 Zhenhong Ye1,2,3,4
Zhenhong Ye1,2,3,4 Yue Zhao
Yue Zhao
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 08 September 2020
Sec. Diabetes: Molecular Mechanisms
Volume 11 - 2020 | https://doi.org/10.3389/fendo.2020.00617
This article is part of the Research Topic Mitochondrial Dynamics in Endocrine Physiology and Disease View all 14 articles
As important metabolic substrates, branched-chain amino acids (BCAAs) and fatty acids (FAs) participate in many significant physiological processes, such as mitochondrial biogenesis, energy metabolism, and inflammation, along with intermediate metabolites generated in their catabolism. The increased levels of BCAAs and fatty acids can lead to mitochondrial dysfunction by altering mitochondrial biogenesis and adenosine triphosphate (ATP) production and interfering with glycolysis, fatty acid oxidation, the tricarboxylic acid cycle (TCA) cycle, and oxidative phosphorylation. BCAAs can directly activate the mammalian target of rapamycin (mTOR) signaling pathway to induce insulin resistance, or function together with fatty acids. In addition, elevated levels of BCAAs and fatty acids can activate the canonical nuclear factor-κB (NF-κB) signaling pathway and inflammasome and regulate mitochondrial dysfunction and metabolic disorders through upregulated inflammatory signals. This review provides a comprehensive summary of the mechanisms through which BCAAs and fatty acids modulate energy metabolism, insulin sensitivity, and inflammation synergistically.
Carbohydrates, lipids, and amino acids are the three major nutrients for humans. They are oxidized, and they supply energy in various ways to maintain activities of the body. When the internal and external environments change, they can interact and transform to each other to adjust various metabolic activities to maintain human health. Once metabolic homeostasis is perturbed, it will cause many endocrine disorders and metabolic diseases, which threaten public health seriously with the increasing morbidity rate and younger trend (1). The pathogeneses of endocrine and metabolic diseases are complex, and the development of multiple disorders are associated with energy metabolism imbalance, chronic low-grade inflammation, and genetic influences (1–4). Recently, alterations in several metabolic pathways have been implicated in the development of metabolic diseases by metabolomic studies (1, 5–9).
Notably, the abnormal levels of branched-chain amino acids (BCAAs) and fatty acids in the circulation are closely related to the progression of metabolic diseases and prognosis, such as obesity, metabolic syndrome, type 2 diabetes mellitus (T2DM), cardiovascular disease, non-alcoholic fatty liver disease, and reproductive endocrine disease (10–19). Insulin resistance (IR) is a common pathophysiological basis of these metabolic diseases (20–24). Mitochondrial dysfunction and inflammation have been considered as key factors of IR (25–27). As important metabolic substrates, BCAAs could induce IR and T2DM by reducing insulin secretion and glucose utilization (28, 29). Moreover, elevated levels of fatty acids could interfere with the insulin signal transduction through regulating inflammation and oxidative stress, leading to the occurrence of IR (30, 31). Thus, both BCAAs and fatty acids play a crucial role in regulating the metabolic homeostasis.
BCAAs include leucine, isoleucine, and valine. In the mitochondrial matrix, BCAAs first undergo reversible transamination to form the corresponding branched-chain α-keto acids (BCKAs) by branched-chain aminotransferase 2 (BCAT2). The branched-chain α-keto acid dehydrogenase complex (BCKDC) then catalyzes the oxidative decarboxylation of BCKAs to form the corresponding acetyl-CoA derivatives (32, 33), which is the first rate-limiting step in BCAA catabolism. BCKDC is a multienzyme complex, consisting of branched-chain α-keto acid dehydrogenase E1α (BCKDHA), branched-chain α-keto acid dehydrogenase E1β (BCKDHB), branched-chain α-keto acid dehydrogenase E2, and branched-chain α-keto acid dehydrogenase E3 (34). Thereafter, the acetyl-CoA derivatives go through several steps to generate the corresponding metabolites and participate in different metabolic processes (Figure 1). BCAAs and their intermediate metabolites such as C3 and C5 acylcarnitine, acetyl-CoA, succinyl-CoA, and 3-hydroxyisobutyrate (3-HIB), for example, are involved in insulin signal transduction, fatty acid oxidation, tricarboxylic acid (TCA) cycle, glycolysis, mitochondrial biogenesis, inflammation, and other physiological processes (35–37).
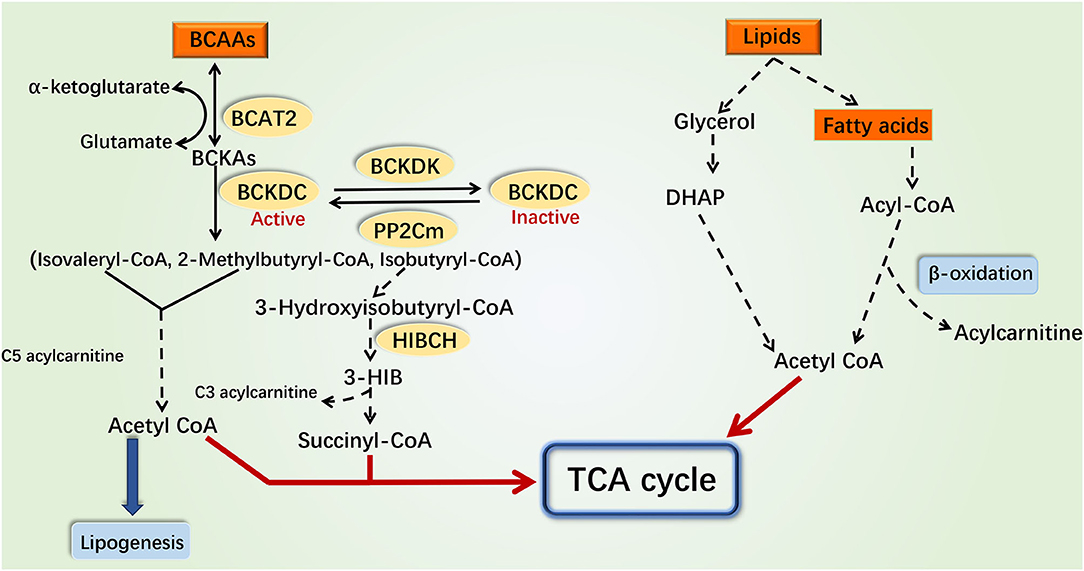
Figure 1. Catabolism pathways of branched-chain amino acids and fatty acids. Various intermediate metabolites produced by the catabolism of BCAAs and fatty acids can participate in the TCA cycle and glycolysis. The catabolism of BCAAs and fatty acids can interplay through these metabolites and ultimately impact the production of mitochondrial ATP through the electronic transport chain. The catabolism of BCAAs in the mitochondria is shown on the left, while the catabolism of fatty acids in cytoplasm and mitochondria is shown on the right. The dotted line indicates a multistep reaction, and the solid line indicates a one-step reaction. DHAP, dihydroxyacetone phosphate; 3-HIB, 3-hydroxyisobuterate; HIBCH, 3-hydroxyisobutyryl-coenzyme A hydrolase.
Lipids is the general term for fats and lipoids. Neutral fats, also called triglycerides, can be broken down into glycerol and fatty acids in the cytoplasm. Glycerol is converted into dihydroxyacetone phosphate that participates in gluconeogenesis or TCA cycle (38). According to chain length, fatty acids can be classified into long-chain fatty acids, medium-chain fatty acids, and short-chain fatty acids (39). Mitochondrial fatty acid oxidation is an important pathway for maintaining energy homeostasis. Medium- and short-chain fatty acids enter the mitochondria directly for oxidation, while long-chain fatty acids are first activated to long-chain acyl-CoA and then transported into mitochondria by the carnitine shuttle (40, 41). Following dehydrogenation, hydration, dehydrogenation, and thiolysis, the long-chain acyl-CoA derivatives are finally decomposed into acetyl-CoA which participates in the TCA cycle. The metabolic by-products NADH and FADH2 enter the electronic respiratory chain to produce ATP [Figure 1; (17)].
Currently, researches indicated that BCAA catabolism and fatty acid oxidation could interact with each other. Disorders of fatty acid oxidation increased the plasma levels of BCAAs (42). Besides, the accumulation of BCAA intermediate metabolites such as C3, C5 acylcarnitine, and acetyl-CoA inhibited the complete oxidation of fatty acids (33, 37). These findings indicated that the coordinated roles between BCAAs and fatty acids may be critical for the pathogenesis of several metabolic diseases. Therefore, in this review, we focused on the function and molecular mechanism of coordinated modulation of mitochondrial function, IR, and inflammation by BCAAs and fatty acids, in order to better understand the pathogenesis of endocrine and metabolic diseases.
The mitochondrial biogenesis is mainly regulated in three aspects (43, 44): first, the changes of mitochondria mass or abundance; second, the expression changes of mitochondrial component genes, including cytochrome C (CytC), adenine nucleotide translocase type 1 (ANT1), and β subunit of the mitochondrial H+-ATP synthase (β-F1-ATPase); and third, the transcriptional modulations of genes related to mitochondrial biogenesis. Peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) has been extensively described as a master regulator of mitochondrial biogenesis in a variety of tissues (43, 45). Previous studies have shown that activated PGC-1α promotes mitochondrial biogenesis in two ways. The elevated PGC-1α protein level induced an increase in transcription activity of estrogen-related receptor α (Errα) and GA repeat-binding protein α (Gabpa) on their own promoters, which facilitated the gene expression of downstream nuclear respiratory factor 1 (NRF-1) and upregulated nuclear genes required for mitochondrial biogenesis (46), such as mitochondria transcription factor B1 (TFB1M) and TFB2M. Besides, it promoted the expression of mitochondrial transcription factor A (TFAM) mRNA to increase mtDNA copy number (43, 44).
