19 treatment.
Infection Mechanism
The mechanisms mediating SARS-CoV-2 infection and viral replication are already defined and will not be described in detail in this review (15–17). Briefly, host cells must express two components that are critical for SARS-CoV-2 infection: (i) angiotensin converting enzyme 2 (ACE2), the surface receptor that mediates viral attachment to the host cell, and (ii) the transmembrane serine protease TMPRSS2, which cleaves the viral spike protein, thereby priming viral fusion to the host cell's membrane (16, 18). All barrier forming cells, including lung epithelial cells (19–21), enteric epithelial cells (22, 23) and vascular endothelial cells (22) express ACE2 and TMPRSS2 in high abundance and therefore, are targeted by the SARS-CoV-2 virus. An additional element, named tumor necrosis factor converting enzyme (TACE; ADAM17), may facilitate viral entry, although the molecular mechanisms mediating the enhanced entry have not been defined (24, 25).
In the absence of an effective vaccine, intervention strategies have primarily focussed on reducing (i) SARS-CoV-2 fusion/entry, (ii) SARS-CoV-2 replication and (iii) excessive inflammation (2, 3). Obviously, preventing SARS-CoV-2 infection is more desirable than reacting to infection: thus, targeting ACE2/SARS-CoV-2 binding and TMPRSS2 activity, the crucial host proteins involved in viral entry, are highly attractive therapeutic strategies. In this regard, a clinical-grade recombinant ACE2 decoy receptor (26) and the clinically available TMPRSS2 inhibitor camostat (18) have displayed positive results in vitro; however, these strategies have yet to be assessed in clinical trials and are currently a long way from the patient's bedside. In fact, after months of intensive study, most medications repurposed to combat COVID-19, including the notable candidates hydroxychloroquine (27) and lopinavir-ritonavir (28), have failed to demonstrate benefit in randomized placebo-controlled clinical trials. At present, remdesivir, an adenosine nucleotide analog that hampers viral replication (29), is the only candidate (30, 31) with an active FDA emergency use authorization (EUA) at present. However, remdesivir is clearly not a magic bullet intervention (31) and may yet fail to demonstrate benefit in properly powered randomized placebo-controlled clinical trials.
Since targeting SARS-CoV-2 viral entry and replication has not been successful to date, “containing” the virus to the respiratory tract is of paramount importance. Since SARS-CoV-2 is predominantly transmitted through the inhalation of airborne droplets and aerosols, epithelial cells within the upper and lower respiratory tract are the first barriers to be attacked. If the virus breaches this barrier and enters the cardiovascular system, the virus will have the opportunity to infect every organ in the body via the microcirculation (32). Indeed, pronounced vascular injury in association with diffuse alveolar damage is a key feature of SARS-CoV, a relative of SARS-CoV-2 that also targets ACE2 (33).
Possible Routes to the Systemic Circulation
In most cases, SARS-CoV-2 remains confined to the upper respiratory tract, favoring mild symptoms. Epithelial cell infection in the upper airways is associated with copious viral shedding, high person-to-person transmissibility, occasional loss of olfaction, sore throat, fever, and a characteristic dry cough. The nasal mucosa potentially provides a highly vascularized entry point to the systemic circulation if the virus can alter the properties of the restrictive tight junctions in the nasopharyngeal epithelium and underlying microvascular endothelium (34).
From the upper respiratory tract, the SARS-CoV-2 may descend down the trachea and infect cells in the lower respiratory tract and alveoli. At the bronchiolar level, SARS-CoV-2 can infect epithelial goblet cells (35), resulting in airway inflammation and mucous secretion. The inflammatory response subsequently impairs mucociliary clearance, which hampers the clearance of the viral particles, and elicits complications such as bronchiectasis and bronchial wall thickening (36). At the alveolar level, infection and subsequent disruption of the “blood-air barrier,” which comprises alveolar epithelial cells and pulmonary microvascular endothelial cells, is a central event in disease progression: in essence, it is a transition point from relatively moderate symptoms to the severe respiratory symptoms and lung injury observed in severe COVID-19 cases. The compromised barrier becomes leaky, permitting alveolar fluid accumulation (edema), and the development of pneumonia and inflammatory cell infiltration. The resulting hypoxia and damage unleashes a “cytokine storm” in a subset of patients that perpetuates a vicious cycle of progressive lung injury, as the inflammatory response further damages pulmonary cells and compromises endothelial function and barrier integrity (37). In addition to driving severe lung injury, the breach of the blood-air barrier permits viral entry into the systemic circulation, where the virus can then cause widespread multi-organ damage (12, 14).
Tace Is a Key Driver of SARS-COV-2 Severity
Not all coronavirus infections elicit severe respiratory system injury and multiorgan damage: for example, HNL63-CoV, a coronavirus that also binds to ACE2 (38), generally causes relatively mild common cold symptoms (39). Although HNL63-CoV and SARS-CoV-2 bind to the same surface receptor, a key difference between the two viruses resides in the activation of TACE: SARS-CoV strongly activates TACE sheddase activity (24, 25), while HNL63-CoV does not (24). This suggests that TACE activation is a key underlying aspect of the SARS-CoV-2 disease severity.
TACE has more than 80 known substrates, including growth factors, cytokines, cell surface receptors and adhesion molecules (40) and hence, plays complex roles in many regulatory processes (40): thus, it is not surprising that perturbing normal TACE function yields a broad spectrum of deleterious effects. In the context of the COVID-19 pathology, three particular TACE substrates stand out: ACE2 (41), tumor necrosis factor (TNF) (4, 42), and the endothelial protein C receptor (EPCR) (43). All three of these proteins play important anti-inflammatory and barrier-stabilization roles: TACE-dependent shedding of these cell surface proteins, therefore, shifts a delicate balance in favor of inflammation and reduced barrier integrity (Figure 1).
