 Brittany A. McKelvey
Brittany A. McKelvey Christopher B. Umbricht
Christopher B. Umbricht Martha A. Zeiger
Martha A. Zeiger
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 31 July 2020
Sec. Cancer Endocrinology
Volume 11 - 2020 | https://doi.org/10.3389/fendo.2020.00485
This article is part of the Research Topic Molecular Mechanisms in Cancer Endocrinology with Clinical Implications View all 12 articles
Telomerase reverse transcriptase (TERT) is the catalytic subunit of the enzyme telomerase and is essential for telomerase activity. Upregulation of TERT expression and resulting telomerase activity occurs in the large majority of malignancies, including thyroid cancer. This upregulation results in continued cellular proliferation and avoidance of cellular senescence and cell death. In this review we will briefly introduce TERT and telomerase activity as it pertains to thyroid cancer and, highlight the effects of TERT on cancer cells. We will also explore in detail the different TERT regulatory strategies and how TERT is reactivated in thyroid cancer cells, specifically. These regulatory mechanisms include both activating single base pair TERT promoter mutations and epigenetic changes at the promoter, including changes in CpG methylation and histone modifications that affect chromatin structure. Further, regulation includes the allele-specific regulation of the TERT promoter in thyroid cancer cells harboring the TERT promoter mutation. These entail allele-specific transcriptional activator binding, DNA methylation, histone modifications, and mono-allelic expression of TERT. Lastly, TERT copy number alterations and alternative splicing are also implicated. Both amplifications of the TERT locus and increased full-length transcripts and decreased inactive and dominant negative isoforms result in active telomerase. Finally, the clinical significance of TERT in thyroid cancer is also reviewed.
In contrast to stem cells, non-transformed somatic cells have a limited capacity to divide in tissue culture before cell division ceases. This is known as cellular senescence and the number of times a normal human somatic cell population will divide before cell division stops is referred to as the Hayflick limit (1). Cellular senescence results from the progressive shortening of chromosomal ends or telomeres, consisting of identical hexamer repeats, with each cell division. This phenomenon is due to the end replication problem, a shortcoming of semiconservative DNA replication, which cannot complete the synthesis of chromosomal ends (2). This critical shortening of telomeres continues until reactivation of the enzyme, telomerase, facilitates the resynthesizing of telomeres of sufficient length to allow continued cellular replication. Telomere lengthening can also occasionally occur through the alternative lengthening of telomeres (ALT) pathway, which extends telomeres without telomerase activity through recombination-dependent pathways. The ALT pathway remains poorly understood and will not be covered in this review of TERT regulation (3).
Telomerase is composed of two subunits: an RNA component, telomerase RNA (TR), serving as a template for telomere hexamer repeat addition onto DNA and, the catalytic component telomerase reverse transcriptase (TERT), responsible for reverse transcribing the hexamer repeats onto chromosomal ends (4). Not only is telomerase active in embryonic and stem cells, it is also upregulated in over 90% of malignancies, including thyroid cancer, enabling the unlimited replication of cancer cells (5–8).
TERT activation in cancer occurs through a variety of mechanisms. These include activating promoter mutations, alterations in promoter DNA methylation, chromatin remodeling, copy number alterations, and alternative splicing of TERT (9–13). Because telomerase plays a key role in carcinogenesis, understanding TERT regulation and telomerase activation in thyroid cancer is critical to understanding its pathogenesis. Indeed, two TERT regulatory mechanisms, TERT promoter mutations and TERT promoter methylation status, are implicated in the stratification of thyroid cancer patient prognosis (14–16).
In this review, we summarize our current understanding of the multiple mechanisms of TERT regulation in thyroid cancer. Activating point mutations in the TERT promoter markedly activate TERT transcription and are associated with epigenetic alterations observed in thyroid cancer cell lines and patient tumors. These epigenetic alterations include promoter methylation patterns and histone tail methylation modifications. The modifications have also been shown to affect the TERT promoter in an allele-specific manner, the details of which we will elucidate. Further, the TERT locus has also been found to have multiple chromosomal copies or, be amplified in cancer with associated increased TERT transcription. Lastly, post-transcription, the TERT transcript undergoes alternative splicing to form either the full-length transcript, which is the active isoform, or other inactive isoforms, which may also have regulatory functions. Finally, we highlight the clinical implications of TERT activation and, describe future potential avenues to explore in order to better understand regulation of this key enzyme as well as its role in carcinogenesis and cancer prognosis.
Activation of TERT transcription can be achieved by heterozygous point mutations at the TERT promoter. The two most common activating mutations are upstream of the TERT translation start site at −124 and −146, respectively, and both are cytosine to thymine mutations, −124C>T and −146 C>T (Figure 1B) (17–19). These TERT promoter mutations are not present in benign thyroid tumors or normal thyroid cells. Their prevalence, however, increases with more aggressive thyroid cancer subtypes and advanced stages of disease. Indeed, TERT mutations are present in only 11.3% of well-differentiated papillary thyroid cancer (PTC) and 17.1% of follicular thyroid cancer (FTC), but present in 32% of widely invasive Hürthle cell carcinoma (HCC), compared to 5% of minimally invasive HCC, and in 43.2% of poorly differentiated thyroid cancer (PDTC) and 40.1% of anaplastic thyroid cancer (ATC) (20, 21). Furthermore, the TERT promoter mutations are found in almost all thyroid cancer cell lines, even those derived from well-differentiated thyroid cancer (DTC) subtypes, in either the heterozygous or homozygous mutant state, indicative of their important role in cellular proliferation (11).
