 Rafiou Agoro
Rafiou Agoro Pu Ni
Pu Ni Megan L. Noonan1
Megan L. Noonan1 Kenneth E. White
Kenneth E. White
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 28 August 2020
Sec. Bone Research
Volume 11 - 2020 | https://doi.org/10.3389/fendo.2020.00592
This article is part of the Research Topic Bone Inside-Out and Outside-In Signals: Control of Body Homeostasis View all 21 articles
Osteocytes, which represent up to 95% of adult skeletal cells, are deeply embedded in bone. These cells exhibit important interactive abilities with other bone cells such as osteoblasts and osteoclasts to control skeletal formation and resorption. Beyond this local role, osteocytes can also influence the function of distant organs due to the presence of their sophisticated lacunocanalicular system, which connects osteocyte dendrites directly to the vasculature. Through these networks, osteocytes sense changes in circulating metabolites and respond by producing endocrine factors to control homeostasis. One critical function of osteocytes is to respond to increased blood phosphate and 1,25(OH)2 vitamin D (1,25D) by producing fibroblast growth factor-23 (FGF23). FGF23 acts on the kidneys through partner fibroblast growth factor receptors (FGFRs) and the co-receptor Klotho to promote phosphaturia via a downregulation of phosphate transporters, as well as the control of vitamin D metabolizing enzymes to reduce blood 1,25D. In the first part of this review, we will explore the signals involved in the positive and negative regulation of FGF23 in osteocytes. In the second portion, we will bridge bone responses with the review of current knowledge on FGF23 endocrine functions in the kidneys.
The mammalian skeleton is formed by several types of bone which interconnect to provide structural support for the body. In adult humans, the reference value for the skeleton weight is 10.5 kg (1, 2) representing up to 15% of the average body weight. Bone mass is dynamically regulated during a lifetime, and subjected to changes with uncontrollable parameters such as age, gender, genetics, and ethnicity; as well as controllable factors such as lifestyle behaviors including physical activity levels, smoking and alcohol consumption patterns, and diet (3, 4). Beyond its important role to enable mobility and provide needed support and structure to the body, bone represents an important reservoir of several minerals including phosphate and calcium, both of which are required for proper mineral metabolism and cellular functions (5).
Bone is a mineralized connective tissue formed with three primary cell types that direct intrinsic skeletal properties: osteoclasts, which resorb mineralized bone; osteoblasts, which form the bone matrix; and osteocytes, which are considered terminally differentiated osteoblasts (6, 7). Although morphologically and functionally distinct in the bone, osteoclasts, osteoblasts, and osteocytes are interdependent and produce growth factors to support each other's functions as well as responding in a coordinated manner to metabolic demands, physical stimuli, and structural duties (8). The three main bone cells are derived from two distinct lineages; osteoblasts and osteocytes derive from pluripotent mesenchymal stem cells and share the same progenitors as fibroblasts and adipocytes (9, 10), whereas osteoclasts are derived from hematopoietic progenitors in the monocyte and macrophage lineage (11–13).
The osteocytes, which represent the majority of adult skeletal cells, are deeply embedded with abilities to communicate locally with osteoblasts and osteoclasts. This function is necessary to control skeletal formation as orchestrated by osteoblasts, and bone resorption dictated by osteoclasts, as well as controlling the physiological function of distant organs such as the kidney. Taking advantage of its dendrites, which connect to the vasculature and give these atypical cells direct access to the circulation, the osteocyte can send and receive signals with the vascularized organs. Among the important osteocyte-secreted factors is fibroblast growth factor-23 (FGF23). Once produced and secreted by osteocytes, this hormone preferentially acts on kidney and parathyroid glands to regulate phosphate and vitamin D homeostasis. FGF23 activity mainly occurs through the binding of FGF23 to FGF receptor (likely FGFR1), which requires the presence of its membrane and/or soluble co-receptor Klotho for a potent FGF23-induced downstream signaling cascade (14). In this review, the stimulative and repressive regulatory mechanisms involved in FGF23 production and processing in osteocytes will be discussed. We will also bridge the control of FGF23 in osteocytes with highlighting the key signaling pathways involved in phosphate, 1,25D, calcium, and sodium metabolism induced by FGF23 in the kidneys.
As the most prevalent cell in bone, osteocytes have important roles both within, and outside the skeleton. The osteocytes are considered as major orchestrators of skeletal activity; these cells can sense and integrate mechanical and chemical stimuli from the microenvironment with the goal to properly regulate bone formation and resorption. Osteocytes derive from mature and matrix-producing terminally differentiated osteoblasts (6). During their last phase of differentiation, mature osteoblasts become embedded in the matrix and generate cellular extensions, which are future osteocyte dendrites. To establish a sophisticated and complex network called the lacunocanalicular system, the dendrites of the newly formed osteocytes are fastened with the dendrites of existing osteocytes through a multitude of canaliculi (15). Even after the terminal differentiation of mature osteoblasts to generate osteocytes, the latter remain active in contributing to bone remodeling. For instance, osteocytes produce sclerostin (SOST), which binds to low-density lipoprotein receptor-related protein (Lrp)5/6, and neutralizes the anabolic Wnt/beta-catenin pathway (16, 17), thus negatively regulating bone formation. Osteocytes also produce the receptor activator of nuclear factor-κB ligand (RANKL) which stimulates osteoclastogenesis, thus promoting bone resorption (18, 19).
In bone, osteocytes are bathed in canalicular fluid that delivers and exchanges nutrients, circulating factors, mechanical signals, and oxygen between the circulation and the “fixed and embedded” osteocytes (20). During the last decade, the osteocyte lacunocanalicular network has gained tremendous attention because of accumulating and convincing evidence describing osteocytes as a major endocrine cell, and its role in the production of critical hormones targeting several organs. One of the most important osteocyte-secreted factors is FGF23. This hormone was first characterized as a mammalian “phosphatonin” by identifying stabilizing mutations in the FGF23 gene in patients with autosomal dominant hypophosphatemic rickets (ADHR), a renal phosphate wasting disorder (21).
FGF23 is a phosphaturic hormone derived and secreted primarily by bone osteocytes. Mature, bioactive FGF23 is physiologically designed to target the kidney to regulate phosphate and vitamin D homeostasis; and, in a feedback mechanism to control bone mineralization and FGF23 production (22, 23). The mechanisms of FGF23 regulation in osteocytes are not fully understood. Several breakthroughs have been made that greatly improved our knowledge on the mechanisms of osteocytic FGF23 upregulation or downregulation through differential signaling pathways, as well as the pathophysiological response to multiple stimuli. In addition to several mechanisms involved in the transcriptional regulation of FGF23, another layer of FGF23 regulation in bone is the ability of the mature protein to be proteolytically cleaved within osteocytes to generate inactive FGF23 fragments before its secretion into the bloodstream (24).
Circulating levels of phosphate control FGF23 production in mammals (25, 26). Although the mechanisms of FGF23 regulation by phosphate are not fully understood, recent studies have implicated the type III sodium phosphate co-transporter PiT2 (Slc20a2) as being required for mediating phosphate-dependent FGF23 production (27). Indeed, in vivo studies using dietary protocols in PiT2 knock out mice showed that the deletion of PiT2 results in “inappropriately” normal intact, bioactive FGF23 in the circulation in response to high or low phosphate diet, which should normally increase or decrease FGF23, respectively (27). Using an ex vivo system of cultured long bone shafts, parallel studies showed that the PiT2 KO bone shafts failed to undergo Pi-induced FGF23 production, illustrating that PiT2 could be required for FGF23 induction in mouse bone (27). These new findings provided interesting insight underlying the phosphate-dependent regulation of FGF23 secretion, perhaps via PiT2 regulating phosphate uptake in osteocytes (27) (Figure 1).
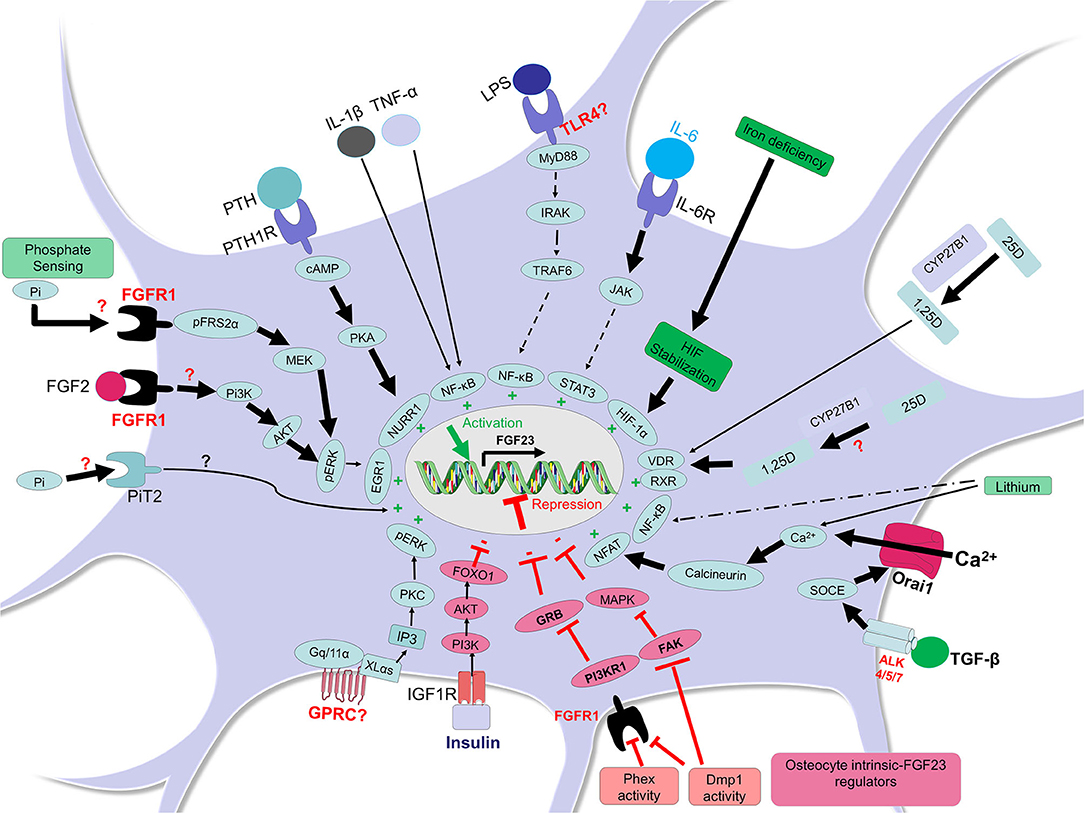
Figure 1. FGF23 regulation in osteocytes. In osteocytes, FGF23 is positively regulated by different factors and pathways (shown in green). In presence of high phosphate, the receptor FGFR1 may sense this condition and the adaptor FRS2α is phosphorylated thus activating FGFR1 and the MEK/pERK/EGR1 pathway leading to FGF23 transcription. It is also possible that the transporter PiT2 senses phosphate increases in osteocyte and responds by inducing FGF23. The activation of FGFR1 by other FGFs such as FGF2 may induce Pi3K/AKT/pERK/EGR1 leading to FGF23 upregulation. The activation of PTH1R by PTH induces cAMP/PKA and the transcription factor NURR1 leading to FGF23 induction. Inflammatory conditions are potent FGF23 inducers; lipopolysaccharide (LPS) may bind to osteocyte toll like receptor 4 (TLR4) and induce MyD88/IRAK/TRAF6/NF-κB to trigger FGF23 transcription (or may act through anemia/hypoxia). The binding of IL-6 to IL-6R in osteocytes may induce JAK/STAT3 and subsequent FGF23 expression. The cytokines IL-1β and TNF-α can induce NF-κB in osteocytes leading to FGF23 transcription. In the condition of iron deficiency, HIF protein is stabilized, an important mechanism involved in FGF23 upregulation. The active form of vitamin D (1,25-dihydroxyvitamin D; 1,25D), which results from the conversion of 25-hydroxyvitamin D (25D) inside or outside the osteocyte, could promote the formation of the complex 1,25D-VDR-RXR which binds the vitamin D receptor element (VDRE, not shown here) and induces FGF23 transcription. The signaling through the TGF-β-Activin Receptors ALK4/5/7 promotes calcium entry in cell through Orai1 and this process activates calcineurin and NFAT and their binding, which induces FGF23. Lithium-induced increased intracellular calcium could also increase FGF23 in osteocytes through calcineurin and NFAT activity. An activation of a yet unknown GPRC could trigger FGF23 transcription through XLαs or Gq/11α and inositol 1,4,5-trisphosphate (IP3) which may activate PKC and stimulate the MAPK pathway. FGF23 can be negatively regulated by other elements and pathways (shown in red). For instance, insulin can bind to IGFR1 and induce PI3K/AKT/FOXO1 pathway which represses FGF23. Phex and Dmp1 activities in osteocytes may control FGFR1/PI3KR1/GRB and cell differentiation to restrain FGF23 production. Dmp1 activity may also neutralize the FAK/MAPK pathway restricting FGF23 expression.
In another study, using proteomic analysis to identify potential upstream sensors in response to elevated phosphate through dietary intervention, the FGFR1 isoform FGFR1c was shown to be activated through the phosphorylation of FGFR substrate 2α (FRS2α, on tyrosine 196) under high phosphate conditions, but in the absence of an FGF ligand. These studies were consistent with the idea that phosphate-sensing mechanisms could control FGF23 production in osteocytes through ligand-independent FGFR1 activation (28, 29). These data suggested that high Pi diet-induced serum FGF23 increases in mice may be mediated by the phosphorylation of FGFR1, a potential “phosphate receptor” upstream of FGF23, which induced the mitogen activated protein kinase (MAPK) and extracellular signal-regulated kinase (ERK) pathways to control FGF23 synthesis (28). Structural and crystallographic studies may be required to understand how an ion can activate an FGFR, as well as whether PiT2 is involved in this regulation (Figure 1).
