 Gisela V. Novack
Gisela V. Novack Pablo Galeano
Pablo Galeano Eduardo M. Castaño
Eduardo M. Castaño Laura Morelli
Laura Morelli
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol. , 11 September 2020
Sec. Cellular Endocrinology
Volume 11 - 2020 | https://doi.org/10.3389/fendo.2020.00600
This article is part of the Research Topic Mitochondrial Dynamics in Endocrine Physiology and Disease View all 14 articles
Several studies suggest that the assembly of mitochondrial respiratory complexes into structures known as supercomplexes (SCs) may increase the efficiency of the electron transport chain, reducing the rate of production of reactive oxygen species. Therefore, the study of the (dis)assembly of SCs may be relevant for the understanding of mitochondrial dysfunction reported in brain aging and major neurodegenerative disorders such as Alzheimer's disease (AD) and Parkinson's disease (PD). Here we briefly reviewed the biogenesis and structural properties of SCs, the impact of mtDNA mutations and mitochondrial dynamics on SCs assembly, the role of lipids on stabilization of SCs and the methodological limitations for the study of SCs. More specifically, we summarized what is known about mitochondrial dysfunction and SCs organization and activity in aging, AD and PD. We focused on the critical variables to take into account when postmortem tissues are used to study the (dis)assembly of SCs. Since few works have been performed to study SCs in AD and PD, the impact of SCs dysfunction on the alteration of brain energetics in these diseases remains poorly understood. The convergence of future progress in the study of SCs structure at high resolution and the refinement of animal models of AD and PD, as well as the use of iPSC-based and somatic cell-derived neurons, will be critical in understanding the biological relevance of the structural remodeling of SCs.
Mitochondria are dynamic organelles that reorganize under stress or variations in the availability of nutrients or oxygen. It was proposed that an equilibrated distribution between individual mitochondrial respiratory complexes (MRC) and supercomplexes (SCs) is relevant to achieve the optimal performance of the electron transfer chain. However, the procedures by which the assembly of SCs can be adapted to the requirements of the cells are still poorly understood. In this mini review we address the organization of SCs in cell cultures and brain tissue from human or animal models of aging, AD and PD to evaluate the possible relevance of the (dis)assembly of SCs in the energetic failures characteristics of these neurodegenerative disorders associated with amyloid deposition.
The respiratory chain in mammals consists of four respiratory complexes (I–IV) encoded by nuclear and mitochondrial DNA (mtDNA) inserted and assembled in the inner mitochondrial membrane (IMM) and two intermediary substrates (coenzyme Q and cytochrome c). According to their enzymatic activities complex I (CI) is known as NADH:ubiquinone oxidoreductase, complex II (CII) as succinate:quinone oxidoreductase; complex III (CIII) as ubiquinol-cytochrome c oxidoreductase and complex IV (CIV) as cytochrome c oxidase (COX). During the last 30 years, the structural organization of the respiratory complexes was intended to be understood in terms of two utmost paradigms: the “fluid” and the “solid” models. The “fluid” model postulates that all redox components are unconstrained diffusible particles with the electron carriers alternating in the middle of the gigantic complexes I–IV (1, 2). This interpretation suggests that mitochondrial electron transfer is a diffusion-based stochastic collision process and that diffusion has an essential and regulating effect on electron transfer (3). The “fluid” model was accepted until 2001 when it was shown that it was possible to purify, by means of Blue-Native polyacrylamide gel electrophoresis (BN-PAGE), stable associations of respiratory complexes (4) and therefore the “solid” model was proposed. In the “solid” model, respiratory complexes establish interactions to form higher-order supramolecular structures called supercomplexes (SCs) formed by two units of CIII and a variable number of CIV in the presence or not of one module of CI, (I1III2IV1−2; III2IV1−2), or even into megacomplex where two units of CI, CIII, and CIV get together (I2III2IV2). Based on BN-PAGE, CII was not detected in SCs (5). However, CII could bind to SCs by weak protein-protein interactions that may dissociate due to dilution during mitochondrial isolation. Recent cryo-EM and cross-linked mass spectrometry studies suggested that CII can be involved in respirasomes (6, 7). By contrast, experiments performed in cell cultures demonstrated that silencing a CII subunit (SDHC) has no effect on the respirasome assembly, although it can play a regulatory role in respirasome formation (8). In the solid model, SCs I1III2IVn that act as single functional entities are known as “respirasomes” which are capable of catalyzing whole reaction pathways by the presence of Q and cyt c associated with these devices. An integrative vision of both models was recently proposed, noted as the “plasticity” model (9). This model consolidates transient assemblies and free lateral diffusion of redox units. Both SCs and the free individual components can perform their actions, with SCs being more efficient in energy production with a lower ROS formation rate. However, this paradigm was suggested based on distinctions in the steady-state amounts of respiratory chain complexes analyzed by BN-PAGE technique, and consequently, the proposed “plasticity” between the individual complexes and the respirasomes in vivo has not yet been proven. A schematic diagram of the three models is depicted in Figures 1A,B. Additionally, the association of single complexes to form the respirasomes can be explained by two mechanisms. The first proposes that CI gets fully-assembled prior to its binding to SCs, while the second favors the sequential binding of CIII2 and CIV to an almost complete CI scaffold (10). Taking into account that “free CI” is underrepresented in mammalian tissues under mild purification conditions, it was proposed that SCs provide a scaffold for the full-assembly and stability of CI (11). 3D maps show extensive and stable interactions between specific CI and CIII2 (12–14) and as CI and CIII2 subunits contain disulfide bonds it was proposed that redox status may modulate CI-CIII2 interactions. A close association between specific CIV and CI subunit (12, 13), and between CIV and CIII2 subunits, was also determined (13). Models of SCs from ovine/bovine/porcine mitochondria suggest that “species-specific” protein sequences are critical for the arrangement of SCs subunits. Moreover, it was demonstrated that different cell types from human and mouse strains show similar SCs composition. However, the composition of the minimal SCs (In + IIIn) was variable in different cell types (15). Since some of the units of single complexes are encoded in mtDNA, the impact of mtDNA mutations on SCs assembly was analyzed in human cybrids containing mutations in CIV and CIII subunits. Based on these studies it was proposed that CIII may be the structural core of SCs while CI, because of its instability, cannot exist as an individual entity (16). The role of SCs assembly proteins COX7A2L and respiratory complex factor (Rcf) was also explored (17, 18). It was postulated that COX7A2L may be a regulatory checkpoint for the biogenesis of CIII2 and CIII-containing SCs (19) while Rcf1 and Rcf2 are CIV-binding proteins that may also interact with CIII2 (20). In contrast, MCJ/DnaJC15 (a co-chaperone localized at the IMM) has been proposed as a negative regulator of SCs formation/stability (21). Even if MCJ is non-essential for mitochondrial function under physiological conditions, MCJ failure impacts on the pathophysiology arising specific metabolic alterations (21).
