 Debapriya Basu
Debapriya Basu Karin E. Bornfeldt
Karin E. Bornfeldt- 1Division of Endocrinology, Diabetes and Metabolism, New York University School of Medicine, New York, NY, United States
- 2Department of Medicine, University of Washington Medicine Diabetes Institute, University of Washington School of Medicine, Seattle, WA, United States
- 3Department of Pathology, University of Washington Medicine Diabetes Institute, University of Washington School of Medicine, Seattle, WA, United States
Human studies support a strong association between hypertriglyceridemia and atherosclerotic cardiovascular disease (CVD). However, whether a causal relationship exists between hypertriglyceridemia and increased CVD risk is still unclear. One plausible explanation for the difficulty establishing a clear causal role for hypertriglyceridemia in CVD risk is that lipolysis products of triglyceride-rich lipoproteins (TRLs), rather than the TRLs themselves, are the likely mediators of increased CVD risk. This hypothesis is supported by studies of rare mutations in humans resulting in impaired clearance of such lipolysis products (remnant lipoprotein particles; RLPs). Several animal models of hypertriglyceridemia support this hypothesis and have provided additional mechanistic understanding. Mice deficient in lipoprotein lipase (LPL), the major vascular enzyme responsible for TRL lipolysis and generation of RLPs, or its endothelial anchor GPIHBP1, are severely hypertriglyceridemic but develop only minimal atherosclerosis as compared with animal models deficient in apolipoprotein (APO) E, which is required to clear TRLs and RLPs. Likewise, animal models convincingly show that increased clearance of TRLs and RLPs by LPL activation (achieved by inhibition of APOC3, ANGPTL3, or ANGPTL4 action, or increased APOA5) results in protection from atherosclerosis. Mechanistic studies suggest that RLPs are more atherogenic than large TRLs because they more readily enter the artery wall, and because they are enriched in cholesterol relative to triglycerides, which promotes pro-atherogenic effects in lesional cells. Other mechanistic studies show that hepatic receptors (LDLR and LRP1) and APOE are critical for RLP clearance. Thus, studies in animal models have provided additional mechanistic insight and generally agree with the hypothesis that RLPs derived from TRLs are highly atherogenic whereas hypertriglyceridemia due to accumulation of very large TRLs in plasma is not markedly atherogenic in the absence of TRL lipolysis products.
Introduction
Whether elevated plasma triglycerides play a causal role in exacerbating cardiovascular disease (CVD) or represent a biomarker of CVD risk has been debated for 4 decades (1). In this comprehensive review organized in four major areas, we focus on how human research has guided mechanistic research in animal models of hypertriglyceridemia and potential effects of triglyceride (TG) modulation on atherosclerosis. These areas include defining (i) hypertriglyceridemia including origin and types of TG-rich lipoproteins (TRLs) and their remnants; (ii) current knowledge on association of hypertriglyceridemia with CVD risk in humans; (iii) animal models of hypertriglyceridemia and effects on atherosclerosis; and (iv) emerging strategies and drug targets to lower TGs and prevent atherosclerosis.
What Is Hypertriglyceridemia?
Hypertriglyceridemia is often a biochemical diagnosis based on the levels of fasting plasma triglycerides (2). Normal fasting plasma TG levels are defined by current clinical guidelines as <1.69 mM (<150 mg/dL). Fasting TG levels of 1.69–2.25 mM (150–199 mg/dL) are considered moderately elevated, while fasting TGs above 2.26 or 2.83 mM (200 or 250 mg/dL) are considered high, and fasting TG levels above 5.65 mM (500 mg/dL) are considered severely elevated (2). An increased risk of acute pancreatitis is a consideration when TGs reach severely elevated levels (typically > 10 mM; close to and above 900 mg/dL) (3). Hypertriglyceridemia can be classified as primary types when a genetic susceptibility contribution is present and secondary types when no genetic component is detected (3). Secondary hypertriglyceridemia associates with poor diet, alcohol use, obesity, metabolic syndrome, and diabetes (2). Hypertriglyceridemia is often multifactorial, caused by genetic factors along with non-genetic factors favoring increased TRL secretion and/or reduced TRL clearance. Some genetic factors lead to hypertriglyceridemia only when associated with a second genetic or environmental factor, such as is the case for remnant removal disease (also known as type III hyperlipoproteinemia, broad band beta disease, or dysbetalipoproteinemia), which is most often caused by homozygosity of the APOE2 genotype in combination with hormonal and/or environmental factors (4). Drugs like thiazides, beta-blockers, oral estrogens, tamoxifen, oral contraceptive pills, corticosteroids, atypical antipsychotics, isotretinoin, bile acid binding resins, anti-retroviral protease inhibitors, and immunosuppressive agents, such as sirolimus (mTOR inhibitors) also increase plasma TG levels (5).
TGs are present in circulation mainly in the cores of the TRLs—chylomicrons and very low-density lipoproteins (VLDL) and their remnant lipoprotein particles [RLPs, which include intermediate-density lipoproteins (IDLs) derived from VLDL]. Chylomicrons are synthesized and secreted from the intestine after a meal, whereas VLDLs are produced by the liver. Chylomicrons contain APOB48, a truncated form of APOB100, as the main apolipoprotein. Elevated TGs or hypertriglyceridemia can be due to greater intestinal and/or hepatic secretion of TRLs or reduced clearance of TRLs from the circulation (Figure 1). TRLs and their RLPs are enriched in apolipoprotein (APO) E, a ligand recognized by receptors of the low-density lipoprotein receptor (LDLR) family, including LDLR and the low-density lipoprotein receptor-related protein 1 (LRP1) receptor in the liver (6–8). While LDLR binds to, and participates in clearance of LDL by binding APOB100 (9), APOE-containing TRL and RLP particles are cleared through both LRP1 and LDLR in the liver.
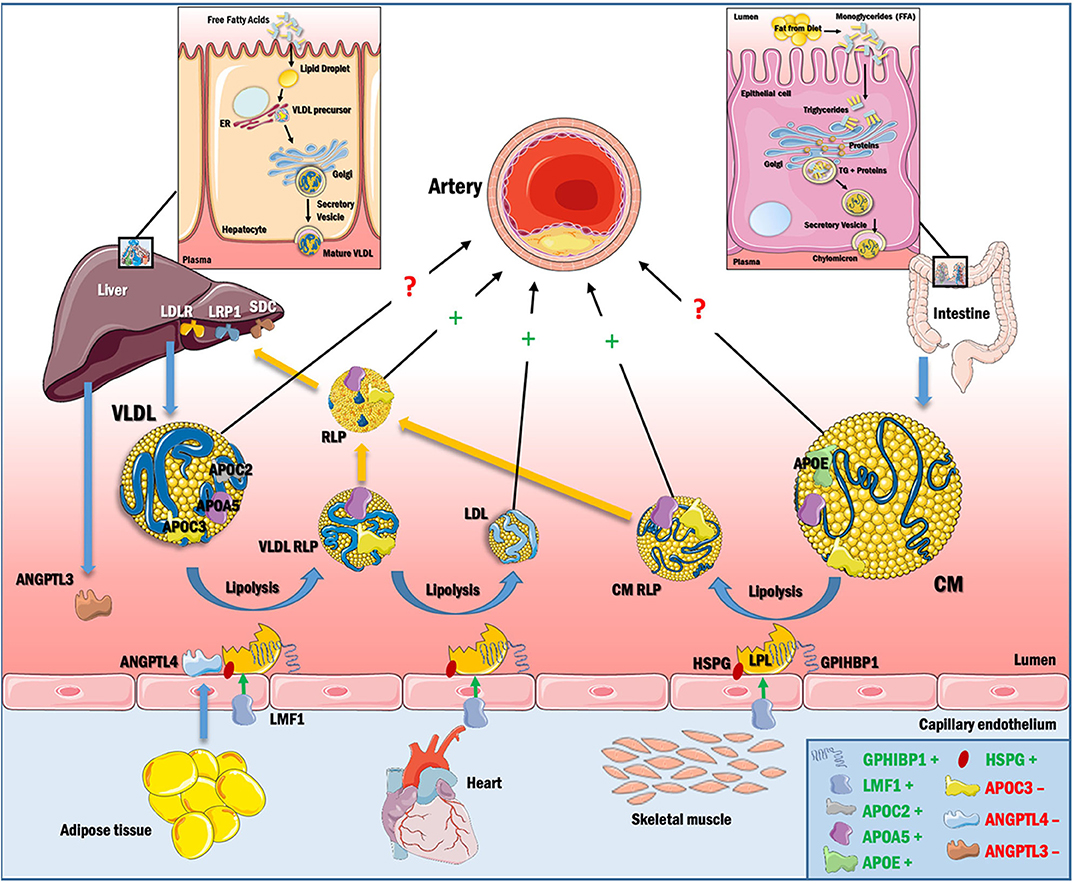
Figure 1. TRL synthesis and catabolism. Dietary TGs are absorbed and packaged as chylomicrons in the intestine, and secreted into the circulation via the lymphatic system. Chylomicrons contain APOB48 and acquire APOC2, APOC3, and APOE, some of which are transferred to other lipoproteins (primarily HDL) during lipolysis. LPL is synthesized in the parenchyma of heart, adipose and skeletal muscle and is further stabilized by LMF1. Active LPL remains anchored to GPIHBP1 and heparan sulfate proteoglycans (HSPGs). APOC2 and APOA5 activate LPL and help in chylomicron hydrolysis, releasing FFA for cellular energy requirements and cholesteryl ester-rich chylomicron remnants (RLPs), which are cleared by hepatic receptors in the LDL receptor family (LDLR and LRP1) with contributions from the HSPG syndecan (SDC) or are trapped in the artery wall. VLDL is synthesized in the liver using free fatty acid (FFA) from adipose tissue or from de novo lipogenesis and is then secreted into circulation. VLDL contains APOB100 on its surface as the major apolipoprotein. VLDL is hydrolyzed by LPL, creating VLDL RLPs, which accumulate in the artery wall or are further converted to LDL, the most atherogenic lipoprotein known. VLDL RLPs can also be removed by hepatic receptors or can be taken up by macrophages in arteries and other tissues. APOC3 reduces VLDL and chylomicron lipolysis by inhibiting LPL and by blocking TRL and RLP uptake by hepatic receptors. Hypertriglyceridemia thus results from increased production or decreased catabolism of chylomicrons and/or VLDL, and has a direct effect on the composition of LDL and HDL. RLPs are capable of penetrating the vascular endothelium and initiate the events of atherogenesis. The direct role of large VLDL and chylomicron in atherogenesis is however unclear, as it appears that these particles are too large to effectively enter the artery wall. In box, +denotes positive regulation of LPL and −denotes negative regulation of LPL.
A combination of human genetic approaches, including the study of genes that encode proteins identified by classical biochemistry, genes identified through genome-wide association studies (GWAS) of cohorts with a range of TGs, and genes identified by studies of families with TG phenotypes, have revealed that genetic factors contribute to a relatively large proportion of variation in plasma TG. In particular, homozygous or compound heterozygous mutations in genes that alter lipoprotein lipase (LPL) activity, like LPL itself, APOC2, LMF1 (lipase maturation factor 1), GPIHBP1 (glycosylphosphatidylinositol anchored high density lipoprotein binding protein 1), APOA5, and GPD1 (encoding glycerol-3-phosphate dehydrogenase 1) are associated with high TG, although both severe forms of hypertriglyceridemia and moderate-to-mild hyperglycemia are usually of polygenic origin (10, 11). Bi-allelic pathogenic mutations in LPL, APOC2, GPIHBP1, APOA5, or LMF1, that lead to reduced LPL action, are used to confirm familial chylomicronemia syndrome, a rare autosomal recessive disorder characterized by severe elevation of TG levels, eruptive cutaneous xanthomas and acute pancreatitis (12). Furthermore, in some patients with chylomicronemia, autoantibodies against GPIHBP1 have been identified. These autoantibodies block the binding of LPL to GPIHBP1, leading to reduced LPL activity (13).
LPL is the major vascular enzyme catalyzing TG hydrolysis, generating free fatty acids and monoacylglycerol for tissue use, and RLPs. Hence, LPL is the principal player in the clearance of circulating TRLs. The binding of LPL to GPIHBP1 focuses the intravascular hydrolysis of TRLs on the surface of capillary endothelial cells in e.g., adipose tissue, skeletal muscle, and heart (14). LPL's action is modulated by activators like APOC2 and APOA5, inhibitors like APOC3 and angiopoietin-like proteins (ANGPTL3 and ANGPTL4), proper trafficking by LMF1 and anchoring by GPIHBP1, as shown schematically in Figure 2.
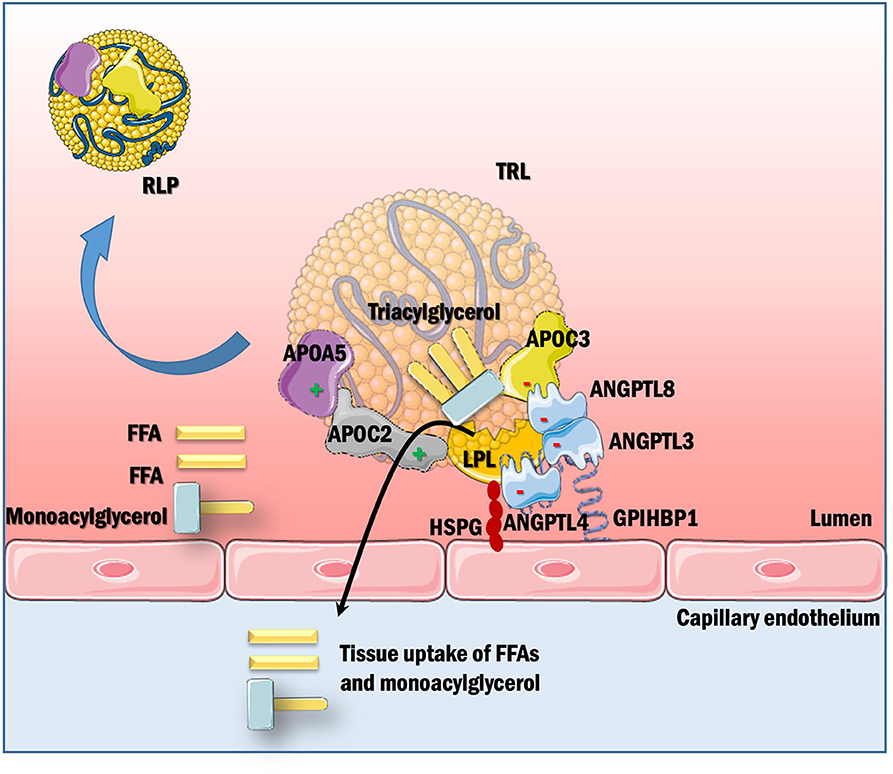
Figure 2. Posttranslational regulation of LPL-mediated TRL hydrolysis. LPL, synthesized in parenchymal cells of certain metabolic tissues, is secreted into the sub-endothelial space. GPIHBPI, expressed solely in capillary endothelial cells, is present on the basolateral side of endothelium where it captures LPL from the interstitial space and shuttles it across the endothelial cells to the luminal side of the capillary, which is the site of LPL action. This interaction with GPIHBP1 helps in TRL margination across the capillaries by enhancing lipolysis. LPL activity is further modulated by apolipoproteins within TRLs as well as secreted factors like angiopoietins. APOC2 is a vital cofactor for LPL activation, whereas APOC3 inhibits lipolysis and uptake of remnants in the liver. APOA5 acts to stabilize the LPL-APOC2 complex by helping TRLs to bind to the endothelial cell surface via HSPGs. ANGPTL3, ANGPTL4, and ANGPTL8 inhibit LPL depending on tissues and nutritional state. LPL hydrolyzes TRLs to generate free fatty acids (FFA) and monoacylglycerol, which are taken up by cells for their energy requirements. This process also generates RLPs, which contain a relative increase in cholesteryl ester and reduction in TGs as compared with their parent TRLs, and are considered to be highly atherogenic.
