 Ming-Hui Zheng
Ming-Hui Zheng Fu-Xing-Zi Li
Fu-Xing-Zi Li Feng Xu1
Feng Xu1 Ling-Qing Yuan
Ling-Qing Yuan- 1Hunan Provincial Key Laboratory of Metabolic Bone Diseases, Department of Endocrinology and Metabolism, National Clinical Research Center for Metabolic Disease, The Second Xiangya Hospital, Central South University, Changsha, China
- 2Department of Radiology, The Second Xiangya Hospital, Central South University, Changsha, China
The renin-angiotensin-aldosterone system (RAAS) is the regulatory system by which renin induces aldosterone production. Angiotensin II (Ang II) is the main effector substance of the RAAS. The RAAS regulates blood pressure and electrolyte balance by controlling blood volume and peripheral resistance. Excessive activation of the RAAS is an important factor in the onset of cardiovascular disease and the deterioration of this disease. The most common RAAS abnormality is primary aldosteronism (PA). Parathyroid hormone (PTH) is a peptide secreted by the main cells of the parathyroid gland, which promotes elevated blood calcium (Ca2+) levels and decreased blood phosphorus (Pi) levels. Excessive secretion of PTH can cause primary hyperparathyroidism (PHPT). Parathyroidism is highly prevalent in postmenopausal women and is often associated with secondary osteoporosis. PA and PHPT are common endocrine system diseases. However, studies have shown a link between the RAAS and PTH, indicating a positive relationship between them. In this review, we explore the complex bidirectional relationship between the RAAS and PTH. We also point out possible future treatment options for related diseases based on this relationship.
Introduction
Primary aldosteronism (PA) is often accompanied by primary hyperparathyroidism (PHPT) (1). In general, the higher the serum aldosterone concentration, the higher the serum Parathyroid hormone (PTH) concentration (2). Patients with PA often present with hypertension, and hypertension patients have a 2- to 8-fold higher risk of developing hyperparathyroidism as individuals with normal blood pressure (3). Hyperparathyroidism increases PTH levels and promotes bone absorption, which increases the risk of osteoporosis, indicating that the renin-angiotensin-aldosterone system (RAAS) is associated with osteoporosis. Meanwhile, RAAS can also cause osteoporosis by other mechanisms. Studies by Hatton et al. and Beavan et al. showed that bone may contain a tissue-renin-angiotensin-aldosterone system. Angiotensin I (Ang I), angiotensin II (Ang II), and aldosterone can effectively stimulate bone resorption of osteoclasts, causing osteolysis, thus leading to osteoporosis (4, 5). Mineralocorticoid receptor antagonism (MRA) may directly or indirectly, via PTH, affect human bone health (6).
It has been reported that PHPT patients have a higher prevalence of cardiovascular abnormalities than the general population (7, 8). PHPT leads to an increased prevalence of cardiovascular diseases and cardiovascular mortality (9–13). PTH is even used to predict cardiovascular risk (13, 14). The increase in cardiovascular mortality is associated with disturbances in the RAAS (15). PTH stimulates aldosterone secretion in human adrenocortical cells (12, 16, 17), which increases blood pressure. PTH can also raise blood pressure in five ways, thus increasing the risk of cardiovascular disease. Firstly, PTH may act as an ionophore for Calcium ion (Ca2+) and promote Ca2+ entry into cells, increasing vasoconstriction and raising blood pressure (18, 19). Secondly, PTH may promote vascular and cardiac remodeling, accelerating the development of cardiovascular disease in patients with PHPT (17, 20), but this is explained by the direct effect of PTH on vascular smooth muscle cells and endothelial cells, instead of the RAAS (21). Thirdly, higher PTH levels increase the degree of vascular stiffness and thereby increase cardiac afterload (22, 23). Fourthly, PTH can also increase plasma renin activity (PRA) and thereby increase blood pressure, while hypertension is associated with vascular calcification and aging (24). Moreover, the PRA level decreases to normal after parathyroidectomy (25, 26). Finally, elevated serum uric acid in PHPT patients may also lead to high blood pressure; the serum uric acid level falls significantly in patients with PHPT after parathyroidectomy (27, 28). Therefore, parathyroidectomy may reduce the risk of cardiovascular disease (11, 29, 30).
Considering that it is clinically important to find out whether a drug that affects one system will affect another system, thereby facilitating clinical diagnosis and treatment, it is interesting to discuss the complex relationship between the RAAS and PTH.
Physiological Mechanism of RAAS Biomarkers and RAAS Inhibitors
The RAAS is an important body fluid regulation system in the human body and one of the most important regulators of sodium retention, potassium (K+) excretion, and blood volume and blood pressure. The RAAS has an important influence on cardiovascular hemodynamics and the development and progression of cardiovascular disease (31). RAAS includes renin, angiotensinogen, Ang I, angiotensin converting enzyme (ACE), Ang II, angiotensin II receptor 1 (AT1), aldosterone, and other components (32). Angiotensinogen is an alpha 2-globulin that is formed primarily by the liver and also by the kidney and other tissues. Renin is mainly formed and secreted by proximal glomerular epithelioid cells of afferent arterioles. The active form of renin has 340 amino acids (33). Decreased sodium intake, decreased extracellular fluid and blood volume, decreased arterial pressure, and increased sympathetic activity can stimulate renin release. Renin is an aspartic protease that cuts 10 peptides (Ang I) from angiotensinogen (34, 35). Renin is specific for angiotensinogen, and the activity of renin determines how much Ang I is produced (36, 37). Ang I is further cleaved to Ang II by ACE in pulmonary capillaries, endothelial cells, and renal epithelial cells (32, 35). Ang II can be converted to Ang III by removing aspartic acid from position 1 of the octapeptide. The role of angiotensin III is modest compared to the effect of angiotensin II (31). ACE2 is a monocarboxypeptidase that converts Ang I to Ang 1-9 and Ang II to Ang 1-7 (38).
Although the renin angiotensin system (RAS) contains multiple peptides, Ang II is the major active metabolite and acts on many different tissues and organs. Ang II promotes vasoconstriction and sympathetic enhancement and increases renal tubular reabsorption of sodium, leading to decreased blood flow and increased vascular resistance. Almost every organ system in the body responds to Ang II (37). Improper elevation of Ang II can lead to high blood pressure and increase the morbidity and mortality of cardiovascular disease (39). AT1 receptor (AT1R) is the primary receptor that mediates Ang II action in the heart and circulatory system (32). AT1R is widely distributed throughout the vasculature. The AT2 receptor (AT2R) can counteract the effects of the AT1R (31) and has a lower expression level in adults, but in some cardiovascular diseases, such as heart failure, the expression level of AT2R may increase.
Ang II and Ang III stimulate adrenal globular zone cells to secrete aldosterone, but Ang IV and Ang 1-7 do not induce adrenal aldosterone secretion. The subtype of angiotensin receptor they act on is not clear (40). Studies suggest that Ang II primarily stimulates aldosterone release by acting on AT1R (41). Mazzocchi et al. suggest that AT2R activation causes local release of catecholamines from chromaffin cells, which in turn enhances aldosterone secretion in a paracrine manner (42). Aldosterone promotes sodium retention and K+ excretion, retains water, and increases fluid volume (32). Excessive secretion of aldosterone causes PA (43). Excess aldosterone is associated with cardiovascular and kidney damage (inflammation, remodeling, and fibrosis) (44, 45). Aldosterone acts on the mineralocorticoid receptor (MR) of the kidney, thereby promoting tissue remodeling and angiogenesis (46).
RAAS inhibitors include renin inhibitors, ACE inhibitors (ACEIs), AT1R antagonists, and MRAs (47). RAS inhibitors (RASIs) mainly contain the first three (44). ACEIs (ramipril, perindopril, captopril) control hypertension by inhibiting ACE to reduce the biosynthesis of Ang II (48). AT1R antagonists can block the action of Ang II on ATR1 and decrease aldosterone release (Figure 1) (40). RASIs inhibit the RAS, increase blood flow, and reduce peripheral vascular resistance, leading to decreased arterial pressure (37). Inhibition of RAS components has been successfully used to treat hypertension, heart failure, and end-organ damage (36). MRA (spironolactone, eplerenone) can be used to treat hypertension, PA, and peripheral edema associated with heart failure and other pathologies associated with aldosteronism (49). In addition, the combined use of ACEI, angiotensin receptor blocker (ARB), MRA, and other drugs can better improve heart failure, especially for patients with a reduced ejection fraction (50).
