 Tongxin Ge
Tongxin Ge Jiawen Yang
Jiawen Yang Shihui Zhou
Shihui Zhou Yuchen Wang
Yuchen Wang Yakui Li
Yakui Li Xuemei Tong
Xuemei Tong- Department of Biochemistry and Molecular Cell Biology, Shanghai Key Laboratory for Tumor Microenvironment and Inflammation, Key Laboratory of Cell Differentiation and Apoptosis of Chinese Ministry of Education, Shanghai Jiao Tong University School of Medicine, Shanghai, China
The pentose phosphate pathway (PPP) branches from glucose 6-phosphate (G6P), produces NADPH and ribose 5-phosphate (R5P), and shunts carbons back to the glycolytic or gluconeogenic pathway. The PPP has been demonstrated to be a major regulator for cellular reduction-oxidation (redox) homeostasis and biosynthesis. Enzymes in the PPP are reported to play important roles in many human diseases. In this review, we will discuss the role of the PPP in type 2 diabetes and cancer.
Introduction
The pentose phosphate pathway (PPP), also known as the pentose phosphate shunt, is an important part of glucose metabolism. The PPP branches after the first step of glycolysis and consumes the intermediate glucose 6-phosphate (G6P) to generate fructose 6-phosphate (F6P) and glyceraldehyde 3-phosphate (G3P) through the oxidative and non-oxidative branches of the PPP. Unlike glycolysis and glucose aerobic oxidation, the PPP does not provide adenosine 5′-triphosphate (ATP) to meet the energy demands of cells. Instead, it supplies NADPH and ribose 5-phosphate (R5P). These two metabolites are vital for the survival and proliferation of cells. R5P is a building block for nucleic acid synthesis. NADPH is the reducing power required for the synthesis of fatty acids, sterols, nucleotides and non-essential amino acids (1, 2). Moreover, NADPH-derived conversion of oxidized glutathione (GSSG) to reduced glutathione (GSH) via glutathione reductase is important for cellular antioxidant defenses. Interestingly, NADPH also serves as the substrate of NADPH oxidases (NOXs) which produce reactive oxygen species (ROS) (3).
Both the oxidative branch and non-oxidative branch of the PPP take place in the cytosol (Figure 1). Glucose 6-phosphate dehydrogenase (G6PD) is the rate-limiting enzyme of the oxidative PPP, determining the flux of G6P directed into the pathway. G6PD catalyzes the conversion of G6P to 6-phosphogluconolactone, accompanied by NADPH production. 6-phosphogluconolactonase (6PGL) is the enzyme that hydrolyses 6-phosphogluconolactone to produce 6-phosphogluconate (6PG). 6-phosphogluconate dehydrogenase (6PGD) converts 6-PG to ribulose 5-phosphate (Ru5P) and generates NAPDH (Figure 1). The largest contributor to cytosolic NADPH is the oxidative PPP in mammalian cells. Moreover, at least 3 other cytoplasmic enzymes including isocitrate dehydrogenase 1 (IDH1), malic enzyme 1 (ME1) and 10-formyltetrahydrofolate dehydrogenase (ALDH1L1) contribute to NADPH synthesis in cytosol. Furthermore, mitochondrial NADPH production is dependent on at least 5 mitochondrial enzymes including nicotinamide nucleotide transhydrogenase (NNT), isocitrate dehydrogenase 2 (IDH2), malic enzyme 3 (ME3), mitochondrial homolog of 10-formyltetrahydrofolate dehydrogenase (ALDH1L2) and methylenetetrahydrofolate dehydrogenase 1 like (MTHFD1L) (4).
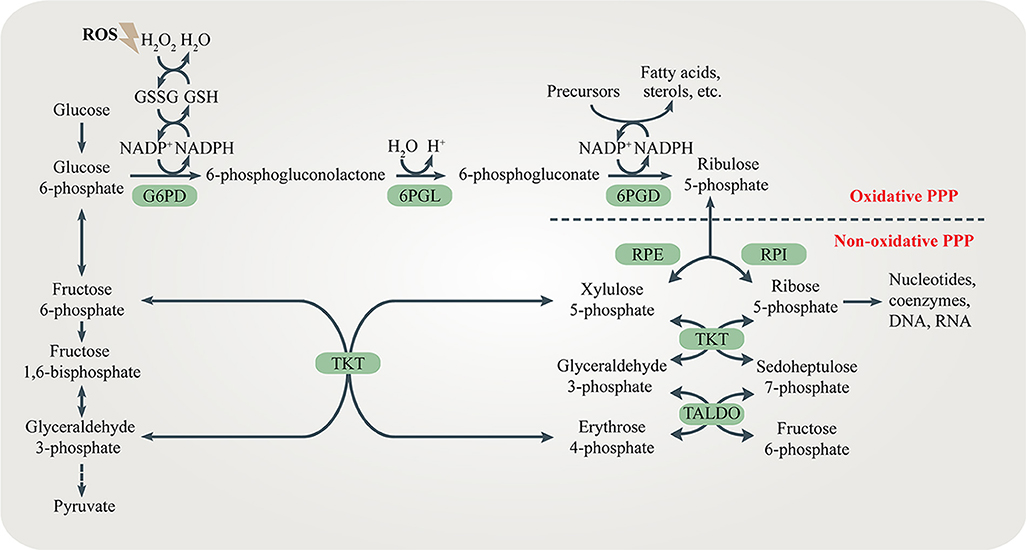
Figure 1. The pentose phosphate pathway (PPP). The PPP branches after the first step of glycolysis and goes back to fructose 6-phosphate and glyceraldehyde 3-phosphate in the glycolytic and gluconeogenic pathway. The PPP produces R5P and NADPH for biosynthesis and redox regulation. Enzymes in the oxidative and non-oxidative PPP are shaded in green.
The non-oxidative branch is composed of a series of reversible transfer reactions of chemical groups. Ribose 5-phosphate isomerase (RPI) and ribulose 5-phosphate epimerase (RPE) catalyze reversible reactions converting Ru5P to R5P and xylulose 5-phosphate (Xu5P), respectively. TKT catalyzes two reversible reactions. One is the conversion of Xu5P and R5P to G3P and sedoheptulose 7-phosphate (S7P). The other is the conversion of Xu5P and erythrose 4-phosphate (E4P) to G3P and F6P. Therefore, TKT can bi-directionally regulate the carbon flux between the non-oxidative PPP and glycolysis or gluconeogenesis. Transaldolase (TALDO) reversibly converts G3P and S7P to E4P and F6P. The non-oxidative branch not only replenishes metabolites of the oxidative branch by the reversible reactions, but also regulates the flux of glycolysis or gluconeogenesis by providing F6P and G3P (5) (Figure 1).
The Role of the PPP in Type 2 Diabetes Mellitus (T2DM)
T2DM is a chronic metabolic disease featured by persistently abnormal hyperglycemia, which can cause serious chronic damage to kidneys, eyes, and nerves. Deregulated insulin secretion and progressive insulin resistance are two main characteristics of T2DM (6). Over the past few decades, studies on the pathogenesis of T2DM have revealed a close relationship between the PPP, obesity-related insulin resistance and T2DM. In this part, we will mainly focus on the role of the PPP in obesity-related insulin resistance, insulin secretion and chronic diabetic complications.
The Role of the PPP in Obesity-Related Insulin Resistance
The term “insulin resistance” indicates that insulin-responsive tissues such as the liver, adipose tissue, and skeletal muscle reduce insulin-mediated glucose uptake, contributing to hyperglycemia (7). Pancreatic islet β cells, therefore, have to secrete more insulin to compensate for insulin resistance, resulting in hyperinsulinemia which leads to dysfunction of β cells and T2DM.
