 Mareike S. Poetsch
Mareike S. Poetsch Anna Strano
Anna Strano Kaomei Guan
Kaomei Guan
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 16 June 2020
Sec. Obesity
Volume 11 - 2020 | https://doi.org/10.3389/fendo.2020.00354
This article is part of the Research Topic Common and Distinct Mechanisms Driving Type 2 Diabetes Mellitus Cardiomyopathy in the Absence and Presence of Obesity View all 5 articles
The adipocyte-derived adipokine leptin exerts pleiotropic effects, which are essential for the regulation of energy balance and cell metabolism, for controlling inflammatory and immune responses, and for the maintenance of homeostasis of the cardiovascular system. Leptin resistance in obese or type 2 diabetes mellitus (T2DM) patients is defined as a decrease in tissue response to leptin. In the cardiovascular system, leptin resistance exhibits the adverse effect on the heart's response to stress conditions and promoting cardiac remodeling due to impaired cardiac metabolism, increased fibrosis, vascular dysfunction, and enhanced inflammation. Leptin resistance or leptin signaling deficiency results in the risk increase of cardiac dysfunction and heart failure, which is a leading cause of obesity- and T2DM-related morbidity and mortality. Animal studies using leptin- and leptin receptor- (Lepr) deficient rodents have provided many useful insights into the underlying molecular and pathophysiological mechanisms of obese- and T2DM-associated metabolic and cardiovascular diseases. However, none of the animal models used so far can fully recapitulate the phenotypes of patients with obese or T2DM. Therefore, the role of leptin in the human cardiovascular system, and whether leptin affects cardiac function directly or acts through a leptin-regulated neurohumoral pathway, remain elusive. As the prevalence of obesity and diabetes is continuously increasing, strategies are needed to develop and apply human cell-based models to better understand the precise role of leptin directly in different cardiac cell types and to overcome the existing translational barriers. The purpose of this review is to discuss the mechanisms associated with leptin signaling deficiency or leptin resistance in the development of metabolic and cardiovascular diseases. We analyzed and comprehensively addressed substantial findings in pathophysiological mechanisms in commonly used leptin- or Lepr-deficient rodent models and highlighted the differences between rodents and humans. This may open up new strategies to develop directly and reliably applicable models, which resemble the human pathophysiology in order to advance health care management of obesity- and T2DM-related cardiovascular complications.
Recent reports of the world health organization (WHO) emphasize the dramatic increase in the prevalence of obesity, which has nearly tripled since 1975. In 2016, almost 39% (1.9 billion) of adults worldwide were overweight and 13% (>650 million) were obese. The prevalence of obesity in children and young adolescents (aged 5–19 years) has also risen from 4% in 1975 to 18% in 2016 (1). A common co-morbidity of excess bodyweight and physical inactivity is type 2 diabetes mellitus (T2DM), which accounts for 90–95% of all diagnosed diabetes cases in adults (2). The global prevalence of diabetes among adults has risen from 4.7% in 1980 to 8.5% in 2014 and by 2014 diabetes mellitus affected more than 422 million people. These escalating healthcare problems of monumental proportions represent a serious hazard of developing cardiovascular diseases with significant morbidity and mortality (2). However, the complex mechanisms underlying the detrimental impact of obesity and diabetes on the cardiovascular system are poorly understood (3). In the last two decades, many studies show that the adipocyte-derived peptide hormone leptin plays an important role in linking obesity, inflammation, metabolic syndrome, and cardiovascular diseases.