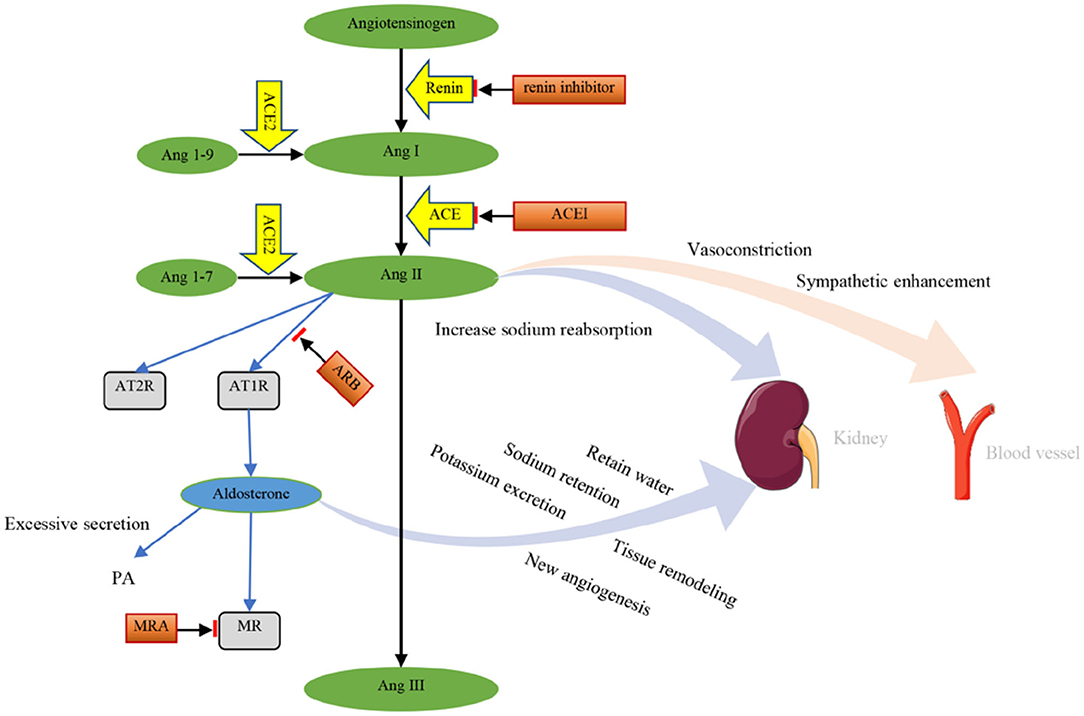
Figure 1. Components and effects of the RAAS. Renin cuts 10 peptides (Ang I) from angiotensinogen. The effect of renin can be inhibited by renin inhibitors. Ang I is further cleaved to Ang II by ACE. ACEI can inhibit the effect of ACE. Ang I and Ang II are converted into Ang1-9 and Ang1-7 by ACE2, respectively. Then, Ang II is converted into Ang III. Ang II acts on AT1R and AT2R. The effects of Ang II on AT1R can be blocked by ARB. Ang II promotes vasoconstriction and sympathetic enhancement and increases renal tubular reabsorption of sodium. Aldosterone retains water and promotes sodium retention, potassium excretion, tissue remodeling, and angiogenesis. Excessive secretion of aldosterone causes PA. Aldosterone acts on MR, which can be blocked by MRA. Ang I, angiotensin I; Ang II, angiotensin II; ACE, angiotensin converting enzyme; ACEI, angiotensin converting enzyme inhibitor; Ang1-9, angiotensin 1-9; Ang1-7, angiotensin 1-7; Ang III, angiotensin III; AT1R, angiotensin II receptor 1; AT2R, angiotensin II receptor 2; ARB, angiotensin receptor blocker; MR, mineralocorticoid receptor; MRA, mineralocorticoid receptor antagonism; PA, primary aldosteronism.
Physiological Role of PTH
PTH is an 84 amino acid single-stranded peptide hormone secreted by the chief cells of the parathyroid gland. The active form of PTH (pth1-84) is inactivated via liver and kidney metabolism (plasma half-life is about 2–4 min) (51). The secretion of PTH is mainly regulated by the concentration of serum Ca2+ (52). When the serum Ca2+ concentration is low, the secretion of PTH increases. When the serum Ca2+ concentration is high, the secretion of PTH decreases (51). This relationship is mediated by the interaction between Ca2+ and the presence of calcium-sensitive receptors (CASR) on the surface of parathyroid cells (52–54). The serum phosphorus (Pi) level is another important factor regulating PTH secretion. Pi in serum can indirectly stimulate the proliferation of parathyroid cells and the secretion of PTH, thereby reducing the concentration of serum Ca2+ (55). Serum Pi can also directly enhance the secretion function of parathyroid cells by increasing the stability of PTH mRNA (56).
The main target organs of PTH are the bones and kidneys (57, 58). PTH functions by binding to the receptor PTH/PTHrP receptor 1 (PTH1R) on target cells. PTH can mobilize bone Ca2+ into the blood and promote renal tubular reabsorption of Ca2+ and Pi excretion (54). PTH promotes the conversion of 25-hydroxyvitamin D (25OHD) in the kidney to 1,25-dihydroxyvitamin D (1,25[OH]2D3), which acts on the vitamin D receptor (VDR) to promote the intestinal reabsorption of Ca2+ and Pi, thereby increasing serum Ca2+ and Pi concentrations (Figure 2) (59, 60).
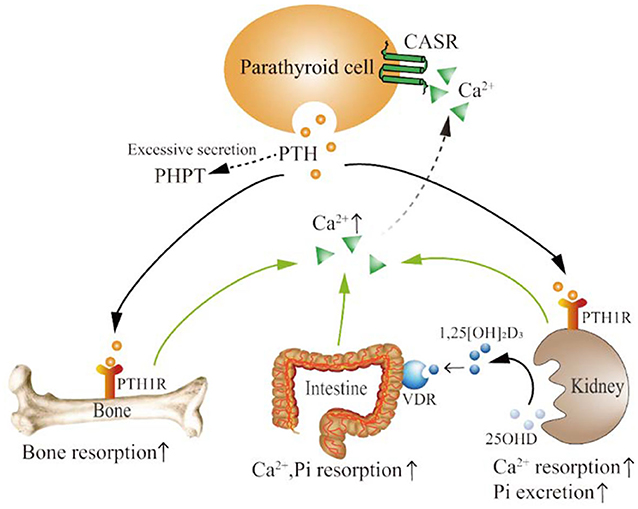
Figure 2. The physiological mechanism of PTH action. PTH is secreted by parathyroid cells. Excessive PTH secretion can cause PHPT. PTH functions by binding to the PTH1R in bone and kidney. PTH promotes bone absorption and the release of bone Ca2+, as well as the reabsorption of Ca2+ and the excretion of Pi in the kidney. PTH can also promote the conversion of 25OHD to 1,25[OH]2D3 in the kidney. 1,25[OH]2D3 binds to VDR, increasing the reabsorption of Ca2+ and Pi in the intestine. Serum Ca2+ regulates PTH secretion by binding to CASR on parathyroid cells. PTH, parathyroid hormone; PHPT, primary hyperparathyroidism; PTH1R, PTH1 receptor; Ca2+, calcium ions; Pi, phosphorus; 25OHD, 25-hydroxyvitamin D; 1,25[OH]2D3, 1,25-dihydroxyvitamin D; VDR, vitamin D receptor; CASR, calcium-sensitive receptors; ↑, upregulated.
Parathyroid adenoma, hyperplasia, or cancer can secrete excessive amounts of PTH, causing PHPT (52). PHPT is the most common manifestation of abnormal PTH levels. In patients with PHPT, long-term increases in PTH levels can lead to bone lesions such as osteoporosis, fragility fractures, and bone pain, as well as kidney lesions such as kidney stones, kidney calcification, and reduced renal function (30, 43, 52, 57, 58). Long-term PHPT can cause a serious condition known as fibrocystic cysts, proximal muscle weakness due to type II fiber atrophy, and even neuropsychiatric symptoms (59). Parathyroidectomy is the most important treatment for PHPT. However, for patients who are not suitable for surgery or who refuse surgery, various drugs, such as bisphosphonates and calcimimetics, can be taken appropriately according to patient symptoms (61).
Chronobiology and Chronotherapy of the RAAS/PTH
Blood pressure and biomarkers of RAAS have been shown to have circadian rhythms in humans (62); this circadian rhythm is also found in dogs (63). Studies have shown that the human circadian rhythm of RAAS is influenced by sodium intake (64), age, sex, and recumbency (65). Cugini et al. showed that limiting sodium intake amplifies the circadian rhythm of PRA and aldosterone. However, ACEIs inhibit this rhythm (66). The administration of ACEI at bedtime is more effective at controlling hypertension as compared to in the morning in hypertensive patients (67) and in a transverse aortic constriction (TAC) mouse model (68). Mochel et al. found that the timing of food intake is critical to the circadian rhythm of RAAS and blood pressure in dogs (69).
Similarly, the secretion of PTH is also rhythmic. In healthy humans, PTH is secreted primarily in a dual fashion. In addition to the most important tonic secretions, PTH can also be secreted in a low-amplitude and high-frequency pulse approximately every 20 min (70, 71). PTH release is affected by the blood Ca2+ concentration (72). This rhythmic secretion of PTH may be very important, as continuous administration of PTH leads to bone damage, while pulsed administration increases bone formation (73). Shinagawa et al. found that osteoporosis can be improved by simulating the pulsatile secretion of PTH to promote bone formation by oral administration of short-acting antagonists of CASR to rats (74).
Therefore, reference to the rhythmic secretion of RAAS and PTH has important diagnostic and therapeutic values for cardiovascular diseases or endocrine diseases in humans and animals.
Effects of RAAS Biomarker Levels on PTH Levels
How Does the RAAS Affect PTH
The RAAS mainly affects the secretion of PTH through AT1R and MR expressed in parathyroid tissue. AT1R and MR were detected in parathyroid adenoma tissue, with a 2- to 4-fold increase in expression when compared to normal parathyroid (75, 76). Chronic elevated aldosterone may increase PTH levels mainly by affecting renal function and serum Ca2+ (77). This can also be explained at the genetic level. Long-term injection of Ang II downregulates the expression of the Klotho gene in the kidney of rats (78), and downregulated Klotho expression will eventually cause an increase in PTH levels (79).