BCAAs can induce mitochondrial biogenesis in different mechanisms according to different treatment conditions (Supplementary Table 1). In human HepG2 cells, BCAAs can affect mitochondrial biogenesis through protein acetylation and phosphorylation modification and transcriptional regulation. A mixture of BCAAs could enhance the phosphorylation of Ser1177 on endothelial nitric oxide synthase (eNOS), resulting in the activation of eNOS, which led to an increase in nitric oxide (NO) production (47). NO could induce nicotinamide adenosine dinucleotide (NAD)-dependent deacetylase sirtuin1 (SIRT1) expression, which could deacetylate eNOS and PGC-1α, thereby promoting mitochondrial biogenesis (47). In addition, NO activated by BCAAs promoted the PGC-1α expression at the transcription level through cGMP (43, 48). Similarly, leucine can also promote mitochondrial biogenesis through protein post-translational modification and transcriptional regulation. First, in skeletal muscle cells, leucine could enhance the gene expression level of SIRT1, then SIRT1 deacetylated and enhanced liver kinase B1 (LKB1) activity, LKB1 phosphorylated AMP-activated protein kinase (AMPK), and activated AMPK increased the level of NAD+ in cells by upregulating nicotinamide phosphoribosyltransferase (Nampt), thereby activating SIRT1. AMPK and SIRT1 further activated PGC-1α through phosphorylation and deacetylation, respectively (49). Secondly, the regulatory-associated protein of mTOR (raptor) is a defining component of the mTOR complex, and leucine could promote the activation of the mTOR–raptor complex (50). mTOR and raptor interacted with the transcription factor Ying-Yang 1 (YY1), and YY1 was recruited to the promoter region of the gene encoding PGC-1α to induce transcription (46). Collectively, BCAA mixture can activate eNOS and promote the gene expression of PGC-1α through two pathways: (1) the eNOS/NO/SIRT1 pathways and (2) the eNOS-Ser1177/NO/cGMP pathway. Leucine can increase the gene expression of PGC-1α through three pathways: (1) leucine induced PGC-1α expression by activation of the SIRT1/LKB1/AMPK pathway; (2) leucine directly activated SIRT1 to promote PGC-1α expression; and (3) leucine activated mTOR, and mTOR controlled mitochondrial gene expression through the direct modulation of YY1-PGC-1α activity. In addition, both BCAA mixture and leucine could increase the mitochondrial content and induce the expression of TFAM and mitochondrial component genes. However, several studies have shown that valine did not affect the mitochondrial biogenesis (49, 51, 52).
Long-chain, medium-chain, and short-chain fatty acids regulate the expression of PGC-1α through different molecular mechanisms (Supplementary Table 1). PGC-1α interplays with peroxisome proliferators-activated receptor γ (PPARγ) as PPARγ coactivator, and some medium-chain fatty acids (MCFAs) and short-chain fatty acids (SCFAs) could activate PPARγ and promote PGC-1α transcription to modulate mitochondrial biogenesis (53–57). Acetate treatment activated G-protein-coupled receptor 43 (GPR43), and GPR43 further activated ERK1/2 and cAMP response element-binding protein (CREB) in differentiated brown adipocytes, significantly increasing the mRNA expression levels of PGC-1α (57, 58). Supplementation of butyrate elevated PGC-1α expression at mRNA and protein levels by activating the ERK1/2-CREB signaling pathway in brown adipocyte (57, 58). Long-chain fatty acids (LCFAs) had a weaker ability to promote mitochondrial biogenesis through PGC-1α, compared to MCFAs (59). However, different LCFAs have opposite effects on regulating PGC-1α expression (Supplementary Table 1). The n-3 long-chain unsaturated fatty acids, such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), enhanced the PGC-1α expression and induced mitochondrial biogenesis in adipocytes (60). EPA stimulated phosphorylation and activation of AMPK signaling and upregulated PGC1-α expression in human subcutaneous adipocytes (61). In contrast, some studies have found that certain LCFAs could reduce or not affect the expression of PGC-1α. A mixture of long-chain saturated fatty acids and unsaturated fatty acids stimulated the production of reactive oxygen species (ROS) in adipocyte, which suppressed the expression level of PGC-1α mRNA (62). A long-chain saturated fatty acid palmitate downregulated PGC-1α expression by activating the extracellular signal-regulated kinases 1 and 2 (ERK1/2), mitogen-activated protein kinase (MAPK), and nuclear factor-kappaB (NF-κB) signaling pathways and inhibited mitochondrial biogenesis in skeletal muscle cells (63). The downregulation of PGC-1α mRNA expression might be caused by binding to transcriptional complex I containing the p65 subunit of NF-κB (64). Moreover, the accumulation of fatty acids in obese patients and animal models led to the lowered secretion of adiponectin (61, 65–68). In skeletal muscle cells, adiponectin increased the expression of calcium/calmodulin-dependent protein kinase (CaMK) by activating AMPK, and CaMK induced the expression of PGC-1α (69). The elevated level of adiponectin also inhibited the activity of BCKDC and the expression of PP2Cm, which suppressed BCAA catabolism, increased the BCAAs levels, and further hindered mitochondrial biogenesis (65). Furthermore, in the animal liver tissues, intervention with a high-protein diet (BCAAs significantly increased) plus high-fat diet induced the expression of PGC-1α and TFAM, and in skeletal muscle tissues, leucine supplementation plus high-fat diet also promoted the expression of PGC-1α and TFAM, which suggested the coordinated effect of BCAAs and fatty acids on mitochondrial biogenesis (70, 71).
Overall, BCAAs and fatty acids can regulate the expression of PGC-1α through different mechanisms to influence mitochondrial biogenesis. Differential expression patterns of key enzymes in BCAA catabolism and fatty acid oxidation in different tissues may be the reason that distinguished the regulatory mechanisms of BCAAs and fatty acids. BCAT, the enzyme that initiates BCAA catabolism, can exist as the cytoplasmic BCAT1 and the mitochondrial BCAT2. BCAT2 is expressed ubiquitously in most tissues and especially in skeletal muscle, whereas for the second step of BCAA catabolism, the key enzyme BCKDH is most active in the liver (72). In fatty acid oxidation, fatty acids are needed to be first activated by the acyl-CoA synthetases (ACS), which express in a tissue-specific manner. For example, long-chain acyl-CoA synthetase 1 (ACSL1) is generally distributed in liver cells, cardiomyocytes, and adipocytes (73), while ACSL6 is more common in skeletal muscle (74). Different ACS isoforms channel different fatty acids into specific downstream pathways that may induce different biological effects (75). Besides, different types of long-chain fatty acids have distinct, and sometimes opposing, effects on mitochondrial biogenesis. Based on the current researches, we speculated that the mitochondrial biogenesis might be inhibited by saturated long-chain fatty acids, and the unsaturated long-chain fatty acids might play a positive role in promoting mitochondrial biogenesis. This hypothesis and possible mechanism need to be further studied and confirmed.
Under aerobic conditions, pyruvate produced by glycolysis enters the TCA cycle to produce carbon dioxide and water. The metabolic by-products NADH and FADH2 undergo oxidative phosphorylation to produce ATP, which maintains the normal energy metabolism of cells. Increased BCAA and fatty acid levels can affect glycolysis (Figure 2). When adding BCAAs to PP2Cm germ-line knockout (KO) mouse heart perfusate, BCAAs inhibited the activity of pyruvate dehydrogenase directly to affect the glycolysis (76). Moreover, enhanced fatty acid oxidation increased the ratio of mitochondrial acetyl-CoA:CoA and NADH:NAD+, leading to the inactivation of pyruvate dehydrogenase. This in turn increased the intracellular level of citrate, thereby inhibiting the accumulation of phosphofructokinase and glucose-6-phosphate, which hindered the normal progress of glycolysis (77).
