 Xiaoyan Chang
Xiaoyan Chang Zelin Li
Zelin Li Xiaosen Ma
Xiaosen Ma Yunying Cui
Yunying Cui Shuchun Chen
Shuchun Chen Anli Tong
Anli Tong
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Endocrinol. , 21 August 2020
Sec. Cancer Endocrinology
Volume 11 - 2020 | https://doi.org/10.3389/fendo.2020.00558
Background: MYC associated factor X (MAX) is a tumor suppressor gene and has been identified as one of the pathogenic genes of hereditary pheochromocytoma (PCC). To date, there have been no reports of ganglioneuroma (GN) with MAX variants.
Case Presentation: The proband was a 45-years-old Chinese female with paroxysmal hypertension and palpitations who had undergone adrenalectomy for PCC 14 years ago. Her plasma free normetanephrine and 24-h urinary norepinephrine excretion were significantly increased, and abdominal computed tomography (CT) revealed an irregular mass in the left adrenal region, suggesting a recurrence of PCC. The mass was surgically removed and pathologically diagnosed as PCC with lymph node metastasis. The proband's son suffered from paroxysmal hypertension and palpitations. His plasma free metanephrine levels were normal. CT revealed a mass in the right adrenal. The tumor was surgically removed, and the pathological diagnosis was GN. Genetic testing of peripheral blood DNA revealed that the proband and her son had germline pathogenic MAX variant c.C97T, p.Arg33Ter, while proband's parents did not have MAX variants. Tumor DNA sequencing showed the same MAX variant (c.C97T, p.Arg33Ter) in PCC of the proband and GN of her son, both with retention of heterozygosity. Immunohistochemistry demonstrated loss of MAX protein expression in most tumor cells in PCC of the proband and some Schwannian cells in GN of the proband's son.
Conclusion: We report a family with a new clinical phenotype of germline pathogenic variants in MAX who developed both PCC and GN. Germline pathogenic variants in MAX may contribute to the development of GN. Our findings suggest that it is not just paternally inherited MAX variants that can cause tumors.
MAX, a tumor suppressor gene encoding MAX protein, locates on chromosome 14q23.3. MAX protein is part of the MYC–MAX–MXD1 complex, which acts as transcription factors to regulate cell proliferation, apoptosis, and differentiation. The MAX–MYC heterodimer activates transcription, while the MAX–MAD heterodimer inhibits transcription and MYC-dependent cell transformation (1). It has been reported that MAX mutations are involved in the pathogenesis of hereditary pheochromocytoma (PCC)/paraganglioma (PGL), but MAX mutations mainly cause PCC (2). MAX mutations lead to the loss of MAX protein expression, which destroys the inhibition of MYC-dependent cell transformation and thus causes tumors (3).
Germline MAX variants have been associated with PCC/PGL and renal oncocytoma, while somatic MAX mutations have been linked to the formation of other tumors such as endometrioid carcinoma and colon cancer (4). Up to now, there have been no reports of ganglioneuroma (GN) with MAX variants. We describe a family with germline MAX variants who developed PCC and GN. To our knowledge, this case is the first report of GN in a patient with germline MAX variants, suggesting the possibility of germline pathogenic MAX variants contributing to the development of GN.
The proband was a 45-years-old-Chinese female who was admitted to Peking Union Medical College hospital for a recurrence of left adrenal PCC. At the age of 31 years, the patient presented with paroxysmal dizziness, sweating, palpitation, and hypertension attacks with a maximal blood pressure of 200/100 mmHg. Abdominal computed tomography (CT) indicated a mass with a size of 10 cm in the left adrenal gland. The patient underwent left adrenalectomy, and the tumor was pathologically diagnosed as PCC. Her symptoms disappeared postoperatively. However, she suffered from paroxysmal palpitation and sweating at age 45. Her blood pressure was 170/100 mmHg at hypertension attacks. Then she was referred to our hospital for further examination and treatment.