Since the discovery of leptin in 1994 (4), extensive research in various murine model systems revealed a complex interplay of genes and environmental factors involved in the development of obesity, which impacts homeostatic control of energy metabolism and feeding in the central nervous system [reviewed in (5)]. As a circulating adipokine, leptin binds to the leptin receptor (LEPR, also known as OB-R) in the arcuate nucleus (ARC) of the hypothalamus where it functions as an anorexigenic (appetite-suppressing) peptide hormone that regulates food intake and energy expenditure. Two types of neurons in the ARC with antagonistic functions are major targets of leptin: (i) pro-opiomelanocortin (POMC) neurons and (ii) neuropeptide-Y (NPY) and agouti-related peptide (AgRP) neurons. In the hypothalamus, both POMC and NPY/AgRP neurons are also major targets of insulin, therefore, the actions of leptin and insulin are interconnected and contribute to optimal metabolic control. While insulin maintains adequate energy storage and utilization, leptin reduces continuous energy intake. It is known that leptin and insulin act on different subpopulations of POMC neurons (6) and both leptin and insulin are required for and play key roles in the central regulation of energy expenditure and glucose homeostasis. Although leptin and insulin are two of the most widely studied peptide hormones, new studies to decipher their precise functions are urgently needed. Recently, a study revealed that leptin increases the expression of the insulin receptor phosphatase PTP1B in the ARC, which leads to reduced insulin sensitivity and impaired insulin-mediated suppression of hepatic glucose production in diet-induced obese mice (7). The central pharmacological inhibition or ARC-targeted deletion of PTP1B restores insulin-mediated suppression of hepatic glucose production in obese mice (7). Moreover, another recent study has shown that hyperleptinemia is a driving force for obesity and its associated metabolic syndrome and that partial deletion, rather than complete elimination of leptin restores hypothalamic leptin sensitivity and increases insulin sensitivity in diet-induced obese mice (8).
This review is focused on leptin and LEPR signaling in the (dys-)regulation of the cardiovascular system in the context of obesity and metabolic syndrome. Since most of the molecular insights regarding leptin signaling are derived from studies in rodent models, we are aiming to highlight the differences in the leptin-LEPR interaction in rodents and humans. Finally, we provide an outlook on recent perspectives on deciphering the underlying pathophysiological mechanisms of leptin-mediated cardiomyopathy and future therapeutic approaches for the treatment of diabetes-mediated cardiovascular disease.
Leptin is primarily produced by adipocytes and released from small vesicles within adipocytes in a diurnal pulsatile manner, with higher rates in the evening and early morning. Although the release of leptin occurs independent of its mRNA regulation (9), increase of leptin mRNA transcription is necessary to retain constant rates of leptin secretion and to prevent rapid exhaustion of leptin vesicle stores (10). Once secreted, leptin circulates in the serum both in a free and in a bound form.
The leptin receptor (LEPR, or OB-R) was identified as a single membrane-spanning receptor that structurally resembles class I cytokine receptors. Interestingly, the gene encoding the leptin receptor, both in mice (db) and in humans (LEPR), comprises a dual promoter: while the B219/OB-R promoter generates db/LEPR transcripts only, the OB-RGRP promoter initiates transcription of both db/LEPR and OB-RGRP/LEPROT (leptin receptor gene-related protein/leptin receptor overlapping transcript) genes (11). LEPR is mainly localized in intracellular compartments, including endosomes, the trans-Golgi apparatus, or the endoplasmic reticulum, and to a lesser extent on the plasma membrane (12). Studies revealed that OB-RGRP/LEPROT (also known as endospanin-1) negatively affected the expression of LEPR on the cell surface (13).
Six isoforms of LEPR (LEPRa, b, c, d, e, and f, also known as OB-Ra, b, c, d, e, and f) with distinct biological activities have been identified (Figure 1). In the mouse, all of them are generated by alternative mRNA splicing (14). In humans, alternative splicing also generates distinct LEPR isoforms with the exception of the soluble isoform LEPRe, which is derived from membrane-spanning receptors by ectodomain shedding (15). All LEPR isoforms share an identical, highly glycosylated extracellular domain of 840 amino acids, which comprises six subdomains: a N-terminal domain of undefined function (NTD), two cytokine receptor homologous domains (CRHI, CRHII) harboring a Trp-Ser-X-Trp-Ser motif, an immunoglobulin-like domain (IgD), and two fibronectin type 3 domains (FNIII). Leptin uses multiple binding sites in the CRHII and IgD domains to engage LEPR, which are fundamental for leptin-binding and LEPR activation (Figure 1).
