 Chelsea DeLeon
Chelsea DeLeon David Q.-H. Wang
David Q.-H. Wang Christopher K. Arnatt
Christopher K. Arnatt
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Endocrinol. , 12 November 2020
Sec. Molecular and Structural Endocrinology
Volume 11 - 2020 | https://doi.org/10.3389/fendo.2020.578536
This article is part of the Research Topic GPER and Human Pathologies View all 11 articles
There are gender differences between men and women in many physiological functions and diseases, which indicates that female sex hormones may be important. Traditionally, estrogen exerts its biological activities by activating two classical nuclear estrogen receptors, ESR1 and ESR2. However, the roles of estrogen in the regulation of physiological functions and the pathogenesis of diseases become more complicated with the identification of the G protein-coupled estrogen receptor (GPER1). Although many GPER1-specific ligands have been developed, the therapeutic mechanisms of exclusively targeting GPER1 are not yet well understood. Translational applications and clinical trial efforts for the identified GPER1 ligands have been focused primarily on the reproductive, cardiovascular, nervous, endocrine, and immune systems. More recently, research found that GPER1 may play an important role in regulating the digestive system. Cholesterol gallstone disease, a major biliary disease, has a higher prevalence in women than in men worldwide. Emerging evidence implies that GPER1 could play an important role, independent of the classical ESR1, in the pathophysiology of cholesterol gallstones in women. This review discusses the complex signaling pathways of three estrogen receptors, highlights the development of GPER1-specific ligands, and summarizes the latest advances in the role of GPER1 in the pathogenesis of gallstone formation.
The prevalence of digestive disease ranges from 10 to 27.8% in the United States (1, 2). Some common chronic digestive diseases include gallstone disease, nonalcoholic fatty liver disease, alcoholic liver disease, gastroesophageal reflux disease, irritable bowel syndrome, inflammatory bowel disease, gastric cancer, pancreatic cancer, and colon cancer. Many digestive disorders exhibit a distinct gender difference in prevalence between women and men (3–5), suggesting that sex hormones are important. Over the past decades, many basic research and clinical investigations have been focused largely on the roles of estrogen, through two classical nuclear estrogen receptors, ESR1 and ESR2 (also called ERα and ERβ), in the regulation of physiological functions and the pathophysiology of diseases such as cardiovascular, kidney, nervous, reproductive, endocrine, and gastrointestinal disorders. However, the discovery of a new estrogen receptor called the G protein-coupled estrogen receptor (GPER1) has made it more complicated to investigate the roles of estrogen in the pathogenesis of numerous diseases because estrogen can produce its biological activities through one of the three nuclear receptor signaling pathways, or a combination of two, or all three. This review discusses the latest advances in the signaling pathways of three estrogen receptors, the development of GPER1-specific ligands, and the roles of GPER1 and its ligands in the pathogenesis of cholesterol gallstone disease.
The identification of three estrogen receptors has implied that estrogen-stimulated receptor signaling is more complex than initially realized (Figure 1). The naturally occurring estrogens are 17β-estradiol (E2), estrone, and estriol, and all of them are C18 steroids. Cellular response to E2 can occur through the activation of the nuclear estrogen receptors, ESR1, and ESR2. The classical ER signaling through the ERs involves the binding of estrogen, receptor dimerization, and subsequent association of coactivator proteins that guide the dimerized ER subunit to estrogen response elements (EREs) that drive transcriptional activity (6, 7). In addition, variants or single nucleotide polymorphisms (SNPs) in the ESR1 and the ESR2 genes increase the complexity and diversity of E2-mediated signaling transduction.