Additionally, in vivo studies have reported that conditional deletion of FGFR1 specifically from the osteocytes of Hyp mice, a mouse model of X-linked hypophosphatemia (XLH) that exhibits elevated FGF23 production, improves the rickets and osteomalacia phenotype of these mice in association with a decrease in both FGF23 bone mRNA and circulating protein. These mice also undergo an alleviation of their hypophosphatemic status, as well as an increase of plasma 1,25D levels (30). Furthermore, promoter studies identified that the activation of FGFR1 signaling through FGF2 is dependent on the PI3K-AKT pathway in the MC3T3-E1 osteoblastic cell line, providing evidence that Fgf23 gene promoter activity is induced by FGFR1 activation (30). In addition to genetic interventions, pharmacologic-based studies have shown that activation of FGFR1 induces FGF23 production and leads to hypophosphatemia, whereas the inhibition of FGFR signaling attenuates FGF23 production (31) (Figure 1).
Another study by Hori et al. investigated whether phosphate could induce reactive oxygen species (ROS) in vitro, leading to increased FGF23 expression. Using the osteoblastic cell line UMR-106, these investigators showed that elevated phosphate in the culture media enhances the production of ROS, and that hydrogen peroxide further boosts FGF23 production in a dose-dependent manner, an effect which can be neutralized by an inhibitor of NADPH oxidase (32). These discoveries suggested that in vitro phosphate directly enhanced FGF23 transcription by stimulating NADPH-induced ROS production and the MEK-ERK pathway. How this translates to the signaling in the osteocyte in vivo will be an interesting facet to this potential regulatory network.
Circulating FGF23 is markedly elevated during chronic kidney disease (CKD), and this is associated with poor long-term outcomes. By investigating the regulation of FGF23 through FGFR1, Hassan et al. demonstrated that the activation of FGFR1 is essential for the high levels of FGF23 observed during both acute and chronic uremia in mice and rats (33). In addition, in mice treated with the receptor FGFR1 inhibitor PD173074 by oral gavage followed with an acute kidney injury induction using folic acid, the prevailing increased FGF23 was reduced by 50% in calvaria and led to a complete prevention of the circulating intact FGF23 rises (33). In a more prolonged uremic condition using a diet with adenine plus high-phosphorus for 14 days to induce CKD, which resulted in high levels of Fgf23 mRNA and serum FGF23 in rats, an oral gavage intervention with PD173074 given during the last 2 days of treatment reduced FGF23 induction by 75% in calvaria and completely normalized circulating FGF23 (33). Therefore, the FGFRs may play an important role in osteocyte-bloodstream communication to control FGF23 production.
As mentioned above, FGF23 signals via the interaction with its receptor FGFR1c and its specific co-receptor Klotho (14, 34). A recent investigation has shown that Klotho could be detected in osteocytes (35) suggesting that FGF23 may be one of the ligands that activates FGFR1 in osteocytes, thereby regulating the FGF23 gene transcription in a positive feedback loop. The expression of Klotho in osteocytes has been suggested to contribute to bone formation and bone volume increases coupled with enhanced osteoblast activity (35). Although Klotho has been detected in bone (35), the kidney, parathyroid glands and brain remain the primary organs with abundant expression of Klotho to the best of our current knowledge (36–38). However, it is possible that in response to pathological circumstances, bone cells could enhance Klotho expression, thus modulating bone FGFR1 signaling via FGF23.
Through proteolytic cleavage of the membrane-bound klotho (mKL) (39–42), a soluble form of Klotho (sKL) can be liberated into the circulation (43, 44). Soluble Klotho has been described to have a direct role to regulate Wnt and MAPK pathways in osteoblastic UMR-106 cells in concert with the presence of FGF23 (45). Indeed, in vitro studies showed that a co-treatment of UMR-106 cells with FGF23 and soluble Klotho activated MAPK signaling, leading to an increase of Dickkopf-1 (DKK1) protein, a soluble inhibitor of Wnt/beta-catenin signaling (45). The induction of Dkk1 through FGF23/FGFR/sKL was shown to be dependent upon the MAPK pathway since the inhibition of this pathway using the MEK inhibitor U0126 completely abrogated p-ERK/ERK induction and abolished downstream Dkk1 expression (45). The binding of the secreted Dkk1 to the receptors Frizzled (Fz) and Lrp5/6 thus promoted the phosphorylation of β-catenin and inactivated the Wnt pathway in osteoblasts (45). Other studies have shown that a treatment of UMR-106 cells with soluble Klotho and FGF23 dose-dependently increased MAPK and Egr1 mRNA responses, an effect which was FGFR- and MEK-dependent, and led to FGF23 upregulation (46). Future studies are needed to confirm potential expression of Klotho in osteocytes and under some circumstances whether FGF23 binding to FGFR1-Klotho complex in osteocytes could induce FGF23 expression.
Inflammation is a complex phenomenon involving multiple immune and non-immune cells which cooperate to respond to endogenous and exogenous events by secreting specific pro-inflammatory and/or anti-inflammatory factors. Pro-inflammatory stimuli such as the cytokines tumor necrosis factor alpha (TNFα), interleukin 1β (IL-1β), the tumor necrosis factor-like weak inducer of apoptosis (TWEAK), and the bacterial component lipopolysaccharide (LPS) have all been shown to dose-dependently upregulate FGF23 in the osteocyte-like cell line IDG-SW3 (47). Particularly, TNF and IL-1β induce FGF23 expression via nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) (47), a major transcription factor complex involved in the control of cytokines and in the mediation of multiple pro-inflammatory cellular responses. The serine/threonine kinase p38 mitogen-activated protein kinase (p38MAPK), which is activated by several cellular stress stimuli and involved in the transcriptional activity of NF-κB, is another positive regulator of FGF23 in bone cells (48). Other investigations have shown that the serine/threonine kinase protein kinase C (PKC), which drives FGF23 expression in response to phorbol ester 12-O-tetradecanoylphorbol-13-acetate (PMA), can be suppressed by 75% in the presence of the PKCα/β inhibitor Go6976. These studies suggested that PKC is a positive regulator of FGF23 synthesis in IDG-SW3 osteocytic cells via NF-κB (49). Furthermore, Notch signaling, which can be mediated by pro-inflammatory cytokines such as TNF-α (50), has been described by Tamamura et al., to positively regulate FGF23 expression which colocalizes with Notch signaling downstream targets, Notch, and Hes1 in mouse osteocytes and UMR-106 osteoblastic cells (51). A study also identified a potential new pathway mediated by G protein–IP3/PKC events to control FGF23 production (52). It was shown that the ablation of the extra-large Gα subunit (XLαs) or Gq/11α in osteocyte-like Ocy454 cells activated IP3/PKC and stimulated the MAPK/ERK1/2 pathway leading to FGF23 induction (52). It is assumed that the trigger of the IP3/PKC/MAPK pathway is controlled by a G protein-coupled receptor which remains to be identified (Figure 1).
More recently, the use of CRISPR/Cas9 technology has helped to identify control regions for the Fgf23 gene in response to various inflammatory stimuli in mice. An enhancer site − 16kb from the transcription start site of the murine Fgf23 gene was deleted via CRISPR, and this study demonstrated that FGF23 expression in bone, bone marrow, spleen, liver, thymus, and intestine was attenuated in response to LPS injection. This deletion was also associated with decreased intact FGF23 compared to normal mice treated with LPS. Similar effects were observed in response to direct TNF-α and IL-1β injections, highlighting the importance of this genomic region in mediating inflammation-induced FGF23 expression (53). Further studies showed that an additional proximal enhancer region in Fgf23 mediates LPS-induced FGF23 expression in vivo through binding motifs to several known transcription factors that mediate inflammation, including NF-κB (54).
Persistent iron deficiency in mice has been shown to induce a pro-inflammatory state (55) that induces a heightened inflammatory response to stimuli such as LPS (55, 56). Both iron deficiency and inflammation increase FGF23 transcription by activating among other signaling pathways, Hif1α, and associated MAPK signaling, in osteocytes (57, 58). Wild type mice fed an iron deficient diet for 8 and 12 weeks showed 5- and 10-fold increases of Fgf23 transcripts, respectively, in femur/tibia samples (57). In vitro, treatment of UMR-106 cells with the iron chelator, deferoxamine (DFO) resulted in marked increases in Fgf23 mRNA, partially dependent on MAPK activity in association with Hif1α stabilization (57, 58) (Figure 1). These discoveries suggest that absolute iron deficiency, characterized by a depletion of both iron stores and circulating iron, promotes FGF23 transcription through Hif1α. Similarly, a functional iron deficiency via a treatment of mice with hepcidin also increased bone Fgf23 mRNA expression (58).
The cytokine IL-6, a known marker of inflammatory states has also been shown to contribute to FGF23 production. In a mouse model of folic acid–induced acute kidney injury, bone Fgf23 mRNA expression increased together with serum FGF23 as well as several circulating cytokines including IL-6 (33). When fed with an adenine diet to induce CKD, IL-6 knock-out mice failed to increase bone Fgf23 mRNA, resulting in an attenuation of circulating FGF23 levels in comparison to wild-type mice with CKD; these data suggest a direct contribution of IL-6 to the increased FGF23 observed during CKD (59). Further, ex vivo and in vitro studies have shown that a treatment of calvaria organ cultures and UMR-106 cells with the IL-6/soluble IL-6 receptor fusion protein induced STAT3 phosphorylation, and increased Fgf23 promoter activity, suggesting a direct effect of IL-6 on the positive regulation of FGF23 expression (59).
Hypoxia inducible factors (HIFs) are constitutively expressed transcription factors that are critical for sensing oxygen and iron levels in the body. Oxygen and iron are important co-factors for the function of HIF-prolyl hydroxylase domain enzymes (HIF-PHDs), which help maintain the constant turnover of HIFs by marking them for degradation. As stated above, it was shown that iron deficiency can drive FGF23 expression in a mouse model of autosomal dominant hypophosphatemic rickets (ADHR) (57). Wild type and ADHR mice (harboring the FGF23-R176Q stabilizing ADHR mutation) fed an iron-deficient diet had elevations in “total” serum FGF23 (intact bioactive FGF23 + proteolytic fragments), however only iron-deficient ADHR mice had increases in intact FGF23 due to their inability to cleave and inactivate the bioactive form of FGF23. This and subsequent studies helped tie together iron handling and phosphate homeostasis, a link that can likely be explained through HIF activity. An iron-deficient state leads to HIF stabilization due to the lack of iron as a co-factor for the HIF-PHDs to tag HIF for degradation. In vitro studies have shown HIF-mediated induction of FGF23 mRNA expression in two different osteoblast cell lines, as well as evidence of HIF binding to the FGF23 promoter (60). More recently, a prospective study in ADHR patients that co-presented with an iron deficiency phenotype demonstrated that iron repletion in these patients reduced their circulating levels of FGF23 and normalized serum phosphate levels. Thus, providing oral iron is a novel therapeutic approach to this disease (61). This finding was supported by studies in iron-deficient women showing that rescue of iron deficiency using iron dextran lowered the prevailing elevated FGF23 (62). It is important to note the type of intravenous iron used, as it has been well-documented that certain types of iron formulations, especially those with carboxymaltose backbones can cause hypophosphatemia due to elevating intact FGF23 (63), potentially through altering FGF23 intracellular proteolysis (see below).
Erythropoietin (EPO) is a hormone made in the kidney that induces new red blood cell production in the bone marrow in response to anemia or blood loss. Recombinant human EPO (rhEPO) is currently used as a therapy to treat anemia related to CKD. Recently, several studies revealed that this hormone can induce FGF23 expression (64–66). WT mice given acute rhEPO treatment after 6 h or after 3–4 days of consecutive injections resulted in elevated total and intact FGF23 and bone marrow Fgf23 mRNA expression (64). To understand the effects of chronically elevated EPO, reports using the transgenic Tg6 mouse model of EPO overexpression showed that these mice have basal elevations in total and intact FGF23 (65, 67). Chronic overexpression of EPO can lead to iron deficiency, so to determine whether the iron deficiency was causing the elevated FGF23, mice were given iron dextran. This only modestly decreased intact FGF23, showing that EPO was primarily driving FGF23 expression (65). In healthy human subjects, total FGF23 was elevated 24 h after a single rhEPO dose with no changes in intact FGF23 (66). In another study, a small population of anemic patients with normal kidney function were given a single dose of rhEPO. This increased total and intact FGF23 over the span of 12–18 h after the injection (64). Few in vivo studies have examined the effect of curing anemia of CKD on mineral metabolism. Most recently, a single rhEPO injection in CKD mice did increase total serum FGF23 after 6 and 24 h but had no effect on intact FGF23 (65).
To mitigate potential adverse effects of high rhEPO treatment, new strategies are leveraging the HIF system by creating inhibitors of the HIF prolyl hydroxylases (HIF-PHDs) called HIF-PHDs inhibitors (HIF-PHI). These therapeutics have become increasingly important for anemic patients with CKD, where they promote endogenous EPO production by stabilizing HIFs. This class of drug also reduce hepcidin levels to increase iron utilization in tissues thereby creating a synergistic effect in providing iron availability to newly forming red blood cells (68). Recent studies have shown in normal mice that several HIF-PHIs can regulate Fgf23 expression. For instance, FG-4592 (Roxadustat) was shown to increase bone Fgf23 mRNA and circulating intact FGF23 (58, 66). Elevations in total and intact FGF23 were observed in WT mice treated with the HIF-PHI BAY 85-3934 (Molidustat) after 6 h that returned to baseline by 24 h. This study showed that elevated EPO precedes increases in FGF23 in response to HIF-PHI BAY85-3934, suggesting that the cause for elevated FGF23 in response to this treatment was due to elevated EPO. This was confirmed by treating HIF-PHI mice with an EPO-neutralizing antibody, which completely attenuated the increased serum total FGF23 (69). A recent paper, however, showed that EPO and HIF-PHI treatment of anemic mice with CKD resulted in suppressed FGF23. This study suggested that under conditions of anemia, as opposed to mice with normal iron homeostasis, rescuing iron utilization during CKD may be a more potent suppressor of FGF23 than EPO and HIF-PHIs are stimulators (70). Although EPO and HIF-PHI can induce FGF23, further studies are required to delineate the mechanisms directing these components on osteocytic FGF23 production.