Laboratory examinations were as follows: 24-h urinary norepinephrine, 325.44 μg/24 h (normal, 16.69–40.65); epinephrine, 4.07 μg/24 h (normal, 1.74–6.42); and dopamine, 235 μg/24 h (normal, 121–331). Contrast-enhanced CT showed multiple masses in the left adrenal, the largest of which was 2.1 × 1.3 cm, wrapping the left renal artery. It also showed multiple enlarged lymph nodes in retroperitoneum and mesenterium. Metaiodobenzylguanidine (MIBG) scintigraphy revealed the abnormally increased uptake in the left adrenal gland mass. These findings indicated recurrent PCC in the left adrenal, but the patient and her family refused surgery. The mass enlarged gradually. Three years later, contrast-enhanced CT showed an irregular left adrenal mass (4.1 × 2.7 × 3.3 cm), which was markedly enhanced and wrapping the left renal artery (Figure 1A). Plasma free normetanephrine was 9.35 nmol/L (normal, <0.9), and plasma free metanephrine was 0.46 nmol/L (normal, <0.5). The mass was surgically removed and pathologically diagnosed as PCC with lymph node metastasis (Figure 2A). Her symptoms disappeared again, and biochemical indexes decreased to normal range postoperatively. The patient had undergone partial thyroidectomy for papillary carcinoma of thyroid at the age of 46 years.
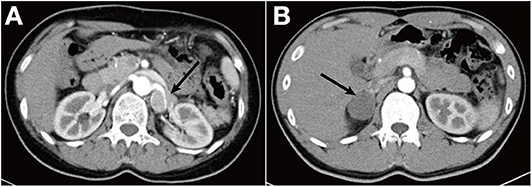
Figure 1. (A) Abdominal contrast-enhanced CT scan demonstrates an irregular left adrenal mass (4.1 × 2.7 × 3.3 cm) with a density of 42 Hounsfield unit (HU), wrapping the left renal artery. (B) CT scan showed a right adrenal mass (3.7 × 3.2 cm) with a density of 28 HU.
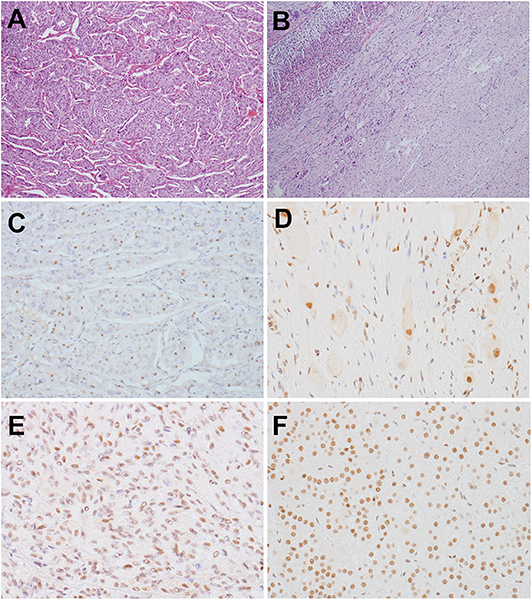
Figure 2. (A,B) HE staining (×60). (A) PCC of the proband. The tumor exhibited an alveolar (Zellballen) pattern. Nests of mild polygonal cells were separated by peripheral capillaries. (B) GN of the proband's son. The tumor located in the adrenal medulla, with a clear border to the adrenal cortex (left). The tumor was composed of scattered ganglion cells in the background of Schwannian stroma. (C–F) Detection of MAX by immunohistochemistry with a MAX C-terminus-specific antibody (×300). (C) Most tumor cells in PCC of the proband were negatively stained, but some tumor cells were positive. (D) The scattered ganglion cells and some Schwannian cells in GN of the son of the proband showed positive staining for MAX, but other Schwannian cells were negatively stained. (E) Positive staining of tumor cells in a RET-mutated PCC. (F) Positive staining of the normal adrenal cortex of the patient with GN.