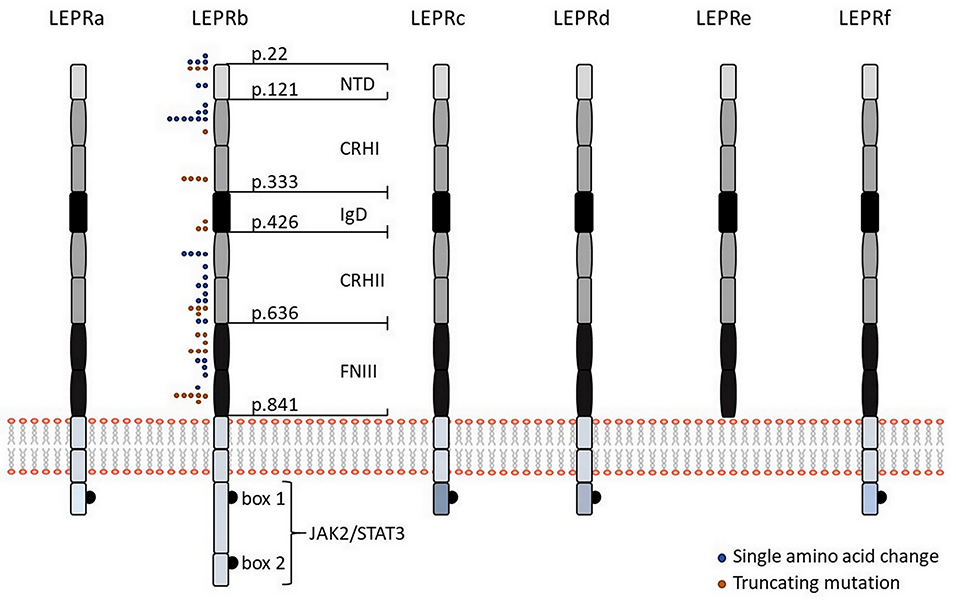
Figure 1. Leptin receptor isoforms and visualization of mutations in the human LEPR protein. Schematic representation of the six different isoforms of LEPR in humans (LEPRa, b, c, d, e, and f). All isoforms share identical extracellular domains as well as the first 29 amino acids containing box 1 motif for binding of JAK2 of the intracellular domain but they differ in the length and sequence of the C-terminal domain. The intracellular domain of LEPRb contains another JAK binding domain (“box 2”) in addition to a STAT binding site, making LEPRb the predominant isoform responsible for signal transduction. Five of the six isoforms have a transmembrane domain and are generated by alternative mRNA splicing, while the shortest isoform LEPRe is derived by ectodomain shedding at the membrane-spanning domain. Colored dots indicate positions of human LEPR mutations, which result in single amino acid changes (blue dots), or a truncated protein (orange dots), as previously described (38). NTD, N-terminal domain of undefined function; CRHI and CRHII, cytokine receptor homologous domain I and II; IgD, immunoglobulin-like domain; FNIII, fibronectin type 3 domains; JAK2, Janus family tyrosine kinase 2; STAT3, signal transducer and activator of transcription 3.
The membrane-bound isoforms LEPRa, b, c, d, and f comprise the same sequence of a 23-amino acid spanning trans-membrane domain as well as the first 29 amino acids containing box 1 motif of the intracellular domain, which is required for binding of Janus family tyrosine kinase 2 (JAK2). Membrane-bound isoforms only differ in the length of the intracellular domains. LEPRa, c, d, and f comprise short intracellular tails of 32–40 amino acids with unique C-termini and are crucial for internalization and lysosomal-dependent degradation of leptin (16). In contrast, the intracellular domain of LEPRb comprises ~306 amino acids (shorter in mice than in humans) and contains another JAK binding domain (“box 2”) in addition to a STAT binding site, making LEPRb the predominant isoform responsible for signal transduction (17). Binding of leptin to LEPRb results in a conformational change that promotes receptor homo-oligomerization and activation of JAK2 through auto-phosphorylation (18). Activated JAK2 phosphorylates LEPRb at three different tyrosine residues (Tyr985, Tyr1077, Tyr1138), each comprising a Src homology 2 (SH2)-binding motif (19–21). Phosphorylation of Tyr1077 promotes recruitment and activation of STAT5 (20, 21), whereas phosphorylation on Tyr1138 results in recruitment and activation of STAT1/5 and STAT3, which permit auto-phosphorylation of STAT proteins (20). Phosphorylated STAT3 dimerizes and translocates to the nucleus to activate transcription of target genes, including suppressor of cytokine signaling 3 (SOCS3), as a negative regulator of LEPRb-induced JAK/STAT signaling. Phosphorylation of Tyr985 likewise activates SOCS3 and thereby inhibits phosphorylation of LEPRb (19). In addition, leptin-mediated activation of LEPRb-associated JAK2 promotes activation of the phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathway by recruiting the insulin receptor substrates IRS1 and IRS2, which are key molecules for regulation of glucose homeostasis, lipid metabolism, protein synthesis as well as cell proliferation and survival (22).
