 Kripa Shankar1
Kripa Shankar1 Deepali Gupta1Bharath K. Mani1Brianna G. Findley1
Deepali Gupta1Bharath K. Mani1Brianna G. Findley1 Sherri Osborne-Lawrence1
Sherri Osborne-Lawrence1 Nathan P. Metzger1Chen Liu1,2Eric D. Berglund1Jeffrey M. Zigman1,3,4*
Nathan P. Metzger1Chen Liu1,2Eric D. Berglund1Jeffrey M. Zigman1,3,4*- 1Department of Internal Medicine, Center for Hypothalamic Research, UT Southwestern Medical Center, Dallas, TX, United States
- 2Department of Neuroscience, UT Southwestern Medical Center, Dallas, TX, United States
- 3Division of Endocrinology, Department of Internal Medicine, UT Southwestern Medical Center, Dallas, TX, United States
- 4Department of Psychiatry, UT Southwestern Medical Center, Dallas, TX, United States
Insulin-induced hypoglycemia is a major limiting factor in maintaining optimal blood glucose in patients with type 1 diabetes and advanced type 2 diabetes. Luckily, a counterregulatory response (1) system exists to help minimize and reverse hypoglycemia, although more studies are needed to better characterize its components. Recently, we showed that the hormone ghrelin is permissive for the normal CRR to insulin-induced hypoglycemia when assessed in mice without diabetes. Here, we tested the hypothesis that ghrelin also is protective against insulin-induced hypoglycemia in the streptozotocin (2) mouse model of type 1 diabetes. STZ-treated ghrelin-knockout (KO) (3) mice as well as STZ-treated wild-type (WT) littermates were subjected to a low-dose hyperinsulinemic-hypoglycemic clamp procedure. The STZ-treated ghrelin-KO mice required a much higher glucose infusion rate than the STZ-treated WT mice. Also, the STZ-treated ghrelin-KO mice exhibited attenuated plasma epinephrine and norepinephrine responses to the insulin-induced hypoglycemia. Taken together, our data suggest that without ghrelin, STZ-treated mice modeling type 1 diabetes are unable to mount the usual CRR to insulin-induced hypoglycemia.
Introduction
Insulin-induced hypoglycemia is prevalent in type 1 diabetes and advanced type 2 diabetes (4). Given the high risk of morbidity and mortality associated with hypoglycemia, mammals have developed a highly integrated counterregulatory response (CRR) system to help prevent, minimize, and reverse hypoglycemia. In individuals without diabetes, this CRR system is mobilized to varying degrees during progression from fasting to starvation. As reviewed by Cryer (5), in humans, the first defense within the traditional CRR involves decreased insulin secretion, which disinhibits glycogenolysis and gluconeogenesis and reduces glucose uptake into muscle and fat. The second defense involves increased glucagon release, stimulating hepatic glycogenolysis and gluconeogenesis. The third defense is an increase in epinephrine, resulting in higher delivery of gluconeogenic substrates to the liver and inhibition of whole body glucose utilization and insulin release. Fourth and fifth defenses include cortisol and growth hormone (GH) rises, which mobilize if hypoglycemia persists and work by limiting glucose utilization and stimulating gluconeogenesis. A sixth defense is activation of the sympathetic nervous system, which, in conjunction with adrenal activation, is linked to a constellation of warning symptoms that include tachycardia, tremors, anxiety, irritability, arousal, sweating, and hunger. Many of these responses are influenced directly or indirectly via glucose-sensing neurons in the central nervous system (6).
Importantly, this CRR system is often compromised in diabetic patients experiencing insulin-induced hypoglycemia (5). For instance, an attenuated sympathoadrenal response may occur, manifesting as hypoglycemia-associated autonomic failure and a markedly increased risk for severe hypoglycemia (7). The normal fine-tuning of insulin release is not an option due to β-cell failure. Also, α-cell dysregulation may occur, due to a lack of functional β-cells in the diabetic islet, and thus loss of normal tonic inhibition of α-cells by intra-islet insulin. This can lead to a reduced glucagon response to insulin-induced hypoglycemia (8).
Although the CRR during insulin-induced hypoglycemia in the context of diabetes has been long appreciated, it is not fully understood and also likely includes other hormones besides those originally described by Cryer. As an example, the stomach-derived hormone ghrelin has many known glucoregulatory actions in both rodent models and humans and seems well-suited to participate in the CRR. Administration of ghrelin (which henceforth mainly refers to the acylated form of ghrelin) reduces insulin sensitivity, restricts insulin secretion, stimulates glucagon secretion and GH release, and raises circulating cortisol (9–12). These interactions, as well as those with GLP-1 (13), and ghrelin receptor (GHSR)-expressing neurons in the hypothalamic arcuate nucleus and caudal brainstem (1, 14), likely contribute to ghrelin's overall glucoregulatory effects, which are emphasized by the actions of administered ghrelin to increase blood glucose (11, 12, 15–20) and conversely, by the blood glucose-lowering effects of ghrelin deletion or blockade, as reviewed in (9, 10).
Regarding the latter, chronic pharmacological blockade of GHSRs or genetic ablation of other ghrelin system components improve glucose tolerance in diet-induced obese mice (21–26). An intact ghrelin system also is required to prevent development of severe hypoglycemia and resulting death in a mouse starvation model. More specifically, ghrelin-KO mice exhibit a progressive decline in fasting blood glucose to the point of near-death following a week-long 60% caloric restriction regimen that depletes body fat to <2% (27). Hypoglycemia under this regimen also occurs in mice with ablated ghrelin cells, mice deficient in ghrelin O-acyltransferase, mice overexpressing the endogenous GHSR antagonist LEAP2, GHSR-null mice, and mice with ghrelin cell-selective deletion of β1-adrenergic receptors (14, 24, 28–30).
A functional ghrelin system also appears to be important in various diabetic models, as reviewed in (10). As examples, ghrelin deletion markedly attenuates hyperglycemia in leptin-deficient (ob/ob) mice, which are hyperphagic, obese, and diabetic (31). GHSR antagonist administration normalizes blood glucose in otherwise hyperglycemic HNF1α-deficient mice – a model of maturity-onset diabetes of the young type 3, which is associated with elevated plasma ghrelin (32, 33). Administration of a GHSR inverse agonist improves glucose tolerance in Zucker diabetic fatty (ZDF) rats (25). Also, type 1 diabetes as modeled in rats and mice by chemical ablation of pancreatic β-cells with streptozotocin (STZ) causes elevation of plasma ghrelin (34–41). Genetic ablation of ghrelin or pharmacological inhibition of GHSR cause significant reductions in STZ-associated hyperphagia (36, 38–40, 42). Furthermore, genetic ablation of GHSR lowers fasting blood glucose in STZ-treated mice (42).
In a recent study, we specifically investigated the contributions of ghrelin to the CRR to insulin-induced hypoglycemia using mice without diabetes (41). We showed that ghrelin-KO mice exhibit more pronounced and prolonged hypoglycemia than WT littermates when administered the same insulin dose in the form of a single bolus (41). Also, ghrelin-KO mice required a much (10-fold) higher glucose infusion rate (GIR) to maintain the same target blood glucose as WT littermates during low-dose hyperinsulinemic-hypoglycemic clamps (41), similar to the findings in GHSR-KO mice (43). Ghrelin-KO mice also exhibited less robust corticosterone and GH responses than their WT counterparts during the clamps (41). Conversely, ghrelin receptor agonist (HM01) administration, which reduced the GIR required by ghrelin-KO mice during the clamps, increased plasma corticosterone and plasma GH (41).
Collectively, these data suggest that endogenously-produced ghrelin not only influences insulin sensitivity, but also is permissive for the normal CRR to insulin-induced hypoglycemia. As the CRR is altered in diabetes (see above), we undertook the current study in order to investigate the protective actions of ghrelin during insulin-induced hypoglycemia in the STZ model of type 1 diabetes.