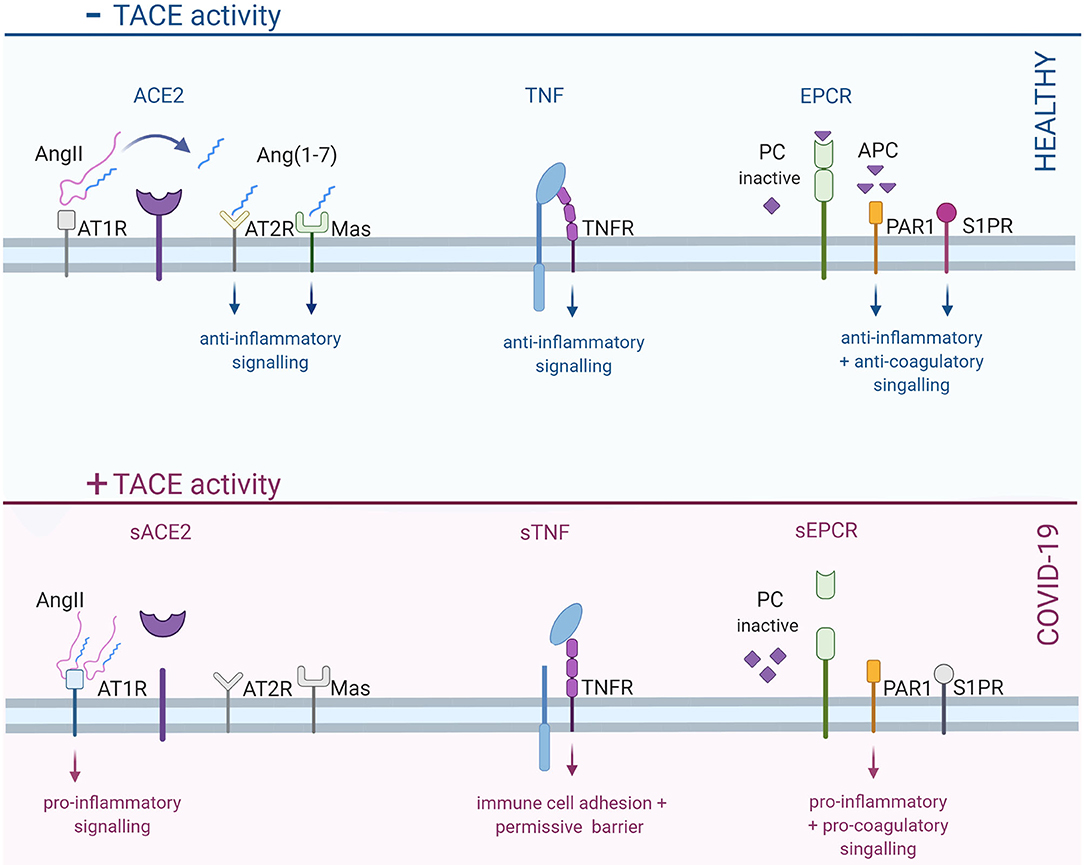
Figure 1. Increased TACE activity in COVID-19 shifts barrier cells toward a pro-inflammatory, pro-coagulatory and permissive state. In healthy cells, TACE is predominantly inactive, thereby permitting anti-inflammatory signals via TACE substrates, including ACE2, TNF, and EPCR. ACE2, a key player in the RAAS system, converts angiotensin II (AngII) into angiotensin-1-7 [Ang (1–7)]: the subsequent activation of AT2R and Mas receptors stimulates anti-inflammatory signals. When ACE2 is cleaved, angiotensin signaling shifts from AT2R/Mas receptors to the pro-inflammatory AT1R. TNF receptors stimulate differential signals, depending on whether the receptor is activated by the membrane-bound or soluble form of TNF: membrane bound TNF elicits important anti-inflammatory signals; in contrast, soluble TNF mediates pro-inflammatory signals. Finally, EPCR plays a critical role in regulating coagulation by converting inactive protein C (PC) into activated protein C (APC). Activated protein C subsequently inactivates pro-coagulatory factors; it also cleaves PAR1 and transactivates S1PR, resulting anti-inflammatory and barrier-stabilizing signals. TACE-dependent EPCR cleavage prevents the activation of PC, leading to reduced capacity to inhibit coagulation; RhoA-dependent signaling via uncleaved PAR1 is barrier destabilizing. TACE, tumor necrosis factor alpha converting enzyme; ACE2, angiotensin converting enzyme 2; TNF, tumor necrosis factor; EPCR, endothelial protein C receptor; RAAS, rennin-angiotensin-aldosterone system; AT2R, angiotensin receptor 2; AT1R, angiotensin receptor 1; PAR1, protease activated receptor 1; S1PR, sphingosine-1-phosphate receptor; TNFR, TNF receptor. Created with BioRender.com.
The physiological functions of ACE2 and the implications of ACE2 shedding in COVID-19 have been extensively reviewed by Gheblawi et al. (41). ACE2 is a central element in the Renin-Angiotensin-Aldosterone System (RAAS) and therefore, has wide ranging effects that intersect with virtually every endocrine and inflammatory mechanism (44). At the molecular level, ACE2 converts angiotensin II into angiotensin (1-7): angiotensin II activates the pro-inflammatory angiotensin II receptor subtype 1 (AT1R) (45), while angiotensin (1-7) preferentially activates the anti-inflammatory angiotensin II receptor subtype 2 (AT2R) (46) and Mas receptors (46–48). Thus, TACE-dependent ACE2 shedding (41, 49) in COVID-19 shifts RAAS signaling in favor of the pro-inflammatory AT1 receptors (50), which favors immune cell adhesion (51), cellular damage (51), and increased vascular permeability (via the modulation of VE-cadherin function) (52, 53).
Although TNF is best known as a pro-inflammatory cytokine, TNF serves many developmental, homeostatic and reparative functions (54–56). In pathological settings, TNF is a critical initiating factor in the immune response; importantly, if the initial pathogenic insult and/or tissue damage is severe, the immune response can spiral out-of-control into the highly damaging “cytokine storm” (54–58). At the level of the endothelium, TNF stimulates two key homeostatic changes: (i) the expression of immune cell adhesion molecules (e.g., VCAM-1 and ICAM1) (59, 60), and (ii) an increase in barrier permeability, via cytoskeletal rearrangement (61, 62) and the regulation of cell-to-cell adhesion junctions (63, 64). These changes allow immune cells to bind to the endothelium at the site of injury/infection and transmigrate into the tissue through the paracellular junctions (54, 58). Among other notable acute effects, TNF also simulates endothelial reactive oxygen species generation and impairs nitric oxide production (54), which can have significant effects on tissue damage and vascular control mechanisms (65, 66). In COVID-19, TACE-dependent TNF shedding favors the rapid breakdown of the endothelial-alveolar barrier, resulting in lung edema, immune cell infiltration, and ultimately, lung tissue damage. This barrier breakdown also opens the gateway to the systemic circulation: once distal endothelial cells are infected; the same inflammatory mechanism provides a means for the virus to escape the systemic circulation and into organ parenchymal cells.
The endothelial protein C receptor (EPCR) (67, 68) is best known for its anti-coagulatory functions (69); however, EPCR signaling also plays an important role in moderating inflammation (70–72), maintaining endothelial barrier function (73, 74) and conferring cytoprotection (75–77). EPCR binds the zymogen protein C and cleaves it into an active protease: this activated form of protein C remains bound to the EPCR and subsequently (i) cleaves protease-activated receptor 1 (PAR1) and (ii) transactivates the sphingosine-1-phosphate receptor 1 subtype (S1PR1) (67, 68). The activation of these receptors inhibits nuclear factor-κB (NF-κB) translocation/signaling, thereby reducing proinflammatory gene expression, the release of cytokines, and the expression of adhesion molecules (67, 68). In addition, activated protein C shifts PAR1 signals from RhoA-dependent, barrier permeabilizing actions to Rac1-dependent, barrier stabilizing actions (67, 68). In the context of COVID-19, TACE-dependent EPCR shedding removes an important molecular brake that dampens immune cell infiltration, edema and tissue damage; by eliminating a barrier stabilization mechanism, the loss of EPCR signaling also undermines viral containment. Finally, deficient EPCR signaling, a key brake element in the coagulation pathway, likely also contributes to the high incidence of thrombotic complications observed in serious COVID-19 cases, including widespread microthrombi, pulmonary embolism, stroke and disseminated intravascular coagulation (12, 78, 79).
In summary, the activation of TACE may significantly augment the inflammatory response following SARS-CoV-2 infection. As a component of the inflammation, endothelial permeability may become severely compromised, permitting the SARS-CoV-2 virus with access to the systemic circulation. Given the remarkably large surface area of the microvascular endothelium (3,000-4,000 m2) and its presence in every tissue, the failure to maintain this barrier eliminates the last line of defense against multi-organ damage and failure.
COVID-19 Risk Factors That Compromise Barrier Function
Several cardiovascular risk factors, including hypertension, obesity, type 2 diabetes mellitus (T2D) and cardiovascular disease, are common underlying conditions in COVID-19 patients (80, 81). Although these risk factors tend to associate with more severe cases, the individual contribution of each risk factor to COVID-19 disease severity/outcome is difficult to define, due to the small sample size of most studies, combined with the fact that these risk factors largely overlap [e.g., 52% of patients with T2D are obese; and 60% of obese patients have metabolic syndrome including hypertension as a leading symptom (82–85)]. To add a further challenge, age is a significant COVID-19 risk factor (86) that has generally not been taken into account in most risk factor studies (81). According to Tadic et al. (81), most risk assessment studies are too small, inconsistent, and fail to account for several confounding factors, most notably age and obesity: consequently, they are ill-equipped to discern important interactions between comorbidities and outcomes. Thus, the data are more epidemiological than analytical and should be viewed cautiously (81).