Parathyroid hormone (PTH) is a hormone secreted by the parathyroid glands that regulates calcium utilization through its effects on bone, kidney, and intestine (71–73). During CKD, a secondary hyperparathyroidism occurs in which PTH is excessively secreted, in response to factors such as hyperphosphatemia, hypocalcemia, and low 1,25D levels, to potentially promote elevated FGF23 (74). Using bioinformatics and chromatin immunoprecipitation assays, studies have reported that Nurr1 is an essential transcription factor involved in FGF23 upregulation in response to PTH in UMR-106 cells. Furthermore, in a mouse model of CKD, the administration of a calcimimetic, which is known to activate the calcium-sensing receptor in different tissues (75) with the goal to attenuate PTH levels and actions, reduced FGF23 concentrations as well as bone Nurr1 mRNA and protein levels (76). To test the relationships between PTH and FGF23, a mouse model with constitutive activation of PTH receptor (PTHR) signaling in osteocytes was used by Rhee et al. in a report that showed that PTHR activation increased FGF23 expression in vivo and in vitro through cAMP and Wnt-dependent mechanisms (77). In addition, Knab et al. showed that PTH-induced increases in FGF23 expression were PKA-dependent in osteocyte-like cells, suggesting that FGF23 production is regulated by the cAMP/PKA/Nurr1 pathway in response to PTH (78) (Figure 1).
TGF-β has been reported to regulate the extracellular matrix by activating ROS, which increases calcium influx and activates calcineurin (79–81). Using UMR-106 cells, TGF-β2 has been described to enhance store-operated Ca2+ entry (SOCE) and induce the stimulation of FGF23, an effect significantly attenuated by both the inhibitor of TGF-β type I receptor activin receptor-like kinases (ALK5, ALK4, and ALK7) SB431542 and SOCE inhibitor 2-APB (82). Recent investigation has shown that the synthesis of FGF23 in UMR-106 cells can be induced by SOCE through Orai1 (83). In addition, the Ca2+ entry activates the phosphatase calcineurin, which dephosphorylates nuclear factor of activated T cells (NFAT) thereby stimulating its transcriptional activity and targeting the Fgf23 gene. This suggested that FGF23 may be positively regulated by calcineurin-NFAT signaling (84). Further analyses confirmed that either the inhibition of calcineurin using ciclosporin A (CsA) and tacrolimus (FK-506) or the blocking of the interaction between calcineurin and NFAT using the inhibitor INCA-6 reduced the abundance of Fgf23 transcripts as well as FGF23 protein (84). Additionally, lithium, a widely used drug for the treatment of mood disorders and known to modify Ca2+ signaling, stimulated the release of FGF23, partially through NF-κB dependent up-regulation of Orai1 transcription and SOCE in UMR-106 cells (85) (Figure 1).
Early FGF23 physiological studies demonstrated that the administration of 1,25D dose-dependently increased both serum FGF23 as well as serum inorganic phosphorus in normal rats (25). In wild type mice with normal renal function, injection of calcitriol increased serum FGF23 levels 1-week post treatment by 15-fold. Calcidiol (25-hydroxyvitamin D), although with a lesser effect could also induce FGF23 in normal mice (86). In a mouse model of adenine diet-induced CKD, a 5 weeks-regimen induced a 40-fold increase of circulating FGF23. However, in the background of a global deletion of Cyp27b1, the gene encoding the enzyme vitamin D 1-α-hydroxylase involved in the formation of calcitriol (1(OH)ase−/− mice), only a 2-fold circulating FGF23 was observed with this treatment, suggesting a contribution of calcitriol to the increased FGF23. At the bone compartment level, a specific deletion of Cyp27b1 in osteoblasts reduced FGF23 induction in long bones from 58-fold in normal mice treated with a 5-weeks adenine diet to a 10-fold induction, suggesting a potential role of a local osteoblastic vitamin D conversion process in the induction of bone FGF23. This attenuation of increased FGF23 was independent of a potential reduction in PTH levels since plasma PTH remained elevated in response to the adenine diet-induced CKD in the mice with global deletion of Cyp27b1, as well as those with osteoblast-specific deletion of Cyp27b1 (86) (Figure 1).
The disease XLH is characterized by hypophosphatemia and impaired mineralization caused by mutations of the phosphate-regulating gene with homologies to endopeptidases on the X chromosome (PHEX). Loss of PHEX leads to the overproduction of FGF23 in osteocytes, causing hypophosphatemia with bone mineralization impairment, and thus bone fragility. Similar to XLH, recessive loss-of-function mutations in the dentin matrix protein-1 (DMP1) gene, a member of small integrin-binding ligand N-linked glycoprotein (SIBLING) proteins, is responsible for a human phosphate wasting and impaired bone mineralization disease termed autosomal recessive hypophosphatemic rickets type 1 (ARHR1). It was shown that a lack of DMP1 in both humans and mice markedly increased FGF23 expression in bone (87).
To gain insight into the mechanisms by which PHEX mutations upregulate FGF23 expression, studies have been designed to investigate the local effects in bone from a mouse model of XLH (Hyp mice) in a normal hormonal environment in comparison to the function of wild type bone in the abnormal metabolic environment of Hyp mice. Using a surgical procedure to perform intramuscular bone cross-transplantations between wild-type and Hyp mice, a study found that increased FGF23 expression in Hyp bone results from intrinsic PHEX deficiency from bone, since FGF23 was increased in Hyp osteocytes before and after explantation into WT mice but was not increased in WT osteocytes after explantation into Hyp mice. This evidence suggested that the mechanisms whereby PHEX mutations lead to increased FGF23 expression in osteocytes is intrinsic to bone (88).
Similar to the phenotype resulting from PHEX inactivation, the inactivation of Dmp1 in mice resulted in equivalent intrinsic bone mineralization defects associated with increased FGF23 expression in osteocytes (89–91). Using cortical bone isolated from 12-days old WT, Hyp, Dmp1−/−, and Hyp/Dmp1−/− mice to perform a genome-wide microarray analysis, the phosphatidylinositol 3-kinase regulatory α subunit (PIK3R1) and growth factor receptor-bound protein 2 (GRB2) pathways were identified as potential common signaling controlled by PHEX and DMP1 to regulate FGF23 promoter activity through FGFs/FGFR in osteocytes (91, 92) (Figure 1). These findings highlight that the activation of FGFRs which contribute to FGF23 production in osteocytes may be independent from the phosphate sensing pathways described above. Additionally, recent in vivo and in vitro studies suggested a direct negative regulation of DMP1 in FGF23 expression in osteocytes by activating FAK-mediated MAPK signaling, thus coordinating the extracellular environment of osteocytic lacunae as well as bone metabolism (93) (Figure 1). The creation of the floxed-Fgf23 mouse has recently emerged as a critical tool to understand FGF23 function in vivo (94). To this end, flox-Fgf23 mice were mated to the global eIIa-cre which mimicked the phenotype of the Fgf23-KO mouse. Fgf23 was also specifically deleted from either the osteoblast lineage using the Col2.3 promoter to drive Cre expression, or from late osteoblasts/osteocytes using the Dmp1-Cre. This resulted in ~50% reduction of iFGF23, with compromised ability to respond to changes in phosphate (94), demonstrating the specificity of osteoblast/osteocyte FGF23 production in response to metabolic changes.
Recent studies reported insulin and insulin-like growth factor 1 (IGF1) as negative regulators of FGF23 production in vitro as well as in mice and humans (95). In vitro, insulin and IGF1 down-regulated FGF23 production by inhibiting the transcription factor forkhead box protein O1 (FOXO1) through phosphoinositide 3-kinase (PI3K)/protein kinase B (PKB)/Akt signaling in UMR-106 cells (95). In vivo, insulin deficiency resulted in an increase of serum FGF23 concentrations in mice, which was reversed by insulin administration. Interestingly, in women subjects, an increase in plasma insulin levels following an oral glucose administration correlated negatively with plasma FGF23 concentrations (95) (Figure 1).
FGF23 is synthesized as a 251-amino acid protein, primarily in osteocytes. The FGF23 signal peptide is represented by the first N-terminal 24 aa. The cleavage of the signal peptide results in a release of a mature peptide that can be secreted as the bioactive hormone, referred to as “intact” FGF23 (“iFGF23”). To control the bioactivity of iFGF23, the protein can be cleaved at the subtilisin-like proprotein convertase (SPC) site R176HTR179/S180AE (RXXR/SAE motif) generating at least two fragments identified as an N-terminal fragment which is structurally similar to other FGF family members and a more unique C-terminal tail (21, 96). The proteolytic cleavage of excess iFGF23 represents a critical secondary regulatory mechanism to maintain stable serum iFGF23 and normal serum phosphate. The absence of this proteolytic activity in humans through the FGF23 gene mutations R176Q, R176W, R179Q, and R179W are causative for autosomal dominant hypophosphatemic rickets (ADHR), characterized by elevated intact FGF23 and hypophosphatemia (21) (Figure 2). In mice carrying the ADHR point mutation R176Q-Fgf23, in response to absolute iron deficiency using dietary intervention, the iFGF23 levels were elevated due to the stabilization of the bioactive FGF23 (57). This elevated iFGF23 condition in response to iron-deficient diet in ADHR mice caused similar phenotypes as observed in ADHR/XLH patients, such as alterations in genes controlling phosphate reabsorption and 1,25D production, and a hypophosphatemic bone disease (57). These findings suggested that iron status is a synergistic factor of the ADHR phenotype, and that ADHR is a disease of gene-environment interactions (57).
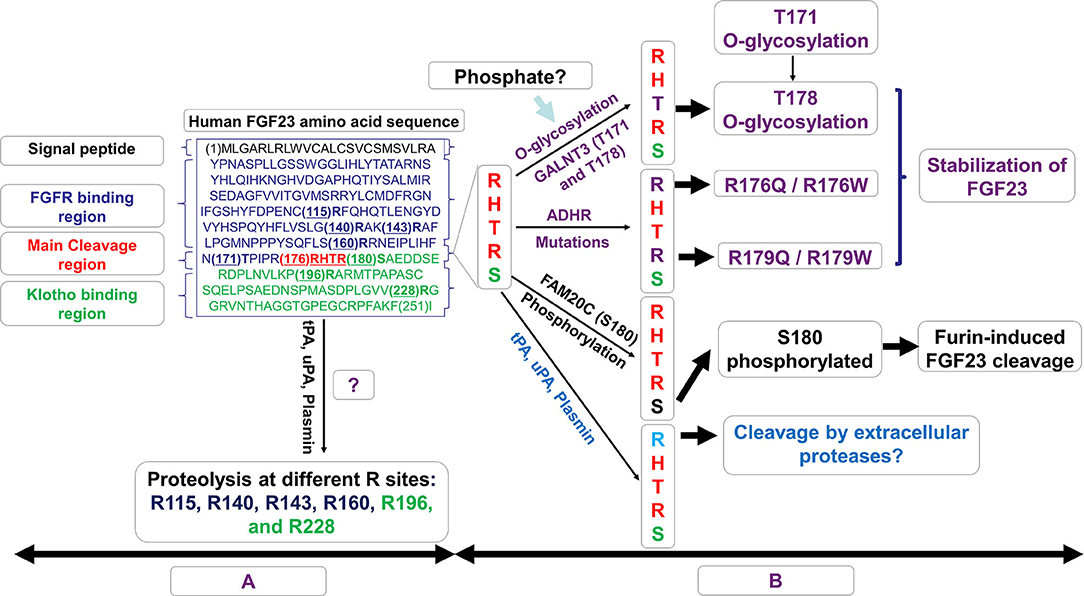
Figure 2. Schematic representation of the possible fates of FGF23. In (A), the amino acid sequence of human FGF23 is shown with its signal peptide, FGFR binding region, the RXXR motif/cleavage sequence, and the Klotho binding region. In (B), the possible fates of mature FGF23 after the loss of its signal peptide sequence is displayed. The O-glycosylation of T178, a process controlled by N-acetylgalactosaminyltransferase 3 (GALNT3) and which requires a precedent glycosylation at T171, stabilizes and prevents FGF23 cleavage. This process may also be stimulated by increased levels of phosphate which induces GALNT3 and the glycosylation. Genetic modifications such as autosomal dominant hypophosphataemic rickets (ADHR) mutations R176Q, R176W, R179Q, and R179W stabilize FGF23 and counteract its natural cleavage. Phosphorylation at position S180 by FAM20C increases FGF23 cleavage controlled by the furin/subtilisin-like proprotein convertase. FGF23 proteolysis may occur at R176 by extracellular proteases of the plasminogen activation system, tissue-type PA (tPA), urokinase-type PA (uPA), and plasmin. This proteolysis could potentially occur at the RXXR motif (R176) as well as at different arginine residue sites at R115, R140, R143, R160, R196, and R228.