Patient 2, son of the proband, was admitted to our hospital with the complaint of paroxysmal palpitation, dizziness, and headache for 2 months. At the onset of symptoms, his blood pressure was 150/90 mmHg, and his heart rate was 130 bpm. His plasma free metanephrine levels were normal. Plasma free normetanephrine was 0.30–0.66 nmol/L, and plasma free metanephrine was 0.07–0.12 nmol/L. CT scan showed a mass (3.7 × 3.2 cm) in the right adrenal with the density of 28 Hounsfield unit (HU) (Figure 1B). Neither MIBG scintigraphy nor somatostatin receptor scintigraphy (SRS) indicated any uptake in the right adrenal gland mass. The mass was removed and pathologically diagnosed as GN (Figure 2B).
The proband's parents suffered from hypertension. But examinations did not find any abdominal mass. The proband had three brothers and a sister. Their abdominal ultrasound did not show any adrenal mass. In addition to a son affected by adrenal GN, the proband had two daughters. Her daughters denied any symptoms such as headache, palpitation, sweating, or paroxysmal hypertension, and abdominal ultrasound showed no mass.
Genomic DNA was extracted from the peripheral blood of the two patients and their family members. The genomic DNA of the proband was examined for potential pathogenic germline variants of PCC by next-generation sequencing covering SDHA, SDHB, SDHC, SDHD, SDHAF2, VHL, RET, MAX, TMEM127, FH, NF1, and KIF1B. A germline MAX variant (c.C97T, p.Arg33Ter) was identified and confirmed by Sanger sequencing (Figure 3A). The proband's son had the same germline variant, which was detected by Sanger sequencing of the coding sequences of exon 3 of MAX. Other familial members including parents of the proband, her husband, and two daughters did not have MAX variants (Figure 3B). The minor allele frequency (MAF) of the MAX variant (c.C97T, p.Arg33Ter) is not specified. Mutation Taster, the Combined Annotation-Dependent Depletion (CADD), and the American College of Medical Genetics and Genomics (ACMG) predict that this variant is pathogenic.
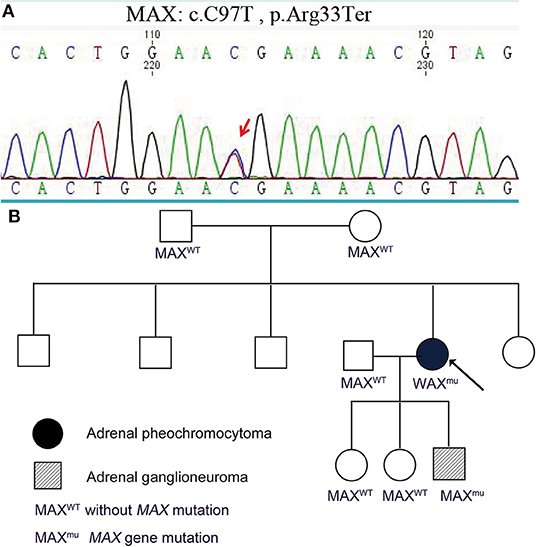
Figure 3. Germline pathogenic MAX variants in this family. (A) Genetic testing indicated that the proband and her son had a germline pathogenic MAX variant (c.C97T, p.Arg33Ter). (B) Pedigree of a family with PCC and GN. Other familial members including parents of the proband, her husband, and two daughters did not have MAX variants.
This study was conducted with informed consent obtained from the patients and approval from the medical ethics committee of the hospital.
DNA extracted from frozen tumor tissues of the proband and her son underwent MAX mutation analysis. A RET-mutant PCC was taken as a control. Genetic analysis of tumor DNA demonstrated that there was a MAX variant (c.C97T, p.Arg33Ter) in PCC of the proband and GN of her son, both with retention of heterozygosity (Figure 4).