Materials and Methods
Animals
All animal experiments performed in this study were approved by the UT Southwestern Medical Center Institutional Animal Care and Use Committee. Ghrelin-KO mice [line GKO1 (41, 44)] and WT littermates on a C57BL/6N background were generated by pairing mice heterozygous for the ghrelin-KO allele. Ghrelin-KO mice contain two copies of the ghrelin-KO allele whereas WT littermates instead contain two copies of the wild-type ghrelin allele. Mice were housed at room temperature (21.5–22.5°C) using a 12 h light-dark cycle and were provided ad lib access to water and regular chow [2016 Teklad Global 16% Protein Rodent Diet (Envigo, Indianapolis, IN)], except as noted.
Jugular Vein Catheterization
Eight to ten week-old male mice were anesthetized using 2% isoflurane, and each was then surgically implanted with a right jugular vein catheter (0.20-inch, Silastic tubing) (Instech Laboratories, Plymouth Meeting, PA). The free end of the catheter was exteriorized from the dorsal intrascapular region, and the incision sites were closed with a 5–0 nylon suture. Mice were fitted with a vascular harness (Instech Laboratories). Mice were provided ketoprofen (5.25 mg/kg s.c.) immediately, 24, and 48 h post-surgically for analgesia. Mice also were closely monitored post-surgically for signs of infection or swelling, neither of which were observed.
Administration of STZ
To model type 1 diabetes mellitus, 2 d following jugular vein catheterization, mice were injected once with STZ (150 mg/kg i.p.; Sigma-Aldrich, St. Louis, MO) freshly dissolved in sodium citrate buffer (0.1 M, pH~4.5). Drinking water containing 10% dextrose was provided for 24 h in petri dishes on the floors of the home cages to preclude development of transient hypoglycemia. Ad lib-fed blood glucose levels were checked before administration of STZ and 3 d later via tail nick using a Contour Next EZ glucometer system (Ascensia Diabetes Care, Parsippany, NJ). Mice with blood glucoses > 200 mg/dL were used for clamp studies. Only two mice (both of which were ghrelin-KO mice) were excluded from the clamp studies due to day 3 post-STZ injection blood glucoses ≤ 200 mg/dL. Blood glucoses at or above the maximum detection limit (600 mg/dL) were noted as 600 mg/dL for quantification purposes.
Low-Dose Hyperinsulinemic–Hypoglycemic Clamp Procedure
Hyperinsulinemic-hypoglycemic clamps were performed in conscious, unrestrained mice 4 d after STZ injection as described previously (41), using modifications that took into account their baseline hyperglycemia in the ad lib-fed state. Mice were fasted overnight for 16 h before starting the clamp procedure at 10:00 AM (with access to water until 9:00 AM). This 16 h fast preceding the clamp studies was longer than that used previously in non-STZ-treated mice (5 h) (41) as it was needed to achieve the target hypoglycemic range using the low-dose insulin. During the clamp, Humulin-R insulin was infused over 2 h at a constant rate of 4 mU/kg/min i.v. Simultaneously, a solution of 20% glucose (prepared using pharmaceutical grade 50% dextrose diluted in sterile saline) was infused i.v. at a variable rate to achieve and maintain blood glucose levels within a target range of 35–45 mg/dL during the final 20 min of the 2-h clamp procedure. Blood glucose was measured via tail nicks every 5 min, as above. Blood samples to measure ghrelin were taken from tail nicks at t = −5 and t = 120 min. At t = 120 min, blood samples to measure insulin, epinephrine, norepinephrine, glucagon, corticosterone, and GH were collected by cardiac puncture from mice that had been anesthetized with isoflurane.
Histologic Assessment of Islets
A separate group of ghrelin-KO and WT littermates (n = 5–7) received a single i.p. injection of vehicle (0.1 M sodium citrate buffer, pH~4.5) or STZ (150 mg/Kg; freshly prepared, as above). Three days later, blood glucose levels of mice in the ad lib-fed state were measured, as above, and blood to assess plasma insulin was collected by quick superficial temporal vein bleed. On the same day, mice were deeply anesthetized with chloral hydrate, transcardially perfused with formalin, and then processed for islet histology. Eight μm-thick pancreatic sections were cut on a cryostat at 50-μm intervals, mounted, and then assessed for both insulin-immunoreactivity (red) and glucagon-immunoreactivity (green). Incubations with primary antibodies and secondary antibodies [guinea pig anti-Insulin (DakoCytomation, Carpinteria, CA; diluted 1:300) followed by Alexa Fluor 594® donkey anti-guinea pig IgG (ThermoFisher Scientific; 1:500); rabbit anti-Glucagon (Millipore, Temecula, CA; diluted 1:300) followed by Alexa Fluor 488® donkey anti-rabbit IgG antibody (ThermoFisher Scientific; 1:500)] were performed as in (42). Three mice of each genotype and treatment were chosen for further analysis. Digital images of labeled islets were taken using the 20X objective of an Olympus BX41. Subjective assessments of islet anatomy were made using 3–4 representative islets from each mouse.
Determination of Plasma Hormone Levels
For ghrelin, blood was collected into ice-cold EDTA-coated microfuge tubes. P-hydroxymercuribenzoic acid (final concentration 1 mmol/L; Sigma-Aldrich) was added, plasma was isolated following centrifugation, and HCl was added to achieve a final concentration of 0.1 nmol/L. For other hormones, blood was collected into 3 different EDTA-coated microfuge tubes. For glucagon, aprotinin (final concentration 250 KIU/ml; Sigma-Aldrich) was added. For catecholamines, EDTA-glutathione solution (9% w/v EDTA and 6% w/v glutathione, pH 7.4; 2 μL per 100 μL blood) was added. For insulin, no reagents were added.
ELISA kits were used for acyl-ghrelin (Millipore-Merck; Burlington, MA), insulin (Crystal Chem, Downers Grove, IL), and glucagon (Mercodia AB, Uppsala, Sweden). Calorimetric assays were performed using a BioTek PowerWave XS Microplate spectrophotometer (Winooski, VT) and BioTek KC4 junior software. Plasma catecholamines were determined using HPLC at the Vanderbilt University Medical Center Hormone Assay and Analytical Services Core.
Statistical Analyses
All statistical analyses and graph preparations were performed using GraphPad Prism 7.0. A Student's t-test, 1-way, or 2-way ANOVA were used to test for significant differences among test groups. Data with significant unequal variance were log transformed prior to performing analyses. Outliers, if any, were detected by the ROUT test.
Results
STZ Induces Diabetes in Both WT and Ghrelin-KO Mice
A single high-dose of STZ (150 mg/Kg, i.p.) was administered to both ghrelin-KO mice and WT littermates in order to model type 1 diabetes mellitus. This administration protocol, which is based on one previously published (45–49) markedly reduced pancreatic β-cell mass (as assessed by comparing insulin-immunoreactivity in representative islets from vehicle-treated mice to insulin-immunoreactivity in islets from STZ-treated mice) in both genotypes without causing any subjective genotype-dependent differences in resulting islet shape, islet size, or patterns of insulin-immunoreactivity and glucagon-immunoreactivity (Figure 1A, Supplementary Figure 1). Plasma insulin levels also were equivalently reduced by STZ in WT and ghrelin-KO littermates (Figure 1B), while corresponding blood glucose levels were raised (Figure 1C). Thus, STZ efficaciously destroyed pancreatic β-cells in both WT and ghrelin-KO littermates—sufficiently enough to induce diabetes in both genotypes but, as expected (42, 49–52), without causing a complete disappearance of β-cells. Of interest, despite the induction of hyperglycemia in both genotypes, the mean blood glucose attained in ghrelin-KO mice was lower than that observed in WT mice (Figure 1C), similar to previously-reported effects of STZ in GHSR-null mice (42).