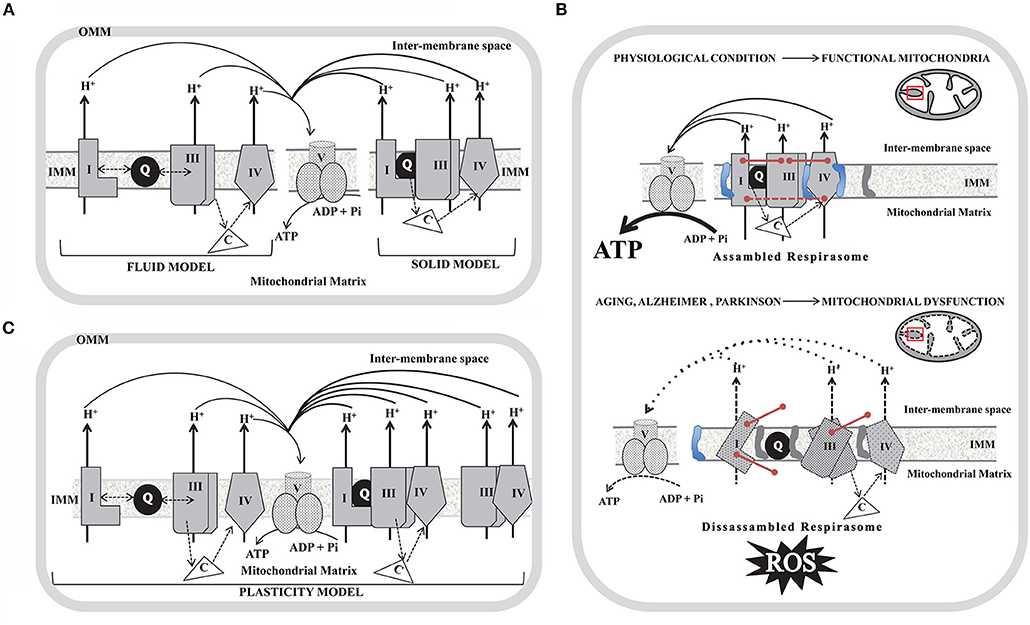
Figure 1. Schematic diagram of the models proposed to explain the organization of the mitochondria respiratory complexes in the inner mitochondrial membrane (IMM) and the impact of the SCs disassembly in aging, Alzheimer and Parkinson disorders. (A) Fluid model (left); Solid model (right). (B) Plasticity model in which respirasomes exist in dynamic equilibrium with randomly organized, enzymatically active, and isolated complexes. In (A) and (B) only individual complexes (CI, CIII, and CIV) that conform respirasomes were depicted and do not show the actual stoichiometry; coenzyme Q (Q) is represented as a black-filled circle in the IMM and cytochrome c (C) as a non-membrane associated white triangle that transfer electrons from CIII to CIV (dotted arrows). Doble head dotted arrows indicate lateral diffusion of Q between membrane embedded complexes in the Fluid and Plasticity models, while in the Solid model, SCs trap the soluble electron carrier and restrict its diffusion. Solid black arrows show the proton pumping activity of each individual complex and the ATP generation by complex V (CV). OMM, outer mitochondrial membrane. (C) In a physiological condition (upper panel) mitochondria show assembled SCs located in intact cristae (depicted as a black line inside the red box in the scheme of mitochondria). SCs integrity is mediated by assembly factors between individual units from each complex (described as pink lines) and by high levels of cardiolipin (represented as a blue structure in the IMM) and by low levels of phosphatidylethanolamine (represented as a gray structure in the IMM). Assembled respirasomes limit ROS generation while improving the efficiency of ATP synthesis. In aging, Alzheimer and Parkinson (lower panel) the dysfunctional mitochondria show dissessembled SCs located in swelled mitochondria cristae (depicted as a dotted black line inside the red box in the scheme of mitochondria). Respirasomes disorganization may be due to impaired assembly factors expression (described as un-connected pink lines) and to low levels of cardiolipin and high levels of phosphatidylethanolamine. Disassembled respirasomes promote ROS generation and decrease ATP levels.
Compared to other sub-cellular membranes, the IMM is highly enriched in phosphatidylethanolamine (PE) and cardiolipin (CL), being the latter an evolutionarily conserved dimeric phospholipid that is precisely located in mitochondria. Data of mammalian samples indicate asymmetric phospholipid location in the IMM leaflets, with particular enrichment of phosphatidylinositol (PI) and CL in the matrix-facing leaflet of the IMM (22). In vitro and in vivo studies highlight the role of PE and CL in the assembly and activity of MRC with the special role of CL in the SCs stabilization and PE in the SCs destabilization (23). In agreement with these remarks, the atomic structure of the respirasomes showed gaps between CI, CIII2, and CIV that were immersed by CL to fix properly the respirasomes (24, 25). The functional importance of CL in SCs stabilization is reinforced by studies on cells from patients with a hereditary disease known as Barth syndrome (BTHS) (an X-linked recessive disorder due to the lack of tafazzin, the transacylase that catalyzes the maturation of CL into its polyunsaturated forms). Mitochondria from BTHS lymphoblasts show reduced abundance and instability of SCs which in turn results in a higher rate of CL degradation (26, 27). CL and PE are synthesized in mitochondria from the precursors Cytidine Diphosphate (CDP)- diacylglycerol and CDP-ethanolamine, respectively, both located in the endoplasmic reticulum (ER). The importation of precursors into the mitochondria requires the juxtaposition of ER-mitochondria membranes. This collocation, known as “lipid-Mitochondria-ER Contacts” (“lipid-MERC”), is needed to be properly assembled and functionally operational to guarantee the correct transport between organelles (28). The gap width needed for lipid transfer is anticipated to be as thin as ≤ 10 nm (29) and it has been proposed that disturbances in the interphase length of apposition and the gap width between these organelles may impair lipid exchange (30). Recently, we experimentally addressed this issue in primary neuronal cultures from McGill-R-Thy1-APP transgenic (Tg) rats, a model of the early stage of Alzheimer-like amyloid pathology (31). We found, using in vivo FRET and transmission electron microscopy (TEM), significant decrements in the average length of the apposition of “lipid-MERC” (gap width ≤ 10 nm) as compared to control cells, suggesting “relaxed” contacts between organelles. In accordance with FRET and TEM results, untargeted lipidomics of isolated mitochondria from neurons exhibited in Tg a significant deviation. The total number of lipids detected was 106 and 12 were significantly different in Tg as compared to control rats. Among them, a 60% decrement in mitochondrial CL and PE was observed (32). These results allow us to speculate that at early stages of AD pathology, there could be a disassembly of the SCs of neuronal mitochondria due to alteration in CL and PE synthesis, which in turn may promote neuronal bioenergetics dysfunction, a characteristic feature of this neurological disorder (33–36).