Obesity is closely related to the onset of insulin resistance. Chronic obesity-induced inflammation is one of the major causes of obesity-related insulin resistance. Adipose tissue macrophages (ATMs) surrounding dead adipocytes cause obesity-induced inflammation and secrete pro-inflammatory cytokines leading to local insulin resistance in adipose tissues (8). The severity of obesity-induced inflammation correlates with the degree of obesity. Abnormally increased number and activity of ATMs as well as higher ratio of pro-inflammatory to anti-inflammatory macrophages are both hallmarks of obesity-induced inflammation (9). Different activity of the oxidative PPP in macrophages contributes to the functional discrepancy of macrophages. Pro-inflammatory M1 macrophages show enhanced glycolysis and PPP flux which provides more energy and NADPH to trigger inflammatory responses, secrete pro-inflammatory cytokines and recruit more immune cells. However, anti-inflammatory M2 macrophages, displaying decreased glycolysis and PPP flux, work adversely to resolute inflammatory responses, secreting anti-inflammatory cytokines to inhibit M1 macrophages (10). The pro-inflammatory cytokines released by M1 macrophages include tumor necrosis factor-α (TNF-α) and interleukin 1β (IL-1β). TNF-α and IL-1β contribute to insulin resistance in adipose tissues by altering the insulin receptor signaling pathway via the stress-responsive c-Jun-NH2-terminal kinase (JNK 1/2), inhibitor of κB kinase (IKK) and mitogen-activated protein kinase (MAPK) p38 (11, 12). In contrast, M2 macrophages secret the anti-inflammatory cytokines including interleukin 10 (IL-10) and arginase1 and maintain insulin sensitivity (12, 13). In addition, pro-inflammatory cytokines recruit more monocytes to adipose tissues, resulting in more severe obesity-induced inflammation. This amplifying feedback loop aggravates local insulin resistance.
Moreover, obesity-induced inflammation can lead to insulin resistance in the skeletal muscle or liver, resulting in systemic insulin resistance (9, 14, 15). Pro-inflammatory cytokines promote lipolysis in adipocytes, leading to elevated levels of circulating free fatty acids (FFAs). In the skeletal muscle, FFAs reduce insulin-stimulated glucose intake, resulting in skeletal muscle insulin resistance (16). On the other hand, lipolysis in adipose tissues increases hepatic acetyl CoA levels and pyruvate carboxylase activity which further promotes hepatic glucose production. Both hepatic glucose production and pro-inflammatory cytokines contribute to hepatic insulin resistance (8, 17, 18). FFAs increase production of ROS in liver, leading to severe insulin resistance and nonalcoholic steatohepatitis (NASH) (19). Furthermore, long-term exposure to elevated levels of FFAs contributes to pancreatic β cells dysfunction and death (20, 21).
Recent studies suggest that the PPP might serve as a novel and promising target for modulating obesity-induced inflammation and insulin sensitivity in different tissues. Over-nutrition causes excessive FFAs released from adipose tissues which up-regulates G6PD expression in ATMs. Elevated levels of G6PD in ATMs can cause obesity-related inflammation (22). Pro-inflammatory cytokines also increase G6PD expression in adipocytes. Accordingly, adipocytes secrete adipocytokines including resistin and TNF-α which further stimulate inflammatory responses and recruit more monocytes to the inflamed adipose tissues (23). This vicious cycle promotes obesity-related insulin resistance, resulting in severe T2DM. The non-oxidative PPP in adipose tissues also plays an important role in regulating insulin sensitivity. TKT deficiency in adipocytes results in R5P accumulation and reduced glycolysis, accompanied by increased lipolysis and fatty acid β-oxidation. Therefore, loss of TKT in adipose tissues alleviates high fat diet (HFD)-induced obesity, leading to reduced hepatic steatosis and improved insulin sensitivity (24). Moreover, increased expression of G6PD in hepatocytes generates more NADPH for de novo lipogenesis (DNL) which promotes hepatic steatosis and insulin resistance (25, 26). Interestingly, excessive NADPH in liver may contribute to oxidative stress via NOXs, leading to liver damage and insulin resistance (27). The peroxisome proliferator-activated receptor δ (PPARδ) reduces hepatic glucose output and improves insulin sensitivity partly by regulating the PPP flux (28). Furthermore, G6PD in skeletal muscle regulates glucose uptake and insulin sensitivity (29). As an excellent intervention in metabolic diseases, exercise increases the level of peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) which promotes G6PD transcription and higher intramyocellular lipid (IMCL) content in skeletal muscles. The combination of increased muscle PGC-1α expression and exercise greatly enhances insulin sensitivity (30, 31).
Carbohydrate kinase-like protein (CARKL), also known as sedoheptulokinase (SHPK), is a carbohydrate kinase catalyzing the phosphorylation of sedoheptulose to S7P (32). Since S7P is the substrate for TKT and TALDO, CARKL is important for regulating the flux through the PPP. High level of S7P generated by CARKL restricts the reversible reaction in the non-oxidative PPP, limiting the flux through the PPP. CARKL is highly expressed in M2 macrophages and its down-regulation is critical for proper M1 polarization (33, 34) (Figure 2). It is reasonable to assume that regulating CARKL may reduce obesity-induced inflammation, leading to increased insulin sensitivity. Furthermore, whether TKT and TALDO can direct macrophage polarization should be studied in the future.
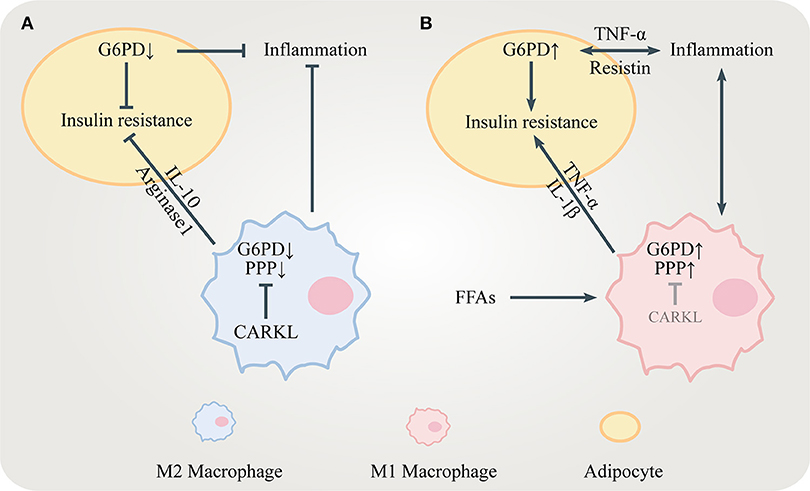
Figure 2. The role of the PPP in insulin resistance. (A) CARKL is highly expressed in M2 macrophages, limiting the PPP flux by inhibiting G6PD. M2 macrophages release anti-inflammatory mediators including IL-10 and arginase 1 to maintain insulin sensitivity. Decreased G6PD in adipocytes suppresses inflammation and ameliorates insulin resistance. (B) FFAs and pro-inflammatory cytokines including TNF-α, IL-1β, and resistin increase G6PD expression in both adipocytes and M1 macrophages, which stimulate inflammatory responses leading to insulin resistance.
The Role of the PPP in Insulin Secretion
Insulin is stored in granules and released via exocytosis from pancreatic islet β cells in response to glucose in a biphasic manner, which is known as the triggering pathway and the amplifying pathway (35). The amplifying pathway accounts for the majority of glucose-stimulated insulin secretion (GSIS). Decreased insulin secretion in the amplifying pathway is often observed in patients with T2DM. Therefore, how to simulate insulin secretion during the amplifying pathway is important for the prevention and treatment of T2DM.
NADPH is one key modulator of the amplifying pathway because it converts GSSG to GSH which elicits insulin granule exocytosis via sentrin/SUMO-specific protease-1 (SENP1) (35, 36) (Figure 3). Being a major source for NADPH, the PPP regulates the GSIS-related NADPH/GSH/SENP1 pathway. Only optimal levels of G6PD and 6PGD, two enzymes generating NADPH in the PPP, are beneficial to GSIS. Patients with G6PD deficiency show decreased insulin secretion (37). Inhibition of G6PD and 6PGD not only blocks GSIS but also increases oxidative stress and β cells apoptosis (38, 39). However, overexpression of G6PD also negatively influences GSIS which is due to increased expression of NADPH oxidases (NOXs) and ROS accumulation (40, 41). In conclusion, the PPP/NADPH/GSH/SENP1 pathway needs to be precisely controlled to achieve beneficial GSIS.