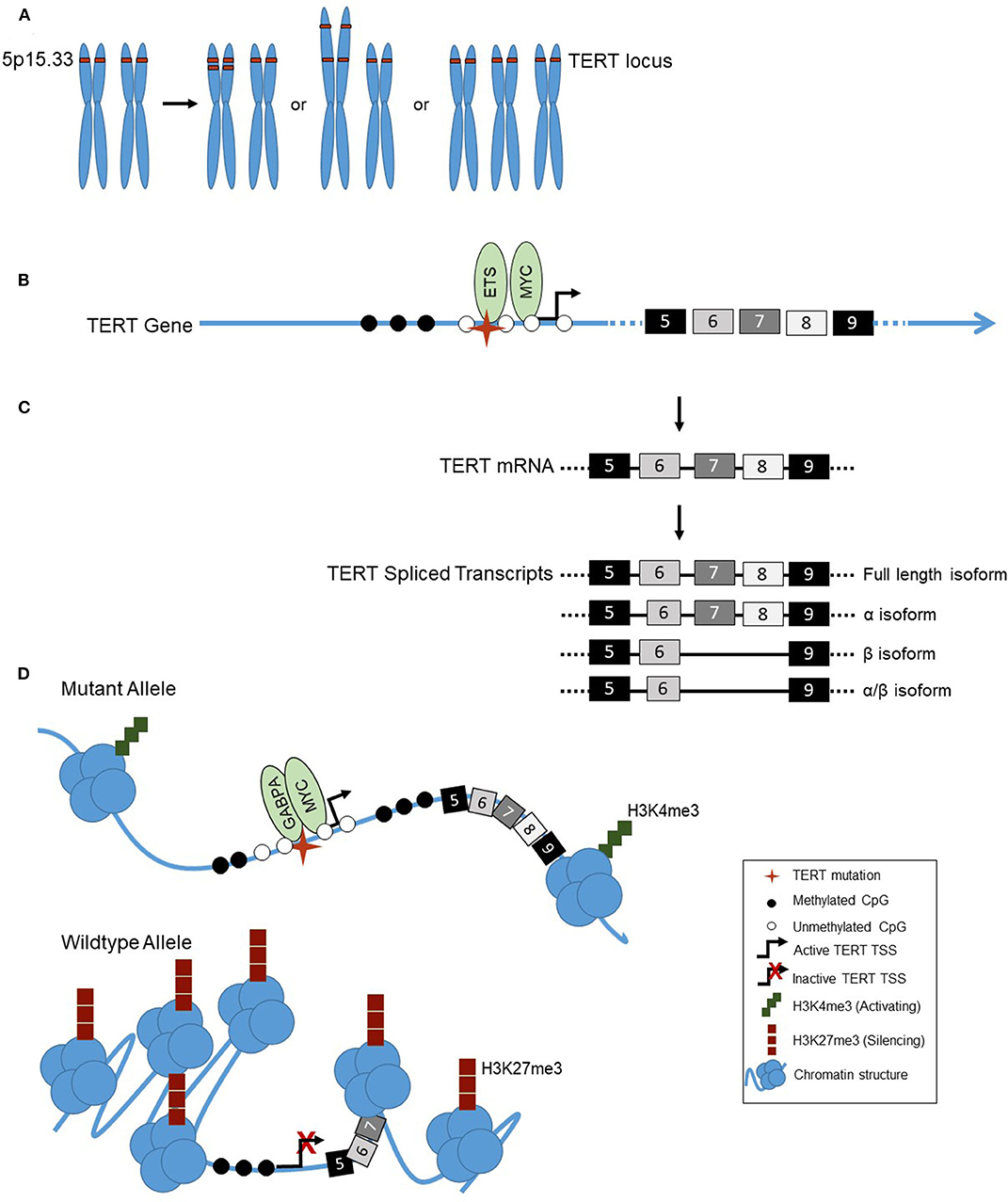
Figure 1. (A) Examples of TERT copy number amplifications (chromosomal location 5p15.33): Focal copy number alterations, arm-level alterations, or chromosomal duplications (trisomy 5). (B) The landscape of the TERT promoter in thyroid cancer shows a lack of methylation (◯) surrounding the transcription start site, TSS, ( ) and TERT promoter mutation (
) and TERT promoter mutation ( ), with hypermethylation further upstream of the TSS (
), with hypermethylation further upstream of the TSS ( ). Transcriptional activators MYC and ETS family factors bind at the TERT promoter, the latter binding in the presence of the TERT mutation only. (C) Alternative splicing of TERT in thyroid cancer. TERT is transcribed into mRNA, containing 16 exons (#5–9 shown). TERT can be alternatively spliced, into either the full length, α, β, or α/β isoforms. (D) Allele-specific regulation of the TERT promoter in thyroid cancer. The TERT mutant allele, with mutation () is in an open chromatin conformation, with histones spread across the DNA, depicted by the cluster of blue circles. The wildtype allele is in the closed chromatin conformation, with compacted histones on the DNA. The mutant TERT allele is associated with activating H3K4me3 histone marks, represented by the green squares, a CpG unmethylated (◯) promoter and methylated () gene body. The transcription factors GABPA and MYC bind at the promoter, and TERT is actively transcribed. The wildtype allele is associated with silencing H3K27me3 histone marks, represented by red squares, and a CpG methylated promoter.
). Transcriptional activators MYC and ETS family factors bind at the TERT promoter, the latter binding in the presence of the TERT mutation only. (C) Alternative splicing of TERT in thyroid cancer. TERT is transcribed into mRNA, containing 16 exons (#5–9 shown). TERT can be alternatively spliced, into either the full length, α, β, or α/β isoforms. (D) Allele-specific regulation of the TERT promoter in thyroid cancer. The TERT mutant allele, with mutation () is in an open chromatin conformation, with histones spread across the DNA, depicted by the cluster of blue circles. The wildtype allele is in the closed chromatin conformation, with compacted histones on the DNA. The mutant TERT allele is associated with activating H3K4me3 histone marks, represented by the green squares, a CpG unmethylated (◯) promoter and methylated () gene body. The transcription factors GABPA and MYC bind at the promoter, and TERT is actively transcribed. The wildtype allele is associated with silencing H3K27me3 histone marks, represented by red squares, and a CpG methylated promoter.
As seen in the TCGA data, the presence a TERT mutation is also strongly associated with high risk of tumor recurrence, older age, higher MACIS (Metastasis, patient Age, Completeness of resection, local Invasion, and tumor Size) scores, and less-differentiated PTC (22). In support of the TCGA data, in a large study of thyroid cancers in which anaplastic carcinoma coexisted with papillary carcinoma, a multivariate comparison between the antecedent papillary carcinoma components and control papillary carcinomas without anaplastic transformation showed that TERT mutations were independently associated with anaplastic transformation (23). Further studies have also highlighted the association of the TERT promoter mutation and non-radioiodine avidity of metastatic disease (24, 25).