The biological significance of respirasomes in oxidative phosphorylation (OXPHOS) has not yet been established. It was proposed that: (1) CIII and CIV association with CI may favor the assembly and stability of CI (37–40). Different studies have shown that the integrity of CIII2 and CIV is crucial for the stability of mammalian CI. Even though, CI dysfunction is infrequent in most of the patients with CIII2 or CIV enzymatic deficiencies, which is consistent with the possibility that major structural alterations of CIII2 and CIV are necessary to induce CI dysfunction (41, 42); (2) The assembly of SCs may reduce the rate of ROS generation (43, 44). CI and CIII2 are the major redox centers in which oxygen is reduced to superoxide and, therefore, their assembly into SCs may minimize ROS production. Studies using bovine heart mitochondria have shown that the disassembly enhanced the release of superoxide from CI, and a direct correlation between free CI and ROS generation was found in neurons and astrocytes (45); (3) SCs may optimize the catalytic activity of the individual complexes (46, 47). In this regard, it was shown that the activity of CI in SCs I+III2 isolated from bovine heart mitochondria was half of the activity of CI in SCs I+III2+IV1, indicating that the full respirasome was the most active unit. It has been proposed that the assembly of CI with CIII2 favors electron transfer through Q by channeling between the two complexes without following a pool behavior. This proposal has been challenged by studies showing that NADH and succinate oxidation include different Q redox steady states, but communicate and converge on a single non-partitioned Q pool (48, 49); (4) SCs can boost the efficiency of electron transfer through substrate channeling (5, 50, 51). However, the lack of a confining structure between CI and CIII (49) is inconsistent with such a mechanism. A recent study showed, by using inverted sealed vesicles from bovine heart IMM, that the SCs do not sequester or channel the Q pool which is freely exchanged within and outside the SCs sets to react with any enzyme in the membrane (52). Although these experiments were performed in a non-physiological solution lacking the mitochondrial environment and interaction pathways, more physiologically relevant studies carried out in Drosophila (53) and mice (54) also agree with these results; (5) SCs may prevent aggregation of their protein subunits. The IMM is characterized by a high protein:lipid ratio and, therefore, the correct assembly of SCs may be crucial in minimizing the possibility of nucleation and non-functional aggregation of their components (48).
During the last 20 years, various techniques have been implemented for the study of SCs. The first evidence for the existence of SCs was supported by BN-PAGE experiments (5) and more recently, data obtained using cryo-EM provided more crucial information about the structural composition of SCs at near-atomic resolution (12, 55, 56). However, these techniques have several limitations to elucidate the interactions between individual complexes and the mechanisms by which SCs are assembled. Major drawbacks are based on the following issues: (1) the procedure of mitochondria isolation and purification may impact on the SCs integrity (57). It appears that detergent solubilization of SCs may affect their native structure. Variations in the type of detergent used and in the ratio of detergent/mitochondrial protein have a significant effect on the characterization of SCs at the quantitative and qualitative levels; (2) the absence of lipids (especially CL) in the isolation buffer for solubilization of mitochondrial proteins is an additional factor that can promote dissociation of poorly linked complexes; (3) interpretation of the bands resolved by BN-PAGE is not an easy task due to overlapping of signals. Moreover, determination of molecular mass of SCs may be difficult using BN-PAGE due to conformational and surface changes. For the resolution of subunits of protein complexes, the most useful technique is to combine BN-PAGE with a second gel dimension (2D)-BN/sodium dodecyl sulfate (SDS)-PAGE (58). It is of note that SCs in BN-PAGE are highly prone to diffusion during the equilibration step before loading onto the second dimension, generating possible distortions in the densitometry analysis of the spots. In addition to BN-PAGE, the CN-PAGE or “clear native gels” (in the absence of the dye Coomassie Blue) are frequently used to assess ATP synthase in-gel activity. CN-PAGE allows better enzymatic preservation and limits the dissociation of labile proteins in high molecular weight SCs and ATP synthase assemblies (59).
Studies that solubilize SCs without detergents by cross-linking and mass spectrometry (MS) have shown the existence of many in situ interactions in proteins throughout electron transfer complexes, ATP synthase, and the mitochondrial contact site and cristae organizing system (60). Recently, it was shown that all respiratory complexes (including CII and CV that were not found by means of cryo-EM) are in close spatial proximity. This provides direct evidence for SCs assembly in the intact mitochondria (7). The heterogeneity in the preparations and particles selected for cryo-EM in different studies may introduce a bias that deserves further clarification (12, 49, 55, 56, 61).
In addition, novel approaches to detect SCs in eukaryotic cells in vivo are being developed. In this regard, by using proximity-dependent labeling followed by MS it is possible to identify potentially interacting proteins and their subcellular spatial localization, overcoming the technical difficulties that are associated with transient protein–protein interactions detection (62–64). Moreover, fluorescence lifetime imaging microscopy (FLIM), that employs fluorescent sensor proteins to assess the assembly of SCs and to monitor SCs plasticity in live cells, is currently implemented (65). Recently, Förster Resonance Energy Transfer (FRET) using CIII subunit k fused to Clover as a donor and CoxVIIIa-mRuby2 as an acceptor showed colocalization of CIII and CIV and SCs formation in live cells (66).