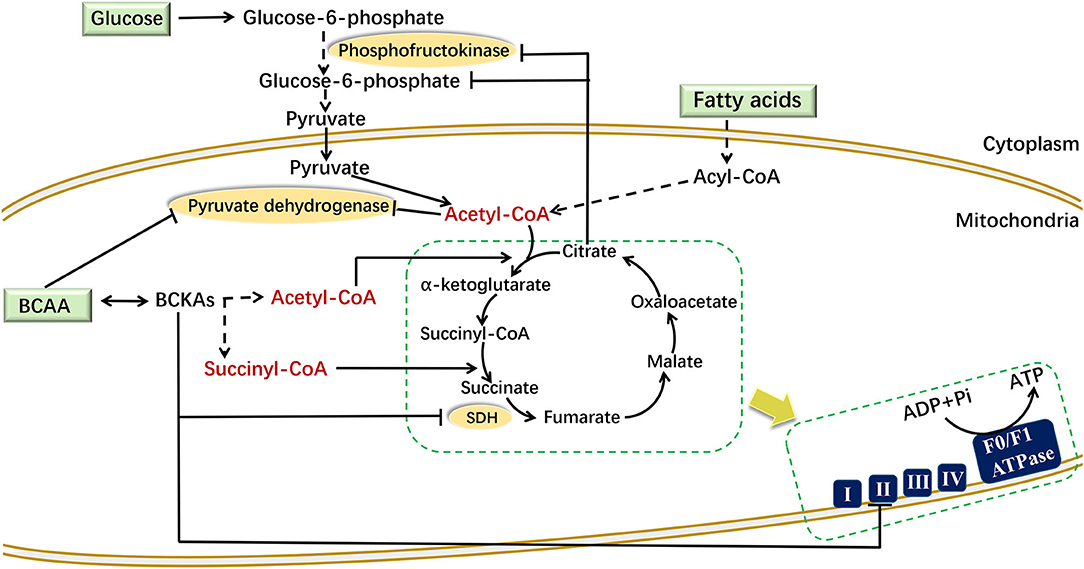
Figure 2. Mechanism of BCAAs and fatty acids regulating energy metabolism. BCAAs and fatty acids affect mitochondrial energy metabolism through different mechanisms in different cells. In mouse heart perfusate, BCAAs inhibited the activity of pyruvate dehydrogenase. In muscle cells, the increased citrate inhibits the accumulation of phosphofructokinase and glucose-6-phosphate. In liver cells, BCKAs can directly suppress the expression of respiratory complex II/SDH to reduce the production of ATP. In the figure, the dotted line indicates a multistep reaction, and the solid line indicates a one-step reaction. SDH, succinate dehydrogenase.
Increased BCAA and fatty acid levels can also disturb the TCA cycle (Figure 2). The carbon produced by BCAA catabolism and fatty acid oxidation enters the TCA cycle in the form of acetyl-CoA or succinyl-CoA. The succinate dehydrogenase catalyzes the conversion from succinate to fumarate in the TCA cycle, and it is also the respiration Complex II in the electron transfer chain. In liver cells, the elevated BCKA levels suppressed the gene expression of succinate dehydrogenase to block the TCA cycle and ATP production (78). In high-fat-diet (HFD) mice, enhanced fatty acid oxidation increased the amount of acetyl-CoA entering the TCA cycle in the early period. While due to the “overload” of the TCA cycle, the flux of acetyl-CoA decreased in the following stage (79).
Increased levels of BCAAs and fatty acids may affect mitochondrial oxidative phosphorylation (OXPHOS) through their common target, PGC-1α. PGC-1α enhanced the expression of Errα and Gabpa and then activated the expression of downstream OXPHOS-related genes (80). These genes mainly included two categories: (1) mitochondrial OXPHOS-related enzyme-encoding genes, such as cytochrome c oxidase 1-5 (COX 1-5), ATP synthase (ATP5G), citrate synthase (CS), ATP synthase peripheral stalk subunit OSCP (Atp5o), succinic dehydrogenase (SDH), and malate dehydrogenase (MDH), and (2) mitochondrial substrate oxidation-related regulators, such as PPARβ/δ and cyclin-dependent kinase 4 (CDK4). In addition, NRF-1 activated by PGC-1α could combine with the promoters of the OXPHOS-related genes to stimulate their transcription levels (45, 62). Therefore, BCAAs and several fatty acids could synergistically enhance mitochondrial OXPHOS through upregulating PGC-1α expression. However, elevated BCAAs and fatty acids can also inhibit ATP production through other mechanisms. In mouse C2C12 myoblast cells, although leucine promoted the expression of PGC-1α and increased OXPHOS, a decrease in glycolysis and ATP content was observed (81). In PP2Cm knockout mouse liver, increased BCKA levels inhibited the expression of respiratory complex II, which subsequently interfered with both mitochondrial OXPHOS and ATP production [Figure 2; (78)]. In addition, a study suggested that treatment of skeletal muscle cells with a low concentration of 0.5–2 μM free fatty acids stimulated ATP production (82), while treatment of adipocytes with a high concentration of free fatty acids >5 μM inhibits ATP production (62). We speculate that a potential reason may be that the condition with low concentration of fatty acids is closer to the physiological state, while the condition with high levels of fatty acids is in the pathological state, therefore having different pathophysiology effects. The mechanism by which different pathophysiology effects of fatty acids on OXPHOS and ATP production requires further research.
BCAAs and fatty acids can induce the inflammation (Figure 3). Supplementation with BCAAs could activate mTORC1 and upregulate the NF-κB signaling pathway, increasing the release of pro-inflammatory cytokines in human peripheral blood mononuclear cells and endothelial cells (83, 84). Treatment with the saturated fatty acid palmitate in C2C12 cells rapidly induced the association of myeloid differentiation factor 88 (MyD88) with the toll-like receptor 2 (TLR2), increased the degradation of IkappaB and NF-κB DNA binding, and enhanced interleukin (IL)-6 production (85). In addition, saturated (lauric acid) and polyunsaturated (DHA) fatty acids reciprocally modulated the activation of TLR4 and its downstream signaling pathways involving PI3K/Akt/NF-κB in macrophages (86), further suggesting the possibility that TLR-mediated target gene expression and cellular inflammatory responses are also differentially mediated by saturated and unsaturated fatty acids. Besides, studies have shown that several types of fatty acids can activate the inflammasome. Saturated fatty acid palmitate, but not unsaturated oleate, activated ROS through inhibiting AMPK, induced the activation of the Nod-like receptor pyrin domain containing 3 (NLRP3) inflammasome, and caused IL-1β and IL-18 production in hematopoietic cells (87). Saturated fatty acids could also directly activate the NLRP3 inflammasome through upregulating thioredoxin-interacting protein (TXNIP) in HFD-induced mice (88). By contrast, omega-3 polyunsaturated fatty acids (ω-3 FAs), including EPA and DHA, alleviated the inflammation by preventing NLPR3 activation in an HFD-induced model. The G protein-coupled receptors (GPR120 and GPR40) and the downstream protein β-arrestin-2 (ARRB2) were shown to be involved in inflammasome inhibition induced by ω-3 FAs (89). Overall, saturated and unsaturated fatty acids may play pro-inflammatory and anti-inflammatory roles, respectively, at the level of transcriptional regulation and protein processing of inflammatory factors.
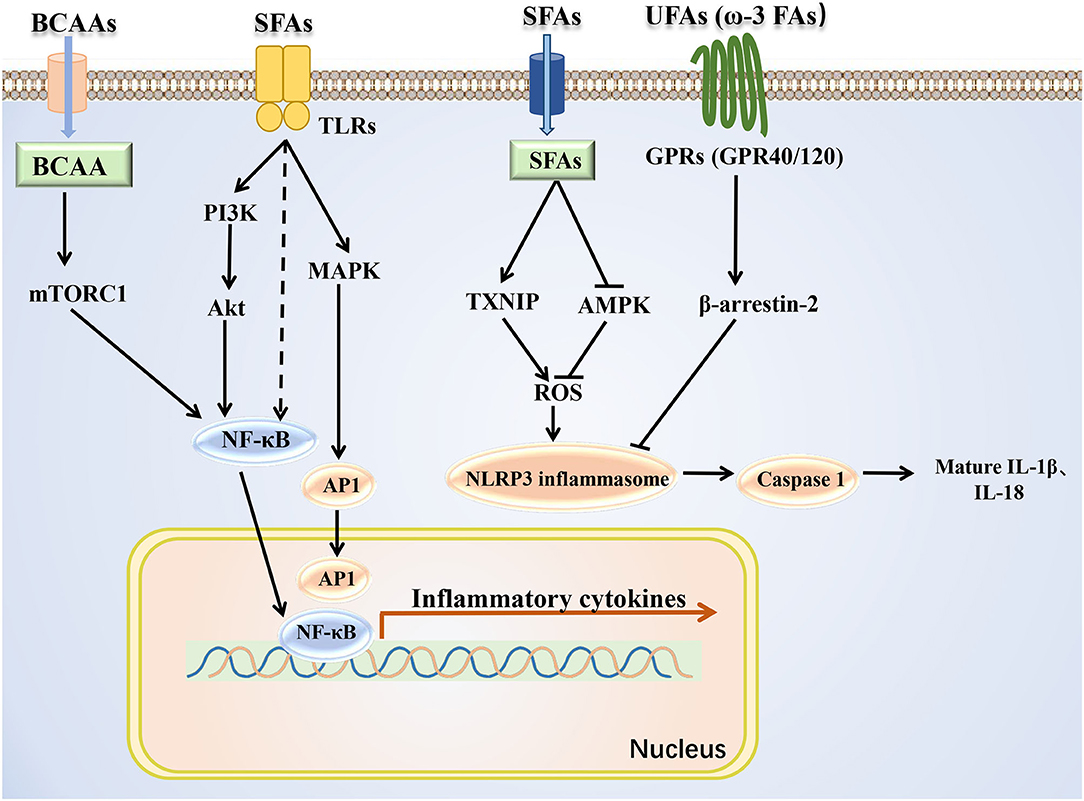
Figure 3. Mechanism of BCAAs and fatty acids regulating inflammatory signals. BCAAs and different types of fatty acids regulate the inflammatory response through the NF-κB pathway and NLRP3. SFAs, saturated fatty acids; UFAs, unsaturated fatty acids; TXNIP, thioredoxin-interacting protein.
