 Nathalie S. Alekos
Nathalie S. Alekos Megan C. Moorer1,2
Megan C. Moorer1,2 Ryan C. Riddle
Ryan C. Riddle
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 23 September 2020
Sec. Bone Research
Volume 11 - 2020 | https://doi.org/10.3389/fendo.2020.578194
This article is part of the Research Topic Bone Inside-Out and Outside-In Signals: Control of Body Homeostasis View all 21 articles
The skeleton is a dynamic and metabolically active organ with the capacity to influence whole body metabolism. This newly recognized function has propagated interest in the connection between bone health and metabolic dysfunction. Osteoblasts, the specialized mesenchymal cells responsible for the production of bone matrix and mineralization, rely on multiple fuel sources. The utilization of glucose by osteoblasts has long been a focus of research, however, lipids and their derivatives, are increasingly recognized as a vital energy source. Osteoblasts possess the necessary receptors and catabolic enzymes for internalization and utilization of circulating lipids. Disruption of these processes can impair osteoblast function, resulting in skeletal deficits while simultaneously altering whole body lipid homeostasis. This article provides an overview of the metabolism of postprandial and stored lipids and the osteoblast's ability to acquire and utilize these molecules. We focus on the requirement for fatty acid oxidation and the pathways regulating this function as well as the negative impact of dyslipidemia on the osteoblast and skeletal health. These findings provide key insights into the nuances of lipid metabolism in influencing skeletal homeostasis which are critical to appreciate the extent of the osteoblast's role in metabolic homeostasis.
Development of the mammalian skeleton and maintenance of its structure for the life of the organism requires the coordinated actions of two specialized cells. Osteoclasts, large multinucleated cells that are derived from the monocyte/macrophage lineage of hematopoietic cells, are responsible for bone resorption. After attaching to an exposed bone surface, osteoclasts acidify a resorption lacuna to dissolve the mineral fraction of bone and then secrete proteolytic enzymes that degrade the organic matrix component (1). During the resorption process, growth factors trapped within bone matrix are released and trigger the recruitment of osteoblasts responsible for new bone formation (2, 3). Derived from mesenchymal stem cells present in the bone marrow stroma, these cells are characterized by their cuboidal shape and abundance of rough endoplasmic reticulum necessary for the production of the collagen-rich bone matrix (4). After building a packet of bone most osteoblasts will die by apoptosis, but small fractions will either become encapsulated within the bone matrix and fulfill regulatory functions as osteocytes or dedifferentiate and line bone surfaces. Known as bone remodeling, this process prevents the accumulation of old or damaged bone that may lead to fracture. In humans, peak bone mass is reached during the second decade of life as a result of net bone accrual during childhood, when bone formation exceeds resorption and osteoblasts and osteoclasts act on different bone surfaces to maintain the overall shape of bones during longitudinal growth (known as modeling). A balance between formation and resorption then occurs in early adulthood. However, with advancing age or as a result of numerous endocrine pathologies, an acceleration of osteoclastic activity leads to bone loss as osteoblastic activity is unable to keep pace. As bone mass decreases and structure integrity deteriorates, the risk of fracture increases (5, 6).
The tremendous economic impact of osteoporotic fractures (7–9) and development of comorbidities after fracture (10–12) highlight the need to understand the genetic, cellular, and endocrine mechanisms that influence bone mass. With the renewed interest in intermediary metabolism in cancer (13–15) and the recognition that bone is not merely a structural organ acting as a reserve of minerals but also an endocrine organ that can influence systemic metabolism (16–21), research in the field of skeletal biology has coalesced over the last few years on the contributions of cellular metabolism to osteoblast function and bone formation. The field reasoned that if bone contributes to the regulation of metabolic homeostasis through the release of osteocalcin and other hormones (16, 21), then the availability of nutrients must be critical to osteoblast function. Indeed, hierarchical analysis of energy requirments of cellular function (22) suggest that the bone remodeling process is energy intensive due to the synthesis of large extracellular matrix proteins and the necessity of concentrating mineral ions for hydroxyapatite crystal formation. Evidence from both the laboratory and the clinic supports this hypothesis as caloric restriction during gestation or during postnatal life strongly influences the trajectory of both the accrual and the maintenance of bone mass (23–25). Additionally, an increase in oxidative phosphorylation and the abundance of mitochondria appears to be a requirement for the differentiation of osteoblasts from marrow stem cells (26–29).
Osteoblasts harvest energy from a number of fuel molecules. Studies performed more than 50 years ago first highlighted the avidity of osteoblasts for glucose. Isolated osteoblasts or bone tissue explants from mice, rats, rabbits, and humans all used glucose to produce lactate even under aerobic conditions (30–34). More contemporary studies indicated that glucose acquisition is mediated by glucose transporter-1 (35) and that metabolic programming of glucose utilization is adjusted according to the stage of differentiation (28, 36). Cells of the osteoblast lineage also consume a significant amount of glutamine which is required for skeletal stem cell specification, can be catabolized by the tricarboxylic acid cycle to generate ATP, and serves as a regulatory signal to maintain endoplasmic reticulum health during stages of heightened protein synthesis (37, 38).
While lipid metabolism yields significantly more ATP than glucose or glutamine catabolism, its role in osteoblast function remains more controversial. Recent studies have highlighted the importance of fatty acid catabolism for normal bone formation (39, 40), but detrimental effects of lipids on osteoblast performance are also well-known (41, 42). In this review, we discuss the dual effects of lipids on osteoblast function and the maintenance of bone mass and strength. We provide a brief overview of the trafficking and metabolism of lipids in target tissues like bone. We then describe studies which highlight the importance of fatty acids metabolism for the accrual of bone mass and the mechanisms that regulate fatty acid utilization. Finally, we discuss the effects of dyslipidemia on osteoblast function and the potential for this condition to desensitize osteoblasts to anabolic signals.
The lipid molecules that support cellular metabolism are primarily derived from three sources: ingested fat, lipoproteins produced by the liver, and non-esterified fatty acids released by white adipose tissue (Figure 1). Postprandial triglycerides and cholesterol esters are broken down in the intestinal lumen by cholesterol esterases, pancreatic lipases, and bile salts. These molecules are then taken up by the enterocyte of the small intestine, re-esterified, and packaged with lipid-soluble vitamins, and apolipoproteins into chylomicrons. Chylomicrons enable water-insoluble fats and cholesterol to move through the lymphatic system and into the circulatory system. Engagement of the chylomicron by lipoprotein lipase (LPL) on capillary endothelium results in the hydrolysis of triglycerides and the delivery of fatty acids to target tissues (43, 44). The chylomicron remnants containing cholesterol and apolipoproteins are then cleared by the liver (45).
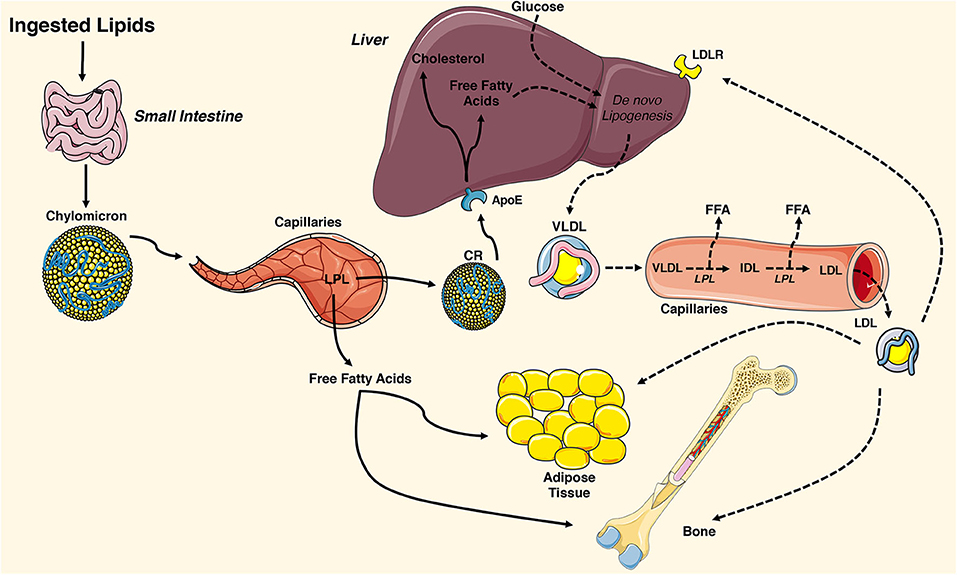
Figure 1. Overview of tissue-targeted lipid metabolism. Ingested lipids are broken down in the intestinal lumen and internalized by enterocytes of the small intestine. The water insoluble triglycerides and cholesterol are repackaged into chylomicrons and travel through the lymphatic system and into the circulatory system where they engage lipoprotein lipase (LPL) on the surface of capillary endothelial cells. The hydrolyzed triglycerides result in release of free fatty acids that are taken up by adipose tissue and the skeleton. The remaining chylomicron remnants (CR) are cleared by the liver via the apolipoprotein E (ApoE) receptor. CR-derived cholesterol and free fatty acids and circulating glucose are used for de novo lipogenesis, generating ATP for the liver, or repackaged into very low-density lipoproteins (VLDL). VLDL particles are released into the circulation where they engage LPL and release free fatty acids, which are also available for uptake. The remaining low-density lipoprotein (LDL) are internalized by cells expressing the low-density lipoprotein receptor (LDLR) including adipocytes and osteoblasts. This figure was created using Servier Medical Art image templates under a Creative Commons Attribution 3.0 Unported License.
In healthy individuals, the liver exhibits a nearly constant lipid flux. Chylomicron remnants and free fatty acids are taken up by the liver, while a portion of the circulating glucose taken up by the organ is used for de novo lipogenesis. Lipid molecules from each of these sources can be used to generate ATP in the liver or they can be packaged along with apolipoprotein (Apo) B-100, ApoC, and ApoE into very low-density lipoproteins (VLDL) on the endoplasmic reticulum. VLDL are released into the circulation and metabolized by target tissues in a manner similar to that of chylomicrons, with LPL hydrolyzing triglycerides to fatty acids that can be imported by cells. In this case, the remaining lipoprotein particle is further metabolized to low density lipoprotein (LDL), which can be taken up by many tissues via the LDL receptor (44).
White adipose tissue is the primary storage depot for excess calories. Non-esterified fatty acids are taken up by adipocytes, esterified and stored as triglycerides, while glucose is metabolized to acetyl-CoA and then used as a substrate for de novo fatty acid synthesis. When energy expenditure exceeds caloric intake or in response to a number of lipolytic hormones, the stored triglycerides can be hydrolyzed to glycerol and free fatty acids that are released into the circulation to be used for β-oxidation in other organs, including the skeleton. Lipolysis is mediated by the stepwise action of three lipases (illustrated in Figure 2). The rate limiting enzyme, adipose triglyceride lipase (ATGL), catalyzes the first reaction by hydrolyzing triacylglycerols at the sn-2 position to diacylglycerol and one fatty acid. Diacylglycerides are then preferentially hydrolyzed by hormone sensitive lipase (HSL) at the sn-3 position to yield a second free fatty acid (46). Monoacyglycerol lipase (MGL) catalyzes the final reaction generating glycerol and a third fatty acid (47). Most free fatty acids released into circulation are bound by albumin (48).