The relationship between the RAAS and PTH is significantly positive, as patients with PA exhibit higher PTH levels compared to secondary aldosteronism and primary hypertension patients (2, 17, 77). Recent studies have suggested that mild hyperparathyroidism is a characteristic of PA (80), irrespective of the subtype of PA (81). Furthermore, PA may even contribute to secondary hyperparathyroidism (77). The regulatory effect of Ang II on PTH is influenced by the injection dose of Ang II and the adequacy status of vitamin D. Supplementation with vitamin D3 can enhance the secretion-promoting effect of low-dose Ang II (1 ng/kg/min for 90 min) on PTH. A high-dose infusion of Ang II (3 ng/kg/min for 90 min) leads to greater increases in aldosterone and a more robust PTH response. Under acute and chronic conditions, the components of the RAAS that affect PTH secretion may also be different. Brown et al. demonstrated that, under acute conditions, it is the acute increase in the Ang II level rather than the acute increase in the aldosterone level that increases the PTH level, indicating that Ang II may be an acute regulator of PTH (76). However, aldosterone may be a chronic regulator of PTH (Figures 3A,B). Studies have shown that chronic long-term increases in aldosterone levels can lead to an increase in PTH levels (2, 17, 77). Chhokar et al. and Rossi et al. reported that an aldosterone infusion led to hyperparathyroidism (82, 83). A study of 11 PA patients and 15 non-PA patients showed that the PTH level in PA patients was elevated and recovered after surgery or spironolactone treatment (43). Another study by Fischer et al. showed that a high aldosterone-to-renin ratio (ARR) was associated with high serum PTH concentrations in the general population, but they found no associations between serum aldosterone or renin and PTH levels (84).
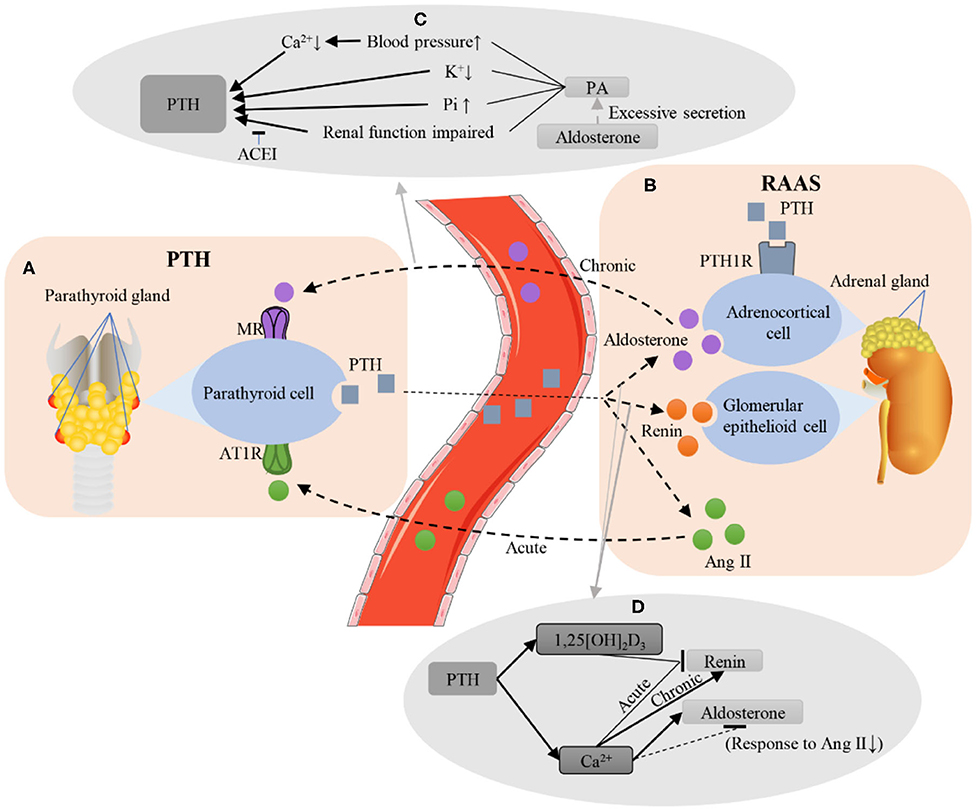
Figure 3. Interactions between the PTH and RAAS. (A,B) Brief illustration of RAAS and PTH. Under chronic conditions, the aldosterone secreted by the adrenal gland binds to the MR of the parathyroid gland. Under acute conditions, Ang II binds to AT1R in the parathyroid gland. PTH can bind to PTH1R in the adrenal gland to promote aldosterone secretion. In addition, PTH secretion can also increase the levels of renin and Ang II. (C) Several possible mechanisms for aldosterone to increase PTH levels. Excessive secretion of aldosterone causes PA, leading to the increase in blood pressure and serum Pi, the decrease in serum Ca2+ and K+, and the impairment of renal function, finally increasing the level of PTH, while ACEI can protect renal function and reduce PTH levels. (D) The possible mechanism of PTH regulating renin and aldosterone levels. PTH increased the serum levels of 1,25 [OH]2D3 and Ca2+. Acute elevation of Ca2+ and 1,25 [OH]2D3 inhibited renin secretion, while chronic elevation of Ca2+ could increase the renin level. Ca2+ can inhibit the diurnal decline of aldosterone, and it may also reduce the level of aldosterone by weakening the response of the kidney to Ang II. PTH, parathyroid hormone; RAAS, renin-angiotensin-aldosterone system; MR, mineralocorticoid receptor; Ang II, angiotensin II; AT1R, angiotensin II receptor 1; PTH1R, PTH1 receptor; PA, primary aldosteronism; Pi, phosphorus; Ca2+, calcium ions; K+, potassium; ACEI, angiotensin converting enzyme inhibitor; 1,25 [OH]2D3, 1,25-dihydroxyvitamin D. ↑, upregulated; ↓, downregulated.
Changes in PTH After the Inhibition of RAAS Function
Several studies have shown that PTH levels return to normal after medical or surgical treatment in PA patients (81). A study by Mizobuchi et al. showed the lowering effect of enalapril on PTH levels in uremic mice (85). A series of human studies also reached similar conclusions. A multi-ethnic study of atherosclerosis showed that the use of ACEI reduces the level of aldosterone, thereby reducing the level of PTH. Moreover, RAAS inhibitor (RAASI) use decreased the level of mean PTH significantly than the use of non-RAASI antihypertensives. In addition, Ca2+-channel blockers were associated with higher PTH levels (2). In a single-arm pilot study, Zaheer et al. (86) found that 1 week of lisinopril therapy titrated to maximally tolerated blood pressure lowering resulted in a modest and marginally statistically significant lowering of PTH levels among participants with PHPT, but did not impact the level of PTH among participants without PHPT. However, there was a decrease in systolic blood pressure, an increase in PRA, and no changes in serum and urine Ca2+ in both groups. Studies showed that RASI use induced a lower PTH level in patients with continuous ambulatory peritoneal dialysis (CAPD) or end-stage renal failure (79, 87).
The use of ARB also significantly reduces the level of PTH (76, 87, 88), although this conclusion remains controversial. A study by Zaheer et al. demonstrated that chronic MRA use modestly lowered PTH levels and raised serum Ca2+ (86). Contrary to the above result, a randomized, placebo-controlled trial found that short-term low-dose valsartan (a kind of ARB) treatment affected neither PTH levels nor the aldosterone levels; however, valsartan increased renin levels (89). A randomized, placebo-controlled trial showed that the level of PTH was not decreased after treatment with eplerenone, a new aldosterone receptor antagonist. However, short-term (1–8 weeks) treatment with RAASI (ACEI and MRA) is unlikely to induce a robust and clinically meaningful reduction in PTH in patients with PHPT (90). Moreover, ACEIs and ARBs do not consistently inhibit the RAAS, which in turn may increase plasma aldosterone levels in some patients. This phenomenon is called “aldosterone escape” or “aldosterone breakthrough” (91). This phenomenon may explain why the use of ACEIs and ARB does not affect the level of PTH. Aldosteronoma resection decreases the PTH level (83). Adrenalectomy not only cures hyperaldosteronism but also corrects hyperparathyroidism (17, 80). Compared with MRA, surgical treatment reduces PTH levels more obviously in PA patients (77). However, adrenalectomy can cause short-term aldosterone deficiency, resulting in hyperkalemia (92), and can even cause adrenal insufficiency, such as Addison's disease, which is rare (93). The findings mentioned above provide convincing evidence for a causal relationship between the RAAS and PTH levels.
The Level of RAAS Biomarker Changes With the Level of PTH
How Does PTH Affect the RAAS
PTH receptor mRNA can be detected in the adrenal gland (94). PTH also autoradiographically binds to the adrenal cortex, suggesting that the adrenal gland is one of the target organs of PTH (95, 96). PHPT may lead to the inactivation of VDR and then increase the level of renin expression (97). The type 1 PTH receptor is expressed in the cytoplasm of aldosterone-producing adenoma cells and nodule cells, indicating that PTH could directly influence the synthesis of aldosterone (17, 75). PTH binds to PTH/PTH-related peptide receptors and voltage-gated l-type calcium channels to initiate multiple signal transduction pathways that activate the RAS (98). PTH can increase the release of cAMP and inositol triphosphate from adrenocortical cells through activating G-protein-coupled signaling cascades (99–101). PTH inhibitors have been shown to inhibit the production of cAMP by adrenocortical cells (95). Both adenylate cyclase inhibitors and phospholipase C blockers block the production of PTH. PTH may function through the PTH receptor coupled with PLC/PKC-dependent signaling cascades (102).