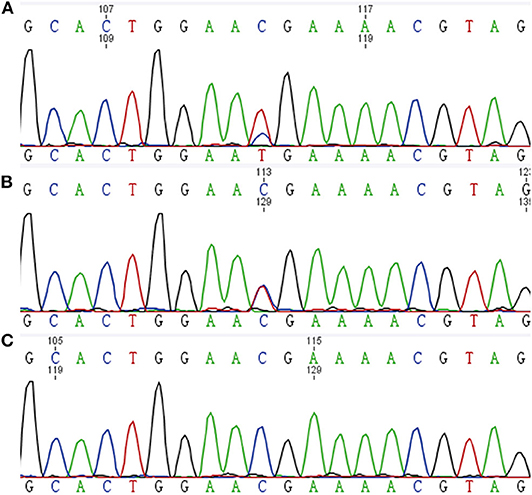
Figure 4. Tumor DNA sequence chromatograms. (A) PCC of the proband. (B) GN of the proband's son. (C) RET-mutated PCC.
Immunohistochemical detection was performed using the EnVision detection kit (Dako). MAX C-terminus-specific antibody was purchased from Abcam (ab101271), and the dilution was 1:1,000. Immunohistochemical staining was performed using 3-μm formalin-fixed paraffin-embedded tumor sections from the proband's PCC and her son's GN, which carry MAX variants. Normal adrenal cortex around GN and a PCC carrying RET mutations was used as controls. Only cases showing nuclear staining were considered as valuable. Immunohistochemical detection of MAX showed that most tumor cells in PCC of the proband were negatively stained, but some tumor cells were positive. And the scattered ganglion cells and some Schwannian cells in GN of proband's son showed positive staining for MAX, but other Schwannian cells were negatively stained (Figures 2C–F).
We describe a germline pathogenic MAX variant family in which the proband had PCC and her son had an adrenal GN. Neither germline MAX-mutated GN nor somatic MAX-mutated GN has been reported. To our knowledge, this is the first report of adrenal GN in a patient with germline pathogenic MAX variants.
PCC is a neuroendocrine tumor of the adrenal medulla, characterized by the production of excessive catecholamines, which can cause a series of symptoms such as hypertension, dizziness, palpitation, and sweating. Approximately 40% of PCCs/PGLs are caused by the germline pathogenic variants in one of almost 20 susceptibility genes (5). These susceptible genes can be classified into two major clusters according to their gene expression profile: pseudohypoxia gene cluster and the kinase signal cluster. Pseudohypoxia gene cluster includes VHL, SDHx, HIF2A, etc. Pathogenic variants in these genes can activate hypoxia-inducible factors and then increases the expression of hypoxia-related growth factors. Kinase signaling cluster includes MAX, NF1, RET, and TMEM127, etc. In this cluster, pathogenic variants in these genes associate with abnormal activation of kinase signaling pathways such as PI3Kinase/AKT and the mTOR pathway (6). In 2011, MAX was identified as a new susceptibility gene of PCC (2). Burnichon et al. reported 19 MAX-mutated cases in 1,694 PCC/PGL patients (1.12%), and all MAX-related tumors were adrenal PCCs (3). Till now, a total of ~40 patients have been reported. The MAX variant c.C97T, p.Arg33Ter detected in this article had been reported previously. It introduces a premature stop codon (7), thus losing the transcriptional repression of MAX protein on MYC-dependent cell transformation. MAX mutations are associated with bilateral PCCs, and about 10% of MAX-mutated patients present with metastases.
In the present case, we found a same heterozygous pathogenic MAX variant (c.C97T, p.Arg33Ter) in the peripheral blood and tumor tissues of both PCC and GN. The tumor DNA sequencing of PCC showed a retention of heterozygosity, which indicated that there still existed a wild-type allele. It is consistent with some tumor cells in PCC which were positive for MAX immunostaining. Nevertheless, immunohistochemistry demonstrated the loss of MAX protein expression in most PCC tumor cells, suggesting a causative role of MAX variants for PCC. In the previously reported cases, MAX-mutated tumors were most often caused by loss of heterozygosity (LOH), which was caused by either chromosomal loss or uniparental disomy and was thought to be a paternal transmission of MAX variants (2). However, Romanet et al. found that there still existed a wild-type allele in the MAX-mutated PCC. And they thought that for MAX-mutated tumors, in addition to chromosomal loss and uniparental disomy, other potential mechanisms can account for the loss of MAX protein expression (8). In the present case, we found that the parents of the proband did not have MAX variants, which suggested that the germline MAX variants of the proband were de novo. Our results are inconsistent with the loss of MAX proteins caused by the LOH related with paternal transmission of MAX variants. For this inconsistent result, we propose a hypothesis that there probably exist other mechanisms that cause PCC in patients with germline pathogenic MAX variants. LOH may occur in the development of the tumor rather than from the beginning.