Likely key to their effect, both TERT promoter mutations result in a novel 11 base pair binding site for the E-twenty-six (ETS) transcription factor family. The −124C>T mutation, however, elicits a higher activation of TERT compared to the −146C>T mutation and, is more common (20, 26). The activating GA Binding Protein Transcription Factor Subunit Alpha (GABPA), a member of the ETS family, binds at the TERT promoter mutation site as a heterotetramer with its counterpart, GABPB, in multiple cancer types, including thyroid cancer (26–29). In PTC cell lines, GABPA knockdown significantly lowered TERT expression in both wildtype and TERT promoter mutant cells, and in a luciferase activity assay, GABPA knockdown led to significant down regulation of the mutant TERT promoter. However, in an analysis of papillary thyroid cancers and TCGA data, an inverse relationship was found between GABPA expression and TERT expression in TERT promoter mutant tumors, highlighting the need for further study of TERT mutation activation (30).
Recently, another ETS factor, ETS Variant 5 (ETV5), was also found to bind the mutant TERT promoter and, bound at higher levels than GABPA in ATC cell lines. Conversely, GABPA bound at higher levels than ETV5 in well-differentiated thyroid cancer-derived cell lines (31), implicating potentially different roles of these specific ETS factors in well-differentiated and poorly differentiated cancers.
The BRAFV600E mutation is also common in thyroid cancer. The BRAFV600E mutation is a single nucleotide mutation at codon 600, resulting in a substitution of glutamic acid for valine in the BRAF protein, a serine/threonine protein kinase that plays a role in the mitogen-activated protein kinase (MAPK) pathway/ERK signaling pathway. The BRAFV600E mutation is more abundant in classic PTC (51%) than in the follicular variant of PTC (FVPTC, 24%) or FTC (1.4%) (32). The BRAFV600E mutation acts synergistically with TERT promoter mutations and the presence of both mutations is associated with greater cancer aggressiveness, lymph node and distant metastasis, advanced tumor stage, recurrence, and increased mortality in patients with PTC (33–35). The mutation may also be responsible for increased activation of the ETS factor family through activation of the MAPK pathway. There are two prevailing proposed mechanisms for the activation of TERT by the BRAFV600E mutation and, the MAPK pathway. In one study of PTC cell lines, the MAPK pathway activates the transcription factor c-FOS, which can in turn bind to the GABPB promoter to increase its expression. This then leads to increase in GABPA-GABPB complex formation, which binds to the mutant TERT promoter to activate TERT (27). Alternatively, another study in PTCs from the TCGA study cohort shows upregulation of ETS factors ETV1, ETV4, and ETV5 in tumors that concomitantly harbored both BRAFV600E and TERT mutations and, demonstrated MAPK pathway activation. In cell lines, these ETS factors were found to bind to the mutant TERT promoter and upregulate transcription (36).
Epigenetic changes are stably inherited during cell division and occur at DNA or chromatin levels, but do not alter the primary base sequence (37). At the DNA level, epigenetic alterations occur by methylation of the cytosine nucleotide in cytosine-guanine dinucleotides (CpGs). CpG methylation surrounding the transcriptional start site (TSS) at gene promoters is usually associated with suppression of gene expression. Conversely, the absence of DNA methylation surrounding the TSS of genes is associated with active transcription. Transcription factors that bind and regulate gene promoters can also be methylation sensitive, where methylation impedes binding (38). The TERT promoter is within a CpG island and has a rich GC content of ~70% (39). Previous studies in a variety of cancer cell lines as well as solid tumors and hematological malignancies show a unique methylation pattern of TERT. Its promoter region surrounding the TSS [−200 to +100 relative to TSS] is unmethylated, while the upstream promoter region [−650 to −200] is hypermethylated (Figure 1B) (13, 40). The unmethylated TSS allows for transcription to occur by availing its binding sites for activators, such as MYC, and when the TERT promoter mutation is present, ETS family factors (41). The role of the hypermethylated upstream promoter, and its association with TERT expression, however, is unknown.
Our recent study in DTC cell lines and normal thyroid tissue confirmed that this dual methylation pattern observed in other cancer types is also seen in thyroid cancer (28). Five DTC cell lines exhibited low levels of methylation surrounding the TERT TSS while the upstream region was largely methylated. Comparatively, normal thyroid tissue showed overall low levels of methylation throughout the TERT promoter compared to the thyroid cancer cell lines (28, 29). Ours is the only study to date that comprehensively characterized TERT promoter methylation, as previous studies have focused on the hypermethylated upstream region only. A 2018 study of follicular neoplasms found the upstream TERT promoter was methylated significantly higher in FTC (13%) than follicular adenoma (8%) (42). Lastly, a study of upstream TERT promoter methylation patterns in 312 PTC, FTC, MTC, and ATC patients found significantly higher upstream DNA methylation in patients with cancer recurrence than in patients whose tumors did not recur. In patients with a recurrence, upstream TERT promoter methylation was also associated with higher tumor stage and the presence of lymph node metastasis (15).
A 2016 study of medullary thyroid cancer (MTC) and normal thyroid tissue samples quantified the TERT upstream promoter methylation at eight CpG sites. Sporadic MTCs had a significantly higher methylation level (12–90.3%) compared to normal thyroid tissues (~10%). And, this methylation pattern positively correlated with expression of TERT and telomerase activation. Further, high methylation of the upstream TERT promoter correlated with shortened survival in patients with MTC (16).
In addition to methylation aberrations, epigenetic changes in the histone code can also modify gene expression by altering the chromatin state from a closed, inactive state to an open, actively transcribed state. The histone mark H3K27me3 maintains heterochromatin and recruitment of the polycomb repressor complex (PRC) for silenced genes (43). Alternatively, the H3K4me3 mark maintains actively transcribed genes in the open confirmation to enable access for transcriptional activators to bind (44). Telomerase negative primary cells exhibit high levels of the silencing H3K27me3 mark at the TERT promoter, compared to telomerase positive cancer cell lines (45), which exhibit the activating H3K4me3 mark (46).
This histone mark pattern was also seen in our recent study. The activating mark H3K4me3 was least abundant in the benign thyroid-derived cell line N-thy-ori-3, as well as a cell line wild type for the TERT mutation. There were intermediate levels of the mark in the cell lines heterozygous for the TERT promoter mutation, and most abundant in the homozygous TERT mutant cell line. The silent mark H3K27me3 displayed the opposite pattern, showing the highest level in the normal and wildtype thyroid cell lines, intermediate in the heterozygous mutant cell lines, and least abundant in the homozygous mutant cell line. The latter phenomenon depicts a pattern of activation at the TERT promoter in cancer cells that is dependent on the TERT promoter mutation status, where activation by chromatin marks also correlated with TERT expression (29).