PTH has been demonstrated to have a positive effect on the RAAS (12, 99), and PTH can induce renin release (103–105). Saussine et al. reported that the renin stimulation of PTH may be mediated by the inhibition of calcium influx (106); however, a study by Hulter et al. showed that PTH infusion resulted in a significant transient increase in aldosterone excretion, but no significant changes in PRA were observed (107). Verheyen et al. proposed that the high incidence of arterial hypertension in PHPT may be partially explained by elevated aldosterone levels (108). PTH promotes aldosterone secretion in a dose-dependent manner with minimal and maximal effective concentrations of 10−10 M and 10−8 M (102); the stimulatory effect of PTH on aldosterone secretion is induced by Ca2+ (109). Jespersen et al. reported that serum Ang II levels are elevated in patients with PHPT (110). PTH alone or in combination with Ang II stimulates adrenal glomerular cells to secrete aldosterone as well as enhancing the role of Ang II in promoting aldosterone secretion (Figures 3A,B) (96). A study by Maniero et al. showed that PTH can trigger and/or maintain hyperaldosteronism in patients with secondary aldosteronism (17). Akmal et al. used a dog model of chronic renal failure (CRF) to cause secondary hyperparathyroidism to demonstrate that increased PTH levels can cause pulmonary hypertension (111), which has been shown to be related to RAAS disorders in humans (112).
Some studies have shown that PTH does not have a significant impact on the RAAS. Bernini et al. compared PRA in hyperparathyroidism patients vs. normal subjects and essential hypertensive (EH) patients, observing no activation of the RAAS, and only found a weak positive correlation between PTH and PRA (113). A study by Richards et al. showed that aldosterone levels in patients with PHPT were not elevated (Table 1) (117).
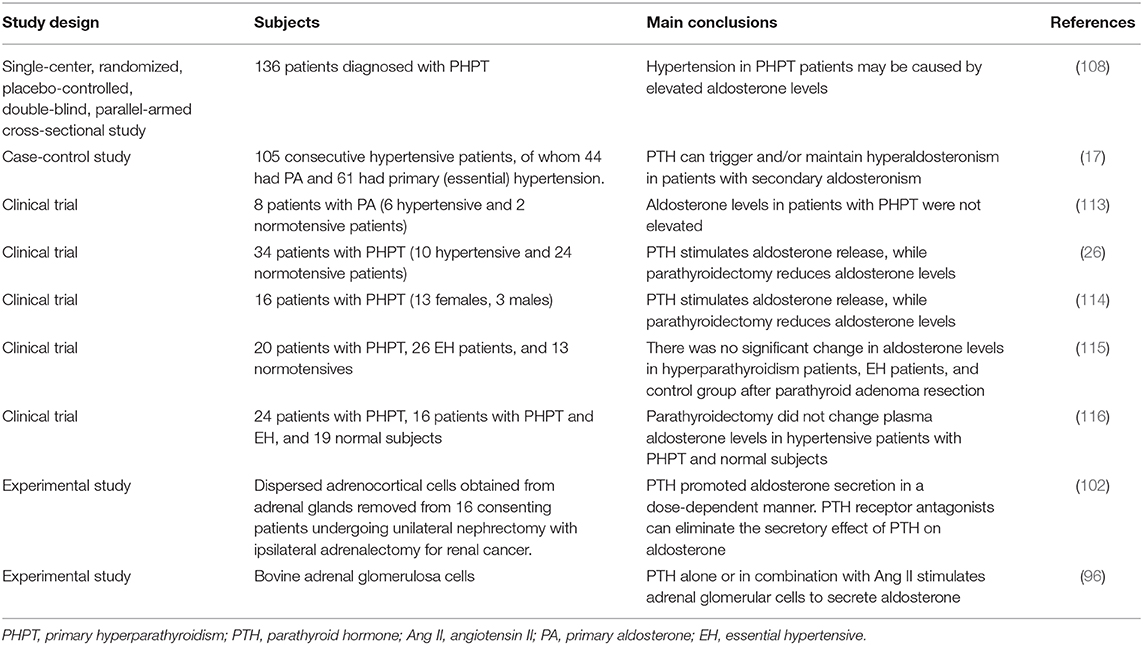
Table 1. Representative studies on the effects of PTH on aldosterone secretion.
Other studies have revealed a link between PTH1R and PRA. In the spontaneous hypertensive rat model driven by somatic human PTH1R (hPTH1R) gene expression, the overexpression of hPTH1R increases PRA, but the blood pressure remains unchanged (118). Interestingly, however, another study in adult rats in which PTH1R was overexpressed in the blood vessels, heart, kidneys, and other organs showed that PRA decreased 3 weeks after plasmid injection, thereby lowering blood pressure and heart rate in the rats (119). These rat models are very important for studying the function of PTH1R, because they have a high degree of genetic similarity with humans. Humans can not only accurately simulate human diseases in rat models but also guide the genetic modification of rats, so as to provide references for the treatment of human diseases.
Changes in the RAAS After the Inhibition of Parathyroid Function
PTH receptor antagonists have been demonstrated to eliminate the secretory effect of PTH on aldosterone (102). Parathyroidectomy has been shown to reduce systolic and diastolic blood pressure in patients with PA and to improve metabolic complications associated with PHPT (120). However, the hypotensive effect of parathyroidectomy was only manifested in PHPT patients with hypertension (114), and this effect was not observed in patients with normal blood pressure (8). Conversely, Kovács et al. found that, in 16 patients with normal blood pressure in PHPT, removal of the parathyroid gland caused a significant decrease in systolic blood pressure (121). Diamond et al. reported that, after parathyroid surgery, systolic blood pressure decreased significantly in patients with normal blood pressure and hypertension (122). There are also studies showing that parathyroidectomy does not lower blood pressure (11, 123, 124). Studies by Kovács et al. and Gennari et al. showed that the direct effect of PTH on the RAAS may explain the role of parathyroidectomy on blood pressure (26, 121).
Studies by Kovács et al. (26), Gennari et al. (121), and Pacifici et al. (115) showed that PTH stimulates the release of aldosterone and that parathyroidectomy reduces blood pressure as well as plasma levels of renin and aldosterone. However, Bernini et al. demonstrated that PH patients are similar to EH patients regarding controls on PRA and aldosterone levels, and there was no significant change after parathyroid adenoma resection (113). Additionally, Salahudeen et al. found that parathyroidectomy did not alter PRA and plasma aldosterone levels in normotensive and hypertensive patients with PHPT (Table 1) (116). The above two studies indicate that PTH may not affect RAAS. Studies have also shown that PTH may have an indirect negative regulatory effect on the RAAS. Zawada et al. used a model of dogs with normal blood pressure and renovascular hypertension to confirm that thyroparathyroidectomy or chelation with EDTA to reduce blood Ca2+ will cause an increase in PRA (125).
Discussion
The parathyroid gland expresses AT1R and MR, which supports the hypothesis that aldosterone contributes to the regulation of PTH secretion. The adrenal gland also expresses the type 1 PTH receptor, which likely explains why human adrenocortical cells respond with aldosterone and cortisol release to either PTH or PTH-related peptide (75). Studies have shown that the RAAS and PTH have a positive effect on each other.
There may be several other mechanisms that explain the increase in PTH as RAAS levels increase. Firstly, patients with PA often present with elevated blood pressure, and high blood pressure may alter the kidney's handling of Ca2+, which may lead to increased 24-h renal Ca2+ excretion and decreased serum Ca2+ (43, 126–129). Several studies have shown that the increase in the PTH level may be caused by a decrease in serum Ca2+ rather than a direct effect of aldosterone in patients with PA, as a low Ca2+ level may contribute to secondary hyperparathyroidism and eventually affect bone mass and strength (17, 77, 82, 88, 126, 130, 131). Secondly, a negative correlation between serum K+ and PTH levels has also been found (81). However, aldosterone can lower serum K+, so the stimulatory effect of aldosterone on PTH secretion may be mediated by K+. Thirdly, early impaired renal function in PA patients may cause parathyroid function enhancement (126), but the renoprotective effect may result in lower PTH levels (85). Enalapril reduces serum creatinine levels in rats, but enalapril reduces PTH levels through a protective effect on the kidney, rather than by directly acting on the parathyroid gland (85). Finally, changes in serum Pi levels in patients with PA and their effects on PTH levels also need to be considered (Figure 3C).
PTH exerts its effects on RAAS biomarkers through several possible mechanisms. PTH can increase the level of 1,25[OH] 2D3 (59, 60), which will inhibit the expression of renin (97). Acute increases in plasma and extracellular and intracellular Ca2+ concentrations also inhibit renin secretion in renal proximal tubular cells, but long-term Ca2+ elevation can cause elevated PRA, which may be a compensation mechanism caused by Ca2+-mediated polyuria (132). PTH increases serum Ca2+ levels, and the increase in Ca2+ may inhibit the diurnal decline in PRA and aldosterone (133). In the case of hypercalcemia, the response of aldosterone to Ang II is weakened (134), which may explain why PTH inhibits the secretion of aldosterone levels (Figure 3D) (132).
As reviewed above, the interaction between RAAS and PTH-Calcium metabolism may be indirectly influenced by secondary factors such as electrolytes, blood pressure, and renal function. However, the direct interaction between the two systems is predominant. Further research is needed to investigate the complex interactions between PTH and RAAS and to assess whether PTH measurements can contribute to the diagnosis of PA. Elucidating the relationship between the RAAS and PTH has important clinical significance and future implications for the treatment of patients with PA or PHPT. RASIs might decrease PTH levels and improve these symptoms. MRA may reduce PTH-induced increases in the fracture rate in PA patients. However, the effect of MRA on PTH in patients without PA is unclear. MRAs may be a promising, novel, and inexpensive therapeutic option for PA patients with elevated PTH levels. Further studies are needed to investigate more medications that mitigate cardiovascular or skeletal diseases mediated by aldosterone and PTH.