The cleavage of FGF23 is controlled by the serine endoprotease furin, a subtilisin-like convertase, at the site R179/S180 in response to several stimuli including PTH (78). Previous investigations have shown that an iron deficient state also promotes iFGF23 cleavage leading to increased secretion of FGF23 fragments but normal iFGF23 in WT mice (57, 58). In HeLa Cells, it was described that an iron deficient state induces furin upregulation via the stabilization of Hif1α (97), a similar mechanism which occurs in osteocytes. Posttranslational modifications of iFGF23 can occur via the O-glycosylation of Thr178 in the furin proprotein processing motif RHT178R179, which stabilized FGF23 (98) (Figure 2). This glycosylation process at Thr178 is controlled by the enzymatic actions of N-acetylgalactosaminyltransferase 3 (GALNT3) which can be upregulated under high phosphate conditions potentially via the control of FGFR1c activation and the induction of the transcriptional activators early growth response 1 (EGR1) and ETS variant 5 (ETV5) (28).
In a recent study, Thr178 was identified as a poor substrate site with limited glycosylation acceptance, likely a protective mechanism to prevent cellular resistances to FGF23 cleavage. Interestingly, Thr178 glycosylation was shown to require a previous glycosylation at Thr171 before generating a furin-resistant and secreted stable iFGF23 (99) (Figure 2). These new discoveries suggest that GALNT3 specificity for FGF23 and its ability to control circulating levels of bioactive FGF23 is a control point achieved by FGF23 being a rather poor substrate for this enzyme (99). In contrast to the O-glycosylation induced by GALNT3 which stabilizes FGF23, the phosphorylation at position S180 by the kinase FAM20C inhibited O-glycosylation of FGF23, thus promoting FGF23 cleavage (98) (Figure 2). Indeed, recessive inactivating mutations in human FAM20C cause ARHR type 3 (ARHR3; Raines syndrome), consistent with its role as an FGF23 de-stabilizer (100, 101).
Besides furin which is thought to cleave FGF23 mainly intracellularly, the extracellular proteases of the plasminogen activation system, tissue-type PA (tPA), and urokinase-type PA (uPA), as well as plasmin may also display proteolytic activity on FGF23 protein at the RXXR motif (R176), with potential additional cleavages at arginine residues R114, R140, R143, R160, R196, and R228 (102). Of note, Klotho knock out mice as well as mice with acute kidney injury, which both exhibit elevated levels of intact FGF23, also display elevated plasminogen activator inhibitor-1 (103, 104). Thus, the proteolysis of FGF23 by tPA, uPA, and plasmin may potentially regulate the levels of active FGF23 thus in part, controlling phosphate and mineral homeostasis.
These collective studies demonstrated that FGF23 protein cleavage is a dynamic process which can be adjusted after production at the cell level with phosphorylation and glycosylation dictating the levels of bioactive FGF23 depending upon the osteocyte cell state. Certainly, future studies are needed to understand the regulation of GALNT3, furin, FAM20C, and potentially the enzymes of the plasminogen activation system in the coordinated control of FGF23 production and bioactivity.
In contrast to paracrine FGFs, such as FGF1 and FGF2, endocrine FGFs such as FGF23 lack the heparin-binding domain in their C-terminus, which enables escape from the osteocyte cell matrix after secretion and their actions on distant target organs including kidney (14, 105). FGF23 acts primarily on kidney to promote phosphaturia and parallel reductions in 1,25D (14, 34). The phosphaturic activity of FGF23 is critical in CKD, preventing and delaying hyperphosphatemia and vascular calcifications as FGF23 progressively rises with the loss of renal function (106). In a study among patients undergoing hemodialysis, high serum phosphate levels across a quartile of patients (>5.5 mg/dl) was associated with a 20% increase in the multivariable adjusted risk of death, as compared with normal levels (3.5–4.5 mg/dl) (107). These findings underscore the importance of controlling circulating phosphate in kidney disease patients. During CKD, as kidney function decreases with a progressive decrease of glomerular filtration rate (GFR), the kidney loses functioning nephrons decreasing the overall excretion capability of the kidney. The dramatic rise of FGF23 during CKD is likely to attempt to boost the excretory capacity of the existing nephrons to maintain normal serum phosphate levels. This is likely the primary reason why early stage CKD patients exhibit normal serum phosphate over much of the disease course [for review (108)].
FGF23 actions are likely mediated by FGF receptors FGFR1c, FGFR3c, and FGFR4 and the co-receptor Klotho, a single-pass transmembrane protein highly expressed in kidney (109–113). FGF23 binds to FGFR1c and the interaction between these two proteins with the co-receptor Klotho (which dramatically increases the binding affinity of the complex FGF23-FGFR-Klotho) triggers potent FGF23 signaling and activity (114). In kidney, FGF23 signaling on the basolateral side of proximal nephron cells causes the internalization of the sodium-dependent phosphate co-transporters NPT2A and NPT2C from the apical surface. These actions decrease phosphate reabsorption processes from the kidney (115) (Figure 3). A global genetic deletion of FGF23 in mice resulted in severe hyperphosphatemia, due to the absence of the FGF23-mediated phosphaturia mechanism (116). In addition, FGF23 signaling regulates vitamin D metabolism via the modulation of the vitamin D metabolic enzymes expression as well as regulating calcium and sodium reabsorption processes (Figure 3).
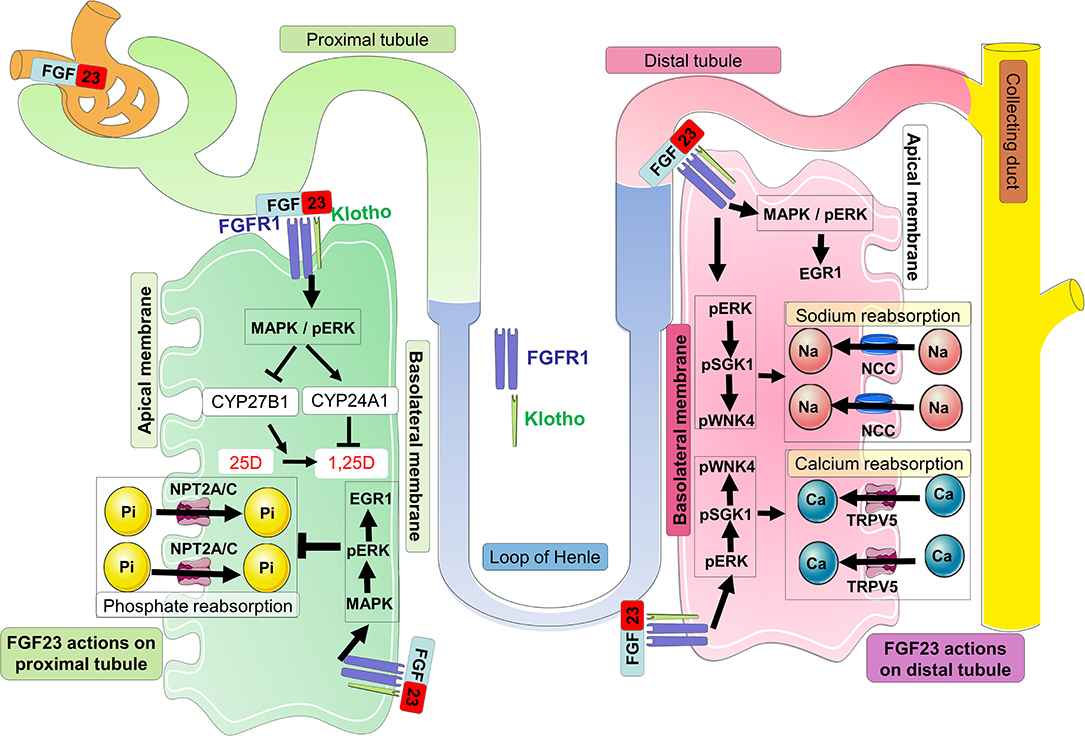
Figure 3. FGF23 signaling in the kidney. In kidney proximal tubules, circulating FGF23 binds to FGFR1-Klotho complexes at the basolateral membrane, and activates the MAPK signaling cascade involving ERK1/2. This signaling leads to the internalization and degradation of NPT2A/C and the decrease of urinary phosphate reabsorption promoting phosphate wasting. In the proximal tubule, FGF23 signaling induces downstream mechanisms which suppress the transcription of the vitamin D 1α-hydroxylase (CYP27B1) and increases the transcription of the vitamin D 24-hydroxylase (CYP24A1); both events work in concert to limit the conversion of 25D to 1,25D as well as degrading 1,25D into inactive metabolites. In the kidney distal tubule, circulating FGF23 binds to the FGFR-Klotho receptor complex at the basolateral membrane, and activates the MAPK/pERK/EGR1 pathway as well as cascade signaling for ERK1/2, SGK1, and the WNK4 complex via their phosphorylation. Activation of WNK signaling increases the expression of TRPV5 and NCC at the apical membrane promoting the reabsorption of calcium and sodium, respectively.
Unbiased studies using large-scale microarray and gene expression approaches from FGF23 transgenic (FGF23 Tg) mice and WT littermates have identified several renal genes associated with increased FGF23. Particularly, genes involved in the phosphate reabsorption process such as Npt2a, Pdzk1 (a scaffolding protein known to interact with Npt2a) and Klotho, the co-receptor for FGF23 were among the genes which showed significantly decreased expression (117). The angiotensin I converting enzyme 2 (Ace2), known to decrease Angiotensin II with potential disruption of the renin-angiotensin system balance (118) was also decreased in FGF23 Tg kidney (117). It was later hypothesized that FGF23 induced the activation of FGFR1, leading to Ace2 reduction, causing hypertension (119). Other genes such as Atp1a2, which has been described to interact with Klotho and regulate calcium metabolism (120), was 2.5-fold- upregulated (117) and lipocalin 2, a neutrophil-derived marker previously described to be involved in CKD progression in mice and humans (121), was also modestly induced (117).
Other approaches using an acute 1-h FGF23 injection revealed that the MAPK related transcription factor Egr1 is typically the most up-regulated gene in microarray datasets testing FGF23 bioactivity. It was also determined that EGR1 binding near genes encoding Npt2a, Npt2c, as well as scaffold proteins (NHERF1-3, EZR and GABARAP), and trafficking proteins (megalin and vacuolar ATPase), known to putatively mediate NPT2A protein internalization and degradation, were increased after FGF23 injection. Collectively, these results suggested a coordinated regulation of renal Pi transport genes through EGR1 (122).
As mentioned above, it has been well-studied that FGF23 delivery induces EGR1, a transcription factor and downstream marker of MAPK signaling, in mice and cells (112, 123). It was shown in the Hyp mouse that MAPK/ERK1/2 signaling is activated in the kidney and this pathway induction drives EGR1 (112, 123). Other studies revealed that an inhibition of ERK1/2 signaling using a selective MEK inhibitor, PD0325901, was associated with a reduction of renal Egr1 upregulation induced by a Phex deletion. This led to improvement in the hypophosphatemia associated with decreased NPT2a protein expression, correction of 1,25D and calcium deficiency, with a reduction of cortical porosity (124, 125).
To further understand the contribution of Egr1 on FGF23-dependent regulation of renal Pi and vitamin D metabolism, the Egr1 knockout mouse (Egr1−/−) has been studied with interesting findings. Reports showed that the effect of FGF23-induced decreases in renal Npt2a and Npt2c proteins is completely abrogated in Egr1−/− mice (122). To potentially attenuate Egr1 activity, FGF23 also upregulates two isoforms of Nab2 which are known to be corepressors of Egr1 (126); however whether this is a direct or an indirect mechanism through Egr1, which is known to induce Nab2 thus establishing a negative feedback loop (127), remains to be investigated.
Four FGFR isoforms (FGFR 1-4) have been described in mammals and an alternative splicing of these isoforms can generate seven major FGFR proteins (FGFRs 1b, 1c, 2b, 2c, 3b, 3c, and 4) (105). FGFR1, FGFR3, and FGFR4 are the most expressed FGFRs in the mouse kidney with potential functional roles in mineral homeostasis (128–130). In mice, a deletion of Fgfr1, Fgfr2, Fgfr3, and Fgfr4 from renal proximal tubules induces hyperphosphatemia, hypercalcemia, hypervitaminosis with elevated FGF23 (128). Other studies using mice with a specific deletion of FGFR1 in distal tubule segments have suggested that FGF23 activates FGFR1/alpha-Klotho complexes in the distal tubule leading to an increase of sodium-chloride symporter (NCC)-dependent sodium (Na) reabsorption, a decrease of Ace2 and renal KIotho, leading to increased blood pressure and hypertension (119). Further, activation of FGFR1 using pharmacologic intervention normalized blood pressure in Hyp mice, a mouse model of elevated FGF23 (119). Although the roles of FGFR3 and FGFR4 in kidney are not completely understood, in vivo studies proposed that FGFR3 and FGFR4 may contribute by acting in concert with FGFR1 to mediate FGF23 effects in kidney. Although their roles may be less substantial at the physiological level, under conditions of high circulating FGF23 in Hyp mice, the deletion of FGFR3 leads to a feedback stimulation of Fgf23 mRNA expression in bone (129, 131), suggesting complex kidney-bone crosstalk.