Additionally, receptor-mediated capture of RLPs produced after lipolysis by hepatic LDLR, LRP1 and syndecan 1 contributes to maintaining plasma TG levels (Figure 1).
Hypertriglyceridemia Associates With Cardiovascular Disease Risk in Humans But Direct Evidence of Causality Is Scarce
Elevated plasma TGs have been known to associate with an increased risk of atherosclerotic CVD for decades. The first clues emerged over 40 years ago from the finding that patients with familial forms of hypertriglyceridemia exhibited an increased risk of coronary artery disease (15). Large epidemiological studies support the association between elevated TG levels and increased CVD risk (16–18), although the risk associated with elevated TGs is attenuated when adjusted for potential confounders, such as HDL-cholesterol (HDL-C) (19). Therefore, a causal link for this association has been challenging to determine, unlike increased LDL-cholesterol (LDL-C), which is a well-established causal risk factor for CVD. The difficulty determining a causal role for TGs in CVD in humans is in part due to the fact that plasma TG levels correlate negatively with HDL-C and can be present with high LDL-C, confounding its independent effect on CVD risk. Some studies however suggest association of elevated TG levels and CVD risk independent of LDL-C. For example, a recent study demonstrated that in older statin-treated subjects, elevated TG levels were associated with higher incidence of recurrent myocardial infarction in patients with LDL <100 mg/dL but not in those with LDL ≥100 mg/dL (20).
Weakening the case for a causal role of hypertriglyceridemia as a CVD risk factor are the clinical trials of TG lowering, which have mostly resulted in a lack of effect on CVD outcomes. Thus, fibrates and omega-3 fatty acids, which lower plasma TGs, have been mostly negative for CVD benefit (18), although subgroup analysis showed protective effects in subjects with high TGs and low HDL-C in some trials (18). So far, the only trials that produced a significantly beneficial effect on CVD outcomes were the REDUCE-IT (21) and JELIS (22) trials, in which subjects were treated with eicosapentaenoic acid (one of the omega-3 fatty acids). However, uncertainty exists regarding the mechanism of action of the eicosapentaenoic acid, as it has been suggested that the cardiobeneficial effects were likely due, at least in part, to mechanisms other than TG lowering (21). The negative results of the TG-lowering trials could have resulted from inclusion of individuals without hypertriglyceridemia in most of the fibrate trials, use of inadequate doses in most of the omega-3 fatty acid trials, or heterogeneity in the atherogenicity of the TG-carrying lipoproteins, at least in terms of TG as a marker of CVD risk (23). It is also possible that elevated TG levels are a general sign of the presence of a more atherogenic lipoprotein particle, such as the RLP. Because RLPs are lipolysis products of TRLs and therefore contain less TG/particle than their parent TRL, total plasma TG levels do not necessarily correlate with RLP levels (23).
Thus, although the association between hypertriglyceridemia and CVD risk in humans is strong, causal evidence is so far scarce. In fact, subjects with familial chylomicronemia syndrome, which is due to pathogenic mutations in genes controlling LPL itself or LPL activity, do not usually exhibit an elevated risk of atherosclerotic CVD despite the very high levels of TGs (12, 24). However, Mendelian randomization analyses indicate that TG-lowering LPL variants and LDL-C-lowering LDLR variants are associated with a similarly lower risk of coronary heart disease per unit difference in APOB (25). Thus, the relationship between plasma TG levels and atherosclerotic CVD is not straightforward, and high TG levels do not necessarily predict an increased CVD risk.
What Are Remnant Lipoprotein Particles and Do They Promote Atherosclerosis?
The complex relationship between elevated TGs and atherosclerosis may be explained by a model in which increased atherosclerosis (and CVD) is not caused by the high TG content/lipoprotein particle, but to the presence of RLPs (chylomicron and VLDL lipolysis products), which are smaller than chylomicrons and VLDL and can more effectively exert pro-atherogenic effects in the artery wall (12). As described above, these RLPs are generated as LPL hydrolyzes TGs in the core of chylomicrons and VLDL, resulting in progressively smaller and TG-depleted particles with an increased ratio of cholesterol to TG. Although there is no universally agreed upon definition of RLPs, there is general agreement that these particles vary in size and composition. A slower clearance rate of RLPs, e.g., due to reduced LPL activity and incomplete conversion of TRLs to lipoproteins that have the greatest affinity for hepatic lipoprotein removal pathways, is thus predicted to result in a prolonged life-time of a spectrum of sizes of RLPs in circulation. These features have made it difficult to accurately quantify RLPs and assess their roles in CVD (23).
The concept that RLPs are highly atherogenic is supported by the finding that patients with remnant removal disease exhibit an increased risk of premature atherosclerotic CVD (26, 27). This condition is due to an impaired ability to clear RLPs through hepatic receptors of the LDLR family, which have a lower affinity for the APOE2 isoform of APOE (23). Because RLPs are larger than LDL, they contain 5–20 times more cholesterol per particle than does LDL (28). Once in the subendothelial space, RLPs are trapped by proteoglycans (18, 29) and can be taken up by lesional macrophages (30, 31). Furthermore, native unmodified RLPs promote rapid cholesterol accumulation in macrophages, in contrast to LDL, which has to be modified by e.g., oxidation to be taken up effectively by macrophages (30, 31). RLPs might therefore be more effective on a per particle basis than LDL at producing macrophage foam cells, a key cell type in atherosclerosis.
While these findings implicate RLPs in promoting atherosclerosis, their exact role in the atherogenic process is still unclear. However, the findings discussed above suggest that lower TG levels might be associated with an increased CVD risk, as compared with severe hypertriglyceridemia, if accompanied by elevated levels of more atherogenic RLPs derived from TRLs.
In order to tackle the issue of whether TRLs or RLPs are causatively involved in exacerbating atherosclerosis, animal models have been used to investigate potential atherogenic effects of TRLs and RLPs and to provide mechanistic insight through an approach that has sometimes been termed “reverse translation,” “human-first,” or “bedside-to-bench” research, which begins with observations in the clinic and works “backwards” to uncover the mechanisms behind the clinical observations (32). In fact, the first genetic mouse model of atherosclerosis with advanced lesions—the APOE-deficient mouse—was generated in the Breslow and Maeda laboratories in the early 1990's in part to mimic the impaired clearance of RLPs and severe premature coronary and peripheral vessel risk observed in patients with remnant removal disease (33, 34).
What Have We Learned From Animal Models?
Like the human studies of subjects with different genetic alterations that are associated with changes in plasma TRLs and RLPs, studies in different animal models generally support the hypothesis that RLPs derived from TRLs are highly atherogenic whereas high TG levels due to accumulation of very large TRLs are much less atherogenic. Thus, the APOE-deficient (Apoe−/−) mouse exhibits a dramatic atherogenic phenotype concomitant with a lipoprotein profile enriched in VLDL and RLPs even when fed a low-fat diet (33), whereas LPL-deficient mice and GPIHBP1-deficient mice, which have very high TG levels due to lack of lipolysis and generation of RLPs exhibit delayed formation of small atherosclerotic lesions (35, 36). Likewise, high-cholesterol diet-fed rabbits support the concept that accumulation of RLPs relatively enriched in cholesterol and depleted of TGs lead to atherosclerosis whereas increased levels of large TRLs cause less atherogenesis (37–39).
The most common animal models of hypertriglyceridemia and the evidence emerging from each of these models related to atherosclerosis are discussed below and are summarized in Table 1. Further research using various animal models and mechanistic ex vivo studies is likely to shed important new light on mechanisms whereby RLPs promote atherosclerosis.

Table 1. Animal models of altered TG levels and effects on atherosclerosis.
Animal Models of Hypertriglyceridemia and Their Susceptibility to Atherosclerosis
Monogenic and Digenic Mouse Models of Hypertriglyceridemia
APOE-Deficiency
The Apoe−/− mouse model has been extensively used as a model of atherosclerosis since its generation (33, 34). APOE, which is enriched in TRLs and RLPs, is recognized by the hepatic LRP1 and LDL receptors, leading to clearance of TRLs and RLPs (Figure 1). Apoe−/− mice fed low-fat chow diet have lipoprotein profiles enriched in VLDL-cholesterol and RLP (IDL)-cholesterol when analyzed by fast phase liquid chromatography (33). The LDL receptor-deficient mouse (Ldlr−/− mouse), which was generated as a model of homozygous familial hypercholesterolemia (caused by LDL receptor mutations in humans), on the other hand, exhibits an increase primarily in LDL-cholesterol when fed a chow diet, reflecting impaired clearance of LDL through hepatic LDL receptors (64). These Ldlr−/− mice are largely free of atherosclerosis when fed a chow diet (64). The differences in severity of atherosclerosis phenotypes between chow-fed Apoe−/− mice and Ldlr−/− mice are most likely due to the elevated levels of TRLs and RLPs in Apoe−/− mice, as compared with Ldlr−/− mice, which in turn is due to the severely impaired hepatic clearance of APOE-containing TRLs and RLPs in Apoe−/− mice. However, APOE also likely prevents atherosclerosis in part through its anti-inflammatory effects (65). Furthermore, the finding that mice deficient in both APOE and LDLR (Apoe−/− Ldlr−/− mice) exhibit higher levels of VLDL and chylomicron RLPs than do Ldlr−/− mice, revealed that TRLs and RLPs are also cleared through a receptor (LRP1) distinct from the LDLR (64). The Apoe−/− mouse also shows higher levels of APOB48 (the truncated form of APOB100 associated primarily with chylomicrons and their remnants) whereas the Ldlr−/− mouse has primarily APOB100 in plasma (64). Thus, both forms of APOB are atherogenic. When fed a Western-style high-fat diet, Apoe−/− mice develop markedly higher levels of VLDL and IDL, and increased atherosclerosis as compared to Apoe−/− mice fed a chow diet (33).
As mentioned above, TRLs and RLPs are cleared by both the LDLR and LRP1 in liver. This conclusion is supported by the finding that inducible liver-specific LRP1-deficiency is sufficient to increase plasma chylomicron RLP levels, but only when the mice are also deficient in LDLR (7, 66, 67). Subsequent studies demonstrated that hepatic expression of LRP1 protects from atherosclerosis through additional mechanisms independent of clearance of APOE-containing RLPs because liver-targeted LRP1 deletion in Apoe−/− Ldlr−/− mice increased atherosclerosis (68).
Together, these studies show that loss of APOE leads to a dramatic increase in atherosclerosis in mouse models, an effect that appears to be mediated in large part by the increased plasma levels of TRLs and RLPs. Furthermore, mouse studies have shown that these APOE-containing TRLs are cleared from circulation through the hepatic receptors LDLR and LRP1.
APOE*3-Leiden Transgenic Mice
APOE has three major alleles; APOE2, APOE3, and APOE4. A mutant APOE3 allele that has a lower LDLR receptor affinity than wildtype APOE3 has been identified. This APOE*3-Leiden gene (* denotes the mutant allele), which is associated with a dominantly inherited form of remnant removal disease was first described in a large Dutch family (69) and is very rare as compared with the APOE2/APOE2 genotype more often associated with remnant removal disease (69). Transgenic mice harboring this mutant allele were generated by introducing a human APOE*3-Leiden gene construct isolated from the APOE*3-Leiden proband, consisting of the APOE*3-Leiden and APOC1 genes and a promoter element that regulates the expression of the APOE and APOC1 genes (70). These mice exhibited significantly elevated levels of total plasma cholesterol and TGs on a regular chow diet. When fed a cholesterol-rich diet, total plasma cholesterol and TG levels increased dramatically. This increase was observed mainly in the VLDL and LDL-sized particles (70). On a high fat/high cholesterol diet, the APOE*3-Leiden mice develop atherosclerotic lesions in the aortic arch, the descending aorta, and the carotid arteries, with characteristics of human vascular pathology, varying from fatty streaks to mild, moderate, and severe plaques (40). In another study, the effect of monocyte/macrophage-expression of APOE and APOE*3-Leiden was investigated by transplanting bone marrow into atherosclerosis-prone Apoe−/− mice. APOE*3-Leiden transplanted bone marrow was less effective in reducing atherosclerosis, as compared with bone marrow cells expressing wildtype murine APOE (41). The APOE*3-Leiden transgenic mouse model is perhaps the most relevant model of impaired RLP removal and resulting increased atherosclerosis. However, the presence of the APOC1 transgene makes data generated by this model as far as the contribution of the APOE3 mutation to atherosclerosis difficult to interpret because APOC1 aggravates atherosclerosis and increases plasma TGs and cholesterol (71), maybe in part through its ability to inhibit LPL activity (72) and reduce hepatic VLDL clearance (73).
An APOE*3-Leiden mouse model lacking the APOC1 region has also been generated (73). However, this mouse model needs further characterization in terms of atherosclerotic lesion development.
Lipoprotein Lipase-Deficiency
As discussed above, LPL is the primary rate-limiting enzyme for hydrolysis of TGs present in the core of chylomicrons, VLDL, and RLPs, generating free fatty acids and monoacylglycerol, which are taken up and used by extrahepatic tissues (Figure 2). Through lipolysis, LPL-mediated catabolism of TRLs in the bloodstream also generates progressively smaller and cholesterol-enriched (TG-depleted) atherogenic lipoprotein particles, such as chylomicron RLPs, VLDL RLPs, and LDL (74, 75). LPL is also vital for generation of HDL (76, 77). This is because as TGs in TRLs are lipolyzed, a significant proportion of the excess phospholipids and cholesterol of the shrinking TRL particle are transferred to HDL, together with APOC apolipoproteins (76) and because of reduced catabolism of APOA1 (the main structural protein of HDL) (78). Therefore, low LPL activity leads to hypertriglyceridemia and low HDL levels.
LPL is synthesized in the parenchymal cells of adipose tissue, skeletal muscle and heart, and is secreted into interstitial spaces, where it is captured by the capillary endothelial cell protein GPIHBP1, and transported to the capillary lumen (2). LPL is also synthesized by lesional macrophages (79). LPL is actively regulated by numerous proteins (Figure 2). Based on previous work, LPL has been assumed to be active as a homodimer, however, a recent study shows that that LPL and GPIHBP1-bound LPL can be active in a monomeric state (80).
Deficiency in LPL activity in humans (type I hyperlipoproteinemia or familial chylomicronemia syndrome) is associated with massive chylomicronemia, low HDL-C levels, and recurrent attacks of pancreatitis when not controlled by a strict diet (81). In contrast to humans, homozygous LPL-deficient mice do not survive suckling and die between 18 and 24 h after birth. Adenovirus-based transient expression of LPL during the suckling period was used in an effort to rescue these LPL-deficient mice. After a single intraperitoneal injection of LPL-expressing virus immediately after birth, more than 90% of the LPL-deficient pups survived the first day of life (82). Furthermore, 3% of these mice survived the entire suckling period and lived for up to 20 months, although LPL activity in mouse tissues and in plasma from mice injected with heparin to liberate LPL tethered to endothelial cells (postheparin plasma) was undetectable in all animals after 6 weeks of age. Adult LPL-deficient mice are smaller than their littermates until 2–3 months of age and exhibit very high TG levels in the fed (5,000 vs. 100 mg/dL in wildtype controls) and fasted state (2,000 vs. 70 mg/dL in wildtype controls). LPL-deficient mice differ from LPL-deficient humans in that they do not develop pancreatitis even when very high plasma TG levels are present.