Author Contributions
L-QY: manuscript writing and approving final version of manuscript. M-HZ: study conduct, data analysis, and manuscript writing. FX, XL, YW, Q-SX, and BG: data analysis. All authors: reviewed the manuscript.
Funding
This work was supported by funding from the National Natural Science Foundation of China (No. 81770881).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Asbach E, Bekeran M, König A, Lang K, Hanslik G, Treitl M, et al. Primary and secondary hyperparathyroidism in patients with primary aldosteronism - findings from the German Conn's registry. Exp Clin Endocrinol Diabetes. (2020) 128:246–54. doi: 10.1055/a-1027-6472
2. Brown J, de Boer IH, Robinson-Cohen C, Siscovick DS, Kestenbaum B, Allison M, et al. Aldosterone, parathyroid hormone, and the use of renin-angiotensin-aldosterone system inhibitors: the multi-ethnic study of atherosclerosis. J Clin Endocrinol Metab. (2015) 100:490–9. doi: 10.1210/jc.2014-3949
3. Boonstra CE, Jackson CE. Serum calcium survey for hyperparathyroidism: results in 50,000 clinic patients. Am J Clin Pathol. (1971) 55:523–6. doi: 10.1093/ajcp/55.5.523
4. Hatton R, Stimpel M, Chambers TJ. Angiotensin II is generated from angiotensin I by bone cells and stimulates osteoclastic bone resorption in vitro. J Endocrinol. (1997) 152:5–10. doi: 10.1677/joe.0.1520005
5. Beavan S, Horner A, Bord S, Ireland D, Compston J. Colocalization of glucocorticoid and mineralocorticoid receptors in human bone. J Bone Miner Res. (2001) 16:1496–504. doi: 10.1359/jbmr.2001.16.8.1496
6. Verheyen N, Grübler MR, Meinitzer A, Trummer C, Schwetz V, Amrein K, et al. Effect of eplerenone on markers of bone turnover in patients with primary hyperparathyroidism - The randomized, placebo-controlled EPATH trial. Bone. (2017) 105:212–7. doi: 10.1016/j.bone.2017.08.030
7. Barletta G, De Feo ML, Del Bene R, Lazzeri C, Vecchiarino S, La Villa G, et al. Cardiovascular effects of parathyroid hormone: a study in healthy subjects and normotensive patients with mild primary hyperparathyroidism. J Clin Endocrinol Metab. (2000) 85:1815–21. doi: 10.1210/jc.85.5.1815
8. Heyliger A, Tangpricha V, Weber C, Sharma J. Parathyroidectomy decreases systolic and diastolic blood pressure in hypertensive patients with primary hyperparathyroidism. Surgery. (2009) 146:1042–7. doi: 10.1016/j.surg.2009.09.024
9. Hedbäck GM, Odén AS. Cardiovascular disease, hypertension and renal function in primary hyperparathyroidism. J Intern Med. (2002) 251:476–83. doi: 10.1046/j.1365-2796.2002.00984.x
10. Garcia de la Torre N, Wass JAH, Turner HE. Parathyroid adenomas and cardiovascular risk. Endocr Relat Cancer. (2003) 10:309–22. doi: 10.1677/erc.0.0100309
11. Nilsson I-L, Aberg J, Rastad J, Lind L. Maintained normalization of cardiovascular dysfunction 5 years after parathyroidectomy in primary hyperparathyroidism. Surgery. (2005) 137:632–8. doi: 10.1016/j.surg.2005.02.001
12. Brunaud L, Germain A, Zarnegar R, Rancier M, Alrasheedi S, Caillard C, et al. Serum aldosterone is correlated positively to parathyroid hormone (PTH) levels in patients with primary hyperparathyroidism. Surgery. (2009) 146:1035–41. doi: 10.1016/j.surg.2009.09.041
13. Hagström E, Hellman P, Larsson TE, Ingelsson E, Berglund L, Sundström J, et al. Plasma parathyroid hormone and the risk of cardiovascular mortality in the community. Circulation. (2009) 119:2765–71. doi: 10.1161/CIRCULATIONAHA.108.808733
14. Pilz S, Tomaschitz A, Drechsler C, Ritz E, Boehm BO, Grammer TB, et al. Parathyroid hormone level is associated with mortality and cardiovascular events in patients undergoing coronary angiography. Eur Heart J. (2010) 31:1591–8. doi: 10.1093/eurheartj/ehq109
15. Kiernan TJ, O'Flynn AM, McDermott JH, Kearney P. Primary hyperparathyroidism and the cardiovascular system. Int J Cardiol. (2006) 113:E89–E92. doi: 10.1016/j.ijcard.2006.05.033
16. Pilz S, Tomaschitz A, März W, Cavalier E, Ritz E. Aldosterone and parathyroid hormone: a complex and clinically relevant relationship. Calcif Tissue Int. (2010) 87:373–4. doi: 10.1007/s00223-010-9409-5
17. Maniero C, Fassina A, Seccia TM, Toniato A, Iacobone M, Plebani M, et al. Mild hyperparathyroidism: a novel surgically correctable feature of primary aldosteronism. J Hypertens. (2012) 30:390–5. doi: 10.1097/HJH.0b013e32834f0451
18. Fardella C, Rodriguez-Portales JA. Intracellular calcium and blood pressure: comparison between primary hyperparathyroidism and essential hypertension. J Endocrinol Invest. (1995) 18:827–32. doi: 10.1007/BF03349828
19. Simonetti G, Mohaupt M. [Calcium and blood pressure]. Ther Umsch. (2007) 64:249–52. doi: 10.1024/0040-5930.64.5.249
20. Rashid G, Bernheim J, Green J, Benchetrit S. Parathyroid hormone stimulates endothelial expression of atherosclerotic parameters through protein kinase pathways. Am J Physiol Renal Physiol. (2007) 292:F1215–F8. doi: 10.1152/ajprenal.00406.2006
21. Schlüter KD, Weber M, Piper HM. Parathyroid hormone induces protein kinase C but not adenylate cyclase in adult cardiomyocytes and regulates cyclic AMP levels via protein kinase C-dependent phosphodiesterase activity. Biochem J. (1995) 310 (Pt 2):439–44. doi: 10.1042/bj3100439
22. Rosa J, Raska I, Wichterle D, Petrak O, Strauch B, Somloova Z, et al. Pulse wave velocity in primary hyperparathyroidism and effect of surgical therapy. Hypertens Res. (2011) 34:296–300. doi: 10.1038/hr.2010.232
23. Schillaci G, Pucci G, Pirro M, Monacelli M, Scarponi AM, Manfredelli MR, et al. Large-artery stiffness: a reversible marker of cardiovascular risk in primary hyperparathyroidism. Atherosclerosis. (2011) 218:96–101. doi: 10.1016/j.atherosclerosis.2011.05.010
24. Xu F, Zhong J-Y, Lin X, Shan S-K, Guo B, Zheng M-H, et al. Melatonin alleviates vascular calcification and ageing through exosomal miR-204/miR-211 cluster in a paracrine manner. J Pineal Res. (2020) 68:e12631. doi: 10.1111/jpi.12631
25. Broulik PD, Horký K, Pacovský V. Effect of parathyroid hormone on plasma renin activity in humans. Horm Metab Res. (1986) 18:490–2. doi: 10.1055/s-2007-1012353
26. Gennari C, Nami R, Gonnelli S. Hypertension and primary hyperparathyroidism: the role of adrenergic and renin-angiotensin-aldosterone systems. Miner Electrolyte Metab. (1995) 21:77–81.
27. Broulik PD, Stepán JJ, Pacovský V. Primary hyperparathyroidism and hyperuricaemia are associated but not correlated with indicators of bone turnover. Clin Chim Acta. (1987) 170:195–200. doi: 10.1016/0009-8981(87)90128-8
28. Feig DI, Kang D-H, Johnson RJ. Uric acid and cardiovascular risk. N Engl J Med. (2008) 359:1811–21. doi: 10.1056/NEJMra0800885
29. Almqvist EG, Bondeson A-G, Bondeson L, Nissborg A, Smedgård P, Svensson S-E. Cardiac dysfunction in mild primary hyperparathyroidism assessed by radionuclide angiography and echocardiography before and after parathyroidectomy. Surgery. (2002) 132:1126–32. doi: 10.1067/msy.2002.128692
30. McMahon DJ, Carrelli A, Palmeri N, Zhang C, DiTullio M, Silverberg SJ, et al. Effect of parathyroidectomy upon left ventricular mass in primary hyperparathyroidism: a meta-analysis. J Clin Endocrinol Metab. (2015) 100:4399–407. doi: 10.1210/jc.2015-3202
31. Davis GK, Roberts DH. Molecular genetics of the renin-angiotensin system: implications for angiotensin II receptor blockade. Pharmacol Ther. (1997) 75:43–50. doi: 10.1016/S0163-7258(97)00021-1
32. Crisan D, Carr J. Angiotensin I-converting enzyme: genotype and disease associations. J Mol Diagn. (2000) 2:105–15. doi: 10.1016/S1525-1578(10)60624-1
33. Bouhnik J, Galen FX, Menard J, Corvol P, Seyer R, Fehrentz JA, et al. Production and characterization of human renin antibodies with region-oriented synthetic peptides. J Biol Chem. (1987) 262:2913–8.