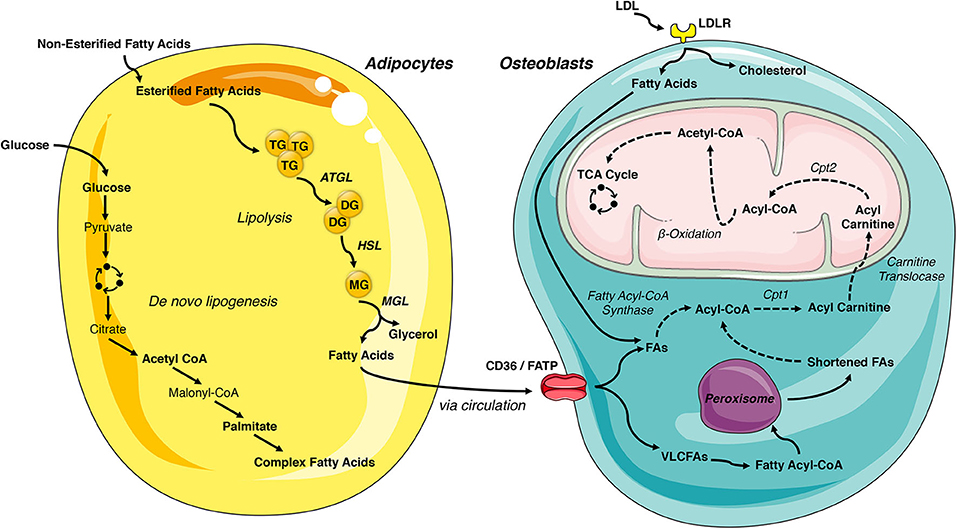
Figure 2. Lipid- flux between the adipocyte and osteoblast. White adipose tissue is the primary storage depot of lipids during excess consumption, which are subsequently released when energy expenditure exceeds caloric intake. Esterified fatty acids are stored in the adipocytes as triglycerides and are hydrolyzed by the rate limiting enzyme, adipose triglyceride lipase (ATGL), into diglycerides. Diglycerides are hydrolyzed into monoglycerides by hormone sensitive lipase (HSL) and further into fatty acids by monoacyglycerol lipase (MGL), which are then released into circulation. Adipocyte uptake of glucose is metabolized to acetyl-CoA and used for de novo fatty acid synthesis. These newly synthesized fatty acids are another lipid source for the osteoblast. LDL-derived fatty acids and uptake of circulating free fatty acids via CD36/FATPs are vital energy sources for the osteoblast. These internalized free fatty acids are converted into acyl-CoA by fatty acyl-CoA synthase. Very long chain fatty acids (VLCFAs) (more than 22 carbons) are first shortened by the peroxisome. Acyl-CoA is transported to the mitochondrial matrix by a carnitine exchange system in order to undergo β-oxidation. The product, acetyl-CoA is transferred to the TCA cycle and electron transport chain for generation of ATP. This figure was created using Servier Medical Art image templates under a Creative Commons Attribution 3.0 Unported License.
The intracellular metabolism of fatty acids taken up by cells depends on chain length. Short-chain (1-6 carbons) and medium-chain (7-12 carbons) fatty acids are produced by the bacterial fermentation of dietary fiber or the ingestion of dairy products. These lipids are primarily metabolized by enterocytes or by hepatocytes and are beyond the scope of this review (47). Long-chain fatty acids (13-21 carbons) are transported into cells by specific transporters (discussed below) but have limited solubility in the cytosol. To increase solubility, trap fatty acids in the cell, and produce a high energy thioester necessary for the next steps of catabolism, long-chain fatty acyl-CoA ligases catalyze the formation of fatty acyl-CoA in a reaction that requires the hydrolysis of 1 ATP to AMP. Acyl-CoA must then be transferred to the mitochondria by a carnitine exchange system to undergo β-oxidation. Carnitine palmitoyltransferase 1 (CPT1), the first and rate-limiting step in this process, is located on the outer mitochondrial membrane and catalyzes the replacement of CoA with carnitine. Acyl-carnitines are recognized and transferred by carnitine-acylcarnitine translocase into the mitochondria matrix where carnitine palmitoyltransferase 2 (CPT2) reverses the reaction of CPT1 and regenerates Acyl-CoA. The four reaction β-oxidation process removes 2 carbons from the carboxy end of the acyl-CoA to generate acetyl-CoA, 1 NADH and 1 FADH2 that are transferred to the TCA cycle and electron transport chain for the generation of ATP (Figure 2). Successive rounds of β-oxidation are necessary to fully metabolize long-chain fatty acids (49).
Very long chain fatty acids (more than 22 carbons) can also be used to generate ATP but must be chain-shortened in peroxisomes before they can enter the mitochondria (50). Multi-functional peroxisomes encase more than 50 enzymes, with more than half involved in fat metabolism, in a single lipid bilayer. As in long chain fatty acid metabolism, very long chain fatty acids are first converted to acyl-CoAs in the cytosol. The fatty acyl-CoA is then transported into the peroxisome by members of the ATP binding cassette transporter D subfamily. Peroxisomal oxidation also involves four reactions but utilizes a separate set of enzymes to shorten the fatty acid chain and is not as efficient at ATP generation as there is no respiratory chain. Indeed, while the FADH2 produced by one round of mitochondrial β-oxidation yields 2 ATP, the electrons from FADH2 produced by peroxisomal oxidation are donated to oxygen to form H2O2. For this reason, chain shortened fatty acids can be shuttled to the mitochondria for further metabolism via β-oxidation.
Although they are smaller than those evident in adipocytes, most cells contain a lipid droplet that can presumably be used to generate ATP via β-oxidation. Histological studies indicate that both mature osteoblasts and differentiating osteoblast progenitors contain stored lipid (51, 52), but these stored lipids do not appear to be a major energy source for mature osteoblast function. Kim et al. (39) ablated the expression of ATGL in cultures of calvarial osteoblasts and mature osteoblasts and osteocytes in vivo (Atglflox/flox; Osteocalcin-Cre), which should eliminate intracellular lipolysis, but did not find a defect in either in vitro osteoblast performance or bone structure in vivo. Therefore, osteoblasts appear to require extracellular lipid sources.
A combination of in vivo and in vitro studies have examined the uptake of circulating lipoproteins and free fatty acids by the osteoblast and the skeleton. In perhaps the most comprehensive study, Neimeier and colleagues (53) modeled postprandial lipoprotein uptake by intravenously injecting fluorescent- or 125I-labeled chylomicron remnants into mice. Skeletal uptake was 17% that of liver but was greater than other catabolic organs including muscle and heart. Importantly, chylomicron remnant uptake by the osteoblast-/osteocyte-enriched femoral diaphysis was greater than that of bone marrow, indicating the skeletal acquisition was not simply carried out by marrow adipocytes. Osteoblasts also appear to take up of LDL and VLDL and acquisition can be enhanced by co-administration with ApoE, but these studies have primarily been performed in cultured osteoblasts (54–56). Skeletal uptake of fatty acids was assessed in vivo by Bartelt et al. (57) and Kim et al. (39) after delivering 3H-linoleic acid and 14C-palmitic acid or 3H-bromo-palmitate, respectively, via oral gavage. Similar to the uptake of chylomicron remnants, these studies revealed that skeletal acquisition of fatty acids is comparable to tissues that are more classically associated with fatty acid metabolism. Together, these studies highlight a potential role of bone in fatty acid metabolism and postprandial clearance of fat from the circulation.
The identity and requirements for specific receptors and transporters that allow osteoblasts to take up fatty acids and lipoproteins (Figure 2) need additional study, but experimental data exists for a number of possible mechanisms. Consistent with osteoblastic uptake of chylomicron remnants and lipoproteins, osteoblasts express the low-density lipoprotein receptor (LDLR) and low-density lipoprotein receptor-related protein-1 (LRP1) (58, 59). Interpretation of the skeletal phenotypes of mice engineered to be deficient for LDLR (LDLR−/−) requires care as studies have reported both reduced (60) and elevated bone volume (61) relative to wildtype mice. Both in vivo (60) and in vitro (62) analyses indicate that the actions of the LDLR are important for osteoblast function as its ablation results in reductions in the expression of gene markers of osteoblastic differentiation. These data accord with the ability of LDL to stimulate cell growth and sustain responsivity to anabolic stimuli in osteoblasts cultured under serum-free conditions (63). The discrepancies in bone volume observed in vivo are likely be related to the requirement for LDLR during osteoclast differentiation (60, 61).
LRP1 can facilitate the endocytosis of triglyceride and cholesterol containing chylomicron remnants in cultures of osteoblastic cell models (58) and polymorphisms in the gene encoding this receptor are associated with bone mineral density (64). However, analysis of an osteoblast-specific knockout mouse (Lrp1flox/flox; Runx2-Cre) revealed an osteopenic phenotype but there was no effect on systemic lipoprotein clearance or osteoblasts' ability to sequester fatty acids (65). While the bone phenotype has been attributed to marked increases in osteoclastogenesis (65), the sustained ability to take up lipoproteins could be due to the engagement of other LDLR family members. LRP5 and LRP6 are most typically associated with the propagation of signaling in response to Wnt ligands (66), but these receptors also have the capacity to bind and mediate the endocytosis of lipoproteins and chylomicron remnants (67, 68). Cultured osteoblasts rendered deficient for LRP5 also retained the ability to take up LDL (56), indicating that combinatorial genetic studies wherein the expression of multiple LRP receptors are simultaneously ablated may be necessary to discern receptor function in lipid particle uptake.
Osteoblasts also take up high density lipoproteins (HDL) and express Scarb1 (also referred to as SR-B1) (55), the major receptor for high-density lipoproteins (69). Some epidemiological studies suggest a positive correlation between BMD and HDL levels, but others have reported contradictory results [see (70) for a comprehensive review]. Interpretation of an association between HDL and bone mass in animal models has been equally challenging. Martineau and colleagues (71) reported that Scarb1 null mice display increases in HDL-associated cholesterol and increases in femoral bone volume and mineralization at 2 and 4 months in association with increases in osteoblast surface and bone formation rate, which suggests a detrimental effect of HDL on skeletal homeostasis. However, it remains possible that the high bone mass phenotype in these mice is due to an increase in serum adrenocorticotropin (ACTH), which has anabolic effects on osteoblasts (72, 73). Futhermore, control and Scab1 deficient osteoblasts exhibited similar levels of HDL-cholesterol uptake in vitro (71). A follow-up study by this same group reported that Scarb1 deletion in MSCs increased osteoblastogenesis but decreased terminal osteocyte differentiation as vertebral osteocyte density was modestly decreased in the mutant mice (74). However, a more recent study contradicted these findings and reported Scarb1 null animals to be osteopenic in the veterbrae at 16 weeks with decreases in resorption and formation markers, and diminished osteoblast differentiation markers both in vitro and in vivo (73). Here too, alterations in bone volume were attributed to dose dependent effects of ACTH on bone. Similarly, mice with impaired HDL synthesis displayed reduced bone mass and impaired differentiation (75) suggesting a necessity for HDL in osteoblast function. Further in vivo studies using genetic models with osteoblast specific deletions are required to further delineate Scarb1 function and a role for reverse cholesterol transport will need to be considered.
Osteoblasts also express the receptors necessary to take up and metabolize free fatty acids. CD36 is a two-transmembrane glycoprotein receptor that binds long-chain fatty acids as well as oxidized low-density lipoprotein (oxLDL) and facilitates their transport into the cell (55, 76). While direct studies of its effect on fatty acid uptake have not yet been completed, CD36 null mice exhibit a low bone mass phenotype secondary to impaired bone formation (77) that implies fatty acid uptake is essential for osteoblast function. The SLC27 family of fatty acid transport proteins (also referred to as FATP1-6), may also contribute to osteoblasts acquisition of long-chain fatty acids for oxidation (76, 78), as multiple family members are expressed by primary osteoblasts (40).
The effects of specific fatty acids on the functions of the major bone cells has recently been reviewed elsewere (79). Direct examination of the requirement for fatty acid oxidation during postnatal bone acquisition and bone repair has been examined in two studies. In the first, Kim and colleagues disrupted the expression of CPT2 in mature osteoblasts and osteocytes (Cpt2flox/flox; Osteocalcin-Cre) (39). As noted above, CPT2 catalyzes an obligate step in fatty acid β-oxidation and was selected for ablation in this model because it is encoded by a single gene (three isoforms of CPT1 are present in mammalian genomes). The skeletal phenotype of the mutant mice was sexually dimorphic, with male mice fed a normal chow diet exhibiting only a transient decrease in trabecular bone volume in the distal femur and L5 vertebrae at 6 weeks of age. By contrast, female mutants exhibited defects in trabecular bone volume in the distal femur and L5 vertebrae and an expansion of cortical bone tissue area at both 6 and 12 weeks. This discrepancy between sexes appears to be related to a greater ability to adjust fuel utilization in males, as male mutants exhibited an increase in femoral glucose uptake that was not evident in female mutants. The greater inhibition of osteoblast performance and inhibition of glucose uptake in CPT2 mutant osteoblasts treated with estrogen may explain the sex differences in metabolic flexibility. Interestingly, both male and female CPT2 mutants exhibited an increase in serum free fatty acid levels, which suggests that disrupting fatty acid utilization by osteoblasts and osteocytes is sufficient to alter lipid homeostasis (39).
