
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Endocrinol. , 25 September 2020
Sec. Pediatric Endocrinology
Volume 11 - 2020 | https://doi.org/10.3389/fendo.2020.540683
 Alessandro Cattoni1*
Alessandro Cattoni1* Alice Spano1
Alice Spano1 Anna Tulone1Annalisa Boneschi2Nicoletta Masera1
Anna Tulone1Annalisa Boneschi2Nicoletta Masera1 Silvia Maitz1Anna Maria Di Blasio3
Silvia Maitz1Anna Maria Di Blasio3 Luca Persani4,5Fabiana Guizzardi3,4
Luca Persani4,5Fabiana Guizzardi3,4 Raffaella Rossetti4
Raffaella Rossetti4Non-syndromic primary ovarian insufficiency due to ovarian dysgenesis in 46,XX patients is an uncommon finding in the general population, even though several monogenic variants have been reported as causative factors. Here, we describe a 15-year-old patient diagnosed with gonadal dysgenesis possibly due to the interaction of three potentially pathogenic variants of genes involved in ovarian maturation, namely factor in the germline alpha (FIGLA), newborn ovary homeobox-encoding (NOBOX) and nuclear receptor subfamily 5 group A member 1 (NR5A1). We also describe a different degree of residual ovarian function within the proband's family, whose female members carry one to three demonstrated variations in the aforementioned genes in a clinical spectrum potentially dependent on the number of alleles involved. Our results support the hypothesis that the severity of the clinical picture of the proband, resulting in complete ovarian dysgenesis, may be due to a synergic detrimental effect of inherited genetic variants.
Primary ovarian insufficiency (POI) is defined as the combination of amenorrhea reported for at least 4 months and enhanced levels of follicle-stimulating hormone (FSH) within the post-menopausal range at two or more subsequent assessments in women younger than 40 years (1). Amenorrhea can be either primary—about 10% of non-iatrogenic cases—or secondary (2). POI can present as an isolated condition, or it can fall within the clinical setting of a defined or undefined syndrome (3). Overall, it affects 1:10,000 women before age of 20, 1:1,000 women before the age of 30, and 1:100 women before the age of 40 years (4).
Several risk factors may contribute to the pathogenesis of POI, with both genetic and non-genetic causes being potentially involved. However, in 50–80% of cases, the underlying mechanism is unknown, and POI is classified as idiopathic. Iatrogenic interventions (i.e., pelvic surgery and anti-cancer treatment), autoimmune conditions, infectious agents (e.g., viral oophoritis) and environmental disruptors (e.g., exposure to 1-bromopropane) can all be listed among the non-genetic factors harmful to ovarian function (5).
Either chromosomal aberrations or monogenic disorders are identified in about 10% of patients diagnosed with POI (6). In addition, it is highly likely that a large percentage of idiopathic POIs are caused by yet unidentified genetic mutations.
Genetic disorders, such as chromosomal abnormalities and monogenic or polygenic mutations involving genes located either on autosomal chromosomes or on the X chromosome, have been described both in syndromic and non-syndromic cases of POI (5). In particular, chromosome aberrations alone (i.e., Turner's syndrome, triple X syndrome, fragile X syndrome and Robertsonian translocations) cause ~9 % of all POI (1, 3, 4). In addition, as the human reproductive phenotype is the result of the structured interaction of the products of several genetic sequences, different combinations of genetic variations may lead to POI.
Although there has been a significant effort in sequencing several POI candidate genes, only few coding mutations have been described so far, which is consistent with the low incidence of this condition. These genes mostly include transcription factors involved in female gonadal embryonal maturation and development. Currently, mutations in FSHR, LHCGR, NR5A1, NOBOX, FOXL2, FIGLA, BMP15, NANOS3, and STAG3 genes have all been formally validated as being causative of non-syndromic POI (5).
Ovarian dysgenesis can be regarded as the most severe clinical picture among those related to POI. In patients affected by this condition, the ovaries are degenerated and completely depleted of follicles before puberty, and only streak gonads are present (3). Ovarian dysgenesis is the most common finding in girls with Turner's syndrome. However, when it occurs in patients with normal sex chromosomes, it gives rise to the “46, XX pure gonadal dysgenesis.” The clinical phenotype of this condition includes primary amenorrhea, lack of breast development, hypoestrogenism, and raised gonadotropins (3, 7).
Clinical suspicion of POI should be raised whenever a girl presents with primary amenorrhea—defined as the absence of menses at the age of 15 years in the presence of normal secondary sexual development—or fails to initiate breast development by the age of 13 years, all conditions where biochemical and radiological investigations are highly recommended (7).