AD and PD are the most frequent neurodegenerative disorders of elderly people with a huge impact on morbidity and mortality. The clinical features of PD are primarily motor deficits, while patients with AD show cognitive impairment. Both conditions are known as proteinopathies, characterized by brain amyloid deposition (abnormal accumulation of misfolded proteins) in specific brain areas and neuronal death. At the histopathological level, the postmortem brain shows similarities and differences in each case. Brains from AD and PD subjects display amyloid deposits (amyloid β and phosphorylated Tau in AD, and α-synuclein in PD), mitochondrial dysfunction and oxidative stress. There is a large amount of experimental evidence indicating that mitochondrial dysfunction is implicated in the pathophysiology of both disorders (67). In this regard, it was proposed that neuronal bioenergetics impairment is a consequence of similar processes in both sporadic disorders. The most relevant alterations include elevated oxidative stress (that can damage mitochondrial respiratory complex expression and/or activity); perturbations in mitochondrial dynamics; alterations in mitochondrial transport within axons; mitophagy; accumulation of somatic mtDNA mutations; impaired quality control mechanisms leading to the accumulation of defective mitochondria; defective calcium (Ca2+) homeostasis and signaling. It was postulated that all of these processes may result in neuronal death (68). In post-mortem substantia nigra from PD patients it was found a decrease in CI activity, reduction of ATP levels, increments of ROS and impaired mitochondrial membrane potential leading to Ca2+-mediated damage (69), while in the caudate nucleus increased variability in mitochondrial morphology was also observed (70). However, a direct link between CI deficiency caused by mtDNA changes and parkinsonism has not been proven (71). Similarly, post-mortem AD brains show decreased levels of ATP (72) and decreased activity of CIV (73–75). Experimental evidence suggests that mtDNA deletions, which accumulate with age, may be responsible for CIV deficiency observed in AD (76). In addition, reduced levels of CI (77, 78) and CIII (79) were described in agreement with a recent report showing decreased expression of subunits from all respiratory complexes in the entorhinal cortex (80). The process of mitochondrial fusion/fission was also reported to be impaired in AD. Drp1, involved in mitochondrial fission, was found to be reduced in hippocampal post-mortem samples as well as the major proteins responsible for mitochondrial fusion (mitofusins and OPA1) (81, 82). However, until today the mechanisms underlying the correlation between the mitochondria dynamics and SCs assembly have not been defined. Studies carried out in AD and PD models show that alterations in mitochondrial dynamics may lead to an increased production of ROS and SCs disassembly, which in turn may further promote ROS generation and mitochondrial dysfunction. This cause/consequence cycle continuously feeds back, and it is possible that the accumulation of misfolded proteins plays a substantial role in triggering such a process. Experimental evidence supports that both, aging and oxidative stress may influence SCs structure and function. However, a systematic approach to address mitochondrial SCs in the brain of subjects affected with these neurodegenerative disorders or in the brain of animal models of AD and/or PD is scarce. The impact of aging on SCs assembly was addressed by analyzing mitochondria isolated from the cortex of young (5-months-old) and aged (30-months-old) Wistar rats (83). It was observed, using two 2D-BN/SDS-PAGE, decrements of SCs I1III2 (58% reduction); I1III2IV2 (40.7% reduction) and I1III2IV1 (30.8% reduction), suggesting that aging alters SCs abundance in the cortex, a brain area associated with cognitive functions and particularly affected in AD brains. In vitro studies of primary cultures of neurons and astrocytes from rats (Wistar strain) and mice (CBA and C57BL/6 strains) (84) using BN-PAGE followed by in-gel CI activity assay showed that Wistar rats and CBA mice exhibit higher CI activity in astrocytes than in neurons. This can be attributed to the capacity to form SCs containing CIV. In addition, in primary cultures of astrocytes from Wistar rats CI abundance in I-III-containing SCs was higher than in I-III2-IV-containing SCs, suggesting that complex IV may determine CI specific activity. Authors suggest that this mechanism could be relevant for neurodegenerative disorders in which lower CI activity has been reported (85). BN-PAGE and the in-gel activity performed in mitochondrial samples isolated from the striatum of 6-hydroxydopamine (6-OHDA) rat model of PD (86), showed that along with the degeneration process, the amount and performance of CI decreased in nearly all forms of SCs. Moreover, CIV activity in SCs (I1III2IV3−1 and I1IV2−1) progressively decreased during the degeneration process. Furthermore, SCs function has been correlated with mitochondrial membrane plasticity in 6-OHDA-induced dopaminergic neuron degeneration (87). PINK1 (a mitochondrial kinase) and DJ-1 (a redox-sensitive chaperone) loss-of-function mutations are associated with early-onset parkinsonism, and both proteins are involved in maintaining normal mitochondrial dynamics and/or oxidative stress responses (88). Experiments using mitochondria isolated from genetically modified Drosophila showed that impairment on mitochondrial fission mediated by the knock-out of PINK1 causes a decrease in the enzymatic activity and defective assembly of CI and CIV, a significant reduction in mitochondrial respiration, and a reduction in ATP synthesis (20). Furthermore, in mitochondria purified from fibroblasts of patients that carry pathogenic PINK1 mutations, it was observed a decrement in free CI, and in free CI vs. SCs-assembled CI. Free CIII was mildly affected, whereas a decrement of the free CIII vs. SCs-assembled CIII was observed. In the same direction, mitochondria from DJ-1 null dopaminergic neurons showed a decrease in CI formation and activity together with a significant reduction in the abundance of SCs (89). Interestingly, CIV was significantly lower in PD human fibroblasts. These results were also observed in primary cultures of neurons from Pink1-/- mice and in the forebrain of mice lacking DJ1 (Dj1-/-) (90). These observations suggest that OXPHOS complex assembly and function may be modulated by oxidative stress and mitochondrial dynamics, probably involving the fission/fusion machinery (91, 92). Altogether, these findings support that mitochondrial dysfunction in PD may require, among other pathways, the structural remodeling of SCs. Recently, it was shown in postmortem samples of the frontal cortex of AD subjects a significant decrease in the levels of CII, CIII, and CV subunits as compared to controls, while a strong tendency was found for decrements on CIV levels and no differences were detected in CI (93). It is of note that the differences observed by Western blotting were not a consequence of alterations in mitochondrial mass between groups. Moreover, BN-PAGE combined with immunoblotting for subunits of CI and CIII showed that the abundance of respirasomes did not differ between AD and control samples. An overview of the literature on SCs organization and activity in studies using aging, AD and PD models is detailed in Table 1 and a schematic representation of the (dis)assembly of SCs in aging, AD, and PD is shown in Figure 1C. Due to the fact that a few works have been performed to study SCs in AD and PD it became difficult to extract conclusions. More studies are needed, particularly in animal models of age-related neurodegenerative disorders, to clarify to what extent bioenergetics dysfunction at the brain level can be attributed to functional impairment of SCs.
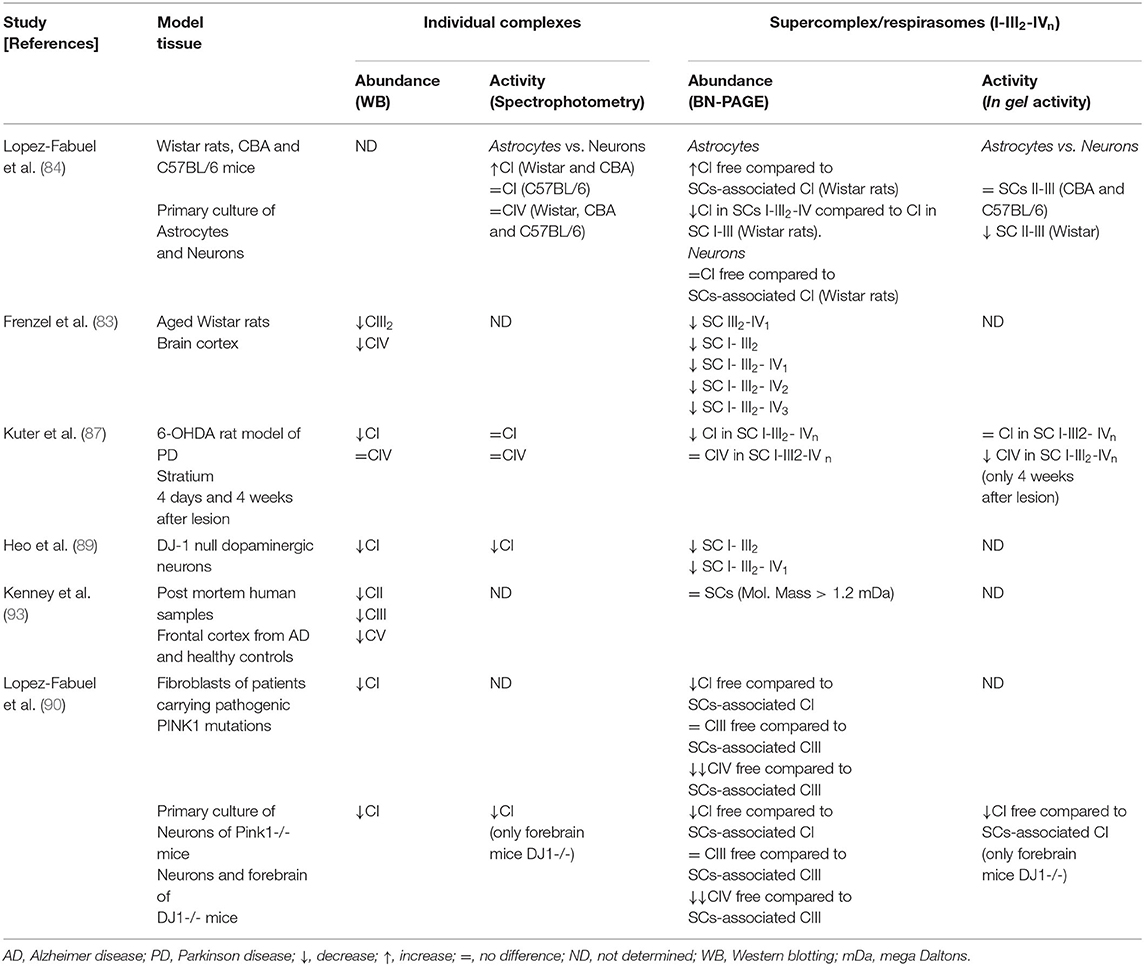
Table 1. Overview of the literature on the individual complexes and supercomplex abundance and activity in aging and neurodegeneration.