Recently, in order to further delineate TERT transcriptional regulation, studies have been conducted in many cancer subtypes to examine allele-specific regulation and the TERT mutation (Figure 1D). Allele-specific regulation, resulting in expression of only one allele, is classically described by the well-known phenomenon, inactivation of one of the X chromosomes in all females. This is due to epigenetic effects, in which one of the two X chromosomes is stably transcriptionally silenced by methylation early in development (47).
A study in 2015 of multiple cancer cell lines showed monoallelic expression of TERT in cell lines heterozygous for the TERT promoter mutation, although the study did not elucidate which allele was transcribed (48). Further studies expanded upon this by showing allele-specific regulation of the alleles, in which the TERT promoter mutant allele exhibits the H3K4me2/3 activating histone modification as well as binding of RNA polymerase II, the polymerase responsible for actively transcribing genes. The activating ETS factor GABPA has also been shown in multiple cell lines to bind in an allele-specific manner to the mutant TERT promoter only. This would be expected since the mutation specifically creates the ETS factor binding site. The wild type TERT allele, however, displays the silencing histone mark H3K27me3 and, is associated with the PRC (45, 49).
Allele-specific regulation in thyroid cancer cell lines has been explored by our group and others. In our recent study of five DTC cell lines we found the mutant allele at the TERT promoter was significantly less methylated than the wildtype allele, while the corresponding gene body associated with the mutant allele was more methylated. Further, a significant drop in methylation at the TSS of the mutant allele, but not the wildtype allele, was observed in the three heterozygous mutant cell lines. The methylation pattern observed on the mutant allele, of decreased methylation at the TSS and increased gene body methylation, is seen in actively transcribed genes. Additionally, we found that the activating transcription factors MYC and GABPA, bound to the mutant TERT allele exclusively. As expected, the activating histone mark H3K4me3 was associated with the mutant allele while the silenced mark H3K27me3 was associated with the wildtype allele. Bullock et al.'s recent study of two heterozygous mutant thyroid cancer cell lines suggested allele-specific regulation of TERT, showing monoallelic expression of TERT in the ATC and PTC cell lines. In the cell lines H3K4me3 was associated with the mutant allele only, while a majority of H3K27me3 was associated with the wildtype allele. In one ATC line, the ETS factor ETV5 bound in an allele-specific manner (31). Furthermore, by exploiting the presence of heterozygous SNPs in the transcripts, our group has definitively demonstrated that this allele-specific regulation results in selective transcription from only the mutant TERT promoter (29).
Previous studies have shown TERT expression is undetectable in normal thyroid tissue, low to absent in benign tumors, and elevated in thyroid cancers (20). TERT promoter mutations also are significantly associated with increased levels of TERT expression (20, 50). In addition, gene expression regulation can occur through alternative splicing (51). Indeed, TERT is regulated by alternative splicing, and thought to be crucial because even small amounts of TERT may have significant cellular consequences (Figure 1C) (9). This is exemplified by the fact that telomerase-positive malignant cells are estimated to contain only an average of 20 TERT transcripts per cell (52). The most common TERT isoforms include the following: (1) full-length transcript, the only isoform resulting in active telomerase (53, 54); (2) the α deletion isoform, an in-frame deletion of 36 base pairs in exon six resulting in a dominant negative inhibitor of telomerase (55); (3) the β deletion isoform, a 182 base pair deletion of exons seven and eight resulting in a reading frame shift forming a truncated TERT transcript that is sent for nonsense mediated decay (56); and (4) the α/β dual deletion isoform. Studies of TERT alternative splicing in multiple cancer types have shown the highest expressed isoform in cancer is the full-length isoform, which correlates with higher telomerase activity (57–59).
A study of 42 MTC tumors found TERT expression and telomerase activity in 50% (21/42) of the tumors. And, over two-thirds of the telomerase positive cancers (15/21) showed expression of the full-length TERT transcript, higher telomerase activity and were associated with a significantly shorter patient survival time (60).
Our group showed, in a study of 60 malignant tumors (PTC, FVPTC, FTC, and HCC) and 73 benign lesions, that malignant tumors showed a higher amount of the full-length isoform than the inactive TERT isoforms, while conversely, the benign tumors showed higher levels of the deletion isoforms (61). Further, only the full-length transcript correlated with telomerase activity (61, 62). By computing the proportion of full-length isoform to the deletion isoforms, we found the percentage score sorted thyroid tumors into subtypes; with the high full-length fractions of TERT found in PTC, HCC, and FTC, while intermediate fractions were found in FVPTC, Hürthle cell adenomas, and follicular adenomas and finally, low fractions in adenomatoid nodules (62).
DNA copy number variations, the gain or loss of chromosomal regions, including whole chromosomal arms (Figure 1A) within the genome, are quite common in cancer. They can provide a tumor cell proliferative advantages by eliminating tumor suppressors, in the case of deletions, or increasing expression of oncogenes, in the case of amplifications. TERT is located on the short arm of chromosome five (5p). 5p is one of the most frequent arm level regions to exhibit amplification with the phenomenon found in 13.2% of solid tumors (63). Further, a variety of high resolution techniques used to examine the TERT locus specifically, including fluorescence in situ hybridization (FISH), Southern blot, and quantitative PCR, have shown copy number amplifications of TERT in both cancer cell lines and tumors (63). Studies have also documented a correlation in cell lines of increased TERT copy number with both increased TERT gene expression and telomerase activity (64).
A recent study by Panebianco et al., of 184 tumors of various thyroid cancer subtypes- PTC, FTC, HCC, MTC, and PDTC/ATC, identified an increased TERT copy number in 4.9% of tumors (65). Specifically, 1/107 (0.9%) PTCs displayed four copies of TERT; while 2/22 (9%) FTCs, 4/29 (13.8%) HCCs, 0/22 (0%) MTCs, and 2/4 (50%) PDTCs/ATCs harbored three copies of TERT. In addition, one HCC and one PDTC/ATC with 3 copies of TERT also harbored the TERT −124 C>T mutation (65). A 2018 study, limited to follicular tumors, found three or more copies of TERT in 6/77 (7.8%) of FTCs, 2/43 (4.7%) of follicular adenomas, and 4/19 (21%) of follicular tumors of uncertain malignant potential (42). In a 2016 study of MTC, 5/42 (11.9%) had three copies of TERT, and no normal thyroid tissue showed copy number variations. In all five tumors with copy number amplifications, TERT expression and telomerase activity was also positive (16). Lastly, the comprehensive TCGA study of ~500 PTCs from multiple institutions showed 5p arm copy number gains in 21/507 (4%). In this study, 5p gain correlated with advanced age, worse pathological stage, extrathyroidal extension, and increased tumor size (22).