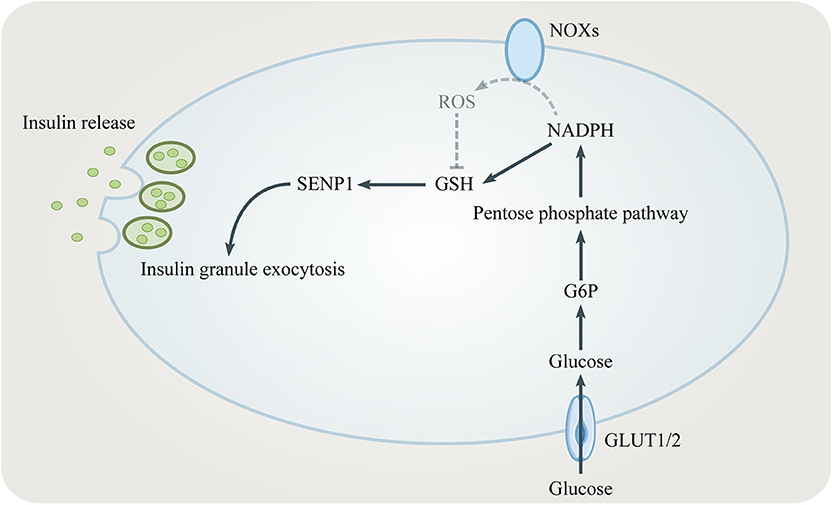
Figure 3. The role of the PPP in insulin secretion. NADPH from the PPP converts oxidized glutathione (GSSG) to reduced glutathione (GSH). GSH elicits insulin granule exocytosis via SENP1. However, NADPH might inhibit insulin secretion by promoting NADPH oxidases (NOXs).
The Role of the PPP in Chronic Diabetic Complications
Diabetes can lead to diabetic nephropathy, diabetic retinopathy, diabetic cardiomyopathy, diabetic macroangiopathy and other chronic complications. Oxidative stress can cause these complications by activating the hexosamine pathway, the advanced glycation end products (AGEs) pathway and the diacylglycerol (DAG)-protein kinase C (PKC) pathway (42).
Hyperglycemia decreases G6PD activity through the activation of protein kinase A (PKA) and increase of intracellular oxidative stress, leading to chronic kidney injury, and diabetic kidney disease (DKD) (43–45). Moreover, overexpression of G6PD in endothelial cells prevents diabetic cardiomyopathy by decreasing ROS accumulation and increasing endothelial cell viability (46).
TKT plays an important role in preventing hyperglycemia-induced vascular cell dysfunction (47). The cofactor of TKT is thiamine diphosphate (TDP), the active form of thiamine. Thiamine deficiency and decreased TKT activity has been reported to contribute to diabetic complications (48). Low plasma thiamine was found in patients with DKD and diabetic rats. After high-dose thiamine therapy, the progression of proteinuria and microalbuminuria was reversed in both patients and animal models, indicating that regulating the activity of TKT may be a promising therapy in treating DKD (47, 49, 50). Benfotiamine, a lipid-soluble thiamine derivative, can prevent diabetic retinopathy and cardiomyopathy as well as accelerate the healing of diabetic limbs by activating TKT (51–53). However, benfotiamine did not show promising efficacy in phase II and IV trials for the treatment of DKD or diabetic peripheral nerve function (54, 55). Therefore, other transketolase activators await further investigation.
The Role of the PPP in Cancer
The PPP is critical for cancer prevention and treatment because NADPH and R5P play important roles in regulating DNA damage response, metabolism, and proliferation in cancer cells. Various enzymes in the PPP have been shown to be potential targets in cancer therapy. These proteins not only function as metabolic enzymes, but also participate in the regulation of other cellular activities. Therefore, we will summarize recent findings in upstream signaling pathways regulating PPP enzymes in cancer initiation and progression (Table 1).
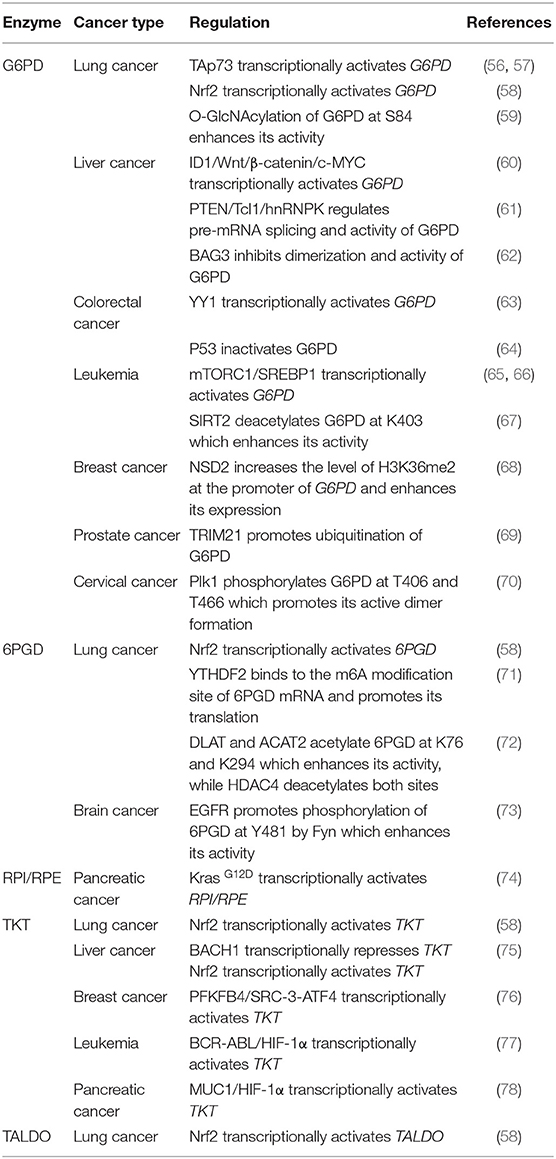
Table 1. The regulation of PPP enzymes in cancer cells.
Enzymes in the Oxidative PPP
Up-regulation of the G6PD level or activity is often observed in many kinds of cancer (79–86). Several signaling pathways have been identified to be responsible for promoting G6PD expression or activity in cancer cells (Figure 4). TP53-induced glycolysis and apoptosis regulator (TIGAR) enhances the PPP flux and biosynthesis (87). The tumor suppressor p53 directly binds to G6PD and prevents formation of the active G6PD dimer whereas the mutant p53 fails to inhibit G6PD in cancer cells (64). The p21-activated kinase 4 (PAK4) regulates G6PD activity by promoting p53 ubiquitination (81). Bcl-2 associated athanogene 3 (BAG3) inhibits dimerization and activity of G6PD (62). TAp73, a member of the p53 family which is often overexpressed in cancers, supports tumor growth by inducing G6PD expression (56, 57). Nuclear factor E2-related factor 2 (NRF2) is a transcription factor regulated by oxidative stress. When the PI3K/Akt signaling pathway is activated, NRF2 directly increases G6PD, 6PGD, TKT, and TALDO expression to enhance metabolic activities and promote cancer cell growth (58). The mammalian target of rapamycin complex 1 (mTORC1) stimulates the oxidative branch of the PPP by enhancing the sterol regulatory element-binding protein (SREBP)-dependent transcription of G6PD (65, 66). Inhibitor of differentiation and DNA binding-1 (ID1) regulates c-MYC through Wnt/β-catenin pathway activation to promote G6PD transcription and activate the PPP (60). Phosphatase and tensin homolog (PTEN) prevents G6PD from activation via the PTEN/Tcl1/hnRNPK/G6PD axis (61). Transcription factor yin yang 1 (YY1) regulates G6PD transcriptional activity by directly binding to the G6PD promoter (63). Histone H3K36 methyltransferase NSD2 methylates H3K36me2 at the G6PD promoter to up-regulate its expression (68). In addition, some post-translational modifications such as phosphorylation, acetylation, O-GlcNAcylation and ubiquitination affect the activity of G6PD (59, 67, 69, 70).
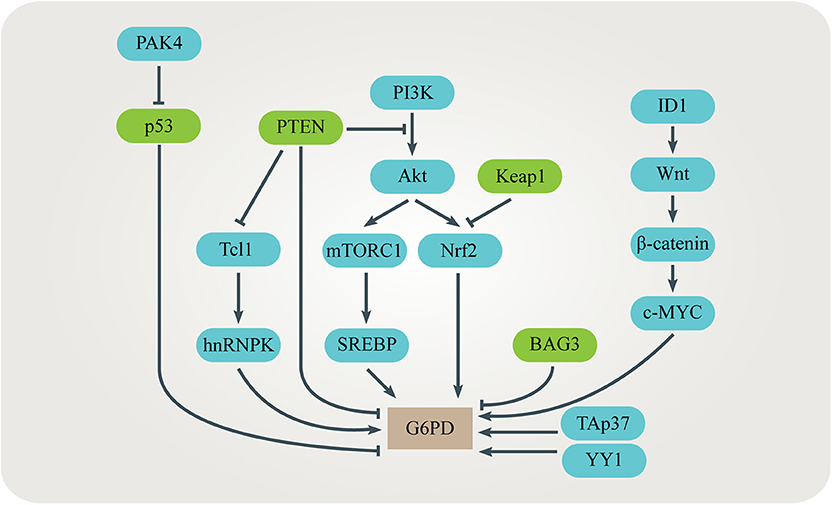
Figure 4. Regulation of G6PD in cancers. Several signaling pathways have been identified to be responsible for promoting G6PD expression or activity in cancer cells. These signaling pathways interact with each other, adding complexity to the regulation of G6PD.