Previous studies have indicated that inflammatory signals could influence the mitochondrial biogenesis by decreasing PGC-1α expression. In human cardiac cells, tumor necrosis factor-α (TNF-α) reduced PGC-1α expression mediated via both p38 MAPK and NF-κB pathways, and PGC-1α downregulation resulted in a reduction in pyruvate dehydrogenase kinase 4 (PDK4) expression and an increase in glucose oxidation rate (64). The excessive TNF-α bound to the p55 subtype of TNF receptor, and then inhibited eNOS activation and reduced NO level in obese rodents, leading to a decrease in the expression of PGC-1α (90). In addition, treatment 3T3-L1 adipocytes with TNF-α induced the downregulation of the mRNA expression of many TCA circulation-related enzyme genes, such as Aco2, Idh2, ogdh, and Fh1 (91). Thus, the coordinated activation of inflammatory signals by BCAAs and some fatty acids may interfere with markers of mitochondrial biogenesis mitochondrial biogenesis and energy metabolism, even leading to metabolic disorders.
Insulin is the key hormone controlling glucose and lipid metabolism. Under normal physiological conditions, secreted insulin binds to the insulin receptor, fostering tyrosine phosphorylation of the insulin receptor substrate (IRS)-1/2 and regulating downstream cascade signals. Several kinases have been implicated in this process, including PI3K, 3-phosphoinositide-dependent protein kinase (PDK), Akt, and S6K1. Feedback control in insulin signaling involves serine phosphorylation of IRS proteins, which inhibits tyrosine phosphorylation and blocks the ectopic expression and translocation of glucose transporters (GLUTs) and ultimately produces IR [Figure 4; (92)]. Some studies have shown that BCAAs, especially leucine, promoted insulin signal transduction. Leucine induced the tyrosine phosphorylation of IRS-1 and improved insulin-stimulated Akt and mTOR phosphorylation, preventing HFD-induced IR in insulin-target tissues (93). The possible mechanism is that BCAAs or leucine-induced protein synthesis were accompanied by energy expenditure, leading to an increase in insulin signal transduction, GLUT4 content, and glucose uptake (94–96). However, some studies have shown that leucine could inhibit insulin signal transduction through other mechanisms (Figure 4). Incubation of rat skeletal muscle with elevated concentrations of leucine suppressed AMPK activation and concomitantly increased mTOR/p70S6K signaling and led to IR (97). Additionally, leucine deprivation could improve hepatic insulin sensitivity by sequentially activating general control non-derepressible (GCN) 2, an amino acid sensor, and decreasing mTOR/ p70S6K signaling (98). The specific mechanisms of BCAAs in regulating the insulin sensitivity and glucose metabolism in different conditions need further exploration.
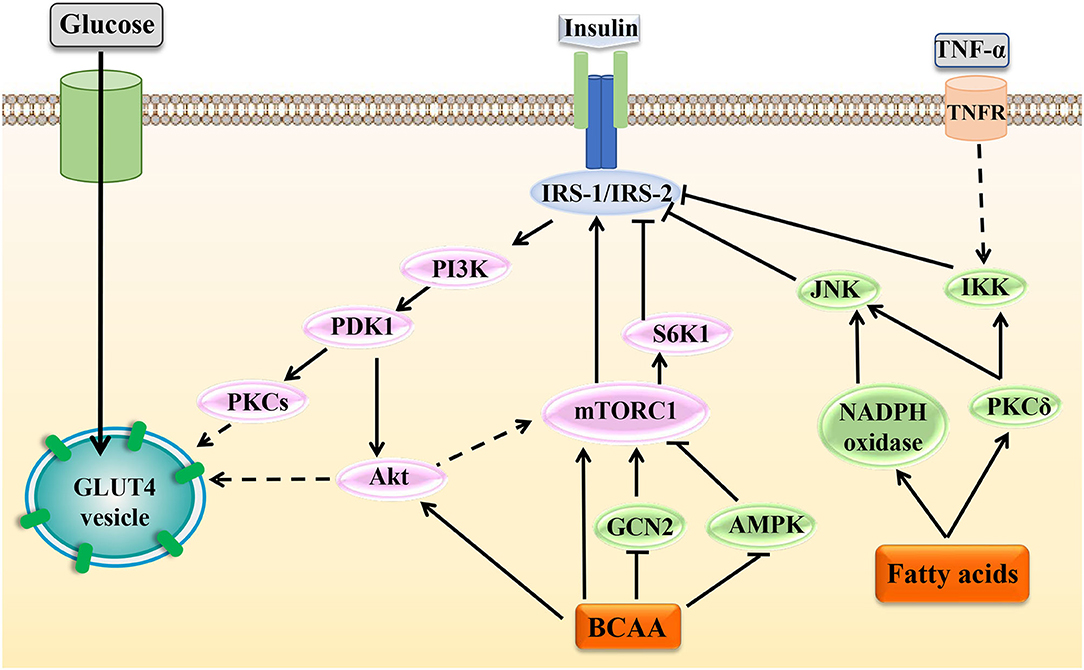
Figure 4. Effects of BCAAs and fatty acids on insulin signal transduction. Under normal circumstances, insulin can activate various molecules such as PI3K, Akt, and mTOR to affect the activation of IRS and regulate the transport and ectopic expression of GLUTs. Increased levels of BCAAs and fatty acids can interfere with normal insulin signaling through various mechanisms and ultimately lead to IR. In the figure, the dotted line indicates a multistep reaction, and the solid line indicates a one-step reaction.
Fatty acids can influence IRS activity through regulating protein phosphorylation of IRS or transcriptional level of IRS via histone acetylation. In adipocytes, fatty acids activated PKCδ, leading to activation of serine kinase inhibitor kappaB kinase (IKK) and c-JUN NH2 terminal kinase (JNK), which catalyzed the phosphorylation of IRS-1, ultimately reducing the insulin-induced glucose uptake (99). Fatty acids induced oxidative stress by activating PKCδ and NADPH oxidase, increased JNK phosphorylation, and thereby enhanced serine phosphorylation of IRS-1 and IRS-2 and impaired hepatic insulin signal transduction (100). However, sodium butyrate, the sodium salt form of butyric acid, is hydrolyzed to form a shortchain fatty acid butyric acid. Butyric acid could act as the histone deacetylase inhibitor and favor insulin sensitivity, and butyrate improved palmitate-induced IR by increasing histone H3 acetylation on chromatin in proximity of the Irs1 transcriptional start site in L6 myotubes, indicating that a certain SLFA-mediated insulin-sensitizing action was dependent on epigenetic effects (101).
PGC-1α- and PGC1α-responsive genes involved in OXPHOS were downregulated in skeletal muscles of patients with IR (45, 80), which suggested that reduced mitochondrial biogenesis and energy metabolism were closely related to IR. Moreover, PGC-1α could induce valine catabolism to produce intermediate 3-Hydroxyisobutyrate (3-HIB), and 3-HIB acted as a paracrine factor to reduce insulin sensitivity by inhibiting Akt phosphorylation in C2C12 myotubes (102). Besides, plasma concentrations of BCAAs and 3-HIB were inversely related to insulin sensitivity in overweight to obese individuals, while changes in 3-HIB rather than changes in BCAAs were associated with metabolic improvements with weight loss (103), supporting a crucial role of 3-HIB in the development of insulin resistance (104, 105). In addition, elevated levels of acylcarnitine, a product of incomplete oxidation of BCAAs and fatty acids, caused mitochondrial stress, which interfered with insulin signal transduction (7, 106, 107). However, recent findings indicated that abnormal mitochondrial function was not an early event in the development of IR but an adaptive response to excess nutrients in the body (71). Further research is needed to clarify the regulatory mechanisms of BCAAs and fatty acids in affecting insulin sensitivity.
An increasing body of evidence has shown that chronic low-grade inflammation participated in the development of IR [Figure 4; (108, 109)]. In skeletal muscle cells, TNF-α activated MAPK, leading to downstream phosphorylation activation of IKK. Then, IKK phosphorylated IRS to cause IR (110). In brown adipocytes, TNF-α could activate MAPK and ERK, which led to the serine phosphorylation of IRS-2 and then caused IR (111). Moreover, in adipocytes, IL-1β inhibited the activation of IRS by phosphorylating JNK or MAPK. Besides, IL-1β could partially inhibit the activation of IRS-1 by activating ERK (112). In adipocytes, IL-6 reduced the protein expression of the insulin receptor β subunit and IRS-1 and simultaneously downregulated the expression of GLUT4. Besides, IL-6 could suppress the insulin-stimulated Akt/PKB and ERK1/2 activation (113). Also, research results showed that in skeletal muscle cells, fatty acids activated the MAPK, JNK, and NF-κB pathways by binding to TLR2, which inhibited IRS tyrosine and Akt phosphorylation, finally inducing IR. At the same time, the activation of this pathway induced the production of the inflammatory cytokine IL-6, which further aggravated the occurrence and development of IR (85). Therefore, as mentioned above, BCAAs and some fatty acids could activate inflammatory signals and increase the release of inflammatory cytokines; thus, they may exacerbate the development of IR by blocking insulin signaling transduction in adipocytes and skeletal muscle cells.
In clinical studies, there was a positive correlation between homeostatic model assessment (HOMA) index, glycated hemoglobin (HbA1c), and increased BCAAs levels in the plasma (106). Furthermore, increased IR and proteolysis could result in elevated plasma levels of BCAAs in patients with NAFLD (114). In addition, there was a significant reduction of the BCAA catabolic enzymes BCKDHA, BCKDHB, and BCAT2 in the visceral white adipose tissue of obese people with metabolic syndrome, leading to an increase in BCAA levels in the circulation (115), while the expression of BCAA catabolic enzymes in the adipose tissue was negatively correlated with IR (116).