34. Verdecchia P, Angeli F, Mazzotta G, Gentile G, Reboldi G. The renin angiotensin system in the development of cardiovascular disease: role of aliskiren in risk reduction. Vasc Health Risk Manag. (2008) 4:971–81. doi: 10.2147/VHRM.S3215
35. Bernstein KE, Gonzalez-Villalobos RA, Giani JF, Shah K, Bernstein E, Janjulia T, et al. Angiotensin-converting enzyme overexpression in myelocytes enhances the immune response. Biol Chem. (2014) 395:1173–8. doi: 10.1515/hsz-2013-0295
36. Wu Z, Cappiello MG, Scott BB, Bukhtiyarov Y, McGeehan GM. Purification and characterization of recombinant human renin for X-ray crystallization studies. BMC Biochem. (2008) 9:19. doi: 10.1186/1471-2091-9-19
37. Navar LG. Physiology: hemodynamics, endothelial function, renin-angiotensin-aldosterone system, sympathetic nervous system. J Am Soc Hypertens. (2014) 8:519–24. doi: 10.1016/j.jash.2014.05.014
38. Patel VB, Zhong J-C, Grant MB, Oudit GY. Role of the ACE2/angiotensin 1-7 axis of the renin-angiotensin system in heart failure. Circ Res. (2016) 118:1313–26. doi: 10.1161/CIRCRESAHA.116.307708
39. Roig E, Perez-Villa F, Morales M, Jiménez W, Orús J, Heras M. Clinical implications of increased plasma angiotensin II despite ACE inhibitor therapy in patients with congestive heart failure. Eur Heart J. (2000) 21:53–7. doi: 10.1053/euhj.1999.1740
40. Yatabe J, Yoneda M, Yatabe MS, Watanabe T, Felder RA, Jose PA, et al. Angiotensin III stimulates aldosterone secretion from adrenal gland partially via angiotensin II type 2 receptor but not angiotensin II type 1 receptor. Endocrinology. (2011) 152:1582–8. doi: 10.1210/en.2010-1070
41. Balla T, Baukal AJ, Eng S, Catt KJ. Angiotensin II receptor subtypes and biological responses in the adrenal cortex and medulla. Mol Pharmacol. (1991) 40:401–6.
42. Mazzocchi G, Gottardo G, Macchi V, Malendowicz LK, Nussdorfer GG. The AT2 receptor-mediated stimulation of adrenal catecholamine release may potentiate the AT1 receptor-mediated aldosterone secretagogue action of angiotensin-II in rats. Endocr Res. (1998) 24:17–28. doi: 10.3109/07435809809031866
43. Salcuni AS, Palmieri S, Carnevale V, Morelli V, Battista C, Guarnieri V, et al. Bone involvement in aldosteronism. J Bone Miner Res. (2012) 27:2217–22. doi: 10.1002/jbmr.1660
44. Schmieder RE, Hilgers KF, Schlaich MP, Schmidt BMW. Renin-angiotensin system and cardiovascular risk. Lancet. (2007) 369:1208–19. doi: 10.1016/S0140-6736(07)60242-6
45. Stowasser M. Update in primary aldosteronism. J Clin Endocrinol Metab. (2015) 100:1–10. doi: 10.1210/jc.2014-3663
46. Mirabito Colafella KM, Bovée DM, Danser AHJ. The renin-angiotensin-aldosterone system and its therapeutic targets. Exp Eye Res. (2019) 186:107680. doi: 10.1016/j.exer.2019.05.020
47. Abraham HMA, White CM, White WB. The comparative efficacy and safety of the angiotensin receptor blockers in the management of hypertension and other cardiovascular diseases. Drug Saf. (2015) 38:33–54. doi: 10.1007/s40264-014-0239-7
48. Hansen ML, Gislason GH, Køber L, Schramm TK, Folke F, Buch P, et al. Different angiotensin-converting enzyme inhibitors have similar clinical efficacy after myocardial infarction. Br J Clin Pharmacol. (2008) 65:217–23. doi: 10.1111/j.1365-2125.2007.02991.x
49. Epstein M, Calhoun DA. Aldosterone blockers (mineralocorticoid receptor antagonism) and potassium-sparing diuretics. J Clin Hypertens. (2011) 13:644–8. doi: 10.1111/j.1751-7176.2011.00511.x
50. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, et al. 2016 ESC Guidelines for the diagnosis treatment of acute chronic heart failure: The Task Force for the diagnosis treatment of acute chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. (2016) 37:2129–200. doi: 10.1093/eurheartj/ehw128
51. Parker VJ, Gilor C, Chew DJ. Feline hyperparathyroidism: pathophysiology, diagnosis and treatment of primary and secondary disease. J Feline Med Surg. (2015) 17:427–39. doi: 10.1177/1098612X15581134
52. Bilezikian JP, Cusano NE, Khan AA, Liu J-M, Marcocci C, Bandeira F. Primary hyperparathyroidism. Nat Rev Dis Primers. (2016) 2:16033. doi: 10.1038/nrdp.2016.33
53. Brown EM. The pathophysiology of primary hyperparathyroidism. J Bone Miner Res. (2002) 17(Suppl 2):N24−9.
54. Rossi GP, Prisco S. Does angiotensin II regulate parathyroid hormone secretion or not? Clin Endocrinol. (2018) 89:568–9. doi: 10.1111/cen.13798
55. Naveh-Many T, Rahamimov R, Livni N, Silver J. Parathyroid cell proliferation in normal and chronic renal failure rats. The effects of calcium, phosphate, vitamin D. J Clin Invest. (1995) 96:1786–93. doi: 10.1172/JCI118224
56. Moallem E, Kilav R, Silver J, Naveh-Many T. RNA-Protein binding and post-transcriptional regulation of parathyroid hormone gene expression by calcium and phosphate. J Biol Chem. (1998) 273:5253–9. doi: 10.1074/jbc.273.9.5253
57. Sitges-Serra A, Bergenfelz A. Clinical update: sporadic primary hyperparathyroidism. Lancet. (2007) 370:468–70. doi: 10.1016/S0140-6736(07)61213-6
58. Silverberg SJ, Clarke BL, Peacock M, Bandeira F, Boutroy S, Cusano NE, et al. Current issues in the presentation of asymptomatic primary hyperparathyroidism: proceedings of the Fourth International Workshop. J Clin Endocrinol Metab. (2014) 99:3580–94. doi: 10.1210/jc.2014-1415
59. Makras P, Anastasilakis AD. Bone disease in primary hyperparathyroidism. Metab Clin Exp. (2018) 80:57–65. doi: 10.1016/j.metabol.2017.10.003
60. Smit MA, van Kinschot CMJ, van der Linden J, van Noord C, Kos S. Clinical guidelines and PTH measurement: does assay generation matter? Endocr Rev. (2019) 40:1468–80. doi: 10.1210/er.2018-00220
61. Bilezikian JP, Brandi ML, Eastell R, Silverberg SJ, Udelsman R, Marcocci C, et al. Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the Fourth International Workshop. J Clin Endocrinol Metab. (2014) 99:3561–69. doi: 10.1210/jc.2014-1413
62. Kawasaki T, Cugini P, Uezono K, Sasaki H, Itoh K, Nishiura M, et al. Circadian rhythms of total renin and active renin concentrations in normotensive young subjects. Prog Clin Biol Res. (1990) 341A:699–708.
63. Mochel JP, Fink M, Peyrou M, Desevaux C, Deurinck M, Giraudel JM, et al. Chronobiology of the renin-angiotensin-aldosterone system in dogs: relation to blood pressure and renal physiology. Chronobiol Int. (2013) 30:1144–59. doi: 10.3109/07420528.2013.807275
64. Cugini P, Scavo D, Cornelissen G, Lee JY, Meucci T, Halberg F. Circadian rhythms of plasma renin, aldosterone and cortisol on habitual and low dietary sodium intake. Horm Res. (1981) 15:7–27. doi: 10.1159/000179430
65. Cugini P, Murano G, Lucia P, Letizia C, Scavo D, Halberg F, et al. Circadian rhythms of plasma renin activity and aldosterone: changes related to age, sex, recumbency and sodium restriction. Chronobiologic specification for reference values. Chronobiol Int. (1985) 2:267–76. doi: 10.3109/07420528509055889
66. Cugini P, Halberg F, Sothern RB, Centanni M, Salandi E, Scavo D. Sodium restriction amplifies and propranolol loading inhibits circadian rhythm of plasma renin-angiotensin and aldosterone. Chronobiologia. (1985) 12:155–65.