The soluble LEPRe isoform, which lacks a trans-membrane domain, circulates in the blood as dimers or oligomers. LEPRe functions as a serum-binding protein that transports leptin across the blood-brain barrier (23). Furthermore, LEPRe modulates steady-state leptin levels and leptin bioactivity by complexing free leptin, which delays its degradation and clearance (24).
Congenital leptin deficiency is a form of monogenic obesity and caused by mutations in the gene encoding leptin (LEP, known as the ob gene in the mouse). Patients with homozygous LEP mutations show undetectable levels of leptin in the serum, are characterized by severe early-onset obesity and marked hyperphagia, and develop glucose intolerance and insulin resistance (25). As leptin has a direct effect on insulin sensitivity, leptin replacement therapy was proposed as a novel therapeutic option in metabolic disorders, including insulin resistance and T2DM. In fact, leptin replacement successfully normalized or improved most of the phenotypes in patients with congenital leptin deficiency, including decrease in insulin resistance, improvement of lipid profile, and remarkable weight loss (25). The leptin-deficient ob/ob mouse is a widely used model to study obesity and largely resembles congenital leptin deficiency in humans. Ob/ob mice have a homozygous nonsense mutation (C to T) in the ob gene, which causes premature termination of leptin translation (R105X). As a result, although there are high levels of leptin mRNA in adipocytes, ob/ob mice completely lack functional leptin (4). Similar to patients with congenital leptin deficiency, ob/ob mice are characterized by hyperphagia, hyperinsulinemia, insulin resistance, and massively increased amounts of white adipose tissue. Exogenous leptin application almost completely reversed these phenotypes. However, as insulin release capacity remains high throughout life and hyperglycemia is reduced after 6 months of age, the ob/ob mouse model is probably not the best study model to cover all manifests of T2DM in humans (26, 27).
In humans, serum leptin levels positively correlate with body mass index (BMI), percentage of body fat, fat mass, and size of adipocytes (28). Obesity-associated enlargement of adipocytes in humans results in accelerated secretion of leptin and therefore higher serum leptin levels, which may also result from chronic hyperinsulinemia and increased cortisol turnover. Additionally, many factors such as free fatty acids, estrogen, tumor necrosis factor-α (TNF-α), or impaired renal clearance, are known to stimulate leptin secretion (29). Previous studies have also shown that hormones and the nutritional status influence serum leptin levels, independent of obesity. Leptin levels usually increase 4–7 h after meal consumption, mainly as a result of insulin-stimulated glucose utilization, leading to up-regulation of mRNA transcription and release of leptin from subcutaneous and omental adipose tissue (29). Similar, glucocorticoids increase leptin mRNA and plasma leptin levels, but as they cause insulin resistance and hyperinsulinemia, the effect of glucocorticoids on leptin levels may be indirect (30). In contrast, low insulin and glucose levels, in concert with elevated levels of catecholamines, are known to decrease serum leptin levels in response to conditions of fasting or starvation (29). In addition, other factors, such as exposure to cold (31), activation of β3-adrenergic receptors (32), and elevated intracellular cAMP levels (33) have been shown to inhibit leptin secretion and expression.