Although the precise relationships between COVID-19 risk factors and disease severity require clarification, there are obvious mechanistic commonalities across these risk factors that permits speculation as to why certain common underlying conditions appear to cluster with more severe outcomes. In this sense, we can predict that the outcomes should be more severe, based on deleterious mechanisms that are already activated prior to infection. As a prime example, metabolic conditions such as obesity and T2D down-regulate tissue inhibitor of metalloproteinase 3 (TIMP3), an important endogenous TACE inhibitor that critically regulates TACE and the release of TNF in metabolic tissues (87, 88). The resulting increase in TACE activity induces a proinflammatory state (e.g., cytokine production, immune cell adhesion molecule expression, and hyperpermeability) and pronounced endothelial dysfunction (e.g., reactive oxygen species generation and impaired autacoid release) (89–92) observed in obese and T2D patients. Aging is a double-hit: in addition to inducing pro-inflammatory endothelial dysfunction (93, 94), it also diminishes infection defense (“immune senescence”) (95, 96).
A common thread across these risk factors is chronic low-level inflammation and endothelial dysfunction: these risk factors facilitate infection, prime an exaggerated immune response and/or compromise viral containment. Thus, it is entirely reasonable to expect that these underlying conditions would synergize with SARS-CoV-2 infection mechanisms to more profoundly erode barrier function and drive more severe disease progression. These underlying conditions also profoundly impact the patient's immunological status, which is an obvious determinant of COVID-19 severity (97). Indeed, deep immune profiling reveals that hospitalized patients fall across a spectrum of immune response patterns (98), with underlying conditions contributing to the diversity in host responses.
GLP-1 Agonists: a Potential Intervention for Mitigating COVID-19
Glucagon-like-peptide 1 (GLP-1) is an enteroendocrine hormone, originally characterized in the orchestration of insulin release in response to ingested nutritional stimuli (99, 100). However, GLP-1 signaling plays a vital role in energy metabolism and cell viability in several tissues: thus, targeting GLP-1 receptors can potentially elicit systems-level effects (101). In this regard, GLP-1 has emerged as an important homeostatic element within the cardiovascular system, where it possesses significant endothelial-protective functions (102, 103). It is also interesting to note that all of the barrier-forming cells in the lung and vascular system express GLP-1 receptors (104–108).
In the lung, GLP-1 tightens barriers via the upregulation of tight junction proteins in barrier-forming cells (108, 109); in alveolar type 2 pneumocytes, GLP-1 stimulates the production of surfactant that, by reducing surface tension, helps to minimize fluid accumulation within alveolar spaces (110). In endothelial cells, GLP-1 inhibits TACE expression and activity (111): it therefore directly opposes key mechanisms that SARS-CoV-2 commandeers to augment inflammation and compromise barrier function. Accordingly, GLP-1 signaling attenuates TACE-dependent EPCR shedding (111); GLP-1 signaling also increases deficient ACE2 levels in pathological settings (112), presumably via reduced ACE2 shedding. Consistent with this anti-inflammatory role, GLP-1 receptor agonists possess several desirable actions, including (i) antagonizing inflammatory NF-κB signaling (103), (ii) reducing immune cell adhesion molecule expression on endothelial cell surfaces (e.g., ICAM-1 and VCAM-1) (103, 113), (iii) reducing immune cells cytokine production (113), and (iv) attenuating endothelial cell oxidative stress (114).
With regard to the risk factors associated with severe COVID-19 cases, it is remarkable to note that GLP-1 receptor agonists (e.g., exendin-4, liraglutide, semaglutide) are either FDA-approved (diabetes) (115) or proposed (obesity, age-related decline) (116, 117) as an intervention for the underlying conditions. GLP-1 receptor agonists were specifically developed to harness GLP-1's potent hypoglycemic effect in the treatment of diabetes (115): the improved glycemic control results in weight loss in T2D patients and consequently, clinical trials are currently assessing their utility as an anti-obesity drug for patients without T2D (116). In addition to its functions as a metabolic hormone, GLP-1 possesses significant anti-inflammatory effects that are independent of glucose homeostasis (118). Consequently, GLP-1 receptor agonists are now being considered for age-related pathologies, including Alzheimer's disease, Parkinson's disease, and cognitive decline, all of which possess a strong inflammatory component (117, 119). Since GLP-1 signaling exerts clear beneficial effects in obesity, T2D and aging, it must potently ameliorate a common pathological thread across these conditions: chronic low-level inflammation and endothelial dysfunction. Since (i) both the risk factors for severe COVID-19 (e.g., obesity, diabetes, age) and the SARS-CoV-2 virus harness similar pro-inflammatory mechanisms and (ii) GLP-1 signaling is anti-inflammatory and has demonstrable benefits in patients with underlying conditions (118), it stands to reason that GLP-1 signaling should directly oppose the inflammatory mechanisms activated in COVID-19. In this context, GLP-1 agonists would be a useful mechanism-based treatment strategy (Figure 2).
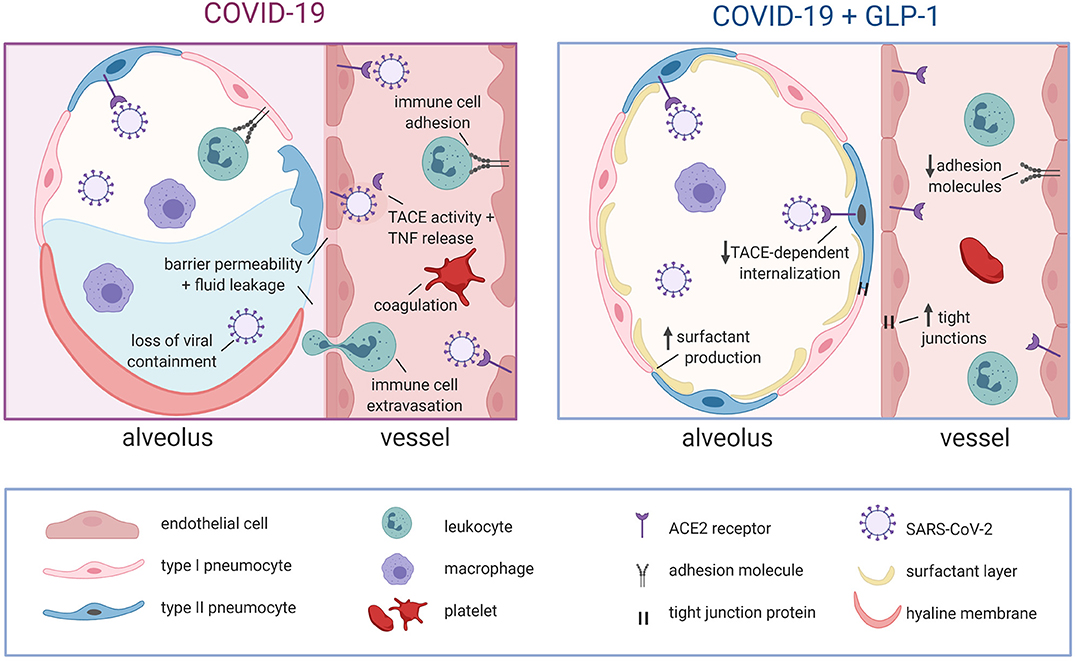
Figure 2. Proposed mechanisms for GLP-1-mediated stabilization of alveolar epithelial and endothelial barriers in COVID-19. In COVID-19, alveolar epithelial cells are severely damaged by direct viral attack and the immune response, leading to hyaline membrane formation, increased barrier permeability, alveolar fluid accumulation and a loss of viral containment. Viral attack on the underlying endothelial cells leads to TACE activation that: augments pro-inflammatory signaling, increases endothelial barrier permeability, promotes immune cell adhesion/extravasation and compromises EPCR-dependent regulation of coagulation. Notably, the loss of endothelial barrier integrity further exacerbates alveolar fluid accumulation. GLP-1 receptor signaling directly opposes these pathological mechanisms by: attenuating TACE expression/activity, initiating counteracting anti-inflammatory signals, promoting barrier function via increased tight junction expression and stimulating surfactant secretion (reduces alveolar fluid accumulation by decreasing surface tension). The GLP-1 mediated attenuation of TACE activity/expression is also expected to hinder viral entry into cells. GLP-1, glucagon like peptide 1; ACE2, angiotensin converting enzyme 2; TACE, tumor necrosis factor alpha converting enzyme; TNF, tumor necrosis factor; EPCR, endothelial protein C receptor. Created with BioRender.com.