In the second study, van Gastel et. al. (40) identified a role for fatty acid utilization during fracture healing and the specification of skeletal cell fate. During the bone healing process, endochondral ossification is initiated by periosteal progenitor cells that differentiate to chondrocytes and form an avascular, cartilaginous callus. The callus is subsequently invaded by the vasculature (80) and replaced by bone (81, 82). Blood vessels are expected to deliver the oxygen, nutrients, and growth factors necessary to drive bone formation. Through biochemical assays and the reanalysis of an existing single cell RNAseq study of skeletal progenitors (83), Van Gastel et. al. (40) reported that chondrocytes express low levels of CPT1a and high Glut1 levels as well as elevated lactate production, which suggests that glycolysis meets the chondrocyte's energy needs. On the other hand, osteoblasts expressed high levels of Glut1 and CPT1a, exhibited higher levels of oxygen consumption, and an increased ability to metabolize palmitate, indicating a reliance on fatty acid oxdiation. Importantly, knocking down the expression of CPT1a prevented the differentiation of skeletal stem cells to osteoblasts while local injection of free fatty acids during fracture repair increased the amount of bone formed in the callus and reduced the amount of cartilage. Mechanistic studies demonstrated that reduced fatty acid availability increased the activation of FOXO3, which in turn activated SOX9 and chondrogenic specification. Taken together, these studies highlight the requirement for fatty acid β-oxidation for bone-forming osteoblasts in bone repair and skeletal development.
Evidence for a role of peroxisomal lipid oxidation in bone is largely based on the phenotypes evident in patients affected by peroxisomal disorders and global knockout models. Human and mouse genetic studies have identified 14 peroxin genes (PEX1- PEX26) that encode proteins necessary for either the formation of peroxisomes or the transport of cargo into the organelle. Loss of function mutations in peroxin genes, which occur at a rate of ~1 in 50,000 births, result in autosomal recessive peroxisomal biogenesis disorders (PBD) that affect a number of organ systems. Individuals with more severe PBD subtypes often exhibit craniofacial anomalies, short stature, and limb length discrepancies. Less severe subtypes have been associated with reductions in bone mineral density and an increased susceptibility for non-traumatic fractures (84–86). In the mouse, hypomorphic alleles for Pex7 leads to a reduction in longitudinal growth and impaired ossification of the digits (87), while a global knockout resulted in delayed ossification at multiple skeletal sites (88). Additional mouse genetic studies will be necessary to fully delineate the role of peroxisomes in skeletal tissue maintenance and function.
If fatty acid metabolism is used to generate the ATP necessary for osteoblast function, then metabolic flux in this pathway should be regulated by the signals that drive bone formation. Indeed, two of the most potently anabolic pathways, Wnt signaling and parathyroid hormone signaling, appear to drive fatty acid oxidation.
The anabolic effects of Wnt signaling on skeletal development, repair, and homeostasis have been well-studied (89, 90), and a number of studies have now demonstrated that the pathway coordinates the intermediary metabolism of the osteoblast with the energetic demands of bone formation (38, 91–93). LRP5 and LRP6 act as co-receptors for the Frizzled receptors that propagate Wnt signals and lead to the stabilization and activation of β-catenin (66). While the osteoblast-specific ablation of either receptor (Lrp5flox/flox; Osteocalcin-Cre and Lrp6flox/flox; Osteocalcin-Cre) results in decreases in bone mineral density and vertebral trabecular bone volume (94), Frey et al. (56) found that the LRP5 mutants also exhibited increases in fat mass and serum triglycerides and free fatty acids, suggestive of a disruption in fatty acid utilization. Indeed, analysis of gene expression in cultured osteoblasts by microarray revealed that LRP5-deficient osteoblasts exhibited a downregulation of multiple genes involved in mitochondrial long-chain fatty acid β-oxidation. The effects of these changes in gene expression on β-oxidation were confirmed by examining the oxidation of oleate, which was reduced in LRP5 deficient osteoblasts when compared to control. Expression of LRP5 with a gain of function mutation (Lrp5G171V) in osteoblasts produced the opposite phenotype, as the transgenic mice exhibited increases in bone volume and oxidative gene expression as well as decreases in fat mass, serum triglycerides, and fatty acids.
Subsequent genetic studies revealed that Wnt-mediated regulation of fatty acid oxidation proceeds via a β-catenin-dependent mechanism. Frey et al. (95) found that only Wnt ligands that increase the abundance of β-catenin in cultured osteoblasts, increase the capacity to fully oxidize oleate to carbon dioxide. Since constitutive ablation of β-catenin in osteoblasts results in early lethality (96) in vivo, the generation of an inducible β-catenin knockout mouse (Ctnnbflox/flox; Osteocalcin-CreERT2) was necessary to examine the transcription factor's effects on fatty acid oxidation. In this model, the temporal ablation of β-catenin resulted in high-turnover bone loss as well as increased fat mass and the development of insulin resistance. Additionally, the expression of genes involved in long-chain fatty acid oxidation and the ability to oxidize oleate were reduced in β-catenin deficient osteoblasts in vitro, while serum fatty acid levels were increased in the mutants in vivo. These studies have expanded the role of canonical Wnt-signaling to influencing fatty acid utilization and coordinating whole-body energy homeostasis.
As indicated above, a number of contemporary studies suggest that Wnt signaling also regulates glucose and glutamine utilization by the osteoblast. Wnt signaling through LRP5 increased aerobic glycolysis in the ST2 bone marrow stromal cell line (93) and mice engineered to overexpress Wnt7b in osteoblasts exhibit dramatic increases in bone volume, but simultaneously ablating the expression of Glut1 completely inhibited the increase in bone accrual (92). Similarly, Wnt signaling induced glutamine catabolism via the TCA cycle (38) which in turn stimulated the expression of genes involved in protein sysnthesis. Interestingly, these effects were mediated by the activation of mTOR and not β-catenin (92, 93, 97). Thus, the metabolic actions of Wnt signaling appear to depend on the specific downstream pathways that are activated.
Parathyroid hormone (PTH) is a master regulator of serum calcium that signals in the bone, kidney, and intestine to increase calcium levels. Intermittent administration of human recombinant PTH (1-34) is now used to reduce the occurrence of vertebral and non-vertebral fractures and increased bone mineral density in postmenopausal osteoporotic women (98). This therapeutic effect is mediated by PTH's ability to decrease apoptosis of mature osteoblasts (99), activate preexisting bone lines cells (100, 101), and stimulate osteoprogenitor recruitment (102).
The first indication that PTH might influence fatty acid oxidation were completed by Adamek et al. (103). In this study, PTH increased palmitate oxidation in specific cell populations isolated from bone by enzymatic digestion, while 1,25-Dihydroxycholecalciferol administration produced a more dramatic effect in multiple cell fractions. A greater reliance of lipids was suggested by Catherwood et al. (63) who demonstrated that the inclusion of LDL or VLDL in a basic medium was sufficient to support the proliferative response of rat ROS17/2.8 to PTH. In a more recent work, Esen et al. (104) used Seahorse technology, radiolabeled metabolites, and MC3T3-E1 cells to examine the effect of PTH on osteoblast metabolism. These studies demonstrated that PTH stimulates glucose uptake and increases lactate production but reduces the shuttling of glucose-derived carbon to the TCA cycle. These findings suggest that the increased rate of oxygen consumption after PTH administration is due to the oxidation of another fuel source, perhaps fatty acids imported from serum. While additional studies will be necessary, this paradigm is congruent with findings from Maridas et. al (105) that tracked the transfer of fatty acids from adipocytes to bone marrow stromal cells as well as the established ability of PTH to induce lipolysis in adipocytes (106). Likewise, the reduction in marrow adipose tissue volume after intermittent PTH treatment suggests that marrow adipocytes represent an energy reserve that provides fatty acids to fuel the anabolic activity of osteoblasts (105, 107). The finding that PTH can increase bone mass even under conditions of caloric restriction suggests that the relationship between PTH activity and metabolism is more complex and worthy of further study (105).
The Centers for Disease Control (CDC) reports that 95 million adults age 20 and older have high cholesterol (>200 ng/dL) while about 25% have elevated triglyceride levels (108). The aforementioned preclinical studies suggest a requirement for fatty acid oxidation for normal skeletal development and homeostasis, but epidemiological studies suggest that dyslipidemia has detrimental effects on bone (109–114). Elevated triglycerides, hypercholesterolemia and increased LDL are associated with higher risk of osteoporosis (111, 114) while increased LDL has been associated with non-vertebral fractures (115). Likewise, the National Health and Nutrition Examination Survey (NHANES III) reports that 63% of osteoporotic patients have hyperlipidemia (116). Studies from elite endurance athletes suggest that even short term exposure to a diet rich in fat can elicit a catabolic state in bone with an increase in markers of bone resorption and decreases in bone formation markers at rest and following high-intensity exercise (117). The inverse relationship between hyperlipidemia and osteoporosis is further noted by the use of statins, a class of drugs used to lower cholesterol by blocking 3-hydroxy-3-methyl-glutaryl-CoA reductase, which was associated with an increase in BMD but no improvement in fracture risk (118–120). The sections below describe effects of dyslipidemia on osteoblast function and skeletal homeostasis in rodent models (Figure 3).
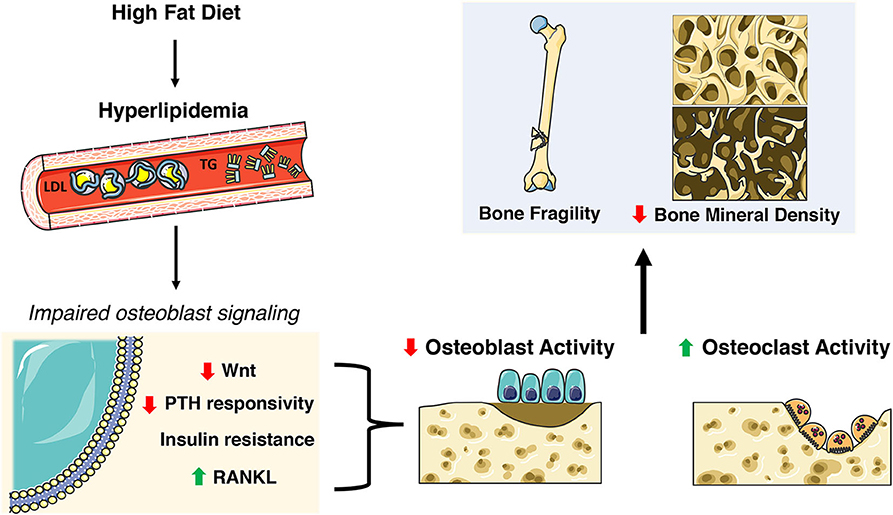
Figure 3. Skeletal deficits elicited by HFD-induced hyperlipidemia. A high fat diet (HFD) induces extensive systemic metabolic and skeletal changes including increases in circulating low density lipoprotein (LDL) and triglycerides (TG). This hyperlipidemic state impacts many nuances of osteoblast function and homeostasis including decreases in Wnt signaling and PTH responsiveness, insulin resistance, and increased RANKL. This results in decreased osteoblast activity and increased osteoclast activity ultimately contributing to poor skeletal health. This figure was created using Servier Medical Art image templates under a Creative Commons Attribution 3.0 Unported License.