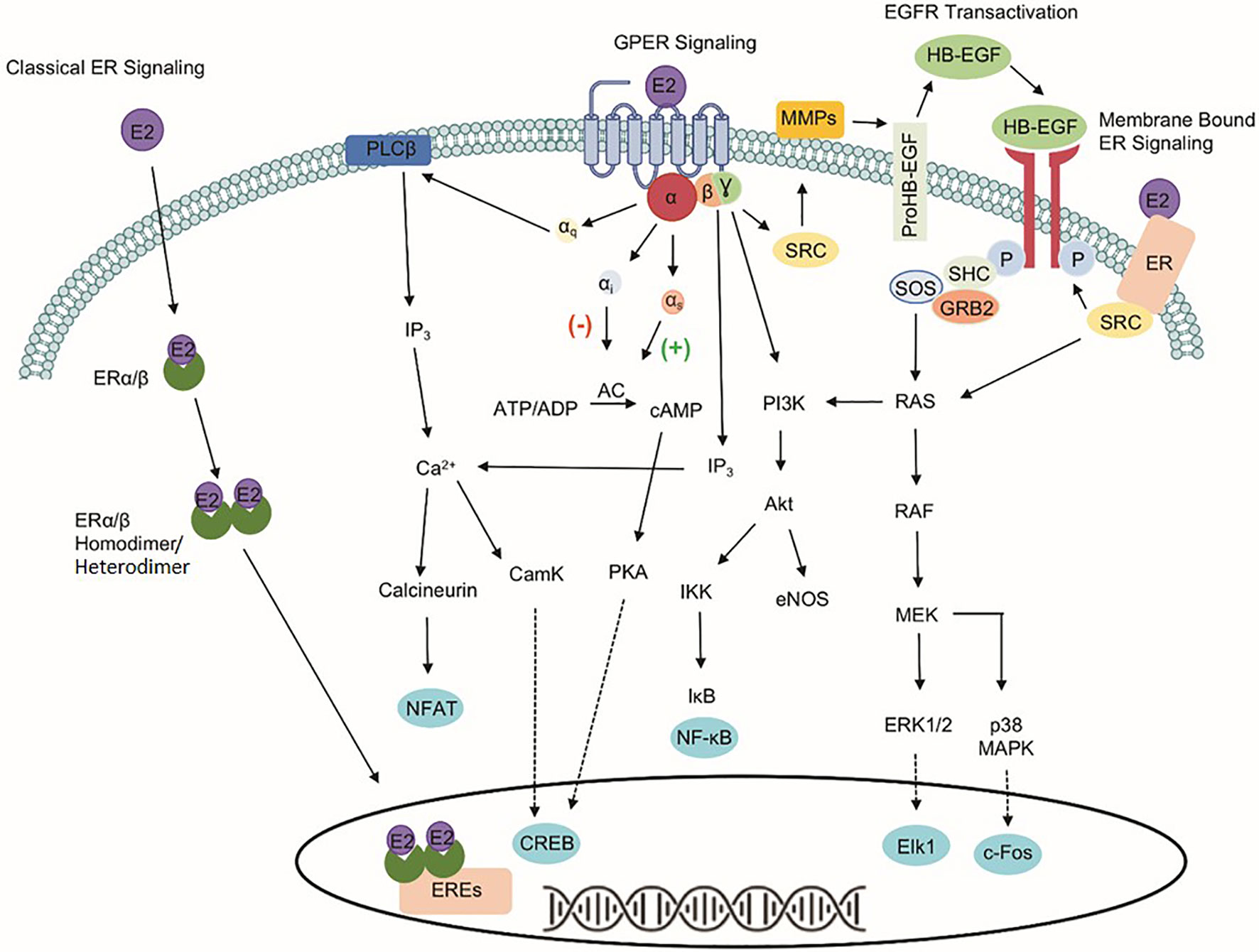
Figure 1 Signaling pathways of three estrogen receptors. The classical estrogen receptors, ESR1 and ESR2, primarily exist within the cytoplasm and nucleus, as well as interact with estrogen response elements (EREs) after dimerization to drive genomic signaling. Unlike the nuclear estrogen receptors, GPER1 signaling pathway occurs through various second messengers. Phospholipase C Beta (PLCβ), inositol triphosphate (IP3), nuclear factor of activated T-cells (NFAT), calcium/calmodulin-dependent protein kinase (CamK), cAMP response element-binding protein (CREB), adenylate cyclase (AC), protein kinase A (PKA), phosphoinositide 3-kinase (PI3K), protein kinase B (Akt), IκB kinase (IKK), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), endothelial nitric oxide synthase (eNOS), non-receptor tyrosine kinase (SRC), matrix metallopeptidases (MMPs), heparin-binding EGF-like growth factor (HB-EGF), son of sevenless (SOS), Src homology 2 domain-containing transforming protein (SHC), growth factor receptor-bound protein 2 (GRB2), RAS protein (RAS), RAF kinase (RAF), mitogen-activated protein kinase kinase (MEK), extracellular signal-regulated kinases 1/2 (ERK 1/2), Elk-1 transcription factor (Elk1), p38 mitogen-activated protein kinase (p38 MAPK), and c-Fos transcription factor (c-Fos).
The identification of GPER1, a 375-amino acid protein known previously as GPR30, makes the well-known ER signaling pathways more complicated. Unlike the classical nuclear estrogen receptors, GPER1 signaling occurs through various second messengers (8–12). Specifically, GPER1 has been shown to activate ERK1/2 phosphorylation through Gβ,γ-dependent transactivation of epidermal growth factor receptor (EGFR), cAMP, calcium mobilization, and protein/lipid kinases (i.e., PKC and PKA) (8, 13–17). Interaction with these signaling pathways influences protein expression, apoptosis, cell proliferation, cell migration, and growth. Despite differences in signaling capabilities, ESR1, ESR2, and GPER1 are expressed ubiquitously throughout the human body and the variability of response in different tissues highlights the importance of understanding the druggability of each target separately due to the downstream signaling events that differ between the proteins (18).
The effects of preferentially targeting GPER1 are not fully understood; therefore, there has been an increased research effort into the development of novel ligands to modulate GPER1 activity. Before the identification of GPER1-specific ligands, the antiestrogens, tamoxifen and fulvestrant, were shown to interact with GPER1 (19). While tamoxifen and fulvestrant block the ability of E2 to signal through ESR1 or ESR2, they also possess the ability to activate the GPER1 signaling pathway similarly to E2. The activity of antiestrogens at GPER1 highlights the cross-reactivity of estrogenic ligands and the difficulty in developing GPER1-specific ligands. In addition to antiestrogens, various non-selective GPER1 agonists have been identified: these include natural products like hydroxytyrosol and oleuropein, as well as phytoestrogens, such as coumestrol, and the endocrine-disrupting compounds Bisphenol A (BPA) (Figure 2) (20–22). Additional studies have identified synthetic polybrominated diphenyl ethers (PBDEs) and hydroxylated PBDEs as potential GPER1 ligands; however, these compounds likely exhibit no selective activity (23).