GLP-1 Secretagogues: a Novel Opportunity for Prophylaxis
Given that GLP-1 signaling confers many positive benefits, especially in individuals with COVID-19 risk factors (118, 120, 121), it is intriguing to hypothesize that the benefits of GLP-1 can be harnessed prophylactically. In essence, the objective would be to activate the endogenous barrier-promoting and anti-inflammatory actions of GLP-1 signaling prior to SARS-CoV-2 infection, with the prospect that this would increase resilience in the event of infection. Based on the rapid and dramatic metabolic effects observed in Roux-en-Y Gastric Bypass (RYGB) patients (122, 123) there is no doubt that the intestinal tract has the enteroendocrine capacity to drive significant beneficial effects on the systemic level. Further, extreme measures are not necessary to elicit systemic effects: even normal, post-prandial GLP-1 secretion is sufficient to stimulate nitric oxide production in the forearm, thereby increasing blood flow and oxygen uptake (124, 125). The fact that “normal” enteroendocrine GLP-1 signaling activates the endothelium is important, because it suggests that the full repertoire of positive GLP-1 effects may be in play. Thus, if endogenous GLP-1 signaling could be stably or perpetually activated, it may be possible to increase endothelial and epithelial “resilience” to SARS-CoV-2 infection.
Eliciting prophylactic GLP-1 release from the intestine is likely mechanistically simple, extremely safe, and in all probability, very low cost. Virtually any carbohydrate, lipid or protein macronutrient could be used as a candidate secretagogue, as these nutrients clearly mobilize GLP-1 secretion mechanisms (126–128). Assuming that secretagogues stimulate sufficient GLP-1 release to positively influence inflammatory and barrier-promoting mechanisms at the systemic level, there would be little argument that these stimuli would: (i) be safe to ingest, as they are basic food components, (ii) not cause the adverse effects associated with the supra-physiological levels of GLP-1 signaling elicited by GLP-1 agonists, such as headache, vomiting or diarrhea (129), (iii) be cost effective to manufacture and purchase and (iv) have immediate worldwide availability. This strategy is obviously speculative and not currently available for use against COVID-19; it is nevertheless worth pursuing, as it could have broad implications for patients with obesity, T2D, cardiovascular disease and other pathologies that target the endothelium, inflammatory mechanisms or barrier function.
Summary and Outlook
In summary, the breakdown of cellular barriers is a key driver of severe SARS-CoV-2 disease progression. The underlying pro-inflammatory mechanisms that disrupt barrier function in COVID-19 are well-characterized and substantially overlap with the disease mechanisms operating in diabetes, obesity and aging, all putative COVID-19 risk factors. Since anti-inflammatory interventions that strongly supress immune function (e.g., anti-TNF therapeutics) are not recommended for treating COVID-19, we need to deploy other options that interfere with these barrier-disrupting mechanisms. GLP-1 is an intriguing candidate, because it possesses both anti-inflammatory and barrier-promoting properties. Indeed, GLP-1 signaling is currently proposed as an intervention for the very risk factors that also drive aggravated COVID-19 severity. It is, therefore, tempting to speculate that GLP-1 signaling could be harnessed to fight COVID-19 on two levels: secretagogues could prophylactically increase the global population's resilience to the infection, and in acute COVID-19, GLP-1 receptor agonists may be useful in supporting acute therapeutic interventions.
Author Contributions
JH, CC-V, and S-SB conceived this review. JH and CC-V contributed equally to the literature research and figure preparation. JH, CC-V, KD, DL, and S-SB all significantly contributed to writing and revising the manuscript.
Funding
S-SB receives stipend support from a Heart and Stroke Foundation of Ontario Mid-Career Investigator award. Aphaia Pharma AG provided funding for publication charges, and had no other involvement in the study.
Conflict of Interest
S-SB and KD are executive board members of Aphaia Pharma AG, JH and CC-V are employees of Aphaia Pharma AG, and DL is a paid consultant for Aphaia Pharma AG.
References
1. Li H, Liu L, Zhang D, Xu J, Dai H, Tang N, et al. SARS-CoV-2 and viral sepsis: observations and hypotheses. Lancet. (2020) 395:1517–20. doi: 10.1016/S0140-6736(20)30920-X
2. Jean S-S, Lee P-I, Hsueh P-R. Treatment options for COVID-19: The reality and challenges. J Microbiol Immunol Infect. (2020) 53:436–43. doi: 10.1016/j.jmii.2020.03.034
3. Sanders JM, Monogue ML, Jodlowski TZ, Cutrell JB. Pharmacologic treatments for coronavirus disease 2019 (COVID-19): a review. JAMA. (2020) 323:1824–36. doi: 10.1001/jama.2020.6019
4. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
5. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. (2020) 8:420–2. doi: 10.1016/S2213-2600(20)30076-X
6. Guan W, Ni Z, Hu Y, Liang W, Ou C, He J, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. (2020) 382:1708–20. doi: 10.1056/NEJMoa2002032
7. World Health Organization. Coronavirus Disease (COVID-19) Situation Report. (2020). Available online at: https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200617-covid-19-sitrep-149.pdf?sfvrsn=3b3137b0_4 (accessed June 17, 2020).
8. The Open SAFELY Collaborative Williamson E, Walker A, Krishnan B, Bacon S, Bates C. OpenSAFELY: factors associated with COVID-19-related hospital death in the linked electronic health records of 17 million adult NHS patients. bioRxiv [Preprint]. (2020). Available at: https://doi.org/10.1101/2020.05.06.20092999 (accessed August 11, 2020).
9. Barron E, Bakhai C, Kar P, Weaver A, Bradley D, Ismail H, et al. Type 1 and type 2 diabete s and COVID-19 relate d mortality in England: a whole population study. EHS England. (2020). Available online at: https://www.england.nhs.uk/publication/type-1-and-type-2-diabetes-and-covid-19-related-mortality-in-england (accessed August 11, 2020)
10. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. (2020) 395:1054–62. doi: 10.1016/S0140-6736(20)30566-3
11. Zhu L, She Z-G, Cheng X, Qin J-J, Zhang X-J, Cai J, et al. Association of blood glucose control and outcomes in patients with COVID-19 and pre-existing type 2 diabetes. Cell Metab. (2020) 31:1068–77. doi: 10.1016/j.cmet.2020.04.021
12. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. (2020) 383:120–8. doi: 10.1056/NEJMoa2015432
13. Teuwen L-A, Geldhof V, Pasut A, Carmeliet P. COVID-19: the vasculature unleashed. Nat Rev Immunol. (2020) 20:389–91. doi: 10.1038/s41577-020-0343-0
14. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. (2020) 395:1417–8. doi: 10.1016/S0140-6736(20)30937-5
15. Fehr AR, Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Coronaviruses. (2015) 1282:1–23. doi: 10.1007/978-1-4939-2438-7_1
16. Hartenian E, Nandakumar D, Lari A, Ly M, Tucker JM, Glaunsinger BA. The molecular virology of coronaviruses. J Biol Chem. (2020). doi: 10.1074/jbc.REV120.013930. [Epub ahead of print].