Research on the impact of normal aging and brain amyloid-associated neurological diseases such as AD and PD upon SCs is at a relatively early stage, and therefore many questions remain to be addressed. It is important to take into account that when postmortem frozen tissues are used to study the (dis)assembly of SCs there are at least four critical variables to be considered: (1) the postmortem delay; (2) the freeze-thaw processes that may fracture mitochondria; (3) the stage of disease at which brain samples are analyzed. In advanced stages changes observed in the (dis)assembly of SCs may correspond to the “surviving” neurons and glia while at early stages a more representative picture of neuronal SCs may be obtained; (4) the brain area analyzed. It is very likely that there are differences in SCs assembly in different brain regions because different brain areas are affected in each disorder (hippocampus and cortex are the most affected in AD while striatum is the most affected in PD). Since studies on SCs assembly in different brain areas within each one of these pathologies have not been carried out, this topic remains inconclusive. The convergence of future progress in the study of SCs structure at high resolution and the refinement of animal models of AD and PD as well as the use of iPSC-based and somatic cell-derived neurons will be critical in understanding the biological relevance of the structural remodeling of SCs.
GN, PG, and LM: conceptualization. GN, PG, EC, and LM: investigation, writing—original draft, and writing—review and editing. PG and LM: resources and funding acquisition. LM: supervision. All authors contributed to the article and approved the submitted version.
This work was supported by funding from the ANPCyT (PICT-2015-0285, PICT-2016-4647, and PIBT/09-2013 to LM), the ISN (CAEN Award 2018 to PG), and the EU-LAC Health Neurodegeneration award (EULAC H16/T01-0131 to LM). GN was supported by CONICET fellowship. PG, EC, and LM are members of the Research Career of CONICET.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Green DE, Tzagoloff A. The mitochondrial electron transfer chain. Arch Biochem Biophys. (1966) 116:293–304. doi: 10.1016/0003-9861(66)90036-1
2. Hochli M, Hackenbrock CR. Fluidity in mitochondrial membranes: thermotropic lateral translational motion of intramembrane particles. Proc Natl Acad Sci USA. (1976) 73:1636–40. doi: 10.1073/pnas.73.5.1636
3. Hackenbrock CR, Chazotte B, Gupte SS. The random collision model and a critical assessment of diffusion and collision in mitochondrial electron transport. J Bioenerget Biomembranes. (1986) 18:331–68. doi: 10.1007/BF00743010
4. Schagger H. Blue-native gels to isolate protein complexes from mitochondria. Method Cell Biol. (2001) 65:231–44. doi: 10.1016/S0091-679X(01)65014-3
5. Schägger H, Pfeiffer K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. (2000) 19:1777–83. doi: 10.1093/emboj/19.8.1777
6. Guo R, Zong S, Wu M, Gu J, Yang M. Architecture of human mitochondrial respiratory megacomplex I2 III2 IV2. Cell. (2017) 170:1247–57. doi: 10.1016/j.cell.2017.07.050
7. Liu F, Lössl P, Rabbitts M, Balaban RS, Heck AJR. The interactome of intact mitochondria by cross-linking mass spectrometry provides evidence for coexisting respiratory supercomplexes. Mol Cell Proteomics. (2018) 17:216–32. doi: 10.1074/mcp.RA117.000470
8. Jang S, Javadov S. Elucidating the contribution of ETC complexes I and II to the respirasome formation in cardiac mitochondria. Sci Rep. (2018) 8:17732. doi: 10.1038/s41598-018-36040-9
9. Acin-Perez R, Fernandez-Silva P, Peleato ML, Perez-Martos A, Enriquez JA. Respiratory active mitochondrial supercomplexes. Mol Cell. (2008) 32:529–39. doi: 10.1016/j.molcel.2008.10.021
10. Guerrero-Castillo S, Baertling F, Kownatzki D, Wessels HJ, Arnold S, Brandt U, et al. The assembly pathway of mitochondrial respiratory chain complex I. Cell Metab. (2017) 25:128–39. doi: 10.1016/j.cmet.2016.09.002
11. Schagger H, Pfeiffer K. The ratio of oxidative phosphorylation complexes I-V in bovine heart mitochondria and the composition of respiratory chain supercomplexes. J Biol Chem. (2001) 276:37861–7. doi: 10.1074/jbc.M106474200
12. Letts JA, Fiedorczuk K, Sazanov LA. The architecture of respiratory supercomplexes. Nature. (2016) 537:644–8. doi: 10.1038/nature19774
13. Letts JA, Sazanov LA. Clarifying the supercomplex: the higher-order organization of the mitochondrial electron transport chain. Nat Struct Mol Biol. (2017) 24:800–8. doi: 10.1038/nsmb.3460
14. Xia D, Yu CA, Kim H, Xia JZ, Kachurin AM, Zhang L, et al. Crystal structure of the cytochrome bc1 complex from bovine heart mitochondria. Science. (1997) 277:60–6. doi: 10.1126/science.277.5322.60
15. Sun D, Li B, Qiu R, Fang H, Lyu J. Cell type-specific modulation of respiratory chain supercomplex organization. Int J Mol Sci. (2016) 17:926. doi: 10.3390/ijms17060926
16. D'Aurelio M, Gajewski CD, Lenaz G, Manfredi G. Respiratory chain supercomplexes set the threshold for respiration defects in human mtDNA mutant cybrids. Hum Mol Genet. (2006) 15:2157–69. doi: 10.1093/hmg/ddl141
17. Perez-Perez R, Lobo-Jarne T, Milenkovic D, Mourier A, Bratic A, Garcia-Bartolome A, et al. COX7A2L is a mitochondrial complex III binding protein that stabilizes the III2+IV supercomplex without affecting respirasome formation. Cell Rep. (2016) 16:2387–98. doi: 10.1016/j.celrep.2016.07.081