In the present study, we describe the case of a 15-year-old girl diagnosed with POI possibly due to a complex pattern of compound monogenic alterations.
The proband is a Caucasian female patient referred to our endocrine outpatient clinic at the age of 15.1 years due to lack of secondary sexual characteristics. The female members of her family had achieved menarche at a suitable age, and no family history consistent with pubertal delay was reported. As the patient's father had prematurely committed suicide, few data about the timing of his pubertal development were available at the time of consultation. The patient was the second child of non-consanguineous parents, and she was born at term following an uneventful pregnancy. She had achieved all the developmental motor and intellectual milestones at an appropriate age.
On physical examination, the proband was classified as Tanner stage B1PH3AH2 and did not present with any signs of estrogenization of genitalia or areolas.
From an auxological perspective, the patient had had a normal growth along the 25th centile (WHO growth charts) until the age of 10 years, but she had subsequently experienced a progressive decrease in height SDS, with a growth velocity of 3.2 cm/years. This clinical picture was deemed consistent with constitutional delay of growth and pubertal development. On first examination, her stature was 152.0 cm (8th centile, −1.41 SDS), while her BMI was 15.37 Kg/m2 (−2.31 SDS), with no weight gain or loss recently reported.
Baseline biochemical and radiological assessment showed normal thyroid function and prolactin levels and negative anti-transglutaminase antibodies, ruling out potential causative factors of pubertal delay. Insulin-like growth factor 1 (IGF-1) levels were within the reference range for chronological and bone age. Cortisol (8.2 μg/dL) and ACTH (42 pg/mL) were normal as well. Nonetheless, the patient presented with high levels of serum gonadotropins (LH 34 U/L, FSH 159.9 U/L) with undetectable estradiol (<5 pg/mL).
Hand and wrist X-ray revealed a remarkably delayed bone age (12.0 vs. a chronological age of 15.1 years), while pelvic ultrasound (US), performed to explore internal genitalia, showed a prepubertal uterus (body 1.1 cm, cervix 1.3 cm) with undetectable ovaries. Pelvic MRI confirmed the finding of infantile internal genitalia, with the typical occurrence of small, streak gonads. Dual X-ray absorptiometry (DXA) scan showed severely reduced bone mineral density (lumbar Z score: −3.9; total body Z score:−3.3). Negative anti-ovary antibodies and autoimmune screening tests (i.e., ANA, ENA, anti-thyroid autoantibodies), previous medical history and normal female karyotype (46,XX) ruled out both non-genetic causes of POI and chromosomal abnormalities.
As all the aforementioned findings, together with the psychological impact of severe pubertal delay, made it necessary to perform immediate pubertal induction, the patient was started on progressively increasing doses of transdermal 17-β-estradiol, which led to subsequent development of secondary sexual characteristics. After 24 months of treatment, she started to experience vaginal spotting. At that point, a new pelvic US showed a post-pubertal appearance of her uterus with thickened endometrial line. Thus, micronized progesterone was administered for 14 every 28 days in order to achieve regular menses.
Dual-energy X-ray absorptiometry (DXA scan), repeated 2 years after the start of hormone therapy, showed a remarkable improvement in both total body and lumbar densitometry (Z scores: −2.4 and −3.1, respectively).
The patient has now achieved a complete pubertal development and has recently been switched to a combined therapy with oral 17ß-estradiol and dydrogesterone in order to improve the compliance to treatment. Her final height of 161.3 cm (39th centile, −0.28 SDS) is entirely within the mid-parental target height. Figure 1 represents the timeline of all diagnostic and therapeutic interventions performed for the proband. Figure 2 represents the pictures captured from the most recently performed pelvic US.
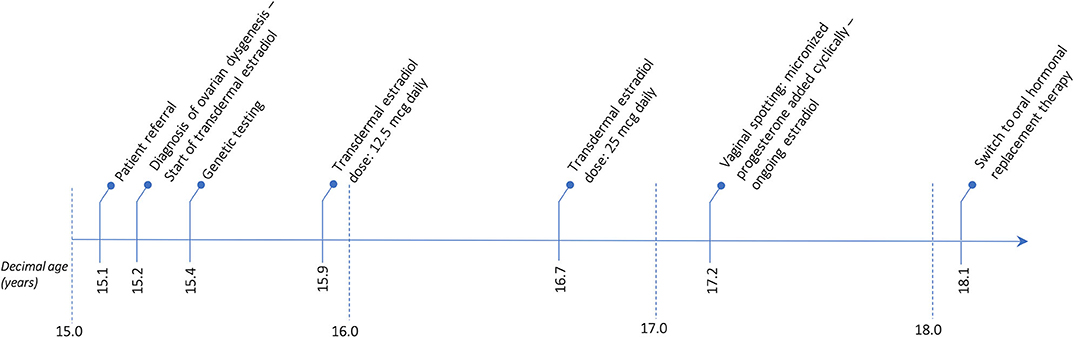
Figure 1. Timetable describing the sequence of the diagnostic and therapeutic interventions undertaken for the proband.