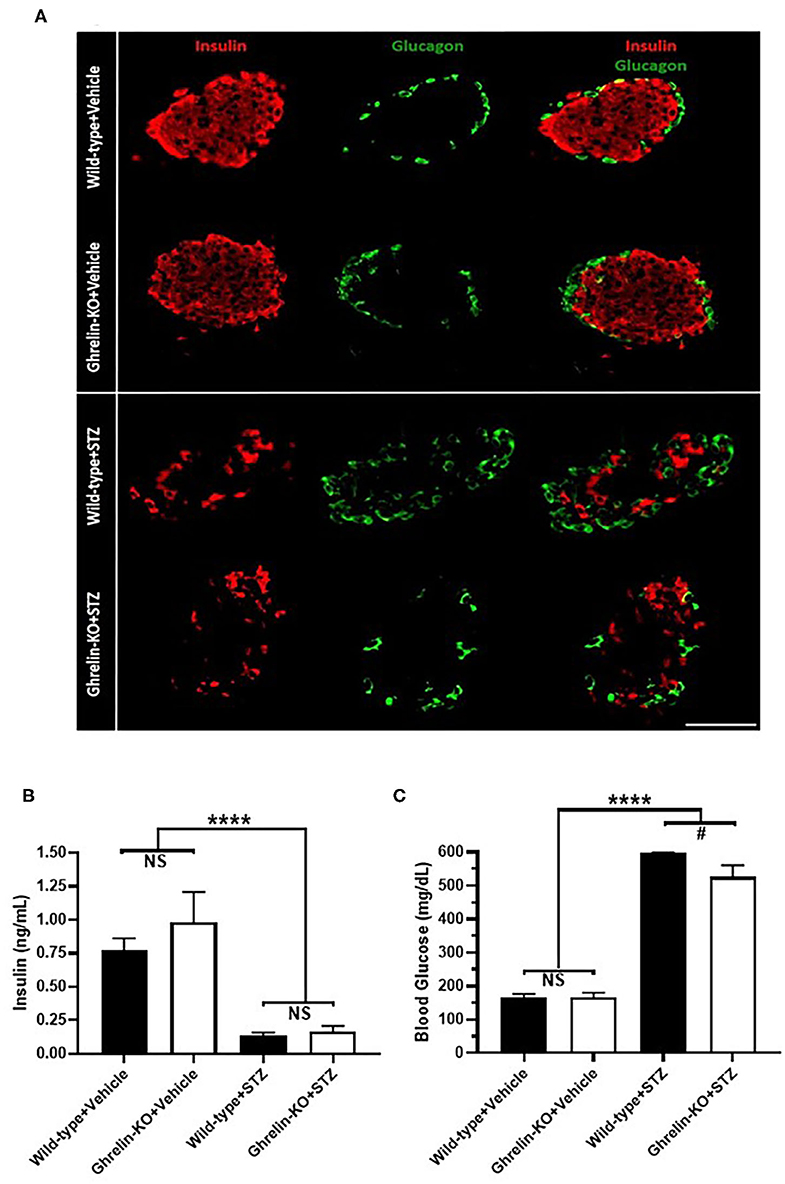
Figure 1. Effects of STZ on islets, plasma insulin, and blood glucose in WT and ghrelin-KO mice. (A) A representative islet co-labeled for insulin-immunoreactivity (red) and glucagon-immunoreactivity (green) from a vehicle-treated WT mouse (1st row), a vehicle-treated ghrelin-KO mouse (2nd row), an STZ-treated WT mouse (3rd row), and an STZ-treated ghrelin-KO mouse (4th row). Scale bar = 100 μm. (B) Ad lib-fed plasma insulin levels and (C) corresponding blood glucose levels in vehicle- and STZ-treated mice. Data are presented as mean ± SEM. n = 5–7. ****P < 0.0001, #P = 0.09, NS, no significant difference.
STZ-Treated Ghrelin-KO Mice Are More Susceptible to Hypoglycemia During Hyperinsulinemic-Hypoglycemic Clamps
Next, we subjected a separate cohort of STZ-treated ghrelin-KO and WT littermates to a hyperinsulinemic-hypoglycemic protocol to assess the CRR. Just as shown in the above cohort (Figure 1C), STZ induced hyperglycemia in both WT and ghrelin-KO mice when assessed 3 d following STZ administration in the ad lib-fed state (Figure 2A). Also similar to the above cohort, the degree of hyperglycemia induced in ghrelin-KO mice was lower than that induced in their WT littermates (Figure 2A). Following a 16 h-fast to prepare the mice for the clamps, blood glucose levels fell in both genotypes, and remained lower in the ghrelin-KO mice than in the WT mice (Figure 2B). A low insulin dose protocol (4 mU/kg/min) was used, as in (41), to minimize direct inhibitory effects of insulin on ghrelin secretion in WT mice, while still achieving hypoglycemia. This low-dose insulin protocol reduced blood glucose in both STZ-treated WT and STZ-treated ghrelin-KO mice, such that within the final 20 min of the procedure, both genotypes were successfully clamped within the target hypoglycemic range (~35–45 mg/dL), with blood glucose levels that were without statistically significant differences (Figure 2B). Notably, a much higher (~5.8-fold) GIR was required by ghrelin-KO mice during the final 20 min of the clamps to maintain a similar blood glucose to that of WT littermates (Figure 2C).
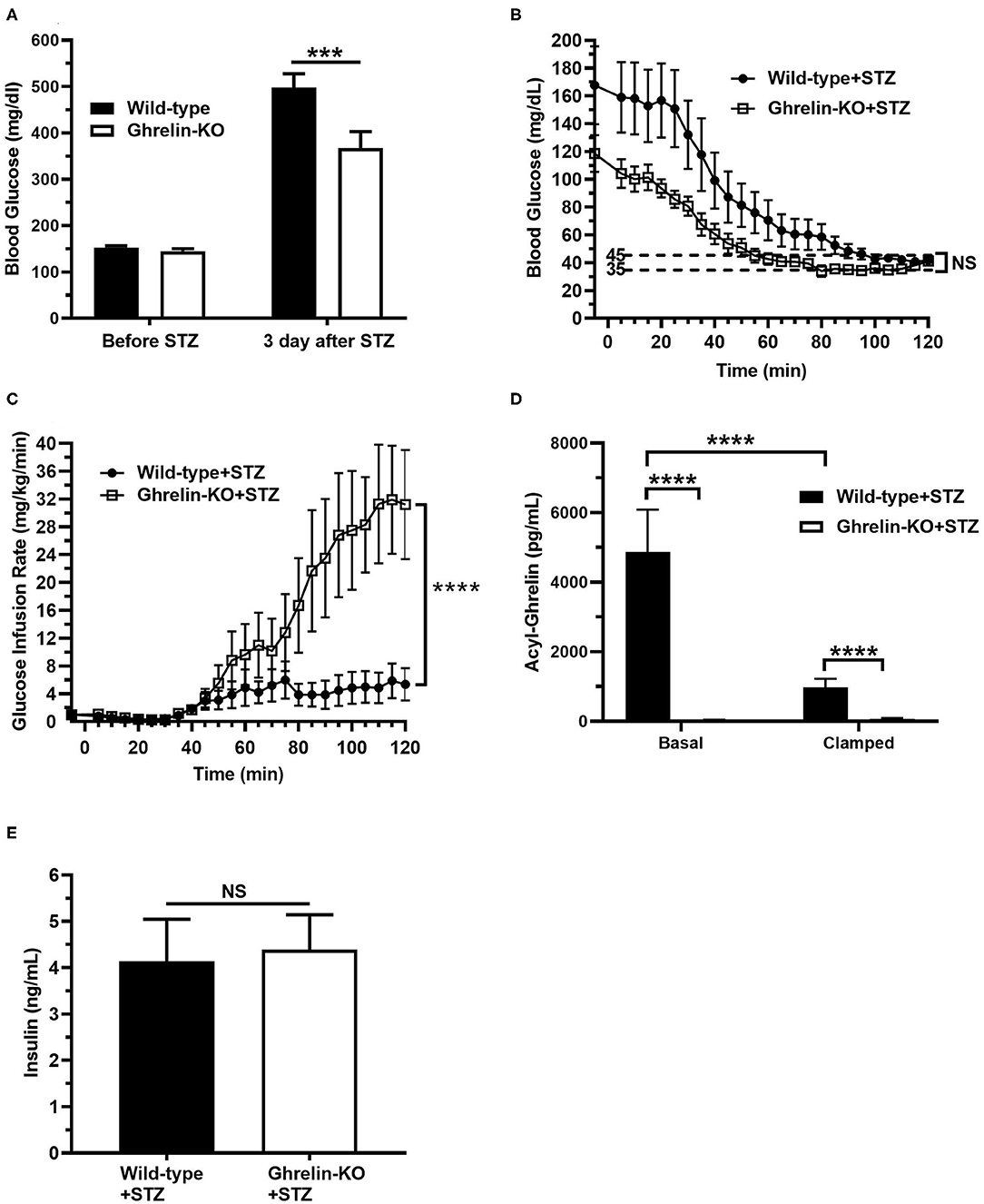
Figure 2. Low-dose hyperinsulinemic–hypoglycemic clamps in STZ-treated mice. (A) Ad lib-fed blood glucose levels before and 3 d after STZ administration. (B) Blood glucose levels during the clamps. (C) Glucose infusion rate (GIR) to achieve the target blood glucose (35–45 mg/dL) by the steady-state period (100–120 min) in mice receiving Humulin-R insulin (4 mU/kg/min) from 0 to 120 min. (D) Plasma ghrelin levels at the start (“Basal”; t = −5 min) and end (“Clamp”; t = 120 min) of the clamps. (E) Plasma insulin levels at the end (“Clamp”; t = 120 min) of the clamps. n = 10–11. Data are presented as mean ± SEM. ***P < 0.001, ****P < 0.0001, NS, no significant difference.