Over the last decade a combination of high fat diet (HFD) feeding models and hyperlipidemic mouse models have been used to investigate the effects of dyslipidemia on skeletal homeostasis. In addition to the development of hypertriglyceridemia, these models exhibit a host of metabolic defects, including but not limited to adipose hyperplasia, hyperinsulinemia, insulin resistance, central leptin resistance, and hepatic steatosis [reviewed in (121)], that can alter the balance of bone remodeling and influence bone strength. The consensus from the majority of these studies is that HFD feeding leads to a deterioration of trabecular bone mass at multiple skeletal sites in the axial and appendicular skeleton (60, 116, 122–126). A hypercholesteremic diet produces a similar effect on trabecular bone parameters (127). Reports on the effects of HFD on cortical bone parameters are more variable. Tencerova and colleagues (125) reported that 12 weeks of HFD increased cortical porosity and decreased cortical thickness in the tibia of male C57Bl/6J mice. These phenotypes would be expected to reduce bone strength and indeed a reduction in maximum force and energy to failure were noted in the femur by Picke et al. (128) when a similar HFD feeding paradigm was employed. By contrast, Silva et al. (129) found that HFD had minimal effects on cortical bone material properties and modestly increased cortical bone area and strength in mice derived from a Large-by-Small advanced intercross, wherein inbred mouse strains with extreme body sizes were crossed. However, this study did note a discrepancy in the relationship between the expansion of femoral tissue area with increasing body mass in HFD fed mice. This finding would appear to be in agreement with the minimal effects of a HFD on cortical bone geometry in female C57Bl/6J until data were normalized to body mass (130). In all likelihood, the differences observed in the cortical bone envelope are due to the balancing of detrimental effects of metabolic dysfunction with increased mechanical loading secondary to weight gain.
Histomorphometric analyses and serum measurements of bone turnover markers consistently demonstrated that trabecular bone loss in HFD and hyperlipidemic mice is secondary to a reduction in osteoblast numbers and function as well as an increase in the abundance of osteoclasts (122, 124, 127, 131, 132). Consistent with this finding, a HFD induces a decrease in the expression of the key osteogenic transcription factors RUNX2 and OSTERIX in the bone (60, 124, 132, 133) and impairments in proliferation and colony forming capacity in bone marrow-derived mesenchymal stem cells (BM-MSCs) (125). Additionally, osteoclast precursors isolated from HFD fed mice exhibit an increased ability to form TRAP-positive osteoclasts after treatment with M-CSF and RANKL (134). The extensive effects of high fat intake were further revealed in gene expression profiling experiments performed by You et al. (135). In this study, 3 months of a high fat/high cholesterol diet led to the down-regulation of 2,200 genes and the up-regulation of 992 genes in RNA samples isolated from whole femur. Downregulated genes were implicated in a number of pathway associated with bone formation including the TGF-ß/BMP2 pathway and the Wnt pathway, while up-regulated genes were associated with the control of bone resorption. Strikingly, comparative cluster analysis of these data with changes in gene expression in ovariectomized rats, a model of osteoporosis, revealed the co-regulation of more than 1,300 genes, suggestive of a convergence of pathogenic pathways.
To dissect the effects of altered lipid metabolism from other metabolic derangements in these models, in vitro culture systems wherein cultures of primary osteoblasts or osteoblast-like cell lines are treated with exogenous lipids have proven helpful (124, 135–139). The common finding in these studies is that the exposure to sufficient quantities of cholesterol, palmitate or oxidized LDL [a product of LDL interaction with reactive oxygen species (140)] reduces the proliferation of osteoblastic cells, induces cell death, and impairs osteoblast differentiation. These same stimuli induce an increase in the expression of RANKL by osteoblasts and enhance osteoclastic differentiation (127). Together these studies suggest that elevated lipid levels or the presence of oxidized lipids alone are sufficient to diminish osteoblast function and in turn lead to an imbalance in anabolic and catabolic processes in the skeleton.
The precise mechanisms by which exogenous or oxidized lipids impair osteoblast function are not completely understood. One potential explanation is the development of an inflammatory state that is thought to contribute to metabolic dysfunction in other tissues. In support of this idea, genetic ablation of the inflammatory cytokine TNFα inhibits bone loss associated with a HFD and the detrimental effects of palmitate on osteoblast differentiation (124). Additionally, a dual impact of lipids on inflammation has been noted. While polyunsaturated omega-3 fatty acids are thought to be beneficial to bone health (79, 141–144), and have anti-inflammatory affects (145), omega-6 fatty acids have been reported to be pro-inflammatory (146), leading to pathological bone remodeling and contributing to bone fracture and osteoporosis (79). In addition to inflammatory effects, a combination of in vitro and in vivo evidence suggests that dyslipidemia desensitizes osteoblasts to anabolic stimuli, including those that regulate lipid utilization.
In additional to regulating the utilization of fatty acids by osteoblasts, Wnt/β-catenin signaling is vulnerable to the detrimental effects of HFD feeding. At the most proximal end of the signaling pathway, dyslipidemia appears to result in an increase in the expression of several secreted antagonists of Wnt signaling. Increases in the abundance of both Dkk1 and Sclerostin in serum have been reported in mice fed a HFD, while the latter was also found to be increased in the serum of ob/ob and db/db mice (128, 147–149). Similar increases have also been noted in obese humans and were accompanied by increases in Dkk-2 and secreted Frizzled-related proteins (150). At the distal end of the pathway, obesity and high fat diet feeding were associated with a reduction in β-catenin protein levels in the femur (151, 152). In a more extreme example, HFD feeding of the ApoE−/− atherosclerosis mouse model, which induces marked decreases in osteoblast numbers and an inhibition of bone formation, resulted in widespread reductions in the expression of Wnt ligands and target genes at multiple skeletal sites (131). The mechanisms underlying these changes in transcription are not yet known.
Aside from changes in gene expression, Wang et al. (153) documented an interaction between the Wnt co-receptor LRP6 and oxidized phospholipids and oxidized LDL, produced as a result of an increase in reactive oxygen species. In this study, HFD fed mice exhibited consistent decreases in the numbers of osteoblast progenitors and the abundance of LRP6 at the cell surface in this cell population. Additional studies revealed that oxidized phospholipids and oxidized LDL induced the endocytosis of LRP6 and rendered cells resistant to the propagation of Wnt signaling. Considering the requirement for LRP6 function for the maintenance of normal bone mass (94), this mechanism may partially explain the ability of antibodies that neutralize oxidized phospholipids to attenuate bone loss due to a HFD (154).
As indicated earlier, supplementation of basal, serum-free medium with LDL is sufficient to rescue responsivity to PTH (63). However, an overabundance of serum lipid can attenuate intermittent PTH-induced bone formation as evidenced by studies in the hyperlipidemic Ldlr−/− and Apoe−/− mouse lines. Intermittent PTH did not increase total bone mineral density or bone mineral content in the femur of these models, and PTH-induced increases in multiple parameters of trabecular bone structure were diminished or abolished in Ldlr−/− mice (155). Later studies suggested that PTH resistance is likely to be due to the accumulation of oxidized lipids as administration of the D-4F peptide, which reduced lipid oxidation products, restored the anabolic effect of PTH (156–158). Given the requirement for LRP6 for normal PTH signaling (157, 159), resistence to PTH may also be mediated by oxidized LDL-induced internalization of LRP6 (157).
The importance of insulin signaling in the osteoblast is revealed by the increased risk of fracture and decreased BMD in type 1 diabetes [reviewed in (160–162)], increased fracture risk despite an increase in BMD in type 2 diabetes (163, 164), and studies utilizing genetic mouse models in which insulin receptor expression is manipulated. The latter demonstrates that insulin receptor signaling is required for proliferation, survival, and osteoblast differentiation, as well as the ability of the osteoblast to contribute to the regulation of whole-body metabolism (165–167). As in skeletal muscle and adipose, dyslipidemia appears to lead to insulin resistance in the osteoblast. Wei and colleagues (168) demonstrated that mice fed a HFD exhibited reduced IRS1/2 phosphorylation in osteoblasts after insulin stimulation in vivo and that stearate treatment in vitro led to SMURF-mediated ubiquitination of the insulin receptor. HFD did not reduce trabecular bone volume in this study (perhaps due to a reduced number of osteoclasts), but multiple markers of bone formation were reduced which suggests that skeletal insulin resistance may contribute to bone loss associated with dyslipidemia.
A final mechanism by which hyperlipidemia could impact osteoblast performance and skeletal homeostasis is through the activation of PPARγ, a transcriptional regulator of adipogenesis that can be activated by elevated lipid levels. In bone, the nuclear receptor influences bone remodeling by stimulating adipogenic differentiation of mesenchymal stem cell at the expense of osteoblastogenesis and by stimulating osteoclastogenesis (169–171). HFD-fed rodents exhibit increased PPARγ gene expression likely leading to defects in osteoblastogenesis (124, 151). Additionally, HFD caused an increase in callus adiposity attributed to increased PPARγ expression and was associated with decreased osteoblast surface during late stages of healing post-fracture (172). One potential explanation for these finds is the ability of PPARγ to interfere with anabolic Wnt/β-catenin signaling (173–175). These effects are critically important for the targeting of PPARγ function in the treatment of type 2 diabetes. Thiazolidinediones (TZDs), synthetic PPARγ ligands, are used to increase insulin senstivity (176–178) but do so at the expense of skeletal health. Long term use of these agonists increased risk of fractures in women (179, 180) and decreased bone formation makers (181) while short term use was sufficient to decrease bone formation markers, total hip bone density, and lumbar spine bone density (182).
It is important to note that genetic ablation of PPARγ has beneficial effects on bone and body composition. Akune and colleagues (169) reported that PPAR+/− exhibit an increase in trabecular bone volume secondary to a doubling of the osteoblast surface. When PPARγ expression was ablated in mature osteoblasts and osteocytes (PPARgflox/flox DMP1-Cre), the mutant mice exhibited increases in femoral bone mineral density and trabecular bone volume as well as reduced fat mass and increased energy expenditure (183). Crosstalk between osteoblasts and adipocytes in this model was indicated by in vitro studies wherein the 3T3-L1 adipocyte cell line was treated with medium conditioned by PPARγ deficient osteoblasts culture media and exhibited reduced Oil Red O staining than those exposed to medium conditioned by wildtype osteoblasts (183). Furthermore, PPARγ ablation in mature osteoblast/osteocytes protected against HFD-induced metabolic affects by improving liver steatosis, increasing lean mass, preventing fat mass increases, maintaining wild-type glycemic control, and improved biomechanical strength (183). Therefore, modulating PPARγ's function in the osteoblast could be a potential target for combating bone loss associated with hyperlipidemia.
In this review, we have attempted to convey the necessity of lipid utilization by the osteoblast for normal skeletal homeostasis as well as the potential for dyslipidemia to impair osteoblast function and lead to an imbalance in bone remodeling. Mitochondrial long chain fatty acid oxidation is of sufficient importance for osteoblast function that [1] genetic impairments in this metabolic pathway lead to alterations in whole body lipid homeostasis and [2] signaling pathways essential to bone mass accrual influence fatty acid metabolism. Future studies should be directed toward more fully delineating the mechanisms of fatty acquisition by osteoblasts. These studies will require the development of new genetic mouse models in which transporters are disrupted specifically in the osteoblasts as global knockout models exhibit disturbances in metabolism that may indirectly influence bone remodeling. Determining the mechanisms by which osteoblasts convey their need for sufficient fatty acid supply to other tissues is equally vital. In this regard, the emergence of bone as a hormone-producing tissue is likely to provide key insights into the responsible endocrine networks. As we noted above, the detrimental effects of dyslipidemia, particularly in response to a high fat feeding in rodent models, on bone mass and the balance of bone formation and resorption are well-known, but the underlying mechanisms are still poorly understood. The increased recognition of bone as a lipid-utilizing tissue is likely to lead to a renewed interest in this area. Together these studies will provide a deeper understanding of the intimate interaction between the skeleton and metabolism and hopefully lead to treatment strategies that simultaneously reduce the burden of obesity and metabolic disease and preserve skeletal homeostasis.
All authors contributed to the drafting of this manuscript and approved the final version.
This work was supported by National Institutes of Health grant DK099134 and Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development grant BX003724.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors are grateful to the many other investigators whose work has not been cited here due to space limitations.