Activated G6PD increases flux through the oxidative branch of PPP. Up-regulation of the PPP flux provides cells with R5P for nucleotide biosynthesis, as well as NADPH for biosynthesis and maintaining redox homeostasis (59, 67, 70). The level of G6PD often negatively correlates to the prognosis of cancer patients (61). Suppression of G6PD induces cellular senescence in hepatocellular carcinoma (HCC) cells and leads to intracellular oxidative stress, making cancer cells sensitive to chemotherapy (60, 61). Interestingly, elevated G6PD is not observed in liver cirrhosis which is a main cause of liver cancer, indicating that G6PD might play an important role in promoting malignant transformation (88).
6PGL is found to be associated with shorter overall survival in breast cancer patients with bone metastases (89). However, the role of 6PGL in cancer remains to be elucidated.
Elevated 6PGD and its product Ru5P inhibit AMPK activation by disrupting the active LKB1 complex, which promotes lipogenesis by abolishing AMPK-dependent acetyl-CoA carboxylase 1 (ACC1) phosphorylation and inactivation (90). 3-phosphoglycerate (3-PG), the intermediate product of glycolysis, inhibits 6PGD enzyme activity. Therefore, elevated glycolytic enzyme phosphoglycerate mutase 1 (PGAM1) keeps its substrate 3-PG to a relatively low level and promotes the PPP in cancer cells (91). In addition to its function as a metabolic enzyme, 6PGD also regulates cell metastasis by promoting phosphorylation of c-Met (92). 6PGD promotes the formation of distant metastatic subclones in pancreatic ductal adenocarcinoma (PDAC) by regulating epigenetic reprogramming. 6PGD inhibitor 6-aminonicotinamide (6AN) selectively and quantitatively reverses several reprogrammed chromatin modifications and blocks the tumorigenic potential of distant metastasis (93). How 6PGD and the PPP regulate epigenetic programs requires further investigation.
Elevated 6PGD expression and/or enzyme activity is also found in liver cancer, cervical cancer, thyroid cancer, breast cancer, ovarian cancer, non-small cell lung cancer (NSCLC) and leukemia (72, 94–99). In lung cancer, YTH domain family 2 (YTHDF2) promotes 6PGD mRNA translation by directly binding to the m6A modification site, leading to increased oxidative PPP flux (71). Post-translational modification of 6PGD is important for cancer cell proliferation and tumor growth. Acetylation of 6PGD plays a key role in coordinating redox homeostasis, lipogenesis and glycolysis (72). In glioma, epidermal growth factor receptor (EGFR) activation increases 6PGD phosphorylation and activation to promote DNA synthesis and resistance to radiation (73). Patients with lower levels of 6PGD Y481 phosphorylation have longer median survival time (73). Aberrant expression of 6PGD can accelerate cancer cell proliferation and induce resistance to chemical or radical therapy (72, 94–99). All these findings suggest that inhibiting the expression or activity of 6PGD might be a promising therapeutic strategy for cancer.
Enzymes in the Non-oxidative PPP
RPI and RPE expression is induced by the oncogenic Kras mutation, which is critical for the initiation of PDAC. KrasG12D regulates the non-oxidative but not oxidative PPP to provide cancer cells with sufficient R5P for nucleotide biosynthesis (74). RPI mRNA and protein levels are elevated in HCC. RPI promotes tumor growth and colony formation by negatively modulating protein phosphatase 2A (PP2A) to activate extracellular signal-regulated kinase (ERK) signaling pathways (100). In zebrafish, overexpression of RPI contributes to fatty liver, liver cirrhosis and cell proliferation (101). Therefore, whether and how RPI plays an important role in HCC development is worthy of further study. Furthermore, RPI increases the stability of β-catenin and promotes colorectal tumorigenesis by inducing Wnt target genes such as Cyclin D1 in zebrafish (102). High level of RPI is reported to predict negative clinical outcomes of colorectal cancer patients. Moreover, miRNA-124 decreases glucose metabolism and cell growth in colorectal cancer by down-regulating RPI (103).
The expression of TKT is elevated in many types of cancer (75–78, 83, 104). BTB and CNC homolog 1 (BACH1) and kelch-like ECH-associated protein 1 (KEAP1) negatively regulate TKT expression while NRF2 positively regulates it (75). 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase 4 (PFKFB4) phosphorylates steroid receptor coactivator-3 (SRC-3) at S857 which increases TKT expression (94). Hypoxia-inducible factor-1α (HIF-1α) induced in leukemia and pancreatic cancer enhances the non-oxidative arm of the PPP by promoting TKT activity, resulting in resistance to chemotherapy (77, 78). In addition to TKT, its two homologs, transketolase like-1 (TKTL1) and transketolase like-2 (TKTL2) are also found in human beings (105). TKT and TKTL1 rather than TKTL2 are essential for promoting cell growth and reducing oxidative stress in cancer cells (106, 107).
Patients with pancreatic cancer have higher levels of serum fructose which induces TKT expression to drive nucleic acid synthesis in cancer cells (104). TKT can maintain redox hemostasis by regulating the level of NADPH in liver cancer cells (75). Inhibition of TKT leads to increased ROS production and decreased glycolytic flux. Despite the accumulation of R5P, knockdown of TKT suppresses tumor growth and sensitizes cancer cells to chemotherapy (75). Recent work suggests that TKT promotes genome instability by regulating nucleotide biosynthesis during liver injury and cancer initiation (108). In addition, TKT can regulate cell cycle and promote the viability and proliferation of cancer cells independent of its enzyme activity. TKT interacting with EGFR and MAPK3 might be the underlying mechanism (109).
TALDO is highly expressed in gastric adenocarcinoma and HCC (110, 111). Moreover, higher TALDO expression often indicates poorer clinical outcomes and more resistance to trastuzumab therapy in breast cancer. When human epidermal growth factor receptor 2 (HER2) signaling is inhibited, breast cancer cells rely on the non-oxidative arm of the PPP to replenish the oxidative arm. Combined with HER2 inhibition, TALDO knockdown can exacerbate the reduction of NADPH and promote cell death (112). Surprisingly, TALDO can protect against cancer initiation. Loss of TALDO reduces GSH and diminishes β-catenin phosphorylation and Fas-dependent apoptosis, promoting hepatocarcinogenesis in mouse models (113).
Could PPP be a Target For T2Dm and Cancer?
The major contributor to cytosolic NADPH is the PPP (114). NADPH, a key intracellular reductant, is required for glutathione system and other ROS scavengers to maintain the redox homeostasis (115). High ROS levels not only damage DNA, proteins and lipids to induce genome instability and activate NF-κB, PI3K, HIF-1α, and MAPK which contributes to carcinogenesis (116), but also result in T2DM (117, 118). Therefore, the PPP serves as an ideal target for regulating the redox homeostasis in metabolic diseases and cancer.
The PPP regulates insulin secretion. When insulin or insulin-like growth factors bind insulin receptor (IR) and insulin-like growth factor-I receptor (IGF-IR), many downstream signaling pathways including the Ras/Raf/Mek/Erk pathway and the PI3K/Akt/mTOR pathway are activated to drive cell growth and proliferation. Insulin promoting the growth and proliferation of cells is one of the mechanisms underlying increased cancer risk in obese and diabetic patients (119, 120). Chronic inflammation is a well-known hallmark of cancer and insulin resistance. Obesity-related inflammation is believed to create a microenvironment contributing to the initiation and progression of cancer (121). In turn, cancer cells secrete cytokines to recruit macrophages, leading to cancer-related inflammation, which plays an important role in cancer cell migration and invasion (122, 123). Therefore, targeting the PPP to block the M1 macrophage function is a possible strategy for both T2DM and cancer.