We also determined plasma ghrelin and insulin levels. Basal plasma ghrelin in STZ-treated WT mice (Figure 2D) was significantly higher than levels usually observed in non-STZ-treated WT mice, as expected (34–41). Also as expected from other studies that examined the effects of insulin on plasma ghrelin or ghrelin secretion (41, 53–57), plasma ghrelin fell (~79%) in STZ-treated WT mice over the course of the clamp (Figure 2D). Nonetheless, plasma ghrelin remained much higher than the essentially undetectable level in ghrelin-KO littermates (Figure 2D). Also, plasma insulin levels were genotype-independent at the end of clamps, reflecting similar levels of endogenous plus infused insulin in the WT and ghrelin-KO mice (Figure 2E).
The CRR Differs in STZ-Treated Ghrelin-KO and WT Littermates
Finally, we assessed the levels of the traditional CRR hormones epinephrine, norepinephrine, glucagon, corticosterone, and GH at the end of the hyperinsulinemic-hypoglycemic clamps. Plasma epinephrine and norepinephrine were significantly lower in ghrelin-KO mice (Figures 3A,B). There were no genotype-dependent differences in end-of-clamp levels of plasma glucagon, corticosterone, and GH (Figures 3C–E).
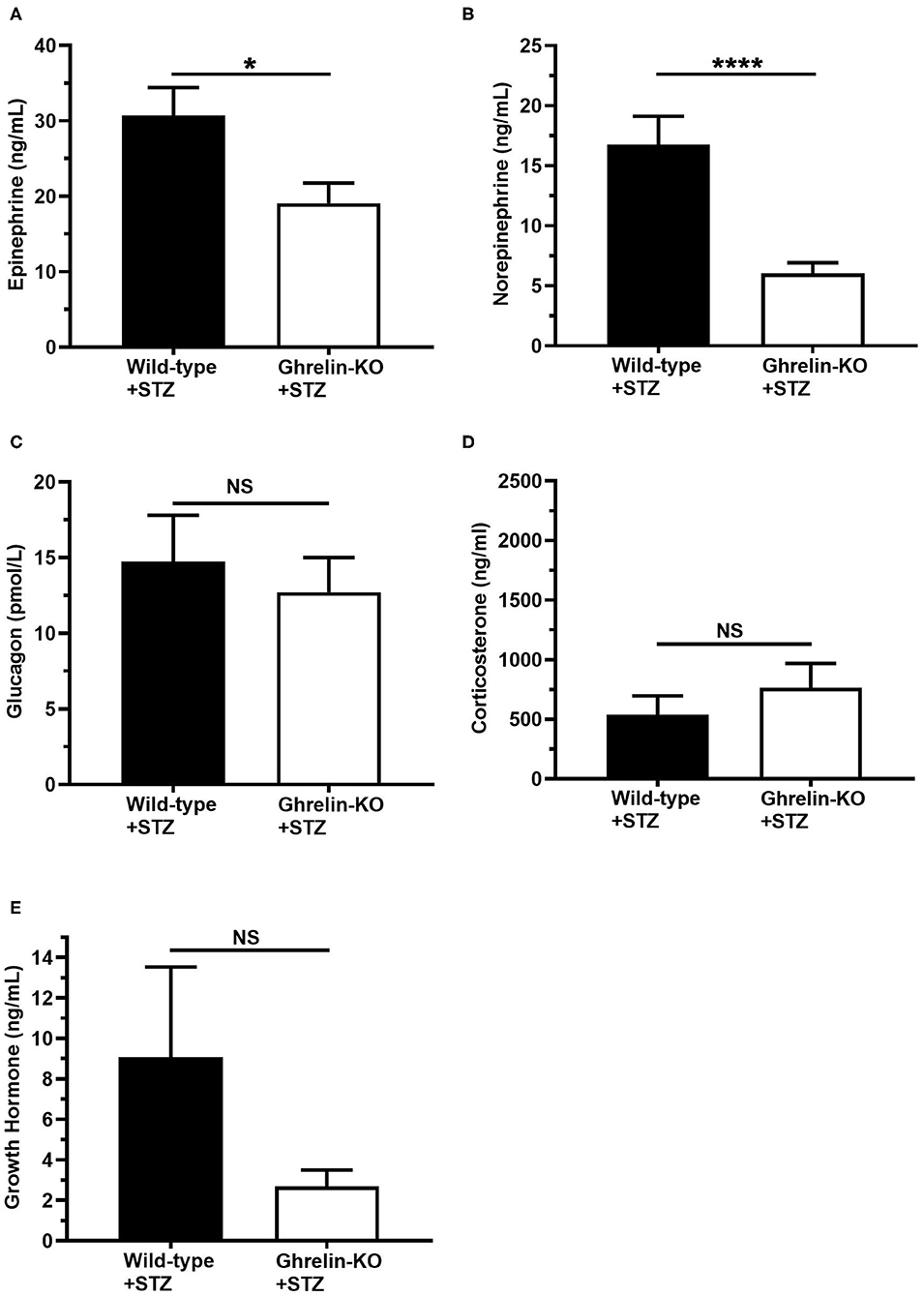
Figure 3. Effects of ghrelin deletion on plasma CRR hormone levels obtained at the end of the hyperinsulinemic-hypoglycemic clamps. Plasma levels of (A) epinephrine, (B) norepinephrine, (C) glucagon, (D) corticosterone, and (E) GH at the end of the clamps (t = 120 min). n = 10–11. Data are presented as mean ± SEM. *P < 0.05, ****P < 0.0001, NS, no significant difference.
Discussion
The current study was designed to characterize the CRR actions of ghrelin during insulin-induced hypoglycemia in the setting of type 1 diabetes. Our new data indicate that ghrelin expression is permissive for the usual CRR to insulin-induced hypoglycemia in mice in which diabetes was induced by STZ. In particular, during hyperinsulinemic-hypoglycemic clamps, diabetic ghrelin-KO mice required a ~5.8-fold higher GIR to maintain blood glucoses in the target hypoglycemic range as compared to WT littermates. Previously, higher GIRs had been shown to be required by both non-diabetic ghrelin-KO and GHSR-KO mice as compared to WT mice during hyperinsulinemic-hypoglycemic clamps (41, 43). Interestingly, as compared to a previous study in which non-diabetic ghrelin-KO mice exhibited less robust corticosterone and GH responses during hyperinsulinemic-hypoglycemic clamps than WT littermates (41), the corticosterone and GH responses observed here in clamped diabetic ghrelin-KO and WT mice were without statistically significant differences. Instead, upon induction of hypoglycemia, the diabetic ghrelin-KO mice exhibited attenuated epinephrine and norepinephrine responses as compared to WT littermates.