17. Tay MZ, Poh CM, Rénia L, MacAry PA, Ng LFP. The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol. (2020) 20:363–74. doi: 10.1038/s41577-020-0311-8
18. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. (2020) 181:271–80.e8. doi: 10.1016/j.cell.2020.02.052
19. Xu H, Zhong L, Deng J, Peng J, Dan H, Zeng X, et al. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int J Oral Sci. (2020) 12:1–5. doi: 10.1038/s41368-020-0074-x
20. Zhao Y, Zhao Z, Wang Y, Zhou Y, Ma Y, Zuo W. Single-cell RNA expression profiling of ACE2, the putative receptor of SARS-CoV-2. Am J Respir Crit Care Med. (2020) 202:756–9. doi: 10.1164/rccm.202001-0179LE
21. Zou X, Chen K, Zou J, Han P, Hao J, Han Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front Med. (2020) 14:185–92. doi: 10.1007/s11684-020-0754-0
22. Hamming I, Timens W, Bulthuis M, Lely A, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. (2004) 203:631–7. doi: 10.1002/path.1570
23. Hikmet F, Méar L, Edvinsson Å, Micke P, Uhlén P, Lindskog C. The protein expression profile of ACE2 in human tissues. Mol Syst Biol. (2020) 16:e9610. doi: 10.15252/msb.20209610
24. Haga S, Yamamoto N, Nakai-Murakami C, Osawa Y, Tokunaga K, Sata T, et al. Modulation of TNF-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF production and facilitates viral entry. Proc Natl Acad Sci USA. (2008) 105:7809–14. doi: 10.1073/pnas.0711241105
25. Haga S, Nagata N, Okamura T, Yamamoto N, Sata T, Yamamoto N, et al. TACE antagonists blocking ACE2 shedding caused by the spike protein of SARS-CoV are candidate antiviral compounds. Antiviral Res. (2010) 85:551–5. doi: 10.1016/j.antiviral.2009.12.001
26. Monteil V, Kwon H, Prado P, Hagelkrüys A, Wimmer RA, Stahl M, et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell. (2020) 181:905–13.e7. doi: 10.1016/j.cell.2020.04.004
27. Boulware DR, Pullen MF, Bangdiwala AS, Pastick KA, Lofgren SM, Okafor EC, et al. A randomized trial of hydroxychloroquine as postexposure prophylaxis for Covid-19. N Engl J Med. (2020) 383:517–25. doi: 10.1056/NEJMoa2016638
28. Cao B, Wang Y, Wen D, Liu W, Wang J, Fan G, et al. A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe Covid-19. N Engl J Med. (2020) 382:1787–99. doi: 10.1056/NEJMoa2001282
29. Agostini ML, Andres EL, Sims AC, Graham RL, Sheahan TP, Lu X, et al. Coronavirus susceptibility to the antiviral remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. mBio. (2018) 9:e00221–18. doi: 10.1128/mBio.00221-18
30. Grein J, Ohmagari N, Shin D, Diaz G, Asperges E, Castagna A, et al. Compassionate use of remdesivir for patients with severe Covid-19. N Engl J Med. (2020) 382:2327–36. doi: 10.1056/NEJMoa2007016
31. Wang Y, Zhang D, Du G, Du R, Zhao J, Jin Y, et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. (2020) 395:1569–78. doi: 10.1016/S0140-6736(20)31022-9
32. Bourgonje AR, Abdulle AE, Timens W, Hillebrands J-L, Navis GJ, Gordijn SJ, et al. Angiotensin-converting enzyme-2 (ACE2), SARS-CoV-2 and pathophysiology of coronavirus disease 2019 (COVID-19). J. Pathol. (2020) 251:228-48. doi: 10.1002/path.5471
33. Gu J, Korteweg C. Pathology and pathogenesis of severe acute respiratory syndrome. Am J Pathol. (2007) 170:1136–47. doi: 10.2353/ajpath.2007.061088
34. Yan Y, Gordon WM, Wang D-Y. Nasal epithelial repair and remodeling in physical injury, infection, and inflammatory diseases. Curr Opin Otolaryngol Head Neck Surg. (2013) 21:263–70. doi: 10.1097/MOO.0b013e32835f80a0
35. Sungnak W, Huang N, Bécavin C, Berg M, Queen R, Litvinukova M, et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat Med. (2020) 26:681–7. doi: 10.1038/s41591-020-0868-6
36. Inui S, Fujikawa A, Jitsu M, Kunishima N, Watanabe S, Suzuki Y, et al. Chest CT findings in cases from the cruise ship “diamond princess” with coronavirus disease 2019 (COVID-19). Radiol Cardiothorac Imag. (2020) 2:e200110. doi: 10.1148/ryct.2020200110
37. Ye Q, Wang B, Mao J. The pathogenesis and treatment of the ‘cytokine storm' in COVID-19. J Infect. (2020) 80:607–13. doi: 10.1016/j.jinf.2020.03.037
38. Hofmann H, Pyrc K, Hoek L, van der Geier M, Berkhout B, Pöhlmann S. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc Natl Acad Sci USA. (2005) 102:7988–93. doi: 10.1073/pnas.0409465102
39. van der Hoek L, Pyrc K, Berkhout B. Human coronavirus NL63, a new respiratory virus. FEMS Microbiol Rev. (2006) 30:760–73. doi: 10.1111/j.1574-6976.2006.00032.x
40. Zunke F, Rose-John S. The shedding protease ADAM17: physiology and pathophysiology. Biochim Biophys Acta BBA - Mol Cell Res. (2017) 1864:2059–70. doi: 10.1016/j.bbamcr.2017.07.001
41. Gheblawi M, Wang K, Viveiros A, Nguyen Q, Zhong J, Turner AJ, et al. Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system. Circ Res. (2020) 126:1456–74. doi: 10.1161/CIRCRESAHA.120.317015
42. Feldmann M, Maini RN, Woody JN, Holgate ST, Winter G, Rowland M, et al. Trials of anti-tumour necrosis factor therapy for COVID-19 are urgently needed. Lancet. 395:1407–9. doi: 10.1016/S0140-6736(20)30858-8
43. Griffin JH, Lyden P. COVID-19 hypothesis: activated protein C for therapy of virus-induced pathologic thromboinflammation. Res Pract Thromb Haemost. (2020) 4:506–9. doi: 10.1002/rth2.12362
44. Patel S, Rauf A, Khan H, Abu-Izneid T. Renin-angiotensin-aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies. Biomed Pharmacother. (2017) 94:317–25. doi: 10.1016/j.biopha.2017.07.091