Excess lipids inhibited the complete oxidation of fatty acids in the mitochondria (107). Different types of fatty acids could regulate insulin sensitivity through distinct mechanisms (17). In patients with metabolic syndrome, enhanced lipolysis of the adipose tissue led to elevated levels of fatty acids in the circulation, disturbing the insulin signals and generating the phenotypes of IR and obesity (117). Ectopic fatty acid accumulation was an early manifestation of NAFLD. As the disease progresses, oxidation of free fatty acid was decreased in the liver, producing toxic metabolites such as diacylglycerol and ceramide (118). Chronic low-grade inflammation was one of the main causes of IR in T2D and obese patients (119, 120). BCAAs and fatty acids could also mediate the occurrence of IR by activating inflammatory cytokines and inflammatory signaling pathways (83, 84, 121).
Additionally, metabolic disorders of BCAAs and fatty acids could also affect the reproductive function. Polycystic ovary syndrome (PCOS) is one of the most common reproductive endocrine diseases and the leading cause of anovulatory infertility. PCOS patients are accompanied by obvious metabolic abnormalities and chronic inflammation and have a higher risk for diabetes and cardiovascular disease compared with the healthy women (16). Previous studies have shown that the levels of BCAAs and fatty acids in both plasma and follicular fluids were significantly increased in PCOS patients (15, 16, 102, 103, 122). Moreover, the increased BCAA levels in the follicular fluid was negatively associated with the pregnancy outcome (103), which suggested that the systemic metabolic disorders of BCAAs could alter metabolic homeostasis of the follicular microenvironment for oocyte maturation and embryo development. Furthermore, there were mitochondrial dysfunction and inflammation in the ovarian granulosa cells of PCOS patients, affecting the microenvironment of follicular development (123–129), but the regulatory mechanism had not been elucidated clearly. Therefore, the modulation of mitochondrial function and inflammation by BCAAs and fatty acids may be helpful for us to comprehensively explore the pathogenesis of PCOS, so as to provide new ideas and targets for clinical diagnosis and treatment.
In summary, elevated levels of BCAAs and fatty acids can regulate cell metabolism by affecting mitochondrial function and inflammation signals. Mitochondrial dysfunction, inflammation, and IR can further lead to the accumulation of BCAAs and fatty acids, thus aggravating the development of metabolic diseases. However, there are still many issues that need further exploration; whether BCAAs and fatty acids synergistically regulate energy metabolism and inflammation through other signaling pathways, whether the mediated mechanisms of BCAAs and fatty acids reported are tissue specific, and whether abnormal levels of BCAAs and fatty acids could be a useful marker for risk prediction and a new target for clinical diagnosis and treatment of metabolic disease. These issues all require further study.
ZY collected the information, designed the pictures, and wrote and submitted the manuscript. SW and CZ collected the information and joined in the critical discussion. YZ critically revised the manuscript and contributed to the conception of the design. All authors contributed to the article and approved the submitted version.
This work was supported by the National Key R&D Program of China (2017YFC1001003), the National Natural Science Funds for general program (81671419, 81871139, and 81601243), and CAMS Innovation Fund for Medical Sciences (2019-I2M-5-001).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2020.00617/full#supplementary-material
1. Li M, Li X, Lu Y. Obstructive sleep apnea syndrome and metabolic diseases. Endocrinology. (2018) 159:2670–5. doi: 10.1210/en.2018-00248
2. Saltiel AR, Olefsky JM. Inflammatory mechanisms linking obesity and metabolic disease. J Clin Invest. (2017) 127:1–4. doi: 10.1172/JCI92035
3. Shokouhi S, Haghani K, Borji P, Bakhtiyari S. Association between PGC-1alpha gene polymorphisms and type 2 diabetes risk: a case-control study of an Iranian population. Can Diabetes J. (2015) 39:65–72. doi: 10.1016/j.jcjd.2014.05.003
4. Schmid AI, Szendroedi J, Chmelik M, Krssak M, Moser E, Roden M. Liver ATP synthesis is lower and relates to insulin sensitivity in patients with: type 2 diabetes. Diabetes Care. (2011) 34:448–53. doi: 10.2337/dc10-1076
5. Brindle JT, Nicholson JK, Schofield PM, Grainger DJ, Holmes E. Application of chemometrics to 1H NMR spectroscopic data to investigate a relationship between human serum metabolic profiles and hypertension. Analyst. (2003) 128:32–6. doi: 10.1039/b209155k
6. Shearer J, Duggan G, Weljie A, Hittel DS, Wasserman DH, Vogel HJ. Metabolomic profiling of dietary-induced insulin resistance in the high fat-fed C57BL/6J mouse. Diabetes Obes Metab. (2008) 10:950–8. doi: 10.1111/j.1463-1326.2007.00837.x
7. Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. (2008) 7:45–56. doi: 10.1016/j.cmet.2007.10.013
8. Yang J, Xu G, Hong Q, Liebich HM, Lutz K, Schmulling RM, et al. Discrimination of Type 2 diabetic patients from healthy controls by using metabonomics method based on their serum fatty acid profiles. J Chromatogr B Analyt Technol Biomed Life Sci. (2004) 813:53–8. doi: 10.1016/j.jchromb.2004.09.023
9. Lu Y, Jiye A, Wang G, Hao H, Huang Q, Yan B, et al. Gas chromatography/time-of-flight mass spectrometry based metabonomic approach to differentiating hypertension- and age-related metabolic variation in spontaneously hypertensive rats. Rapid Commun Mass Spectrom. (2008) 22:2882–8. doi: 10.1002/rcm.3670
10. Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, et al. Metabolite profiles and the risk of developing diabetes. Nat Med. (2011) 17:448–53. doi: 10.1038/nm.2307
11. Sun H, Olson KC, Gao C, Prosdocimo DA, Zhou M, Wang Z, et al. Catabolic defect of branched-chain amino acids promotes heart failure. Circulation. (2016) 133:2038–49. doi: 10.1161/CIRCULATIONAHA.115.020226
12. Wiklund PK, Pekkala S, Autio R, Munukka E, Xu L, Saltevo J, et al. Serum metabolic profiles in overweight and obese women with and without metabolic syndrome. Diabetol Metab Syndr. (2014) 6:40. doi: 10.1186/1758-5996-6-40
13. van den Berg E, Flores-Guerrero J, Gruppen E, de Borst M, Wolak-Dinsmore J, Connelly M, et al. Non-alcoholic fatty liver disease and risk of incident type 2 diabetes: role of circulating branched-chain amino acids. Nutrients. (2019) 11:705. doi: 10.3390/nu11030705
14. She P, Van Horn C, Reid T, Hutson SM, Cooney RN, Lynch CJ. Obesity-related elevations in plasma leucine are associated with alterations in enzymes involved in branched-chain amino acid metabolism. Am J Physiol Endocrinol Metab. (2007) 293:E1552–63. doi: 10.1152/ajpendo.00134.2007
15. Zhao Y, Fu L, Li R, Wang LN, Yang Y, Liu NN, et al. Metabolic profiles characterizing different phenotypes of polycystic ovary syndrome: plasma metabolomics analysis. BMC Med. (2012) 10:153. doi: 10.1186/1741-7015-10-153
16. Mu L, Li R, Lai Y, Zhao Y, Qiao J. Adipose insulin resistance is associated with cardiovascular risk factors in polycystic ovary syndrome. J Endocrinol Investig. (2019) 42:541–8. doi: 10.1007/s40618-018-0949-2
17. Yang Q, Vijayakumar A, Kahn BB. Metabolites as regulators of insulin sensitivity and metabolism. Nat Rev Mol Cell Biol. (2018) 19:654–72. doi: 10.1038/s41580-018-0044-8
18. Jackson KH, Harris WS. Blood fatty acid profiles: new biomarkers for cardiometabolic disease risk. Curr Atheroscler Rep. (2018) 20:22. doi: 10.1007/s11883-018-0722-1
19. Guo X, Yang B, Tang J, Li D. Fatty acid and non-alcoholic fatty liver disease: meta-analyses of case-control and randomized controlled trials. Clin Nutr. (2018) 37:113–22. doi: 10.1016/j.clnu.2017.01.003
20. Hesselink MKC, Schrauwen-Hinderling V, Schrauwen P. Skeletal muscle mitochondria as a target to prevent or treat type 2 diabetes mellitus. Nat Rev Endocrinol. (2016) 12:633–45. doi: 10.1038/nrendo.2016.104
21. Greenwood EA, Huddleston HG. Insulin resistance in polycystic ovary syndrome: concept versus cutoff. Fertil Steril. (2019) 112:827–8. doi: 10.1016/j.fertnstert.2019.08.100
22. Mu W, Cheng X, Liu Y, Lv Q, Liu G, Zhang J, et al. Potential nexus of non-alcoholic fatty liver disease and type 2 diabetes mellitus: insulin resistance between hepatic and peripheral tissues. Front Pharmacol. (2019) 9:1566. doi: 10.3389/fphar.2018.01566
23. Wu H, Ballantyne CM. Metabolic inflammation and insulin resistance in obesity. Circ Res. (2020) 126:1549–64. doi: 10.1161/CIRCRESAHA.119.315896
24. Palomba S, Falbo A, Chiossi G, Orio F, Tolino A, Colao A, et al. Low-grade chronic inflammation in pregnant women with polycystic ovary syndrome: a prospective controlled clinical study. J Clini Endocrinol Metab. (2014) 99:2942–51. doi: 10.1210/jc.2014-1214
25. Wu H, Ballantyne CM. Skeletal muscle inflammation and insulin resistance in obesity. J Clin Invest. (2017) 127:43–54. doi: 10.1172/JCI88880
26. Gonzalez-Franquesa A, Patti M. Insulin resistance and mitochondrial dysfunction. Adv Exp Med Biol. (2017) 982:465. doi: 10.1007/978-3-319-55330-6_25
27. Di Meo S, Iossa S, Venditti P. Skeletal muscle insulin resistance: role of mitochondria and other ROS sources. J Endocrinol. (2017) 233:R15–42. doi: 10.1530/JOE-16-0598
28. Gannon NP, Schnuck JK, Vaughan RA. BCAA metabolism and insulin sensitivity - dysregulated by metabolic status? Mol Nutr Food Res. (2018) 62:e1700756. doi: 10.1002/mnfr.201700756
29. Horiuchi M, Takeda T, Takanashi H, Ozaki-Masuzawa Y, Taguchi Y, Toyoshima Y, et al. Branched-chain amino acid supplementation restores reduced insulinotropic activity of a low-protein diet through the vagus nerve in rats. Nutr Metab. (2017) 14:59. doi: 10.1186/s12986-017-0215-1
30. Ringseis R, Eder K, Mooren FC, Krüger K. Metabolic signals and innate immune activation in obesity and exercise. Exerc Immunol Rev. (2015) 21:58–68.