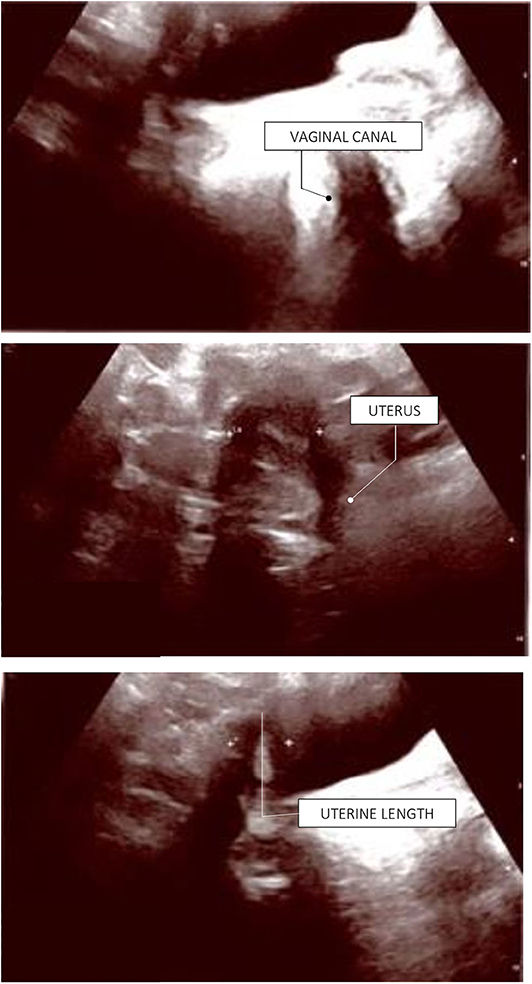
Figure 2. Proband's last pelvic ultrasound, performed at the end of the pubertal induction. Ovaries were not detected due to ovarian dysgenesis. The uterus has reached a mature post-pubertal shape and volume (9.5 mL). Endometrial thickness: 4 mm (secretive phase).
Due to complaints of impaired self-esteem and perceived low levels of social interaction with peers, the patient has also been under the care of a psychologist since her initial diagnosis of POI. Hormonal treatment and subsequent development of secondary sexual features have helped the patient overcome her psychological issues. Currently, her self-esteem and own perception of physical attributes have remarkably improved.
Blood samples from both the proband and her relatives were collected after informed consent and analyzed by the IRCCS Istituto Auxologico Italiano, Laboratories of Endocrine and Metabolic Research and Molecular Biology. All coding and flanking intronic regions of a wide panel of genes potentially involved in POI were analyzed via Next Generation Sequencing. These included the BMP15, FIGLA, FOXL2, FSHR, GDF9, NOBOX, NR5A1, and GALT genes. The library was generated through Nextera Rapid Capture Custom Enrichment (Illumina). As a result, a complex pattern of genetic variations in the proband was detected. Firstly, we identified a rare frameshift homozygous variant within exon 2 of the FIGLA (factor in the germline alpha) gene [NM_001004311.3(FIGLA_v001):c.364del,NM_001004311.3 (FIGLA_i001):p.(Glu122Lysfs*45)], generating a truncated protein. Secondly, we found a novel heterozygous frameshift variant within exon 9 of the NOBOX (newborn ovary homeobox-encoding) gene [NM_001080413.3(NOBOX_v001):c.1626del, NM_001080413.3 (NOBOX_i001):p.(Phe543Serfs*7)], encoding for a truncated protein as well. Both deletions involved a well-known functional domain and were predicted to likely be disease-causing by a bioinformatic pathogenicity prediction tool (Table 1). Lastly, we detected a rare missense variant of the NR5A1 (nuclear receptor subfamily 5 group A member 1) gene NM_004959.5(NR5A1_v001):c.1063G>A, NM_004959.5(NR5A1_i001):p.(Val355Met), which was predicted to be pathogenic by 5 out of 7 prediction tools available online (Table 1).