The co-receptor KL is required for high-affinity FGF23 activity in the kidney. Whole-nephron deletion of Klotho in mice results in renal FGF23 resistance, characterized by hypervitaminosis D, hyperphosphatemia, and other phenotypes that may resemble premature aging (39, 132). However, provision of a low phosphate diet to KL-null mice reversed the severe phenotypes, showing that the majority of KL-null phenotypes are due to extreme phosphate imbalances (133, 134). Klotho is highly expressed in the distal tubule segments of the nephron in comparison to its relatively modest expression in proximal tubules (123) although the phosphate reabsorption occurs primarily in the proximal tubules due to an abundant expression of the sodium phosphate transporters Npt2a and Npt2c (135–137). To delineate and identify the main effector sites of FGF23 actions in kidney, recent elegant studies have been performed using mouse models with nephron segment-specific deletion of Klotho in concert with a full characterization of mineral metabolism of these mice (138, 139). Olauson et al. generated a mouse model with deletion of Klotho in distal tubular segments (Ksp-KL2/2) which was characterized as fertile with a normal gross phenotype despite a disrupted mineral metabolism. These phenotypes were in contrast to Klotho-null mice which are not fertile in addition to undergoing premature death and severe vascular calcifications (138). By using immunohistochemistry analysis, investigators showed that partial deletion of Klotho in distal tubule resulted in hyperphosphatemia with elevated plasma FGF23 and increased Npt2a protein expression in the proximal tubule apical membrane (138). In other studies where Klotho was conditionally deleted from renal proximal tubule, the mineral metabolism phenotype was variable depending upon the strategies used to perform specific Klotho deletion. Indeed, in the studies of Ide et al., only a mild phenotype on mineral metabolism with a decrease of urinary phosphate was observed when Klotho was deleted from proximal tubules using three different proximal tubule specific Cre transgenic mice: Kap-Cre (kidney androgen-regulated protein), Slc34a1-Cre (sodium phosphate cotransporter-2a1) or Pepck-Cre (phosphoenolpyruvate carboxykinase) (139). The latter, harbors elevated serum iFGF23 and a slight increase in Npt2a protein (139). In contrast, other studies using an inducible promoter-Cre, Ndrg1-Cre, to delete Klotho from proximal tubules revealed a more pronounced effect on mineral metabolism with markedly elevated iFGF23 and hyperphosphatemia (128). The phenotypic differences across these mouse models could be explained by the variability in Cre-mediated recombination efficiency, which can be factored by the cell-type specificity of Cre expression, the Cre expression efficiency in specific cell types, and the recombination feasibility from different genetic modification strategies.
It was later shown that FGF23 signaling can cross-talk with PTH signaling to control mineral metabolism. A deletion of both PTH1R and Klotho from the kidney proximal tubule (PT-PTH1R/KL−/− mice) led to a severe disturbance of mineral metabolism including hyperphosphatemia at baseline and increased circulating phosphate in response to high phosphate diet (140). The hyperphosphatemia observed in PT-PTH1RKL−/− mice was associated with elevated circulating FGF23, PTH, decreased circulating 1,25D and increased Npt2a (140). These new data underscore that FGF23 and PTH signaling pathways can interact in kidney thus coordinating renal phosphate handling in the proximal tubule (140, 141).
Using animal models, studies confirmed over recent years that FGF23 is a negative regulator of 1,25D production. Indeed, FGF23 potently inhibits the expression of the 25(OH)D-1α-hydroxylase CYP27B1 in the renal proximal tubule while stimulating in contrario the expression of the vitamin D catabolic enzyme CYP24A1 at the mRNA level (23, 142–144). Mice with global deletion of the Fgf23 gene displayed elevations in serum 1,25D by 2-4 fold (144). Since FGF23 acts as a requisite partner with its co-receptor Klotho to control mineral metabolism, Klotho ablation in mice resulted in a strikingly similar phenotype to the Fgf23-null mice, including increased serum levels of 1,25D associated with increased renal Cyp27b1 expression (39, 145). Interestingly, the premature aging-like phenotype of Fgf23−/− and Klotho−/− mice can be completely rescued using a genetic approach to ablate 1,25D synthesis through the generation of double mutant Fgf23−/−/1α(OH)ase−/− and Klotho−/−/1α(OH)ase−/− mice (146, 147). These data suggested that increased vitamin D played a major role in the abnormal mineral ion metabolism and soft-tissue anomalies observed in Fgf23−/− and Klotho−/− mice. Although Klotho deletion results in hypervitaminosis, and the kidney is the predominant organ expressing Klotho, studies using targeted deletion of Klotho in the proximal or distal tubule segment of the nephron have shown an overall modest effects on circulating vitamin D levels (138, 139), likely due to endocrine compensation.
It has also been described that the deletion of vitamin D receptor (VDR) in Fgf23−/− and Klotho−/− mice rescued these mice from an early lethality phenotype due to the absence of vitamin D signaling causing reduced phosphate absorption (148, 149). Therefore, Fgf23−/−/VDRΔ/Δ and Kl−/−/VDRΔ/Δ double mutant mice can be used to examine the roles of FGF23 and Klotho at older ages by keeping these mice on a rescue diet rich in calcium, phosphorus, and lactose (150, 151) with the goal of preventing hypocalcemia and severe hyperparathyroidism due to the non-functioning VDR status.
Study of the Fgf23−/−/VDRΔ/Δ and Kl−/−/VDRΔ/Δ mice showed that the deletion of Fgf23 or Klotho leads to a decrease in the membrane abundance of NCC (the sodium chloride cotransporter) in the kidney distal tubule and subsequently to decreased Na+ reabsorption (152). In contrast, treatment of WT mice with FGF23 over 5 days upregulated distal tubular NCC resulting in increased Na+ reabsorption and increased blood Na+ concentrations (152). Using Hyp mice with elevated FGF23, studies have also shown that these mice have increased distal tubular Na+ uptake and membrane abundance of NCC (152). It was explored whether the effects of FGF23 on NCC expression in kidney may potentially drive physiological changes including hypertension and heart hypertrophy in a αKlotho-dependent manner. The inhibition of NCC using chlorothiazide abrogated FGF23-induced heart hypertrophy suggesting that FGF23 may act as a potential regulator of renal Na+ reabsorption with downstream consequences, although patients with FGF23-related gain or loss of function mutations primarily show more severe defects in phosphate metabolism. Another mineral that may be regulated by FGF23 in distal tubule is calcium (153). In this regard, studies have shown that renal calcium reabsorption and renal membrane abundance of TRPV5 are reduced in Fgf23−/−/VDRΔ/Δ and Kl−/−/VDRΔ/Δ double mutant mice (153).
Although FGFR1c, 3c, and 4 are ubiquitously expressed, Klotho expression is predominantly expressed in specific tissues such as kidney renal tubules, parathyroid gland, and choroid plexus of brain, suggesting that these organs are the physiological targets for FGF23-mediated endocrine actions (39, 154, 155). FGF23 preferentially binds to FGFR1c and Klotho, and this complex initiates FGFR1c signal transduction via the cytoplasmic adaptor FRS2α (156), which activates the FRS2α/Ras/MAPK pathway (157) (Figure 3). In vitro studies using human embryonic kidney cells (HEK293), which endogenously express FGFRs but not Klotho (158), showed that the presence of soluble Klotho (sKL) or membrane-bound Klotho (mKL) is required for FGF23-induced MAPK activity, which was assessed by pERK1/2 induction and EGR1 mRNA expression (157, 159). Although initial studies suggested that mKL and sKL share a common function of mediating FGF23-induced FRS2α/Ras/MAPK signaling, recent findings suggested that FGF23 responses were quantitatively different depending on mKL or sKL availability (159). In vivo studies, potentially using genetic targeting to isolate the biological effects of mKL from sKL will be required to deepen our understanding of these interactions.
In the absence of Klotho, in HEK293 cells, FGF23 alone can activate pPLCγ and pAKT, and these activities are completely neutralized by the presence of Klotho (159, 160). These studies further suggested that FGF23 preferentially induced FGFR1c signaling via Klotho. However, in the absence of Klotho, high concentrations of FGF23 can activate FGFR4 (157). The activation of FGFR4 was shown to induce PLCγ-catalyzed production of diacylglycerol and inositol 1,4,5-triphosphate that increased cytoplasmic calcium levels, thereby activating several calcium-sensing signal mediators, including the protein phosphatase calcineurin (157). The activation of calcineurin dephosphorylates the transcription factor NFAT, which permits its translocation into the nucleus to modulate the expression of specific target genes (161). This FGFR4-mediated effect may play a key role in cardiac hypertrophy through FGFR4 during highly elevated FGF23 in CKD (157, 162).
The phosphaturic action of FGF23 in kidney proximal tubule and actions in the distal tubule may occur primarily through FGFR1c, the main “phosphaturic” FGFR expressed in both segments and colocalized with Klotho (123, 129). A C-terminal FGF23 peptide antagonist has been developed recently to block FGF23 signaling by inhibiting tyrosine phosphorylation of FRS2α and downstream activation of the MAPK cascades (34). Studies in Hyp mice using this novel peptide confirmed that the inhibition of FGF23 signaling in kidney upregulates the expression of the sodium-phosphate cotransporters Npt2a and Npt2c, coupled with the alleviation of the observed hypophosphatemia (34). In a mouse model of CKD, this FGF23 antagonist peptide has been shown to rescue the prevailing anemia (163).
Beside the effects of FGF23 on phosphate homeostasis, FGF23 signaling has been described to promote renal calcium reabsorption through the TRPV5 channel. Indeed, the apical membrane abundance of TRPV5 in renal distal tubules could be regulated by the binding of FGF23 to FGFR-Klotho complexes which activated a signaling pathway implicating ERK1/2, SGK1, and WNK4. This signaling pathway led to the increase of intracellular transport of fully glycosylated TRPV5 from the Golgi apparatus to the apical plasma membrane, thus decreasing the renal loss of calcium (153). In distal convoluted tubule, the ERK1/2-SGK1-WNK4 signaling pathway leads to WNK4 serine phosphorylation at residue 71 and kinase activation. FGF23 promoted the physical interaction between NCC and WNK4, increasing NCC membrane abundance, and would promote sodium reabsorption (152). Additional studies of the actions of FGF23 and Klotho specifically within the kidney distal tubule are required to determine the full extent of kidney FGF23 bioactivity.
The hormone FGF23 is mainly produced by osteocytes with the ability to target distant organs such as kidney. In late osteoblasts and osteocytes, FGF23 can be upregulated by elevated phosphate, anemia, inflammation, PTH and 1,25D; and downregulated by hypophosphatemia, insulin, and insulin-like growth factor 1. Although not covered here, studies have shown that FGF23 can be produced at lower levels by other cells such as immune cells, bone marrow erythroid cells and other tissues such as liver in response to diverse stimuli (106, 164). The posttranslational modifications of FGF23 protein via O-glycosylation and phosphorylation controls the proteolytic cleavage of mature FGF23 protein which dictates biologically active FGF23 concentrations.
The binding of FGF23 to FGFR1-Klotho complexes in the kidney has been shown to induce a signaling cascade through MAPK which controls mineral metabolism. The signals induced by FGF23 in kidney downregulate the expression of Npt2a/c leading to decreased phosphate reabsorption in proximal tubules, and upregulation of TPRV5 and NCC, potentially promoting calcium and sodium reabsorption, respectively, in the distal tubule. Alterations of FGF23 expression in osteocytes, FGF23 processing, and FGF23 activity cause severe endocrine pathologies resulting in rare and common diseases. Thus, further understanding the mechanisms controlling FGF23 production in osteocytes and bioactivity in kidney will lead to improved patient outcomes.
In summary, although much is known regarding FGF23 regulation and actions, gaps in our knowledge exist. These include the potential contributions of bone cells such as osteoblasts and osteoclasts in the regulation of osteocytic FGF23, and it remains unclear whether aged osteocytes (mature cells and deeply embedded in the mineralized bone matrix) are more effective in terms of upregulating FGF23 in response to physiological and pathological changes vs. early osteocytes. Finally, whether FGF23 can target other cell-types in the kidney beyond its defined actions on proximal and distal tubules remains unknown, thus future investigation could examine the effects of FGF23 on renal immune cells such as macrophages and regulatory T cells, critical for the control of renal inflammation and kidney remodeling during acute kidney injury and CKD.
RA, PN, MN, and KW wrote and edited the manuscript and figures. All authors contributed to the article and approved the submitted version.
The authors would like to acknowledge NIH grants R21-AR059278, R01-DK112958, and R01- HL145528 (KW); T32-HL007910 and F31-DK122679 (MN); and The David Weaver Professorship (KW).
KW receives royalties from Kyowa-Hakko-Kirin Pharmaceutics, Inc., and research funding from Akebia-Keryx, both of which had no part in the study design, data collection and analysis, decision to publish, or preparation of this manuscript.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. ICRP. Basic Anatomical and Physiological Data for use in Radiological Protection—The Skeleton. Kidlington; Oxford; Elsevier Science (1995).
2. Avtandilashvili M, Tolmachev SY. Modeling the skeleton weight of an adult caucasian man. Health Phys. (2019) 117:149–55. doi: 10.1097/HP.0000000000000881
3. Mosekilde L. Age-related changes in bone mass, structure, and strength–effects of loading. Z Rheumatol. (2000) 59(Suppl.1):1–9. doi: 10.1007/s003930070031
4. Cahman KDGF. Age-Related Changes in Bone Mass. Encyclopedia of Food Sciences and Nutrition. 2nd ed. Cambridge, MA: Academic Press (2003). p. 6,000.