31. Lepretti M, Martucciello S, Burgos Aceves M, Putti R, Lionetti L. Omega-3 fatty acids and insulin resistance: focus on the regulation of mitochondria and endoplasmic reticulum stress Nutrients. (2018) 10:350. doi: 10.3390/nu10030350
32. Brosnan JT, Brosnan ME. Branched-chain amino acids: enzyme and substrate regulation. J Nutr. (2006) 136(Suppl. 1):207–11S. doi: 10.1093/jn/136.1.207S
33. Zhou M, Shao J, Wu CY, Shu L, Dong W, Liu Y, et al. Targeting BCAA catabolism to treat obesity-associated insulin resistance. Diabetes. (2019) 68:1730–46. doi: 10.2337/db18-0927
34. Adeva-Andany MM, López-Maside L, Donapetry-García C, Fernández-Fernández C, Sixto-Leal C. Enzymes involved in branched-chain amino acid metabolism in humans. Amino Acids. (2017) 49:1005–28. doi: 10.1007/s00726-017-2412-7
35. Valerio A, D'Antona G, Nisoli E. Branched-chain amino acids, mitochondrial biogenesis, and healthspan: an evolutionary perspective. Aging. (2011) 3:464–78. doi: 10.18632/aging.100322
36. Jang C, Oh SF, Wada S, Rowe GC, Liu L, Chan MC, et al. A branched-chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat Med. (2016) 22:421–6. doi: 10.1038/nm.4057
37. Newgard CB. Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab. (2012) 15:606–14. doi: 10.1016/j.cmet.2012.01.024
38. Kompare M, Rizzo WB. Mitochondrial fatty-acid oxidation disorders. Semin Pediatr Neurol. (2008) 15:140–9. doi: 10.1016/j.spen.2008.05.008
39. Tvrzicka E, Kremmyda LS, Stankova B, Zak A. Fatty acids as biocompounds. their role in human metabolism, health and disease–a review. Part 1: classification, dietary sources and biological functions. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. (2011) 155:117–30. doi: 10.5507/bp.2011.038
40. Glatz JF, Luiken JJ, Bonen A. Membrane fatty acid transporters as regulators of lipid metabolism: implications for metabolic disease Physiol Rev. (2010) 90:367–417. doi: 10.1152/physrev.00003.2009
41. Rector RS, Ibdah JA. Fatty acid oxidation disorders: maternal health and neonatal outcomes. Semin Fetal Neonatal Med. (2010) 15:122–8. doi: 10.1016/j.siny.2009.10.006
42. Duan Y, Li F, Li Y, Tang Y, Kong X, Feng Z, et al. The role of leucine and its metabolites in protein and energy metabolism. Amino Acids. (2016) 48:41–51. doi: 10.1007/s00726-015-2067-1
43. Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, et al. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. (2003) 299:896–9. doi: 10.1126/science.1079368
44. Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. (1999) 98:115–24. doi: 10.1016/S0092-8674(00)80611-X
45. Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. (2003) 34:267–73. doi: 10.1038/ng1180
46. Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1–PGC-1α transcriptional complex. Nature. (2007) 450:736–40. doi: 10.1038/nature06322
47. Tedesco L, Corsetti G, Ruocco C, Ragni M, Rossi F, Carruba MO, et al. A specific amino acid formula prevents alcoholic liver disease in rodents. Am J Physiol Gastr Liver Physiol. (2018) 314:G566–G582. doi: 10.1152/ajpgi.00231.2017
48. Nisoli E, Falcone S, Tonello C, Cozzi V, Palomba L, Fiorani M, et al. Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Process Natl Acad Sci USA. (2004) 101:16507–12. doi: 10.1073/pnas.0405432101
49. Liang C, Curry BJ, Brown PL, Zemel MB. Leucine modulates mitochondrial biogenesis and SIRT1-AMPK signaling in C2C12 myotubes. J Nutr Metab. (2014) 2014:1–11. doi: 10.1155/2014/239750
50. Kimball SR, Jefferson LS. Signaling pathways and molecular mechanisms through which branched-chain amino acids mediate translational control of protein synthesis. J Nutr. (2006) 136(Suppl. 1):227–31S. doi: 10.1093/jn/136.1.227S
51. Schnuck JK, Sunderland KL, Gannon NP, Kuennen MR, Vaughan RA. Leucine stimulates PPARβ/δ-dependent mitochondrial biogenesis and oxidative metabolism with enhanced GLUT4 content and glucose uptake in myotubes. Biochimie. (2016) 128–29:1–7. doi: 10.1016/j.biochi.2016.06.009
52. Chen X, Xiang L, Jia G, Liu G, Zhao H, Huang Z. Leucine regulates slow-twitch muscle fibers expression and mitochondrial function by Sirt1/AMPK signaling in porcine skeletal muscle satellite cells. Anim Sci J. (2018) 90:255–63. doi: 10.1111/asj.13146
53. Manio MC, Matsumura S, Inoue K. Low-fat diet, and medium-fat diets containing coconut oil and soybean oil exert different metabolic effects in untrained and treadmill-trained mice. J Int Soc Sport Nutr. (2018) 15:29. doi: 10.1186/s12970-018-0234-y
54. Augustin K, Khabbush A, Williams S, Eaton S, Orford M, Cross JH, et al. Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol. (2018) 17:84–93. doi: 10.1016/S1474-4422(17)30408-8
55. Hughes SD, Kanabus M, Anderson G, Hargreaves IP, Rutherford T, Donnell MO, et al. The ketogenic diet component decanoic acid increases mitochondrial citrate synthase and complex I activity in neuronal cells. J Neurochem. (2014) 129:426–33. doi: 10.1111/jnc.12646
56. Kanabus M, Fassone E, Hughes SD, Bilooei SF, Rutherford T, Donnell MO, et al. The pleiotropic effects of decanoic acid treatment on mitochondrial function in fibroblasts from patients with complex I deficient Leigh syndrome. J Inherit Metab Dis. (2016) 39:415–26. doi: 10.1007/s10545-016-9930-4
57. Hu J, Kyrou I, Tan BK, Dimitriadis GK, Ramanjaneya M, Tripathi G, et al. Short-chain fatty acid acetate stimulates adipogenesis and mitochondrial biogenesis via GPR43 in brown adipocytes. Endocrinology. (2016) 157:1881–94. doi: 10.1210/en.2015-1944
58. Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, et al. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes. (2009) 58:1509–17. doi: 10.2337/db08-1637
59. Montgomery MK, Osborne B, Brown SHJ, Small L, Mitchell TW, Cooney GJ, et al. Contrasting metabolic effects of medium- versus long-chain fatty acids in skeletal muscle. J Lipid Res. (2013) 54:3322–33. doi: 10.1194/jlr.M040451
60. Kopecky J, Rossmeisl M, Flachs P, Kuda O, Brauner P, Jilkova Z, et al. n-3 PUFA: bioavailability and modulation of adipose tissue function Proc Nutr Soc. (2009) 68:361–9. doi: 10.1017/S0029665109990231
61. Weyer C, Funahashi T, Tanaka S, Hotta K, Matsuzawa Y, Pratley RE, et al. Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab. (2001) 86:1930–5. doi: 10.1210/jcem.86.5.7463
62. Gao CL, Zhu C, Zhao YP, Chen XH, Ji CB, Zhang CM, et al. Mitochondrial dysfunction is induced by high levels of glucose and free fatty acids in 3T3-L1 adipocytes. Mol Cell Endocrinol. (2010) 320:25–33. doi: 10.1016/j.mce.2010.01.039
63. Coll T, Jove M, Rodriguez-Calvo R, Eyre E, Palomer X, Sanchez RM, et al. Palmitate-mediated downregulation of peroxisome proliferator-activated receptor- coactivator 1. in skeletal muscle cells involves MEK1/2 and nuclear factor- B activation. Diabetes. (2006) 55:2779–87. doi: 10.2337/db05-1494
64. Palomer X, Álvarez-Guardia D, Rodríguez-Calvo R, Coll T, Laguna JC, Davidson MM, et al. TNF-α reduces PGC-1α expression through NF-κB and p38 MAPK leading to increased glucose oxidation in a human cardiac cell model. Cardiovasc Res. (2009) 81:703–12. doi: 10.1093/cvr/cvn327
65. Lian K, Du C, Liu Y, Zhu D, Yan W, Zhang H, et al. Impaired adiponectin signaling contributes to disturbed catabolism of branched-chain amino acids in diabetic mice. Diabetes. (2015) 64:49–59. doi: 10.2337/db14-0312
66. Turer AT, Scherer PE. Adiponectin: mechanistic insights and clinical implications. Diabetologia. (2012) 55:2319–26. doi: 10.1007/s00125-012-2598-x