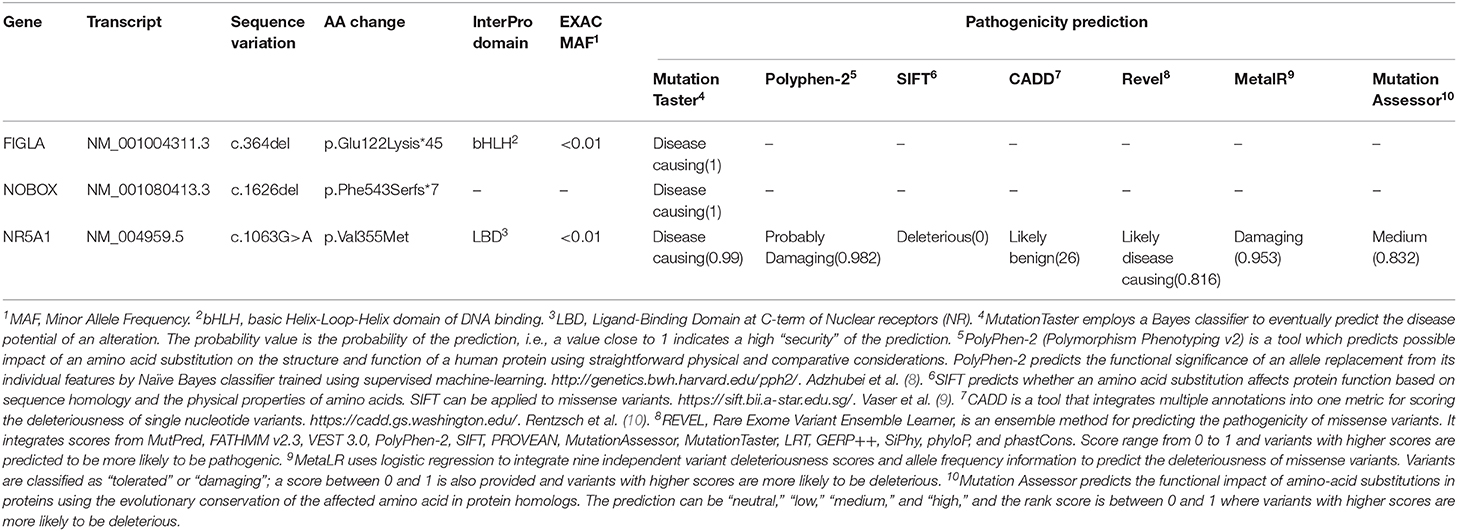
Table 1. Pathogenicity prediction for the three genic variants detected in the family described.
In order to further assess the pathogenic role of these variants, the analysis was extended to the patient's first-degree relatives, namely her mother and sister. While the proband's mother was a heterozygous carrier of the FIGLA nucleotide deletion, her sister—aged 19 years at that time—harbored heterozygous variants in both FIGLA and NR5A1 genes. Thus, it is likely that both sisters inherited the NR5A1 missense variant from their father. All the identified variants of the FIGLA, NOBOX, and NR5A1 genes were confirmed in the proband and assessed in her mother and sister by Sanger sequencing.
Despite having non-consanguineous parents, the patient was homozygous for an extremely rare FIGLA variant. In order to explain this peculiarity, additional analyses were conducted (see Supplementary files for an in-depth explanation). Real-time PCR ruled out a deletion involving the long arm of chromosome 2. Furthermore, a microsatellite analysis was performed, and it excluded uniparental isodisomy (UPD) for chromosome 2. Nevertheless, we were not able to exclude segmental UPD, albeit rare. Figure 3 represents the results relative to the NGS profile detected.