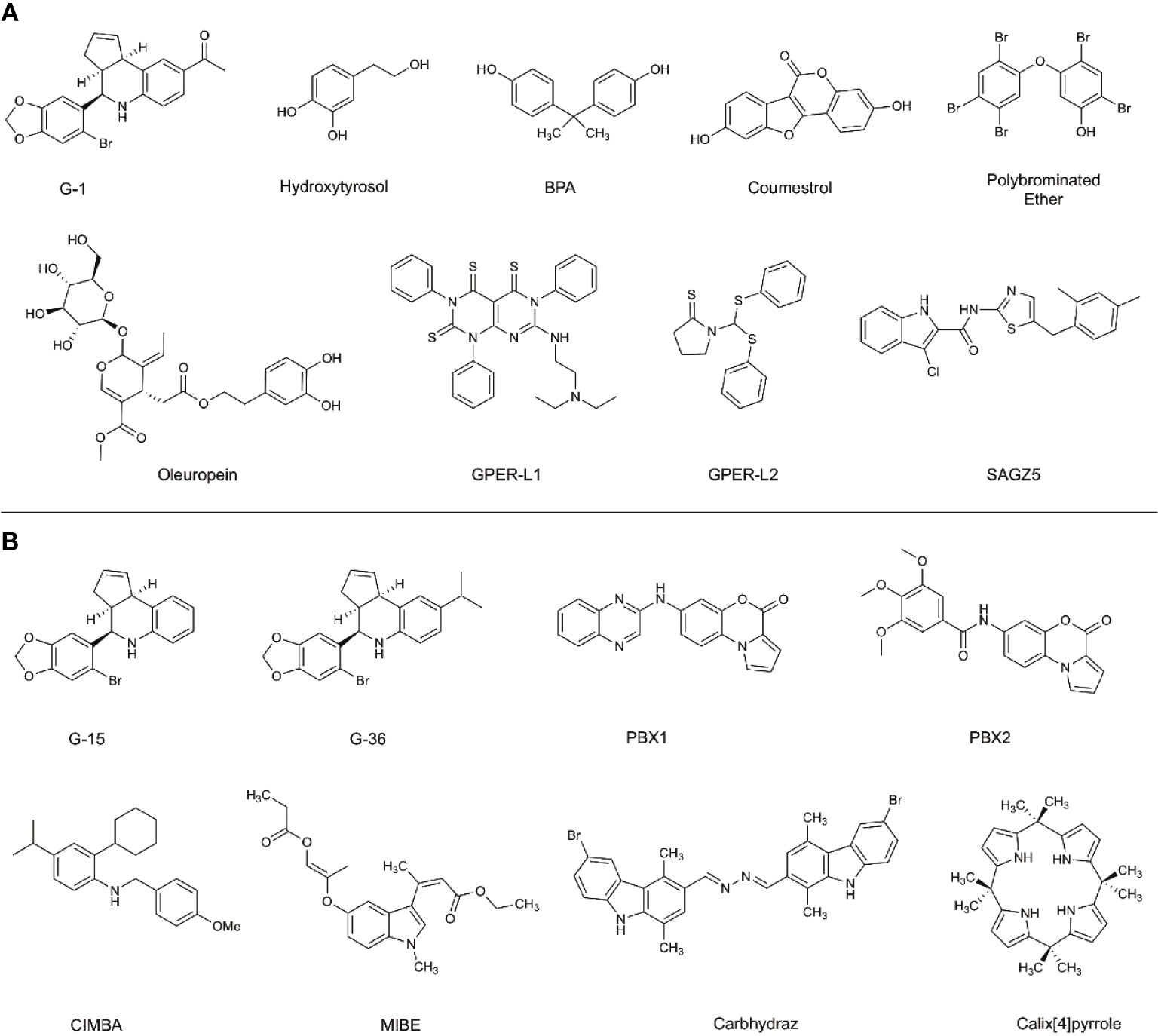
Figure 2 Selective and non-selective GPER1 agonists (A) and antagonists (B). (A) Various GPER1 agonists have been identified. This includes non-selective natural products like hydroxytyrosol and oleuropein, phytoestrogens such as coumestrol, as well as endocrine-disrupting compounds like BPA. Various synthetic GPER1 agonists have been identified. A series of polybrominated ethers have been identified; however, these compounds likely do not exhibit specificity for GPER1. Several GPER1-specific agonists have been identified. These compounds include G-1, GPER-L1, GPER-L2, and SAGZ5. (B) Currently, there are no known naturally occurring GPER1 antagonists. Modifications were made to the tetrahydroquinoline scaffold of G-1 to create G-15 and G-36. These alterations modified the activity of the compounds to antagonists. Since the identification of G-15 and G-36, there have been a limited number of GPER1-specific antagonists identified. These include PBX1, PBX2, CIMBA, carbhydraz, and calix[4]pyrrole. MIBE has been identified as an antagonist for GPER1 and ESR1. In certain circumstances, there may be a therapeutic benefit in jointly targeting GPER1 and ESR1.
A hallmark challenge in the identification and discovery of GPER1-specific ligands has been the difficulty in achieving a crystalized structure of the receptor. Presently, a crystallized structure of GPER1 does not exist. For this reason, the identification and optimization of ligands has relied upon large-library virtual screening techniques and homology modeling (24–27). Due to the cross-reactivity of estrogenic ligands, a limited number of GPER1-specific ligands have been identified. The current benchmark for GPER1-specific ligands were identified through virtual screening of 10,000 into a model of GPER1 based on 2D- and 3D-similarity approaches and GPER-privileged substructures (24). From the screening, a substituted dihydroquinoline was identified and named GPR30-specific compound 1, G-1 (24). Binding studies revealed no appreciable binding to ESR1 or ESR2 below 100 nM (24). Subsequent functional bioassays with GPER1-transfected COS-7 cell and G-1 showed that E2 and G-1 exhibit an increase in calcium mobilization at 1 nM; however, a closer analysis of the data suggests that the kinetic profile of the calcium mobilization differs between the compounds such that G-1 exhibits slow receptor occupancy and an asymptotic curve and E2 exhibits fast receptor occupancy with a quick peak in calcium release (24). Medicinal chemistry approaches to modify the dihydroquinoline of G-1 altered the pharmacological activity of the scaffold from an agonist to an antagonist (28). While the identified antagonist, G-15, inhibited G-1 activity at GPER1, off-target binding to ESR1 and activation of EREs persisted (28). The reduction of binding and ERE activation was accomplished with the addition of an isopropyl group to the scaffold to make G-36 (29). While the G-series has become the standard for GPER1 agonists and antagonists, the success with the compounds has been variable and may be related to the tissue-specific signaling events of GPER1 (30, 31).
Since the development of the G-series of ligands, a limited number of groups have published data on synthesized ligands for GPER1. Lappano et al. proposed two tricyclic tetrahydroquinolines, GPER-L1 and GPER-L2 (32). These compounds were shown to bind exclusively to GPER1 without significant ESR1 binding above 100 μM (Figure 2) (33). Previously, we identified a series of N-thiazol-2-yl-1H-indole-2-carboxamide derivatives as GPER1 agonists (30). These compounds exhibited a similar effect on breast cancer proliferation as reported in the literature in response to the GPER1-selective agonist, G-1 (Figure 2) (30). Based on that work and further computational modeling, we have since reported the first structure-activity relationship for GPER1 antagonists and discovered CIMBA (2-cyclohexyl-4-isopropyl-N-(4-methoxybenzyl)aniline) (34). In addition to our group, Maggiolini et al. developed two selective GPER1 antagonists (PBX1 and PBX2) based on a benzo[b]pyrrolo[1,2-d][1,4]oxazin-4-one scaffold (Figure 2) (35). Both PBX1 and PBX2 effectively blocked agonist-induced GPER1 activity without transcriptional activation of the classical ERs. Additional non-selective GPER1 ligands have also been described in the literature. Unlike the GPER1 antagonists identified by DeLeon et al. and Maggioloini et al., Lappano et al. identified MIBE (ethyl 3-[5-(2-ethoxycarbonyl-1-methylvinyloxy)-1-methyl-1H-indol-3-yl]but-2-enoate) and demonstrated that MIBE blocks agonist activity at both GPER1 and ESR1 (36, 37). In addition to MIBE novel ligands, such as calixpyrrole derivatives that include a cyclic structure and resemble a porphyrin ring system, have been proposed as GPER1 antagonists (Figure 2) (36, 37).