There are several notable topics of discussion that these new data bring to mind. One relates to the demonstration of important actions by ghrelin during the CRR to insulin-induced hypoglycemia, now newly shown using the STZ model of type 1 diabetes. Just as had been observed in non-diabetic mice (41, 43), the elevated GIR required during the hyperinsulinemic-hypoglycemic clamps in STZ-treated ghrelin-KO mice as compared to WT littermates again suggests that the presence of circulating ghrelin is permissive for the usual CRR to insulin-induced hypoglycemia. This role for ghrelin during the CRR to insulin-induced hypoglycemia aligns with its other well-described glucoregulatory actions. These include effects of administered ghrelin and endogenous plasma ghrelin elevations to increase blood glucose (9, 10) and numerous examples of endogenously-produced ghrelin preventing falls in blood glucose—including life-threatening falls in blood glucose in certain settings, as reviewed in (10).
Our new data also highlight another observation previously made in insulin-induced hypoglycemic, non-diabetic mice—namely, that despite ghrelin expression allowing the usual CRR to insulin-induced hypoglycemia, plasma ghrelin levels do not elevate in STZ-treated WT mice upon hypoglycemia induction, as occurs with the traditional CRR hormones. Instead, plasma ghrelin levels fall over the course of the hyperinsulinemic-hypoglycemic clamps in the STZ-treated WT mice, just as occurs in non-STZ-treated mice (41). Nevertheless, despite this drop in plasma ghrelin, the remaining circulating ghrelin continues to serve a crucial permissive function during the CRR, as emphasized by the requirement for a markedly higher GIR and the attenuated changes to epinephrine and norepinephrine in clamped STZ-treated ghrelin-KO mice.
Another noteworthy topic of discussion relates to these lower epinephrine and norepinephrine levels. This finding suggests that the sympathoadrenal arms of CRR in the STZ-treated ghrelin-KO mice are blunted and potentially might contribute to their requirement for a higher GIR during the clamps. This blunted catecholamine response may result, at least in part, from decreased stimulation of hypothalamic corticotropin releasing factor (CRF) neurons. Notably, ghrelin induces c-fos expression and increases CRF expression in hypothalamic CRF neurons, induces CRF release from hypothalamic explants, and increases plasma ACTH and epinephrine in rodent and/or human subjects (58–63). Also relevant, epinephrine and norepinephrine are both potent ghrelin secretagogues, acting via ghrelin cell-expressed β1-adrenergic receptors to stimulate ghrelin secretion (30, 64). As mentioned, mice with ghrelin cell-selective deletion of β1-adrenergic receptors exhibit marked hypoglycemia when exposed to a week-long, severe caloric restriction regimen (30). Given their attenuated epinephrine and norepinephrine responses, hypoglycemic ghrelin-KO mice likely experience a reduction both in the effects that epinephrine and norepinephrine usually exert during hypoglycemia [namely, inhibition of whole body glucose utilization, increased delivery of gluconeogenic substrates to the liver, and enhanced hepatic and renal glucose production (65)] and in other protective glucoregulatory processes that would otherwise be initiated if epinephrine and norepinephrine could stimulate the release of more ghrelin [for instance, activation of brainstem or hypothalamic neurons, which in turn, raise blood glucose, as reviewed in (10)].
As mentioned, although the CRR response in ghrelin-KO mice during the hyperinsulinemic-hypoglycemic clamps had previously been shown to be attenuated in non-STZ-treated subjects (41), the deficits were different than those observed here. Specifically, attenuated plasma corticosterone and GH responses but normal epinephrine and norepinephrine responses had previously been observed in hypoglycemia-clamped non-STZ-treated ghrelin-KO mice (41). These contrast with the observations here of attenuated plasma epinephrine and norepinephrine responses but normal corticosterone and GH responses in hypoglycemia-clamped STZ-treated ghrelin-KO mice. These different patterns of traditional CRR hormone responses in clamped ghrelin-KO mice treated with STZ vs. non-STZ-treated animals might result from one or more of the known differential CRR responses in individuals with and without diabetes. For instance, loss of functional β-cells in the diabetic islet causes α-cell dysregulation, and in turn, loss of normal tonic inhibition of α-cells by intra-islet insulin (8). Also, mRNA levels of adrenal tyrosine hydroxylase, which is the rate-limiting enzyme in catecholamine biosynthesis, are decreased in STZ-induced diabetic rats (66). Interestingly, the observed catecholamine deficits observed here in the clamped STZ-treated ghrelin-KO mice are reminiscent of the attenuated sympathoadrenal system response associated with diabetes in the form of hypoglycemia-associated autonomic dysfunction (5). More studies are needed to determine if reconstitution of the epinephrine and norepinephrine responses in STZ-treated ghrelin-KO mice could rescue their deficient CRR, and also to determine if an aberrant ghrelin response might contribute to the occurrence of hypoglycemia-associated autonomic dysfunction.
There are some caveats to our study that should be kept in mind when interpreting the results. First, the STZ mouse model adapted for the current study from several studies in the literature (45–49) does not completely mimic the pathophysiology or presentation of type 1 diabetes in humans. For instance, although STZ is toxic to pancreatic β-cells, reducing their numbers enough to lower circulating plasma insulin and raise blood glucose into the hyperglycemic range, some β-cells remain (Figure 1A, Supplementary Figure 1) (67), unlike that observed in humans with long-standing type 1 diabetes. Although plasma ghrelin elevates upon STZ administration (Figure 2D) (36, 38, 40, 42) and also although this elevated ghrelin is thought to contribute to STZ-induced hyperphagia (38), changes to plasma ghrelin have not been consistently reported in the literature in type 1 diabetes in humans (33, 68–73) nor is hyperphagia a usual feature of type 1 diabetes in humans. Also, following STZ induction of diabetes, the mice did not receive insulin to treat the hyperglycemia until the clamp procedure, which is unlike the optimal situation in humans with type 1 diabetes in which normoglycemia would be the goal. Additionally, these mice have new-onset diabetes as opposed to most individuals with type 1 diabetes who have been living with the disorder for years. Nonetheless, STZ as administered here reduced plasma insulin substantially (by 82–84%), via efficacious reduction in pancreatic β cell mass, and induced hyperglycemia, which are key hallmarks of type 1 diabetes.
A second limitation of the study relates to the potential influence of the collection method for the blood samples used to measure epinephrine and norepinephrine. Specifically, the samples for CRR hormones were collected by cardiac puncture from isoflurane-anesthetized mice at the end of the clamps. Although handling of the mice was minimized during the clamps, frequent sampling of blood from tail nicks to assess blood glucose was performed throughout the 2 h clamp procedure. Prior work has shown that blood sampling from tail nicks as compared to indwelling arterial cannulas induces a rise in plasma catecholamines (74). Thus, the relatively high levels of catecholamines observed here [in the ~20–30 ng/mL range for epinephrine and the ~5–17 ng/mL range for norepinephrine] as compared to those in another hypoglycemic clamp study (74) [in the ~0–1.2 ng/mL range for epinephrine and the ~0.21–0.42 ng/mL range for norepinphrine], in which blood sampling from non-STZ-treated mice was performed via indwelling arterial cannulas, may have been influenced by the blood collection methods. Other potential stressors, including the recent STZ treatment, the days-long period of hyperglycemia, the 16 h fast preceding the induction of hypoglycemia, and background genetic strain of the mice also likely impacted the magnitude of the detected catecholamine levels (41, 75). That said, ghrelin-KO and WT mice were exposed to the same stressors and were handled exactly the same during the study, and yet, epinephrine and norepinephrine levels were lower in the ghrelin-KO mice. Thus, we predict the differences observed in catecholamine levels between ghrelin-KO and wild-type littermates reflect a genotype-dependent, differential response to hypoglycemia.
A third caveat relates to the use of ghrelin-KO mice, which, because of long-term absence of ghrelin, could exhibit a phenotype reflective of potential compensatory developmental changes that alter the true effects of absent ghrelin action in the setting of insulin-induced hypoglycemia. That said, at least when assessed in non-STZ-treated subjects, ghrelin receptor agonist administration has been shown to rescue the deficits observed in ghrelin-KO mice under the hyperinsulinemic-hypoglycemic protocol (41).