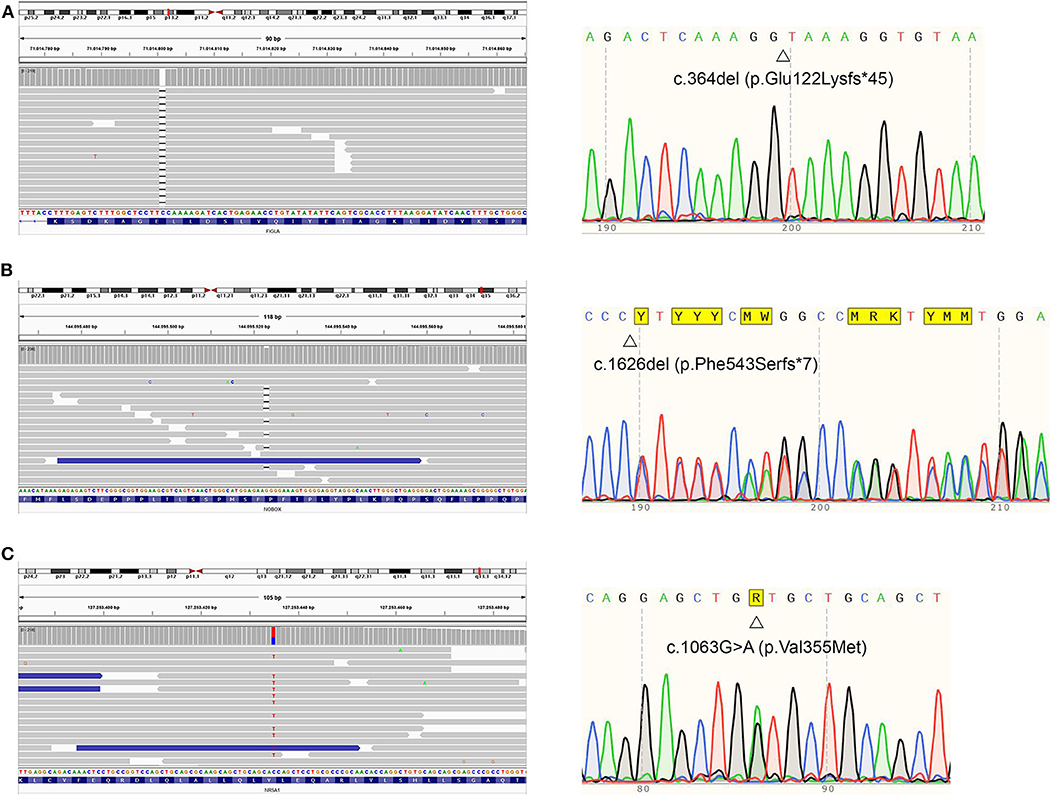
Figure 3. Proband's NGS profiles (right) and sequence (left) of FIGLA, NOBOX, and NR5A1 genes. The identified variants c.364del (A), c.1626del (B), and c.1063G>A (C) are indicated.
While in the proband the described complex interaction of multiple genetic alterations was associated with overt ovarian dysgenesis, infertility and ensuing need for hormonal replacement therapy, the patient's sister presented with normal pubertal development, achieving menarche at the age of 13 years, never reporting any clinical signs consistent with menstrual irregularity. As she requested safe contraception at the age of 19, she was started on estroprogestin pill, a therapy currently ongoing. However, when we assessed her residual ovarian function before prescribing hormonal therapy, we found decreased levels of anti-Müllerian hormone (AMH) (0.9 μg/L; reference value 1.5–13.3 μg/L), despite adequate gonadotropin (LH 4.5 U/L, FSH 7.0 U/L) and estradiol (42 pg/mL) levels and normal appearance of internal genitalia on pelvic US. This clinical picture was consistent with a diagnosis of diminished ovarian reserve (11), where a reduction in AMH concomitant with normal estradiol and gonadotropins levels may be predictive of progressive depletion of ovarian follicles, leading to a menopausal status in young adulthood. Conversely, the heterozygous variation of FIGLA found in the mother did not appear to have affected ovarian function, as she presented regular menses, with no clinical or biochemical signs of menopause at the age of 43 years—gonadotropins and AMH levels were in fact within the reference ranges for her age.
The present study reports on three family members (i.e., the proband and her sister and mother), carriers of one to three genetic variations, experiencing different degrees of residual ovarian function in a clinical spectrum potentially dependent on the number of genes and alleles involved. Specifically, the proband had early-onset ovarian dysgenesis associated with a homozygous variant of FIGLA together with heterozygous variants of NOBOX and NR5A1 genes—the proband underwent hormonal therapy to induce puberty. An asymptomatic phenotype was instead observed for both the proband's mother, who carried a heterozygous variant of the FIGLA gene but retained normal ovarian function, and the proband's sister, who harbored heterozygous variants of both FIGLA and NR5A1 genes but no signs of POI other than biochemical results consistent with diminished ovarian reserve. Importantly, except for the NR5A1 variant, all the aforementioned genetic variants have not previously been described in the literature (ClinVar database).
The FIGLA gene encodes for a germ-cell specific transcription factor playing a major role in the regulation of multiple oocyte-specific genes, including those initiating folliculogenesis and those regulating the expression of various zona pellucida genes (12). Mutations involving this gene are mostly reported as responsible for POI with an autosomal dominant inheritance pattern (OMIM #612310) (13). Thus, it is somewhat surprising that in our family the POI phenotype was associated with a biallelic recessive loss-of-function of the FIGLA gene, an occurrence that, to date, has only been shown in two other families (14, 15).