Because of the drawbacks of the whole-body LPL-deficient mouse model discussed above, several tissue-selective LPL-deletion models and other approaches to reduce LPL expression have been used. For example, skeletal muscle specific- or endothelial cell-specific overexpression of an LPL minigene has been shown to rescue Lpl−/− mice from neonatal lethality (83, 84). Heart-specific LPL expression to levels found in wildtype mice prevents lethality and leads to almost complete normalization of circulating TG levels in mice deficient in skeletal muscle and adipose tissue LPL (85) whereas cardiac-specific deletion of LPL leads to only mildly elevated plasma TGs (86). Whole-body inducible LPL-deficient mice have also been generated. These mice are viable when LPL deletion is induced in adult mice (87). On a chow diet, these mice develop high TGs (~500 mg/dL) and when fed a high-fat diet, TG levels can reach 3,000 mg/dL (67, 87).
Aged LPL-deficient mice (15 month-old) spontaneously develop early fatty streaks when fed a chow diet (35), suggesting that the high levels of TRLs in these mice can promote some atherosclerosis even in the absence of RLP generation, consistent with the hypothesis that large TRLs can be modestly atherogenic. Whole-body LPL-deficient mice have never been crossed to Ldlr−/− or Apoe−/− mice, so the phenotype is still unknown. Recently, mice with inducible LPL-deficiency have been used to study regression of lesions of atherosclerosis. These studies showed that near-complete loss of LPL caused elevated TG levels of ~500 mg/dL, but did not impede regression of atherosclerosis measured as lesional macrophage content (42), supporting the hypothesis that large TRLs in the absence of RLP generation are not markedly atherogenic. Consistently, heterozygous LPL-deficient mice crossed with Ldlr−/− mice and fed a high-fat diet are hypertriglyceridemic, as compared with Ldlr−/− controls but exhibit no increase in atherosclerosis progression (88). The authors suggested that the results could be explained by the loss of LPL in the vascular wall, which would prevent the action of pro-atherogenic lipolysis products, including atherogenic RLPs, locally in the lesion (89–91). Thus, macrophage-targeted deficiency of LPL in mice reduces atherosclerotic lesions (90, 91).
The direct relationship between LPL and atherosclerosis is complex. Ldlr−/− mice as well as Apoe−/− mice overexpressing LPL are resistant to diet-induced atherosclerosis, due to the efficient clearance of TRLs and RLPs in these mice (43, 44). Furthermore, it is probable that the RLPs are different in Ldlr−/− mice vs. Apoe−/− mice with normal LPL or supraphysiological levels of LPL. In Ldlr−/− mice, which express APOE, the increased lipolysis due to LPL overexpression generates smaller, TG-poor and cholesterol-poor VLDL RLPs, which are cleared faster. In Apoe−/− mice with LPL overexpression, VLDL remnant TG is lower but cholesterol content is unchanged. The exact reason for this difference is unknown. However, lack of APOE could interfere with RLP clearance. Although LPL overexpression is atheroprotective in both models, Ldlr−/− mice with LPL overexpression exhibit suppressed lesion formation 10 times greater than Apoe−/− mice with LPL overexpression. Another interesting perspective is that in Apoe−/− mice with or without LPL overexpression, non-HDL cholesterol levels are similar, yet the atherosclerosis is suppressed. This model represents one mouse model showing that lowering TRL and RLP levels is atheroprotective, independent of cholesterol levels. A similar example of such a model is the diabetes model in which APOC3 has been silenced, discussed below. These findings are consistent with the notion that TRLs/RLPs are pro-atherogenic even without changes in LDL.
The results from LPL-deficient mice and mice with induced LPL-deficiency, which do not readily develop atherosclerosis or exhibit impaired lesion regression, appear to be consistent with studies on homozygous LPL-deficient human subjects, whereof very few develop premature atherosclerosis (92–94), supporting the hypothesis that homozygous LPL deficiency causes accumulation of large TRLs which cannot effectively promote atherosclerosis. On the other hand, heterozygous LPL-deficiency is associated with increased ischemic heart disease, elevated risk of coronary atherosclerosis and diminished clinical event–free survival (95, 96). This evidence supports a second hypothesis, which is not incompatible with the first hypothesis, that partial loss of LPL is pro-atherogenic, possibly because lipolysis of TRLs is less efficient than when LPL is normally expressed, causing RLPs to circulate for longer periods of time and accumulate in the artery wall.
The difficulty in interpreting the phenotype of LPL-deficiency and partial LPL-deficiency stems from the known role of LPL in reduction of overall TG levels through increased RLP levels and subsequent clearance of TRLs and RLPs. Therefore, complete LPL-deficiency leads to increased levels of very large TRLs and reduced atherogenic RLPs, whereas a partial LPL-deficiency is likely to result in increased levels of atherogenic RLPs with an increased retention time in circulation and increased accumulation in the artery wall, as compared to wild type levels of LPL. Moreover, LPL expressed in macrophages has detrimental effects on atherosclerosis, which are distinct from its effects on TRL levels. There is no animal model as of yet, in which LPL-generated RLPs can be altered independently of TRLs.
Glycosylphosphatidylinositol-Anchored HDL-Binding Protein 1 (GPIHBP1)-Deficiency
GPIHBP1 is a GPI-anchored protein expressed in the capillaries of heart, adipose tissue, and skeletal muscle (97–99) (Figure 1). GPIHBP1, present at the basolateral side of the endothelial cell, captures LPL from the subendothelial spaces and transports it across the endothelial cell to the capillary lumen (100, 101). GPIHBP1 is critically important for the margination (the partitioning of large TRLs along the capillary endothelium) of lipoproteins by facilitating TRL hydrolysis by LPL (102). In addition, GPIHBP1 stabilizes the structure and catalytic activity of LPL (103, 104). Recently, GPIHBP1 proved to be crucial for solving the structure of LPL (105, 106).
Mice lacking Gpihbp1 have severe chylomicronemia due to a decreased ability of LPL to efficiently metabolize TRLs (97). Nearly all of the cholesterol and TGs in plasma of Gpihbp1−/− mice are associated with large TRLs 50–135 nm in diameter, leading to plasma TG levels close to 3,000 mg/dL, and both male and female Gpihbp1−/− mice spontaneously develop small atherosclerotic lesions in the aortic root when fed a chow diet (36), phenocopying the spontaneous atherosclerosis in chow-fed Lpl−/− mice (35). These lesions are much smaller than those that develop in Apoe−/− mice, which have an accumulation of RLPs. A recent study reported that combined GPIHBP1- and LDLR-deficiency results in more extensive and advanced atherosclerosis as well as rare lesions in coronary arteries in diabetic mice, as compared to Ldlr−/− controls (107). Therefore, the combination of severe hypertriglyceridemia, LDLR-deficiency and diabetes appears to worsen the effects of hypertriglyceridemia on atherogenesis.
Data obtained from studies of GPIHBP1-deficient mice support the proposal that large TRLs can be atherogenic but are much less atherogenic than RLPs and LDL.
Combined Lipase-Deficiency (Lipase Maturation Factor 1)
Combined lipase deficiency (cld) was identified in a spontaneous mouse mutant deficient in both LPL and hepatic lipase (HL) activity (108). These mice display postpartum chylomicronemia and death after nursing (108, 109). Although LPL protein is normally expressed in cld mutant mice, it fails to attain the catalytically active conformation and is subject to retention in the ER and subsequent degradation (110). The cld mutation was mapped to the Lmf1 (lipase maturation factor 1; formerly known as Tmem112) gene in murine chromosome 17. Subsequent studies showed that LMF1 localizes to the ER membrane and facilitates maturation of LPL, HL, and endothelial lipase (EL) (109).
Overexpression of LMF1 in adipose tissue, heart, and muscle tissues resulted in increased LPL activity in all of these tissues (111). Other naturally occurring murine mutations in Lmf1 have also been demonstrated to compromise LMF1 function in vitro (112). The hypertriglyceridemia in cld mice suggested that the human ortholog LMF1 on chromosome 16 could be a candidate gene for regulating TG metabolism, and various LMF1 mutations have been identified in patients with hypertriglyceridemia (113). Thus, a role for LMF1 in regulating TG metabolism is indicated by phenotypic and functional consequences of rare large-effect mutations in humans and mice. However, its common variants interrogated using GWAS failed to produce an association signal with hypertriglyceridemia (114).
The effect of LMF1 on atherosclerosis in mouse models is unknown. One would predict that LMF1 loss-of-function mutations would have effects on atherosclerosis surpassing those of LPL-deficiency because LMF1 is also needed for maturation of HL and EL.
Apolipoprotein C2 (APOC2)-Deficiency
APOC2 is a crucial cofactor for LPL activity. Mechanistically, APOC2 binds LPL and has been proposed to promote its activation by helping guide the TG substrate into the active site of LPL (115, 116). APOC2 is primarily expressed by the liver and secreted into plasma, but it is also produced at lower levels by other tissues, like the intestine, macrophages, adipose tissue, brain, skin, lungs, retina, and retinal pigment epithelium (115). Like other APOC apolipoproteins, APOC2 moves from APOB-containing lipoproteins to HDL during lipolysis (117, 118). In humans, APOC2 loss-of-function mutations can promote a prodigious rise in plasma TG levels due to chylomicronemia and consequently can provoke acute pancreatitis (119). Likewise, mice expressing a homozygous loss-of-function Apoc2 mutant (120) have increased plasma TGs (~700 mg/dL) and low HDL-C (~30 mg/dL) compared with wildtype mice (TGs; 60 mg/dL; HDL-C, 60 mg/dL), consistent with its role as an LPL cofactor (Figure 2).
Whether the effect of APOC2-deficiency on atherosclerosis phenocopies the effect of LPL-deficiency in mice needs further investigation.
Apolipoprotein C3 (APOC3) Overexpression
Human studies have firmly linked APOC3 to plasma TG levels (121–123). APOC3 is mainly produced by the liver and to a lesser extent, by the intestine. Like APOC2, it shuttles between APOB-containing lipoproteins and HDL (124, 125). Thus, while APOC3 is associated with chylomicrons, VLDL, and their RLPs, lipolysis of these particles is associated with transfer of APOC3 to HDL. Some APOC3 is also associated with LDL. APOC3 prevents TRL and RLP clearance by at least two different mechanisms: (i) it inhibits the action of LPL through a mechanism believed to be due to blocking of LPL's access to its triacylglycerol substrates when LPL is bound to GPIHBP1 (126); and (ii) it interferes with the binding of APOE to hepatic receptors of the LDL receptor family, thus resulting in a delayed catabolism and clearance of TRLs and RLPs (67). Early studies showed that at high concentrations, APOC3 also inhibits HL, thus further reducing lipolysis (127). However, no changes in HL activity were observed in a recent study of mice treated with an APOC3 antisense oligonucleotide (ASO) (128), suggesting that HL inhibition is not a significant mechanism of APOC3 in vivo. Overexpression of APOC3 in mice leads to increased TG levels because of the low LPL catalytic activity and delayed catabolism of VLDL and chylomicrons (129, 130).
When mice carrying the human APOC3 transgene were crossed with Ldlr−/− mice, significant increases in VLDL and RLPs were observed concomitant with increased atherosclerosis, as compared with Ldlr−/− controls (46). Later studies confirmed that APOC3 overexpression promotes restenosis and atherosclerosis (47, 131).
Together, there is strong evidence to suggest that APOC3 overexpression promotes atherosclerosis in mouse models. This effect is likely due, at least in part, to increased retention of RLPs in the artery wall due to the slower clearance rate of RLPs. Whether APOC3 also has direct effects on lesional cells independent of its effects on TRL and RLP catabolism, as has been suggested (47, 132–134), requires further study.
Apolipoprotein A5 (APOA5)-Deficiency
The APOA5 gene is located in the APOA1/APOC3/APOA4/APOA5 gene cluster on chromosome 11q23 in humans, a locus well-known to play a major role in regulating plasma cholesterol and TG levels (135). APOA5 is produced by the liver and circulates in plasma at very low concentrations (0.1–0.4 mg/mL). Mice lacking APOA5 exhibit TG levels in the 800 mg/dL range (136). In mice with hepatic APOA5 downregulation using an ASO, plasma TG levels were increased modestly (to ~150 mg/dL) associated with reduced LPL activity (137). It has been suggested that the mechanism whereby APOA5 increases LPL activity (135) is by guiding VLDL and chylomicrons to endothelial cell-bound LPL for lipolysis (138). Consistently, metabolic studies in vivo show that APOA5 enhances the catabolism of TRL rather than reducing TRL production.
The atherosclerosis phenotype of Apoa5−/− mice has not been reported. However, consistent with the increased TRL/RLP clearance by APOA5 (139), overexpression of human APOA5 in Apoe−/− mice resulted in decreased atherosclerotic lesion formation (48).
Together, these results show that APOA5 plays a critical role in promoting TRL and RLP clearance, an effect associated with protection from atherosclerosis.
Cyclic AMP-Responsive Element-Binding Protein H (CREB-H)-Deficiency
The transcription factor CREB-H (CREB3L3) is required for the maintenance of normal plasma TG concentrations, and loss-of-function mutations in the CREB3L3 gene are associated with severe hypertriglyceridemia in humans (140). CREB-H most likely works through indirectly increasing LPL activation, because its target genes include APOC2 and APOA5, although it also regulates other genes of importance in cardiometabolic diseases (140). CREB-H deficiency in Ldlr−/− mice leads to increased VLDL-TG levels and increased atherosclerosis (49).
Mouse Population Models and Models of Secondary Hypertriglyceridemia
The genetic mouse models of hypertriglyceridemia described above are primarily based on monogenic alterations on a single genetic background. The significant contributions of complex traits and polygenic contributions to hypertriglyceridemia in humans (141, 142) are therefore difficult to study. To tackle this issue, a panel of over 100 inbred strains of mice, known as the Hybrid Mouse Diversity Panel was generated by the Lusis laboratory (143). This panel offers important insights into genetically-derived differences in phenotype among inbred mice, and has been used to show that plasma TGs in mice fed a high fat/high sucrose diet are regulated by different quantitative trait loci (QTLs) on mouse chromosome 7 in male and female mice (144). Mice made hyperlipidemic by expression of human APOE-Leiden and human cholesteryl ester transfer protein (CETP) revealed that plasma TG levels varied widely in the different genetic backgrounds (from ~100 to 1,500 mg/dL) and highlighted a TG locus on chromosome 1, containing several genes with unknown functions in TG metabolism (145). Plasma TG levels were not associated with atherosclerosis in these studies. It would be interesting to perform similar studies to identify loci associated with elevated RLP levels to test the hypothesis that RLPs, rather than TGs, confer pro-atherogenic effects.