18. Strogolova V, Furness A, Robb-McGrath M, Garlich J, Stuart RA. Rcf1 and rcf2, members of the hypoxia-induced gene 1 protein family, are critical components of the mitochondrial cytochrome bc1-cytochrome c oxidase supercomplex. Mol Cell Biol. (2012) 32:1363–73. doi: 10.1128/MCB.06369-11
19. Hatle KM, Gummadidala P, Navasa N, Bernardo E, Dodge J, Silverstrim B, et al. MCJ/DnaJC15, an endogenous mitochondrial repressor of the respiratory chain that controls metabolic alterations. Mol Cell Biol. (2013) 33:2302–14. doi: 10.1128/MCB.00189-13
20. Liu W, Acin-Perez R, Geghman KD, Manfredi G, Lu B, Li C. Pink1 regulates the oxidative phosphorylation machinery via mitochondrial fission. Proc Natl Acad Sci USA. (2011) 108:12920–4. doi: 10.1073/pnas.1107332108
21. Baker N, Patel J, Khacho M. Linking mitochondrial dynamics, cristae remodeling and supercomplex formation: how mitochondrial structure can regulate bioenergetics. Mitochondrion. (2019) 49:259–68. doi: 10.1016/j.mito.2019.06.003
22. Horvath SE, Daum G. Lipids of mitochondria. Progr Lipid Res. (2013) 52:590–614. doi: 10.1016/j.plipres.2013.07.002
23. Basu Ball W, Neff JK, Gohil VM. The role of non-bilayer phospholipids in mitochondrial structure and function. FEBS Lett. (2018) 592:1273–90. doi: 10.1002/1873-3468.12887
24. Dudek J, Cheng IF, Balleininger M, Vaz FM, Streckfuss-Bomeke L, Hubscher D, et al. Cardiolipin deficiency affects respiratory chain function and organization in an induced pluripotent stem cell model of barth syndrome. Stem Cell Res. (2013) 11:806–19. doi: 10.1016/j.scr.2013.05.005
25. Althoff T, Mills DJ, Popot JL, Kuhlbrandt W. Arrangement of electron transport chain components in bovine mitochondrial supercomplex I1III2IV1. EMBO J. (2011) 30:4652–64. doi: 10.1038/emboj.2011.324
26. McKenzie M, Lazarou M, Thorburn DR, Ryan MT. Mitochondrial respiratory chain supercomplexes are destabilized in Barth Syndrome patients. J Mol Biol. (2006) 361:462–9. doi: 10.1016/j.jmb.2006.06.057
27. Xu Y, Phoon CK, Berno B, D'Souza K, Hoedt E, Zhang G, et al. Loss of protein association causes cardiolipin degradation in Barth Syndrome. Nat Chem Biol. (2016) 12:641–7. doi: 10.1038/nchembio.2113
28. Flis VV, Daum G. Lipid transport between the endoplasmic reticulum and mitochondria. Cold Spring Harbor Perspect Biol. (2013) 5:a013235. doi: 10.1101/cshperspect.a013235
29. Schauder CM, Wu X, Saheki Y, Narayanaswamy P, Torta F, Wenk MR, et al. Structure of a lipid-bound extended synaptotagmin indicates a role in lipid transfer. Nature. (2014) 510:552–5. doi: 10.1038/nature13269
30. Giacomello M, Pellegrini L. The coming of age of the mitochondria-ER contact: a matter of thickness. Cell Death Differ. (2016) 23:1417–27. doi: 10.1038/cdd.2016.52
31. Leon WC, Canneva F, Partridge V, Allard S, Ferretti MT, DeWilde A, et al. A novel transgenic rat model with a full Alzheimer's-like amyloid pathology displays pre-plaque intracellular amyloid-beta-associated cognitive impairment. J Alzheimers Dis. (2010) 20:113–26. doi: 10.3233/JAD-2010-1349
32. Martino Adami PV, Nichtova Z, Weaver DB, Bartok A, Wisniewski T, Jones DR, et al. Perturbed mitochondria-ER contacts in live neurons that model the amyloid pathology of Alzheimer's disease. J Cell Sci. (2019) 132:229906. doi: 10.1242/jcs.229906
33. Leuner K, Hauptmann S, Abdel-Kader R, Scherping I, Keil U, Strosznajder JB, et al. Mitochondrial dysfunction: the first domino in brain aging and Alzheimer's disease? Antioxid Redox Signal. (2007) 9:1659–75. doi: 10.1089/ars.2007.1763
34. Mancuso M, Coppede F, Murri L, Siciliano G. Mitochondrial cascade hypothesis of Alzheimer's disease: myth or reality? Antioxid Redox Signal. (2007) 9:1631–46. doi: 10.1089/ars.2007.1761
35. Mosconi L, Mistur R, Switalski R, Tsui WH, Glodzik L, Li Y, et al. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer's disease. Eur J Nucl Med Mol Imaging. (2009) 36:811–22. doi: 10.1007/s00259-008-1039-z
36. Vukotic M, Oeljeklaus S, Wiese S, Vogtle FN, Meisinger C, Meyer HE, et al. Rcf1 mediates cytochrome oxidase assembly and respirasome formation, revealing heterogeneity of the enzyme complex. Cell Metab. (2012) 15:336–47. doi: 10.1016/j.cmet.2012.01.016
37. Enriquez JA. Supramolecular organization of respiratory complexes. Ann Rev Physiol. (2016) 78:533–61. doi: 10.1146/annurev-physiol-021115-105031
38. Genova ML, Lenaz G. Functional role of mitochondrial respiratory supercomplexes. Biochim Biophys Acta. (2014) 1837:427–43. doi: 10.1016/j.bbabio.2013.11.002
39. Hirst J. Mitochondrial complex I. Ann Rev Biochem. (2013) 82:551–75. doi: 10.1146/annurev-biochem-070511-103700
40. Lamantea E, Carrara F, Mariotti C, Morandi L, Tiranti V, Zeviani M, et al. A novel nonsense mutation (Q352X) in the mitochondrial cytochrome b gene associated with a combined deficiency of complexes I and III. Neuromuscul Disord. (2002) 12:49–52. doi: 10.1016/S0960-8966(01)00244-9
41. Fernandez-Vizarra E, Zeviani M. Nuclear gene mutations as the cause of mitochondrial complex III deficiency. Front Genet. (2015) 6:134. doi: 10.3389/fgene.2015.00134
42. Rak M, Benit P, Chretien D, Bouchereau J, Schiff M, El-Khoury R, et al. Mitochondrial cytochrome c oxidase deficiency. Clin Sci. (2016) 130:393–407. doi: 10.1042/CS20150707
43. Lapuente-Brun E, Moreno-Loshuertos R, Acin-Perez R, Latorre-Pellicer A, Colas C, Balsa E, et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science. (2013) 340:1567–70. doi: 10.1126/science.1230381
44. Maranzana E, Barbero G, Falasca AI, Lenaz G, Genova ML. Mitochondrial respiratory supercomplex association limits production of reactive oxygen species from complex I. Antioxid Redox Signal. (2013) 19:1469–80. doi: 10.1089/ars.2012.4845