5. Bonjour JP. Calcium and phosphate: a duet of ions playing for bone health. J Am Coll Nutr. (2011) 30(5Suppl.1):438S−48S. doi: 10.1080/07315724.2011.10719988
6. Dallas SL, Bonewald LF. Dynamics of the transition from osteoblast to osteocyte. Ann N Y Acad Sci. (2010) 1192:437–43. doi: 10.1111/j.1749-6632.2009.05246.x
7. Downey PA, Siegel MI. Bone biology and the clinical implications for osteoporosis. Phys Ther. (2006) 86:77–91. doi: 10.1093/ptj/86.1.77
8. Lerner UH. Osteoblasts, osteoclasts, and osteocytes: unveiling their intimate-associated responses to applied orthodontic forces. Semin Orthodontics. (2012) 18:237–48. doi: 10.1053/j.sodo.2012.06.002
9. Aubin JE. Bone stem cells. J Cell Biochem Suppl. (1998) 30–1:73–82. doi: 10.1002/(SICI)1097-464472:30/31+<73::AID-JCB11>3.0.CO;2-L
10. Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, et al. Multilineage potential of adult human mesenchymal stem cells. Science. (1999) 284:143–7. doi: 10.1126/science.284.5411.143
11. Ash P, Loutit JF, Townsend KM. Osteoclasts derived from haematopoietic stem cells. Nature. (1980) 283:669–70. doi: 10.1038/283669a0
12. Scheven BA, Visser JW, Nijweide PJ. In vitro osteoclast generation from different bone marrow fractions, including a highly enriched haematopoietic stem cell population. Nature. (1986) 321:79–81. doi: 10.1038/321079a0
13. Tondravi MM, McKercher SR, Anderson K, Erdmann JM, Quiroz M, Maki R, et al. Osteopetrosis in mice lacking haematopoietic transcription factor PU.1. Nature. (1997) 386:81–4. doi: 10.1038/386081a0
14. Goetz R, Mohammadi M. Exploring mechanisms of FGF signalling through the lens of structural biology. Nat Rev Mol Cell Biol. (2013) 14:166–80. doi: 10.1038/nrm3528
15. Dallas SL, Veno PA, Tiede-Lewis LM. Live cell imaging of bone cell and organ cultures. Methods Mol Biol. (2019) 1914:467–506. doi: 10.1007/978-1-4939-8997-3_27
16. Balemans W, Ebeling M, Patel N, Van Hul E, Olson P, Dioszegi M, et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet. (2001) 10:537–43. doi: 10.1093/hmg/10.5.537
17. Brunkow ME, Gardner JC, Van Ness J, Paeper BW, Kovacevich BR, Proll S, et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet. (2001) 68:577–89. doi: 10.1086/318811
18. Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. (2011) 17:1231–4. doi: 10.1038/nm.2452
19. Xiong J, Piemontese M, Thostenson JD, Weinstein RS, Manolagas SC, O'Brien CA. Osteocyte-derived RANKL is a critical mediator of the increased bone resorption caused by dietary calcium deficiency. Bone. (2014) 66:146–54. doi: 10.1016/j.bone.2014.06.006
20. Knothe Tate ML, Niederer P, Knothe U. In vivo tracer transport through the lacunocanalicular system of rat bone in an environment devoid of mechanical loading. Bone. (1998) 22:107–17. doi: 10.1016/S8756-3282(97)00234-2
21. Consortium A. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet. (2000) 26:345–8. doi: 10.1038/81664
22. Perwad F, Zhang MY, Tenenhouse HS, Portale AA. Fibroblast growth factor 23 impairs phosphorus and vitamin D metabolism in vivo and suppresses 25-hydroxyvitamin D-1alpha-hydroxylase expression in vitro. Am J Physiol Renal Physiol. (2007) 293:F1577–83. doi: 10.1152/ajprenal.00463.2006
23. Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. (2004) 19:429–35. doi: 10.1359/JBMR.0301264
24. Wolf M, White KE. Coupling fibroblast growth factor 23 production and cleavage: iron deficiency, rickets, and kidney disease. Curr Opin Nephrol Hypertens. (2014) 23:411–9. doi: 10.1097/01.mnh.0000447020.74593.6f
25. Saito H, Maeda A, Ohtomo S, Hirata M, Kusano K, Kato S, et al. Circulating FGF-23 is regulated by 1alpha,25-dihydroxyvitamin D3 and phosphorus in vivo. J Biol Chem. (2005) 280:2543–9. doi: 10.1074/jbc.M408903200
26. Perwad F, Azam N, Zhang MY, Yamashita T, Tenenhouse HS, Portale AA. Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1,25-dihydroxyvitamin D metabolism in mice. Endocrinology. (2005) 146:5358–64. doi: 10.1210/en.2005-0777
27. Bon N, Frangi G, Sourice S, Guicheux J, Beck-Cormier S, Beck L. Phosphate-dependent FGF23 secretion is modulated by PiT2/Slc20a2. Mol Metab. (2018) 11:197–204. doi: 10.1016/j.molmet.2018.02.007
28. Takashi Y, Kosako H, Sawatsubashi S, Kinoshita Y, Ito N, Tsoumpra MK, et al. Activation of unliganded FGF receptor by extracellular phosphate potentiates proteolytic protection of FGF23 by its O-glycosylation. Proc Natl Acad Sci USA. (2019) 116:11418–27. doi: 10.1073/pnas.1815166116
29. Takashi Y, Fukumoto S. Phosphate-sensing and regulatory mechanism of FGF23 production. J Endocrinol Invest. (2020) 43:877–83. doi: 10.1007/s40618-020-01205-9
30. Xiao Z, Huang J, Cao L, Liang Y, Han X, Quarles LD. Osteocyte-specific deletion of Fgfr1 suppresses FGF23. PLoS ONE. (2014) 9:e104154. doi: 10.1371/journal.pone.0104154
31. Wu AL, Feng B, Chen MZ, Kolumam G, Zavala-Solorio J, Wyatt SK, et al. Antibody-mediated activation of FGFR1 induces FGF23 production and hypophosphatemia. PLoS ONE. (2013) 8:e57322. doi: 10.1371/journal.pone.0057322
32. Hori M, Kinoshita Y, Taguchi M, Fukumoto S. Phosphate enhances Fgf23 expression through reactive oxygen species in UMR-106 cells. J Bone Miner Metab. (2016) 34:132–9. doi: 10.1007/s00774-015-0651-9
33. Hassan A, Durlacher K, Silver J, Naveh-Many T, Levi R. The fibroblast growth factor receptor mediates the increased FGF23 expression in acute and chronic uremia. Am J Physiol Renal Physiol. (2016) 310:F217–21. doi: 10.1152/ajprenal.00332.2015
34. Goetz R, Nakada Y, Hu MC, Kurosu H, Wang L, Nakatani T, et al. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc Natl Acad Sci USA. (2010) 107:407–12. doi: 10.1073/pnas.0902006107
35. Komaba H, Kaludjerovic J, Hu DZ, Nagano K, Amano K, Ide N, et al. Klotho expression in osteocytes regulates bone metabolism and controls bone formation. Kidney Int. (2017) 92:599–611. doi: 10.1016/j.kint.2017.02.014
36. Avin KG, Coen PM, Huang W, Stolz DB, Sowa GA, Dube JJ, et al. Skeletal muscle as a regulator of the longevity protein, Klotho. Front Physiol. (2014) 5:189. doi: 10.3389/fphys.2014.00189
37. Drueke TB, Massy ZA. Circulating Klotho levels: clinical relevance and relationship with tissue Klotho expression. Kidney Int. (2013) 83:13–5. doi: 10.1038/ki.2012.370
38. Lim K, Groen A, Molostvov G, Lu T, Lilley KS, Snead D, et al. α-klotho expression in human tissues. J Clin Endocrinol Metab. (2015) 100:E1308–18. doi: 10.1210/jc.2015-1800
39. Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. (1997) 390:45–51. doi: 10.1038/36285
40. Matsumura Y, Aizawa H, Shiraki-Iida T, Nagai R, Kuro-o M, Nabeshima Y. Identification of the human klotho gene and its two transcripts encoding membrane and secreted klotho protein. Biochem Biophys Res Commun. (1998) 242:626–30.
41. Bloch L, Sineshchekova O, Reichenbach D, Reiss K, Saftig P, Kuro-o M, et al. Klotho is a substrate for alpha-, beta- and gamma-secretase. FEBS Lett. (2009) 583:3221–4. doi: 10.1016/j.febslet.2009.09.009
42. Chen CD, Podvin S, Gillespie E, Leeman SE, Abraham CR. Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17. Proc Natl Acad Sci USA. (2007) 104:19796–801. doi: 10.1073/pnas.0709805104
43. Lindberg K, Amin R, Moe OW, Hu MC, Erben RG, Ostman Wernerson A, et al. The kidney is the principal organ mediating klotho effects. J Am Soc Nephrol. (2014) 25:2169–75. doi: 10.1681/ASN.2013111209
44. Hu MC, Shi M, Zhang J, Addo T, Cho HJ, Barker SL, et al. Renal production, uptake, and handling of circulating αKlotho. J Am Soc Nephrol. (2016) 27:79–90. doi: 10.1681/ASN.2014101030
45. Carrillo-Lopez N, Panizo S, Alonso-Montes C, Roman-Garcia P, Rodriguez I, Martinez-Salgado C, et al. Direct inhibition of osteoblastic Wnt pathway by fibroblast growth factor 23 contributes to bone loss in chronic kidney disease. Kidney Int. (2016) 90:77–89. doi: 10.1016/j.kint.2016.01.024
46. Hum JM, O'Bryan LM, Tatiparthi AK, Cass TA, Clinkenbeard EL, Cramer MS, et al. Chronic hyperphosphatemia and vascular calcification are reduced by stable delivery of soluble klotho. J Am Soc Nephrol. (2017) 28:1162–74. doi: 10.1681/ASN.2015111266
47. Ito N, Wijenayaka AR, Prideaux M, Kogawa M, Ormsby RT, Evdokiou A, et al. Regulation of FGF23 expression in IDG-SW3 osteocytes and human bone by pro-inflammatory stimuli. Mol Cell Endocrinol. (2015) 399:208–18. doi: 10.1016/j.mce.2014.10.007
48. Ewendt F, Foller M. p38MAPK controls fibroblast growth factor 23 (FGF23) synthesis in UMR106-osteoblast-like cells and in IDG-SW3 osteocytes. J Endocrinol Invest. (2019) 42:1477–83. doi: 10.1007/s40618-019-01073-y
49. Bar L, Hase P, Foller M. PKC regulates the production of fibroblast growth factor 23 (FGF23). PLoS ONE. (2019) 14:e0211309. doi: 10.1371/journal.pone.0211309
50. Maniati E, Bossard M, Cook N, Candido JB, Emami-Shahri N, Nedospasov SA, et al. Crosstalk between the canonical NF-κB and Notch signaling pathways inhibits Pparγ expression and promotes pancreatic cancer progression in mice. J Clin Invest. (2011) 121:4685–99. doi: 10.1172/JCI45797
51. Tamamura Y, Sakamoto K, Katsube KI, Yamaguchi A. Notch signaling is involved in Fgf23 upregulation in osteocytes. Biochem Biophys Res Commun. (2019) 518:233–8. doi: 10.1016/j.bbrc.2019.08.038
52. He Q, Shumate LT, Matthias J, Aydin C, Wein MN, Spatz JM, et al. A G protein-coupled, IP3/protein kinase C pathway controlling the synthesis of phosphaturic hormone FGF23. JCI Insight. (2019) 4:125007. doi: 10.1172/jci.insight.125007
53. Onal M, Carlson AH, Thostenson JD, Benkusky NA, Meyer MB, Lee SM, et al. A novel distal enhancer mediates inflammation-, PTH-, and early onset murine kidney disease-induced expression of the mouse Fgf23 gene. JBMR Plus. (2018) 2:32–47. doi: 10.1002/jbm4.10023
54. Lee SM, Carlson AH, Onal M, Benkusky NA, Meyer MB, Pike JW. A control region near the fibroblast growth factor 23 gene mediates response to phosphate, 1,25(OH)2D3, and LPS in vivo. Endocrinology. (2019) 160:2877–91. doi: 10.1210/en.2019-00622
55. Pagani A, Nai A, Corna G, Bosurgi L, Rovere-Querini P, Camaschella C, et al. Low hepcidin accounts for the pro-inflammatory status associated with iron deficiency. Blood. (2011) 118:736–46. doi: 10.1182/blood-2011-02-337212
56. Agoro R, Taleb M, Quesniaux VFJ, Mura C. Cell iron status influences macrophage polarization. PLoS ONE. (2018) 13:e0196921. doi: 10.1371/journal.pone.0196921
57. Farrow EG, Yu X, Summers LJ, Davis SI, Fleet JC, Allen MR, et al. Iron deficiency drives an autosomal dominant hypophosphatemic rickets (ADHR) phenotype in fibroblast growth factor-23 (Fgf23) knock-in mice. Proc Natl Acad Sci USA. (2011) 108:E1146–55. doi: 10.1073/pnas.1110905108
58. David V, Martin A, Isakova T, Spaulding C, Qi L, Ramirez V, et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int. (2016) 89:135–46. doi: 10.1038/ki.2015.290
59. Durlacher-Betzer K, Hassan A, Levi R, Axelrod J, Silver J, Naveh-Many T. Interleukin-6 contributes to the increase in fibroblast growth factor 23 expression in acute and chronic kidney disease. Kidney Int. (2018) 94:315–25. doi: 10.1016/j.kint.2018.02.026
60. Zhang Q, Doucet M, Tomlinson RE, Han X, Quarles LD, Collins MT, et al. The hypoxia-inducible factor-1α activates ectopic production of fibroblast growth factor 23 in tumor-induced osteomalacia. Bone Res. (2016) 4:16011. doi: 10.1038/boneres.2016.11
61. Imel EA, Liu Z, Coffman M, Acton D, Mehta R, Econs MJ. Oral iron replacement normalizes fibroblast growth factor 23 in iron-deficient patients with autosomal dominant hypophosphatemic rickets. J Bone Miner Res. (2019) 35:231–8. doi: 10.1002/jbmr.3878
62. Wolf M, Koch TA, Bregman DB. Effects of iron deficiency anemia and its treatment on fibroblast growth factor 23 and phosphate homeostasis in women. J Bone Miner Res. (2013) 28:1793–803. doi: 10.1002/jbmr.1923
63. Klein K, Asaad S, Econs M, Rubin JE. Severe FGF23-based hypophosphataemic osteomalacia due to ferric carboxymaltose administration. BMJ Case Rep. (2018) 2018:222851. doi: 10.1136/bcr-2017-222851
64. Clinkenbeard EL, Hanudel MR, Stayrook KR, Appaiah HN, Farrow EG, Cass TA, et al. Erythropoietin stimulates murine and human fibroblast growth factor-23, revealing novel roles for bone and bone marrow. Haematologica. (2017) 102:e427–30. doi: 10.3324/haematol.2017.167882
65. Hanudel MR, Eisenga MF, Rappaport M, Chua K, Qiao B, Jung G, et al. Effects of erythropoietin on fibroblast growth factor 23 in mice and humans. Nephrol Dial Transplant. (2018) 34:2057–65. doi: 10.1093/ndt/gfy189
66. Daryadel A, Bettoni C, Haider T, Imenez Silva PH, Schnitzbauer U, Pastor-Arroyo EM, et al. Erythropoietin stimulates fibroblast growth factor 23 (FGF23) in mice and men. Pflugers Arch. (2018) 470:1569–82. doi: 10.1007/s00424-018-2171-7
67. Daryadel A, Natale L, Seebeck P, Bettoni C, Schnitzbauer U, Gassmann M, et al. Elevated FGF23 and disordered renal mineral handling with reduced bone mineralization in chronically erythropoietin over-expressing transgenic mice. Sci Rep. (2019) 9:14989. doi: 10.1038/s41598-019-51577-z
68. Haase VH. HIF-prolyl hydroxylases as therapeutic targets in erythropoiesis and iron metabolism. Hemodial Int. (2017) 21(Suppl.1):S110–24. doi: 10.1111/hdi.12567
69. Flamme I, Ellinghaus P, Urrego D, Kruger T. FGF23 expression in rodents is directly induced via erythropoietin after inhibition of hypoxia inducible factor proline hydroxylase. PLoS ONE. (2017) 12:e0186979. doi: 10.1371/journal.pone.0186979
70. Noonan ML, Clinkenbeard EL, Ni P, Swallow EA, Tippen SP, Agoro R, et al. Erythropoietin and a hypoxia-inducible factor prolyl hydroxylase inhibitor (HIF-PHDi) lowers FGF23 in a model of chronic kidney disease (CKD). Physiol Rep. (2020) 8:e14434. doi: 10.14814/phy2.14434
71. Qin L, Raggatt LJ, Partridge NC. Parathyroid hormone: a double-edged sword for bone metabolism. Trends Endocrinol Metab. (2004) 15:60–5. doi: 10.1016/j.tem.2004.01.006
72. Hou YC, Lu CL, Lu KC. Mineral bone disorders in chronic kidney disease. Nephrology. (2018) 23(Suppl.4):88–94. doi: 10.1111/nep.13457
73. Murray TM, Rao LG, Divieti P, Bringhurst FR. Parathyroid hormone secretion and action: evidence for discrete receptors for the carboxyl-terminal region and related biological actions of carboxyl- terminal ligands. Endocr Rev. (2005) 26:78–113. doi: 10.1210/er.2003-0024
74. Hruska KA, Seifert M. Pathophysiology of chronic kidney disease mineral bone disorder (CKD-MBD). In: Rosen JC, editor. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. Hoboken, NJ: Wiley-Blackwell (2013). p. 632–9.