Other mouse models have been used to study the effects of non-genetic conditions that elevate plasma TG levels (referred to as secondary hypertriglyceridemia in humans). For example, mice made diabetic using the beta cell toxin streptozotocin or a virus develop elevated plasma TG levels. Interestingly, in a virally-induced diabetes model (a model of poorly controlled type 1 diabetes), plasma TG levels are elevated without a significant increase in plasma APOB (which is most abundant in LDL), suggesting a selective effect on TRLs (45). The mechanisms behind the elevated TG levels include reduced LPL activity (146) and increased APOC3 levels (45), leading to reduced TRL and RLP clearance. Furthermore, intensive insulin therapy but not blood glucose lowering by a sodium-glucose cotransporter 2 inhibitor was shown to prevent the effects of diabetes on APOC3 elevation (45). The accelerated atherosclerosis in diabetic mice was prevented by APOC3 silencing as discussed below. Thus, elevated APOC3 appears to explain the effect of diabetes on TGs and atherosclerosis in this model. Mouse models of type 2 diabetes can also exhibit elevated plasma TG levels. For example, in diabetic KKAy mice (a model of polygenic diabetes) elevated plasma TG levels have been shown to be due to reduced hepatic TRL clearance through syndecan 1 and flotillin 1-mediated endocytosis (147). Together, these studies suggest that the principal cause of the elevated TG levels in diabetes models is ineffective TRL clearance rather than increased hepatic VLDL production (146).
Rat Models
The rat was used for some of the early studies on TRL and RLP metabolism. For example, studies in hepatectomized rats demonstrated the importance of the liver in RLP removal (148) and studies in rats injected with chylomicrons demonstrated that phospholipids and some apolipoproteins are transferred from chylomicrons to HDL during lipolysis (149).
There are also rat models of hypertriglyceridemia. Rats homozygous for the corpulent gene (cp/cp) become obese, insulin resistant, and hypertriglyceridemic, primarily due to hepatic hypersecretion of VLDL (150). The cp trait results from a premature stop codon in the extracellular domain of the leptin receptor and therefore these rats lack a functional leptin receptor (151). Among the different cp strains, the JCR:LA-cp rat strain displays hyperlipidemia associated with APOB48-containing lipoprotein particles, particularly in the early post-prandial phase (152), and vasculopathy with atherosclerotic lesions and associated ischemic myocardial lesions (54), consistent with a pro-atherogenic effect of VLDL or its lipolysis products. However, other mechanisms, including leptin-related effects could have contributed to the atherosclerosis phenotype in this model.
Different models of APOE-deficiency have been generated by various gene editing strategies in rats. These Apoe−/− rats display a range of atherosclerosis phenotypes, ranging from no detectable lesions, to very early signs of lipid deposition and atherosclerosis (55, 56, 153, 154), at least as compared with Apoe−/− mice, to more advanced lesions (57). Thus, one Apoe−/− rat model exhibits an atherosclerosis phenotype similar to that of the Apoe−/− mice described above but lesion progression appears to be generally slower than that in the mouse. The reason for the differences in atherosclerosis burden in the different Apoe−/− rat models is so far unclear.
Finally, overfeeding rats with sucrose or fructose induces hypertriglyceridemia. Whereas sucrose favors hepatic fatty acid esterification and VLDL synthesis, fructose-rich diets deteriorate VLDL-TG catabolism (155). Wistar rats fed a high-sucrose (68%) or fructose (60%) diet have a 4-fold increase in TG levels after 3 and 8 weeks, respectively (155, 156).
So far, studies in rats support the more detailed mechanistic studies on TRLs and atherosclerosis in mouse models. Certain characteristics of lipid metabolism in rats are closer to those of humans as compared to mice (157), hence development of rat atherosclerosis models might provide advantages over mouse models. In addition, rats, due to their size have the benefit of easier blood collection, dissection of blood vessels, and other tissues, and thus are beneficial for atherosclerosis studies. Further analysis of different rat models and the role of TRLs and RLPs in atherosclerosis is needed as technologies for gene manipulation in rats are becoming more widely used.
Rabbit Models
Lipoprotein metabolism of rabbits resembles that of humans in composition of APOB-containing lipoproteins (158), production of APOB100-containing VLDL by the liver (159), presence of plasma CETP (which is lacking in mice and rats) (160), and high absorption rate of dietary cholesterol (161). One difference is the relatively low expression of HL in rabbits (162). Watanabe Heritable Hyperlipidemic (WHHL) rabbits with a natural defect in LDLR function, when fed high-fat diet, exhibit elevated plasma lipid levels and aortic lesions within 8 weeks. At 16 weeks, the lesions are advanced, with lipid core formation and calcification (58, 59). Selective crossing of WHHL rabbits with Japanese White (JW) rabbits and screening for blood TG levels led to the identification of two hypertriglyceridemic strains (163). The high hereditary hypertriglyceridemia (TGH) rabbits show autosomal recessive inheritance, and exhibit TG values >500 mg/dL. In addition, physical examination and necropsy of TGH rabbits revealed xanthomas and lesions of aortic atherosclerosis similar to those in human disease (163). A cross-breed between TGH rabbits and JW rabbits resulted in a post-prandial hypertriglyceridemic rabbit model (PHT) that exhibits remarkably high levels of postprandial TG (500–3,000 mg/dL) (163, 164) and increased atherosclerosis (60). In addition, the St. Thomas Hospital (STH) rabbit displays a Mendelian form of hypertriglyceridemia accompanied by increased APOB lipoprotein production and accelerated atherosclerosis (61).
The rabbit models described above confer advantages over mice and rats, but mechanistic studies on the role of TRLs in atherosclerosis lag behind those of the mouse models.
Pig Models
Lipid metabolism and cardiovascular physiology in pigs share several similarities with humans, and pigs develop atherosclerosis and a human-like lipoprotein profile without genetic manipulation. Hence, some pig models of hypertriglyceridemia have been developed. Göttingen minipigs under a dietary intervention consisting of a high-fat/high-energy diet have significantly elevated plasma TG levels (62) and develop atherosclerosis. Transgenic pigs expressing human APOC3 in the liver and intestine have also been developed, and these animals have increased plasma TG levels (2.5-fold), but normal total cholesterol and HDL-C levels. They also exhibit delayed TG absorbance and clearance (165), consistent with the known role of APOC3 in impeding TRL catabolism.
Zebrafish Model
The zebrafish is an emerging animal model for the study of abnormalities of lipid metabolism and associated diseases. In 2015, zebrafish expressing a non-functional APOC2 mutant were developed (63). These mutant zebrafish displayed decreased plasma TG lipase activity and severe hypertriglyceridemia, mainly due to buildup of chylomicrons. The hypertriglyceridemia was then rescued by injection of plasma from wildtype zebrafish with functional APOC2 or by injection of a human APOC2 mimetic peptide. The zebrafish mutants accumulated lipid and lipid-laden macrophages in the vasculature even on a normal diet—early events in atherosclerosis progression. However, fish do not express all genes contributing to hypertriglyceridemia in humans. For example, GPIHBP1 is not expressed in fish. It is believed that GPIHBP1 originated in eutherian mammals from a gene duplication event of an ancestral LY6-like gene (166). Nevertheless, the zebrafish model could be used to test factors affecting hypertriglyceridemia in a time and cost-effective manner.
Non-human Primate Models
Rhesus macaques, when fed a high-fructose diet, develop hypertriglyceridemia along with many other features of the metabolic syndrome, including central obesity, dyslipidemia, and inflammation observed in humans. In these insulin resistant monkeys, fasting TG levels were increased by 60% at 6 months and by almost 90% at 12 months (167). Fish oil supplementation reduced hypertriglyceridemia in this model of fructose-fed rhesus monkeys (168). In another study aimed at testing the effect of APOC3 inhibition using an ASO, rhesus monkeys were made hypertriglyceridemic via administration of a high-fructose supplement for 16 weeks and were then treated with APOC3 ASO for 12 weeks as the high-fructose diet was maintained (169). With fructose supplementation, plasma TG levels were increased by at least 3-fold over initial baseline levels in all treatment groups (40 mg/dL at day 1 vs. 140 mg/dL at day 112), just before initiation of dosing. APOC3 ASO significantly reduced fasting TG levels and postprandial TGs, as compared with monkeys receiving control ASO. Since non-human primates are more closely related and have similar lipid profiles to humans (170), the fructose-fed rhesus monkeys can represent a translatable model for studying the effects of TGs and novel TG-lowering strategies on atherosclerosis in humans.
Emerging Strategies to Lower Hypertriglyceridemia and Their Effects on Atherosclerosis in Animal Models
APOC3 Inhibition
The physiological role of APOC3 is likely to ensure delivery of fatty acids derived from TGs to adipose tissue, muscle and the heart both after a meal and between meals by slowing the clearance of TRLs and RLPs. Because human studies convincingly show that loss-of-function mutations in APOC3 are associated with lower TG levels and cardioprotection (121, 122, 171–174), APOC3 has recently emerged as a drug target for the treatment of hypertriglyceridemia and possibly the associated increase in CVD risk (18, 175–177).
The human studies spurred pharmaceutical development of APOC3 inhibitors in the form of ASOs, interference RNAs and monoclonal antibodies (122, 168, 178). Volanesorsen (IONIS-APOCIII Rx) is a second-generation ASO drug targeted to APOC3 mRNA, which effectively reduces plasma TG levels in human clinical trials (175–177). However, this ASO was not approved by the FDA for the treatment of familial chylomicronemia syndrome due to adverse effects related to thrombocytopenia. The Committee for Medicinal Products for Human Use of the European Medicines Agency however, authorized conditional marketing for Waylivra (volanesorsen) as an adjunct to diet in adult patients with genetically confirmed familial chylomicronemia syndrome who are at high risk for pancreatitis, in whom response to diet and TG-lowering therapy has not worked (123). New formulations of APOC3 inhibitory drugs are likely to reduce side-effects. CVD outcomes studies have not yet been performed in humans using APOC3-lowering therapies.
Animal models have provided additional mechanistic insight into the protective effects of APOC3 inhibition. APOC3 ASOs similar to volanesorsen have been effective in reducing APOC3 expression and plasma TGs in several rodent models and in non-human primates (45, 169). Mechanistic studies in mice using the APOC3 ASO showed that plasma TG levels were lowered by increasing activation of LPL as well as furthering the clearance of TRLs by the liver, via the LDLR family of receptors (67). Importantly, inhibition of APOC3 lowers TRLs without significantly affecting LDL levels or APOB100 levels in both humans and mice (45, 174).
A recent study analyzing both human samples and a mouse model showed that elevated serum levels of APOC3 predict coronary artery disease events in human subjects with type 1 diabetes independent of LDL-C and several other risk factors, and that plasma levels of APOC3 were significantly increased in a mouse model of type 1 diabetes (45). There was a striking protective effect of APOC3 inhibition by ASO on both early and more advanced atherosclerosis in this mouse model of type 1 diabetes-accelerated atherosclerosis (45). This effect was associated with protection from accumulation of APOB, APOE, and APOC3 in the artery wall (most likely associated with reduced RLPs retention) as well as protection from cholesteryl ester accumulation in macrophages from diabetic mice.
Together, these studies strongly suggest that APOC3 inhibition could be effective in treating hypertriglyceridemia and preventing atherosclerotic CVD, especially in conditions in which APOC3 levels are increased.
APOC2 Mimetic Peptide
As discussed above, APOC2 is an essential cofactor for LPL activation and APOC2 therefore promotes TRL hydrolysis. Current treatment options for severely hypertriglyceridemic patients with loss-of-function mutations in APOC2 include a stringent low-fat diet and plasma exchange with APOC2-containing donor plasma as a direct and lifesaving procedure during severe pancreatitis episodes (179). Recently, APOC2 mimetic peptides have been developed for treatment of hypertriglyceridemia, especially in APOC2-deficient patients (115). These peptides, which incorporate the third helix of APOC2, are promising as potential future therapy. Intravenous injection of one such peptide normalized TGs in a mouse model of APOC2-deficiency (120). Furthermore, the APOC2 mimetic peptide potentiated LPL activity in other non-APOC2-deficient hypertriglyceridemic conditions, like APOE-deficiency (180).
The most potent peptide (D6PV), developed on the basis of structural insights gained from all-atom molecular dynamics simulations, directly activates LPL and also inhibits APOC3's action. D6PV treatment drastically lowers TGs in APOC2-deficient mice and in human APOC3-transgenic mice (181). This peptide shows some LPL-independent TG lowering activity, probably because it has dual APOC2 and APOC3 antagonistic effects, as it partially decreases plasma TG in whole-body inducible Lpl−/− mice. It also lowers TG levels in non-human primates (181). Its effect on atherosclerosis in animal models (and humans) is so far unknown.
Angiopoietin-Like 3 (ANGPTL3) Inhibition
ANGPTL3 is a hepatically secreted protein, which inhibits the vascular lipases LPL and EL. Loss-of-function mutations in ANGPTL3 lead to familial combined hypolipidemia, characterized by low circulating levels of LDL-C, HDL-C, and TGs (182) and apparent protection against atherosclerotic CVD (182). ANGPTL3 acts to increase TG levels by catalyzing the irreversible unfolding of LPL's hydrolase domain, resulting in a loss of both TG hydrolase activity and esterase activity (104). A complex consisting of ANGPTL3 and ANGPTL8 (another protein in the ANGPTL family) has a markedly increased ability to inhibit LPL activity, likely by increasing the binding of ANGPTL3 to LPL (183).
The favorable lipid effects seen in animal models and humans with loss of ANGPTL3 function have led to a spurt in development of ANGPTL3 inhibitors. Evinacumab is a monoclonal antibody, which binds to ANGPTL3 and completely reverses its inhibitory activity on LPL and EL (184). In vivo studies on normolipidemic mice showed a dose-dependent reduction in TG, total cholesterol, LDL-C and HDL-C serum levels after subcutaneous injections of evinacumab. An increase in LPL and EL activity has been recorded in normolipidemic wildtype mice as well as in dyslipidemic wildtype (C57BL/6) mice, in db/db mice and in APOE*3-Leiden mice in which ANGPTL3 is inhibited (184). The same results have been obtained by treating dyslipidemic cynomolgus monkeys (185). Mice on evinacumab treatment showed a significant decrease in total cholesterol and TG levels as well as in atherosclerotic lesion size. ASO-mediated ANGPTL3 inhibition in mice with different lipid phenotypes (wildtype C57BL/6 mice, Ldlr−/− mice, Apoc3−/− Ldlr−/− mice, heterozygous Apoc3+/− Ldlr−/− mice, diet-induced obese mice, and mice over-expressing human APOC3) resulted in lowered plasma TGs, LDL-C and HDL-C and reduced atherosclerosis (50). Another group targeted ANGPTL3 using a modified CRISPR-Cas9 platform. Wildtype mice and hyperlipidemic Ldlr−/− mice targeted with this ANGPTL3 silencing strategy showed reduced plasma TG and total cholesterol levels (186).
Together, these findings suggest that ANGPTL3 is a promising target for TG lowering and perhaps prevention of atherosclerotic CVD.
Angiopoietin-Like 4 (ANGPTL4) Inhibition
ANGPTL4 was identified as a fasting-induced adipose factor and was found to act as a potent LPL inhibitor (187–189). ANGPTL4 is more widely expressed than ANGPTL3, and is highly expressed in the liver, adipose tissue, hematopoietic cells, and other tissues. Studies in ANGPTL4 transgenic mice and in mice with adenoviral ANGPTL4 overexpression show decreased LPL activity, delayed TG clearance, and increased plasma TG levels (190, 191). The increase in plasma TGs and free fatty acids associated with ANGPTL4 overexpression is thought to be independent of food intake and hepatic VLDL secretion. Instead, ANGPTL4 has recently been shown to inactivate LPL by catalyzing LPL unfolding (14). Conversely, ANGPTL4-deficient mice exhibit increased plasma LPL activity, increased TG clearance, decreased plasma TG levels, and reduced atherosclerosis (51, 190). Several GWAS in humans corroborate the findings of ANGPTL4-mediated regulation of lipid metabolism in mice (192, 193). As a result, ANGPTL4 was targeted for TG lowering using monoclonal antibodies. Humanized mice treated with ANGPTL4 blocking antibodies display reduced plasma TG levels (194). Moreover, cynomolgus monkeys and hyperlipidemic rhesus monkeys also display reduced TGs when treated with ANGPTL4-neutralizing antibodies (194). However, mice lacking ANGPTL4, when fed a high-fat diet, demonstrate a severe inflammatory phenotype and increased mortality (195).