The limited number of available GPER1-specific ligands may be attributed to a lack of clarity in the localization as well as the complex pharmacology associated with GPER1. The localization and expression of GPER1 has been long debated. Numerous studies have shown that GPER1 is expressed both along the cell membrane surface as well as intracellularly within the endoplasmic reticulum and Golgi apparatus (38–40). After several decades, it is now recognized that even though GPER1 expression exists within the cell membrane, the expression level is substantially less than the subcellular expression (40). This has important implications for drug discovery in that GPER1 ligands may need to be lipophilic and able to cross the cell membrane to access the receptor. The data achieved relating to the pharmacology of the G-series has varied among groups and has posed challenges to defining G protein coupling (31, 40). Together, the localization and varied success with currently available probes substantiate the need for novel GPER1-specific ligands to better understand the pharmacology associated with GPER1 and the clinical implications for the receptor.
Cholesterol gallstone disease is one of the most prevalent and costly digestive diseases in the United States, with at least 20 million Americans (12% of adults) being affected (41). Clinical and epidemiological investigations have demonstrated that women are twice as likely as men to form cholesterol gallstones in every population that has been studied (42). Oral contraceptives and conjugated estrogens significantly increase gallstone prevalence in premenopausal and postmenopausal women (43–53). Similar lithogenic effects are also found in men with prostate cancer during postoperative estrogen therapy (54–56). All these studies show that E2 is a critical risk factor for gallstone disease and a high predisposition to gallstones in women than in men is related to differences in how the liver metabolizes cholesterol in response to E2 (57). Although both ESR1 and ESR2 are expressed in the liver of mice and humans, ESR1 expression is approximately 50-fold higher compared to ESR2 expression (58). Despite these observations, the mechanism by which ESR1 plays a key role in mediating E2-induced lithogenic actions at a cellular and molecular level is not yet fully understood. Exciting results show that E2 enhances cholelithogenesis by increasing hepatic expression of ESR1 but not ESR2, and the lithogenic actions of E2 can be blocked completely by the antiestrogenic agent, ICI 182,780 (58). Furthermore, the ESR1-selective agonist propylpyrazole, but not the ESR2-selective agonist diarylpropionitrile, promotes hepatic cholesterol output, leading to cholesterol-supersaturated bile and gallstones (58). Similar to E2 treatment, tamoxifen significantly increased biliary cholesterol secretion and gallstone prevalence (58, 59). These results indicate that the hepatic ESR1, but not ESR2, plays a critical role in E2-induced gallstones in female mice. More importantly, ESR1 stimulated by E2 dramatically increases hepatic expression of sterol regulatory element-binding protein-2 (SREBP-2), activating SREBP-2-responsive genes in the cholesterol biosynthetic pathway (60). Thus, the E2-treated mice continue to synthesize cholesterol despite its excess availability from high dietary cholesterol, which reflects a loss in controlling the negative feedback regulation of cholesterol synthesis. As a result, more newly synthesized cholesterol determined by the estrogen-ESR1-SREBP-2 pathway is secreted into bile, leading to biliary cholesterol hypersecretion and enhancing the lithogenicity of bile (60).
More interestingly, the deletion of Esr1 diminishes susceptibility to E2-induced gallstones by reducing hepatic cholesterol secretion and desaturating gallbladder bile; however, this cannot completely protect against gallstone formation in mice treated with high doses of E2 and fed the lithogenic diet (61). As found by a powerful genetic quantitative trait locus (QTL) analysis, Gper1 is a new gallstone gene, Lith18, on chromosome 5 in mice (62–66). GPER1 activated by its agonist, G-1, enhances cholelithogenesis by deterring expression of cholesterol 7α-hydroxylase, the rate-limiting enzyme for the classical pathway of bile salt synthesis (67). These metabolic abnormalities greatly increase biliary cholesterol concentrations in company with hepatic hyposecretion of biliary bile salts, leading to cholesterol-supersaturated gallbladder bile and accelerating cholesterol crystallization (68). Moreover, E2 activates GPER1 and ESR1 toproduce liquid crystalline versus anhydrous crystalline metastable intermediates evolving to cholesterol monohydrate crystals from supersaturated bile (69). However, cholesterol crystallization is drastically retarded in Gper1/Esr1 double knockout mice. This indicates that GPER1 produces a synergistic lithogenic action with ESR1 to enhance E2-induced gallstone formation.
Impaired gallbladder motility is often a distinctive clinical feature of pregnant women and subjects received high doses of E2, which promotes the formation of biliary sludge, the precursor of gallstones (70–75). Immunohistochemical studies find that GPER1 is expressed predominately in the epithelial cells of the gallbladder (69). By contrast, ESR1 is expressed mainly in the smooth muscle of the gallbladder (69). This suggests that GPER1 could impair gallbladder motility, working independently of ESR1, as both can cause sluggish gallbladder contractility from different mechanisms. Indeed, G-1 impairs gallbladder emptying through the GPER1 pathway in mice, leading to sluggish gallbladder motility and accelerating the development of biliary sludge in the early stage of gallstone formation (67).
More recently, exciting evidence shows that a novel, potent GPER1-selective antagonist, CIMBA, reduces the prevalence of E2-induced gallstones in a dose-dependent manner by impeding the GPER1 signaling pathway in female wild-type mice (76). However, gallstones can be completely prevented in E2-treated ESR1 knockout mice even on the lithogenic diet (76). These results are consistent with the findings that the deletion of either Esr1 or Gper1 significantly reduces the prevalence of E2-induced gallstones but could not abolish it completely.
Overall, these studies have established a novel concept that GPER1 is involved in E2-dependent lithogenic actions, working independently of ESR1, as both GPER1 and ESR1 can promote the formation of E2-induced gallstones through different pathways. Thus, both GPER1 and ESR1 are potential therapeutic targets for cholesterol gallstone disease, particularly in women and patients exposed to high levels of E2 (77).