GN is a rare sympathetic nervous system tumor; it occurs anywhere along the sympathetic nerve chain, about 20% of which occur in the adrenal gland (9). The pathogenesis of GN is still unclear. Most GNs are primary, but it has been reported that some GNs also arise after treatment for neuroblastoma or as a result of spontaneous neuroblastoma differentiation (10). GNs, just like PCCs, originate from neural crest cells in the neuroectoderm. In our study, there was a heterozygous pathogenic MAX variant (c.C97T, p.Arg33Ter) in both the peripheral blood and tumor tissues in the GN. And the scattered ganglion cells and some Schwannian cells in GN showed positive staining for MAX, but other Schwannian cells were negatively stained. These findings suggest that the pathogenic MAX variants may be involved in the development of GNs.
There are several reports of GN pedigrees with pathogenic variants in RET, NF1, and SDHB. RET, NF1, and SDHB genes have been implicated as causes of familial PCCs. NF1-mutated GNs occurred mainly in the neck, mediastinum, and retroperitoneum (11–14). NF1 is a tumor suppressor gene, encoding neurofibromin. Loss of neurofibromin may induce abnormal proliferation of Schwann cells and PCC cells, thus causing a composite PCC–GN (15). And two studies found that RET variants can cause adrenal GNs in transgenic mice (16, 17). Besides RET-mutated adrenal GN, composite PCC–GN patients have been reported (8, 18). The RET gene encodes a tyrosine kinase receptor, which regulates intracellular signaling in neural crest-derived tissues. RET mutations could lead to neural crest-derived tumors, including PCC and GN. In addition, Niemeijer et al. reported an abdominal GN with SDHB negative immunoreaction, and the GN showed retention of heterozygosity (ROH) (19). These cases suggest that pathogenic variants in one of the PCC/PGL susceptibility genes can cause the GN.
Dysregulation of the MAX–MYC pathway can lead to the development of many tumors, including neuroblastoma (3). Approximately 20% of neuroblastomas have a deletion of chromosome 14q23–q32 (20), and the MAX gene located on chromosome 14q23.3. MAX may be the target of chromosome 14q loss. However, two studies did not find any MAX mutations in a total of 327 neuroblastoma patients (21, 22). Whether MAX mutations can cause neuroblastoma is still uncertain. Neuroblastoma, ganglioneuroblastoma, and GN originated from primitive sympathogonia and are collectively known as neuroblastic tumors. Neuroblastoma can mature and differentiate into ganglioneuroblastoma and GN spontaneously. Wu et al. used exome sequencing and found two 3′-UTR variants in MAX in ganglioneuroblastoma, suggesting that MAX variants were responsible for ganglioneuroblastoma (23). We reported a GN patient with germline MAX variants. Based on the above, we have a hypothesis that pathogenic MAX variants might be associated with neuroblastoma and that neuroblastoma then differentiates into an adrenal GN spontaneously.
In conclusion, loss of the MAX protein expression would be sufficient to suggest the causative role of germline MAX variants for PCC and GN. But the mechanism behind this requires more research to verify. We report a family with germline pathogenic MAX variants who developed both PCC and GN. This family is important in that, to our knowledge, it is the first report of a GN in a patient with a germline MAX variant. This case suggests the possibility of germline pathogenic MAX variants also contributing to the development of GN in addition to PCC. Because the MAX variants and GN are rare entities, the case report is important as it identifies a new clinical phenotype of germline MAX variants.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Written informed consent was obtained from the individuals for the publication of any potentially identifiable images or data included in this article.