Although ANGPTL4 inhibition is effective in lowering TGs in mice and primates and also reduces atherosclerosis progression, an important question remains regarding the safety of this approach due to the mesenteric lymphadenopathy seen in high fat-fed animals in which ANGPTL4 is blocked.
Summary
We have reviewed the most common animal models of hypertriglyceridemia and the critical mechanistic insights provided by these models. One important conclusion is that these models, in general, are consistent with the proposal that RLPs derived from TRLs are highly atherogenic whereas hypertriglyceridemia due to accumulation of very large TRLs is not markedly atherogenic in the absence of TRL lipolysis products. Combination of mechanistic animal studies and studies on isolated lesional cells indicate that RLPs are more atherogenic than large TRLs, perhaps because they more readily enter the artery wall, and because they are enriched in cholesterol relative to TGs, which promotes pro-atherogenic effects in lesional cells. Animal studies have also revealed that RLPs are cleared through hepatic LDLR and LRP1 receptors, and that LPL has effects additional to its role in TG hydrolysis in capillaries, including by acting locally in the lesion. However, definite proof that RLPs rather than nascent TRLs are the cause of greater CVD in patients with hypertriglyceridemia and/or defects in genes leading to reduced LPL actions is still missing and will require further research.
Future Directions
Elevated plasma TG concentrations appear to be a predictor of CVD risk in humans. GWAS have greatly widened the repertoire of TG-associated genes linked to CVD risk. Clinical hypertriglyceridemia has a complex genetic basis that includes a burden of both common variants, such that each has a relatively small effect on TG levels, and rare variants, such that each has a relatively large effect on TG levels. It is quite clear that LPL and factors affecting its activity or function play a major role in atherogenesis, at least in animal models.
The multifactorial nature of hypertriglyceridemia makes it challenging to draw clear conclusions concerning its atherogenicity. A more complete understanding of the genes and variants that modulate plasma TGs requires strong functional validation at all stages: in vitro, in vivo, and ultimately in clinical trials, and will be crucial for identifying key players in TG metabolism, which might help specifying new directions for therapeutic interventions. Many such studies are ongoing. For instance, studies of the effect of hypertriglyceridemia in the context of atherosclerosis regression in animal models, in which the confounding factor of hypercholesterolemia is absent, are likely to provide important insights. Furthermore, studies of the role of RLPs in CVD associated with diabetes or other conditions associated with increased CVD risk will be important. Additionally, the role of LPL in peripheral TG metabolism vs. its effect locally in the artery wall and on macrophage lipid metabolism can be dissected using newer genetic models. Moreover, the mechanistic role of how fish oils (in particular eicosapentaenoic acid) protect against CVD risk, and whether this protection is related to TG lowering or other mechanisms should be assessed in animal models of hypertriglyceridemia. New tools and methods to measure and evaluate TRLs and RLPs will also be critical for understanding if TRLs and RLPs have distinct effects on atherosclerotic lesions and as risk factors for CVD (23).
The last decade saw the discovery of new drug targets and the early phase clinical trials testing these targets, including APOC3 and ANGPTL3, which both inhibit LPL activity. The development of several different strategies for reducing TG levels is likely to be more specific and effective than drugs currently on the market. Animal models will help to provide mechanistic insight through reverse translation of clinical observations. There is hope that the current decade will reveal whether such strategies will also prevent CVD risk in patients with hypertriglyceridemia and others with impaired TRL and RLP catabolism.
Author Contributions
DB wrote the manuscript. KB wrote parts of the manuscript and edited the full manuscript. All authors contributed to the article and approved the submitted version.
Funding
Research in the authors' laboratories was supported by the National Institutes of Health grants DP3DK108209, R01HL127694, R01HL126028, P01HL092969, and R35HL150754 (KB), and the Diabetes Research Center at the University of Washington, P30DK017047, and an American Heart Association Postdoctoral fellowship 17POST33660283 (DB) and Russell Berrie Fellowship in Diabetes Research (DB).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Ms. Julie De Bermont for help generating the figures.
References
1. Hulley SB, Rosenman RH, Bawol RD, Brand RJ. Epidemiology as a guide to clinical decisions. The association between triglyceride and coronary heart disease. N Engl J Med. (1980) 302:1383–9. doi: 10.1056/NEJM198006193022503
3. Yuan G, Al-Shali KZ, Hegele RA. Hypertriglyceridemia: its etiology, effects and treatment. CMAJ. (2007) 176:1113–20. doi: 10.1503/cmaj.060963
4. Chait A, Brunzell JD, Albers JJ, Hazzard WR. Type-III Hyperlipoproteinaemia (“remnant removal disease”). Insight into the pathogenetic mechanism. Lancet. (1977) 1:1176–8. doi: 10.1016/S0140-6736(77)92717-9
5. Rhainds D, Brodeur MR, Tardif JC. Investigational drugs in development for hypertriglyceridemia: a coming-of-age story. Expert Opin Investig Drugs. (2019) 28:1059–79. doi: 10.1080/13543784.2019.1696772
6. Kowal RC, Herz J, Goldstein JL, Esser V, Brown MS. Low density lipoprotein receptor-related protein mediates uptake of cholesteryl esters derived from apoprotein E-enriched lipoproteins. Proc Natl Acad Sci USA. (1989) 86:5810–4. doi: 10.1073/pnas.86.15.5810
7. Rohlmann A, Gotthardt M, Hammer RE, Herz J. Inducible inactivation of hepatic LRP gene by cre-mediated recombination confirms role of LRP in clearance of chylomicron remnants. J Clin Invest. (1998) 101:689–95. doi: 10.1172/JCI1240
8. Yu KC, Chen W, Cooper AD. LDL receptor-related protein mediates cell-surface clustering and hepatic sequestration of chylomicron remnants in LDLR-deficient mice. J Clin Invest. (2001) 107:1387–94. doi: 10.1172/JCI11750
9. Brown MS, Goldstein JL. Lipoprotein receptors in the liver. Control signals for plasma cholesterol traffic. J Clin Invest. (1983) 72:743–7. doi: 10.1172/JCI111044
10. Dron JS, Hegele RA. Genetics of triglycerides and the risk of atherosclerosis. Curr Atheroscler Rep. (2017) 19:31. doi: 10.1007/s11883-017-0667-9
11. Dron JS, Wang J, McIntyre AD, Cao H, Hegele RA. The polygenic nature of mild-to-moderate hypertriglyceridemia. J Clin Lipidol. (2020) 14:28–34.e2. doi: 10.1016/j.jacl.2020.01.003
12. Baass A, Paquette M, Bernard S, Hegele RA. Familial chylomicronemia syndrome: an under-recognized cause of severe hypertriglyceridaemia. J Intern Med. (2019) 287:340–8. doi: 10.1111/joim.13016
13. Beigneux AP, Miyashita K, Ploug M, Blom DJ, Ai M, Linton MRF, et al. Autoantibodies against GPIHBP1 as a cause of hypertriglyceridemia. N Engl J Med. (2017) 376:1647–58. doi: 10.1056/NEJMoa1611930
14. Kristensen KK, Leth-Espensen KZ, Mertens HDT, Birrane G, Meiyappan M, Olivecrona G, et al. Unfolding of monomeric lipoprotein lipase by ANGPTL4: insight into the regulation of plasma triglyceride metabolism. Proc Natl Acad Sci USA. (2020) 117:4337–46. doi: 10.1073/pnas.1920202117
15. Goldstein JL, Hazzard WR, Schrott HG, Bierman EL, Motulsky AG. Hyperlipidemia in coronary heart disease. I. Lipid levels in 500 survivors of myocardial infarction. J Clin Invest. (1973) 52:1533–43. doi: 10.1172/JCI107331
16. Miller M, Stone NJ, Ballantyne C, Bittner V, Criqui MH, Ginsberg HN, et al. Triglycerides and cardiovascular disease: a scientific statement from the American heart association. Circulation. (2011) 123:2292–333. doi: 10.1161/CIR.0b013e3182160726
17. Nordestgaard BG, Benn M, Schnohr P, Tybjaerg-Hansen A. Nonfasting triglycerides and risk of myocardial infarction, ischemic heart disease, and death in men and women. JAMA. (2007) 298:299–308. doi: 10.1001/jama.298.3.299
18. Sandesara PB, Virani SS, Fazio S, Shapiro MD. The forgotten lipids: triglycerides, remnant cholesterol, and atherosclerotic cardiovascular disease risk. Endocr Rev. (2019) 40:537–57. doi: 10.1210/er.2018-00184
19. Chapman MJ, Ginsberg HN, Amarenco P, Andreotti F, Boren J, Catapano AL, et al. Triglyceride-rich lipoproteins and high-density lipoprotein cholesterol in patients at high risk of cardiovascular disease: evidence and guidance for management. Eur Heart J. (2011) 32:1345–61. doi: 10.1093/eurheartj/ehr112
20. Suzuki K, Oikawa T, Nochioka K, Miura M, Kasahara S, Sato M, et al. Elevated serum non-hdl (high-density lipoprotein) cholesterol and triglyceride levels as residual risks for myocardial infarction recurrence under statin treatment. Arteriosc Thromb Vasc Biol. (2019) 39:934–44. doi: 10.1161/ATVBAHA.119.312336
21. Bhatt DL, Steg PG, Miller M, Brinton EA, Jacobson TA, Jiao L, et al. Reduction in first and total ischemic events with icosapent ethyl across baseline triglyceride tertiles. J Am Coll Cardiol. (2019) 74:1159–61. doi: 10.1016/j.jacc.2019.06.043
22. Yokoyama M, Origasa H, Matsuzaki M, Matsuzawa Y, Saito Y, Ishikawa Y, et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): a randomised open-label, blinded endpoint analysis. Lancet. (2007) 369:1090–8. doi: 10.1016/S0140-6736(07)60527-3
23. Chait A, Ginsberg HN, Vaisar T, Heinecke JW, Goldberg IJ, Bornfeldt KE. Remnants of the triglyceride-rich lipoproteins, diabetes and cardiovascular disease. Diabetes. (2020) 69:508–16. doi: 10.2337/dbi19-0007
24. Brahm AJ, Hegele RA. Chylomicronaemia–current diagnosis and future therapies. Nat Rev Endocrinol. (2015) 11:352–62. doi: 10.1038/nrendo.2015.26
25. Ference BA, Kastelein JJP, Ray KK, Ginsberg HN, Chapman MJ, Packard CJ, et al. Association of triglyceride-lowering LPL variants and LDL-C-lowering LDLR variants with risk of coronary heart disease. JAMA. (2019) 321:364–73. doi: 10.1001/jama.2018.20045
26. Morganroth J, Levy RI, Fredrickson DS. The biochemical, clinical, and genetic features of type III hyperlipoproteinemia. Ann Intern Med. (1975) 82:158–74. doi: 10.7326/0003-4819-82-2-158
27. Koopal C, Retterstol K, Sjouke B, Hovingh GK, Ros E, de Graaf J, et al. Vascular risk factors, vascular disease, lipids and lipid targets in patients with familial dysbetalipoproteinemia: a European cross-sectional study. Atherosclerosis. (2015) 240:90–7. doi: 10.1016/j.atherosclerosis.2015.02.046
28. Rosenson RS, Davidson MH, Hirsh BJ, Kathiresan S, Gaudet D. Genetics and causality of triglyceride-rich lipoproteins in atherosclerotic cardiovascular disease. J Am Coll Cardiol. (2014) 64:2525–40. doi: 10.1016/j.jacc.2014.09.042
29. Zilversmit DB. A proposal linking atherogenesis to the interaction of endothelial lipoprotein lipase with triglyceride-rich lipoproteins. Circ Res. (1973) 33:633–8. doi: 10.1161/01.RES.33.6.633
30. Van Lenten BJ, Fogelman AM, Jackson RL, Shapiro S, Haberland ME, Edwards PA. Receptor-mediated uptake of remnant lipoproteins by cholesterol-loaded human monocyte-macrophages. J Biol Chem. (1985) 260:8783–8.
31. Whitman SC, Miller DB, Wolfe BM, Hegele RA, Huff MW. Uptake of type III hypertriglyceridemic VLDL by macrophages is enhanced by oxidation, especially after remnant formation. Arteriosc Thromb Vasc Biol. (1997) 17:1707–15. doi: 10.1161/01.ATV.17.9.1707
32. Shakhnovich V. It's time to reverse our thinking: the reverse translation research paradigm. Clin Transl Sci. (2018) 11:98–9. doi: 10.1111/cts.12538
33. Plump AS, Smith JD, Hayek T, Aalto-Setala K, Walsh A, Verstuyft JG, et al. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. (1992) 71:343–53. doi: 10.1016/0092-8674(92)90362-G
34. Zhang SH, Reddick RL, Piedrahita JA, Maeda N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science. (1992) 258:468–71. doi: 10.1126/science.1411543
35. Zhang XH, Qi R, Xian XD, Yang F, Blackstein M, Deng XM, et al. Spontaneous atherosclerosis in aged lipoprotein lipase-deficient mice with severe hypertriglyceridemia on a normal chow diet. Circul Res. (2008) 102:250–6. doi: 10.1161/CIRCRESAHA.107.156554
36. Weinstein MM, Yin L, Tu Y, Wang X, Wu X, Castellani LW, et al. Chylomicronemia elicits atherosclerosis in mice–brief report. Arteriosc Thromb Vasc Biol. (2010) 30:20–3. doi: 10.1161/ATVBAHA.109.196329
37. Minnich A, Zilversmit DB. Impaired triacylglycerol catabolism in hypertriglyceridemia of the diabetic, cholesterol-fed rabbit: a possible mechanism for protection from atherosclerosis. Biochim Biophys Acta. (1989) 1002:324–32. doi: 10.1016/0005-2760(89)90346-9
38. Duff GL, Mc MG. The effect of alloxan diabetes on experimental cholesterol atherosclerosis in the rabbit. J Exp Med. (1949) 89:611–30. doi: 10.1084/jem.89.6.611
39. Nordestgaard BG, Agerholm-Larsen B, Mortensen A, Fischer Hansen B, Fischer Hansen J, Ibsen P, et al. Intermediate density lipoprotein cholesterol as the best lipoprotein predictor of atherosclerosis severity in the watanabe heritable hyperlipidemic rabbit. Atherosclerosis. (1997) 132:119–22. doi: 10.1016/S0021-9150(97)00051-8
40. van Vlijmen BJ, van den Maagdenberg AM, Gijbels MJ, van der Boom H, HogenEsch H, Frants RR, et al. Diet-induced hyperlipoproteinemia and atherosclerosis in apolipoprotein E3-Leiden transgenic mice. J Clin Invest. (1994) 93:1403–10. doi: 10.1172/JCI117117
41. Van Eck M, Herijgers N, Van Dijk KW, Havekes LM, Hofker MH, Groot PH, et al. Effect of macrophage-derived mouse ApoE, human ApoE3-Leiden, and human ApoE2 (Arg158–>Cys) on cholesterol levels and atherosclerosis in ApoE-deficient mice. Arteriosc Thromb Vasc Biol. (2000) 20:119–27. doi: 10.1161/01.ATV.20.1.119
42. Basu D, Josefs T, Fisher EA, Goldberg IJ. Effect Of Lipoprotein Lipase (Lpl) deletion on atherosclerosis regression. Atherosclerosis. (2019) 287:e61. doi: 10.1016/j.atherosclerosis.2019.06.174
43. Shimada M, Ishibashi S, Inaba T, Yagyu H, Harada K, Osuga JI, et al. Suppression of diet-induced atherosclerosis in low density lipoprotein receptor knockout mice overexpressing lipoprotein lipase. Proc Natl Acad Sci USA. (1996) 93:7242–6. doi: 10.1073/pnas.93.14.7242
44. Yagyu H, Ishibashi S, Chen Z, Osuga J, Okazaki M, Perrey S, et al. Overexpressed lipoprotein lipase protects against atherosclerosis in apolipoprotein E knockout mice. J Lipid Res. (1999) 40:1677–85.