Summary
The PPP plays a critical role in type 2 diabetes and cancer. Being the major source for NADPH, the PPP serves an ideal target for regulating the redox homeostasis in metabolic diseases and cancer. In addition, the intermediate R5P in the PPP is a precursor for nucleotide biosynthesis, which is essential for DNA replication and DNA damage repair. G6PD and 6PGD are the two enzymes in the PPP which catalyze the reactions to produce NADPH. Although TKT in the non-oxidative PPP does not directly catalyze the formation of NADPH, recent study has revealed its role in regulating cellular NADPH and R5P levels by balancing the flux between glycolysis and the PPP. Therefore, G6PD, 6PGD, and TKT are promising targets in the PPP for prevention and treatment of metabolic diseases and cancer.
Author Contributions
XT designed and revised the manuscript. TG, JY, YW, SZ, and YL wrote the manuscript and made the figures.
Funding
This work was supported by grants from the 12th Undergraduate Training Program for Innovation of Shanghai Jiao Tong University School of Medicine (1218033), from National Natural Science Foundation of China (81672322, 81972210, and 31601118); the Shanghai Municipal Science and Technology Major Project (19JC1410200); National Key R&D Program of China (2019YFA0906100); National Key Research and Development Program of China (No. 2016YFC1304800); The Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning and Construction Plan of Laboratory Technical Team in Shanghai Universities (SYjdyx18007).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor is currently co-organizing a Research Topic with one of the authors XT, and confirms the absence of any other collaboration.
Acknowledgments
We would like to thank all members in the Tong laboratory for their helpful suggestions. We apologize to those researchers whose work could not be cited or discussed in detail due to the space limitation.
References
1. Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci. (2014) 39:347–54. doi: 10.1016/j.tibs.2014.06.005
2. Wamelink MM, Struys EA, Jakobs C. The biochemistry, metabolism and inherited defects of the pentose phosphate pathway: a review. J Inherit Metab Dis. (2008) 31:703–17. doi: 10.1007/s10545-008-1015-6
3. Yen WC, Wu YH, Wu CC, Lin HR, Stern A, Chen SH, et al. Impaired inflammasome activation and bacterial clearance in G6PD deficiency due to defective NOX/p38 MAPK/AP-1 redox signaling. Redox Biol. (2020) 28:101363. doi: 10.1016/j.redox.2019.101363
4. Bradshaw PC. Cytoplasmic and mitochondrial NADPH-coupled redox systems in the regulation of aging. Nutrients. (2019) 11:504. doi: 10.3390/nu11030504
5. Stincone A, Prigione A, Cramer T, Wamelink MM, Campbell K, Cheung E, et al. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev Camb Philos Soc. (2015) 90:927–63. doi: 10.1111/brv.12140
6. Zheng Y, Ley SH, Hu FB. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol. (2018) 14:88–98. doi: 10.1038/nrendo.2017.151
7. Czech MP. Insulin action and resistance in obesity and type 2 diabetes. Nat Med. (2017) 23:804–14. doi: 10.1038/nm.4350
8. Zatterale F, Longo M, Naderi J, Raciti GA, Desiderio A, Miele C, et al. Chronic adipose tissue inflammation linking obesity to insulin resistance and type 2 diabetes. Front Physiol. (2020) 10:1607. doi: 10.3389/fphys.2019.01607
9. Saltiel AR, Olefsky JM. Inflammatory mechanisms linking obesity and metabolic disease. J Clin Invest. (2017) 127:1–4. doi: 10.1172/JCI92035
10. Viola A, Munari F, Sanchez-Rodriguez R, Scolaro T, Castegna A. The metabolic signature of macrophage responses. Front Immunol. (2019) 10:1462. doi: 10.3389/fimmu.2019.01462
11. Donath MY, Dinarello CA, Mandrup-Poulsen T. Targeting innate immune mediators in type 1 and type 2 diabetes. Nat Rev Immunol. (2019) 19:734–46. doi: 10.1038/s41577-019-0213-9
12. Lackey DE, Olefsky JM. Regulation of metabolism by the innate immune system. Nat Rev Endocrinol. (2016) 12:15–28. doi: 10.1038/nrendo.2015.189
13. Reilly SM, Saltiel AR. Adapting to obesity with adipose tissue inflammation. Nat Rev Endocrinol. (2017) 13:633–43. doi: 10.1038/nrendo.2017.90
14. Kim OK, Jun W, Lee J. Mechanism of ER stress and inflammation for hepatic insulin resistance in obesity. Ann Nutr Metab. (2015) 67:218–27. doi: 10.1159/000440905
15. Petersen KF, Dufour S, Savage DB, Bilz S, Solomon G, Yonemitsu S, et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci USA. (2007) 104:12587–94. doi: 10.1073/pnas.0705408104
16. Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol. (2008) 9:367–77. doi: 10.1038/nrm2391
17. Perry RJ, Camporez JPG, Kursawe R, Titchenell PM, Zhang D, Perry CJ, et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell. (2015) 160:745–58. doi: 10.1016/j.cell.2015.01.012
18. Li Y, Ding L, Hassan W, Abdelkader D, Shang J. Adipokines and hepatic insulin resistance. J Diabetes Res. (2013) 2013:170532. doi: 10.1155/2013/170532
19. Manne V, Handa P, Kowdley KV. Pathophysiology of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Clin Liver Dis. (2018) 22:23–37. doi: 10.1016/j.cld.2017.08.007
20. Gerber PA, Rutter GA. The role of oxidative stress and hypoxia in pancreatic beta-cell dysfunction in diabetes mellitus. Antioxid Redox Signal. (2017) 26:501–18. doi: 10.1089/ars.2016.6755
21. Lytrivi M, Castell AL, Poitout V, Cnop M. Recent insights into mechanisms of β-cell lipo- and glucolipotoxicity in type 2 diabetes. J Mol Biol. (2020) 432:1514–34. doi: 10.1016/j.jmb.2019.09.016
22. Ham M, Lee JW, Choi AH, Jang H, Choi G, Park J, et al. Macrophage glucose-6-phosphate dehydrogenase stimulates proinflammatory responses with oxidative stress. Mol Cell Biol. (2013) 33:2425–35. doi: 10.1128/MCB.01260-12
23. Park YJ, Choe SS, Sohn JH, Kim JB. The role of glucose-6-phosphate dehydrogenase in adipose tissue inflammation in obesity. Adipocyte. (2017) 6:147–53. doi: 10.1080/21623945.2017.1288321
24. Tian N, Liu Q, Li Y, Tong L, Lu Y, Zhu Y, et al. Transketolase deficiency in adipose tissues protects mice from diet-induced obesity by promoting lipolysis. Diabetes. (2020). doi: 10.2337/db19-1087. [Epub ahead of print].