XC, XM, YC, and AT managed the case. ZL drafted the manuscript. XC, ZL, YC, SC, and AT reviewed the manuscript. XC prepared histopathological results. All authors contributed to manuscript revision, read, and approved the submitted version.
The project was supported by the Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Sciences (CIFMS) 2017-I2M-1-001.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Grandori C, Cowley SM, James LP, Eisenman RN. The Myc/Max/Mad network and the transcriptional control of cell behavior. Ann Rev Cell Develop Biol. (2000) 16:653–99. doi: 10.1146/annurev.cellbio.16.1.653
2. Comino-Méndez I, Gracia-Aznárez F, Schiavi F, Landa I, Leandro-García L, Letón R, et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet. (2011) 43:663–7. doi: 10.1038/ng.861
3. Burnichon N, Cascón A, Schiavi F, Morales N, Comino-Méndez I, Abermil N, et al. MAX mutations cause hereditary and sporadic pheochromocytoma and paraganglioma. Clin Cancer Res. (2012) 18:2828–37. doi: 10.1158/1078-0432.CCR-12-0160
4. Roszko KL, Blouch E, Blake M, Powers JF, Tischler AS, Hodin R, et al. Case report of a prolactinoma in a patient with a novel MAX mutation and bilateral pheochromocytomas. J Endocrine Soc. (2017) 1:1401–7. doi: 10.1210/js.2017-00135
5. Khatami F, Mohammadamoli M, Tavangar S. Genetic and epigenetic differences of benign and malignant pheochromocytomas and paragangliomas (PPGLs). Endocr Regul. (2018) 52:41–54. doi: 10.2478/enr-2018-0006
6. Pillai S, Gopalan V, Smith R, Lam A. Updates on the genetics and the clinical impacts on phaeochromocytoma and paraganglioma in the new era. Crit Rev Oncol Hematol. (2016) 100:190–208. doi: 10.1016/j.critrevonc.2016.01.022
7. Bausch B, Schiavi F, Ni Y, Welander J, Patocs A, Ngeow J, et al. Clinical characterization of the pheochromocytoma and paraganglioma susceptibility genes SDHA, TMEM127, MAX, and SDHAF2 for gene-informed prevention. JAMA Oncol. (2017) 3:1204–12. doi: 10.1001/jamaoncol.2017.0223
8. Romanet P, Guerin C, Pedini P, Essamet W, Castinetti F, Sebag F, et al. Pathological and genetic characterization of bilateral adrenomedullary hyperplasia in a patient with germline MAX mutation. Endocr Pathol. (2017) 28:302–7. doi: 10.1007/s12022-016-9460-5
9. Lora M, Waguespack S, Moley J, Walvoord E. Adrenal ganglioneuromas in children with multiple endocrine neoplasia type 2: a report of two cases. J Clin Endocrinol Metab. (2005) 90:4383–7. doi: 10.1210/jc.2004-2526
10. Alexander N, Sullivan K, Shaikh F, Irwin M. Characteristics and management of ganglioneuroma and ganglioneuroblastoma-intermixed in children and adolescents. Pediatr Blood Cancer. (2018) 65:e26964. doi: 10.1002/pbc.26964
11. Kannu P, Nour M, Irving M, Xie J, Loder D, Lai J, et al. Paraspinal ganglioneuroma in the proband of a large family with mild cutaneous manifestations of NF1, carrying a deep NF1 intronic mutation. Clin Genet. (2013) 83:191–4. doi: 10.1111/j.1399-0004.2012.01882.x
12. Miyakoshi N, Hongo M, Kasukawa Y, Misawa A, Shimada Y. Bilateral and symmetric C1-C2 dumbbell ganglioneuromas associated with neurofibromatosis type 1 causing severe spinal cord compression. Spine J. (2010) 10:e11–5. doi: 10.1016/j.spinee.2010.01.023