45. Kanter JE, Shao B, Kramer F, Barnhart S, Shimizu-Albergine M, Vaisar T, et al. Increased apolipoprotein C3 drives cardiovascular risk in type 1 diabetes. J Clin Invest. (2019) 130:4165–79. doi: 10.1172/JCI127308
46. Masucci-Magoulas L, Goldberg IJ, Bisgaier CL, Serajuddin H, Francone OL, Breslow JL, et al. A mouse model with features of familial combined hyperlipidemia. Science. (1997) 275:391–4. doi: 10.1126/science.275.5298.391
47. Li H, Han Y, Qi R, Wang Y, Zhang X, Yu M, et al. Aggravated restenosis and atherogenesis in ApoCIII transgenic mice but lack of protection in ApoCIII knockouts: the effect of authentic triglyceride-rich lipoproteins with and without ApoCIII. Cardiovasc Res. (2015) 107:579–89. doi: 10.1093/cvr/cvv192
48. Mansouri RM, Bauge E, Gervois P, Fruchart-Najib J, Fievet C, Staels B, et al. Atheroprotective effect of human apolipoprotein A5 in a mouse model of mixed dyslipidemia. Circ Res. (2008) 103:450–3. doi: 10.1161/CIRCRESAHA.108.179861
49. Park JG, Xu X, Cho S, Lee AH. Loss of transcription factor CREBH accelerates diet-induced atherosclerosis in Ldlr-/- Mice. Arteriosc Thromb Vasc Biol. (2016) 36:1772–81. doi: 10.1161/ATVBAHA.116.307790
50. Graham MJ, Lee RG, Brandt TA, Tai LJ, Fu W, Peralta R, et al. Cardiovascular and metabolic effects of ANGPTL3 antisense oligonucleotides. N Engl J Med. (2017) 377:222–32. doi: 10.1056/NEJMoa1701329
51. Adachi H, Fujiwara Y, Kondo T, Nishikawa T, Ogawa R, Matsumura T, et al. Angptl 4 deficiency improves lipid metabolism, suppresses foam cell formation and protects against atherosclerosis. Biochem Biophys Res Commun. (2009) 379:806–11. doi: 10.1016/j.bbrc.2008.12.018
52. Johansson F, Kramer F, Barnhart S, Kanter JE, Vaisar T, Merrill RD, et al. Type 1 diabetes promotes disruption of advanced atherosclerotic lesions in LDL receptor-deficient mice. Proc Natl Acad Sci USA. (2008) 105:2082–7. doi: 10.1073/pnas.0709958105
53. Renard CB, Kramer F, Johansson F, Lamharzi N, Tannock LR, von Herrath MG, et al. Diabetes and diabetes-associated lipid abnormalities have distinct effects on initiation and progression of atherosclerotic lesions. J Clin Invest. (2004) 114:659–68. doi: 10.1172/JCI200417867
54. Russell JC, Graham SE, Richardson M. Cardiovascular disease in the JCR:LA-cp rat. Mol Cell Biochem. (1998) 188:113–26. doi: 10.1007/978-1-4615-5763-0_13
55. Ekuni D, Yoneda T, Endo Y, Kasuyama K, Irie K, Mizutani S, et al. Occlusal disharmony accelerates the initiation of atherosclerosis in apoE knockout rats. Lipids Health Dis. (2014) 13:144. doi: 10.1186/1476-511X-13-144
56. Rune I, Rolin B, Lykkesfeldt J, Nielsen DS, Krych L, Kanter JE, et al. Long-term Western diet fed apolipoprotein E-deficient rats exhibit only modest early atherosclerotic characteristics. Sci Rep. (2018) 8:5416. doi: 10.1038/s41598-018-23835-z
57. Zhao Y, Yang Y, Xing R, Cui X, Xiao Y, Xie L, et al. Hyperlipidemia induces typical atherosclerosis development in Ldlr and Apoe deficient rats. Atherosclerosis. (2018) 271:26–35. doi: 10.1016/j.atherosclerosis.2018.02.015
58. Havel RJ, Yamada N, Shames DM. Watanabe heritable hyperlipidemic rabbit. Animal model for familial hypercholesterolemia. Arteriosclerosis. (1989) 9(1 Suppl):I33–8.
59. Ning B, Wang X, Yu Y, Waqar AB, Yu Q, Koike T, et al. High-fructose and high-fat diet-induced insulin resistance enhances atherosclerosis in Watanabe heritable hyperlipidemic rabbits. Nutr Metab. (2015) 12:30. doi: 10.1186/s12986-015-0024-3
60. Matsumoto S, Gotoh N, Hishinuma S, Abe Y, Shimizu Y, Katano Y, et al. The role of hypertriglyceridemia in the development of atherosclerosis and endothelial dysfunction. Nutrients. (2014) 6:1236–50. doi: 10.3390/nu6031236
61. Beaty TH, Prenger VL, Virgil DG, Lewis B, Kwiterovich PO, Bachorik PS. A genetic model for control of hypertriglyceridemia and apolipoprotein B levels in the Johns Hopkins colony of St. Thomas Hospital rabbits. Genetics. (1992) 132:1095–104.
62. Johansen T, Hansen HS, Richelsen B, Malmlof R. The obese gottingen minipig as a model of the metabolic syndrome: dietary effects on obesity, insulin sensitivity, and growth hormone profile. Comp Med. (2001) 51:150–5. Available online at: https://www.ingentaconnect.com/content/aalas/cm/2001/00000051/00000002/art00009
63. Liu C, Gates KP, Fang L, Amar MJ, Schneider DA, Geng H, et al. Apoc2 loss-of-function zebrafish mutant as a genetic model of hyperlipidemia. Dis Model Mech. (2015) 8:989–98. doi: 10.1242/dmm.019836
64. Ishibashi S, Goldstein JL, Brown MS, Herz J, Burns DK. Massive xanthomatosis and atherosclerosis in cholesterol-fed low density lipoprotein receptor-negative mice. J Clin Invest. (1994) 93:1885–93. doi: 10.1172/JCI117179
65. Getz GS, Reardon CA. Apoprotein E as a lipid transport and signaling protein in the blood, liver, and artery wall. J Lipid Res. (2009) 50(Suppl):S156–61. doi: 10.1194/jlr.R800058-JLR200
66. Basford JE, Wancata L, Hofmann SM, Silva RA, Davidson WS, Howles PN, et al. Hepatic deficiency of low density lipoprotein receptor-related protein-1 reduces high density lipoprotein secretion and plasma levels in mice. J Biol Chem. (2011) 286:13079–87. doi: 10.1074/jbc.M111.229369
67. Gordts PL, Nock R, Son NH, Ramms B, Lew I, Gonzales JC, et al. ApoC-III inhibits clearance of triglyceride-rich lipoproteins through LDL family receptors. J Clin Invest. (2016) 126:2855–66. doi: 10.1172/JCI86610
68. Espirito Santo SM, Pires NM, Boesten LS, Gerritsen G, Bovenschen N, van Dijk KW, et al. Hepatic low-density lipoprotein receptor-related protein deficiency in mice increases atherosclerosis independent of plasma cholesterol. Blood. (2004) 103:3777–82. doi: 10.1182/blood-2003-11-4051
69. Havekes L, de Wit E, Leuven JG, Klasen E, Utermann G, Weber W, et al. Apolipoprotein E3-Leiden. A new variant of human apolipoprotein E associated with familial type III hyperlipoproteinemia. Hum Genet. (1986) 73:157–63. doi: 10.1007/BF00291607
70. van den Maagdenberg AM, Hofker MH, Krimpenfort PJ, de Bruijn I, van Vlijmen B, van der Boom H, et al. Transgenic mice carrying the apolipoprotein E3-Leiden gene exhibit hyperlipoproteinemia. J Biol Chem. (1993) 268:10540–5.
71. Westerterp M, Van Eck M, de Haan W, Offerman EH, Van Berkel TJ, Havekes LM, et al. Apolipoprotein CI aggravates atherosclerosis development in ApoE-knockout mice despite mediating cholesterol efflux from macrophages. Atherosclerosis. (2007) 195:e9–16. doi: 10.1016/j.atherosclerosis.2007.01.015
72. Berbee JF, van der Hoogt CC, Sundararaman D, Havekes LM, Rensen PC. Severe hypertriglyceridemia in human APOC1 transgenic mice is caused by apoC-I-induced inhibition of LPL. J Lipid Res. (2005) 46:297–306. doi: 10.1194/jlr.M400301-JLR200
73. Jong MC, Dahlmans VE, van Gorp PJ, Breuer ML, Mol MJ, van der Zee A, et al. Both lipolysis and hepatic uptake of VLDL are impaired in transgenic mice coexpressing human apolipoprotein E*3Leiden and human apolipoprotein C1. Arteriosc Thromb Vasc Biol. (1996) 16:934–40. doi: 10.1161/01.ATV.16.8.934
74. Goldberg IJ. Triglyceride: one molecule at the center of health and disease. Biochim Biophys Acta. (2012) 1821:719–20. doi: 10.1016/j.bbalip.2012.02.005
75. Goldberg IJ. Lipoprotein lipase and lipolysis: central roles in lipoprotein metabolism and atherogenesis. J Lipid Res. (1996) 37:693–707.
77. Goldberg IJ, Le NA, Ginsberg HN, Krauss RM, Lindgren FT. Lipoprotein metabolism during acute inhibition of lipoprotein lipase in the cynomolgus monkey. J Clin Invest. (1988) 81:561–8. doi: 10.1172/JCI113354
78. Goldberg IJ, Blaner WS, Vanni TM, Moukides M, Ramakrishnan R. Role of lipoprotein lipase in the regulation of high density lipoprotein apolipoprotein metabolism. Studies in normal and lipoprotein lipase-inhibited monkeys. J Clin Invest. (1990) 86:463–73. doi: 10.1172/JCI114732
79. O'Brien KD, Gordon D, Deeb S, Ferguson M, Chait A. Lipoprotein lipase is synthesized by macrophage-derived foam cells in human coronary atherosclerotic plaques. J Clin Invest. (1992) 89:1544–50. doi: 10.1172/JCI115747
80. Beigneux AP, Allan CM, Sandoval NP, Cho GW, Heizer PJ, Jung RS, et al. Lipoprotein lipase is active as a monomer. Proc Natl Acad Sci USA. (2019) 116:6319–28. doi: 10.1073/pnas.1900983116
81. Burnett JR, Hooper AJ, Hegele RA. Familial lipoprotein lipase deficiency. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington (1993). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK1308/
82. Strauss JG, Frank S, Kratky D, Hammerle G, Hrzenjak A, Knipping G, et al. Adenovirus-mediated rescue of lipoprotein lipase-deficient mice. Lipolysis of triglyceride-rich lipoproteins is essential for high density lipoprotein maturation in mice. J Biol Chem. (2001) 276:36083–90. doi: 10.1074/jbc.M104430200
83. Takahashi M, Hiyama Y, Yokoyama M, Yu S, Hu Y, Melford K, et al. In vivo arterial lipoprotein lipase expression augments inflammatory responses and impairs vascular dilatation. Arteriosc Thromb Vasc Biol. (2008) 28:455–62. doi: 10.1161/ATVBAHA.107.153239
84. Levak-Frank S, Weinstock PH, Hayek T, Verdery R, Hofmann W, Ramakrishnan R, et al. Induced mutant mice expressing lipoprotein lipase exclusively in muscle have subnormal triglycerides yet reduced high density lipoprotein cholesterol levels in plasma. J Biol Chem. (1997) 272:17182–90. doi: 10.1074/jbc.272.27.17182
85. Levak-Frank S, Hofmann W, Weinstock PH, Radner H, Sattler W, Breslow JL, et al. Induced mutant mouse lines that express lipoprotein lipase in cardiac muscle, but not in skeletal muscle and adipose tissue, have normal plasma triglyceride and high-density lipoprotein-cholesterol levels. Proc Natl Acad Sci USA. (1999) 96:3165–70. doi: 10.1073/pnas.96.6.3165
86. Augustus A, Yagyu H, Haemmerle G, Bensadoun A, Vikramadithyan RK, Park SY, et al. Cardiac-specific knock-out of lipoprotein lipase alters plasma lipoprotein triglyceride metabolism and cardiac gene expression. J Biol Chem. (2004) 279:25050–7. doi: 10.1074/jbc.M401028200
87. Bharadwaj KG, Hiyama Y, Hu Y, Huggins LA, Ramakrishnan R, Abumrad NA, et al. Chylomicron- and VLDL-derived lipids enter the heart through different pathways: in vivo evidence for receptor- and non-receptor-mediated fatty acid uptake. J Biol Chem. (2010) 285:37976–86. doi: 10.1074/jbc.M110.174458
88. Semenkovich CF, Coleman T, Daugherty A. Effects of heterozygous lipoprotein lipase deficiency on diet-induced atherosclerosis in mice. J Lipid Res. (1998) 39:1141–51.