In summary, TERT is regulated by a variety of mechanisms, indicative of the complex regulatory effort cells exert to control telomerase activity. The majority of malignancies reactivate TERT expression, which in turn activates telomerase, to allow for continued proliferation. TERT reactivation is important for cancer cells, as cancer cells have significantly shorter telomeres. Shortened telomeres are found in PTC, FTC, HCC, and MTC compared to normal thyroid tissue and benign thyroid nodules (60, 62, 66–68). In PTC tumors, short telomeres were significantly correlated with TERT promoter mutations and older age (69).
In this review, we have summarized the current knowledge of TERT regulation, including TERT promoter mutations, CpG promoter methylation, histone tail modifications, copy number amplifications, and alternative splicing. Furthermore, we have described the allele-specific effects of the TERT mutation on transcriptional activator binding, promoter methylation, histone marks, and TERT expression. While all of these regulatory strategies are altered in thyroid cancer, and may play a significant role in TERT activation, these altered regulatory mechanisms have not yet been leveraged clinically.
TERT mRNA expression has been proposed as a prognostic marker, independent of the TERT promoter mutation. A study in PTC found a subset of wildtype TERT tumors with high TERT expression, which correlated with a higher recurrence rate (70). However, other studies have shown that TERT immunohistochemistry (IHC), or measurement of protein expression is not a useful clinical tool for prognostication. A study in FTC showed no correlation between TERT mRNA expression and TERT immunoreactivity (71), and a study in PTC showed no correlation with TERT IHC and clinicopathological traits (72).
Presence of the TERT promoter mutation, however, may be promising for future use to guide clinical decision making and treatment. The presence of the TERT promoter mutation is indicative of worse clinicopathological factors in thyroid cancer. The thyroid research field has recently begun to embrace the use of molecular markers to help in making clinical decisions. For example, post-operative screening for the TERT promoter mutation in follicular tumors of uncertain malignant potential (FT-UMP) was found to inform follow-up and treatment, as the TERT mutation was a predictive marker of distant metastases (73). Further, current molecular testing does include the TERT promoter mutations in some platforms, including ThyroSeq and ThyGenX. Current guidelines, however, do not support using the TERT mutation status for initial risk stratification (74). In the new American Association of Endocrine Surgeons Guidelines for the Definitive Surgical Management of Thyroid Disease in Adults, there is acknowledgment of inclusion of the TERT promoter mutation in assessment of the overall mutational burden in thyroid cancers (74). But for many molecular markers, the utility has not been tested given the newly published guidelines, nor has there been an incorporation of the new reclassification of the Non-Invasive Follicular Thyroid neoplasm with Papillary-like nuclear features (NIFTP). The 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer listed TERT, alone or in combination with BRAF, as potentially helpful to risk stratify patients in conjunction with other clinicopathological risk factors. However, this was considered a weak recommendation with low-quality evidence only (75).
In conclusion, we know much about telomerase regulation. Capitalizing upon our deep understanding of these regulatory mechanisms may lead to crucial and needed therapeutic options in the future. Nevertheless, major gaps in our understanding of telomerase regulation remain.
Understanding how TERT splicing is regulated is essential to a better understanding of the activation of telomerase. Characterization of TERT promoter methylation as well as ETS transcriptional activator binding in specific thyroid cancer subtypes, especially in PDTC and ATC, may lead to potential treatment options for these fatal diseases. Additionally, the study of chromosomal architecture surrounding TERT, and the effect of the TERT mutation on 3D chromatin structure in thyroid cancer have yet to be explored. One aspect that remains to be fully understood is how the higher levels of TERT expression and activity found in TERT-mutant cells provides a proliferative or other competitive advantage, since cancer cells typically have activated TERT expression to overcome the Hayflick limit long before TERT promoter mutations arise. Further understanding of how cancer cells that do not harbor the TERT mutation activate TERT through various regulatory methods still needs to be elucidated. Further studies are needed to determine the true utility of the TERT mutation status in a clinical setting and, how best to target TERT regulation.
BM, CU, and MZ designed, wrote, and edited the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by NSF grant DGE-1746891 (BM) and NIH training grant #T32 GM007445 (BM).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer KB declared a shared affiliation, with no collaboration, with the authors, BM and CU to the handling editor at the time of the review.
ALT, alternative lengthening of telomeres; ATC, anaplastic thyroid cancer; ATRX, ATRX chromatin remodeler; CpG, cytosine-guanine dinucleotides; DAXX, death domain associated protein; DTC, differentiated thyroid cancer; ETV5, ETS variant 5; FISH, fluorescence in situ hybridization; FTC, follicular thyroid cancer; GABPA, GA binding protein transcription factor subunit alpha; HCC, Hürthle cell carcinoma; MAPK, mitogen-activated protein kinase; MTC, medullary thyroid cancer; PDTC, poorly differentiated thyroid cancer; PRC, polycomb repressor complex; PTC, papillary thyroid cancer; TERT, telomerase reverse transcriptase; TR, telomerase RNA; TSS, transcription start site.
1. Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. (1965) 37:614–36. doi: 10.1016/0014-4827(65)90211-9
2. de Lange T. How telomeres solve the end-protection problem. Science. (2009) 326:948–52. doi: 10.1126/science.1170633
3. Donati B, Ciarrocchi A. Telomerase and telomeres biology in thyroid cancer. Int J Mol Sci. (2019) 20:2887. doi: 10.3390/ijms20122887
4. Greider CW, Blackburn EH. The telomere terminal transferase of tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity. Cell. (1987) 51:887–98. doi: 10.1016/0092-8674(87)90576-9
5. Koziel JE, Fox MJ, Steding CE, Sprouse AA, Herbert B-S. Medical genetics and epigenetics of telomerase. J Cell Mol Med. (2011) 15:457–67. doi: 10.1111/j.1582-4934.2011.01276.x
6. Haugen BR, Nawaz S, Markham N, Hashizumi T, Shroyer AL, Werness B, et al. Telomerase activity in benign and malignant thyroid tumors. Thyroid. (1997) 7:337–42. doi: 10.1089/thy.1997.7.337
7. Yuan X, Larsson C, Xu D. Mechanisms underlying the activation of TERT transcription and telomerase activity in human cancer: old actors and new players. Oncogene. (2019) 38:6172–83. doi: 10.1038/s41388-019-0872-9
8. Yoo S-K, Song YS, Lee EK, Hwang J, Kim HH, Jung G, et al. Integrative analysis of genomic and transcriptomic characteristics associated with progression of aggressive thyroid cancer. Nat Commun. (2019) 10:2764. doi: 10.1038/s41467-019-10680-5
9. Wong MS, Chen L, Foster C, Kainthla R, Shay JW, Wright WE. Regulation of telomerase alternative splicing: a target for chemotherapy. Cell Rep. (2013) 3:1028–35. doi: 10.1016/j.celrep.2013.03.011
10. Castelo-Branco P, Choufani S, Mack S, Gallagher D, Zhang C, Lipman T, et al. Methylation of the TERT promoter and risk stratification of childhood brain tumours: an integrative genomic and molecular study. Lancet Oncol. (2013) 14:534–42. doi: 10.1016/S1470-2045(13)70110-4
11. Liu X, Bishop J, Shan Y, Pai S, Liu D, Murugan AK, et al. Highly prevalent TERT promoter mutations in aggressive thyroid cancers. Endocr Relat Cancer. (2013) 20:603–10. doi: 10.1530/ERC-13-0210
12. Kyo S, Takakura M, Fujiwara T, Inoue M. Understanding and exploiting hTERT promoter regulation for diagnosis and treatment of human cancers. Cancer Sci. (2008) 99:1528–38. doi: 10.1111/j.1349-7006.2008.00878.x
13. Avin B, Umbricht C, Zeiger M. Human telomerase reverse transcriptase regulation by DNA methylation, transcription factor binding and alternative splicing. Int J Oncol. (2016) 49:2199–205. doi: 10.3892/ijo.2016.3743
14. Cipriani NA. Prognostic parameters in differentiated thyroid carcinomas. Surg Pathol Clin. (2019) 12:883–900. doi: 10.1016/j.path.2019.07.001
15. Li J-J, Zheng PCJ-R, Wang Y-Z. The correlations between DNA methylation and polymorphisms in the promoter region of the human telomerase reverse transcriptase (hTERT) gene with postoperative recurrence in patients with thyroid carcinoma (TC). World J Surg Oncol. (2017) 15:114. doi: 10.1186/s12957-017-1170-z
16. Wang N, Kjellin H, Sofiadis A, Fotouhi O, Juhlin CC, Bäckdahl M, et al. Genetic and epigenetic background and protein expression profiles in relation to telomerase activation in medullary thyroid carcinoma. Oncotarget. (2016) 7:21332–46. doi: 10.18632/oncotarget.7237
17. Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. (2013) 339:957–9. doi: 10.1126/science.1229259
18. Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, et al. TERT promoter mutations in familial and sporadic melanoma. Science. (2013) 339:959–61. doi: 10.1126/science.1230062
19. Chiba K, Johnson JZ, Vogan JM, Wagner T, Boyle JM, Hockemeyer D, et al. Cancer-associated TERT promoter mutations abrogate telomerase silencing. Elife. (2015) 4:e07918. doi: 10.7554/eLife.07918
20. Liu R, Xing M. TERT promoter mutations in thyroid cancer. Endocr Relat Cancer. (2016) 23:R143–55. doi: 10.1530/ERC-15-0533
21. Ganly I, Makarov V, Deraje S, Dong Y, Reznik E, Seshan V, et al. Integrated genomic analysis of hürthle cell cancer reveals oncogenic drivers, recurrent mitochondrial mutations, and unique chromosomal landscapes. Cancer Cell. (2018) 34:256–70.e5. doi: 10.1016/j.ccell.2018.07.002
22. Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell. (2014) 159:676–90. doi: 10.1016/j.cell.2014.09.050
23. Oishi N, Kondo T, Ebina A, Sato Y, Akaishi J, Hino R, et al. Molecular alterations of coexisting thyroid papillary carcinoma and anaplastic carcinoma: identification of TERT mutation as an independent risk factor for transformation. Mod Pathol. (2017) 30:1527–37. doi: 10.1038/modpathol.2017.75
24. Yang X, Li J, Li X, Liang Z, Gao W, Liang J, et al. TERT promoter mutation predicts radioiodine-refractory character in distant metastatic differentiated thyroid cancer. J Nucl Med. (2017) 58:258–65. doi: 10.2967/jnumed.116.180240
25. Meng Z, Matsuse M, Saenko V, Yamashita S, Ren P, Zheng X, et al. TERT promoter mutation in primary papillary thyroid carcinoma lesions predicts absent or lower 131I uptake in metastases. IUBMB Life. (2019) 71:iub.2056. doi: 10.1002/iub.2056
26. Bell RJA, Rube HT, Kreig A, Mancini A, Fouse SD, Nagarajan RP, et al. Cancer. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science. (2015) 348:1036–9. doi: 10.1126/science.aab0015
27. Liu R, Zhang T, Zhu G, Xing M. Regulation of mutant TERT by BRAF V600E/MAP kinase pathway through FOS/GABP in human cancer. Nat Commun. (2018) 9:579. doi: 10.1038/s41467-018-03033-1
28. Avin BA, Wang Y, Gilpatrick T, Workman RE, Lee I, Timp W, et al. Characterization of human telomerase reverse transcriptase promoter methylation and transcription factor binding in differentiated thyroid cancer cell lines. Genes Chromosom Cancer. (2019) 58:530–40. doi: 10.1002/gcc.22735
29. McKelvey BA, Gilpatrick T, Wang Y, Timp W, Umbricht CB, Zeiger MA. Characterization of allele-specific regulation of telomerase reverse transcriptase in promoter mutant thyroid cancer cell lines. Thyroid. (2020). doi: 10.1089/thy.2020.0055. [Epub ahead of print].