75. Goodman WG. Calcimimetic agents and secondary hyperparathyroidism: treatment and prevention. Nephrol Dial Transplant. (2002) 17:204–7. doi: 10.1093/ndt/17.2.204
76. Meir T, Durlacher K, Pan Z, Amir G, Richards WG, Silver J, et al. Parathyroid hormone activates the orphan nuclear receptor Nurr1 to induce FGF23 transcription. Kidney Int. (2014) 86:1106–15. doi: 10.1038/ki.2014.215
77. Rhee Y, Bivi N, Farrow E, Lezcano V, Plotkin LI, White KE, et al. Parathyroid hormone receptor signaling in osteocytes increases the expression of fibroblast growth factor-23 in vitro and in vivo. Bone. (2011) 49:636–43. doi: 10.1016/j.bone.2011.06.025
78. Knab VM, Corbin B, Andrukhova O, Hum JM, Ni P, Rabadi S, et al. Acute parathyroid hormone injection increases C-terminal but not intact fibroblast growth factor 23 levels. Endocrinology. (2017) 158:1130–9. doi: 10.1210/en.2016-1451
79. Ishiyama N, Shibata H, Kanzaki M, Shiozaki S, Miyazaki J, Kobayashi I, et al. Calcium as a second messenger of the action of transforming growth factor-β on insulin secretion. Mol Cell Endocrinol. (1996) 117:1–6. doi: 10.1016/0303-7207(95)03726-8
80. McGowan TA, Madesh M, Zhu Y, Wang L, Russo M, Deelman L, et al. TGF-beta-induced Ca(2+) influx involves the type III IP(3) receptor and regulates actin cytoskeleton. Am J Physiol Renal Physiol. (2002) 282:F910–20. doi: 10.1152/ajprenal.00252.2001
81. Gooch JL, Gorin Y, Zhang BX, Abboud HE. Involvement of calcineurin in transforming growth factor-β-mediated regulation of extracellular matrix accumulation. J Biol Chem. (2004) 279:15561–70. doi: 10.1074/jbc.M308759200
82. Feger M, Hase P, Zhang B, Hirche F, Glosse P, Lang F, et al. The production of fibroblast growth factor 23 is controlled by TGF-β2. Sci Rep. (2017) 7:4982. doi: 10.1038/s41598-017-05226-y
83. Zhang B, Yan J, Umbach AT, Fakhri H, Fajol A, Schmidt S, et al. NFκB-sensitive Orai1 expression in the regulation of FGF23 release. J Mol Med. (2016) 94:557–66. doi: 10.1007/s00109-015-1370-3
84. Bar L, Grossmann C, Gekle M, Foller M. Calcineurin inhibitors regulate fibroblast growth factor 23 (FGF23) synthesis. Naunyn Schmiedebergs Arch Pharmacol. (2017) 390:1117–23. doi: 10.1007/s00210-017-1411-2
85. Zhang B, Yan J, Schmidt S, Salker MS, Alexander D, Foller M, et al. Lithium-sensitive store-operated Ca2+ entry in the regulation of FGF23 release. Neurosignals. (2015) 23:34–48. doi: 10.1159/000442602
86. Nguyen-Yamamoto L, Karaplis AC, St-Arnaud R, Goltzman D. Fibroblast growth factor 23 regulation by systemic and local osteoblast-synthesized 1,25-dihydroxyvitamin D. J Am Soc Nephrol. (2017) 28:586–97. doi: 10.1681/ASN.2016010066
87. Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet. (2006) 38:1310–5. doi: 10.1038/ng1905
88. Liu S, Tang W, Zhou J, Vierthaler L, Quarles LD. Distinct roles for intrinsic osteocyte abnormalities and systemic factors in regulation of FGF23 and bone mineralization in Hyp mice. Am J Physiol Endocrinol Metab. (2007) 293:E1636–44. doi: 10.1152/ajpendo.00396.2007
89. Liu S, Zhou J, Tang W, Jiang X, Rowe DW, Quarles LD. Pathogenic role of Fgf23 in Hyp mice. Am J Physiol Endocrinol Metab. (2006) 291:E38–49. doi: 10.1152/ajpendo.00008.2006
90. Liu S, Zhou J, Tang W, Menard R, Feng JQ, Quarles LD. Pathogenic role of Fgf23 in Dmp1-null mice. Am J Physiol Endocrinol Metab. (2008) 295:E254–61. doi: 10.1152/ajpendo.90201.2008
91. Martin A, Liu S, David V, Li H, Karydis A, Feng JQ, et al. Bone proteins PHEX and DMP1 regulate fibroblastic growth factor Fgf23 expression in osteocytes through a common pathway involving FGF receptor (FGFR) signaling. FASEB J. (2011) 25:2551–62. doi: 10.1096/fj.10-177816
92. Liu S, Tang W, Fang J, Ren J, Li H, Xiao Z, et al. Novel regulators of Fgf23 expression and mineralization in Hyp bone. Mol Endocrinol. (2009) 23:1505–18. doi: 10.1210/me.2009-0085
93. Lee JW, Yamaguchi A, Iimura T. Functional heterogeneity of osteocytes in FGF23 production: the possible involvement of DMP1 as a direct negative regulator. Bonekey Rep. (2014) 3:543. doi: 10.1038/bonekey.2014.38
94. Clinkenbeard EL, Cass TA, Ni P, Hum JM, Bellido T, Allen MR, et al. Conditional deletion of murine Fgf23: interruption of the normal skeletal responses to phosphate challenge and rescue of genetic hypophosphatemia. J Bone Miner Res. (2016) 31:1247–57. doi: 10.1002/jbmr.2792
95. Bar L, Feger M, Fajol A, Klotz LO, Zeng S, Lang F, et al. Insulin suppresses the production of fibroblast growth factor 23 (FGF23). Proc Natl Acad Sci USA. (2018) 115:5804–9. doi: 10.1073/pnas.1800160115
96. Gribaa M, Younes M, Bouyacoub Y, Korbaa W, Ben Charfeddine I, Touzi M, et al. An autosomal dominant hypophosphatemic rickets phenotype in a Tunisian family caused by a new FGF23 missense mutation. J Bone Miner Metab. (2010) 28:111–5. doi: 10.1007/s00774-009-0111-5
97. Silvestri L, Pagani A, Camaschella C. Furin-mediated release of soluble hemojuvelin: a new link between hypoxia and iron homeostasis. Blood. (2008) 111:924–31. doi: 10.1182/blood-2007-07-100677
98. Tagliabracci VS, Engel JL, Wiley SE, Xiao J, Gonzalez DJ, Nidumanda Appaiah H, et al. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc Natl Acad Sci USA. (2014) 111:5520–5. doi: 10.1073/pnas.1402218111
99. de Las Rivas M, Paul Daniel EJ, Narimatsu Y, Companon I, Kato K, Hermosilla P, et al. Molecular basis for fibroblast growth factor 23 O-glycosylation by GalNAc-T3. Nat Chem Biol. (2020) 16:351–60. doi: 10.1038/s41589-019-0444-x
100. Rafaelsen SH, Raeder H, Fagerheim AK, Knappskog P, Carpenter TO, Johansson S, et al. Exome sequencing reveals FAM20c mutations associated with fibroblast growth factor 23-related hypophosphatemia, dental anomalies, and ectopic calcification. J Bone Miner Res. (2013) 28:1378–85. doi: 10.1002/jbmr.1850
101. Liu P, Ma S, Zhang H, Liu C, Lu Y, Chen L, et al. Specific ablation of mouse Fam20C in cells expressing type I collagen leads to skeletal defects and hypophosphatemia. Sci Rep. (2017) 7:3590. doi: 10.1038/s41598-017-03960-x
102. Eren M, Place AT, Thomas PM, Flevaris P, Miyata T, Vaughan DE. PAI-1 is a critical regulator of FGF23 homeostasis. Sci Adv. (2017) 3:e1603259. doi: 10.1126/sciadv.1603259
103. Takeshita K, Yamamoto K, Ito M, Kondo T, Matsushita T, Hirai M, et al. Increased expression of plasminogen activator inhibitor-1 with fibrin deposition in a murine model of aging, “Klotho” mouse. Semin Thromb Hemost. (2002) 28:545–54. doi: 10.1055/s-2002-36699
104. Eren M, Boe AE, Murphy SB, Place AT, Nagpal V, Morales-Nebreda L, et al. PAI-1-regulated extracellular proteolysis governs senescence and survival in Klotho mice. Proc Natl Acad Sci USA. (2014) 111:7090–5. doi: 10.1073/pnas.1321942111
105. Itoh N, Nakayama Y, Konishi M. Roles of FGFs as paracrine or endocrine signals in liver development, health, and disease. Front Cell Dev Biol. (2016) 4:30. doi: 10.3389/fcell.2016.00030
106. Clinkenbeard EL, Noonan ML, Thomas JC, Ni P, Hum JM, Aref M, et al. Increased FGF23 protects against detrimental cardio-renal consequences during elevated blood phosphate in CKD. JCI Insight. (2019) 4:123817. doi: 10.1172/jci.insight.123817
107. Gutierrez OM, Mannstadt M, Isakova T, Rauh-Hain JA, Tamez H, Shah A, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med. (2008) 359:584–92. doi: 10.1056/NEJMoa0706130
108. John GB, Cheng CY, Kuro-o M. Role of Klotho in aging, phosphate metabolism, and CKD. Am J Kidney Dis. (2011) 58:127–34. doi: 10.1053/j.ajkd.2010.12.027
109. Gattineni J, Twombley K, Goetz R, Mohammadi M, Baum M. Regulation of serum 1,25(OH)2 vitamin D3 levels by fibroblast growth factor 23 is mediated by FGF receptors 3 and 4. Am J Physiol Renal Physiol. (2011) 301:F371–7. doi: 10.1152/ajprenal.00740.2010
110. Gattineni J, Alphonse P, Zhang Q, Mathews N, Bates CM, Baum M. Regulation of renal phosphate transport by FGF23 is mediated by FGFR1 and FGFR4. Am J Physiol Renal Physiol. (2014) 306:F351–8. doi: 10.1152/ajprenal.00232.2013
111. Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, et al. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem. (2006) 281:6120–3. doi: 10.1074/jbc.C500457200
112. Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. (2006) 444:770–4. doi: 10.1038/nature05315
113. Nabeshima Y. [Discovery of alpha-Klotho and FGF23 unveiled new insight into calcium and phosphate homeostasis]. Clin Calcium. (2008) 18:923–34.