1. Teitelbaum SL. Bone resorption by osteoclasts. Science. (2000) 289:1504–8. doi: 10.1126/science.289.5484.1504
2. Tang Y, Wu X, Lei W, Pang L, Wan C, Shi Z, et al. TGF-B1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat Med. (2009) 15:757–65. doi: 10.1038/nm.1979
3. Xian L, Wu X, Pang L, Lou M, Rosen CJ, Qiu T, et al. Matrix IGF-1 maintains bone mass by activation of mTOR in mesenchymal stem cells. Nat Med. (2012) 18:1095–101. doi: 10.1038/nm.2793
4. Dudley HR, Spiro D. The fine structure of bone cells. J Biophys Biochem Cytol. (1961) 11:627–49. doi: 10.1083/jcb.11.3.627
5. Khosla S, Amin S, Orwoll E. Osteoporosis in men. Endocr Rev. (2008) 29:441–64. doi: 10.1210/er.2008-0002
6. Syed FA, Ng AC. The pathophysiology of the aging skeleton. Curr Osteoporos Rep. (2010) 8:235–40. doi: 10.1007/s11914-010-0035-y
7. Borgström F, Karlsson L, Ortsäter G, Norton N, Halbout P, Cooper C, et al. Fragility fractures in Europe: burden, management and opportunities. Arch Osteoporos. (2020) 15:59. doi: 10.1007/s11657-020-0706-y
8. Lewiecki EM, Ortendahl JD, Vanderpuye-Orgle J, Grauer A, Arellano J, Lemay J, et al. Healthcare policy changes in osteoporosis can improve outcomes and reduce costs in the United States. JBMR Plus. (2019) 3:e10192. doi: 10.1002/jbm4.10192
9. Tatangelo G, Watts J, Lim K, Connaughton C, Abimanyi-Ochom J, Borgström F, et al. The cost of osteoporosis, osteopenia, and associated fractures in Australia in 2017. J Bone Miner Res. (2019) 34:616–25. doi: 10.1002/jbmr.3640
10. Edelmuth SVCL, Sorio GN, Sprovieri FAA, Gali JC, Peron SF. Comorbidities, clinical intercurrences, and factors associated with mortality in elderly patients admitted for a hip fracture. Rev Bras Ortop. (2018) 53:543–51. doi: 10.1016/j.rboe.2018.07.014
11. Pedersen AB, Ehrenstein V, Szépligeti SK, Sørensen HT. Hip fracture, comorbidity, and the risk of myocardial infarction and stroke: a danish nationwide cohort study, 1995–2015. J Bone Miner Res. (2017) 32:2339–46. doi: 10.1002/jbmr.3242
12. Roche JJW, Wenn RT, Sahota O, Moran CG. Effect of comorbidities and postoperative complications on mortality after hip fracture in elderly people: Prospective observational cohort study. Br Med J. (2005) 331:1374–6. doi: 10.1136/bmj.38643.663843.55
13. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. (2016) 23:27–47. doi: 10.1016/j.cmet.2015.12.006
14. Yi M, Li J, Chen S, Cai J, Ban Y, Peng Q, et al. Emerging role of lipid metabolism alterations in Cancer stem cells. J Exp Clin Cancer Res. (2018) 37:1–18. doi: 10.1186/s13046-018-0784-5
15. Currie E, Schulze A, Zechner R, Walther TC, Farese R V. Cellular fatty acid metabolism and cancer. Cell Metab. (2013) 18:153–61. doi: 10.1016/j.cmet.2013.05.017
16. Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, Confavreux C, et al. Endocrine regulation of energy metabolism by the skeleton. Cell. (2007) 130:456–69. doi: 10.1016/j.cell.2007.05.047
17. Oury F, Sumara G, Sumara O, Ferron M, Chang H, Smith CE, et al. Endocrine regulation of male fertility by the skeleton. Cell. (2011) 144:796–809. doi: 10.1016/j.cell.2011.02.004
18. Wei J, Karsenty G. An overview of the metabolic functions of osteocalcin. Rev Endocr Metab Disord. (2015) 16:93–8. doi: 10.1007/s11154-014-9307-7
19. Guntur AR, Rosen CJ. Bone as an endocrine organ. Endocr Pract. (2012) 18:758–62. doi: 10.4158/EP12141.RA
20. Lee W-C, Guntur AR, Long F, Rosen CJ. Energy metabolism of the osteoblast: implications for osteoporosis. Endocr Rev. (2017) 38:255–66. doi: 10.1210/er.2017-00064
21. Dirckx N, Moorer MC, Clemens TL, Riddle RC. The role of osteoblasts in energy homeostasis. Nat Rev Endocrinol. (2019) 15:651–65. doi: 10.1038/s41574-019-0246-y
22. Buttgereit F, Brand MD. A hierarchy of ATP-consuming processes in mammalian cells. Biochem J. (1995) 312:(Pt 1):163–7. doi: 10.1042/bj3120163
23. Knight BS, Pennell CE, Adamson SL, Lye SJ. The impact of murine strain and sex on postnatal development after maternal dietary restriction during pregnancy. J Physiol. (2007) 581:873–81. doi: 10.1113/jphysiol.2006.126573
24. Devlin MJ, Cloutier AM, Thomas NA, Panus DA, Lotinun S, Pinz I, et al. Caloric restriction leads to high marrow adiposity and low bone mass in growing mice. J Bone Miner Res. (2010) 25:2078–88. doi: 10.1002/jbmr.82
25. Miller KK, Lee EE, Lawson EA, Misra M, Minihan J, Grinspoon SK, et al. Determinants of skeletal loss and recovery in anorexia nervosa. J Clin Endocrinol Metab. (2006) 91:2931–7. doi: 10.1210/jc.2005-2818
26. Shum LC, White NS, Mills BN, Bentley KL de M, Eliseev RA. Energy metabolism in mesenchymal stem cells during osteogenic differentiation. Stem Cells Dev. (2016) 25:114–22. doi: 10.1089/scd.2015.0193
27. Shares BH, Busch M, White N, Shum L, Eliseev RA. Active mitochondria support osteogenic differentiation by stimulating β-catenin acetylation. J Biol Chem. (2018) 293:16019–27. doi: 10.1074/jbc.RA118.004102
28. Guntur AR, Le PT, Farber CR, Rosen CJ. Bioenergetics during calvarial osteoblast differentiation reflect strain differences in bone mass. Endocrinology. (2014) 155:1589–95. doi: 10.1210/en.2013-1974
29. Chen C-T, Shih Y-RV, Kuo TK, Lee OK, Wei YH. Coordinated changes of mitochondrial biogenesis and antioxidant enzymes during osteogenic differentiation of human mesenchymal stem cells. Stem Cells. (2008) 26:960–8. doi: 10.1634/stemcells.2007-0509
30. Nichols FC, Neuman WF. Lactic acid production in mouse calvaria in vitro with and without parathyroid hormone stimulation: lack of acetazolamide effects. Bone. (1987) 8:105–9. doi: 10.1016/8756-3282(87)90078-0
31. Neuman WF, Neuman MW, Brommage R. Aerobic glycolysis in bone: lactate production and gradients in calvaria. Am J Physiol. (1978) 234:C41–50. doi: 10.1152/ajpcell.1978.234.1.C41
32. Borle AB, Nichols N, Nichols G. Metabolic studies of bone in vitro. J Biol Chem. (1960) 235:1206–10.
34. Felix R, Neuman WF, Fleisch H. Aerobic glycolysis in bone: lactic acid production by rat calvaria cells in culture. Am J Physiol. (1978) 234:C51–5. doi: 10.1152/ajpcell.1978.234.1.C51
35. Wei J, Shimazu J, Makinistoglu MP, Maurizi A, Kajimura D, Zong H, et al. Glucose uptake and Runx2 synergize to orchestrate osteoblast differentiation and bone formation. Cell. (2015) 161:1576–91. doi: 10.1016/j.cell.2015.05.029
36. Komarova SV, Ataullakhanov FI, Globus RK. Bioenergetics and mitochondrial transmembrane potential during differentiation of cultured osteoblasts. Am J Physiol Cell Physiol. (2000) 279:C1220–9. doi: 10.1152/ajpcell.2000.279.4.C1220
37. Yu Y, Newman H, Shen L, Sharma D, Hu G, Mirando AJ, et al. Glutamine metabolism regulates proliferation and lineage allocation in skeletal stem cells. Cell Metab. (2019) 29:966–78.e4. doi: 10.1016/j.cmet.2019.01.016
38. Karner CM, Esen E, Okunade AL, Patterson BW, Long F. Increased glutamine catabolism mediates bone anabolism in response to WNT signaling. J Clin Invest. (2015) 125:551–62. doi: 10.1172/JCI78470
39. Kim SP, Li Z, Zoch ML, Frey JL, Bowman CE, Kushwaha P, et al. Fatty acid oxidation by the osteoblast is required for normal bone acquisition in a sex- and diet-dependent manner. JCI insight. (2017) 2:e92704. doi: 10.1172/jci.insight.92704
40. van Gastel N, Stegen S, Eelen G, Schoors S, Carlier A, Daniëls VW, et al. Lipid availability determines fate of skeletal progenitor cells via SOX9. Nature. (2020) 579:111–7. doi: 10.1038/s41586-020-2050-1
41. Singh L, Tyagi S, Myers D, Duque GG. Bad or Ugly: the biological roles of bone marrow fat. Curr Osteoporos Rep. (2018) 16:130–7. doi: 10.1007/s11914-018-0427-y
42. Tintut Y, Demer LL. Effects of bioactive lipids and lipoproteins on bone. Trends Endocrinol Metab. (2014) 25:53–9. doi: 10.1016/j.tem.2013.10.001
43. Tso P, Balint JA. Formation and transport of chylomicrons by enterocytes to the lymphatics. Am J Physiol. (1986) 250:G715–26. doi: 10.1152/ajpgi.1986.250.6.G715
44. Kersten S. Physiological regulation of lipoprotein lipase. Biochim Biophys Acta. (2014) 1841:919–33. doi: 10.1016/j.bbalip.2014.03.013
45. Windler E, Chao Y, Havel RJ. Determinants of hepatic uptake of triglyceride-rich lipoproteins and their remnants in the rat. J Biol Chem. (1980) 255:5475–80.