30. Yuan X, Mu N, Wang N, Strååt K, Sofiadis A, Guo Y, et al. GABPA inhibits invasion/metastasis in papillary thyroid carcinoma by regulating DICER1 expression. Oncogene. (2019) 38:965–79. doi: 10.1038/s41388-018-0483-x
31. Bullock M, Lim G, Zhu Y, Åberg H, Kurdyukov S, Clifton-Bligh RJ. The ETS factor ETV5 activates the mutant TERT promoter in thyroid cancer. Thyroid. (2019) 29:1623–33. doi: 10.1089/thy.2018.0314
32. Kebebew E, Weng J, Bauer J, Ranvier G, Clark OH, Duh Q-Y, et al. The prevalence and prognostic value of BRAF mutation in thyroid cancer. Ann Surg. (2007) 246:466–70. doi: 10.1097/SLA.0b013e318148563d
33. Song YS, Lim JA, Choi H, Won J-K, Moon JH, Cho SW, et al. Prognostic effects of TERT promoter mutations are enhanced by coexistence with BRAF or RAS mutations and strengthen the risk prediction by the ATA or TNM staging system in differentiated thyroid cancer patients. Cancer. (2016) 122:1370–9. doi: 10.1002/cncr.29934
34. Xing M, Liu R, Liu X, Murugan AK, Zhu G, Zeiger MA, et al. BRAF V600E and TERT promoter mutations cooperatively identify the most aggressive papillary thyroid cancer with highest recurrence. J Clin Oncol. (2014) 32:2718–26. doi: 10.1200/JCO.2014.55.5094
35. Liu R, Bishop J, Zhu G, Zhang T, Ladenson PW, Xing M. Mortality risk stratification by combining BRAF V600E and TERT promoter mutations in papillary thyroid cancer. JAMA Oncol. (2017) 3:202–8. doi: 10.1001/jamaoncol.2016.3288
36. Song YS, Yoo S-K, Kim HH, Jung G, Oh A-R, Cha J-Y, et al. Interaction of BRAF-induced ETS factors with mutant TERT promoter in papillary thyroid cancer. Endocr Relat Cancer. (2019) 26:629–41. doi: 10.1530/ERC-17-0562
37. Nebbioso A, Tambaro FP, Dell'Aversana C, Altucci L. Cancer epigenetics: moving forward. PLoS Genet. (2018) 14:e1007362. doi: 10.1371/journal.pgen.1007362
38. Jang HS, Shin WJ, Lee JE, Do JT. CpG and non-CpG methylation in epigenetic gene regulation and brain function. Genes (Basel). (2017) 8:148. doi: 10.3390/genes8060148
39. Choi J-H, Park SH, Park J, Park BG, Cha S-J, Kong K-H, et al. Site-specific methylation of CpG nucleotides in the hTERT promoter region can control the expression of hTERT during malignant progression of colorectal carcinoma. Biochem Biophys Res Commun. (2007) 361:615–20. doi: 10.1016/j.bbrc.2007.07.051
40. Zinn RL, Pruitt K, Eguchi S, Baylin SB, Herman JG. hTERT is expressed in cancer cell lines despite promoter DNA methylation by preservation of unmethylated DNA and active chromatin around the transcription start site. Cancer Res. (2007) 67:194–201. doi: 10.1158/0008-5472.CAN-06-3396
41. Lopatina NG, Poole JC, Saldanha SN, Hansen NJ, Key JS, Pita MA, et al. Control mechanisms in the regulation of telomerase reverse transcriptase expression in differentiating human teratocarcinoma cells. Biochem Biophys Res Commun. (2003) 306:650–9. doi: 10.1016/S0006-291X(03)01033-7
42. Paulsson JO, Mu N, Shabo I, Wang N, Zedenius J, Larsson C, et al. TERT aberrancies: a screening tool for malignancy in follicular thyroid tumours. Endocr Relat Cancer. (2018) 25:723–33. doi: 10.1530/ERC-18-0050
43. Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. (2011) 21:381–95. doi: 10.1038/cr.2011.22
44. Salgado C, Roelse C, Nell R, Gruis N, van Doorn R, van der Velden P. Interplay between TERT promoter mutations and methylation culminates in chromatin accessibility and TERT expression. bioRxiv. (2019) 859892. doi: 10.1101/859892
45. Stern JL, Theodorescu D, Vogelstein B, Papadopoulos N, Cech TR. Mutation of the TERT promoter, switch to active chromatin, and monoallelic TERT expression in multiple cancers. Genes Dev. (2015) 29:2219–24. doi: 10.1101/gad.269498.115
46. Zhao Y, Cheng D, Wang S, Zhu J. Dual roles of c-Myc in the regulation of hTERT gene. Nucleic Acids Res. (2014) 42:10385–98. doi: 10.1093/nar/gku721
47. Panning B. X-chromosome inactivation: the molecular basis of silencing. J Biol. (2008) 7:30. doi: 10.1186/jbiol95
48. Huang FW, Bielski CM, Rinne ML, Hahn WC, Sellers WR, Stegmeier F, et al. TERT promoter mutations and monoallelic activation of TERT in cancer. Oncogenesis. (2015) 4:e176. doi: 10.1038/oncsis.2015.39
49. Stern JL, Paucek RD, Huang FW, Ghandi M, Nwumeh R, Costello JC, et al. Allele-specific DNA methylation and its interplay with repressive histone marks at promoter-mutant TERT genes. Cell Rep. (2017) 21:3700–7. doi: 10.1016/j.celrep.2017.12.001
50. Vinagre J, Almeida A, Pópulo H, Batista R, Lyra J, Pinto V, et al. Frequency of TERT promoter mutations in human cancers. Nat Commun. (2013) 4:2185. doi: 10.1038/ncomms3185
51. Sebestyén E, Zawisza M, Eyras E. Detection of recurrent alternative splicing switches in tumor samples reveals novel signatures of cancer. Nucleic Acids Res. (2015) 43:1345–56. doi: 10.1093/nar/gku1392
52. Wong MS, Wright WE, Shay JW. Alternative splicing regulation of telomerase: a new paradigm? Trends Genet. (2014) 30:430–8. doi: 10.1016/j.tig.2014.07.006
53. Liu L, Liu C, Fotouhi O, Fan Y, Wang K, Xia C, et al. TERT promoter hypermethylation in gastrointestinal cancer: a potential stool biomarker. Oncologist. (2017) 22:1178–88. doi: 10.1634/theoncologist.2017-0064
54. Ulaner GA, Hu JF, Vu TH, Giudice LC, Hoffman AR. Telomerase activity in human development is regulated by human telomerase reverse transcriptase (hTERT) transcription and by alternate splicing of hTERT transcripts. Cancer Res. (1998) 58:4168–72.