Another important aspect emerging form this study is that the proband presented with three alterations of genes involved in oocyte differentiation and folliculogenesis (i.e., FIGLA, NOBOX and NR5A1). This, to the best of our knowledge, is the first report of a non-syndromic patient presenting with compound alterations of the aforementioned genes. However, despite the potential functional relevance of our genetic findings, we are well aware that from our data we cannot conclusively establish a causative link between gene expression of the three aforementioned variants and the overall endocrine and reproductive phenotype of the proband. It is in fact possible that the homozygous frameshift variant of the FIGLA gene alone may have been responsible for the early-onset POI, which would then call into question the synergic effect of the other genetic variants. Contrary to this view and in support to our central hypothesis, a previous NGS analysis of a panel of genes potentially involved in POI has shown that 42% of patients with POI may actually present with pathogenic variants of two or more genes involved in ovarian function or maturation (16). It is therefore possible that POI in patients with heterozygous variants in genes reported as causative factors for sporadic POI—identified through a traditional gene approach—may actually be the result of an interaction with additional variants involving different genes. Similar considerations have also been proposed by two other groups (14, 15), independently describing two couples of sisters with primary amenorrhea bearing a biologically inactive full-length FIGLA protein due homozygous missense mutations, but with unaffected heterozygous mothers. Moreover, the patients presented by Yuan et al. had short stature, while our proband's final height fell within normal Italian centiles (15). According to these authors, the normal ovarian function observed in these two mothers, as in our case, supported the hypothesis that FIGLA haploinsufficiency alone is not sufficient to cause POI. From this standpoint, overall in disagreement with other studies describing POI due to heterozygous mutations involving FIGLA (13), the different biochemical phenotypes that we have described in proband's mother and sister, harboring the same heterozygous FIGLA variant, could be regarded as the result of concomitant genetic variants. Even though from our data we cannot rule out that the phenotypic spectrum of the family described may be the result of an incomplete penetrance of the FIGLA variant, it is highly likely that the additional variants described in this study may act as modifier genes and, as such, may play a role in the pathogenesis and clinical severity of POI. However, as no other patients with this specific substitution have been reported so far, it is not quite possible at this stage to generalize our observation suggesting a cause-effect link between the expression of the two NOBOX and NR5A1 heterozygous variants and POI.
The oocyte-specific homeobox gene encoding the newborn ovary homeobox protein (NOBOX) is expressed in germ cells and primordial oocytes. It belongs to a group of tissue-specific homeobox genes contributing to ovarian development. NOBOX is mostly expressed in primordial and growing oocytes and, when mutated, exerts a detrimental effect on ovarian function. Although pathogenic events have been most frequently associated with homozygous mutations in the NOBOX gene, a large study by Bouilly and colleagues has shown heterozygous mutation of the NOBOX gene (OMIM #610934) to be present in 5.6% of 213 unrelated patients with POI (17). In particular, the authors found five missense heterozygous variants with a detrimental effect on NOBOX transcriptional activity, even though functional studies ruled out a dominant-negative effect of those variants. On the other hand, two sisters described by França et al. (18) and a single patient reported by Li et al. (19) presented with POI due to a homozygous deletion involving this gene, with their heterozygous mothers being unaffected. In most patients reported, heterozygous mutations were associated with milder phenotypes, with spontaneous puberty progression, retrieved fertility, and premature menopause, while primary amenorrhea was mainly found in girls with homozygous variants (20). The presence of heterogeneous phenotypes associated with the same mutation in NOBOX as described in several cohorts suggests that also in this case no clear genotype-phenotype correlation can be defined for NOBOX variants. Although loss-of-function variants have been more frequently found in recessive forms and missense variants in dominant cases (18), more studies are warranted to confirm this association. It is possible in fact that additional detrimental variants involving different genes may result in POI in patients with NOBOX haploinsufficiency. In this regard, the reported NOBOX frameshift variant detected in our proband may have played a role in the precocious depletion of primordial oocytes, leading to ovarian dysgenesis in combination with the other reported variations.