46. Eichmann TO, Kumari M, Haas JT, Farese RV, Zimmermann R, Lass A, et al. Studies on the substrate and stereo/regioselectivity of adipose triglyceride lipase, hormone-sensitive lipase, and diacylglycerol-O-acyltransferases. J Biol Chem. (2012) 287:41446–57. doi: 10.1074/jbc.M112.400416
47. Schönfeld P, Wojtczak L. Short- and medium-chain fatty acids in energy metabolism: the cellular perspective. J Lipid Res. (2016) 57:943–54. doi: 10.1194/jlr.R067629
48. Richieri G V, Anel A, Kleinfeld AM. Interactions of long-chain fatty acids and albumin: determination of free fatty acid levels using the fluorescent probe ADIFAB. Biochemistry. (1993) 32:7574–80. doi: 10.1021/bi00080a032
49. Adeva-Andany MM, Carneiro-Freire N, Seco-Filgueira M, Fernández-Fernández C, Mouriño-Bayolo D. Mitochondrial β-oxidation of saturated fatty acids in humans. Mitochondrion. (2019) 46:73–90. doi: 10.1016/j.mito.2018.02.009
50. Reddy JK, Hashimoto T. Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: an adaptive metabolic system. Annu Rev Nutr. (2001) 21:193–230. doi: 10.1146/annurev.nutr.21.1.193
51. Enlow DH, Conklin JL, Bang S. Observations on the occurrence and the distribution of lipids in compact bone. Clin Orthop Relat Res. (1965) 38:157–69. doi: 10.1097/00003086-196500380-00022
52. Rendina-Ruedy E, Guntur AR, Rosen CJ. Intracellular lipid droplets support osteoblast function. Adipocyte. (2017) 6:250–8. doi: 10.1080/21623945.2017.1356505
53. Niemeier A, Niedzielska D, Secer R, Schilling A, Merkel M, Enrich C, et al. Uptake of postprandial lipoproteins into bone in vivo: impact on osteoblast function. Bone. (2008) 43:230–7. doi: 10.1016/j.bone.2008.03.022
54. Newman P, Bonello F, Wierzbicki AS, Lumb P, Savidge GF, Shearer MJ. The uptake of lipoprotein-borne phylloquinone. (vitamin K1) by osteoblasts and osteoblast-like cells: role of heparan sulfate proteoglycans and apolipoprotein E. J Bone Miner Res. (2002) 17:426–33. doi: 10.1359/jbmr.2002.17.3.426
55. Brodeur MR, Brissette L, Falstrault L, Luangrath V, Moreau R. Scavenger receptor of class B expressed by osteoblastic cells are implicated in the uptake of cholesteryl ester and estradiol from LDL and HDL3. J Bone Miner Res. (2008) 23:326–37. doi: 10.1359/jbmr.071022
56. Frey JL, Li Z, Ellis JM, Zhang Q, Farber CR, Aja S, et al. Wnt-Lrp5 signaling regulates fatty acid metabolism in the osteoblast. Mol Cell Biol. (2015) 35:1979–91. doi: 10.1128/MCB.01343-14
57. Bartelt A, Koehne T, Tödter K, Reimer R, Müller B, Behler-Janbeck F, et al. Quantification of bone fatty acid metabolism and its regulation by adipocyte lipoprotein lipase. Int J Mol Sci. (2017) 18:1264. doi: 10.3390/ijms18061264
58. Niemeier A, Kassem M, Toedter K, Wendt D, Ruether W, Beisiegel U, et al. Expression of LRP1 by human osteoblasts: a mechanism for the delivery of lipoproteins and vitamin K1 to bone. J Bone Miner Res. (2005) 20:283–93. doi: 10.1359/JBMR.041102
59. Grey A, Banovic T, Zhu Q, Watson M, Callon K, Palmano K, et al. The low-density lipoprotein receptor-related protein 1 is a mitogenic receptor for lactoferrin in osteoblastic cells. Mol Endocrinol. (2004) 18:2268–78. doi: 10.1210/me.2003-0456
60. Chen X, Wang C, Zhang K, Xie Y, Ji X, Huang H, et al. Reduced femoral bone mass in both diet-induced and genetic hyperlipidemia mice. Bone. (2016) 93:104–12. doi: 10.1016/j.bone.2016.09.016
61. Okayasu M, Nakayachi M, Hayashida C, Ito J, Kaneda T, Masuhara M, et al. Low-density lipoprotein receptor deficiency causes impaired osteoclastogenesis and increased bone mass in mice because of defect in osteoclastic cell-cell fusion. J Biol Chem. (2012) 287:19229–41. doi: 10.1074/jbc.M111.323600
62. Zhang N, Zhang Y, Lin J, Qiu X, Chen L, Pan X, et al. Low-density lipoprotein receptor deficiency impaired mice osteoblastogenesis in vitro. Biosci Trends. (2018) 11:658–66. doi: 10.5582/bst.2017.01267
63. Catherwood BD, Addison J, Chapman G, Contreras S, Lorang M. Growth of rat osteoblast-like cells in a lipid-enriched culture medium and regulation of function by parathyroid hormone and 1,25-dihydroxyvitamin D. J Bone Miner Res. (1988) 3:431–8. doi: 10.1002/jbmr.5650030410
64. Sims A-M, Shephard N, Carter K, Doan T, Dowling A, Duncan EL, et al. Genetic analyses in a sample of individuals with high or low BMD shows association with multiple Wnt pathway genes. J Bone Miner Res. (2008) 23:499–506. doi: 10.1359/jbmr.071113
65. Bartelt A, Behler-Janbeck F, Beil FT, Koehne T, Müller B, Schmidt T, et al. Lrp1 in osteoblasts controls osteoclast activity and protects against osteoporosis by limiting PDGF-RANKL signaling. Bone Res. (2018) 6:4. doi: 10.1038/s41413-017-0006-3
66. Teufel S, Hartmann C. Wnt-signaling in skeletal development. Curr Top Dev Biol. (2019) 133:235–79. doi: 10.1016/bs.ctdb.2018.11.010
67. Kim DH, Inagaki Y, Suzuki T, Ioka RX, Yoshioka SZ, Magoori K, et al. A new low density lipoprotein receptor related protein, LRP5, is expressed in hepatocytes and adrenal cortex, and recognizes apolipoprotein E. J Biochem. (1998) 124:1072–6. doi: 10.1093/oxfordjournals.jbchem.a022223
68. Fujino T, Asaba H, Kang M-J, Ikeda Y, Sone H, Takada S, et al. Low-density lipoprotein receptor-related protein 5. (LRP5) is essential for normal cholesterol metabolism and glucose-induced insulin secretion. Proc Natl Acad Sci USA. (2003) 100:229–34. doi: 10.1073/pnas.0133792100
69. Shen W-J, Hu J, Hu Z, Kraemer FB, Azhar S. Scavenger receptor class B type I. (SR-BI): a versatile receptor with multiple functions and actions. Metabolism. (2014) 63:875–86. doi: 10.1016/j.metabol.2014.03.011
70. Ackert-Bicknell CL. HDL cholesterol and bone mineral density: is there a genetic link? Bone. (2012) 50:525–33. doi: 10.1016/j.bone.2011.07.002
71. Martineau C, Martin-Falstrault L, Brissette L, Moreau R. The atherogenic Scarb1 null mouse model shows a high bone mass phenotype. Am J Physiol Endocrinol Metab. (2014) 306:E48–57. doi: 10.1152/ajpendo.00421.2013
72. Isales CM, Zaidi M, Blair HC. ACTH is a novel regulator of bone mass. Ann N Y Acad Sci. (2010) 1192:110–6. doi: 10.1111/j.1749-6632.2009.05231.x
73. Tourkova IL, Dobrowolski SF, Secunda C, Zaidi M, Papadimitriou-Olivgeri I, Papachristou DJ, et al. The high-density lipoprotein receptor Scarb1 is required for normal bone differentiation in vivo and in vitro. Lab Invest. (2019) 99:1850–60. doi: 10.1038/s41374-019-0311-0
74. Martineau C, Kevorkova O, Brissette L, Moreau R. Scavenger receptor class B, type I. (Scarb1) deficiency promotes osteoblastogenesis but stunts terminal osteocyte differentiation. Physiol Rep. (2014) 2:1–12. doi: 10.14814/phy2.12117
75. Blair HC, Kalyvioti E, Papachristou NI, Tourkova IL, Syggelos SA, Deligianni D, et al. Apolipoprotein A-1 regulates osteoblast and lipoblast precursor cells in mice. Lab Invest. (2016) 96:763–72. doi: 10.1038/labinvest.2016.51
76. Glatz JFC, Luiken JJFP, Bonen A. Membrane fatty acid transporters as regulators of lipid metabolism: implications for metabolic disease. Physiol Rev. (2010) 90:367–417. doi: 10.1152/physrev.00003.2009
77. Kevorkova O, Martineau C, Martin-Falstrault L, Sanchez-Dardon J, Brissette L, Moreau R. Low-bone-mass phenotype of deficient mice for the cluster of differentiation 36. (CD36). PLoS ONE. (2013) 8:e77701. doi: 10.1371/journal.pone.0077701
78. Anderson CM, Stahl A. SLC27 fatty acid transport proteins. Mol Aspects Med. (2013) 34:516–28. doi: 10.1016/j.mam.2012.07.010
79. Bao M, Zhang K, Wei Y, Hua W, Gao Y, Li X, Ye L. Therapeutic potentials and modulatory mechanisms of fatty acids in bone. Cell Prolif . (2020) 53:e12735. doi: 10.1111/cpr.12735
80. Mark H, Penington A, Nannmark U, Morrison W, Messina A. Microvascular invasion during endochondral ossification in experimental fractures in rats. Bone. (2004) 35:535–42. doi: 10.1016/j.bone.2004.04.010
81. Thompson Z, Miclau T, Hu D, Helms JA. A model for intramembranous ossification during fracture healing. J Orthop Res. (2002) 20:1091–8. doi: 10.1016/S0736-0266(02)00017-7
82. Kronenberg HM. Developmental regulation of the growth plate. Nature. (2003) 423:332–6. doi: 10.1038/nature01657
83. Baryawno N, Przybylski D, Kowalczyk MS, Kfoury Y, Severe N, Gustafsson K, et al. A cellular taxonomy of the bone marrow stroma in homeostasis and leukemia. Cell. (2019) 177:1915–32.e16. doi: 10.1016/j.cell.2019.04.040
84. Steinberg SJ, Raymond G V, Braverman NE, Moser AB. Zellweger Spectrum Disorder. (1993). Available at: https://www.ncbi.nlm.nih.gov/books/NBK1448/
85. Brosius U, Gärtner J. Cellular and molecular aspects of Zellweger syndrome and other peroxisome biogenesis disorders. Cell Mol Life Sci. (2002) 59:1058–69. doi: 10.1007/s00018-002-8486-7
86. Heymans HS, Oorthuys JW, Nelck G, Wanders RJA, Schutgens RBH. Rhizomelic chondrodysplasia punctata: another peroxisomal disorder. N Engl J Med. (1985) 313:187–8. doi: 10.1056/NEJM198507183130322
87. Braverman N, Zhang R, Chen L, Nimmo G, Scheper S, Tran T, et al. A Pex7 hypomorphic mouse model for plasmalogen deficiency affecting the lens and skeleton. Mol Genet Metab. (2010) 99:408–16. doi: 10.1016/j.ymgme.2009.12.005
88. Brites P, Motley AM, Gressens P, Mooyer PAW, Ploegaert I, Everts V, et al. Impaired neuronal migration and endochondral ossification in Pex7 knockout mice: a model for rhizomelic chondrodysplasia punctata. Hum Mol Genet. (2003) 12:2255–67. doi: 10.1093/hmg/ddg236
89. Huybrechts Y, Mortier G, Boudin E, Van Hul W. WNT signaling and bone: lessons from skeletal dysplasias and disorders. Front Endocrinol. (2020) 11:165. doi: 10.3389/fendo.2020.00165
90. Zhong Z, Ethen NJ, Williams BO. WNT signaling in bone development and homeostasis. Wiley Interdiscip Rev Dev Biol. (2014) 3:489–500. doi: 10.1002/wdev.159
91. Moorer MC, Riddle RC. Regulation of Osteoblast Metabolism by Wnt Signaling. Endocrinol Metab. (2018) 33:318–30. doi: 10.3803/EnM.2018.33.3.318
92. Chen H, Ji X, Lee W-C, Shi Y, Li B, Abel ED, et al. Increased glycolysis mediates Wnt7b-induced bone formation. FASEB J. (2019) 33:7810–21. doi: 10.1096/fj.201900201RR
93. Esen E, Chen J, Karner CM, Okunade AL, Patterson BW, Long F. WNT-LRP5 signaling induces Warburg effect through mTORC2 activation during osteoblast differentiation. Cell Metab. (2013) 17:745–55. doi: 10.1016/j.cmet.2013.03.017
94. Riddle RC, Diegel CR, Leslie JM, Van Koevering KK, Faugere M-C, Clemens TL, et al. Lrp5 and Lrp6 exert overlapping functions in osteoblasts during postnatal bone acquisition. PLoS ONE. (2013) 8:e63323. doi: 10.1371/journal.pone.0063323
95. Frey JL, Kim SP, Li Z, Wolfgang MJ, Riddle RC. β-catenin directs long-chain fatty acid catabolism in the osteoblasts of male mice. Endocrinology. (2018) 159:272–84. doi: 10.1210/en.2017-00850
96. Holmen SL, Zylstra CR, Mukherjee A, Sigler RE, Faugere M-C, Bouxsein ML, et al. Essential role of beta-catenin in postnatal bone acquisition. J Biol Chem. (2005) 280:21162–8. doi: 10.1074/jbc.M501900200
97. Chen J, Tu X, Esen E, Joeng KS, Lin C, Arbeit JM, Rüegg MA, et al. WNT7B promotes bone formation in part through mTORC1. PLoS Genet. (2014) 10:e1004145. doi: 10.1371/journal.pgen.1004145
98. Neer RM, Arnaud CD, Zanchetta JR, Prince R, Gaich GA, Reginster JY, et al. Effect of parathyroid hormone. (1-34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med. (2001) 344:1434–41. doi: 10.1097/00006254-200110000-00018
99. Jilka RL, Weinstein RS, Bellido T, Roberson P, Parfitt AM, Manolagas SC. Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J Clin Invest. (1999) 104:439–46. doi: 10.1172/JCI6610
100. Dobnig H, Turner RT. Evidence that intermittent treatment with parathyroid hormone increases bone formation in adult rats by activation of bone lining cells. Endocrinology. (1995) 136:3632–8. doi: 10.1210/endo.136.8.7628403
101. Kim SW, Pajevic PD, Selig M, Barry KJ, Yang J-Y, Shin CS, et al. Intermittent parathyroid hormone administration converts quiescent lining cells to active osteoblasts. J Bone Miner Res. (2012) 27:2075–84. doi: 10.1002/jbmr.1665
102. Wu X, Pang L, Lei W, Lu W, Li J, Li Z, et al. Inhibition of Sca-1-positive skeletal stem cell recruitment by alendronate blunts the anabolic effects of parathyroid hormone on bone remodeling. Cell Stem Cell. (2010) 7:571–80. doi: 10.1016/j.stem.2010.09.012
103. Adamek G, Felix R, Guenther HL, Fleisch H. Fatty acid oxidation in bone tissue and bone cells in culture. Characterization and hormonal influences. Biochem J. (1987) 248:129–37. doi: 10.1042/bj2480129
104. Esen E, Lee S-Y, Wice BM, Long F. PTH promotes bone anabolism by stimulating aerobic glycolysis via IGF signaling. J Bone Miner Res. (2015) 30:2137. doi: 10.1002/jbmr.2714
105. Maridas DE, Rendina-Ruedy E, Helderman RC, DeMambro VE, Brooks D, Guntur AR, et al. Progenitor recruitment and adipogenic lipolysis contribute to the anabolic actions of parathyroid hormone on the skeleton. FASEB J. (2019) 33:2885–98. doi: 10.1096/fj.201800948RR
106. Larsson S, Jones HA, Göransson O, Degerman E, Holm C. Parathyroid hormone induces adipocyte lipolysis via PKA-mediated phosphorylation of hormone-sensitive lipase. Cell Signal. (2016) 28:204–13. doi: 10.1016/j.cellsig.2015.12.012
107. Fan Y, Hanai J-I, Le PT, Bi R, Maridas D, DeMambro V, et al. Parathyroid hormone directs bone marrow mesenchymal cell fate. Cell Metab. (2017) 25:661–72. doi: 10.1016/j.cmet.2017.01.001
108. Carroll M, Kit B, Lacher D. Trends in elevated triglyceride in adults: United States, 2001-2012. NCHS Data Brief . (2015) 198.