45. Kawai T, Forrester SJ, O'Brien S, Baggett A, Rizzo V, Eguchi S. AT1 receptor signaling pathways in the cardiovascular system. Pharmacol Res. (2017) 125:4–13. doi: 10.1016/j.phrs.2017.05.008
46. Matavelli L, Siragy H. AT2 receptor activities and pathophysiological implications. J Cardiovasc Pharmacol. (2015) 65:226–32. doi: 10.1097/FJC.0000000000000208
47. Passos-Silva DG, Verano-Braga T, Santos RAS. Angiotensin-(1–7): beyond the cardio-renal actions. Clin Sci. (2013) 124:443–56. doi: 10.1042/CS20120461
48. Villela D, Leonhardt J, Patel N, Joseph J, Kirsch S, Hallberg A, et al. Angiotensin type 2 receptor (AT2R) and receptor mas: a complex liaison. Clin Sci. (2015) 128:227–34. doi: 10.1042/CS20130515
49. Lambert DW, Yarski M, Warner FJ, Thornhill P, Parkin ET, Smith AI, et al. Tumor necrosis factor-α convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J Biol Chem. (2005) 280:30113–9. doi: 10.1074/jbc.M505111200
50. Tseng Y-H, Yang R-C, Lu T-S. Two hits to the renin-angiotensin system may play a key role in severe COVID-19. Kaohsiung J Med Sci. (2020) 36:389–92. doi: 10.1002/kjm2.12237
51. Zhang Y-H, Zhang Y, Dong X-F, Hao Q-Q, Zhou X-M, Yu Q-T, et al. ACE2 and Ang- (1–7) protect endothelial cell function and prevent early atherosclerosis by inhibiting inflammatory response. Inflamm Res. (2015) 64:253–60. doi: 10.1007/s00011-015-0805-1
52. Guo S, Som AT, Arai K, Lo EH. Effects of angiotensin-II on brain endothelial cell permeability via PPARalpha regulation of para- and trans-cellular pathways. Brain Res. (2019) 1722:146353. doi: 10.1016/j.brainres.2019.146353
53. Li Y, Yao Y, Li J, Chen Q, Zhang L, Wang QK. Losartan protects against myocardial ischemia and reperfusion injury via vascular integrity preservation. FASEB J. (2019) 33:8555–64. doi: 10.1096/fj.201900060R
54. Urschel K, Cicha I. TNF-α in the cardiovascular system: from physiology to therapy. Int J Interferon Cytokine Mediat Res. (2015) 7:9–25. doi: 10.2147/IJICMR.S64894
55. Kalliolias GD, Ivashkiv LB. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheumatol. (2016) 12:49–62. doi: 10.1038/nrrheum.2015.169
56. Clark IA, Vissel B. The meteorology of cytokine storms, and the clinical usefulness of this knowledge. Semin Immunopathol. (2017) 39:505–16. doi: 10.1007/s00281-017-0628-y
57. Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the eye of the cytokine storm. Microbiol Mol Biol Rev. (2012) 76:16–32. doi: 10.1128/MMBR.05015-11
58. D'Elia RV, Harrison K, Oyston PC, Lukaszewski RA, Clark GC. Targeting the “cytokine storm” for therapeutic benefit. Clin Vaccine Immunol. (2013) 20:319–27. doi: 10.1128/CVI.00636-12
59. Bara H, Sambanis A. Insulin-secreting L-cells for the treatment of insulin-dependent diabetes. Biochem Biophys Res Commun. (2008) 371:39–43. doi: 10.1016/j.bbrc.2008.03.154
60. Lin C-C, Pan C-S, Wang C-Y, Liu S-W, Hsiao L-D, Yang C-M. Tumor necrosis factor-alpha induces VCAM-1-mediated inflammation via c-Src-dependent transactivation of EGF receptors in human cardiac fibroblasts. J Biomed Sci. (2015) 22:53. doi: 10.1186/s12929-015-0165-8
61. Goldblum SE, Ding X, Campbell-Washington J. TNF-alpha induces endothelial cell F-actin depolymerization, new actin synthesis, and barrier dysfunction. Am J Physiol - Cell Physiol. (1993) 264:C894–905. doi: 10.1152/ajpcell.1993.264.4.C894
62. Petrache I, Birukova A, Ramirez SI, Garcia JGN, Verin AD. The role of the microtubules in tumor necrosis factor-α-induced endothelial cell permeability. Am J Respir Cell Mol Biol. (2003) 28:574–81. doi: 10.1165/rcmb.2002-0075OC
63. Angelini DJ, Hyun S-W, Grigoryev DN, Garg P, Gong P, Singh IS. TNF-α increases tyrosine phosphorylation of vascular endothelial cadherin and opens the paracellular pathway through fyn activation in human lung endothelia. Am J Physiol-Lung Cell Mol Physiol. (2006) 291:L1232–45. doi: 10.1152/ajplung.00109.2006
64. Ni Y, Teng T, Li R, Simonyi A, Sun GY, Lee JC. TNFα alters occludin and cerebral endothelial permeability: role of p38MAPK. PLoS ONE. (2017) 12:e0170346. doi: 10.1371/journal.pone.0170346
65. Armour J, Tyml K, Lidington D, Wilson JX. Ascorbate prevents microvascular dysfunction in the skeletal muscle of the septic rat. J Appl Physiol. (2001) 90:795–803. doi: 10.1152/jappl.2001.90.3.795
66. Lidington D, Li F, Tyml K. Deletion of neuronal NOS prevents impaired vasodilation in septic mouse skeletal muscle. Cardiovasc Res. (2007) 74:151–8. doi: 10.1016/j.cardiores.2006.12.022
67. Esmon CT. Protein C anticoagulant system—anti-inflammatory effects. Semin Immunopathol. (2012) 34:127–32. doi: 10.1007/s00281-011-0284-6
68. Mohan Rao LV, Esmon CT, Pendurthi UR. Endothelial cell protein C receptor: a multiliganded and multifunctional receptor. Blood. (2014) 124:1553–62. doi: 10.1182/blood-2014-05-578328
69. Stearns-Kurosawa DJ, Kurosawa S, Mollica JS, Ferrell GL, Esmon CT. The endothelial cell protein C receptor augments protein C activation by the thrombin-thrombomodulin complex. Proc Natl Acad Sci USA. (1996) 93:10212–6. doi: 10.1073/pnas.93.19.10212
70. Grey ST, Tsuchida A, Hau H, Orthner CL, Salem HH, Hancock WW. Selective inhibitory effects of the anticoagulant activated protein C on the responses of human mononuclear phagocytes to LPS, IFN-gamma, or phorbol ester. J Immunol Baltim. (1994) 153:3664–72.