The similarity between estrogenic compounds poses significant challenges in the design of new, selective ligands due to the promiscuous binding of estrogenic compounds to different types of ERs and is a particular challenge for designing new compounds. While estrogen binding is frequently associated with the nuclear ERs, GPER1 has been recognized as a new ER. A frequently neglected aspect of ER signaling is the ability of E2 and estrogenic compounds to directly activate calcium channels, specifically L-type calcium channels and calcium-activated BK (big potassium) channels (78, 79). The activation of ion channels by estrogenic compounds adds another level of complexity to studying ER signaling pathways and the design of GPER1-specific compounds.
The signaling pathways of ERs are complex and multifaceted. For this reason, studies that aim to examine a singular ER signaling pathway should not neglect existence of the three ERs. The therapeutic implications of targeting multiple ER signaling pathways are not well understood; however, evidence exists that cross-reactivity may severely limit the application of certain therapeutics. For instance, even though selective estrogen receptor modulators (SERMs) exhibit antiestrogen effects at the classical ERs, the cross-reactivity and activation of GPER1 may contribute to therapeutic resistance, which renders the therapeutic ineffective (80, 81). This limitation has been observed with tamoxifen. Alternatively, there may be some therapeutic opportunities for cross-reactivity, specifically in the gallbladder. In this circumstance, previous evidence has shown that inhibition of ESR1 or GPER1 alone is not sufficient to completely prevent gallstone formation (49). In this instance, the cross-reactivity of a compound, such as MIBE (Figure 2), which acts as an antiestrogen at both ESR1 and GPER1 may be a useful tool. The identification of new agonists has largely occurred in breast cancer cell lines that endogenously express GPER1. The pharmacology associated with GPER1 may be tissue-specific since GPER1 is expressed ubiquitously throughout the body. The use of additional cell lines may lead to a greater number of potent and efficacious ligands.
In most areas of the digestive system, there are still opportunities for further understanding the impact of exclusively targeting GPER1 and understanding the potential pharmacological implications of targeting multiple ERs. While G-1 has served as a valuable tool for understanding the role of GPER1 in health and disease associated with the digestive system in animals, and development of further GPER1 agonists and antagonists will lead to potential therapeutics with greater activity, specificity, and solubility in water or oil. The role of GPER1 in cholesterol gallstone disease presented in this review highlights the potential importance of GPER1 in hepatobiliary diseases. Overall, the prevention of lithogensis via a GPER1 antagonist represents a novel treatment option for high-risk populations and may prove to be an adjunct therapy to nonsurgical gallstone treatments.
All authors equally contributed to writing this perspective. All authors contributed to the article and approved the submitted version.
This work was supported in part by the start-up funds from Saint Louis University (to CKA), as well as by research grants DK106249, DK114516, and DK126369 (to DQ-HW), as well as P30 DK041296 (to Marion Bessin Liver Research Center), all from the National Institutes of Health (US Public Health Service).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
E2, 17β-estradiol; ER, estrogen receptor; ERE, estrogen response element; ESR1, estrogen receptor α; ESR2, estrogen receptor β; 2-ME, 2-methoxy-estradiol; GPER1, G protein-coupled estrogen receptor; QTL, quantitative trait locus.
1. El-Serag HB, Sweet S, Winchester CC, Dent J. Update on the epidemiology of gastro-oesophageal reflux disease: a systematic review. Gut (2014) 63:871–80. doi: 10.1136/gutjnl-2012-304269
2. Saito YA, Schoenfeld P, Locke RG. III, The Epidemiology of Irritable Bowel Syndrome in North America: A Systematic Review. Am J Gastroenterol (2002) 97:1910–5. doi: 10.1016/S0002-9270(02)04270-3
3. Kim YS, Kim N, Kim GH. Sex and Gender Differences in Gastroesophageal Reflux Disease. J Neurogastroenterol Motil (2016) 22:575–88. doi: 10.5056/jnm16138
4. Kim YS, Kim N. Sex-Gender Differences in Irritable Bowel Syndrome. J Neurogastroenterol Motil (2018) 24:544–58. doi: 10.5056/jnm18082
5. Stinton LM, Shaffer EA. Epidemiology of Gallbladder Disease: Cholelithiasis and Cancer. Gut Liver (2012) 6:172–87. doi: 10.5009/gnl.2012.6.2.172
6. Nilsson S, Koehler KF. Oestrogen receptors and selective oestrogen receptor modulators: molecular and cellular pharmacology. Basic Clin Pharmacol Toxicol (2005) 96:15–25. doi: 10.1111/j.1742-7843.2005.pto960103.x
7. Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engström O, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature (1997) 389:753–8. doi: 10.1038/39645
8. Filardo EJ, Quinn JA, Bland KI, Frackelton ARJ. Estrogen-Induced Activation of Erk-1 and Erk-2 Requires the G Protein-Coupled Receptor Homolog, GPR30, and Occurs via Trans-Activation of the Epidermal Growth Factor Receptor through Release of HB-EGF. Mol Cell Endocrinol (2000) 14:1649–60. doi: 10.1210/mend.14.10.0532
9. Filardo EJ, Quinn JA, Frackelton A, Bland KI. Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol (2002) 16:70–84. doi: 10.1210/mend.16.1.0758
10. Maggiolini M, Vivacqua A, Fasanella G, Recchia AG, Sisci D, Pezzi V, et al. The G protein-coupled receptor GPR30 mediates c-fos up-regulation by 17beta-estradiol and phytoestrogens in breast cancer cells. J Biol Chem (2004) 279:27008–16. doi: 10.1074/jbc.M403588200
11. Filardo EJ, Thomas P. GPR30: a seven-transmembrane-spanning estrogen receptor that triggers EGF release. Trends Endocrinol Metab (2005) 16:362–7. doi: 10.1016/j.tem.2005.08.005
12. Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science (2005) 307:1625–30. doi: 10.1126/science.1106943