25. Jin ES, Lee MH, Murphy RE, Malloy CR. Pentose phosphate pathway activity parallels lipogenesis but not antioxidant processes in rat liver. Am J Physiol Endocrinol Metab. (2018) 314:E543–51. doi: 10.1152/ajpendo.00342.2017
26. Roumans KHM, Lindeboom L, Veeraiah P, Remie CME, Phielix E, Havekes B, et al. Hepatic saturated fatty acid fraction is associated with de novo lipogenesis and hepatic insulin resistance. Nat Commun. (2020) 11:1891. doi: 10.1038/s41467-020-15684-0
27. Gupte RS, Floyd BC, Kozicky M, George S, Ungvari ZI, Neito V, et al. Synergistic activation of glucose-6-phosphate dehydrogenase and NAD(P)H oxidase by Src kinase elevates superoxide in type 2 diabetic, Zucker fa/fa, rat liver. Free Radical Biol Med. (2009) 47:219–28. doi: 10.1016/j.freeradbiomed.2009.01.028
28. Lee CH, Olson P, Hevener A, Mehl I, Chong LW, Olefsky JM, et al. PPARdelta regulates glucose metabolism and insulin sensitivity. Proc Natl Acad Sci USA. (2006) 103:3444–9. doi: 10.1073/pnas.0511253103
29. Lee-Young RS, Hoffman NJ, Murphy KT, Henstridge DC, Samocha-Bonet D, Siebel AL, et al. Glucose-6-phosphate dehydrogenase contributes to the regulation of glucose uptake in skeletal muscle. Mol Metab. (2016) 5:1083–91. doi: 10.1016/j.molmet.2016.09.002
30. Summermatter S, Baum O, Santos G, Hoppeler H, Handschin C. Peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) promotes skeletal muscle lipid refueling in vivo by activating de novo lipogenesis and the pentose phosphate pathway. J Bio Chem. (2010) 285:32793–800. doi: 10.1074/jbc.M110.145995
31. Summermatter S, Shui G, Maag D, Santos G, Wenk MR, Handschin C. PGC-1α improves glucose homeostasis in skeletal muscle in an activity-dependent manner. Diabetes. (2013) 62:85–95. doi: 10.2337/db12-0291
32. O'Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. (2016) 16:553–65. doi: 10.1038/nri.2016.70
33. Haschemi A, Kosma P, Gille L, Evans CR, Burant CF, Starkl P, et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab. (2012) 15:813–26. doi: 10.1016/j.cmet.2012.04.023
34. Nagy C, Haschemi A. Time and demand are two critical dimensions of immunometabolism: the process of macrophage activation and the pentose phosphate pathway. Front Immunol. (2015) 6:164. doi: 10.3389/fimmu.2015.00164
35. Kalwat MA, Cobb MH. Mechanisms of the amplifying pathway of insulin secretion in the beta cell. Pharmacol Ther. (2017) 179:17–30. doi: 10.1016/j.pharmthera.2017.05.003
36. Ferdaoussi M, Dai X, Jensen MV, Wang R, Peterson BS, Huang C, et al. Isocitrate-to-SENP1 signaling amplifies insulin secretion and rescues dysfunctional β cells. J Clin Invest. (2015) 125:3847–60. doi: 10.1172/JCI82498
37. Monte Alegre S, Saad ST, Delatre E, Saad MJ. Insulin secretion in patients deficient in glucose-6-phosphate dehydrogenase. Horm Metab Res. (1991) 23:171–3. doi: 10.1055/s-2007-1003644
38. Spegel P, Sharoyko VV, Goehring I, Danielsson AP, Malmgren S, Nagorny CL, et al. Time-resolved metabolomics analysis of beta-cells implicates the pentose phosphate pathway in the control of insulin release. Biochem J. (2013) 450:595–605. doi: 10.1042/BJ20121349
39. Zhang Z, Liew CW, Handy DE, Zhang Y, Leopold JA, Hu J, et al. High glucose inhibits glucose-6-phosphate dehydrogenase, leading to increased oxidative stress and beta-cell apoptosis. Faseb J. (2010) 24:1497–505. doi: 10.1096/fj.09-136572
40. Lee JW, Choi AH, Ham M, Kim JW, Choe SS, Park J, et al. G6PD up-regulation promotes pancreatic beta-cell dysfunction. Endocrinology. (2011) 152:793–803. doi: 10.1210/en.2010-0606
41. Riganti C, Gazzano E, Polimeni M, Aldieri E, Ghigo D. The pentose phosphate pathway: an antioxidant defense and a crossroad in tumor cell fate. Free Radic Biol Med. (2012) 53:421–36. doi: 10.1016/j.freeradbiomed.2012.05.006
42. Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circulation Res. (2010) 107:1058–70. doi: 10.1161/CIRCRESAHA.110.223545
43. Xu Y, Osborne BW, Stanton RC. Diabetes causes inhibition of glucose-6-phosphate dehydrogenase via activation of PKA. which contributes to oxidative stress in rat kidney cortex. Am J Physiol Renal Physiol. (2005) 289:F1040–7. doi: 10.1152/ajprenal.00076.2005
44. Wang M, Hu J, Yan L, Yang Y, He M, Wu M, et al. High glucose-induced ubiquitination of G6PD leads to the injury of podocytes. Faseb J. (2019) 33:6296–310. doi: 10.1096/fj.201801921R
45. Stanton RC. Glucose-6-phosphate dehydrogenase, NADPH and cell survival. IUBMB Life. (2012) 64:362–9. doi: 10.1002/iub.1017
46. Knapp M, Tu X, Wu R. Vascular endothelial dysfunction, a major mediator in diabetic cardiomyopathy. Acta Pharmacol Sin. (2019) 40:1–8. doi: 10.1038/s41401-018-0042-6
47. Rabbani N, Thornalley PJ. Emerging role of thiamine therapy for prevention and treatment of early-stage diabetic nephropathy. Diabetes Obes Metab. (2011) 13:577–83. doi: 10.1111/j.1463-1326.2011.01384.x
48. Maguire D, Talwar D, Shiels PG, McMillan D. The role of thiamine dependent enzymes in obesity and obesity related chronic disease states: a systematic review. Clin Nutr ESPEN. (2018) 25:8–17. doi: 10.1016/j.clnesp.2018.02.007
49. Babaei-Jadidi R. Karachalias N, Ahmed N, Battah S, Thornalley PJ. Prevention of incipient diabetic nephropathy by high-dose thiamine and benfotiamine. Diabetes. (2003) 52:2110–20. doi: 10.2337/diabetes.52.8.2110
50. Rabbani N, Alam SS, Riaz S, Larkin JR, Akhtar MW, Shafi T, et al. High-dose thiamine therapy for patients with type 2 diabetes and microalbuminuria: a randomised, double-blind placebo-controlled pilot study. Diabetologia. (2009) 52:208–12. doi: 10.1007/s00125-008-1224-4
51. Hammes HP, Du X, Edelstein D, Taguchi T, Matsumura T, Ju Q, et al. Brownlee. Benfotiamine blocks three major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy. Nat Med. (2003) 9:294–9. doi: 10.1038/nm834
52. Katare RG, Caporali A, Oikawa A, Meloni M, Emanueli C, Madeddu P. Vitamin B1 analog benfotiamine prevents diabetes-induced diastolic dysfunction and heart failure through Akt/Pim-1-mediated survival pathway. Circ Heart Fail. (2010) 3:294–305. doi: 10.1161/CIRCHEARTFAILURE.109.903450
53. Gadau S, Emanueli C, van Linthout S, Graiani G, Todaro M, Meloni M, et al. Benfotiamine accelerates the healing of ischaemic diabetic limbs in mice through protein kinase B/Akt-mediated potentiation of angiogenesis and inhibition of apoptosis. Diabetologia. (2006) 49:405–20. doi: 10.1007/s00125-005-0103-5
54. Alkhalaf A, Klooster A, van Oeveren W, Achenbach U, Kleefstra N, Slingerland RJ, et al. A double-blind, randomized, placebo-controlled clinical trial on benfotiamine treatment in patients with diabetic nephropathy. Diabetes Care. (2010) 33:1598–601. doi: 10.2337/dc09-2241
55. Fraser DA, Diep LM, Hovden IA, Nilsen KB, Sveen KA, Seljeflot I, et al. The effects of long-term oral benfotiamine supplementation on peripheral nerve function and inflammatory markers in patients with type 1 diabetes: a 24-month, double-blind, randomized, placebo-controlled trial. Diabetes Care. (2012) 35:1095–7. doi: 10.2337/dc11-1895
56. Du W, Jiang P, Mancuso A, Stonestrom A, Brewer MD, Minn AJ, et al. TAp73 enhances the pentose phosphate pathway and supports cell proliferation. Nat Cell Biol. (2013) 15:991–1000. doi: 10.1038/ncb2789
57. Jiang P, Du W, Yang X. A critical role of glucose-6-phosphate dehydrogenase in TAp73-mediated cell proliferation. Cell Cycle. (2013) 12:3720–6. doi: 10.4161/cc.27267
58. Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. (2012) 22:66–79. doi: 10.1016/j.ccr.2012.05.016
59. Rao X, Duan X, Mao W, Li X, Li Z, Li Q, et al. O-GlcNAcylation of G6PD promotes the pentose phosphate pathway and tumor growth. Nat Commun. (2015) 6:8468. doi: 10.1038/ncomms9468
60. Yin X, Tang B, Li JH, Wang Y, Zhang L, Xie XY, et al. ID1 promotes hepatocellular carcinoma proliferation and confers chemoresistance to oxaliplatin by activating pentose phosphate pathway. J Exp Clin Cancer Res. (2017) 36:166. doi: 10.1186/s13046-017-0637-7
61. Hong X, Song R, Song H, Zheng T, Wang J, Liang Y, et al. PTEN antagonises Tcl1/hnRNPK-mediated G6PD pre-mRNA splicing which contributes to hepatocarcinogenesis. Gut. (2014) 63:1635–47. doi: 10.1136/gutjnl-2013-305302
62. Kong DH, Li S, Du ZX, Liu C, Liu BQ, Li C, et al. BAG3 elevation inhibits cell proliferation via direct interaction with G6PD in hepatocellular carcinomas. Oncotarget. (2016) 7:700–11. doi: 10.18632/oncotarget.6396
63. Wu S, Wang H, Li Y, Xie Y, Huang C, Zhao H, et al. Transcription factor YY1 promotes cell proliferation by directly activating the pentose phosphate pathway. Cancer Res. (2018) 78:4549–62. doi: 10.1158/0008-5472.CAN-17-4047
64. Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M, et al. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol. (2011) 13:310–6. doi: 10.1038/ncb2172
65. Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. (2010) 39:171–83. doi: 10.1016/j.molcel.2010.06.022
66. Poulain L, Sujobert P, Zylbersztejn F, Barreau S, Stuani L, Lambert M, High mTORC1 activity drives glycolysis addiction and sensitivity to G6PD inhibition in acute myeloid leukemia cells. Leukemia. (2017) 31:2326–35. doi: 10.1038/leu.2017.81
67. Xu SN, Wang TS, Li X, Wang YP. SIRT2 activates G6PD to enhance NADPH production and promote leukaemia cell proliferation. Sci Rep. (2016) 6:32734. doi: 10.1038/srep32734
68. Wang J, Duan Z, Nugent Z, Zou JX, Borowsky AD, Zhang Y, et al. Reprogramming metabolism by histone methyltransferase NSD2 drives endocrine resistance via coordinated activation of pentose phosphate pathway enzymes. Cancer Lett. (2016) 378:69–79. doi: 10.1016/j.canlet.2016.05.004
69. Cheng J, Huang Y, Zhang X, Yu Y, Wu S, Jiao J, et al. TRIM21 and PHLDA3 negatively regulate the crosstalk between the PI3K/AKT pathway and PPP metabolism. Nat Commun. (2020) 11:1880. doi: 10.1038/s41467-020-15819-3
70. Ma X, Wang L, Huang D, Li Y, Yang D, Li T, et al. Polo-like kinase 1 coordinates biosynthesis during cell cycle progression by directly activating pentose phosphate pathway. Nat Commun. (2017) 8:1506. doi: 10.1038/s41467-017-01647-5
71. Sheng H, Li Z, Su S, Sun W, Zhang X, Li L, et al. YTH domain family 2 promotes lung cancer cell growth by facilitating 6-phosphogluconate dehydrogenase mRNA translation. Carcinogenesis. (2019). doi: 10.1093/carcin/bgz152. [Epub ahead of print].