55. Colgin LM, Wilkinson C, Englezou A, Kilian A, Robinson MO, Reddel RR. The hTERTalpha splice variant is a dominant negative inhibitor of telomerase activity. Neoplasia. (2000) 2:426–32. doi: 10.1038/sj.neo.7900112
56. Bounacer A, Wicker R, Caillou B, Cailleux AF, Sarasin A, Schlumberger M, et al. High prevalence of activating ret proto-oncogene rearrangements, in thyroid tumors from patients who had received external radiation. Oncogene. (1997) 15:1263–73. doi: 10.1038/sj.onc.1200206
57. Lincz LF, Mudge L-M, Scorgie FE, Sakoff JA, Hamilton CS, Seldon M. Quantification of hTERT splice variants in melanoma by SYBR green real-time polymerase chain reaction indicates a negative regulatory role for the β deletion variant. Neoplasia. (2008) 10:1131–7. doi: 10.1593/neo.08644
58. Mavrogiannou E, Strati A, Stathopoulou A, Tsaroucha EG, Kaklamanis L, Lianidou ES. Real-time RT-PCR quantification of human telomerase reverse transcriptase splice variants in tumor cell lines and non-small cell lung cancer. Clin Chem. (2007) 53:53–61. doi: 10.1373/clinchem.2006.073015
59. Rha SY, Jeung HC, Park KH, Kim JJ, Chung HC. Changes of telomerase activity by alternative splicing of full-length and β variants of hTERT in breast cancer patients. Oncol Res Featur Preclin Clin Cancer Ther. (2009) 18:213–20. doi: 10.3727/096504009X12596189659123
60. Wang N, Xu D, Sofiadis A, Höög A, Vukojević V, Bäckdahl M, et al. Telomerase-dependent and independent telomere maintenance and its clinical implications in medullary thyroid carcinoma. J Clin Endocrinol Metab. (2014) 99:E1571–9. doi: 10.1210/jc.2014-1158
61. Wang Y, Kowalski J, Tsai H-L, Marik R, Prasad N, Somervell H, et al. Differentiating alternative splice variant patterns of human telomerase reverse transcriptase in thyroid neoplasms. Thyroid. (2008) 18:1055–63. doi: 10.1089/thy.2008.0101
62. Wang Y, Meeker AK, Kowalski J, Tsai H-L, Somervell H, Heaphy C, et al. Telomere length is related to alternative splice patterns of telomerase in thyroid tumors. Am J Pathol. (2011) 179:1415–24. doi: 10.1016/j.ajpath.2011.05.056
63. Cao Y, Bryan TM, Reddel RR. Increased copy number of the TERT and TERC telomerase subunit genes in cancer cells. Cancer Sci. (2008) 99:1092–9. doi: 10.1111/j.1349-7006.2008.00815.x
64. Zhang A, Zheng C, Lindvall C, Hou M, Ekedahl J, Lewensohn R, et al. Frequent amplification of the telomerase reverse transcriptase gene in human tumors. Cancer Res. (2000) 60:6230–5.
65. Panebianco F, Nikitski AV, Nikiforova MN, Nikiforov YE. Spectrum of TERT promoter mutations and mechanisms of activation in thyroid cancer. Cancer Med. (2019) 8:5831–9. doi: 10.1002/cam4.2467
66. Capezzone M, Cantara S, Marchisotta S, Busonero G, Formichi C, Benigni M, et al. Telomere length in neoplastic and nonneoplastic tissues of patients with familial and sporadic papillary thyroid cancer. J Clin Endocrinol Metab. (2011) 96:E1852–6. doi: 10.1210/jc.2011-1003
67. Kammori M, Takubo K, Nakamura K, Furugouri E, Endo H, Kanauchi H, et al. Telomerase activity and telomere length in benign and malignant human thyroid tissues. Cancer Lett. (2000) 159:175–81. doi: 10.1016/S0304-3835(00)00547-4
68. Sugishita Y, Kammori M, Yamada O, Yamazaki K, Ito K, Fukumori T, et al. Biological differential diagnosis of follicular thyroid tumor and Hürthle cell tumor on the basis of telomere length and hTERT expression. Ann Surg Oncol. (2014) 21:2318–25. doi: 10.1245/s10434-014-3552-6
69. Liu T, Wang N, Cao J, Sofiadis A, Dinets A, Zedenius J, et al. The age- and shorter telomere-dependent TERT promoter mutation in follicular thyroid cell-derived carcinomas. Oncogene. (2014) 33:4978–84. doi: 10.1038/onc.2013.446
70. Tanaka A, Matsuse M, Saenko VA, Nakao T, Yamanouchi K, Sakimura C, et al. TERT mRNA expression as a novel prognostic marker in papillary thyroid carcinomas. Thyroid. (2019) 29:1105–14. doi: 10.1089/thy.2018.0695
71. Paulsson JO, Olander A, Haglund F, Zedenius J, Juhlin CC. TERT immunohistochemistry is a poor predictor of TERT promoter mutations and gene expression in follicular thyroid carcinoma. Endocr Pathol. (2018) 29:380–3. doi: 10.1007/s12022-018-9551-6
72. Cacciato Insilla A, Proietti A, Borrelli N, Macerola E, Niccoli C, Vitti P, et al. TERT promoter mutations and their correlation with BRAF and RAS mutations in a consecutive cohort of 145 thyroid cancer cases. Oncol Lett. (2017) 15:2763–70. doi: 10.3892/ol.2017.7675
73. Hysek M, Paulsson JO, Jatta K, Shabo I, Stenman A, Höög A, et al. Clinical routine TERT promoter mutational screening of follicular thyroid tumors of uncertain malignant potential (FT-UMPs): a useful predictor of metastatic disease. Cancers (Basel). (2019) 11:1443. doi: 10.3390/cancers11101443
74. Patel KN, Yip L, Lubitz CC, Grubbs EG, Miller BS, Shen W, et al. The American association of endocrine surgeons guidelines for the definitive surgical management of thyroid disease in adults. Ann Surg. (2020) 271:e21–93. doi: 10.1097/SLA.0000000000003580
Keywords: TERT, telomerase, thyroid cancer, transcription, epigenetics, copy number variation, alternative splice variants
Citation: McKelvey BA, Umbricht CB and Zeiger MA (2020) Telomerase Reverse Transcriptase (TERT) Regulation in Thyroid Cancer: A Review. Front. Endocrinol. 11:485. doi: 10.3389/fendo.2020.00485
Received: 19 May 2020; Accepted: 22 June 2020;
Published: 31 July 2020.
Edited by:
Roberta Malaguarnera, University of Catanzaro, ItalyReviewed by:
Karen Beemon, Johns Hopkins University, United StatesCopyright © 2020 McKelvey, Umbricht and Zeiger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martha A. Zeiger, bWFydGhhLnplaWdlckBuaWguZ292
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.