Finally, although NR5A1 was initially considered as non-pathogenic in women given the maternal transmission of its mutated form in males with sex differentiation disorders (21), a growing body of literature has more recently demonstrated a potential role of this gene in ovarian insufficiency (22). NR5A1 is a nuclear receptor pivotal in the transcription of multiple target genes involved in adrenal and gonadal development, steroidogenesis, and reproduction. Homozygous mutations of this gene may alter both gonadal and adrenal steroidogenesis, leading to primary adrenal failure, while heterozygous mutations have been shown to cause POI in women (OMIM #612964) (21). The specific mutation found in our patient (p.Val355Met) had already been described by Philibert and colleagues in one boy with micropenis and testicular vanishing syndrome (21). As his mother, heterozygous for the same variant, had developed ovarian cysts and multiple miscarriages—potentially regarded as indirect signs of ovarian insufficiency—, it may be inferred that also heterozygous mutations in NR5A1 can mildly impair ovarian function. Although this conclusion cannot be systematically drawn from available data, it is tempting to speculate that heterozygous NR5A1 variant may play a detrimental role on ovarian function in both our proband and sister. Nevertheless, as in the family described this variant was inherited by both the daughters but absent in the mother, we hypothesize paternal inheritance. Intriguingly, the father had not been diagnosed with any specific gonadal issues. Probably, the ovarian impact of the reported genetic variation in NR5A1 will become clearer in the future, by comparing the reproductive and endocrine outcomes (i.e., age at onset of menopause, pregnancies) of the proband's sister to those of the mother. Indeed, as they share the same heterozygous FIGLA variant, an eventual deleterious effect of aberrant NR5A1 would be proven in case of ovarian insufficiency and/or reproductive disorders in the sister, as the mother had achieved two pregnancies without any medically-assisted procreation techniques and had not presented any sign of menopause at the age of 43 years. The biochemical finding of suppressed AMH levels in the proband's sister may suggest a more compromised reproductive function. However, as NR5A1 transcriptionally regulates AMH gene expression along with that of other transcription factors, the decreased AMH expression levels detected in the proband may not reflect the actual residual ovarian reserve.
To the best of our knowledge, this is the first evidence in the literature of a family where a variable combination of three variants of genes involved in oocyte maturation may have played a synergic effect on clinically different phenotypes in relation to ovarian reserve and endocrine function.
Overall, this case outlines that the clinical phenotypic variability experienced by different patients carrying the same mutation may be the result of the detrimental effect of concurrent genetic factors.
Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
AC conceived the idea of describing the present case report, drafted the paper, critically revised its contents and provided the final revision: in addition, AC produced the pictures of the paper. AT and AB drafted the first version of the paper. SM and AS performed the genetic counseling for the family and critically revised the final version of the paper. NM critically revised the contents of the paper and approved the final outcome. RR, LP, AD, and FG performed the genetic testing, analyzed the potential pathogenicity of the variants found, and actively contributed to the drafting of the paper. RR produced Table 1, for pathogenicity assessment of the genetic variants described, and Figure 3. All authors contributed to the article and approved the submitted version.
This work has been partially supported by Italian Ministry of Health ‘Ricerca Finalizzata' grant (GR-2011-02351636, BIOEFFECT) and by IRCCS Istituto Auxologico Italiano (RICCOR).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Patiño LC, Beau I, Carlosama C, Buitrago JC, González R, Suárez CF, et al. New mutations in non-syndromic primary ovarian insufficiency patients identified via whole-exome sequencing. Hum Reprod. (2017) 32:1512–20. doi: 10.1093/humrep/dex089
2. Nelson LM. Primary ovarian insufficiency. N Engl J Med. (2009) 360:606–14. doi: 10.1056/NEJMcp0808697