The majority of obese patients are hyperleptinaemic. High levels of leptin are associated with insulin resistance, hypothalamic inflammation, and disturbances in hemostatic factors, which are all risk factors for the development of hypertension, metabolic syndrome, or other cardiovascular diseases (34). Opposite to the benefits of leptin replacement therapy in patients with congenital leptin deficiency, treatment of common obese patients with recombinant leptin did not result in reduced food intake and weight loss (25), leading to the spread of the concept of leptin resistance. Leptin resistance is a condition by which hypothalamic neurons are less sensitive to leptin or do not respond to leptin despite the presence of copious amounts of leptin (35). Mechanisms underlying the development of leptin resistance have been discussed previously [reviewed in (35–37)]. Leptin resistance is mainly caused by mutations in the genes encoding leptin and LEPR, reduced expression of LEPR at the plasma membrane level, deterioration of LEPR function and signaling, or alterations in the transport of leptin across the blood-brain barrier (35–37). Furthermore, elevated leptin levels increase the predisposition of patients to diet-induced obesity, which results in a vicious circle leading to a further increase in leptin levels and aggravation of existing leptin resistance, indicating that leptin itself plays an important role in the development of its resistance, as termed “leptin-induced leptin resistance” (37). Importantly, two recent studies have proposed that hyperleptinemia-associated leptin resistance is indeed a driving force for obesity and its associated various metabolic disorders, suggesting that new therapeutic tools to restore leptin sensitivity could offer a new strategy to promote weight loss in obese patients and to treat obesity-associated complications (7, 8).
Obesity-causing LEPR mutations are usually inherited in an autosomal-recessive manner. The majority of LEPR mutations have been reported in populations in which consanguineous marriage is common. Patients carrying a LEPR mutation are hyperphagic and show early-onset morbid obesity. To date, 57 cases with 38 distinct LEPR mutations have been described with most mutations occurring in the extracellular domain, particularly in the leptin-binding or the LEPR activation domain (Figure 1). Not a single LEPR mutation has been identified in the intracellular domain (38). The first LEPR mutation linked to severe obesity was described as a G to A transversion in the splice site of exon 16 (c.2597 +1G>A), which results in the exon 16 skipping and premature termination of protein synthesis (39). Functional in silico analyses are available for 35 LEPR mutations, but functional in vitro data have only been described for four mutations (38, 40). Within these four mutations, only patients carrying the compound heterozygous LEPR mutation R612H (together with a non-sense mutation) revealed residual ability to phosphorylate STAT3 in response to leptin. The missense mutations A409E, W664R, and H684P caused a complete loss of leptin signaling. However, all patients with these four mutations were characterized by hyperphagia, severe early-onset obesity, alterations in immune function, and delayed puberty (40). In future, functional in vitro and/or in vivo analyses of other LEPR mutations are essential to assess the genotype and phenotype correlation, which will help to understand the precise regulation of leptin signaling.
Obesity-causing LEPR mutations have also been linked to insulin resistance and development of T2DM, which due to its slow progression, often remains asymptomatic for many years. In fact, all patients carrying an obesity-causing LEPR mutation (aged 4 to 55 years) show hyperinsulinemia to an extent consistent with the degree of obesity, but T2DM was only reported in two adults (41- and 55-year-old) (40). These data, together with other published data in humans, indicate that subjects with obesity-causing LEPR mutations may be at high risk for early-onset insulin resistance and T2DM (38).
Lepr-deficient db/db mice are one of the most useful models to study the pathophysiology of obesity-associated T2DM. Db/db mice display an autosomal recessive G>T mutation at Arg 890 in the db gene, which results in abnormal splicing and the replacement of the full-length leptin receptor (Ob-Rb) by Ob-Ra (14). As Ob-Ra lacks the most part of the cytoplasmic domain, leptin-mediated signal transduction is severely impaired in db/db mice. Similar to ob/ob mice, homozygous db/db mice display severe and early-onset obesity, associated with severe hyperinsulinemia, but normal glucose tolerance by 4 weeks of age. By 8 weeks of age, db/db mice develop insulin resistance and hyperglycemia. However, db/db mice cannot fully recapitulate the obesogenic phenotype of T2DM patients, which are characterized by elevated triglycerides and very low-density lipoprotein (VLDL) cholesterol, normal or even increased low-density lipoprotein (LDL) cholesterol and total cholesterol, but reduced high-density lipoprotein (HDL) cholesterol. In contrast, db/db mice show elevated cholesterol levels due to an increase in HDL cholesterol (41).