13. Filardo EJ, Thomas P. Minireview: G Protein-Coupled Estrogen Receptor-1, GPER-1: Its Mechanism of Action and Role in Female Reproductive Cancer, Renal and Vascular Physiology. Endocrinology (2012) 153:2953–62. doi: 10.1210/en.2012-1061
14. Björnström L, Sjöberg M. Mechanisms of Estrogen Receptor Signaling: Convergence of Genomic and Nongenomic Actions on Target Genes. Mol Endocrinol (2005) 19:833–42. doi: 10.1210/me.2004-0486
15. Levin ER. Invited Review: Cell localization, physiology, and nongenomic actions of estrogen receptors. J Appl Physiol (2001) 91:1860. doi: 10.1152/jappl.2001.91.4.1860
16. Teskarik J, Mendoza C. Nongenomic effects of 17 beta-estradiol on maturing human oocytes: relationship to oocyte developmental potential. J Clin Endocrinol Metab (1995) 80:1438–43. doi: 10.1210/jc.80.4.1438
17. Kelly MJ, Wagner EJ. Estrogen Modulation of G-protein-coupled Receptors. Trends Endocrinol Metab (1999) 10:369–74. doi: 10.1016/S1043-2760(99)00190-3
18. Nilsson S, Gustafsson JÅ. Estrogen Receptors: Therapies Targeted to Receptor Subtypes. Clin Pharmacol Ther (2011) 89:44–55. doi: 10.1038/clpt.2010.226
19. Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology (2005) 146:624–32. doi: 10.1210/en.2004-1064
20. Xu F, Wang X, Wu N, He S, Yi W, Xiang S, et al. Bisphenol A induces proliferative effects on both breast cancer cells and vascular endothelial cells through a shared GPER-dependent pathway in hypoxia. Environ Pollut (2017) 231:1609–20. doi: 10.1016/j.envpol.2017.09.069
21. Chimento A, Casaburi I, Rosano C, Avena P, De Luca A, Campana C, et al. Oleuropein and hydroxytyrosol activate GPER/ GPR30-dependent pathways leading to apoptosis of ER-negative SKBR3 breast cancer cells. Mol Nutr Food Res (2014) 58:478–89. doi: 10.1002/mnfr.201300323
22. Molina L, Bustamante FA, Bhoola KD, Figueroa CD, Ehrenfeld P. Possible role of phytoestrogens in breast cancer via GPER-1/GPR30 signaling. Clin Sci (2018) 132:2583–98. doi: 10.1042/CS20180885
23. Cao L-Y, Ren X-M, Yang Y, Wan B, Guo L-H, Chen D, et al. Hydroxylated Polybrominated Diphenyl Ethers Exert Estrogenic Effects via Non-Genomic G Protein-Coupled Estrogen Receptor Mediated Pathways. Environ Health Perspect (2018) 126:057005–5. doi: 10.1289/EHP2387
24. Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol (2006) 2:207–12. doi: 10.1038/nchembio775
25. Arnatt CK, Zhang Y. G Protein-Coupled Estrogen Receptor (GPER) Agonist Dual Binding Mode Analyses Toward Understanding of Its Activation Mechanism: A Comparative Homology Modeling Approach. Mol Inf (2013) 32:647–58. doi: 10.1002/minf.201200136
26. Bruno A, Aiello F, Costantino G, Radi M. Homology Modeling, Validation and Dynamics of the G Protein-coupled Estrogen Receptor 1 (GPER-1). Mol Inf (2016) 35:333–9. doi: 10.1002/minf.201501024
27. Méndez-Luna D, Martínez-Archundia M, Maroun RC, Ceballos-Reyes G, Fragoso-Vázquez MJ, González-Juárez DE, et al. Deciphering the GPER/GPR30-agonist and antagonists interactions using molecular modeling studies, molecular dynamics, and docking simulations. J Biomol Struct Dynamics (2015) 33:2161–72. doi: 10.1080/07391102.2014.994102
28. Dennis MK, Burai R, Ramesh C, Petrie WK, Alcon SN, Nayak TK, et al. In vivo Effects of a GPR30 Antagonist. Nat Chem Biol (2009) 5:421–7. doi: 10.1038/nchembio.168
29. Dennis MK, Field AS, Burai R, Ramesh C, Petrie WK, Bologa CG, et al. Identification of a GPER/GPR30 Antagonist with Improved Estrogen Receptor Counterselectivity. J Steroid Biochem Mol Biol (2011) 127:358–66. doi: 10.1016/j.jsbmb.2011.07.002
30. O’Dea A, Sondergard C, Sweeney P, Arnatt CK. A Series of Indole-Thiazole Derivatives Act as GPER Agonists and Inhibit Breast Cancer Cell Growth. ACS Med Chem Lett (2018) 9:901–6. doi: 10.1021/acsmedchemlett.8b00212
31. Feldman RD, Limbird LE. GPER (GPR30): A Nongenomic Receptor (GPCR) for Steroid Hormones with Implications for Cardiovascular Disease and Cancer. Annu Rev Pharmacol Toxicol (2017) 57:567–84. doi: 10.1146/annurev-pharmtox-010716-104651
32. Lappano R, Rosano C, Santolla MF, Pupo M, De Francesco EM, De Marco P, et al. Two novel GPER agonists induce gene expression changes and growth effects in cancer cells. Curr Cancer Drug Targets (2012) 12:531–42. doi: 10.2174/156800912800673284
33. Cerra B, Mostarda S, Custodi C, Macchiarulo A, Gioiello A. Integrating multicomponent flow synthesis and computational approaches for the generation of a tetrahydroquinoline compound based library. Med Chem Comm 7 (2016) 7:439–46. doi: 10.1039/C5MD00455A
34. DeLeon C, Wang HH, Gunn J, Wilhelm M, Cole A, Arnett S, et al. Novel GPER Antagonist Protects Against the Formation of Estrogen-Induced Cholesterol Gallstones in Female Mice. J Lipid Res (2020) 61:767–77. doi: 10.1194/jlr.RA119000592
35. Maggiolini M, Santolla MF, Avino S, Aiello F, Rosano C, Garofalo A, et al. Identification of two benzopyrroloxazines acting as selective GPER antagonists in breast cancer cells and cancer-associated fibroblasts. Future Med Chem (2015) 7:437–48. doi: 10.4155/fmc.15.3
36. Lappano R, Rosano C, Pisano A, Santolla MF, De Francesco EM, De Marco P, et al. A calixpyrrole derivative acts as an antagonist to GPER, a G-protein coupled receptor: mechanisms and models. Dis Models Mech (2015) 8:1237. doi: 10.1242/dmm.021071
37. Lappano R, Santolla MF, Pupo M, Sinicropi MS, Caruso A, Rosano C, et al. MIBE acts as antagonist ligand of both estrogen receptor α and GPER in breast cancer cells. Breast Cancer Res (2012) 14:R12. doi: 10.1186/bcr3096
38. Zimmerman MA, Budish RA, Kashyap S, Lindsey SH. GPER-novel membrane oestrogen receptor. Clin Sci (Lond) (2016) 130:1005–16. doi: 10.1042/CS20160114
39. Cheng S-B, Quinn JA, Graeber CT, Filardo EJ. Down-modulation of the G-protein-coupled estrogen receptor, GPER, from the cell surface occurs via a trans-Golgi-proteasome pathway. J Biol Chem (2011) 286:22441–55. doi: 10.1074/jbc.M111.224071
40. Gaudet HM, Cheng SB, Christensen EM, Filardo EJ. The G-protein coupled estrogen receptor, GPER: The inside and inside-out story. Mol Cell Endocrinol (2015) 418 Pt 3:207–19. doi: 10.1016/j.mce.2015.07.016
41. Wang DQ, Portincasa P. Gallstones: Recent advances in epidemiology, pathogenesis, diagnosis and management. New York, NY: Nova Science Publishers (2017).