45. Lopez-Fabuel I, Le douce J, Logan A, James AM, Bonvento G, Murphy MP, et al. Complex I assembly into supercomplexes determines differential mitochondrial ROS production in neurons and astrocytes. Proc Natl Acad Sci USA. (2016) 113:13063–8. doi: 10.1073/pnas.1613701113
46. Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature. (1961) 191:144–8. doi: 10.1038/191144a0
47. Moreno-Lastres D, Fontanesi F, Garcia-Consuegra I, Martin MA, Arenas J, Barrientos A, et al. Mitochondrial complex I plays an essential role in human respirasome assembly. Cell Metab. (2012) 15:324–35. doi: 10.1016/j.cmet.2012.01.015
48. Blaza JN, Serreli R, Jones AJ, Mohammed K, Hirst J. Kinetic evidence against partitioning of the ubiquinone pool and the catalytic relevance of respiratory-chain supercomplexes. Proc Natl Acad Sci USA. (2014) 111:15735–40. doi: 10.1073/pnas.1413855111
49. Milenkovic D, Blaza JN, Larsson NG, Hirst J. The enigma of the respiratory chain supercomplex. Cell Metab. (2017) 25:765–776. doi: 10.1016/j.cmet.2017.03.009
50. Schafer E, Seelert H, Reifschneider NH, Krause F, Dencher NA, Vonck J. Architecture of active mammalian respiratory chain supercomplexes. J Biol Chem. (2006) 281:15370–5. doi: 10.1074/jbc.M513525200
51. Smirnov AY, Nori F. Modeling the Q-cycle mechanism of transmembrane energy conversion. Phys Biol. (2012) 9:016011. doi: 10.1088/1478-3975/9/1/016011
52. Fedor JG, Hirst J. Mitochondrial supercomplexes do not enhance catalysis by quinone channeling. Cell Metab. (2018) 28:525–531 e4. doi: 10.1016/j.cmet.2018.05.024
53. Fernandez-Ayala DJ, Sanz A, Vartiainen S, Kemppainen KK, Babusiak M, Mustalahti E, et al. Expression of the Ciona intestinalis alternative oxidase (AOX) in Drosophila complements defects in mitochondrial oxidative phosphorylation. Cell Metab. (2009) 9:449–60. doi: 10.1016/j.cmet.2009.03.004
54. El-Khoury R, Dufour E, Rak M, Ramanantsoa N, Grandchamp N, Csaba Z, et al. Alternative oxidase expression in the mouse enables bypassing cytochrome c oxidase blockade and limits mitochondrial ROS overproduction. PLoS Genet. (2013) 9:e1003182. doi: 10.1371/journal.pgen.1003182
55. Gu J, Wu M, Guo R, Yan K, Lei J, Gao N, et al. The architecture of the mammalian respirasome. Nature. (2016) 537:639–43. doi: 10.1038/nature19359
56. Sousa JS, Mills DJ, Vonck J, Kuhlbrandt W. Functional asymmetry and electron flow in the bovine respirasome. eLife. (2016) 5:21290. doi: 10.7554/eLife.21290
57. Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. (2013) 155:160–71. doi: 10.1016/j.cell.2013.08.032
58. Sunderhaus S, Eubel H, Braun HP. Two-dimensional blue native/blue native polyacrylamide gel electrophoresis for the characterization of mitochondrial protein complexes and supercomplexes. Methods Mol Biol. (2007) 372:315–24. doi: 10.1007/978-1-59745-365-3_23
59. Cuillerier A, Burelle Y. Hybrid clear/blue native electrophoresis for the separation and analysis of mitochondrial respiratory chain supercomplexes. J Visualized Exp. (2019) 147:e59294. doi: 10.3791/59294
60. Schweppe DK, Chavez JD, Lee CF, Caudal A, Kruse SE, Stuppard R, et al. Mitochondrial protein interactome elucidated by chemical cross-linking mass spectrometry. Proc Natl Acad Sci USA. (2017) 114:1732–7. doi: 10.1073/pnas.1617220114
61. Wu M, Gu J, Guo R, Huang Y, Yang M. Structure of mammalian respiratory supercomplex I1III2IV1. Cell. (2016) 167:1598–609 e10. doi: 10.1016/j.cell.2016.11.012
62. Roux KJ. Marked by association: techniques for proximity-dependent labeling of proteins in eukaryotic cells. Cell Mol Life Sci. (2013) 70:3657–64. doi: 10.1007/s00018-013-1287-3
63. Rees JS, Li XW, Perrett S, Lilley KS, Jackson AP. Protein neighbors and proximity proteomics. Mol Cell Proteomics. (2015) 14:2848–56. doi: 10.1074/mcp.R115.052902
64. Varnaite R, MacNeill SA. Meet the neighbors: mapping local protein interactomes by proximity-dependent labeling with BioID. Proteomics. (2016) 16:2503–18. doi: 10.1002/pmic.201600123
65. Jang S, Javadov S. Current challenges in elucidating respiratory supercomplexes in mitochondria: methodological obstacles. Front Physiol. (2018) 9:238. doi: 10.3389/fphys.2018.00238
66. Rieger B, Shalaeva DN, Sohnel AC, Kohl W, Duwe P, Mulkidjanian AY, et al. Lifetime imaging of GFP at CoxVIIIa reports respiratory supercomplex assembly in live cells. Sci Rep. (2017) 7:46055. doi: 10.1038/srep46055
67. Moran M, Moreno-Lastres D, Marin-Buera L, Arenas J, Martin MA, Ugalde C. Mitochondrial respiratory chain dysfunction: implications in neurodegeneration. Free Radic Biol Med. (2012) 53:595–609. doi: 10.1016/j.freeradbiomed.2012.05.009
68. Tapias V. Mitochondrial dysfunction and neurodegeneration. Front Neurosci. (2019) 13:1372. doi: 10.3389/fnins.2019.01372
69. Greenamyre JT, Sherer TB, Betarbet R, Panov SV. Complex I and Parkinson's disease. IUBMB Life. (2001) 52:135–41. doi: 10.1080/15216540152845939
70. Lach B, Grimes D, Benoit B, Minkiewicz-Janda A. Caudate nucleus pathology in Parkinson's disease: ultrastructural and biochemical findings in biopsy material. Acta Neuropathol. (1992) 83:352–60. doi: 10.1007/BF00713525
71. Palin EJH, Hakonen AH, Korpela M, Paetau A, Suomalainen A. Mitochondrial recessive ataxia syndrome mimicking dominant spinocerebellar ataxia. J Neurol Sci. (2012) 315:160–3. doi: 10.1016/j.jns.2011.11.028
72. Hoyer S. Oxidative energy metabolism in Alzheimer brain: studies in early-onset and late-onset cases. Mol Chem Neuropathol. (1992) 16:207–24. doi: 10.1007/BF03159971