67. Hermida RC, Ayala DE. Chronotherapy with the angiotensin-converting enzyme inhibitor ramipril in essential hypertension: improved blood pressure control with bedtime dosing. Hypertension. (2009) 54:40–6. doi: 10.1161/HYPERTENSIONAHA.109.130203
68. Martino TA, Tata N, Simpson JA, Vanderlaan R, Dawood F, Kabir MG, et al. The primary benefits of angiotensin-converting enzyme inhibition on cardiac remodeling occur during sleep time in murine pressure overload hypertrophy. J Am Coll Cardiol. (2011) 57:2020–8. doi: 10.1016/j.jacc.2010.11.022
69. Mochel JP, Fink M, Bon C, Peyrou M, Bieth B, Desevaux C, et al. Influence of feeding schedules on the chronobiology of renin activity, urinary electrolytes and blood pressure in dogs. Chronobiol Int. (2014) 31:715–30. doi: 10.3109/07420528.2014.897711
70. Samuels MH, Veldhuis J, Cawley C, Urban RJ, Luther M, Bauer R, et al. Pulsatile secretion of parathyroid hormone in normal young subjects: assessment by deconvolution analysis. J Clin Endocrinol Metab. (1993) 77:399–403. doi: 10.1210/jcem.77.2.8345044
71. Chiavistelli S, Giustina A, Mazziotti G. Parathyroid hormone pulsatility: physiological and clinical aspects. Bone Res. (2015) 3:14049. doi: 10.1038/boneres.2014.49
72. Schaefer F. Pulsatile parathyroid hormone secretion in health and disease. Novartis Found Symp. (2000) 227:225–39. doi: 10.1002/0470870796.ch13
73. Potter LK, Greller LD, Cho CR, Nuttall ME, Stroup GB, Suva LJ, et al. Response to continuous and pulsatile PTH dosing: a mathematical model for parathyroid hormone receptor kinetics. Bone. (2005) 37:159–69. doi: 10.1016/j.bone.2005.04.011
74. Shinagawa Y, Inoue T, Katsushima T, Kiguchi T, Ikenogami T, Ogawa N, et al. Discovery of a potent and short-acting oral calcilytic with a pulsatile secretion of parathyroid hormone. ACS Med Chem Lett. (2011) 2:238–42. doi: 10.1021/ml100268k
75. Maniero C, Fassina A, Guzzardo V, Lenzini L, Amadori G, Pelizzo MR, et al. Primary hyperparathyroidism with concurrent primary aldosteronism. Hypertension. (2011) 58:341–6. doi: 10.1161/HYPERTENSIONAHA.111.173948
76. Brown JM, Williams JS, Luther JM, Garg R, Garza AE, Pojoga LH, et al. Human interventions to characterize novel relationships between the renin-angiotensin-aldosterone system and parathyroid hormone. Hypertension. (2014) 63:273–80. doi: 10.1161/HYPERTENSIONAHA.113.01910
77. Pilz S, Kienreich K, Drechsler C, Ritz E, Fahrleitner-Pammer A, Gaksch M, et al. Hyperparathyroidism in patients with primary aldosteronism: cross-sectional and interventional data from the GECOH study. J Clin Endocrinol Metab. (2012) 97:E75–9. doi: 10.1210/jc.2011-2183
78. Mitani H, Ishizaka N, Aizawa T, Ohno M, Usui S-i, Suzuki T, et al. In vivo klotho gene transfer ameliorates angiotensin II-induced renal damage. Hypertension. (2002) 39:838–43. doi: 10.1161/01.HYP.0000013734.33441.EA
79. Gao L, Wang Z, Feng S, Jiang S, Ouyang H, Shi Y, et al. The effect of renin-angiotensin system inhibitors on intact parathyroid hormone levels in peritoneal dialysis patients. J Renin Angiotensin Aldosterone Syst. (2015) 16:1260–5. doi: 10.1177/1470320314548868
80. Lenzini L, Prisco S, Vanderriele PE, Lerco S, Torresan F, Maiolino G, et al. PTH modulation by aldosterone and angiotensin II is blunted in hyperaldosteronism and rescued by adrenalectomy. J Clin Endocrinol Metab. (2019) 104:3726–34. doi: 10.1210/jc.2019-00143
81. Jiang Y, Zhang C, Ye L, Su T, Zhou W, Jiang L, et al. Factors affecting parathyroid hormone levels in different types of primary aldosteronism. Clin Endocrinol. (2016) 85:267–74. doi: 10.1111/cen.12981
82. Chhokar VS, Sun Y, Bhattacharya SK, Ahokas RA, Myers LK, Xing Z, et al. Hyperparathyroidism and the calcium paradox of aldosteronism. Circulation. (2005) 111:871–8. doi: 10.1161/01.CIR.0000155621.10213.06
83. Rossi GP, Ragazzo F, Seccia TM, Maniero C, Barisa M, Calò LA, et al. Hyperparathyroidism can be useful in the identification of primary aldosteronism due to aldosterone-producing adenoma. Hypertension. (2012) 60:431–6. doi: 10.1161/HYPERTENSIONAHA.112.195891
84. Fischer E, Hannemann A, Rettig R, Lieb W, Nauck M, Pallauf A, et al. A high aldosterone to renin ratio is associated with high serum parathyroid hormone concentrations in the general population. J Clin Endocrinol Metab. (2014) 99:965–71. doi: 10.1210/jc.2013-3214
85. Mizobuchi M, Morrissey J, Finch JL, Martin DR, Liapis H, Akizawa T, et al. Combination therapy with an angiotensin-converting enzyme inhibitor and a vitamin D analog suppresses the progression of renal insufficiency in uremic rats. J Am Soc Nephrol. (2007) 18:1796–806. doi: 10.1681/ASN.2006091028
86. Zaheer S, Brown JM, Connors M, Williams JS, Adler GK, Vaidya A. Angiotensin-converting enzyme inhibition and parathyroid hormone secretion. Int J Endocrinol. (2017) 2017:4138783. doi: 10.1155/2017/4138783
87. Koiwa F, Komukai D, Hirose M, Yoshimura A, Ando R, Sakaguchi T, et al. Influence of renin-angiotensin system on serum parathyroid hormone levels in uremic patients. Clin Exp Nephrol. (2012) 16:130–5. doi: 10.1007/s10157-011-0534-x
88. Rossi E, Sani C, Perazzoli F, Casoli MC, Negro A, Dotti C. Alterations of calcium metabolism and of parathyroid function in primary aldosteronism, and their reversal by spironolactone or by surgical removal of aldosterone-producing adenomas. Am J Hypertens. (1995) 8:884–93. doi: 10.1016/0895-7061(95)00182-O
89. Bislev LS, Langagergaard Rødbro L, Nørgaard Bech J, Bjerregaard Pedersen E, Rolighed L, Sikjaer T, et al. Effects of treatment with an angiotensin 2 receptor blocker and/or vitamin D3 on parathyroid hormone and aldosterone: a randomized, placebo-controlled trial. Clin Endocrinol. (2018) 89:656–66. doi: 10.1111/cen.13734
90. Tomaschitz A, Verheyen N, Meinitzer A, Pieske B, Belyavskiy E, Brussee H, et al. Effect of eplerenone on parathyroid hormone levels in patients with primary hyperparathyroidism: results from the EPATH randomized, placebo-controlled trial. J Hypertens. (2016) 34:1347–56. doi: 10.1097/HJH.0000000000000927
91. Bomback AS, Klemmer PJ. The incidence and implications of aldosterone breakthrough. Nat Clin Pract Nephrol. (2007) 3:486–92. doi: 10.1038/ncpneph0575
92. Shariq OA, Bancos I, Cronin PA, Farley DR, Richards ML, Thompson GB, et al. Contralateral suppression of aldosterone at adrenal venous sampling predicts hyperkalemia following adrenalectomy for primary aldosteronism. Surgery. (2018) 163:183–90. doi: 10.1016/j.surg.2017.07.034
93. Elikowski W, Małek-Elikowska M, Greberska W, Słomczynski M, Marchlewska J, Korol L. Takotsubo syndrome in a patient with adrenal insufficiency, severe hyponatremia and coexistent coronary artery disease. Pol Merkur Lekarski. (2019) 46:182−6.
94. Ureña P, Kong XF, Abou-Samra AB, Jüppner H, Kronenberg HM, Potts JT, et al. Parathyroid hormone (PTH)/PTH-related peptide receptor messenger ribonucleic acids are widely distributed in rat tissues. Endocrinology. (1993) 133:617–23. doi: 10.1210/endo.133.2.8393771
95. Rosenberg J, Pines M, Hurwitz S. Response of adrenal cells to parathyroid hormone stimulation. J Endocrinol. (1987) 112:431–7. doi: 10.1677/joe.0.1120431
96. Isales CM, Barrett PQ, Brines M, Bollag W, Rasmussen H. Parathyroid hormone modulates angiotensin II-induced aldosterone secretion from the adrenal glomerulosa cell. Endocrinology. (1991) 129:489–95. doi: 10.1210/endo-129-1-489
97. Li YC, Kong J, Wei M, Chen Z-F, Liu SQ, Cao L-P. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest. (2002) 110:229–38. doi: 10.1172/JCI0215219
98. Rossier MF, Burnay MM, Vallotton MB, Capponi AM. Distinct functions of T- and L-type calcium channels during activation of bovine adrenal glomerulosa cells. Endocrinology. (1996) 137:4817–26. doi: 10.1210/endo.137.11.8895352
99. Rosenberg J, Pines M, Hurwitz S. Stimulation of chick adrenal steroidogenesis by avian parathyroid hormone. J Endocrinol. (1988) 116:91–5. doi: 10.1677/joe.0.1160091
100. Nussdorfer GG, Malendowicz LK. Role of VIP, PACAP, and related peptides in the regulation of the hypothalamo-pituitary-adrenal axis. Peptides. (1998) 19:1443–67. doi: 10.1016/S0196-9781(98)00102-8
101. Schwindinger WF, Fredericks J, Watkins L, Robinson H, Bathon JM, Pines M, et al. Coupling of the PTH/PTHrP receptor to multiple G-proteins. Direct demonstration of receptor activation of Gs, Gq/11, and Gi(1) by [alpha-32P]GTP-gamma-azidoanilide photoaffinity labeling. Endocrine. (1998) 8:201–9. doi: 10.1385/ENDO:8:2:201
102. Mazzocchi G, Aragona F, Malendowicz LK, Nussdorfer GG. PTH and PTH-related peptide enhance steroid secretion from human adrenocortical cells. Am J Physiol Endocrinol Metab. (2001) 280:E209–E13. doi: 10.1152/ajpendo.2001.280.2.E209
103. Smith JM, Mouw DR, Vander AJ. Effect of parathyroid hormone on plasma renin activity and sodium excretion. Am J Physiol. (1979) 236:F311–F9. doi: 10.1152/ajprenal.1979.236.3.F311
104. Smith JM, Mouw DR, Vander AJ. Effect of parathyroid hormone on renin secretion. Proc Soc Exp Biol Med. (1983) 172:482–7. doi: 10.3181/00379727-172-41591
105. Helwig JJ, Musso MJ, Judes C, Nickols GA. Parathyroid hormone and calcium: interactions in the control of renin secretion in the isolated, nonfiltering rat kidney. Endocrinology. (1991) 129:1233–42. doi: 10.1210/endo-129-3-1233
106. Saussine C, Judes C, Massfelder T, Musso MJ, Simeoni U, Hannedouche T, et al. Stimulatory action of parathyroid hormone on renin secretion in vitro: a study using isolated rat kidney, isolated rabbit glomeruli and superfused dispersed rat juxtaglomerular cells. Clin Sci. (1993) 84:11–9. doi: 10.1042/cs0840011
107. Hulter HN, Melby JC, Peterson JC, Cooke CR. Chronic continuous PTH infusion results in hypertension in normal subjects. J Clin Hypertens. (1986) 2:360–70.