72. Shan C, Elf S, Ji Q, Kang HB, Zhou L, Hitosugi T, et al. Lysine acetylation activates 6-phosphogluconate dehydrogenase to promote tumor growth. Mol Cell. (2014) 55:552–65. doi: 10.1016/j.molcel.2014.06.020
73. Liu R, Li W, Tao B, Wang X, Yang Z, Zhang Y, et al. Tyrosine phosphorylation activates 6-phosphogluconate dehydrogenase and promotes tumor growth and radiation resistance. Nat Commun. (2019) 10:991. doi: 10.1038/s41467-019-08921-8
74. Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. (2012) 149:656–70. doi: 10.1016/j.cell.2012.01.058
75. Xu IM, Lai RK, Lin SH, Tse AP, Chiu DK, Koh HY, et al. Transketolase counteracts oxidative stress to drive cancer development. Proc Natl Acad Sci USA. (2016) 113:E725–34. doi: 10.1073/pnas.1508779113
76. Dasgupta S, Rajapakshe K, Zhu B, Nikolai BC, Yi P, Putluri N, et al. Metabolic enzyme PFKFB4 activates transcriptional coactivator SRC-3 to drive breast cancer. Nature. (2018) 556:249–54. doi: 10.1038/s41586-018-0018-1
77. Zhao F, Mancuso A, Bui TV, Tong X, Gruber JJ, Swider CR, et al. Thompson. Imatinib resistance associated with BCR-ABL upregulation is dependent on HIF-1alpha-induced metabolic reprograming. Oncogene. (2010) 29:2962–72. doi: 10.1038/onc.2010.67
78. Shukla SK, Purohit V, Mehla K, Gunda V, Chaika NV, Vernucci E, et al. Singh. MUC1 and HIF-1alpha signaling crosstalk induces anabolic glucose metabolism to impart gemcitabine resistance to pancreatic cancer. Cancer Cell. (2017) 32:71–87.e7. doi: 10.1016/j.ccell.2017.06.004
79. Wang X, Li X, Zhang X, Fan R, Gu H, Shi Y, et al. Glucose-6-phosphate dehydrogenase expression is correlated with poor clinical prognosis in esophageal squamous cell carcinoma. Eur J Surg Oncol. (2015) 41:1293–9. doi: 10.1016/j.ejso.2015.08.155
80. Wang J, Yuan W, Chen Z, Wu S, Chen J, Ge J, et al. Overexpression of G6PD is associated with poor clinical outcome in gastric cancer. Tumour Biol. (2012) 33:95–101. doi: 10.1007/s13277-011-0251-9
81. Zhang X, Li Y, Shao Y, Xiao J, Zhu G, Li F. PAK4 regulates G6PD activity by p53 degradation involving colon cancer cell growth. Cell Death Dis. (2017) 8:e2820. doi: 10.1038/cddis.2017.85
82. Chen X, Xu Z, Zhu Z, Chen A, Fu G, Wang Y, et al. Modulation of G6PD affects bladder cancer via ROS accumulation and the AKT pathway in vitro. Int J Oncol. (2018) 53:1703–12. doi: 10.3892/ijo.2018.4501
83. Benito A, Polat IH, Noe V, Ciudad CJ, Marin S, Cascante M. Glucose-6-phosphate dehydrogenase and transketolase modulate breast cancer cell metabolic reprogramming and correlate with poor patient outcome. Oncotarget. (2017) 8:106693–706. doi: 10.18632/oncotarget.21601
84. Ding Y, Yang M, She S, Min H, Xv X, Ran X, et al. iTRAQ-based quantitative proteomic analysis of cervical cancer. Int J Oncol. (2015) 46:1748–58. doi: 10.3892/ijo.2015.2859
85. Zhang Q, Yang Z, Han Q, Bai H, Wang Y, Yi X, et al. G6PD promotes renal cell carcinoma proliferation through positive feedback regulation of p-STAT3. Oncotarget. (2017) 8:109043–60. doi: 10.18632/oncotarget.22566
86. Nagashio R, Oikawa S, Yanagita K, Hagiuda D, Kuchitsu Y, Igawa S, et al. Prognostic significance of G6PD expression and localization in lung adenocarcinoma. Biochim Biophys Acta Proteins Proteom. (2019) 1867:38–46. doi: 10.1016/j.bbapap.2018.05.005
87. Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, et al. TIGAR. a p53-inducible regulator of glycolysis and apoptosis. Cell. (2006) 126:107–20. doi: 10.1016/j.cell.2006.05.036
88. Kowalik MA, Guzzo G, Morandi A, Perra A, Menegon S, Masgras I, et al. Metabolic reprogramming identifies the most aggressive lesions at early phases of hepatic carcinogenesis. Oncotarget. (2016) 7:32375–93. doi: 10.18632/oncotarget.8632
89. Cha YJ, Jung WH, Koo JS. Differential site-based expression of pentose phosphate pathway-related proteins among breast cancer metastases. Dis Markers. (2017) 2017:7062517. doi: 10.1155/2017/7062517
90. Lin R, Elf S, Shan C, Kang HB, Ji Q, Zhou L, et al. 6-Phosphogluconate dehydrogenase links oxidative PPP. lipogenesis and tumour growth by inhibiting LKB1-AMPK signalling. Nat Cell Biol. (2015) 17:1484–96. doi: 10.1038/ncb3255
91. Hitosugi T, Zhou L, Elf S, Fan J, Kang HB, Seo JH, et al. Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell. (2012) 22:585–600. doi: 10.1016/j.ccr.2012.09.020
92. Chan B, VanderLaan PA, Sukhatme VP. 6-Phosphogluconate dehydrogenase regulates tumor cell migration in vitro by regulating receptor tyrosine kinase c-Met. Biochem Biophys Res Commun. (2013) 439:247–51. doi: 10.1016/j.bbrc.2013.08.048
93. McDonald OG, Li X, Saunders T, Tryggvadottir R, Mentch SJ, Warmoes MO, et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat Genet. (2017) 49:367. doi: 10.1038/ng.3753
94. Chen H, Wu D, Bao L, Yin T, Lei D, Yu J 6PGD inhibition sensitizes hepatocellular carcinoma to chemotherapy via AMPK activation and metabolic reprogramming. Biomed Pharmacother. (2019) 111:1353–8. doi: 10.1016/j.biopha.2019.01.028
95. Guo H, Xiang Z, Zhang Y, Sun D. Inhibiting 6-phosphogluconate dehydrogenase enhances chemotherapy efficacy in cervical cancer via AMPK-independent inhibition of RhoA and Rac1. Clin Transl Oncol. (2019) 21:404–11. doi: 10.1007/s12094-018-1937-x
96. Giusti L, Iacconi P, Ciregia F, Giannaccini G, Donatini GL, Basolo F, et al. Fine-needle aspiration of thyroid nodules: proteomic analysis to identify cancer biomarkers. J Proteome Res. (2008) 7:4079–88. doi: 10.1021/pr8000404
97. Yang X, Peng X, Huang J. Inhibiting 6-phosphogluconate dehydrogenase selectively targets breast cancer through AMPK activation. Clin Transl Oncol. (2018) 20:1145–52. doi: 10.1007/s12094-018-1833-4
98. Zheng W, Feng Q, Liu J, Guo Y, Gao L, Li R, et al. Inhibition of 6-phosphogluconate dehydrogenase reverses cisplatin resistance in ovarian and lung cancer. Front Pharmacol. (2017) 8:421. doi: 10.3389/fphar.2017.00421