109. Saoji R, Das RS, Desai M, Pasi A, Sachdeva G, Das TK, et al. Association of high-density lipoprotein, triglycerides, and homocysteine with bone mineral density in young Indian tribal women. Arch Osteoporos. (2018) 13:108. doi: 10.1007/s11657-018-0525-6
110. Chi JH, Shin MS, Lee BJ. Identification of hypertriglyceridemia based on bone density, body fat mass, and anthropometry in a Korean population. BMC Cardiovasc Disord. (2019) 19:66. doi: 10.1186/s12872-019-1050-2
111. Hsu Y-H, Venners SA, Terwedow HA, Feng Y, Niu T, Li Z, et al. Relation of body composition, fat mass, and serum lipids to osteoporotic fractures and bone mineral density in Chinese men and women. Am J Clin Nutr. (2006) 83:146–54. doi: 10.1093/ajcn/83.1.146
112. Cui L-H, Shin M-H, Chung E-K, Lee Y-H, Kweon S-S, Park K-S, et al. Association between bone mineral densities and serum lipid profiles of pre- and post-menopausal rural women in South Korea. Osteoporos Int. (2005) 16:1975–81. doi: 10.1007/s00198-005-1977-2
113. Nuzzo V, de Milita AM, Ferraro T, Monaco A, Florio E, Miano P, et al. Analysis of skeletal status by quantitative ultrasonometry in a cohort of postmenopausal women with high blood cholesterol without documented osteoporosis. Ultrasound Med Biol. (2009) 35:717–22. doi: 10.1016/j.ultrasmedbio.2008.11.003
114. Bijelic R, Balaban J, Milicevic S. Correlation of the lipid profile, BMI and bone mineral density in postmenopausal women. Mater Sociomed. (2016) 28:412–5. doi: 10.5455/msm.2016.28.412-415
115. Yamauchi M, Yamaguchi T, Nawata K, Tanaka K, Takaoka S, Sugimoto T. Increased low-density lipoprotein cholesterol level is associated with non-vertebral fractures in postmenopausal women. Endocrine. (2015) 48:279–86. doi: 10.1007/s12020-014-0292-0
116. Pirih F, Lu J, Ye F, Bezouglaia O, Atti E, Ascenzi M-G, et al. Adverse effects of hyperlipidemia on bone regeneration and strength. J Bone Miner Res. (2012) 27:309–18. doi: 10.1002/jbmr.541
117. Heikura IA, Burke LM, Hawley JA, Ross ML, Garvican-Lewis L, Sharma AP, et al. A Short-term ketogenic diet impairs markers of bone health in response to exercise. Front Endocrinol. (2019) 10:880. doi: 10.3389/fendo.2019.00880
118. Edwards CJ, Hart DJ, Spector TD. Oral statins and increased bone-mineral density in postmenopausal women. Lancet. (2000) 355:2218–9. doi: 10.1016/S0140-6736(00)02408-9
119. Wang Z, Li Y, Zhou F, Piao Z, Hao J. Effects of statins on bone mineral density and fracture risk: a PRISMA-compliant systematic review and meta-analysis. Medicine. (2016) 95:e3042. doi: 10.1097/MD.0000000000003042
120. Zheng J, Brion M-J, Kemp JP, Warrington NM, Borges M-C, Hemani G, et al. The effect of plasma lipids and lipid-lowering interventions on bone mineral density: a mendelian randomization study. J Bone Miner Res. (2020) 35:1224–35. doi: 10.1002/jbmr.3989
121. Wali JA, Jarzebska N, Raubenheimer D, Simpson SJ, Rodionov RN, O'Sullivan JF. Cardio-metabolic effects of high-fat diets and their underlying mechanisms-a narrative review. Nutrients. (2020) 12:1–18. doi: 10.3390/nu12051505
122. Montalvany-Antonucci CC, Zicker MC, Ferreira AVM, Macari S, Ramos-Junior ES, Gomez RS, et al. High-fat diet disrupts bone remodeling by inducing local and systemic alterations. J Nutr Biochem. (2018) 59:93–103. doi: 10.1016/j.jnutbio.2018.06.006
123. Shu L, Beier E, Sheu T, Zhang H, Zuscik MJ, Puzas EJ, et al. High-fat diet causes bone loss in young mice by promoting osteoclastogenesis through alteration of the bone marrow environment. Calcif Tissue Int. (2015) 96:313–23. doi: 10.1007/s00223-015-9954-z
124. Zhang K, Wang C, Chen Y, Ji X, Chen X, Tian L, et al. Preservation of high-fat diet-induced femoral trabecular bone loss through genetic target of TNF-α. Endocrine. (2015) 50:239–49. doi: 10.1007/s12020-015-0554-5
125. Tencerova M, Figeac F, Ditzel N, Taipaleenmäki H, Nielsen TK, Kassem M. High-fat diet-induced obesity promotes expansion of bone marrow adipose tissue and impairs skeletal stem cell functions in mice. J Bone Miner Res. (2018) 33:1154–65. doi: 10.1002/jbmr.3408
126. McCabe LR, Irwin R, Tekalur A, Evans C, Schepper JD, Parameswaran N, et al. Exercise prevents high fat diet-induced bone loss, marrow adiposity and dysbiosis in male mice. Bone. (2019) 118:20–31. doi: 10.1016/j.bone.2018.03.024
127. Sul O-J, Kim J-E, Ke K, Suh J-H, Choi H-S. Atherogenic diet-induced bone loss is primarily due to increased osteoclastogenesis in mice. J Nutr Biochem. (2020) 79:108337. doi: 10.1016/j.jnutbio.2019.108337
128. Picke A-K, Sylow L, Møller LLV, Kjøbsted R, Schmidt FN, Steejn MW, et al. Differential effects of high-fat diet and exercise training on bone and energy metabolism. Bone. (2018) 116:120–34. doi: 10.1016/j.bone.2018.07.015
129. Silva MJ, Eekhoff JD, Patel T, Kenney-Hunt JP, Brodt MD, Steger-May K, et al. Effects of high-fat diet and body mass on bone morphology and mechanical properties in 1100 advanced intercross mice. J Bone Miner Res. (2019) 34:711–25. doi: 10.1002/jbmr.3648
130. Devlin MJ, Robbins A, Cosman MN, Moursi CA, Cloutier AM, Louis L, et al. Differential effects of high fat diet and diet-induced obesity on skeletal acquisition in female C57BL/6J vs. FVB/NJ Mice. Bone Rep. (2018) 8:204–14. doi: 10.1016/j.bonr.2018.04.003
131. Liu Y, Almeida M, Weinstein RS, O'Brien CA, Manolagas SC, Jilka RL. Skeletal inflammation and attenuation of Wnt signaling, Wnt ligand expression, and bone formation in atherosclerotic ApoE-null mice. Am J Physiol Endocrinol Metab. (2016) 310:E762–73. doi: 10.1152/ajpendo.00501.2015
132. Colditz J, Picke A, Hofbauer LC, Rauner M. Contributions of Dickkopf-1 to obesity-induced bone loss and marrow adiposity. JBMR plus. (2020) 4:e10364. doi: 10.1002/jbm4.10364
133. Xiao Y, Cui J, Li YX, Shi YH, Le GW. Expression of genes associated with bone resorption is increased and bone formation is decreased in mice fed a high-fat diet. Lipids. (2010) 45:345–55. doi: 10.1007/s11745-010-3397-0
134. Cao JJ, Sun L, Gao H. Diet-induced obesity alters bone remodeling leading to decreased femoral trabecular bone mass in mice. Ann N Y Acad Sci. (2010) 1192:292–7. doi: 10.1111/j.1749-6632.2009.05252.x
135. You L, Sheng Z, Tang C, Chen L, Pan L, Chen J. High cholesterol diet increases osteoporosis risk via inhibiting bone formation in rats. Acta Pharmacol Sin. (2011) 32:1498–504. doi: 10.1038/aps.2011.135
136. Brodeur MR, Brissette L, Falstrault L, Ouellet P, Moreau R. Influence of oxidized low-density lipoproteins. (LDL) on the viability of osteoblastic cells. Free Radic Biol Med. (2008) 44:506–17. doi: 10.1016/j.freeradbiomed.2007.08.030
137. Parhami F, Morrow AD, Balucan J, Leitinger N, Watson AD, Tintut Y, et al. Lipid oxidation products have opposite effects on calcifying vascular cell and bone cell differentiation. A possible explanation for the paradox of arterial calcification in osteoporotic patients. Arterioscler Thromb Vasc Biol. (1997) 17:680–7. doi: 10.1161/01.ATV.17.4.680
138. Kim J-E, Ahn M-W, Baek S-H, Lee IK, Kim Y-W, Kim J-Y, et al. AMPK activator, AICAR, inhibits palmitate-induced apoptosis in osteoblast. Bone. (2008) 43:394–404. doi: 10.1016/j.bone.2008.03.021
139. Yang L, Guan G, Lei L, Lv Q, Liu S, Zhan X, et al. Palmitic acid induces human osteoblast-like Saos-2 cell apoptosis via endoplasmic reticulum stress and autophagy. Cell Stress Chaperones. (2018) 23:1283–94. doi: 10.1007/s12192-018-0936-8
140. Kita T, Kume N, Yokode M, Ishii K, Arai H, Horiuchi H, et al. Oxidized-LDL and atherosclerosis. Role of LOX-1. Ann N Y Acad Sci. (2000) 902:95–100; discussion 100–2. doi: 10.1111/j.1749-6632.2000.tb06304.x
141. Li Y, Greiner RS, Salem N, Watkins BA. Impact of dietary n-3 FA deficiency on rat bone tissue FA composition. Lipids. (2003) 38:683–6. doi: 10.1007/s11745-003-1115-8
142. Watkins BA, Li Y, Lippman HE, Feng S. Modulatory effect of omega-3 polyunsaturated fatty acids on osteoblast function and bone metabolism. Prostaglandins Leukot Essent Fatty Acids. (2003) 68:387–98. doi: 10.1016/S0952-3278(03)00063-2
143. Reinwald S, Li Y, Moriguchi T, Salem N, Watkins BA. Repletion with. (n-3) fatty acids reverses bone structural deficits in. (n-3)-deficient rats. J Nutr. (2004) 134:388–94. doi: 10.1093/jn/134.2.388
144. Kuroda T, Ohta H, Onoe Y, Tsugawa N, Shiraki M. Intake of omega-3 fatty acids contributes to bone mineral density at the hip in a younger Japanese female population. Osteoporos Int. (2017) 28:2887–91. doi: 10.1007/s00198-017-4128-7
145. Saini RK, Keum Y-S. Omega-3 and omega-6 polyunsaturated fatty acids: Dietary sources, metabolism, and significance - A review. Life Sci. (2018) 203:255–67. doi: 10.1016/j.lfs.2018.04.049
146. Innes JK, Calder PC. Omega-6 fatty acids and inflammation. Prostaglandins Leukot Essent Fatty Acids. (2018) 132:41–8. doi: 10.1016/j.plefa.2018.03.004