89. Babaev VR, Fazio S, Gleaves LA, Carter KJ, Semenkovich CF, Linton MF. Macrophage lipoprotein lipase promotes foam cell formation and atherosclerosis in vivo. J Clin Invest. (1999) 103:1697–705. doi: 10.1172/JCI6117
90. Babaev VR, Patel MB, Semenkovich CF, Fazio S, Linton MF. Macrophage lipoprotein lipase promotes foam cell formation and atherosclerosis in low density lipoprotein receptor-deficient mice. J Biol Chem. (2000) 275:26293–9. doi: 10.1074/jbc.M002423200
91. Takahashi M, Yagyu H, Tazoe F, Nagashima S, Ohshiro T, Okada K, et al. Macrophage lipoprotein lipase modulates the development of atherosclerosis but not adiposity. J Lipid Res. (2013) 54:1124–34. doi: 10.1194/jlr.M035568
92. Ebara T, Okubo M, Horinishi A, Adachi M, Murase T, Hirano T. No evidence of accelerated atherosclerosis in a 66-yr-old chylomicronemia patient homozygous for the nonsense mutation (Tyr61 -> Stop) in the lipoprotein lipase gene. Atherosclerosis. (2001) 159:375–9. doi: 10.1016/S0021-9150(01)00510-X
93. Ebara T, Endo Y, Yoshiike S, Tsuji M, Taguchi S, Murase T, et al. A 60-y-old chylomicronemia patient homozygous for missense mutation (G188E) in the lipoprotein lipase gene showed no accelerated atherosclerosis. Clin Chim Acta. (2007) 386:100–4. doi: 10.1016/j.cca.2007.08.011
94. Kawashiri M, Higashikata T, Mizuno M, Takata M, Katsuda S, Miwa K, et al. Long-term course of lipoprotein lipase (LPL) deficiency due to homozygous LPLArita in a patient with recurrent pancreatitis, retained glucose tolerance, and atherosclerosis. J Clin Endocrinol Metabol. (2005) 90:6541–4. doi: 10.1210/jc.2005-1098
95. Nordestgaard BG, Abildgaard S, Wittrup HH, Steffensen R, Jensen G, Tybjaerg-Hansen A. Heterozygous lipoprotein lipase deficiency: frequency in the general population, effect on plasma lipid levels, and risk of ischemic heart disease. Circulation. (1997) 96:1737–44. doi: 10.1161/01.CIR.96.6.1737
96. Jukema JW, van Boven AJ, Groenemeijer B, Zwinderman AH, Reiber JH, Bruschke AV, et al. The Asp9 Asn mutation in the lipoprotein lipase gene is associated with increased progression of coronary atherosclerosis. REGRESS study group, interuniversity cardiology institute, utrecht, The Netherlands. Regression growth evaluation statin study. Circulation. (1996) 94:1913–8. doi: 10.1161/01.CIR.94.8.1913
97. Beigneux AP, Davies BS, Gin P, Weinstein MM, Farber E, Qiao X, et al. Glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 plays a critical role in the lipolytic processing of chylomicrons. Cell Metab. (2007) 5:279–91. doi: 10.1016/j.cmet.2007.02.002
98. Young SG, Fong LG, Beigneux AP, Allan CM, He C, Jiang H, et al. GPIHBP1 and lipoprotein lipase, partners in plasma triglyceride metabolism. Cell Metab. (2019) 30:51–65. doi: 10.1016/j.cmet.2019.05.023
99. Meng X, Zeng W, Young SG, Fong LG. GPIHBP1, a partner protein for lipoprotein lipase, is expressed only in capillary endothelial cells. J Lipid Res. (2020) 61:591. doi: 10.1194/jlr.ILR120000735
100. Davies BS, Beigneux AP, Barnes RH 2nd, Tu Y, Gin P, Weinstein MM, et al. GPIHBP1 is responsible for the entry of lipoprotein lipase into capillaries. Cell Metab. (2010) 12:42–52. doi: 10.1016/j.cmet.2010.04.016
101. Davies BS, Goulbourne CN, Barnes RH 2nd, Turlo KA, Gin P, Vaughan S, et al. Assessing mechanisms of GPIHBP1 and lipoprotein lipase movement across endothelial cells. J Lipid Res. (2012) 53:2690–7. doi: 10.1194/jlr.M031559
102. Goulbourne CN, Gin P, Tatar A, Nobumori C, Hoenger A, Jiang H, et al. The GPIHBP1-LPL complex is responsible for the margination of triglyceride-rich lipoproteins in capillaries. Cell Metab. (2014) 19:849–60. doi: 10.1016/j.cmet.2014.01.017
103. Mysling S, Kristensen KK, Larsson M, Beigneux AP, Gardsvoll H, Fong LG, et al. The acidic domain of the endothelial membrane protein GPIHBP1 stabilizes lipoprotein lipase activity by preventing unfolding of its catalytic domain. Elife. (2016) 5:e12095. doi: 10.7554/eLife.12095
104. Mysling S, Kristensen KK, Larsson M, Kovrov O, Bensadouen A, Jorgensen TJ, et al. The angiopoietin-like protein ANGPTL4 catalyzes unfolding of the hydrolase domain in lipoprotein lipase and the endothelial membrane protein GPIHBP1 counteracts this unfolding. eLife. (2016) 5:e20958. doi: 10.7554/eLife.20958
105. Arora R, Nimonkar AV, Baird D, Wang C, Chiu CH, Horton PA, et al. Structure of lipoprotein lipase in complex with GPIHBP1. Proc Natl Acad Sci USA. (2019) 116:10360–5. doi: 10.1073/pnas.1820171116
106. Birrane G, Beigneux AP, Dwyer B, Strack-Logue B, Kristensen KK, Francone OL, et al. Structure of the lipoprotein lipase-GPIHBP1 complex that mediates plasma triglyceride hydrolysis. Proc Natl Acad Sci USA. (2019) 116:1723–32. doi: 10.1073/pnas.1817984116
107. Liu X, Li J, Liao J, Wang H, Huang X, Dong Z, et al. Gpihbp1 deficiency accelerates atherosclerosis and plaque instability in diabetic Ldlr(-/-) mice. Atherosclerosis. (2019) 282:100–9. doi: 10.1016/j.atherosclerosis.2019.01.025
108. Paterniti JR Jr, Brown WV, Ginsberg HN, Artzt K. Combined lipase deficiency (cld): a lethal mutation on chromosome 17 of the mouse. Science. (1983) 221:167–9. doi: 10.1126/science.6857276
109. Peterfy M, Ben-Zeev O, Mao HZ, Weissglas-Volkov D, Aouizerat BE, Pullinger CR, et al. Mutations in LMF1 cause combined lipase deficiency and severe hypertriglyceridemia. Nat Genet. (2007) 39:1483–7. doi: 10.1038/ng.2007.24
110. Briquet-Laugier V, Ben-Zeev O, White A, Doolittle MH. cld and lec23 are disparate mutations that affect maturation of lipoprotein lipase in the endoplasmic reticulum. J Lipid Res. (1999) 40:2044–58.
111. Hosseini M, Ehrhardt N, Weissglas-Volkov D, Lai CM, Mao HZ, Liao JL, et al. Transgenic expression and genetic variation of Lmf1 affect LPL activity in mice and humans. Arteriosc Thromb Vasc Biol. (2012) 32:1204–10. doi: 10.1161/ATVBAHA.112.245696
112. Yin F, Doolittle MH, Peterfy M. A quantitative assay measuring the function of lipase maturation factor 1. J Lipid Res. (2009) 50:2265–9. doi: 10.1194/jlr.M900196-JLR200
113. Cefalu AB, Noto D, Arpi ML, Yin F, Spina R, Hilden H, et al. Novel LMF1 nonsense mutation in a patient with severe hypertriglyceridemia. J Clin Endocrinol Metab. (2009) 94:4584–90. doi: 10.1210/jc.2009-0594
114. Johansen CT, Wang J, McIntyre AD, Martins RA, Ban MR, Lanktree MB, et al. Excess of rare variants in non-genome-wide association study candidate genes in patients with hypertriglyceridemia. Circ Cardiovasc Genet. (2012) 5:66–72. doi: 10.1161/CIRCGENETICS.111.960864
115. Wolska A, Dunbar RL, Freeman LA, Ueda M, Amar MJ, Sviridov DO, et al. Apolipoprotein C-II: New findings related to genetics, biochemistry, and role in triglyceride metabolism. Atherosclerosis. (2017) 267:49–60. doi: 10.1016/j.atherosclerosis.2017.10.025
116. Zdunek J, Martinez GV, Schleucher J, Lycksell PO, Yin Y, Nilsson S, et al. Global structure and dynamics of human apolipoprotein CII in complex with micelles: evidence for increased mobility of the helix involved in the activation of lipoprotein lipase. Biochemistry. (2003) 42:1872–89. doi: 10.1021/bi0267184
117. Brown WV, Levy RI, Fredrickson DS. Further characterization of apolipoproteins from the human plasma very low density lipoproteins. J Biol Chem. (1970) 245:6588–94.
118. LaRosa JC, Levy RI, Herbert P, Lux SE, Fredrickson DS. A specific apoprotein activator for lipoprotein lipase. Biochem Biophys Res Commun. (1970) 41:57–62. doi: 10.1016/0006-291X(70)90468-7
119. Ueda M, Dunbar RL, Wolska A, Sikora TU, Escobar MDR, Seliktar N, et al. A novel APOC2 missense mutation causing apolipoprotein c-ii deficiency with severe triglyceridemia and pancreatitis. J Clin Endocrinol Metab. (2017) 102:1454–7. doi: 10.1210/jc.2016-3903
120. Sakurai T, Sakurai A, Vaisman BL, Amar MJ, Liu C, Gordon SM, et al. Creation of Apolipoprotein C-II (ApoC-II) mutant mice and correction of their hypertriglyceridemia with an ApoC-II Mimetic Peptide. J Pharmacol Exp Ther. (2016) 356:341–53. doi: 10.1124/jpet.115.229740
121. Pollin TI, Damcott CM, Shen H, Ott SH, Shelton J, Horenstein RB, et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science. (2008) 322:1702–5. doi: 10.1126/science.1161524
122. Khetarpal SA, Zeng X, Millar JS, Vitali C, Somasundara AVH, Zanoni P, et al. A human APOC3 missense variant and monoclonal antibody accelerate apoC-III clearance and lower triglyceride-rich lipoprotein levels. Nat Med. (2017) 23:1086–94. doi: 10.1038/nm.4390
123. Taskinen MR, Packard CJ, Boren J. Emerging evidence that apoc-iii inhibitors provide novel options to reduce the residual CVD. Curr Atheroscler Rep. (2019) 21:27. doi: 10.1007/s11883-019-0791-9
124. Eisenberg S, Olivecrona T. Very low density lipoprotein. Fate of phospholipids, cholesterol, and apolipoprotein C during lipolysis in vitro. J Lipid Res. (1979) 20:614–23.
125. Marcoux C, Tremblay M, Fredenrich A, Davignon J, Cohn JS. Lipoprotein distribution of apolipoprotein C-III and its relationship to the presence in plasma of triglyceride-rich remnant lipoproteins. Metabolism. (2001) 50:112–9. doi: 10.1053/meta.2001.19452
126. Larsson M, Allan CM, Jung RS, Heizer PJ, Beigneux AP, Young SG, et al. Apolipoprotein C-III inhibits triglyceride hydrolysis by GPIHBP1-bound LPL. J Lipid Res. (2017) 58:1893–902. doi: 10.1194/jlr.M078220
127. Kinnunen PK, Ehnolm C. Effect of serum and C-apoproteins from very low density lipoproteins on human postheparin plasma hepatic lipase. FEBS Lett. (1976) 65:354–7. doi: 10.1016/0014-5793(76)80145-7
128. Ramms B, Patel S, Nora C, Pessentheiner AR, Chang MW, Green CR, et al. ApoC-III ASO promotes tissue LPL activity in the absence of apoE-mediated TRL clearance. J Lipid Res. (2019) 60:1379–95. doi: 10.1194/jlr.M093740
129. Jong MC, van Ree JH, Dahlmans VE, Frants RR, Hofker MH, Havekes LM. Reduced very-low-density lipoprotein fractional catabolic rate in apolipoprotein C1-deficient mice. Biochem J. (1997) 321 (Pt 2):445–50. doi: 10.1042/bj3210445
130. Ito Y, Azrolan N, O'Connell A, Walsh A, Breslow JL. Hypertriglyceridemia as a result of human apo CIII gene expression in transgenic mice. Science. (1990) 249:790–3. doi: 10.1126/science.2167514
131. Yingchun H, Yahong M, Jiangping W, Xiaokui H, Xiaohong Z. Increased inflammation, endoplasmic reticulum stress and oxidative stress in endothelial and macrophage cells exacerbate atherosclerosis in ApoCIII transgenic mice. Lipids Health Dis. (2018) 17:220. doi: 10.1186/s12944-018-0867-5
132. Zewinger S, Reiser J, Jankowski V, Alansary D, Hahm E, Triem S, et al. Apolipoprotein C3 induces inflammation and organ damage by alternative inflammasome activation. Nat Immunol. (2020) 21:30–41. doi: 10.1038/s41590-019-0548-1
133. Kawakami A, Aikawa M, Libby P, Alcaide P, Luscinskas FW, Sacks FM. Apolipoprotein CIII in apolipoprotein B lipoproteins enhances the adhesion of human monocytic cells to endothelial cells. Circulation. (2006) 113:691–700. doi: 10.1161/CIRCULATIONAHA.105.591743
134. Kawakami A, Aikawa M, Alcaide P, Luscinskas FW, Libby P, Sacks FM. Apolipoprotein CIII induces expression of vascular cell adhesion molecule-1 in vascular endothelial cells and increases adhesion of monocytic cells. Circulation. (2006) 114:681–7. doi: 10.1161/CIRCULATIONAHA.106.622514
135. Pennacchio LA, Olivier M, Hubacek JA, Cohen JC, Cox DR, Fruchart JC, et al. An apolipoprotein influencing triglycerides in humans and mice revealed by comparative sequencing. Science. (2001) 294:169–73. doi: 10.1126/science.1064852
136. Nelbach L, Shu X, Konrad RJ, Ryan RO, Forte TM. Effect of apolipoprotein A-V on plasma triglyceride, lipoprotein size, and composition in genetically engineered mice. J Lipid Res. (2008) 49:572–80. doi: 10.1194/jlr.M700281-JLR200
137. Camporez JP, Kanda S, Petersen MC, Jornayvaz FR, Samuel VT, Bhanot S, et al. ApoA5 knockdown improves whole-body insulin sensitivity in high-fat-fed mice by reducing ectopic lipid content. J Lipid Res. (2015) 56:526–36. doi: 10.1194/jlr.M054080
138. Merkel M, Loeffler B, Kluger M, Fabig N, Geppert G, Pennacchio LA, et al. Apolipoprotein AV accelerates plasma hydrolysis of triglyceride-rich lipoproteins by interaction with proteoglycan-bound lipoprotein lipase. J Biol Chem. (2005) 280:21553–60. doi: 10.1074/jbc.M411412200
139. Kluger M, Heeren J, Merkel M. Apoprotein A-V: an important regulator of triglyceride metabolism. J Inherit Metab Dis. (2008) 31:281–8. doi: 10.1007/s10545-008-0863-4
140. Lee JH, Giannikopoulos P, Duncan SA, Wang J, Johansen CT, Brown JD, et al. The transcription factor cyclic AMP-responsive element-binding protein H regulates triglyceride metabolism. Nat Med. (2011) 17:812–5. doi: 10.1038/nm.2347
141. Dron JS, Wang J, Cao H, McIntyre AD, Iacocca MA, Menard JR, et al. Severe hypertriglyceridemia is primarily polygenic. J Clin Lipidol. (2019) 13:80–8. doi: 10.1016/j.jacl.2018.10.006
142. Hegele RA, Ginsberg HN, Chapman MJ, Nordestgaard BG, Kuivenhoven JA, Averna M, et al. The polygenic nature of hypertriglyceridaemia: implications for definition, diagnosis, and management. Lancet Diabetes Endocrinol. (2014) 2:655–66. doi: 10.1016/S2213-8587(13)70191-8
143. Bennett BJ, Farber CR, Orozco L, Kang HM, Ghazalpour A, Siemers N, et al. A high-resolution association mapping panel for the dissection of complex traits in mice. Genome Res. (2010) 20:281–90. doi: 10.1101/gr.099234.109
144. Norheim F, Hasin-Brumshtein Y, Vergnes L, Chella Krishnan K, Pan C, Seldin MM, et al. Gene-by-sex interactions in mitochondrial functions and cardio-metabolic traits. Cell Metab. (2019) 29:932–49 e4. doi: 10.1016/j.cmet.2018.12.013
145. Bennett BJ, Davis RC, Civelek M, Orozco L, Wu J, Qi H, et al. Genetic architecture of atherosclerosis in mice: a systems genetics analysis of common inbred strains. PLoS Genet. (2015) 11:e1005711. doi: 10.1371/journal.pgen.1005711
146. Willecke F, Scerbo D, Nagareddy P, Obunike JC, Barrett TJ, Abdillahi ML, et al. Lipolysis, and not hepatic lipogenesis, is the primary modulator of triglyceride levels in streptozotocin-induced diabetic mice. Arteriosc Thromb Vasc Biol. (2015) 35:102–10. doi: 10.1161/ATVBAHA.114.304615
147. Chen K, Wu Q, Hu K, Yang C, Wu X, Cheung P, et al. Suppression of hepatic FLOT1 (Flotillin-1) by type 2 diabetes mellitus impairs the disposal of remnant lipoproteins via syndecan-1. Arteriosc Thromb Vasc Biol. (2018) 38:102–13. doi: 10.1161/ATVBAHA.117.310358
148. Redgrave TG, Small DM. Quantitation of the transfer of surface phospholipid of chylomicrons to the high density lipoprotein fraction during the catabolism of chylomicrons in the rat. J Clin Invest. (1979) 64:162–71. doi: 10.1172/JCI109435
149. Tall AR, Green PH, Glickman RM, Riley JW. Metabolic fate of chylomicron phospholipids and apoproteins in the rat. J Clin Invest. (1979) 64:977–89. doi: 10.1172/JCI109564
150. Dolphin PJ, Stewart B, Amy RM, Russell JC. Serum lipids and lipoproteins in the atherosclerosis prone LA/N corpulent rat. Biochim Biophys Acta. (1987) 919:140–8. doi: 10.1016/0005-2760(87)90200-1
151. Diane A, Pierce WD, Kelly SE, Sokolik S, Borthwick F, Jacome-Sosa M, et al. Mechanisms of comorbidities associated with the metabolic syndrome: insights from the JCR:LA-cp corpulent rat strain. Front Nutr. (2016) 3:44. doi: 10.3389/fnut.2016.00044
152. Mangat R, Su J, Scott PG, Russell JC, Vine DF, Proctor SD. Chylomicron and apoB48 metabolism in the JCR:LA corpulent rat, a model for the metabolic syndrome. Biochem Soc Trans. (2007) 35:477–81. doi: 10.1042/BST0350477
153. Wei S, Zhang Y, Su L, He K, Wang Q, Zhang Y, et al. Apolipoprotein E-deficient rats develop atherosclerotic plaques in partially ligated carotid arteries. Atherosclerosis. (2015) 243:589–92. doi: 10.1016/j.atherosclerosis.2015.10.093
154. Lee JG, Ha CH, Yoon B, Cheong SA, Kim G, Lee DJ, et al. Knockout rat models mimicking human atherosclerosis created by Cpf1-mediated gene targeting. Sci Rep. (2019) 9:2628. doi: 10.1038/s41598-019-38732-2
155. Hirano T, Mamo JCL, Poapst ME, Kuksis A, Steiner G. Impaired very low-density lipoprotein-triglyceride catabolism in acute and chronic fructose-fed rats. Am J Physiol. (1989) 256:E559–E65. doi: 10.1152/ajpendo.1989.256.4.E559
156. Kawashima Y, Eguchi Y, Yamazaki T, Karahashi M, Kawai H, Kudo N. Reduction in secretion of very low density lipoprotein-triacylglycerol by a matrix metalloproteinase inhibitor in a rat model of diet-induced hypertriglyceridemia. J Pharmacol Exp Ther. (2018) 366:194–204. doi: 10.1124/jpet.117.246165
157. Heinonen SE, Genove G, Bengtsson E, Hubschle T, Akesson L, Hiss K, et al. Animal models of diabetic macrovascular complications: key players in the development of new therapeutic approaches. J Diabetes Res. (2015) 2015:404085. doi: 10.1155/2015/404085