71. Murakami K, Okajima K, Uchiba M, Johno M, Nakagaki T, Okabe H, et al. Activated protein C prevents LPS-induced pulmonary vascular injury by inhibiting cytokine production. Am J Physiol. (1997) 272:L197–202. doi: 10.1152/ajplung.1997.272.2.L197
72. White B, Schmidt M, Murphy C, Livingstone W, O'Toole D, Lawler M, et al. Activated protein C inhibits lipopolysaccharide-induced nuclear translocation of nuclear factor κB (NF-κB) and tumour necrosis factor α (TNF-α) production in the THP-1 monocytic cell line. Br J Haematol. (2000) 110:130–4. doi: 10.1046/j.1365-2141.2000.02128.x
73. Feistritzer C, Riewald M. Endothelial barrier protection by activated protein C through PAR1-dependent sphingosine 1–phosphate receptor-1 crossactivation. Blood. (2005) 105:3178–84. doi: 10.1182/blood-2004-10-3985
74. Finigan JH, Dudek SM, Singleton PA, Chiang ET, Jacobson JR, Camp SM, et al. Activated protein C mediates novel lung endothelial barrier enhancement: role of sphingosine 1-phosphate receptor transactivation. J Biol Chem. (2005) 280:17286–93. doi: 10.1074/jbc.M412427200
75. Joyce DE, Gelbert L, Ciaccia A, DeHoff B, Grinnell BW. Gene expression profile of antithrombotic protein c defines new mechanisms modulating inflammation and apoptosis. J Biol Chem. (2001) 276:11199–203. doi: 10.1074/jbc.C100017200
76. Cheng T, Liu D, Griffin JH, Fernández JA, Castellino F, Rosen ED, et al. Activated protein C blocks p53-mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nat Med. (2003) 9:338–42. doi: 10.1038/nm826
77. Mosnier LO, Griffin JH. Inhibition of staurosporine-induced apoptosis of endothelial cells by activated protein C requires protease-activated receptor-1 and endothelial cell protein C receptor. Biochem J. (2003) 373:65–70. doi: 10.1042/BJ20030341
78. Jain R, Young M, Dogra S, Kennedy H, Nguyen V, Jones S, et al. COVID-19 related neuroimaging findings: A signal of thromboembolic complications and a strong prognostic marker of poor patient outcome. J Neurol Sci. (2020) 414:116923. doi: 10.1016/j.jns.2020.116923
79. Klok FA, Kruip MJHA, van der Meer NJM, Arbous MS, Gommers D, Kant KM, et al. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: An updated analysis. Thromb Res. (2020) 191:148–50. doi: 10.1016/j.thromres.2020.04.041
80. Liu H, Chen S, Liu M, Nie H, Lu H. Comorbid chronic diseases are strongly correlated with disease severity among COVID-19 patients: a systematic review and meta-analysis. Aging Dis. (2020) 11:668–78. doi: 10.14336/AD.2020.0502
81. Tadic M, Cuspidi C, Sala C. COVID-19 and diabetes: Is there enough evidence? J Clin Hypertens. (2020) 22:943–8. doi: 10.1111/jch.13912
82. Daousi C, Casson IF, Gill GV, MacFarlane IA, Wilding JPH, Pinkney JH. Prevalence of obesity in type 2 diabetes in secondary care: association with cardiovascular risk factors. Postgrad Med J. (2006) 82:280–4. doi: 10.1136/pmj.2005.039032
83. Bradshaw PT, Monda KL, Stevens J. Metabolic syndrome in healthy obese, overweight, and normal weight individuals: the atherosclerosis risk in communities study. Obesity. (2013) 21:203–9. doi: 10.1002/oby.20248
84. Hales C, Caroll M, Fryar C, Ogden C. Prevalence of Obesity Among Adults and Youth : United States, 2015–2016. (2017). Available online at: https://stacks.cdc.gov/view/cdc/49223 (accessed May 26, 2020).
85. Mendrick DL, Diehl AM, Topor LS, Dietert RR, Will Y, La Merrill MA, et al. Metabolic syndrome and associated diseases: from the bench to the clinic. Toxicol Sci. (2018) 162:36–42. doi: 10.1093/toxsci/kfx233
86. Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. (2020) 180:1–11. doi: 10.1001/jamainternmed.2020.0994
87. Chavey C, Mari B, Monthouel M-N, Bonnafous S, Anglard P, Van Obberghen E, et al. Matrix metalloproteinases are differentially expressed in adipose tissue during obesity and modulate adipocyte differentiation. J Biol Chem. (2003) 278:11888–96. doi: 10.1074/jbc.M209196200
88. Menghini R, Fiorentino L, Casagrande V, Lauro R, Federici M. The role of ADAM17 in metabolic inflammation. Atherosclerosis. (2013) 228:12–7. doi: 10.1016/j.atherosclerosis.2013.01.024
89. Hadi HA, Suwaidi JA. Endothelial dysfunction in diabetes mellitus. Vasc Health Risk Manag. (2007) 3:853–76. Available online at: https://www.dovepress.com/getfile.php?fileID=1800
90. Engin A. Endothelial dysfunction in obesity. In: Engin AB, Engin A, editors. Obesity and Lipotoxicity Advances in Experimental Medicine and Biology. Cham: Springer International Publishing (2017). p. 345–79. doi: 10.1007/978-3-319-48382-5_15
91. Cole SA, Laviada-Molina HA, Serres-Perales JM, Rodriguez-Ayala E, Bastarrachea RA. The COVID-19 pandemic during the time of the diabetes pandemic: likely fraternal twins? Pathogens. (2020) 9:389. doi: 10.3390/pathogens9050389
92. Petrakis D, Margină D, Tsarouhas K, Tekos F, Stan M, Nikitovic D, et al. Obesity - a risk factor for increased COVID-19 prevalence, severity and lethality. Mol Med Rep. (2020) 22:9–19. doi: 10.3892/mmr.2020.11127
93. Matz RL, Andriantsitohaina R. Age-related endothelial dysfunction: potential implications for pharmacotherapy. Drugs Aging. (2003) 20:527–50. doi: 10.2165/00002512-200320070-00005
94. El Assar De La Fuente M, Angulo Frutos J, Vallejo Fernán S, Peiró Vallejo C, Sánchez-Ferrer CF, Rodríguez-Mañas L. Mechanisms involved in the aging-induced vascular dysfunction. Front Physiol. (2012) 3:132. doi: 10.3389/fphys.2012.00132
95. Nikolich-Žugich J. The twilight of immunity: emerging concepts in aging of the immune system. Nat Immunol. (2018) 19:10–9. doi: 10.1038/s41590-017-0006-x
96. Chen J, Kelley WJ, Goldstein DR. Role of aging and the immune response to respiratory viral infections: potential implications for COVID-19. J Immunol. (2020) 205:313–20. doi: 10.4049/jimmunol.2000380
97. Blanco-Melo D, Nilsson-Payant BE, Liu W-C, Uhl S, Hoagland D, Møller R, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. (2020) 181:1036-45.e9. doi: 10.1016/j.cell.2020.04.026
98. Mathew D, Giles JR, Baxter AE, Greenplate AR, Wu JE, Alanio C, et al. Deep immune profiling of COVID-19 patients reveals patient heterogeneity and distinct immunotypes with implications for therapeutic interventions. bioRxiv [Preprint]. (2020). Available online at: https://doi.org/10.1101/2020.05.20.106401 (accessed August 11, 2020).
99. Gribble FM, Reimann F. Enteroendocrine cells: chemosensors in the intestinal epithelium. Annu Rev Physiol. (2016) 78:277–99. doi: 10.1146/annurev-physiol-021115-105439
100. Paternoster S, Falasca M. Dissecting the physiology and pathophysiology of glucagon-like peptide-1. Front Endocrinol. (2018) 9:584. doi: 10.3389/fendo.2018.00584
101. Janssens J, Etienne H, Idriss S, Azmi A, Martin B, Maudsley S. Systems-level G protein-coupled receptor therapy across a neurodegenerative continuum by the GLP-1 receptor system. Front Endocrinol. (2014) 5:142. doi: 10.3389/fendo.2014.00142
102. Sun Y-H, He L, Yan M-Y, Zhao R-Q, Li B, Wang F, et al. Overexpression of GLP-1 receptors suppresses proliferation and cytokine release by airway smooth muscle cells of patients with chronic obstructive pulmonary disease via activation of ABCA1. Mol Med Rep. (2017) 16:929–36. doi: 10.3892/mmr.2017.6618
103. Helmstädter J, Frenis K, Filippou K, Grill A, Dib M, Kalinovic S, et al. Endothelial GLP-1 (glucagon-like peptide-1) receptor mediates cardiovascular protection by liraglutide in mice with experimental arterial hypertension. Arterioscler Thromb Vasc Biol. (2020) 40:145–58. doi: 10.1161/atv.0000615456.97862.30
104. Campos RV, Lee YC, Drucker DJ. Divergent tissue-specific and developmental expression of receptors for glucagon and glucagon-like peptide-1 in the mouse. Endocrinology. (1994) 134:2156–64. doi: 10.1210/en.134.5.2156
105. Dai Y, Mehta JL, Chen M. Glucagon-like peptide-1 receptor agonist liraglutide inhibits endothelin-1 in endothelial cell by repressing nuclear factor-kappa B activation. Cardiovasc Drugs Ther. (2013) 27:371–80. doi: 10.1007/s10557-013-6463-z
106. Fujita H, Morii T, Fujishima H, Sato T, Shimizu T, Hosoba M, et al. The protective roles of GLP-1R signaling in diabetic nephropathy: possible mechanism and therapeutic potential. Kidney Int. (2014) 85:579–89. doi: 10.1038/ki.2013.427
107. Travaglini KJ, Nabhan AN, Penland L, Sinha R, Gillich A, Sit RV, et al. A molecular cell atlas of the human lung from single cell RNA sequencing. bioRxiv [Preprint]. (2019). Available online at: https://doi.org/10.1101/742320 (accessed August 11, 2020).