99. Bhanot H, Weisberg EL, Reddy MM, Nonami A, Neuberg D, Stone RM, et al. Acute myeloid leukemia cells require 6-phosphogluconate dehydrogenase for cell growth and NADPH-dependent metabolic reprogramming. Oncotarget. (2017) 8:67639–50. doi: 10.18632/oncotarget.18797
100. Ciou SC, Chou YT, Liu YL, Nieh YC, Lu JW, Huang SF, et al. Ribose-5-phosphate isomerase A regulates hepatocarcinogenesis via PP2A and ERK signaling. Int J Cancer. (2015) 137:104–15. doi: 10.1002/ijc.29361
101. Chou YT, Chen LY, Tsai SL, Tu HC, Lu JW, Ciou SC, et al. Ribose-5-phosphate isomerase A overexpression promotes liver cancer development in transgenic zebrafish via activation of ERK and beta-catenin pathways. Carcinogenesis. (2019) 40:461–73. doi: 10.1093/carcin/bgy155
102. Chou YT, Jiang JK, Yang MH, Lu JW, Lin HK, Wang HD, et al. Identification of a noncanonical function for ribose-5-phosphate isomerase A promotes colorectal cancer formation by stabilizing and activating beta-catenin via a novel C-terminal domain. PLoS Biol. (2018) 16:e2003714. doi: 10.1371/journal.pbio.2003714
103. Qiu Z, Guo W, Wang Q, Chen Z, Huang S, Zhao F, et al. MicroRNA-124 reduces the pentose phosphate pathway and proliferation by targeting PRPS1 and RPIA mRNAs in human colorectal cancer cells. Gastroenterology. (2015) 149:1587–98.e11. doi: 10.1053/j.gastro.2015.07.050
104. Liu H, Huang D, McArthur DL, Boros LG, Nissen N, Heaney AP. Fructose induces transketolase flux to promote pancreatic cancer growth. Cancer Res. (2010) 70:6368–76. doi: 10.1158/0008-5472.CAN-09-4615
105. Kochetov GA, Solovjeva ON. Structure and functioning mechanism of transketolase. Biochim Biophys Acta. (2014) 1844:1608–18. doi: 10.1016/j.bbapap.2014.06.003
106. Vizan P, Alcarraz-Vizan G, Diaz-Moralli S, Solovjeva ON, Frederiks WM, Cascante M. Modulation of pentose phosphate pathway during cell cycle progression in human colon adenocarcinoma cell line HT29. Int J Cancer. (2009) 124:2789–96. doi: 10.1002/ijc.24262
107. Xu X, Zur Hausen A, Coy JF, Lochelt M. Transketolase-like protein 1 (TKTL1) is required for rapid cell growth and full viability of human tumor cells. Int J Cancer. (2009) 124:1330–7. doi: 10.1002/ijc.24078
108. Li M, Lu Y, Tong L, Gu XC, Meng J, Zhu Y, et al. Transketolase deficiency protects the liver from DNA damage by increasing levels of ribose 5-phosphate and nucleotides. Cancer Res. (2019) 79:3689–701. doi: 10.1158/0008-5472.CAN-18-3776
109. Qin Z, Xiang C, Zhong F, Liu Y, Dong Q, Li K, et al. Transketolase (TKT) activity and nuclear localization promote hepatocellular carcinoma in a metabolic and a non-metabolic manner. J Exp Clin Cancer Res. (2019) 38:154. doi: 10.1186/s13046-019-1131-1
110. Kocevar N, Odreman F, Vindigni A, Grazio SF, Komel R. Proteomic analysis of gastric cancer and immunoblot validation of potential biomarkers. World J Gastroenterol. (2012) 18:1216–28. doi: 10.3748/wjg.v18.i11.1216
111. Wang C, Guo K, Gao D, Kang X, Jiang K, Li Y, et al. Identification of transaldolase as a novel serum biomarker for hepatocellular carcinoma metastasis using xenografted mouse model and clinic samples. Cancer Lett. (2011) 313:154–66. doi: 10.1016/j.canlet.2011.08.031
112. Ding Y, Gong C, Huang D, Chen R, Sui P, Lin KH, et al. Synthetic lethality between HER2 and transaldolase in intrinsically resistant HER2-positive breast cancers. Nat Commun. (2018) 9:4274. doi: 10.1038/s41467-018-06651-x
113. Hanczko R, Fernandez DR, Doherty E, Qian Y, Vas G, Niland B, et al. Prevention of hepatocarcinogenesis and increased susceptibility to acetaminophen-induced liver failure in transaldolase-deficient mice by N-acetylcysteine. J Clin Invest. (2009) 119:1546–57. doi: 10.1172/JCI35722
114. Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, Rabinowitz JD. Quantitative flux analysis reveals folate-dependent NADPH production. Nature. (2014) 510:298–302. doi: 10.1038/nature13236
115. Traverso N, Ricciarelli R, Nitti M, Marengo B, Furfaro AL, Pronzato MA, et al. Role of glutathione in cancer progression and chemoresistance. Oxid Med Cell Longev. (2013) 2013:972913. doi: 10.1155/2013/972913
116. Assi M. The differential role of reactive oxygen species in early and late stages of cancer. Am J Physiol Regul Integr Comp Physiol. (2017) 313:R646–53. doi: 10.1152/ajpregu.00247.2017
117. Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive oxygen species in metabolic and inflammatory signaling. Circulation Res. (2018) 122:877–902. doi: 10.1161/CIRCRESAHA.117.311401
118. Newsholme P, Cruzat VF, Keane KN, Carlessi R, de Bittencourt PIH Jr. Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem J. (2016) 473:4527–50. doi: 10.1042/BCJ20160503C
119. Malaguarnera R, Belfiore A. The insulin receptor: a new target for cancer therapy. Front Endocrinol. (2011) 2:93. doi: 10.3389/fendo.2011.00093
120. Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. (2008) 8:915–28. doi: 10.1038/nrc2536
121. Andersen DK, Korc M, Petersen GM, Eibl G, Li D, Rickels MR, et al. Diabetes, pancreatogenic diabetes, and pancreatic cancer. Diabetes. (2017) 66:1103–10. doi: 10.2337/db16-1477
122. Poh AR, Ernst M. Targeting macrophages in cancer: from bench to bedside. Front Oncol. (2018) 8:49. doi: 10.3389/fonc.2018.00049
Keywords: the pentose phosphate pathway, metabolism, obesity, diabetes, cancer
Citation: Ge T, Yang J, Zhou S, Wang Y, Li Y and Tong X (2020) The Role of the Pentose Phosphate Pathway in Diabetes and Cancer. Front. Endocrinol. 11:365. doi: 10.3389/fendo.2020.00365
Received: 27 October 2019; Accepted: 11 May 2020;
Published: 09 June 2020.
Edited by:
Debbie C. Thurmond, Beckman Research Institute, City of Hope, United StatesReviewed by:
Lei Jiang, City of Hope National Medical Center, United StatesMatthew J. Brady, University of Chicago, United States
Copyright © 2020 Ge, Yang, Zhou, Wang, Li and Tong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuemei Tong, eHVlbWVpdG9uZ0BzaHNtdS5lZHUuY24=
†These authors have contributed equally to this work