158. Chapman MJ. Animal lipoproteins: chemistry, structure, and comparative aspects. J Lipid Res. (1980) 21:789–853.
159. Greeve J, Altkemper I, Dieterich JH, Greten H, Windler E. Apolipoprotein B mRNA editing in 12 different mammalian species: hepatic expression is reflected in low concentrations of apoB-containing plasma lipoproteins. J Lipid Res. (1993) 34:1367–83.
160. Nagashima M, McLean JW, Lawn RM. Cloning and mRNA tissue distribution of rabbit cholesteryl ester transfer protein. J Lipid Res. (1988) 29:1643–9.
161. Yang BC, Phillips MI, Mohuczy D, Meng H, Shen L, Mehta P, et al. Increased angiotensin II type 1 receptor expression in hypercholesterolemic atherosclerosis in rabbits. Arteriosc Thromb Vasc Biol. (1998) 18:1433–9. doi: 10.1161/01.ATV.18.9.1433
162. Clay MA, Hopkins GJ, Ehnholm CP, Barter PJ. The rabbit as an animal model of hepatic lipase deficiency. Biochim Biophys Acta. (1989) 1002:173–81. doi: 10.1016/0005-2760(89)90284-1
163. Mitsuguchi Y, Ito T, Ohwada K. Pathologic findings in rabbit models of hereditary hypertriglyceridemia and hereditary postprandial hypertriglyceridemia. Comp Med. (2008) 58:465–80. Available online at: https://www.ingentaconnect.com/content/aalas/cm/2008/00000058/00000005/art00007
164. Kawai T, Ito T, Ohwada K, Mera Y, Matsushita M, Tomoike H. Hereditary postprandial hypertriglyceridemic rabbit exhibits insulin resistance and central obesity - A novel model of metabolic syndrome. Arteriosc Thrombosis Vasc Biol. (2006) 26:2752–7. doi: 10.1161/01.ATV.0000245808.12493.40
165. Wei J, Ouyang H, Wang Y, Pang D, Cong NX, Wang T, et al. Characterization of a hypertriglyceridemic transgenic miniature pig model expressing human apolipoprotein CIII. FEBS J. (2012) 279:91–9. doi: 10.1111/j.1742-4658.2011.08401.x
166. Holmes RS, Cox LA. Comparative studies of glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1: evidence for a eutherian mammalian origin for the GPIHBP1 gene from an LY6-like gene. 3 Biotech. (2012). 2:37–52. doi: 10.1007/s13205-011-0026-4
167. Bremer AA, Stanhope KL, Graham JL, Cummings BP, Wang W, Saville BR, et al. Fructose-fed rhesus monkeys: a nonhuman primate model of insulin resistance, metabolic syndrome, and type 2 diabetes. Clin Transl Sci. (2011) 4:243–52. doi: 10.1111/j.1752-8062.2011.00298.x
168. Butler AA, Price CA, Graham JL, Stanhope KL, King S, Hung YH, et al. Fructose-induced hypertriglyceridemia in rhesus macaques is attenuated with fish oil or ApoC3 RNA interference. J Lipid Res. (2019) 60:805–18. doi: 10.1194/jlr.M089508
169. Graham MJ, Lee RG, Bell TA, 3rd Fu W, Mullick AE, Alexander VJ, et al. Antisense oligonucleotide inhibition of apolipoprotein C-III reduces plasma triglycerides in rodents, nonhuman primates, and humans. Circ Res. (2013) 112:1479–90. doi: 10.1161/CIRCRESAHA.111.300367
170. Havel PJ, Kievit P, Comuzzie AG, Bremer AA. Use and importance of nonhuman primates in metabolic disease research: current state of the field. ILAR J. (2017) 58:251–68. doi: 10.1093/ilar/ilx031
171. Saleheen D, Natarajan P, Armean IM, Zhao W, Rasheed A, Khetarpal SA, et al. Human knockouts and phenotypic analysis in a cohort with a high rate of consanguinity. Nature. (2017) 544:235–9. doi: 10.1038/nature22034
172. Tg, Hdl Working Group of the Exome Sequencing Project NHL, Blood I, Crosby J, Peloso GM, Auer PL, et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med. (2014) 371:22–31. doi: 10.1056/NEJMoa1307095
173. Jorgensen AB, Frikke-Schmidt R, Nordestgaard BG, Tybjaerg-Hansen A. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med. (2014) 371:32–41. doi: 10.1056/NEJMoa1308027
174. Pechlaner R, Tsimikas S, Yin X, Willeit P, Baig F, Santer P, et al. Very-low-density lipoprotein-associated apolipoproteins predict cardiovascular events and are lowered by inhibition of APOC-III. J Am Coll Cardiol. (2017) 69:789–800. doi: 10.1016/j.jacc.2016.11.065
175. Gaudet D, Alexander VJ, Baker BF, Brisson D, Tremblay K, Singleton W, et al. Antisense Inhibition of apolipoprotein C-III in patients with hypertriglyceridemia. N Engl J Med. (2015) 373:438–47. doi: 10.1056/NEJMoa1400283
176. Gaudet D, Brisson D, Tremblay K, Alexander VJ, Singleton W, Hughes SG, et al. Targeting APOC3 in the familial chylomicronemia syndrome. N Engl J Med. (2014) 371:2200–6. doi: 10.1056/NEJMoa1400284
177. Witztum JL, Gaudet D, Freedman SD, Alexander VJ, Digenio A, Williams KR, et al. Volanesorsen and triglyceride levels in familial chylomicronemia syndrome. N Engl J Med. (2019) 381:531–42. doi: 10.1056/NEJMoa1715944
178. Gouni-Berthold I. The role of antisense oligonucleotide therapy against apolipoprotein-CIII in hypertriglyceridemia. Atheroscler Suppl. (2017) 30:19–27. doi: 10.1016/j.atherosclerosissup.2017.05.003
179. Joglekar K, Brannick B, Kadaria D, Sodhi A. Therapeutic plasmapheresis for hypertriglyceridemia-associated acute pancreatitis: case series and review of the literature. Ther Adv Endocrinol Metab. (2017) 8:59–65. doi: 10.1177/2042018817695449
180. Amar MJ, Sakurai T, Sakurai-Ikuta A, Sviridov D, Freeman L, Ahsan L, et al. A novel apolipoprotein C-II mimetic peptide that activates lipoprotein lipase and decreases serum triglycerides in apolipoprotein E-knockout mice. J Pharmacol Exp Ther. (2015) 352:227–35. doi: 10.1124/jpet.114.220418
181. Wolska A, Lo L, Sviridov DO, Pourmousa M, Pryor M, Ghosh SS, et al. A dual apolipoprotein C-II mimetic-apolipoprotein C-III antagonist peptide lowers plasma triglycerides. Sci Transl Med. (2020) 12(528). doi: 10.1126/scitranslmed.aaw7905
182. Stitziel NO, Khera AV, Wang X, Bierhals AJ, Vourakis AC, Sperry AE, et al. ANGPTL3 deficiency and protection against coronary artery disease. J Am Coll Cardiol. (2017) 69:2054–63. doi: 10.1016/j.jacc.2017.02.030
183. Chi X, Britt EC, Shows HW, Hjelmaas AJ, Shetty SK, Cushing EM, et al. ANGPTL8 promotes the ability of ANGPTL3 to bind and inhibit lipoprotein lipase. Mol Metab. (2017) 6:1137–49. doi: 10.1016/j.molmet.2017.06.014
184. Dewey FE, Gusarova V, Dunbar RL, O'Dushlaine C, Schurmann C, Gottesman O, et al. Genetic and pharmacologic inactivation of ANGPTL3 and cardiovascular disease. N Engl J Med. (2017) 377:211–21. doi: 10.1056/NEJMoa1612790
185. Gusarova V, Alexa CA, Wang Y, Rafique A, Kim JH, Buckler D, et al. ANGPTL3 blockade with a human monoclonal antibody reduces plasma lipids in dyslipidemic mice and monkeys. J Lipid Res. (2015) 56:1308–17. doi: 10.1194/jlr.M054890
186. Chadwick AC, Evitt NH, Lv W, Musunuru K. Reduced blood lipid levels with in vivo CRISPR-Cas9 base editing of ANGPTL3. Circulation. (2018) 137:975–7. doi: 10.1161/CIRCULATIONAHA.117.031335
187. Kersten S, Mandard S, Tan NS, Escher P, Metzger D, Chambon P, et al. Characterization of the fasting-induced adipose factor FIAF, a novel peroxisome proliferator-activated receptor target gene. J Biol Chem. (2000) 275:28488–93. doi: 10.1074/jbc.M004029200
188. Yoon JC, Chickering TW, Rosen ED, Dussault B, Qin Y, Soukas A, et al. Peroxisome proliferator-activated receptor gamma target gene encoding a novel angiopoietin-related protein associated with adipose differentiation. Mol Cell Biol. (2000) 20:5343–9. doi: 10.1128/MCB.20.14.5343-5349.2000
189. Kim I, Kim HG, Kim H, Kim HH, Park SK, Uhm CS, et al. Hepatic expression, synthesis and secretion of a novel fibrinogen/angiopoietin-related protein that prevents endothelial-cell apoptosis. Biochem J. (2000) 346:603–10. doi: 10.1042/bj3460603
190. Koster A, Chao YB, Mosior M, Ford A, Gonzalez-DeWhitt PA, Hale JE, et al. Transgenic angiopoietin-like (angptl)4 overexpression and targeted disruption of angptl4 and angptl3: regulation of triglyceride metabolism. Endocrinology. (2005) 146:4943–50. doi: 10.1210/en.2005-0476
191. Yoshida K, Shimizugawa T, Ono M, Furukawa H. Angiopoietin-like protein 4 is a potent hyperlipidemia-inducing factor in mice and inhibitor of lipoprotein lipase. J Lipid Res. (2002) 43:1770–2. doi: 10.1194/jlr.C200010-JLR200
192. Klarin D, Damrauer SM, Cho K, Sun YV, Teslovich TM, Honerlaw J, et al. Genetics of blood lipids among similar to 300,000 multi-ethnic participants of the million veteran program. Nat Genet. (2018) 50:1514. doi: 10.1038/s41588-018-0222-9
193. Yang Q, Yin RX, Cao XL, Huang F, Zhou YJ, Chen WX. ANGPTL4 variants and their haplotypes are associated with serum lipid levels, the risk of coronary artery disease and ischemic stroke and atorvastatin cholesterol-lowering responses. Nutr Metabol. (2018) 15:70. doi: 10.1186/s12986-018-0308-5
194. Dewey FF, Gusarova V, O'Dushlaine C, Gottesman O, Trejos J, Hunt C, et al. Inactivating variants in ANGPTL4 and risk of coronary artery disease. N Engl J Med. (2016) 374:1123–33. doi: 10.1056/NEJMoa1510926
195. Lichtenstein L, Mattijssen F, de Wit NJ, Georgiadi A, Hooiveld GJ, van der Meer R, et al. Angptl4 protects against severe proinflammatory effects of saturated fat by inhibiting fatty acid uptake into mesenteric lymph node macrophages. Cell Metab. (2010) 12:580–92. doi: 10.1016/j.cmet.2010.11.002
Keywords: angiopoietin-like 3, animal model, apolipoprotein, atherosclerosis, lipoprotein lipase, hypertriglyceridemia
Citation: Basu D and Bornfeldt KE (2020) Hypertriglyceridemia and Atherosclerosis: Using Human Research to Guide Mechanistic Studies in Animal Models. Front. Endocrinol. 11:504. doi: 10.3389/fendo.2020.00504
Received: 22 February 2020; Accepted: 23 June 2020;
Published: 06 August 2020.
Edited by:
Marja-Riitta Taskinen, University of Helsinki, FinlandReviewed by:
Aldons Jake Lusis, University of California, United StatesGodfrey Getz, University of Chicago, United States
Copyright © 2020 Basu and Bornfeldt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karin E. Bornfeldt, Ym9ybmZAdXcuZWR1