67. Asterholm IW, Scherer PE. Enhanced metabolic flexibility associated with elevated adiponectin levels. Am J Pathol. (2010) 176:1364–76. doi: 10.2353/ajpath.2010.090647
68. Tumminia A, Vinciguerra F, Parisi M, Graziano M, Sciacca L, Baratta R, et al. Adipose tissue, obesity and adiponectin: role in endocrine cancer risk. Int J Mol Sci. (2019) 20:2863. doi: 10.3390/ijms20122863
69. Wu H, Kanatous SB, Thurmond FA, Gallardo T, Isotani E, Bassel-Duby R, et al. Regulation of mitochondrial biogenesis in skeletal muscle by CaM. Science. (2002) 296:349–52. doi: 10.1126/science.1071163
70. Garcia Caraballo SC, Comhair TM, Houten SM, Dejong CHC, Lamers WH, Koehler SE. High-protein diets prevent steatosis and induce hepatic accumulation of monomethyl branched-chain fatty acids. J Nutr Biochem. (2014) 25:1263–74. doi: 10.1016/j.jnutbio.2014.07.005
71. Liu R, Li H, Fan W, Jin Q, Chao T, Wu Y, et al. Leucine supplementation differently modulates branched-chain amino acid catabolism, mitochondrial function and metabolic profiles at the different stage of insulin resistance in rats on high-fat diet. Nutrients. (2017) 9:565. doi: 10.3390/nu9060565
72. Holeček M. Branched-chain amino acids in health and disease: metabolism, alterations in blood plasma, and as supplements. Nutr Metab. (2018) 15:33. doi: 10.1186/s12986-018-0271-1
73. Grevengoed TJ, Klett EL, Coleman RA. Acyl-CoA metabolism and partitioning. Annu Rev Nutr. (2014) 34:1–30. doi: 10.1146/annurev-nutr-071813-105541
74. Teodoro BG, Sampaio IH, Bomfim LHM, Queiroz AL, Silveira LR, Souza AO, et al. Long-chain acyl-CoA synthetase 6 regulates lipid synthesis and mitochondrial oxidative capacity in human and rat skeletal muscle. J Physiol. (2017) 595:677–93. doi: 10.1113/JP272962
75. Ellis JM, Frahm JL, Li LO, Coleman RA. Acyl-coenzyme A synthetases in metabolic control. Curr Opin Lipidol. (2010) 21:212–7. doi: 10.1097/mol.0b013e32833884bb
76. Li T, Zhang Z, Kolwicz SC, Abell L, Roe ND, Kim M, et al. Defective branched-chain amino acid catabolism disrupts glucose metabolism and sensitizes the heart to ischemia-reperfusion injury. Cell Metab. (2017) 25:374–85. doi: 10.1016/j.cmet.2016.11.005
77. Savage DB, Petersen KF, Shulman GI. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol Rev. (2007) 87:507–20. doi: 10.1152/physrev.00024.2006
78. Wang J, Liu Y, Lian K, Shentu X, Fang J, Shao J, et al. BCAA catabolic defect alters glucose metabolism in lean mice. Front Physiol. (2019) 10:1140. doi: 10.3389/fphys.2019.01140
79. Boulangé CL, Claus SP, Chou CJ, Collino S, Montoliu I, Kochhar S, et al. Early metabolic adaptation in C57BL/6 mice resistant to high fat diet induced weight gain involves an activation of mitochondrial oxidative pathways. J Proteome Res. (2013) 12:1956–68. doi: 10.1021/pr400051s
80. Mootha VK, Handschin C, Arlow D, Xie X, St PJ, Sihag S, et al. Erralpha and Gabpa/b specify PGC-1alpha-dependent oxidative phosphorylation gene: expression that is altered in diabetic muscle Proc Natl Acad Sci U S A. (2004) 101:6570–5. doi: 10.1073/pnas.0401401101
81. Vaughan RA, Garcia-Smith R, Gannon NP, Bisoffi M, Trujillo KA, Conn CA. Leucine treatment enhances oxidative capacity through complete carbohydrate oxidation and increased mitochondrial density in skeletal muscle cells. Amino Acids. (2013) 45:901–11. doi: 10.1007/s00726-013-1538-5
82. Abdul-Ghani MA, Muller FL, Liu Y, Chavez AO, Balas B, Zuo P, et al. Deleterious action of FA metabolites on ATP synthesis: possible link between lipotoxicity, mitochondrial dysfunction, and insulin resistance. Am J Physiol Endocrinol Metab. (2008) 295:E678–85. doi: 10.1152/ajpendo.90287.2008
83. Zhenyukh O, González Amor M, Rodrigues Diez RR, Esteban V, Ruiz Ortega M, Salaices M, et al. Branched-chain amino acids promote endothelial dysfunction through increased reactive oxygen species generation and inflammation. J Cell Mol Med. (2018) 22:4948–62. doi: 10.1111/jcmm.13759
84. Zhenyukh O, Civantos E, Ruiz-Ortega M, Sanchez MS, Vazquez C, Peiro C, et al. High concentration of branched-chain amino acids promotes oxidative stress, inflammation and migration of human peripheral blood mononuclear cells via mTORC1 activation. Free Radic Biol Med. (2017) 104:165–77. doi: 10.1016/j.freeradbiomed.2017.01.009
85. Senn JJ. Toll-like receptor-2 is essential for the development of palmitate-induced insulin resistance in myotubes. J Biol Chem. (2006) 281:26865–75. doi: 10.1074/jbc.M513304200
86. Lee JY, Ye J, Gao Z, Youn HS, Lee WH, Zhao L, et al. Reciprocal modulation of Toll-like receptor-4 signaling pathways involving MyD88: and phosphatidylinositol 3-kinase/AKT by saturated and polyunsaturated fatty acids. J Biol Chem. (2003) 278:37041–51. doi: 10.1074/jbc.M305213200
87. Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. (2011) 12:408–15. doi: 10.1038/ni.2022
88. Xu L, Lin X, Guan M, Zeng Y, Liu Y. Verapamil attenuated prediabetic neuropathy in high-fat diet-fed mice through inhibiting TXNIP-mediated apoptosis and inflammation. Oxid Med Cell Longev. (2019) 2019:1896041. doi: 10.1155/2019/1896041
89. Yan Y, Jiang W, Spinetti T, Tardivel A, Castillo R, Bourquin C, et al. Omega-3 fatty acids prevent inflammation and metabolic disorder through inhibition of NLRP3 inflammasome activation. Immunity. (2013) 38:1154–63. doi: 10.1016/j.immuni.2013.05.015
90. Valerio A. TNF- downregulates eNOS expression and mitochondrial biogenesis in fat and muscle of obese rodents. J Clin Invest. (2006) 116:2791–8. doi: 10.1172/JCI28570
91. Burrill JS, Long EK, Reilly B, Deng Y, Armitage IM, Scherer PE, et al. Inflammation and ER stress regulate branched-chain amino acid uptake and metabolism in adipocytes. Mol Endocrinol. (2015) 29:411–20. doi: 10.1210/me.2014-1275
92. Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. (2001) 414:799–806. doi: 10.1038/414799a
93. Li X, Wang X, Liu R, Ma Y, Guo H, Hao L, et al. Chronic leucine supplementation increases body weight and insulin sensitivity in rats on high-fat diet likely by promoting insulin signaling in insulin-target tissues Mol Nutr Food Res. (2013) 57:1067–79. doi: 10.1002/mnfr.201200311
94. Balage M, Dupont J, Mothe-Satney I, Tesseraud S, Mosoni L, Dardevet D. Leucine supplementation in rats induced a delay in muscle IR/PI3K signaling pathway associated with overall impaired glucose tolerance. J Nutr Biochem. (2011) 22:219–26. doi: 10.1016/j.jnutbio.2010.02.001
95. Zhang Y, Guo K, LeBlanc RE, Loh D, Schwartz GJ, Yu YH. Increasing dietary leucine intake reduces diet-induced obesity and improves glucose and cholesterol metabolism in mice via multimechanisms. Diabetes. (2007) 56:1647–54. doi: 10.2337/db07-0123
96. Bernard JR, Liao Y, Hara D, Ding Z, Chen C, Nelson JL, et al. An amino acid mixture improves glucose tolerance and insulin signaling in Sprague-Dawley rats. Am J Physiol Endocrinol Metab. (2011) 300:E752–60. doi: 10.1152/ajpendo.00643.2010
97. Saha AK, Xu XJ, Lawson E, Deoliveira R, Brandon AE, Kraegen EW, et al. Downregulation of AMPK accompanies leucine- and glucose-induced increases in protein synthesis and insulin resistance in rat skeletal muscle. Diabetes. (2010) 59:2426–34. doi: 10.2337/db09-1870
98. Xiao F, Huang Z, Li H, Yu J, Wang C, Chen S, et al. Leucine deprivation increases hepatic insulin sensitivity via GCN2/mTOR/S6K1 and. AMPK pathways Diabetes. (2011) 60:746–56. doi: 10.2337/db10-1246