147. Kim SP, Frey JL, Li Z, Kushwaha P, Zoch ML, Tomlinson RE, et al. Sclerostin influences body composition by regulating catabolic and anabolic metabolism in adipocytes. Proc Natl Acad Sci USA. (2017) 114:E11238–47. doi: 10.1073/pnas.1707876115
148. Mabilleau G, Perrot R, Flatt PR, Irwin N, Chappard D. High fat-fed diabetic mice present with profound alterations of the osteocyte network. Bone. (2016) 90:99–106. doi: 10.1016/j.bone.2016.06.008
149. Baek K, Hwang HR, Park H-J, Kwon A, Qadir AS, Ko S-H, et al. TNF-α upregulates sclerostin expression in obese mice fed a high-fat diet. J Cell Physiol. (2014) 229:640–50. doi: 10.1002/jcp.24487
150. Razny U, Polus A, Goralska J, Zdzienicka A, Gruca A, Kapusta M, et al. Effect of insulin resistance on whole blood mRNA and microRNA expression affecting bone turnover. Eur J Endocrinol. (2019) 181:525–37. doi: 10.1530/EJE-19-0542
151. Chen J-R, Lazarenko OP, Wu X, Tong Y, Blackburn ML, Shankar K, et al. Obesity reduces bone density associated with activation of PPARγ and suppression of Wnt/β-catenin in rapidly growing male rats. PLoS ONE. (2010) 5:e13704. doi: 10.1371/journal.pone.0013704
152. Zahoor M, Cha P-H, Choi K-Y. Indirubin-3′-oxime, an activator of Wnt/β-catenin signaling, enhances osteogenic commitment of ST2 cells and restores bone loss in high-fat diet-induced obese male mice. Bone. (2014) 65:60–8. doi: 10.1016/j.bone.2014.05.003
153. Wang L, Chai Y, Li C, Liu H, Su W, Liu X, et al. Oxidized phospholipids are ligands for LRP6. Bone Res. (2018) 6:22. doi: 10.1038/s41413-018-0023-x
154. Ambrogini E, Que X, Wang S, Yamaguchi F, Weinstein RS, Tsimikas S, et al. Oxidation-specific epitopes restrain bone formation. Nat Commun. (2018) 9:2193. doi: 10.1038/s41467-018-04047-5
155. Huang MS, Lu J, Ivanov Y, Sage AP, Tseng W, Demer LL, et al. Hyperlipidemia impairs osteoanabolic effects of PTH. J Bone Miner Res. (2008) 23:1672–9. doi: 10.1359/jbmr.080513
156. Sage AP, Lu J, Atti E, Tetradis S, Ascenzi M-G, Adams DJ, et al. Hyperlipidemia induces resistance to PTH bone anabolism in mice via oxidized lipids. J Bone Miner Res. (2011) 26:1197–206. doi: 10.1002/jbmr.312
157. Li C, Xing Q, Yu B, Xie H, Wang W, Shi C, et al. Disruption of LRP6 in osteoblasts blunts the bone anabolic activity of PTH. J Bone Miner Res. (2013) 28:2094–108. doi: 10.1002/jbmr.1962
158. Huang MS, Morony S, Lu J, Zhang Z, Bezouglaia O, Tseng W, et al. Atherogenic phospholipids attenuate osteogenic signaling by BMP-2 and parathyroid hormone in osteoblasts. J Biol Chem. (2007) 282:21237–43. doi: 10.1074/jbc.M701341200
159. Wan M, Yang C, Li J, Wu X, Yuan H, Ma H, et al. Parathyroid hormone signaling through low-density lipoprotein-related protein 6. Genes Dev. (2008) 22:2968–79. doi: 10.1101/gad.1702708
160. Palermo A, D'Onofrio L, Buzzetti R, Manfrini S, Napoli N. Pathophysiology of bone fragility in patients with diabetes. Calcif Tissue Int. (2017) 100:122–32. doi: 10.1007/s00223-016-0226-3
161. Shah VN, Carpenter RD, Ferguson VL, Schwartz AV. Bone health in type 1 diabetes. Curr Opin Endocrinol Diabetes Obes. (2018) 25:231–6. doi: 10.1097/MED.0000000000000421
162. Keenan HA, Maddaloni E. Bone microarchitecture in type 1 diabetes: it is complicated. Curr Osteoporos Rep. (2016) 14:351–8. doi: 10.1007/s11914-016-0338-8
163. Strotmeyer ES, Cauley JA, Schwartz AV, Nevitt MC, Resnick HE, Zmuda JM, et al. Diabetes is associated independently of body composition with BMD and bone volume in older white and black men and women: the health, aging, and body composition study. J Bone Miner Res. (2004) 19:1084–91. doi: 10.1359/JBMR.040311
164. Vestergaard P. Discrepancies in bone mineral density and fracture risk in patients with type 1 and type 2 diabetes–a meta-analysis. Osteoporos Int. (2007) 18:427–44. doi: 10.1007/s00198-006-0253-4
165. Fulzele K, Riddle RC, DiGirolamo DJ, Cao X, Wan C, Chen D, et al. Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell. (2010) 142:309–19. doi: 10.1016/j.cell.2010.06.002
166. Ferron M, Wei J, Yoshizawa T, Del Fattore A, DePinho RA, Teti A, et al. Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell. (2010) 142:296–308. doi: 10.1016/j.cell.2010.06.003
167. Thrailkill K, Bunn RC, Lumpkin C, Wahl E, Cockrell G, Morris L, et al. Loss of insulin receptor in osteoprogenitor cells impairs structural strength of bone. J Diabetes Res. (2014) 2014:703589. doi: 10.1155/2014/703589
168. Wei J, Ferron M, Clarke CJ, Hannun YA, Jiang H, Blaner WS, et al. Bone-specific insulin resistance disrupts whole-body glucose homeostasis via decreased osteocalcin activation. J Clin Invest. (2014) 124:1–13. doi: 10.1172/JCI72323
169. Akune T, Ohba S, Kamekura S, Yamaguchi M, Chung U-I, Kubota N, et al. PPARgamma insufficiency enhances osteogenesis through osteoblast formation from bone marrow progenitors. J Clin Invest. (2004) 113:846–55. doi: 10.1172/JCI200419900
170. Sun H, Kim JK, Mortensen R, Mutyaba LP, Hankenson KD, Krebsbach PH. Osteoblast-targeted suppression of PPARγ increases osteogenesis through activation of mTOR signaling. Stem Cells. (2013) 31:2183–92. doi: 10.1002/stem.1455
171. Muruganandan S, Ionescu AM, Sinal CJ. At the crossroads of the adipocyte and osteoclast differentiation programs: future therapeutic perspectives. Int J Mol Sci. (2020) 21:2277. doi: 10.3390/ijms21072277
172. Brown ML, Yukata K, Farnsworth CW, Chen D-G, Awad H, Hilton MJ, et al. Delayed fracture healing and increased callus adiposity in a C57BL/6J murine model of obesity-associated type 2 diabetes mellitus. PLoS ONE. (2014) 9:e99656. doi: 10.1371/journal.pone.0099656
173. Rahman S, Czernik PJ, Lu Y, Lecka-Czernik B. β-catenin directly sequesters adipocytic and insulin sensitizing activities but not osteoblastic activity of PPARγ2 in marrow mesenchymal stem cells. PLoS ONE. (2012) 7:e51746. doi: 10.1371/journal.pone.0051746
174. Almeida M, Ambrogini E, Han L, Manolagas SC, Jilka RL. Increased lipid oxidation causes oxidative stress, increased peroxisome proliferator-activated receptor-gamma expression, and diminished pro-osteogenic Wnt signaling in the skeleton. J Biol Chem. (2009) 284:27438–48. doi: 10.1074/jbc.M109.023572
175. Shockley KR, Lazarenko OP, Czernik PJ, Rosen CJ, Churchill GA, Lecka-Czernik B. PPARgamma2 nuclear receptor controls multiple regulatory pathways of osteoblast differentiation from marrow mesenchymal stem cells. J Cell Biochem. (2009) 106:232–46. doi: 10.1002/jcb.21994
176. Nolan JJ, Ludvik B, Beerdsen P, Joyce M, Olefsky J. Improvement in glucose tolerance and insulin resistance in obese subjects treated with troglitazone. N Engl J Med. (1994) 331:1188–93. doi: 10.1056/NEJM199411033311803
177. Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma. (PPAR gamma). J Biol Chem. (1995) 270:12953–6. doi: 10.1074/jbc.270.22.12953
178. Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. (2008) 77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829
179. Kahn SE, Haffner SM, Heise MA, Herman WH, Holman RR, Jones NP, et al. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med. (2006) 355:2427–43. doi: 10.1056/NEJMoa066224
180. Home PD, Pocock SJ, Beck-Nielsen H, Curtis PS, Gomis R, Hanefeld M, et al. RECORD Study Team. Rosiglitazone evaluated for cardiovascular outcomes in oral agent combination therapy for type 2 diabetes. (RECORD): a multicentre, randomised, open-label trial. Lancet. (2009) 373:2125–35. doi: 10.1016/S0140-6736(09)60953-3
181. Zinman B, Haffner SM, Herman WH, Holman RR, Lachin JM, Kravitz BG, et al. Effect of rosiglitazone, metformin, and glyburide on bone biomarkers in patients with type 2 diabetes. J Clin Endocrinol Metab. (2010) 95:134–42. doi: 10.1210/jc.2009-0572
182. Grey A, Bolland M, Gamble G, Wattie D, Horne A, Davidson J, et al. The peroxisome proliferator-activated receptor-gamma agonist rosiglitazone decreases bone formation and bone mineral density in healthy postmenopausal women: a randomized, controlled trial. J Clin Endocrinol Metab. (2007) 92:1305–10. doi: 10.1210/jc.2006-2646
Keywords: osteoblast, fatty acid metabolism, dyslipidemia, bone mass, lipoproteins
Citation: Alekos NS, Moorer MC and Riddle RC (2020) Dual Effects of Lipid Metabolism on Osteoblast Function. Front. Endocrinol. 11:578194. doi: 10.3389/fendo.2020.578194
Received: 30 June 2020; Accepted: 25 August 2020;
Published: 23 September 2020.
Edited by:
Lilian Irene Plotkin, Indiana University Bloomington, United StatesReviewed by:
Ahmed Al Saedi, The University of Melbourne, AustraliaCopyright © 2020 Alekos, Moorer and Riddle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ryan C. Riddle, cnJpZGRsZTFAamhtaS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.