108. Verheyen N, Fahrleitner-Pammer A, Pieske B, Meinitzer A, Belyavskiy E, Wetzel J, et al. Parathyroid hormone, aldosterone-to-renin ratio and fibroblast growth factor-23 as determinants of nocturnal blood pressure in primary hyperparathyroidism: the eplerenone in primary hyperparathyroidism trial. J Hypertens. (2016) 34:1778–86. doi: 10.1097/HJH.0000000000001004
109. Olgaard K, Lewin E, Bro S, Daugaard H, Egfjord M, Pless V. Enhancement of the stimulatory effect of calcium on aldosterone secretion by parathyroid hormone. Miner Electrolyte Metab. (1994) 20:309–14.
110. Jespersen B, Pedersen EB, Charles P, Danielsen H, Juhl H. Elevated angiotensin II and vasopressin in primary hyperparathyroidism. Angiotensin II infusion studies before and after removal of the parathyroid adenoma. Acta Endocrinol. (1989) 120:362–8. doi: 10.1530/acta.0.1200362
111. Akmal M, Barndt RR, Ansari AN, Mohler JG, Massry SG. Excess PTH in CRF induces pulmonary calcification, pulmonary hypertension and right ventricular hypertrophy. Kidney Int. (1995) 47:158–63. doi: 10.1038/ki.1995.18
112. de Man FS, Tu L, Handoko ML, Rain S, Ruiter G, François C, et al. Dysregulated renin-angiotensin-aldosterone system contributes to pulmonary arterial hypertension. Am J Respir Crit Care Med. (2012) 186:780–9. doi: 10.1164/rccm.201203-0411OC
113. Bernini G, Moretti A, Lonzi S, Bendinelli C, Miccoli P, Salvetti A. Renin-angiotensin-aldosterone system in primary hyperparathyroidism before and after surgery. Metab Clin Exp. (1999) 48:298–300. doi: 10.1016/S0026-0495(99)90075-6
114. Broulik PD, Horký K, Pacovský V. Blood pressure in patients with primary hyperparathyroidism before and after parathyroidectomy. Exp Clin Endocrinol. (1985) 86:346–52. doi: 10.1055/s-0029-1210507
115. Pacifici R, Perry HM, Shieber W, Biglieri E, Droke DM, Avioli LV. Adrenal responses to subtotal parathyroidectomy for primary hyperparathyroidism. Calcif Tissue Int. (1987) 41:119–23. doi: 10.1007/BF02563790
116. Salahudeen AK, Thomas TH, Sellars L, Tapster S, Keavey P, Farndon JR, et al. Hypertension and renal dysfunction in primary hyperparathyroidism: effect of parathyroidectomy. Clin Sci. (1989) 76:289–96. doi: 10.1042/cs0760289
117. Richards AM, Espiner EA, Nicholls MG, Ikram H, Hamilton EJ, Maslowski AH. Hormone, calcium and blood pressure relationships in primary hyperparathyroidism. J Hypertens. (1988) 6:747–52. doi: 10.1097/00004872-198809000-00009
118. Massfelder T, Taesch N, Fritsch S, Eichinger A, Barthelmebs M, Stewart AF, et al. Type 1 parathyroid hormone receptor expression level modulates renal tone and plasma renin activity in spontaneously hypertensive rat. J Am Soc Nephrol. (2002) 13:639–48.
119. Fritsch S, Lindner V, Welsch S, Massfelder T, Grima M, Rothhut S, et al. Intravenous delivery of PTH/PTHrP type 1 receptor cDNA to rats decreases heart rate, blood pressure, renal tone, renin angiotensin system, and stress-induced cardiovascular responses. J Am Soc Nephrol. (2004) 15:2588–600. doi: 10.1097/01.ASN.0000141040.77536.AF
120. Broulik PD, Brouliková A, Adámek S, Libanský P, Tvrdon J, Broulikova K, et al. Improvement of hypertension after parathyroidectomy of patients suffering from primary hyperparathyroidism. Int J Endocrinol. (2011) 2011:309068. doi: 10.1155/2011/309068
121. Kovács L, Góth MI, Szabolcs I, Dohán O, Ferencz A, Szilágyi G. The effect of surgical treatment on secondary hyperaldosteronism and relative hyperinsulinemia in primary hyperparathyroidism. Eur J Endocrinol. (1998) 138:543–7. doi: 10.1530/eje.0.1380543
122. Diamond TW, Botha JR, Wing J, Meyers AM, Kalk WJ. Parathyroid hypertension. A reversible disorder. Arch Intern Med. (1986) 146:1709–12. doi: 10.1001/archinte.1986.00360210073012
123. Stefenelli T, Abela C, Frank H, Koller-Strametz J, Globits S, Bergler-Klein J, et al. Cardiac abnormalities in patients with primary hyperparathyroidism: implications for follow-up. J Clin Endocrinol Metab. (1997) 82:106–12. doi: 10.1210/jcem.82.1.3666
124. Piovesan A, Molineri N, Casasso F, Emmolo I, Ugliengo G, Cesario F, et al. Left ventricular hypertrophy in primary hyperparathyroidism. Effects of successful parathyroidectomy. Clin Endocrinol. (1999) 50:321–8. doi: 10.1046/j.1365-2265.1999.00651.x
125. Zawada ET, Johnson M. Effects of changes in serum calcium and parathyroid hormone on plasma renin in intact mongrel dogs. Nephron. (1985) 40:368–71. doi: 10.1159/000183495
126. McCarron DA, Pingree PA, Rubin RJ, Gaucher SM, Molitch M, Krutzik S. Enhanced parathyroid function in essential hypertension: a homeostatic response to a urinary calcium leak. Hypertension. (1980) 2:162–8. doi: 10.1161/01.HYP.2.2.162
127. Blackwood AM, Sagnella GA, Cook DG, Cappuccio FP. Urinary calcium excretion, sodium intake and blood pressure in a multi-ethnic population: results of the Wandsworth Heart and Stroke Study. J Hum Hypertens. (2001) 15:229–37. doi: 10.1038/sj.jhh.1001171
128. Chhokar VS, Sun Y, Bhattacharya SK, Ahokas RA, Myers LK, Xing Z, et al. Loss of bone minerals and strength in rats with aldosteronism. Am J Physiol Heart Circ Physiol. (2004) 287:H2023–6. doi: 10.1152/ajpheart.00477.2004
129. Kesteloot H, Tzoulaki I, Brown IJ, Chan Q, Wijeyesekera A, Ueshima H, et al. Relation of urinary calcium and magnesium excretion to blood pressure: the international study of macro- and micro-nutrients and blood pressure and the international cooperative study on salt, other factors, and blood pressure. Am J Epidemiol. (2011) 174:44–51. doi: 10.1093/aje/kwr049
130. Resnick LM, Laragh JH. Calcium metabolism and parathyroid function in primary aldosteronism. Am J Med. (1985) 78:385–90. doi: 10.1016/0002-9343(85)90328-6
131. Grant FD, Mandel SJ, Brown EM, Williams GH, Seely EW. Interrelationships between the renin-angiotensin-aldosterone and calcium homeostatic systems. J Clin Endocrinol Metab. (1992) 75:988–92. doi: 10.1210/jcem.75.4.1400892
132. Atchison DK, Beierwaltes WH. The influence of extracellular and intracellular calcium on the secretion of renin. Pflugers Arch. (2013) 465:59–69. doi: 10.1007/s00424-012-1107-x
133. Porter L, Conlin PR, Scott J, Brown EM, El-Hajj Fuleihan G. Calcium modulation of the renin-aldosterone axis. J Endocrinol Invest. (1999) 22:115–21. doi: 10.1007/BF03350890
Keywords: renin-angiotensin-aldosterone system, aldosterone, angiotensin II, primary aldosteronism, parathyroid hormone, primary hyperparathyroidism
Citation: Zheng M-H, Li F-X-Z, Xu F, Lin X, Wang Y, Xu Q-S, Guo B and Yuan L-Q (2020) The Interplay Between the Renin-Angiotensin-Aldosterone System and Parathyroid Hormone. Front. Endocrinol. 11:539. doi: 10.3389/fendo.2020.00539
Received: 04 February 2020; Accepted: 03 July 2020;
Published: 20 August 2020.
Edited by:
Marcus M. Seldin, University of California, Irvine, United StatesReviewed by:
Jonathan Paul Mochel, Iowa State University, United StatesLuigi Petramala, Sapienza University of Rome, Italy
Claudio Letizia, Sapienza University of Rome, Italy
Copyright © 2020 Zheng, Li, Xu, Lin, Wang, Xu, Guo and Yuan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ling-Qing Yuan, YWxsZW55bHFAY3N1LmVkdS5jbg==