13. Sinha P, Sharma S, Agarwal S, Gupta S. Parapharyngeal ganglioneuroma with neurofibromatosis: an unusual presentation. Ann Otol Rhinol Laryngol. (2011) 120:769–74. doi: 10.1177/000348941112001201
14. Onozawa M, Fukuhara T, Minoguchi M, Takahata M, Yamamoto Y, Miyake T, et al. Hypokalemic rhabdomyolysis due to WDHA syndrome caused by VIP-producing composite pheochromocytoma: a case in neurofibromatosis type 1. Jpn J Clin Oncol. (2005) 35:559–63. doi: 10.1093/jjco/hyi139
15. Kimura N, Watanabe T, Fukase M, Wakita A, Noshiro T, Kimura I. Neurofibromin and NF1 gene analysis in composite pheochromocytoma and tumors associated with von Recklinghausen's disease. Mod Pathol. (2002) 15:183–8. doi: 10.1038/modpathol.3880513
16. Sweetser D, Froelick G, Matsumoto A, Kafer K, Marck B, Palmiter R, et al. Ganglioneuromas and renal anomalies are induced by activated RET(MEN2B) in transgenic mice. Oncogene. (1999) 18:877–86. doi: 10.1038/sj.onc.1202376
17. Smith-Hicks C, Sizer K, Powers J, Tischler A, Costantini F. C-cell hyperplasia, pheochromocytoma and sympathoadrenal malformation in a mouse model of multiple endocrine neoplasia type 2B. EMBO J. (2000) 19:612–22. doi: 10.1093/emboj/19.4.612
18. Efared B, Atsame-Ebang G, Tahirou S, Mazaz K, Hammas N, El Fatemi H, et al. Bilateral pheochromocytoma with ganglioneuroma component associated with multiple neuroendocrine neoplasia type 2A: a case report. J Med Case Rep. (2017) 11:208. doi: 10.1186/s13256-017-1364-6
19. Niemeijer ND, Papathomas TG, Korpershoek E, de Krijger RR, Oudijk L, Morreau H, et al. Succinate Dehydrogenase (SDH)-deficient pancreatic neuroendocrine tumor expands the SDH-related tumor spectrum. J Clin Endocrinol Metab. (2015) 100:E1386–93. doi: 10.1210/jc.2015-2689
20. Thompson P, Seifried B, Kyemba S, Jensen S, Guo C, Maris J, et al. Loss of heterozygosity for chromosome 14q in neuroblastoma. Med Pediatr Oncol. (2001) 36:28–31. doi: 10.1002/1096-911X(20010101)36:1<28::AID-MPO1008>3.0.CO;2-0
21. Molenaar J, Koster J, Zwijnenburg D, van Sluis P, Valentijn L, van der Ploeg I, et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature. (2012) 483:589–93. doi: 10.1038/nature10910
22. Pugh T, Morozova O, Attiyeh E, Asgharzadeh S, Wei J, Auclair D, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. (2013) 45:279–84. doi: 10.1038/ng.2529
Keywords: MAX, pheochromocytoma, ganglioneuroma, adrenal, neurogenic tumor
Citation: Chang X, Li Z, Ma X, Cui Y, Chen S and Tong A (2020) A Novel Phenotype of Germline Pathogenic Variants in MAX: Concurrence of Pheochromocytoma and Ganglioneuroma in a Chinese Family and Literature Review. Front. Endocrinol. 11:558. doi: 10.3389/fendo.2020.00558
Received: 12 April 2020; Accepted: 07 July 2020;
Published: 21 August 2020.
Edited by:
Antongiulio Faggiano, Sapienza University of Rome, ItalyReviewed by:
Letizia Canu, University of Florence, ItalyCopyright © 2020 Chang, Li, Ma, Cui, Chen and Tong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anli Tong, dG9uZ2FubGlAaG90bWFpbC5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.