Materials Availability
All unique reagents, mouse lines generated in this study will be made available on request, but we may require a payment and/or a completed Materials Transfer Agreement if there is potential for commercial application.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Ethics Statement
The animal study was reviewed and approved by UT Southwestern Medical Center Institutional Animal Care and Use Committee.
Author Contributions
KS conceptualized and performed the experiments, analyzed and interpreted the data, and helped write the manuscript. BF, SO-L, and NM performed the experiments. DG performed the experiments and helped analyze and interpret the data. BM conceptualized and performed some experiments. CL helped design and generate the ghrelin-KO mice. EB conceptualized the experiments, secured funding, interpreted the data, and supervised the research activity. JZ conceptualized the experiments, secured funding, interpreted the data, supervised the research activity, and helped write the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported through research grants from the NIH (R56 DK071320 to JZ, R01 DK119341 to JZ and EB, and R01 DK109408 to EB), a gift from the David and Teresa Disiere Foundation (to JZ), the Diana and Richard C. Strauss Professorship in Biomedical Research, the Mr. and Mrs. Bruce G. Brookshire Professorship in Medicine, and the Kent and Jodi Foster Distinguished Chair in Endocrinology, in Honor of Daniel Foster, M.D (to JZ).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the Vanderbilt University Medical Center Hormone Assay and Analytical Services Core for performing the catecholamines assays.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2020.00606/full#supplementary-material
References
1. Scott MM, Perello M, Chuang JC, Sakata I, Gautron L, Lee CE, et al. Hindbrain ghrelin receptor signaling is sufficient to maintain fasting glucose. PLoS ONE. (2012) 7:e44089. doi: 10.1371/journal.pone.0044089
2. Asakawa A, Inui A, Kaga T, Yuzuriha H, Nagata T, Fujimiya M, et al. A role of ghrelin in neuroendocrine and behavioral responses to stress in mice. Neuroendocrinology. (2001) 74:143–7. doi: 10.1159/000054680
3. Kawakami A, Okada N, Rokkaku K, Honda K, Ishibashi S, Onaka T. Leptin inhibits and ghrelin augments hypothalamic noradrenaline release after stress. Stress. (2008) 11:363–9. doi: 10.1080/10253890701820257
4. U. K. Hypoglycaemia Study Group. Risk of hypoglycaemia in types 1 and 2 diabetes: effects of treatment modalities and their duration. Diabetologia. (2007) 50:1140–7. doi: 10.1007/s00125-007-0599-y
5. Cryer PE. Physiologic response to hypoglycemia in normal subjects and patients with diabetes mellitus. [Internet], (2015). Available online at: https://www.uptodate.com/contents/physiologic-response-to-hypoglycemia-in-normal-subjectsand-patients-with-diabetes-mellitus?search5physiologic%20response%20to%20hypoglycemia&source5search_result&selectedTitle51;150&usage_type5default&display_rank51 (accessed October 26, 2015).
6. Donovan CM, Watts AG. Peripheral and central glucose sensing in hypoglycemic detection. Physiology. (2014) 29:314–24. doi: 10.1152/physiol.00069.2013
7. Schopman JE, Geddes J, Frier BM. Frequency of symptomatic and asymptomatic hypoglycaemia in Type 1 diabetes: effect of impaired awareness of hypoglycaemia. Diabet Med. (2011) 28:352–5. doi: 10.1111/j.1464-5491.2010.03203.x
8. Raju B, Cryer PE. Loss of the decrement in intraislet insulin plausibly explains loss of the glucagon response to hypoglycemia in insulin-deficient diabetes: documentation of the intraislet insulin hypothesis in humans. Diabetes. (2005) 54:757–64. doi: 10.2337/diabetes.54.3.757
9. Gray SM, Page LC, Tong J. Ghrelin regulation of glucose metabolism. J Neuroendocrinol. (2019) 31:e12705. doi: 10.1111/jne.12705
10. Mani BK, Shankar K, Zigman JM. Ghrelin's relationship to blood glucose. Endocrinology. (2019) 160:1247–61. doi: 10.1210/en.2019-00074
11. Chuang JC, Sakata I, Kohno D, Perello M, Osborne-Lawrence S, Repa JJ, et al. Ghrelin directly stimulates glucagon secretion from pancreatic alpha-cells. Mol Endocrinol. (2011) 25:1600–11. doi: 10.1210/me.2011-1001
12. Heppner KM, Tong J. Mechanisms in endocrinology: regulation of glucose metabolism by the ghrelin system: multiple players and multiple actions. Eur J Endocrinol. (2014) 171:R21–32. doi: 10.1530/EJE-14-0183
13. Page LC, Gastaldelli A, Gray SM, D'Alessio DA, Tong J. Interaction of GLP-1 and ghrelin on glucose tolerance in healthy humans. Diabetes. (2018) 67:1976–85. doi: 10.2337/db18-0451
14. Wang Q, Liu C, Uchida A, Chuang JC, Walker A, Liu T, et al. Arcuate AgRP neurons mediate orexigenic and glucoregulatory actions of ghrelin. Mol Metab. (2014) 3:64–72. doi: 10.1016/j.molmet.2013.10.001
15. Dezaki K, Hosoda H, Kakei M, Hashiguchi S, Watanabe M, Kangawa K, et al. Endogenous ghrelin in pancreatic islets restricts insulin release by attenuating Ca2+ signaling in beta-cells: implication in the glycemic control in rodents. Diabetes. (2004) 53:3142–51. doi: 10.2337/diabetes.53.12.3142
16. Dezaki K, Kakei M, Yada T. Ghrelin uses Galphai2 and activates voltage-dependent K+ channels to attenuate glucose-induced Ca2+ signaling and insulin release in islet beta-cells: novel signal transduction of ghrelin. Diabetes. (2007) 56:2319–27. doi: 10.2337/db07-0345
17. Broglio F, Arvat E, Benso A, Gottero C, Muccioli G, Papotti M, et al. Ghrelin, a natural GH secretagogue produced by the stomach, induces hyperglycemia and reduces insulin secretion in humans. J Clin Endocrinol Metab. (2001) 86:5083–6. doi: 10.1210/jcem.86.10.8098
18. Reed JA, Benoit SC, Pfluger PT, Tschop MH, D'Alessio DA, Seeley RJ. Mice with chronically increased circulating ghrelin develop age-related glucose intolerance. Am J Physiol Endocrinol Metab. (2008) 294:E752–60. doi: 10.1152/ajpendo.00463.2007
19. Colombo M, Gregersen S, Xiao J, Hermansen K. Effects of ghrelin and other neuropeptides (CART, MCH, orexin A and B, and GLP-1) on the release of insulin from isolated rat islets. Pancreas. (2003) 27:161–6. doi: 10.1097/00006676-200308000-00009
20. Tong J, Prigeon RL, Davis HW, Bidlingmaier M, Kahn SE, Cummings DE, et al. Ghrelin suppresses glucose-stimulated insulin secretion and deteriorates glucose tolerance in healthy humans. Diabetes. (2010) 59:2145–51. doi: 10.2337/db10-0504
21. Dezaki K, Sone H, Koizumi M, Nakata M, Kakei M, Nagai H, et al. Blockade of pancreatic islet-derived ghrelin enhances insulin secretion to prevent high-fat diet-induced glucose intolerance. Diabetes. (2006) 55:3486–93. doi: 10.2337/db06-0878