42. Wang DQ, Portincasa P, Wang HH. “Bile formation and pathophysiology of gallstones”. In: Kuipers EJ, editor. Encyclopedia of Gastroenterology. New York, NY: Elsevier (2020). p. 287–306.
43. Bennion LJ, Ginsberg RL, Gernick MB, Bennett PH. Effects of oral contraceptives on the gallbladder bile of normal women. N Engl J Med (1976) 294:189–92. doi: 10.1056/NEJM197601222940403
44. Grodstein F, Colditz GA, Hunter DJ, Manson JE, Willett WC, Stampfer MJ. A prospective study of symptomatic gallstones in women: relation with oral contraceptives and other risk factors. Obstetr Gynecol (1994) 84:207–14. doi: 10.1093/jnci/86.19.1466
45. Grodstein F, Colditz GA, Stampfer MJ. Postmenopausal hormone use and cholecystectomy in a large prospective study. Obstetr Gynecol (1994) 83:5–11. doi: 10.7326/0003-4819-128-9-199805010-00001
46. Uhler ML, Marks JW, Judd HL. Estrogen replacement therapy and gallbladder disease in postmenopausal women. Menopause (2000) 7:162–7. doi: 10.1097/00042192-200007030-00006
47. Simon JA, Hunninghake DB, Agarwal SK, Lin F, Cauley JA, Ireland CC, et al. Effect of estrogen plus progestin on risk for biliary tract surgery in postmenopausal women with coronary artery disease. The Heart and Estrogen/progestin Replacement Study. Ann Internal Med (2001) 135:493–501. doi: 10.7326/0003-4819-135-7-200110020-00008
48. Hart AR, Luben R, Welch A, Bingham S, Khaw KT. Hormone replacement therapy and symptomatic gallstones - a prospective population study in the EPIC-Norfolk cohort. Digestion (2008) 77:4–9. doi: 10.1159/000113897
49. Everson GT, McKinley C, Kern F Jr. Mechanisms of gallstone formation in women. Effects of exogenous estrogen (Premarin) and dietary cholesterol on hepatic lipid metabolism. J Clin Invest (1991) 87:237–46. doi: 10.1172/JCI114977
50. Petitti DB. Estrogen use and gallstone disease. Am J Public Health (1988) 78:1365. doi: 10.2105/AJPH.78.10.1365
51. Dhiman RK, Chawla YK. Is there a link between oestrogen therapy and gallbladder disease? Expert Opin Drug Saf (2006) 5:117–29. doi: 10.1517/14740338.5.1.117
52. Cirillo DJ, Wallace RB, Rodabough RJ, Greenland P, LaCroix AZ, Limacher MC, et al. Effect of estrogen therapy on gallbladder disease. J Am Med Assoc (2005) 293:330–9. doi: 10.1001/jama.293.3.330
53. Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. J Am Med Assoc (2002) 288:321–33. doi: 10.1001/jama.288.3.321
54. Angelin B, Olivecrona H, Reihner E, Rudling M, Stahlberg D, Eriksson M, et al. Hepatic cholesterol metabolism in estrogen-treated men. Gastroenterology (1992) 103:1657–63. doi: 10.1016/0016-5085(92)91192-7
55. Henriksson P, Einarsson K, Eriksson A, Kelter U, Angelin B. Estrogen-induced gallstone formation in males. Relation to changes in serum and biliary lipids during hormonal treatment of prostatic carcinoma. J Clin Invest (1989) 84:811–6. doi: 10.1172/JCI114240
56. Sweeting J. Estrogen-induced gallstones in males. Gastroenterology (1990) 98:800–1. doi: 10.1016/0016-5085(90)90309-O
57. Wang HH, Liu M, Clegg DJ, Portincasa P, Wang DQ. New insights into the molecular mechanisms underlying effects of estrogen on cholesterol gallstone formation. Biochim Biophys Acta (2009) 1791:1037–47. doi: 10.1016/j.bbalip.2009.06.006
58. Wang HH, Afdhal NH, Wang DQ. Estrogen receptor alpha, but not beta, plays a major role in 17beta-estradiol-induced murine cholesterol gallstones. Gastroenterology (2004) 127:239–49. doi: 10.1053/j.gastro.2004.03.059
59. Akin ML, Uluutku H, Erenoglu C, Karadag A, Gulluoglu BM, Sakar B, et al. Tamoxifen and gallstone formation in postmenopausal breast cancer patients: retrospective cohort study. World J Surg (2003) 27:395–9. doi: 10.1007/s00268-002-6468-4
60. Wang HH, Afdhal NH, Wang DQ. Overexpression of estrogen receptor alpha increases hepatic cholesterogenesis, leading to biliary hypersecretion in mice. J Lipid Res (2006) 47:778–86. doi: 10.1194/jlr.M500454-JLR200
61. de Bari O, Wang HH, Portincasa P, Liu M, Wang DQ. The deletion of the estrogen receptor alpha gene reduces susceptibility to estrogen-induced cholesterol cholelithiasis in female mice. Biochim Biophys Acta (2015) 1852:2161–9. doi: 10.1016/j.bbadis.2015.07.020
62. Lyons MA, Korstanje R, Li R, Sheehan SM, Walsh KA, Rollins JA, et al. Single and interacting QTLs for cholesterol gallstones revealed in an intercross between mouse strains NZB and SM. Mamm Genome (2005) 16:152–63. doi: 10.1007/s00335-004-2446-5