99. Gao Z, Zhang X, Zuberi A, Hwang D, Quon MJ, Lefevre M, et al. Inhibition of insulin sensitivity by free fatty acids requires activation of multiple serine kinases in 3T3-L1 adipocytes. Mol Endocrinol. (2004) 18:2024–34. doi: 10.1210/me.2003-0383
100. Pereira S, Park E, Mori Y, Haber CA, Han P, Uchida T, et al. FFA-induced hepatic insulin resistance in vivo is mediated by PKCδ, NADPH oxidase, and oxidative stress. Am J Physiol Endocrinol Metab. (2014) 307:E34–46. doi: 10.1152/ajpendo.00436.2013
101. Chriett S, Zerzaihi O, Vidal H, Pirola L. The histone deacetylase inhibitor sodium butyrate improves insulin signalling in palmitate-induced insulin resistance in L6 rat muscle cells through epigenetically-mediated up-regulation of Irs1. Mol Cell Endocrinol. (2017) 439:224–32. doi: 10.1016/j.mce.2016.09.006
102. Holte J, Bergh T, Berne C, Lithell H. Serum lipoprotein lipid profile in women with the polycystic ovary syndrome: relation to anthropometric, endocrine and metabolic variables. Clin Endocrinol. (1994) 41:463–71. doi: 10.1111/j.1365-2265.1994.tb02577.x
103. Zhang CM, Zhao Y, Li R, Yu Y, Yan LY, Li L, et al. Metabolic heterogeneity of follicular amino acids in polycystic ovary syndrome is affected by obesity and related to pregnancy outcome. BMC Pregnancy Childbirth. (2014) 14:11. doi: 10.1186/1471-2393-14-11
104. Lyon ES, Rivera ME, Johnson MA, Sunderland KL, Vaughan RA. Actions of chronic physiological 3-hydroxyisobuterate treatment on mitochondrial metabolism and insulin signaling in myotubes. Nutr Res. (2019) 66:22–31. doi: 10.1016/j.nutres.2019.03.012
105. Haufe S, Engeli S, Kaminski J, Witt H, Rein D, Kamlage B, et al. Branched-chain amino acid catabolism rather than amino acids plasma concentrations is associated with diet-induced changes in insulin resistance in overweight to obese individuals. Nutr Metab Cardiovasc Dis. (2017) 27:858–64. doi: 10.1016/j.numecd.2017.07.001
106. Lynch CJ, Adams SH. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat Rev Endocrinol. (2014) 10:723–36. doi: 10.1038/nrendo.2014.171
107. Muoio DM, Newgard CB. Molecular and metabolic mechanisms of insulin resistance and β-cell failure in type 2 diabetes. Nat Rev Mol Cell Bio. (2008) 9:193–205. doi: 10.1038/nrm2327
108. Yaribeygi H, Farrokhi FR, Butler AE, Sahebkar A. Insulin resistance: review of the underlying molecular mechanisms. J Cell Physiol. (2018) 234:8152–61. doi: 10.1002/jcp.27603
109. Matulewicz N, Karczewska-Kupczewska M. Insulin resistance and chronic inflammation. Postepy Hig Med Dosw. (2016) 70:1245–58. doi: 10.5604/17322693.1226662
110. de Alvaro C, Teruel T, Hernandez R, Lorenzo M. Tumor necrosis factor α produces insulin resistance in skeletal muscle by activation of inhibitor κB kinase in a p38 MAPK-dependent manner. J Biol Chem. (2004) 279:17070–8. doi: 10.1074/jbc.M312021200
111. Hernandez R, Teruel T, de Alvaro C, Lorenzo M. Rosiglitazone ameliorates insulin resistance in brown adipocytes of Wistar rats by impairing TNF-α induction of p38 and p42/p44 mitogen-activated protein kinases. Diabetologia. (2004) 47:1615–24. doi: 10.1007/s00125-004-1503-7
112. Jager J, Grémeaux T, Cormont MY, Le Marchand-Brustel Y, Tanti J. Interleukin-1β-induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology. (2007) 148:241–51. doi: 10.1210/en.2006-0692
113. Lagathu C, Bastard JP, Auclair M, Maachi M, Capeau J, Caron M. Chronic interleukin-6 (IL-6) treatment increased IL-6 secretion and induced insulin resistance in adipocyte: prevention by rosiglitazone. Biochem Biophys Res Commun. (2003) 311:372–9. doi: 10.1016/j.bbrc.2003.10.013
114. Gaggini M, Carli F, Rosso C, Buzzigoli E, Marietti M, Della Latta V, et al. Altered amino acid concentrations in NAFLD: impact of obesity and insulin resistance. Hepatology. (2018) 67:145–58. doi: 10.1002/hep.29465
115. Lackey DE, Lynch CJ, Olson KC, Mostaedi R, Ali M, Smith WH, et al. Regulation of adipose branched-chain amino acid catabolism enzyme expression and cross-adipose amino acid flux in human obesity Am J Physiol Endocrinol Metab. (2013) 304:E1175–87. doi: 10.1152/ajpendo.00630.2012
116. White PJ, McGarrah RW, Grimsrud PA, Tso S, Yang W, Haldeman JM, et al. The BCKDH kinase and phosphatase integrate BCAA and lipid metabolism via regulation of ATP-citrate lyase. Cell Metab. (2018) 27:1281–93.e7. doi: 10.1016/j.cmet.2018.04.015
117. Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. (2005) 365:1415–28. doi: 10.1016/S0140-6736(05)66378-7
118. Bessone F, Razori MV, Roma MG. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell Mol Life Sci. (2019) 76:99–128. doi: 10.1007/s00018-018-2947-0
119. Luft VC, Schmidt MI, Pankow JS, Couper D, Ballantyne CM, Young JH, et al. Chronic inflammation role in the obesity-diabetes association: a case-cohort study. Diabetol Metab Syndr. (2013) 5:31. doi: 10.1186/1758-5996-5-31
120. Emanuela F, Grazia M, Marco DR, Maria PL, Giorgio F, Marco B. Inflammation as a link between obesity and metabolic syndrome. J Nutr Metab. (2012) 2012:476380. doi: 10.1155/2012/476380
121. Calder PC. Functional roles of fatty acids and their effects on human health. JPEN J Parenter Enteral Nutr. (2015) 39(Suppl. 1):18–32S. doi: 10.1177/0148607115595980
122. Niu Z, Lin N, Gu R, Sun Y, Feng Y. Associations between insulin resistance, free fatty acids, and oocyte quality in polycystic ovary syndrome during in vitro fertilization. J Clin Endocrinol Metab. (2014) 99:E2269–76. doi: 10.1210/jc.2013-3942
123. Zhao H, Zhao Y, Li T, Li M, Li J, Li R, et al. Metabolism alteration in follicular niche: the nexus among intermediary metabolism, mitochondrial function, and classic polycystic ovary syndrome Free Radic Biol Med. (2015) 86:295–307. doi: 10.1016/j.freeradbiomed.2015.05.013
124. Serra D, Mera P, Malandrino MI, Mir JF, Herrero L. Mitochondrial fatty acid oxidation in obesity. Antioxid Redox Signal. (2013) 19:269–84. doi: 10.1089/ars.2012.4875
125. Cheng Z, Tseng Y, White MF. Insulin signaling meets mitochondria in metabolism. Trends Endocrinol Metab. (2010) 21:589–98. doi: 10.1016/j.tem.2010.06.005
126. Hu W, Qiao J, Yang Y, Wang L, Li R. Elevated C-reactive protein and monocyte chemoattractant protein-1 in patients with polycystic ovary syndrome. Eur J Obstet Gynecol Reprod Biol. (2011) 157:53–6. doi: 10.1016/j.ejogrb.2011.03.015
127. Kelly CC, Lyall H, Petrie JR, Gould GW, Connell JM, Sattar N. Low grade chronic inflammation in women with polycystic ovarian syndrome. J Clin Endocrinol Metab. (2001) 86:2453–5. doi: 10.1210/jcem.86.6.7580
128. Zhao Y, Zhang C, Huang Y, Yu Y, Li R, Li M, et al. Up-regulated expression of WNT5a increases inflammation and oxidative stress via. PI3K/AKT/NF-kappaB signaling in the granulosa cells of PCOS patients. J Clin Endocrinol Metab. (2015) 100:201–11. doi: 10.1210/jc.2014-2419
Keywords: branched-amino acids, fatty acids, energy metabolism, inflammation, insulin resistance, mitochondrial biogenesis
Citation: Ye Z, Wang S, Zhang C and Zhao Y (2020) Coordinated Modulation of Energy Metabolism and Inflammation by Branched-Chain Amino Acids and Fatty Acids. Front. Endocrinol. 11:617. doi: 10.3389/fendo.2020.00617
Received: 05 May 2020; Accepted: 28 July 2020;
Published: 08 September 2020.
Edited by:
Maria Clara Franco, Oregon State University, United StatesReviewed by:
Charles E. McCall, Wake Forest Baptist Medical Center, United StatesCopyright © 2020 Ye, Wang, Zhang and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yue Zhao, emhhb3l1ZTA2MzBAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.