22. Longo KA, Charoenthongtrakul S, Giuliana DJ, Govek EK, McDonagh T, Qi Y, et al. Improved insulin sensitivity and metabolic flexibility in ghrelin receptor knockout mice. Regul Pept. (2008) 150:55–61. doi: 10.1016/j.regpep.2008.03.011
23. Wortley KE, del Rincon JP, Murray JD, Garcia K, Iida K, Thorner MO, et al. Absence of ghrelin protects against early-onset obesity. J Clin Invest. (2005) 115:3573–8. doi: 10.1172/JCI26003
24. Zhao TJ, Liang G, Li RL, Xie X, Sleeman MW, Murphy AJ, et al. Ghrelin O-acyltransferase (GOAT) is essential for growth hormone-mediated survival of calorie-restricted mice. Proc Natl Acad Sci USA. (2010) 107:7467–72. doi: 10.1073/pnas.1002271107
25. Abegg K, Bernasconi L, Hutter M, Whiting L, Pietra C, Giuliano C, et al. Ghrelin receptor inverse agonists as a novel therapeutic approach against obesity-related metabolic disease. Diabetes Obes Metab. (2017) 19:1740–50. doi: 10.1111/dom.13020
26. Pfluger PT, Kirchner H, Gunnel S, Schrott B, Perez-Tilve D, Fu S, et al. Simultaneous deletion of ghrelin and its receptor increases motor activity and energy expenditure. Am J Physiol Gastrointest Liver Physiol. (2008) 294:G610–8. doi: 10.1152/ajpgi.00321.2007
27. Li RL, Sherbet DP, Elsbernd BL, Goldstein JL, Brown MS, Zhao TJ. Profound hypoglycemia in starved, ghrelin-deficient mice is caused by decreased gluconeogenesis and reversed by lactate or fatty acids. J Biol Chem. (2012) 287:17942–50. doi: 10.1074/jbc.M112.358051
28. McFarlane MR, Brown MS, Goldstein JL, Zhao TJ. Induced ablation of ghrelin cells in adult mice does not decrease food intake, body weight, or response to high-fat diet. Cell Metab. (2014) 20:54–60. doi: 10.1016/j.cmet.2014.04.007
29. Ge X, Yang H, Bednarek MA, Galon-Tilleman H, Chen P, Chen M, et al. LEAP2 Is an endogenous antagonist of the ghrelin receptor. Cell Metab. (2018) 27:461–9.e6. doi: 10.1016/j.cmet.2017.10.016
30. Mani BK, Osborne-Lawrence S, Vijayaraghavan P, Hepler C, Zigman JM. beta1-Adrenergic receptor deficiency in ghrelin-expressing cells causes hypoglycemia in susceptible individuals. J Clin Invest. (2016) 126:3467–78. doi: 10.1172/JCI86270
31. Sun Y, Asnicar M, Saha PK, Chan L, Smith RG. Ablation of ghrelin improves the diabetic but not obese phenotype of ob/ob mice. Cell Metab. (2006) 3:379–86. doi: 10.1016/j.cmet.2006.04.004
32. Brial F, Lussier CR, Belleville K, Sarret P, Boudreau F. Ghrelin inhibition restores glucose homeostasis in hepatocyte nuclear factor-1α (MODY3)-deficient mice. Diabetes. (2015) 64:3314–20. doi: 10.2337/db15-0124
33. Nowak N, Hohendorff J, Solecka I, Szopa M, Skupien J, Kiec-Wilk B, et al. Circulating ghrelin level is higher in HNF1A-MODY and GCK-MODY than in polygenic forms of diabetes mellitus. Endocrine. (2015) 50:643–9. doi: 10.1007/s12020-015-0627-5
34. Verhulst PJ, De Smet B, Saels I, Thijs T, Ver Donck L, Moechars D, et al. Role of ghrelin in the relationship between hyperphagia and accelerated gastric emptying in diabetic mice. Gastroenterology. (2008) 135:1267–76. doi: 10.1053/j.gastro.2008.06.044
35. Ariga H, Imai K, Chen C, Mantyh C, Pappas TN, Takahashi T. Does ghrelin explain accelerated gastric emptying in the early stages of diabetes mellitus? Am J Physiol Regul Integr Comp Physiol. (2008) 294:R1807–12. doi: 10.1152/ajpregu.00785.2007
36. Dong J, Peeters TL, De Smet B, Moechars D, Delporte C, Vanden Berghe P, et al. Role of endogenous ghrelin in the hyperphagia of mice with streptozotocin-induced diabetes. Endocrinology. (2006) 147:2634–42. doi: 10.1210/en.2005-1335
37. Tsubone T, Masaki T, Katsuragi I, Tanaka K, Kakuma T, Yoshimatsu H. Leptin downregulates ghrelin levels in streptozotocin-induced diabetic mice. Am J Physiol Regul Integr Comp Physiol. (2005) 289:R1703–6. doi: 10.1152/ajpregu.00773.2004
38. Ishii S, Kamegai J, Tamura H, Shimizu T, Sugihara H, Oikawa S. Role of ghrelin in streptozotocin-induced diabetic hyperphagia. Endocrinology. (2002) 143:4934–7. doi: 10.1210/en.2002-220612
39. Gelling RW, Overduin J, Morrison CD, Morton GJ, Frayo RS, Cummings DE, et al. Effect of uncontrolled diabetes on plasma ghrelin concentrations and ghrelin-induced feeding. Endocrinology. (2004) 145:4575–82. doi: 10.1210/en.2004-0605
40. Masaoka T, Suzuki H, Hosoda H, Ota T, Minegishi Y, Nagata H, et al. Enhanced plasma ghrelin levels in rats with streptozotocin-induced diabetes. FEBS Lett. (2003) 541:64–8. doi: 10.1016/S0014-5793(03)00306-5
41. Shankar K, Gupta D, Mani BK, Findley BG, Lord CC, Osborne-Lawrence S, et al. Acyl-ghrelin is permissive for the normal counterregulatory response to insulin-induced hypoglycemia. Diabetes. (2020) 69:228–37. doi: 10.2337/db19-0438
42. Mani BK, Uchida A, Lee Y, Osborne-Lawrence S, Charron MJ, Unger RH, et al. Hypoglycemic effect of combined ghrelin and glucagon receptor blockade. Diabetes. (2017) 66:1847–57. doi: 10.2337/db16-1303
43. Lin L, Saha PK, Ma X, Henshaw IO, Shao L, Chang BH, et al. Ablation of ghrelin receptor reduces adiposity and improves insulin sensitivity during aging by regulating fat metabolism in white and brown adipose tissues. Aging Cell. (2011) 10:996–1010. doi: 10.1111/j.1474-9726.2011.00740.x
44. Rodriguez JA, Bruggeman EC, Mani BK, Osborne-Lawrence S, Lord CC, Roseman HF, et al. Ghrelin receptor agonist rescues excess neonatal mortality in a prader-willi syndrome mouse model. Endocrinology. (2018) 159:4006–22. doi: 10.1210/en.2018-00801
45. Le May C, Chu K, Hu M, Ortega CS, Simpson ER, Korach KS, et al. Estrogens protect pancreatic beta-cells from apoptosis and prevent insulin-deficient diabetes mellitus in mice. Proc Natl Acad Sci USA. (2006) 103:9232–7. doi: 10.1073/pnas.0602956103
46. Mani BK, Puzziferri N, He Z, Rodriguez JA, Osborne-Lawrence S, Metzger NP, et al. LEAP2 changes with body mass and food intake in humans and mice. J Clin Invest. (2019) 129:3909–23. doi: 10.1172/JCI125332
47. Tsantoulas C, Lainez S, Wong S, Mehta I, Vilar B, McNaughton PA. Hyperpolarization-activated cyclic nucleotide-gated 2 (HCN2) ion channels drive pain in mouse models of diabetic neuropathy. Sci Transl Med. (2017) 9:eaam6072. doi: 10.1126/scitranslmed.aam6072
48. Wei J, Shimazu J, Makinistoglu MP, Maurizi A, Kajimura D, Zong H, et al. Glucose uptake and Runx2 synergize to orchestrate osteoblast differentiation and bone formation. Cell. (2015) 161:1576–91. doi: 10.1016/j.cell.2015.08.018
49. Yamaguchi T, Sato H, Kato-Itoh M, Goto T, Hara H, Sanbo M, et al. Interspecies organogenesis generates autologous functional islets. Nature. (2017) 542:191–6. doi: 10.1038/nature21070