3. Cordts EB, Christofolini DM, Dos Santos AA, Bianco B, Barbosa CP. Genetic aspects of premature ovarian failure: a literature review. Arch Gynecol Obstet. (2011) 283:635–43. doi: 10.1007/s00404-010-1815-4
4. Fortuño C, Labarta E. Genetics of primary ovarian insufficiency: a review. J Assist Reprod Genet. (2014) 31:1573–85. doi: 10.1007/s10815-014-0342-9
5. Laissue P. Aetiological coding sequence variants in non-syndromic premature ovarian failure: from genetic linkage analysis to next generation sequencing. Mol Cell Endocrinol. (2015) 411:243–57. doi: 10.1016/j.mce.2015.05.005
6. Lakhal B, Braham R, Berguigua R, Bouali N, Zaouali M, Chaieb M, et al. Cytogenetic analyses of premature ovarian failure using karyotyping and interphase fluorescence in situ hybridization (FISH) in a group of 1000 patients. Clin Genet. (2010) 78:181–5. doi: 10.1111/j.1399-0004.2009.01359.x
8. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. (2010) 7:248–9. doi: 10.1038/nmeth0410-248
9. Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protoc. (2016) 11:1–9. doi: 10.1038/nprot.2015.123
10. Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47:D886–94. doi: 10.1093/nar/gky1016
11. Broekmans FJ, Visser JA, Laven JSE, Broer SL, Themmen APN, Fauser BC. AntiMüllerian hormone and ovarian dysfunction. Trends Endocrinol Metab. (2008) 19:340–7. doi: 10.1016/j.tem.2008.08.002
12. Bayne RA, Martins da Silva SJ, Anderson RA. Increased expression of the FIGLA transcription factor is associated with primordial follicle formation in the human fetal ovary. Mol Hum Reprod. (2004) 10:373–81. doi: 10.1093/molehr/gah056
13. Tosh D, Rani HS, Murty US, Deenadayal A, Grover P. Mutational analysis of the FIGLA gene in women with idiopathic premature ovarian failure. Menopause. (2015) 22:520–6. doi: 10.1097/GME.0000000000000340
14. Chen B, Li L, Wang J, Li T, Pan H, Liu B, et al. Consanguineous familial study revealed biallelic FIGLA mutation associated with premature ovarian insufficiency. J Ovarian Res. (2018) 11:48. doi: 10.1186/s13048-018-0413-0
15. Yuan P, He Z, Sun S, Li Y, Wang W, Liang X, et al. Bi-allelic recessive loss-of-function mutations in FIGLA cause premature ovarian insufficiency with short stature. Clin Genet. (2019) 95:409–14. doi: 10.1111/cge.13486
16. Jiao X, Ke H, Qin Y, Chen ZJ. Molecular genetics of premature ovarian insufficiency. Trends Endocrinol Metab. (2018) 29:795–807. doi: 10.1016/j.tem.2018.07.002
17. Bouilly J, Roucher-Boulez F, Gompel A, Bry-Gauillard H, Azibi K, Beldjord C, et al. New NOBOX mutations identified in a large cohort of women with primary ovarian insufficiency decrease KIT-L expression. J Clin Endocrinol Metab. (2015) 100:994–1001. doi: 10.1210/jc.2014-2761
18. França M, Funari MFA, Lerario AL, Nishi MY, Pita CC, Fontenele EGP, et al. A novel homozygous 1-bp deletion in the NOBOX gene in two Brazilian sisters with primary ovarian failure. Endocrine. (2017) 58:442–47. doi: 10.1007/s12020-017-1459-2
19. Li L, Wang B, Zhang W, Chen B, Luo M, Wang J, et al. A homozygous NOBOX truncating variantcauses defective reanscriptional activation and leads to primary ovarian insufficiency. Hum Reprod. (2017) 32:248–55. doi: 10.1093/humrep/dew271
20. Qin Y, Choi Y, Zhao H, Simpson JL, Chen ZI, Rajkovic A. NOBOX homeobox mutation causes premature ovarian failure. Am J Hum Genet. (2007) 81:576–81. doi: 10.1086/519496
21. Philibert P, Zenaty D, Lin L, Soskin S, Audran F, Léger J, et al. Mutational analysis of steroidogenic factor 1 (NR5a1) in 24 boys with bilateral anorchia: a French collaborative study. Hum Reprod. (2007) 22:3255–61. doi: 10.1093/humrep/dem278
Keywords: primary ovarian insufficiency (POI), gonadal dysgenesis (GD), antimullerian hormone (AMH), FIGLA gene, NOBOX gene, NR5A1 gene, fertility, gene variants
Citation: Cattoni A, Spano A, Tulone A, Boneschi A, Masera N, Maitz S, Di Blasio AM, Persani L, Guizzardi F and Rossetti R (2020) The Potential Synergic Effect of a Complex Pattern of Multiple Inherited Genetic Variants as a Pathogenic Factor for Ovarian Dysgenesis: A Case Report. Front. Endocrinol. 11:540683. doi: 10.3389/fendo.2020.540683
Received: 05 March 2020; Accepted: 25 August 2020;
Published: 25 September 2020.
Edited by:
Amit V. Pandey, University of Bern, SwitzerlandReviewed by:
Maki Fukami, National Center for Child Health and Development (NCCHD), JapanCopyright © 2020 Cattoni, Spano, Tulone, Boneschi, Masera, Maitz, Di Blasio, Persani, Guizzardi and Rossetti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessandro Cattoni, YWxlc3NhbmRyby5jYXR0b25pQHVuaW1pYi5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.