73. Kish SJ. Bergeron C, Rajput A, Dozic S, Mastrogiacomo F, Chang JL, et al. Brain cytochrome oxidase in Alzheimer's disease. J Neurochem. (1992) 59:776–9. doi: 10.1111/j.1471-4159.1992.tb09439.x
74. Mutisya EM, Bowling AC, Beal MF. Cortical cytochrome oxidase activity is reduced in Alzheimer's disease. J Neurochem. (1994) 63:2179–84. doi: 10.1046/j.1471-4159.1994.63062179.x
75. Maurer I, Zierz S, Möller HJ. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol Aging. (2000) 21:455–62. doi: 10.1016/S0197-4580(00)00112-3
76. Krishnan KJ, Ratnaike TE, De Gruyter HLM, Jaros E, Turnbull DM. Mitochondrial DNA deletions cause the biochemical defect observed in Alzheimer's disease. Neurobiol Aging. (2012) 33:2210–14. doi: 10.1016/j.neurobiolaging.2011.08.009
77. Fukuyama R, Hatanpää K, Rapoport SI, Chandrasekaran K. Gene expression of ND4, a subunit of complex I of oxidative phosphorylation in mitochondria, is decreased in temporal cortex of brains of Alzheimer's disease patients. Brain Res. (1996) 713:290–3. doi: 10.1016/0006-8993(95)01517-5
78. Kim SH, Vlkolinsky R, Cairns N, Fountoulakis M, Lubec G. The reduction of NADH: Ubiquinone oxidoreductase 24- and 75-kDa subunits in brains of patients with Down syndrome and Alzheimer's disease. Life Sci. (2001) 68:2741–50. doi: 10.1016/S0024-3205(01)01074-8
79. Kim SH, Vlkolinsky R, Cairns N, Lubec G. Decreased levels of complex III core protein 1 and complex V β chain in brains from patients with Alzheimer's disease and down syndrome. Cell Mol Life Sci. (2000) 57:1810–6. doi: 10.1007/PL00000661
80. Armand-Ugon M, Ansoleaga B, Berjaoui S, Ferrer I. Reduced mitochondrial activity is early and steady in the entorhinal cortex but it is mainly unmodified in the frontal cortex in Alzheimer's disease. Curr Alzheimer Res. (2017) 14:1327–34. doi: 10.2174/1567205014666170505095921
81. Wang X, Su B, Lee H-g, Li X, Perry GM. Smith Zhu AX. Impaired balance of mitochondrial fission fusion in Alzheimer's disease. J Neurosci. (2009) 29:9090–103. doi: 10.1523/JNEUROSCI.1357-09.2009
82. Manczak M, Calkins MJ, Reddy PH. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: implications for neuronal damage. Hum Mol Genet. (2011) 20:2495–509. doi: 10.1093/hmg/ddr139
83. Frenzel M, Rommelspacher H, Sugawa MD, Dencher NA. Ageing alters the supramolecular architecture of OxPhos complexes in rat brain cortex. Exp Gerontol. (2010) 45:563–72. doi: 10.1016/j.exger.2010.02.003
84. Lopez-Fabuel I, Resch-Beusher M, Carabias-Carrasco M, Almeida A, Bolanos JP. Mitochondrial complex I activity is conditioned by supercomplex I-III2-IV assembly in brain cells: relevance for Parkinson's disease. Neurochem Res. (2017) 42:1676–82. doi: 10.1007/s11064-017-2191-2
85. Papa S, De rasmo D. Complex i deficiencies in neurological disorders. Trends Mol Med. (2013) 19:61–9. doi: 10.1016/j.molmed.2012.11.005
86. Jeon BS, Jackson-Lewis V, Burke RE. 6-Hydroxydopamine lesion of the rat substantia nigra: time course and morphology of cell death. Neurodegen J Neurodegenerat Disord Neuroprotect Neuroregeneration. (1995) 4:131–7. doi: 10.1006/neur.1995.0016
87. Kuter K, Kratochwil M, Berghauzen-Maciejewska K, Glowacka U, Sugawa MD, Ossowska K, et al. Adaptation within mitochondrial oxidative phosphorylation supercomplexes and membrane viscosity during degeneration of dopaminergic neurons in an animal model of early Parkinson's disease. Biochim Biophys Acta. (2016) 1862:741–53. doi: 10.1016/j.bbadis.2016.01.022
88. Cookson MR. Parkinsonism due to mutations in PINK1, parkin, and DJ-1 and oxidative stress and mitochondrial pathways. Cold Spring Harbor Perspect Med. (2012) 2:a009415. doi: 10.1101/cshperspect.a009415
89. Heo JY, Park JH, Kim SJ, Seo KS, Han JS, Lee SH, et al. DJ-1 null dopaminergic neuronal cells exhibit defects in mitochondrial function and structure: involvement of mitochondrial complex I assembly. PLoS ONE. (2012) 7:e32629. doi: 10.1371/journal.pone.0032629
90. Lopez-Fabuel I, Martin-Martin L, Resch-Beusher M, Azkona G, Sanchez-Pernaute R, Bolanos JP. Mitochondrial respiratory chain disorganization in Parkinson's disease-relevant PINK1 and DJ1 mutants. Neurochem Int. (2017) 109:101–5. doi: 10.1016/j.neuint.2017.03.023
91. Kapogiannis D, Mattson MP. Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer's disease. Lancet Neurol. (2011) 10:187–98. doi: 10.1016/S1474-4422(10)70277-5
92. Chen YC, Taylor EB, Dephoure N, Heo JM, Tonhato A, Papandreou I, et al. Identification of a protein mediating respiratory supercomplex stability. Cell Metab. (2012) 15:348–60. doi: 10.1016/j.cmet.2012.02.006
Keywords: supercomplexes organization, respirasome structure, neurodegeneration, Alzheimer's disease, Parkinson's disease, mitochondrial dysfunction, brain bioenergetics
Citation: Novack GV, Galeano P, Castaño EM and Morelli L (2020) Mitochondrial Supercomplexes: Physiological Organization and Dysregulation in Age-Related Neurodegenerative Disorders. Front. Endocrinol. 11:600. doi: 10.3389/fendo.2020.00600
Received: 22 May 2020; Accepted: 22 July 2020;
Published: 11 September 2020.
Edited by:
Beatrice Maria Filippi, University of Leeds, United KingdomReviewed by:
Renato Xavier Coelho dos Santos, University of Aberdeen, United KingdomCopyright © 2020 Novack, Galeano, Castaño and Morelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura Morelli, bG1vcmVsbGlAbGVsb2lyLm9yZy5hcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.