50. Luo M, Guan X, Luczak ED, Lang D, Kutschke W, Gao Z, et al. Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. J Clin Invest. (2013) 123:1262–74. doi: 10.1172/JCI65268
51. McEvoy RC, Andersson J, Sandler S, Hellerstrom C. Multiple low-dose streptozotocin-induced diabetes in the mouse. Evidence for stimulation of a cytotoxic cellular immune response against an insulin-producing beta cell line. J Clin Invest. (1984) 74:715–22. doi: 10.1172/JCI111487
52. Hedbacker K, Birsoy K, Wysocki RW, Asilmaz E, Ahima RS, Farooqi IS. Antidiabetic effects of IGFBP2, a leptin-regulated gene. Cell Metab. (2010) 11:11–22. doi: 10.1016/j.cmet.2010.02.010
53. Sakata I, Park WM, Walker AK, Piper PK, Chuang JC, Osborne-Lawrence S, et al. Glucose-mediated control of ghrelin release from primary cultures of gastric mucosal cells. Am J Physiol Endocrinol Metab. (2012) 302:E1300–10. doi: 10.1152/ajpendo.00041.2012
54. Gagnon J, Anini Y. Insulin and norepinephrine regulate ghrelin secretion from a rat primary stomach cell culture. Endocrinology. (2012) 153:3646–56. doi: 10.1210/en.2012-1040
55. Lucidi P, Murdolo G, Di Loreto C, De Cicco A, Parlanti N, Fanelli C, et al. Ghrelin is not necessary for adequate hormonal counterregulation of insulin-induced hypoglycemia. Diabetes. (2002) 51:2911–4. doi: 10.2337/diabetes.51.10.2911
56. Flanagan DE, Evans ML, Monsod TP, Rife F, Heptulla RA, Tamborlane WV, et al. The influence of insulin on circulating ghrelin. Am J Physiol Endocrinol Metab. (2003) 284:E313–6. doi: 10.1152/ajpendo.00569.2001
57. Lauritzen ES, Voss T, Kampmann U, Mengel A, Vendelbo MH, Jorgensen JO, et al. Circulating acylghrelin levels are suppressed by insulin and increase in response to hypoglycemia in healthy adult volunteers. Eur J Endocrinol. (2015) 172:357–62. doi: 10.1530/EJE-14-0880
58. Bando M, Iwakura H, Ariyasu H, Koyama H, Hosoda K, Adachi S, et al. Overexpression of intraislet ghrelin enhances beta-cell proliferation after streptozotocin-induced beta-cell injury in mice. Am J Physiol Endocrinol Metab. (2013) 305:E140–8. doi: 10.1152/ajpendo.00112.2013
59. Wren AM, Small CJ, Fribbens CV, Neary NM, Ward HL, Seal LJ, et al. The hypothalamic mechanisms of the hypophysiotropic action of ghrelin. Neuroendocrinology. (2002) 76:316–24. doi: 10.1159/000066629
60. Cabral A, Portiansky E, Sanchez-Jaramillo E, Zigman JM, Perello M. Ghrelin activates hypophysiotropic corticotropin-releasing factor neurons independently of the arcuate nucleus. Psychoneuroendocrinology. (2016) 67:27–39. doi: 10.1016/j.psyneuen.2016.01.027
61. Mozid AM, Tringali G, Forsling ML, Hendricks MS, Ajodha S, Edwards R, et al. Ghrelin is released from rat hypothalamic explants and stimulates corticotrophin-releasing hormone and arginine-vasopressin. Horm Metab Res. (2003) 35:455–9. doi: 10.1055/s-2003-41801
62. Nagaya N, Miyatake K, Uematsu M, Oya H, Shimizu W, Hosoda H, et al. Hemodynamic, renal, and hormonal effects of ghrelin infusion in patients with chronic heart failure. J Clin Endocrinol Metab. (2001) 86:5854–9. doi: 10.1210/jcem.86.12.8115
63. Stevanovic D, Milosevic V, Starcevic VP, Severs WB. The effect of centrally administered ghrelin on pituitary ACTH cells and circulating ACTH and corticosterone in rats. Life Sci. (2007) 80:867–72. doi: 10.1016/j.lfs.2006.11.018
64. Gupta D, Chuang JC, Mani BK, Shankar K, Rodriguez JA, Osborne-Lawrence S, et al. β1-adrenergic receptors mediate plasma acyl-ghrelin elevation and depressive-like behavior induced by chronic psychosocial stress. Neuropsychopharmacology. (2019) 44:1319–27. doi: 10.1038/s41386-019-0334-7
65. Cryer PE. Banting lecture. Hypoglycemia: the limiting factor in the management of IDDM. Diabetes. (1994) 43:1378–89. doi: 10.2337/diabetes.43.11.1378
66. Inouye KE, Chan O, Yue JT, Matthews SG, Vranic M. Effects of diabetes and recurrent hypoglycemia on the regulation of the sympathoadrenal system and hypothalamo-pituitary-adrenal axis. Am J Physiol Endocrinol Metab. (2005) 288:E422–9. doi: 10.1152/ajpendo.00389.2004
67. Lu WT, Juang JH, Hsu BR, Huang HS. Effects of high or low dose of streptozocin on pancreatic islets in C57BL/6 and C.B17-SCID mice. Transplant Proc. (1998) 30:609–10. doi: 10.1016/S0041-1345(97)01425-5
68. Bideci A, Camurdan MO, Cinaz P, Demirel F. Ghrelin, IGF-I and IGFBP-3 levels in children with type 1 diabetes mellitus. J Pediatr Endocrinol Metab. (2005) 18:1433–9. doi: 10.1515/JPEM.2005.18.12.1433
69. Soriano-Guillen L, Barrios V, Lechuga-Sancho A, Chowen JA, Argente J. Response of circulating ghrelin levels to insulin therapy in children with newly diagnosed type 1 diabetes mellitus. Pediatr Res. (2004) 55:830–5. doi: 10.1203/01.PDR.0000120679.92416.70
70. Holdstock C, Ludvigsson J, Karlsson FA. Abnormal ghrelin secretion in new onset childhood Type 1 diabetes. Diabetologia. (2004) 47:150–1. doi: 10.1007/s00125-003-1258-6
71. Prodam F, Cadario F, Bellone S, Trovato L, Moia S, Pozzi E, et al. Obestatin levels are associated with C-peptide and antiinsulin antibodies at the onset, whereas unacylated and acylated ghrelin levels are not predictive of long-term metabolic control in children with type 1 diabetes. J Clin Endocrinol Metab. (2014) 99:E599–607. doi: 10.1210/jc.2013-3294
72. Murdolo G, Lucidi P, Di Loreto C, Parlanti N, De Cicco A, Fatone C, et al. Insulin is required for prandial ghrelin suppression in humans. Diabetes. (2003) 52:2923–7. doi: 10.2337/diabetes.52.12.2923
73. Huml M, Kobr J, Siala K, Varvarovska J, Pomahacova R, Karlikova M, et al. Gut peptide hormones and pediatric type 1 diabetes mellitus. Physiol Res. (2011) 60:647–58. doi: 10.33549/physiolres.931995
74. Ayala JE, Bracy DP, McGuinness OP, Wasserman DH. Considerations in the design of hyperinsulinemic-euglycemic clamps in the conscious mouse. Diabetes. (2006) 55:390–7. doi: 10.2337/diabetes.55.02.06.db05-0686
Keywords: ghrelin, type 1 diabetes, glucose clamp, hypoglycemia, glucose counterregulation
Citation: Shankar K, Gupta D, Mani BK, Findley BG, Osborne-Lawrence S, Metzger NP, Liu C, Berglund ED and Zigman JM (2020) Ghrelin Protects Against Insulin-Induced Hypoglycemia in a Mouse Model of Type 1 Diabetes Mellitus. Front. Endocrinol. 11:606. doi: 10.3389/fendo.2020.00606
Received: 22 May 2020; Accepted: 27 July 2020;
Published: 11 September 2020.
Edited by:
Åke Sjöholm, Gävle Hospital, SwedenReviewed by:
Mark L. Evans, University of Cambridge, United KingdomCraig Beall, University of Exeter, United Kingdom
Copyright © 2020 Shankar, Gupta, Mani, Findley, Osborne-Lawrence, Metzger, Liu, Berglund and Zigman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeffrey M. Zigman, amVmZnJleS56aWdtYW5AdXRzb3V0aHdlc3Rlcm4uZWR1