114. Chen G, Liu Y, Goetz R, Fu L, Jayaraman S, Hu MC, et al. α-Klotho is a non-enzymatic molecular scaffold for FGF23 hormone signalling. Nature. (2018) 553:461–6. doi: 10.1038/nature25451
115. Larsson T, Marsell R, Schipani E, Ohlsson C, Ljunggren O, Tenenhouse HS, et al. Transgenic mice expressing fibroblast growth factor 23 under the control of the α1(I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology. (2004) 145:3087–94. doi: 10.1210/en.2003-1768
116. Sitara D, Razzaque MS, Hesse M, Yoganathan S, Taguchi T, Erben RG, et al. Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol. (2004) 23:421–32. doi: 10.1016/j.matbio.2004.09.007
117. Marsell R, Krajisnik T, Goransson H, Ohlsson C, Ljunggren O, Larsson TE, et al. Gene expression analysis of kidneys from transgenic mice expressing fibroblast growth factor-23. Nephrol Dial Transplant. (2008) 23:827–33. doi: 10.1093/ndt/gfm672
118. Wysocki J, Gonzalez-Pacheco FR, Batlle D. Angiotensin-converting enzyme 2: possible role in hypertension and kidney disease. Curr Hypertens Rep. (2008) 10:70–7. doi: 10.1007/s11906-008-0014-1
119. Han X, Ross J, Kolumam G, Pi M, Sonoda J, King G, et al. Cardiovascular effects of renal distal tubule deletion of the FGF receptor 1 gene. J Am Soc Nephrol. (2018) 29:69–80. doi: 10.1681/ASN.2017040412
120. Imura A, Tsuji Y, Murata M, Maeda R, Kubota K, Iwano A, et al. α-Klotho as a regulator of calcium homeostasis. Science. (2007) 316:1615–8. doi: 10.1126/science.1135901
121. Viau A, El Karoui K, Laouari D, Burtin M, Nguyen C, Mori K, et al. Lipocalin 2 is essential for chronic kidney disease progression in mice and humans. J Clin Invest. (2010) 120:4065–76. doi: 10.1172/JCI42004
122. Portale AA, Zhang MY, David V, Martin A, Jiao Y, Gu W, et al. Characterization of FGF23-dependent Egr-1 cistrome in the mouse renal proximal tubule. PLoS ONE. (2015) 10:e0142924. doi: 10.1371/journal.pone.0142924
123. Farrow EG, Davis SI, Summers LJ, White KE. Initial FGF23-mediated signaling occurs in the distal convoluted tubule. J Am Soc Nephrol. (2009) 20:955–60. doi: 10.1681/ASN.2008070783
124. Zhang MY, Ranch D, Pereira RC, Armbrecht HJ, Portale AA, Perwad F. Chronic inhibition of ERK1/2 signaling improves disordered bone and mineral metabolism in hypophosphatemic (Hyp) mice. Endocrinology. (2012) 153:1806–16. doi: 10.1210/en.2011-1831
125. Ranch D, Zhang MY, Portale AA, Perwad F. Fibroblast growth factor 23 regulates renal 1,25-dihydroxyvitamin D and phosphate metabolism via the MAP kinase signaling pathway in Hyp mice. J Bone Miner Res. (2011) 26:1883–90. doi: 10.1002/jbmr.401
126. Fukuda T, Kanomata K, Nojima J, Urakawa I, Suzawa T, Imada M, et al. FGF23 induces expression of two isoforms of NAB2, which are corepressors of Egr-1. Biochem Biophys Res Commun. (2007) 353:147–51. doi: 10.1016/j.bbrc.2006.12.011
127. Kumbrink J, Gerlinger M, Johnson JP. Egr-1 induces the expression of its corepressor nab2 by activation of the nab2 promoter thereby establishing a negative feedback loop. J Biol Chem. (2005) 280:42785–93. doi: 10.1074/jbc.M511079200
128. Takeshita A, Kawakami K, Furushima K, Miyajima M, Sakaguchi K. Central role of the proximal tubular αKlotho/FGF receptor complex in FGF23-regulated phosphate and vitamin D metabolism. Sci Rep. (2018) 8:6917. doi: 10.1038/s41598-018-25087-3
129. Liu S, Vierthaler L, Tang W, Zhou J, Quarles LD. FGFR3 and FGFR4 do not mediate renal effects of FGF23. J Am Soc Nephrol. (2008) 19:2342–50. doi: 10.1681/ASN.2007121301
130. Gattineni J, Bates C, Twombley K, Dwarakanath V, Robinson ML, Goetz R, et al. FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am J Physiol Renal Physiol. (2009) 297:F282–91. doi: 10.1152/ajprenal.90742.2008
131. Li H, Martin A, David V, Quarles LD. Compound deletion of Fgfr3 and Fgfr4 partially rescues the Hyp mouse phenotype. Am J Physiol Endocrinol Metab. (2011) 300:E508–17. doi: 10.1152/ajpendo.00499.2010
132. Nakatani T, Ohnishi M, Razzaque MS. Inactivation of klotho function induces hyperphosphatemia even in presence of high serum fibroblast growth factor 23 levels in a genetically engineered hypophosphatemic (Hyp) mouse model. FASEB J. (2009) 23:3702–11. doi: 10.1096/fj.08-123992
133. Morishita K, Shirai A, Kubota M, Katakura Y, Nabeshima Y, Takeshige K, et al. The progression of aging in klotho mutant mice can be modified by dietary phosphorus and zinc. J Nutr. (2001) 131:3182–8. doi: 10.1093/jn/131.12.3182
134. Ohnishi M, Razzaque MS. Dietary and genetic evidence for phosphate toxicity accelerating mammalian aging. FASEB J. (2010) 24:3562–71. doi: 10.1096/fj.09-152488
135. Murer H, Hernando N, Forster I, Biber J. Proximal tubular phosphate reabsorption: molecular mechanisms. Physiol Rev. (2000) 80:1373–409. doi: 10.1152/physrev.2000.80.4.1373
136. Ohkido I, Segawa H, Yanagida R, Nakamura M, Miyamoto K. Cloning, gene structure and dietary regulation of the type-IIc Na/Pi cotransporter in the mouse kidney. Pflugers Arch. (2003) 446:106–15. doi: 10.1007/s00424-003-1010-6
137. Prie D, Urena Torres P, Friedlander G. Latest findings in phosphate homeostasis. Kidney Int. (2009) 75:882–9. doi: 10.1038/ki.2008.643
138. Olauson H, Lindberg K, Amin R, Jia T, Wernerson A, Andersson G, et al. Targeted deletion of Klotho in kidney distal tubule disrupts mineral metabolism. J Am Soc Nephrol. (2012) 23:1641–51. doi: 10.1681/ASN.2012010048
139. Ide N, Olauson H, Sato T, Densmore MJ, Wang H, Hanai JI, et al. In vivo evidence for a limited role of proximal tubular Klotho in renal phosphate handling. Kidney Int. (2016) 90:348–62. doi: 10.1016/j.kint.2016.04.009
140. Ide N, Ye R, Courbebaisse M, Olauson H, Densmore MJ, Larsson TE, et al. In vivo evidence for an interplay of FGF23/Klotho/PTH axis on the phosphate handling in renal proximal tubules. Am J Physiol Renal Physiol. (2018) 315:F1261–70. doi: 10.1152/ajprenal.00650.2017
141. Andrukhova O, Streicher C, Zeitz U, Erben RG. Fgf23 and parathyroid hormone signaling interact in kidney and bone. Mol Cell Endocrinol. (2016) 436:224–39. doi: 10.1016/j.mce.2016.07.035
142. Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci USA. (2001) 98:6500–5. doi: 10.1073/pnas.101545198
143. Saito H, Kusano K, Kinosaki M, Ito H, Hirata M, Segawa H, et al. Human fibroblast growth factor-23 mutants suppress Na+-dependent phosphate co-transport activity and 1α,25-dihydroxyvitamin D3 production. J Biol Chem. (2003) 278:2206–11. doi: 10.1074/jbc.M207872200
144. Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, et al. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. (2004) 113:561–8. doi: 10.1172/JCI200419081
145. Nakatani T, Sarraj B, Ohnishi M, Densmore MJ, Taguchi T, Goetz R, et al. In vivo genetic evidence for klotho-dependent, fibroblast growth factor 23 (Fgf23) -mediated regulation of systemic phosphate homeostasis. FASEB J. (2009) 23:433–41. doi: 10.1096/fj.08-114397
146. Razzaque MS, Sitara D, Taguchi T, St-Arnaud R, Lanske B. Premature aging-like phenotype in fibroblast growth factor 23 null mice is a vitamin D-mediated process. FASEB J. (2006) 20:720–2. doi: 10.1096/fj.05-5432fje
147. Ohnishi M, Nakatani T, Lanske B, Razzaque MS. Reversal of mineral ion homeostasis and soft-tissue calcification of klotho knockout mice by deletion of vitamin D 1α-hydroxylase. Kidney Int. (2009) 75:1166–72. doi: 10.1038/ki.2009.24
148. Hesse M, Frohlich LF, Zeitz U, Lanske B, Erben RG. Ablation of vitamin D signaling rescues bone, mineral, and glucose homeostasis in Fgf-23 deficient mice. Matrix Biol. (2007) 26:75–84. doi: 10.1016/j.matbio.2006.10.003
149. Anour R, Andrukhova O, Ritter E, Zeitz U, Erben RG. Klotho lacks a vitamin D independent physiological role in glucose homeostasis, bone turnover, and steady-state PTH secretion in vivo. PLoS ONE. (2012) 7:e31376. doi: 10.1371/journal.pone.0031376
150. Li YC, Amling M, Pirro AE, Priemel M, Meuse J, Baron R, et al. Normalization of mineral ion homeostasis by dietary means prevents hyperparathyroidism, rickets, and osteomalacia, but not alopecia in vitamin D receptor-ablated mice. Endocrinology. (1998) 139:4391–6. doi: 10.1210/endo.139.10.6262
151. Erben RG, Soegiarto DW, Weber K, Zeitz U, Lieberherr M, Gniadecki R, et al. Deletion of deoxyribonucleic acid binding domain of the vitamin D receptor abrogates genomic and non-genomic functions of vitamin D. Mol Endocrinol. (2002) 16:1524–37. doi: 10.1210/mend.16.7.0866
152. Andrukhova O, Slavic S, Smorodchenko A, Zeitz U, Shalhoub V, Lanske B, et al. FGF23 regulates renal sodium handling and blood pressure. EMBO Mol Med. (2014) 6:744–59. doi: 10.1002/emmm.201303716
153. Andrukhova O, Smorodchenko A, Egerbacher M, Streicher C, Zeitz U, Goetz R, et al. FGF23 promotes renal calcium reabsorption through the TRPV5 channel. EMBO J. (2014) 33:229–46. doi: 10.1002/embj.201284188
154. Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, Goetz R, Kuro-o M, Mohammadi M, et al. The parathyroid is a target organ for FGF23 in rats. J Clin Invest. (2007) 117:4003–8. doi: 10.1172/JCI32409
155. Olauson H, Mencke R, Hillebrands JL, Larsson TE. Tissue expression and source of circulating αKlotho. Bone. (2017) 100:19–35. doi: 10.1016/j.bone.2017.03.043
156. Xu Y, Sun Z. Regulation of S-formylglutathione hydrolase by the anti-aging gene klotho. Oncotarget. (2017) 8:88259–75. doi: 10.18632/oncotarget.19111
157. Grabner A, Amaral AP, Schramm K, Singh S, Sloan A, Yanucil C, et al. Activation of cardiac fibroblast growth factor receptor 4 causes left ventricular hypertrophy. Cell Metab. (2015) 22:1020–32. doi: 10.1016/j.cmet.2015.09.002
158. Xiao Z, Riccardi D, Velazquez HA, Chin AL, Yates CR, Carrick JD, et al. A computationally identified compound antagonizes excess FGF-23 signaling in renal tubules and a mouse model of hypophosphatemia. Sci Signal. (2016) 9:ra113. doi: 10.1126/scisignal.aaf5034
159. Xiao Z, King G, Mancarella S, Munkhsaikhan U, Cao L, Cai C, et al. FGF23 expression is stimulated in transgenic α-Klotho longevity mouse model. JCI Insight. (2019) 4:132820. doi: 10.1172/jci.insight.132820
160. Han X, Cai C, Xiao Z, Quarles LD. FGF23 induced left ventricular hypertrophy mediated by FGFR4 signaling in the myocardium is attenuated by soluble Klotho in mice. J Mol Cell Cardiol. (2020) 138:66–74. doi: 10.1016/j.yjmcc.2019.11.149
161. Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell. (2002) 109(Suppl.):S67–79. doi: 10.1016/S0092-8674(02)00699-2
162. Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. (2011) 121:4393–408. doi: 10.1172/JCI46122
163. Agoro R, Montagna A, Goetz R, Aligbe O, Singh G, Coe LM, et al. Inhibition of fibroblast growth factor 23 (FGF23) signaling rescues renal anemia. FASEB J. (2018) 32:3752–64. doi: 10.1096/fj.201700667R
Keywords: osteocyte, FGF23, FGF23 signaling, Klotho, kidney
Citation: Agoro R, Ni P, Noonan ML and White KE (2020) Osteocytic FGF23 and Its Kidney Function. Front. Endocrinol. 11:592. doi: 10.3389/fendo.2020.00592
Received: 15 June 2020; Accepted: 20 July 2020;
Published: 28 August 2020.
Edited by:
Lilian Irene Plotkin, School of Medicine, Indiana University Bloomington, United StatesReviewed by:
Paola Divieti Pajevic, Boston University, United StatesCopyright © 2020 Agoro, Ni, Noonan and White. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kenneth E. White, a2VuZXdoaXRAaXUuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.