63. Lyons MA, Wittenburg H. Cholesterol gallstone susceptibility loci: a mouse map, candidate gene evaluation, and guide to human LITH genes. Gastroenterology (2006) 131:1943–70. doi: 10.1053/j.gastro.2006.10.024
64. Wang HH, Portincasa P, Afdhal NH, Wang DQ. Lith genes and genetic analysis of cholesterol gallstone formation. Gastroenterol Clinics North Am (2010) 39:185–207. doi: 10.1016/j.gtc.2010.02.007
65. Krawczyk M, Wang DQ, Portincasa P, Lammert F. Dissecting the genetic heterogeneity of gallbladder stone formation. Semin Liver Dis (2011) 31:157–72. doi: 10.1055/s-0031-1276645
66. Wang TY, Portincasa P, Liu M, Tso P, Wang DQ. Mouse models of gallstone disease. Curr Opin Gastroenterol (2018) 34:59–70. doi: 10.1097/MOG.0000000000000417
67. Wang HH, de Bari O, Arnatt CK, Liu M, Portincasa P, Wang DQ. Activation of a novel estrogen receptor GPR30 enhances cholesterol cholelithogenesis in female mice. Hepatology (2020) (in press). doi: 10.1002/hep.31212
68. Di Ciaula A, Wang DQ, Portincasa P. An update on the pathogenesis of cholesterol gallstone disease. Curr Opin Gastroenterol (2018) 34:71–80. doi: 10.1097/MOG.0000000000000423
69. de Bari O, Wang TY, Liu M, Portincasa P, Wang DQ. Estrogen induces two distinct cholesterol crystallization pathways by activating ERalpha and GPR30 in female mice. J Lipid Res (2015) 56:1691–700. doi: 10.1194/jlr.M059121
70. de Bari O, Wang TY, Liu M, Paik CN, Portincasa P, Wang DQ. Cholesterol cholelithiasis in pregnant women: pathogenesis, prevention and treatment. Ann Hepatol (2014) 13:728–45. doi: 10.1016/S1665-2681(19)30975-5
71. Lammert F, Gurusamy K, Ko CW, Miquel JF, Mendez-Sanchez N, Portincasa P, et al. Gallstones. Nat Rev Dis Primers (2016) 2:16024. doi: 10.1038/nrdp.2016.24
72. Portincasa P, Di Ciaula A, Wang HH, Palasciano G, van Erpecum KJ, Moschetta A, et al. Coordinate regulation of gallbladder motor function in the gut-liver axis. Hepatology (2008) 47:2112–26. doi: 10.1002/hep.22204
73. Ko CW, Beresford SA, Schulte SJ, Matsumoto AM, Lee SP. Incidence, natural history, and risk factors for biliary sludge and stones during pregnancy. Hepatology (2005) 41:359–65. doi: 10.1002/hep.20534
74. Ko CW. Risk factors for gallstone-related hospitalization during pregnancy and the postpartum. Am J Gastroenterol (2006) 101:2263–8. doi: 10.1111/j.1572-0241.2006.00730.x
75. Ko CW, Sekijima JH, Lee SP. Biliary sludge. Ann Internal Med (1999) 130:301–11. doi: 10.7326/0003-4819-130-4-199902160-00016
76. DeLeon C, Wang HH, Gunn J, Wilhelm M, Cole A, Arnett S, et al. A novel GPER antagonist protects against the formation of estrogen-induced cholesterol gallstones in female mice. J Lipid Res (2020) 61:767–77. doi: 10.1194/jlr.RA119000592
77. Wang DQ, Afdhal NH, Brandt L. Gallstone Disease. In: Feldman M, Friedman LS, editors. Sleisenger and Fordtrans Gastrointestinal and Liver Disease. Philadelphia: Elsevier Saunders (2020). p. 1016–46.
78. Vega-Vela NE, Osorio D, Avila-Rodriguez M, Gonzalez J, García-Segura LM, Echeverria V, et al. L-Type Calcium Channels Modulation by Estradiol. Mol Neurobiol (2017) 54:4996–5007. doi: 10.1007/s12035-016-0045-6
79. Kow L-M, Pfaff DW. Rapid estrogen actions on ion channels: A survey in search for mechanisms. Steroids (2016) 111:46–53. doi: 10.1016/j.steroids.2016.02.018
80. Ignatov T, Claus M, Nass N, Haybaeck J, Seifert B, Kalinski T, et al. G-protein-coupled estrogen receptor GPER-1 expression in hormone receptor-positive breast cancer is associated with poor benefit of tamoxifen. Breast Cancer Res Treat (2019) 174:121–7. doi: 10.1007/s10549-018-5064-8
Keywords: biliary sludge, bile salts, cholesterol gallstone disease, estrogen, estrogen receptors, gallbladder hypomotility, GPER1, GPER1 antagonists
Citation: DeLeon C, Wang DQ-H and Arnatt CK (2020) G Protein-Coupled Estrogen Receptor, GPER1, Offers a Novel Target for the Treatment of Digestive Diseases. Front. Endocrinol. 11:578536. doi: 10.3389/fendo.2020.578536
Received: 30 June 2020; Accepted: 06 October 2020;
Published: 12 November 2020.
Edited by:
Yves Jacquot, Université Paris Descartes, FranceReviewed by:
Damian Jacenik, University of Lodz, PolandCopyright © 2020 DeLeon, Wang and Arnatt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher K. Arnatt, Y2hyaXMuYXJuYXR0QHNsdS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.