108. Xu J, Wei G, Wang J, Zhu J, Yu M, Zeng X, et al. Glucagon-like peptide-1 receptor activation alleviates lipopolysaccharide-induced acute lung injury in mice via maintenance of endothelial barrier function. Lab Invest. (2019) 99:577–87. doi: 10.1038/s41374-018-0170-0
109. Wang J, Yu M, Xu J, Cheng Y, Li X, Wei G, et al. Glucagon-like peptide-1 (GLP-1) mediates the protective effects of dipeptidyl peptidase IV inhibition on pulmonary hypertension. J Biomed Sci. (2019) 26:6. doi: 10.1186/s12929-019-0496-y
110. Vara E, Arias-Díaz J, Garcia C, Balibrea JL, Blázquez E. Glucagon-like peptide-1 (7-36) amide stimulates surfactant secretion in human type II pneumocytes. Am J Respir Crit Care Med. (2001) 163:840–6. doi: 10.1164/ajrccm.163.4.9912132
111. Ku S-K, Han M-S, Park EJ, Na DH, Bae J-S. Exendin-4 inhibits endothelial protein C receptor shedding in vitro and in vivo. Pharmacol Res. (2014) 84:18–25. doi: 10.1016/j.phrs.2014.04.005
112. Romaní-Pérez M, Outeiriño-Iglesias V, Moya CM, Santisteban P, González-Matías LC, Vigo E, et al. Activation of the GLP-1 receptor by liraglutide increases ACE2 expression, reversing right ventricle hypertrophy, and improving the production of SP-A and SP-B in the lungs of type 1 diabetes rats. Endocrinology. (2015) 156:3559–69. doi: 10.1210/en.2014-1685
113. Arakawa M, Mita T, Azuma K, Ebato C, Goto H, Nomiyama T, et al. Inhibition of monocyte adhesion to endothelial cells and attenuation of atherosclerotic lesion by a glucagon-like peptide-1 receptor agonist, exendin-4. Diabetes. (2010) 59:1030–7. doi: 10.2337/db09-1694
114. Ceriello A, Novials A, Ortega E, Canivell S, Sala LL, Pujadas G, et al. Glucagon-like peptide 1 reduces endothelial dysfunction, inflammation, and oxidative stress induced by both hyperglycemia and hypoglycemia in type 1 diabetes. Diabetes Care. (2013) 36:2346–50. doi: 10.2337/dc12-2469
115. Gilbert MP, Pratley RE. GLP-1 analogs and DPP-4 inhibitors in type 2 diabetes therapy: review of head-to-head clinical trials. Front Endocrinol. (2020) 11:178. doi: 10.3389/fendo.2020.00178
116. Christou GA, Katsiki N, Blundell J, Fruhbeck G, Kiortsis DN. Semaglutide as a promising antiobesity drug. Obes Rev. (2019) 20:805–15. doi: 10.1111/obr.12839
117. Yaribeygi H, Ashrafizadeh M, Henney NC, Sathyapalan T, Jamialahmadi T, Sahebkar A. Neuromodulatory effects of anti-diabetes medications: a mechanistic review. Pharmacol Res. (2020) 152:104611. doi: 10.1016/j.phrs.2019.104611
118. Lee Y-S, Jun H-S. Anti-inflammatory effects of GLP-1-based therapies beyond glucose control. Mediat Inflamm. (2016) 2016:1–11. doi: 10.1155/2016/3094642
119. Salcedo I, Tweedie D, Li Y, Greig NH. Neuroprotective and neurotrophic actions of glucagon-like peptide-1: an emerging opportunity to treat neurodegenerative and cerebrovascular disorders. Br J Pharmacol. (2012) 166:1586–99. doi: 10.1111/j.1476-5381.2012.01971.x
120. Drucker DJ. The cardiovascular biology of glucagon-like peptide-1. Cell Metab. (2016) 24:15–30. doi: 10.1016/j.cmet.2016.06.009
121. Drucker DJ. Coronavirus infections and type 2 diabetes-shared pathways with therapeutic implications. Endocr Rev. (2020) 41:bnaa011. doi: 10.1210/endrev/bnaa011
122. Falkén Y, Hellström PM, Holst JJ, Näslund E. Changes in glucose homeostasis after Roux-en-Y gastric bypass surgery for obesity at day three, two months, and one year after surgery: role of gut peptides. J Clin Endocrinol Metab. (2011) 96:2227–35. doi: 10.1210/jc.2010-2876
123. Gimeno RE, Briere DA, Seeley RJ. Leveraging the gut to treat metabolic disease. Cell Metab. (2020) 31:679–98. doi: 10.1016/j.cmet.2020.02.014
124. Chai W, Dong Z, Wang N, Wang W, Tao L, Cao W, et al. Glucagon-like peptide 1 recruits microvasculature and increases glucose use in muscle via a nitric oxide–dependent mechanism. Diabetes. (2012) 61:888–96. doi: 10.2337/db11-1073
125. Dong Z, Chai W, Wang W, Zhao L, Fu Z, Cao W, et al. Protein kinase A mediates glucagon-like peptide 1-induced nitric oxide production and muscle microvascular recruitment. Am J Physiol - Endocrinol Metab. (2013) 304:E222–8. doi: 10.1152/ajpendo.00473.2012
126. Beglinger S, Drewe J, Schirra J, Göke B, D'Amato M, Beglinger C. Role of fat hydrolysis in regulating glucagon-like peptide-1 secretion. J Clin Endocrinol Metab. (2010) 95:879–86. doi: 10.1210/jc.2009-1062
127. Avesaat M, van Troost FJ, Ripken D, Hendriks HF, Masclee AM. Ileal brake activation: macronutrient-specific effects on eating behavior? Int J Obes. (2015) 39:235–43. doi: 10.1038/ijo.2014.112
128. Poppitt SD, Shin HS, McGill A-T, Budgett SC, Lo K, Pahl M, et al. Duodenal and ileal glucose infusions differentially alter gastrointestinal peptides, appetite response, and food intake: a tube feeding study. Am J Clin Nutr. (2017) 106:725–35. doi: 10.3945/ajcn.117.157248
Keywords: glucagon like peptide 1 (GLP-1), enteroendocrine, lung, immune cells, tumor necrosis factor (TNF), tumor necrosis factor converting enzyme (TACE), endothelial barrier disruption, acute respiratory and circulatory disruption
Citation: Hanchard J, Capó-Vélez CM, Deusch K, Lidington D and Bolz S-S (2020) Stabilizing Cellular Barriers: Raising the Shields Against COVID-19. Front. Endocrinol. 11:583006. doi: 10.3389/fendo.2020.583006
Received: 13 July 2020; Accepted: 24 August 2020;
Published: 30 September 2020.
Edited by:
Pieter de Lange, University of Campania Luigi Vanvitelli, ItalyReviewed by:
Stuart Maudsley, University of Antwerp, BelgiumMarta Letizia Hribal, University of Catanzaro, Italy
Copyright © 2020 Hanchard, Capó-Vélez, Deusch, Lidington and Bolz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Steffen-Sebastian Bolz, c3RzLmJvbHpAdXRvcm9udG8uY2E=
†These authors have contributed equally to this work and share first authorship