Similar to mouse models, Zucker fatty (fa/fa) rats and Zucker diabetic fatty (ZDF) rats exhibit overt obesity by 5 weeks of age. The fa/fa rat harbors the homozygous missense mutation Q269P in the CRH1 domain in all Lepr isoforms, leading to loss of receptor function due to defective Lepr dimerization. The ZDF rat is an inbred sub-strain of the fa/fa rat, which, in addition to the Lepr mutation, carries an inherited autosomal recessive genetic defect in β-cell transcription. Phenotypically, both fa/fa and ZDF rats show consistently elevated levels of LDL and HDL cholesterol as well as increased activity of lipoprotein lipase, which are different from those of T2DM patients. Additionally, both transgenic rat models develop insulin resistance at different age. However, fa/fa rats are not hyperglycemic although they reveal hyperinsulinemia at 3–4 weeks of age, but plasma insulin levels return to normal by 30 weeks of age. In contrast, plasma insulin levels of ZDF rats dramatically increase from 6 to 8 weeks of age and decline afterwards. ZDF rats become insulinopenic (insufficient secretion of insulin) at 14 weeks of age due to pancreatic β-cell insufficiency and the male rats develop gender-specific hyperglycemia by 10–12 weeks of age (27, 42).
Studies have shown that obesity is associated with chronic low-grade systemic inflammation, which results from changes in both the innate and the adaptive immune system (43, 44). In children and adults who are overweight or obese, the inflammatory state was reflected by elevated levels of circulating pro-inflammatory cytokines, including TNF-α, interleukin (IL)-6, and C-reactive protein, and by the activation of pro-inflammatory signaling pathways in the adipose tissue. Higher concentrations of pro-inflammatory cytokines have also been detected in individuals who are insulin resistant and obese and are used to predict the development of T2DM and cardiovascular diseases (43, 45). Leptin production is regulated by inflammatory stimuli, including lipopolysaccharide, TNF-α, IL-6, and IL-1β, and plasma leptin levels increase during acute infection, inflammation, or sepsis. LEPR has been detected in all cell types of innate and adaptive immunity, including macrophages, dendritic cells, nature killer (NK) cells, neutrophils, and lymphocytes. Leptin modulates the immune response and inflammation at various levels, which have been detailed reviewed previously (46, 47) and are discussed here only briefly.
Leptin exerts various functions in the innate immune system. In monocytes, leptin stimulates the release of pro-inflammatory cytokines, including TNF-α, IL-6, and IL-12 and induces expression of cell surface markers important for activation of resting monocytes. It promotes proliferation and differentiation of monocytes into macrophages, but on macrophages, leptin acts as an anti-apoptotic cytokine by stimulating phagocytosis. Moreover, leptin stimulates chemotactic activity and function of neutrophils by enhancing production of oxygen species crucial for host cell defense against infections (48), and by promoting an anti-apoptotic cellular response (49). Importantly, leptin-mediated stimulation of neutrophils requires TNF-α release from monocytes (50). In dendritic cells, leptin upregulates the production of IL-1β, IL-6, IL-12, TNF-α, and macrophage inflammatory protein (MIP)-1α, and protects dendritic cells from apoptosis. In addition, leptin downregulates IL-10 production and drives polarization of naïve T cell toward the Th1 phenotype (51). Previous studies have shown that leptin sustains cytotoxic activities of NK cells by inducing phosphorylation of STAT3 and transcription of IL-2 and perforin (52).
In the adaptive immune system, leptin promotes lymphopoiesis by stimulating maturation of CD4+CD8+ and CD4+CD8− T cells. Leptin induces proliferation of naïve CD4+CD45RA+ T cells, but inhibits proliferation of memory CD4+CD45RO+ T cells by increasing the production of the Th1 cytokines interferon (IFN)-γ and IL-2, and suppressing the production of the Th2 cytokine IL-4 (53). Beyond this, it has been shown that leptin promotes differentiation of CD4+ cells into IL-17 producing Th17 cells, while suppressing formation of CD4+CD25+ Treg cells, in addition to prevention of stress-induced T cell apoptosis (54–56). In malnourished individuals with extremely low BMI, a dramatic reduction in leptin levels was observed, which was associated with thymic atrophy, reduced T-cell function, and increased susceptibility to infection (57). Ob/ob mice, fa/fa rats, and humans with congenital leptin deficiency or with LEPR mutations are more susceptible to infections, which go along with a decrease in the number of total lymphocytes and circulating CD4+ T cells, apoptosis of thymocytes, and a shift toward a Th2 immune response. Administration of recombinant leptin substantially increased thymic cellularity and restored abnormalities in proliferating T cells and cytokine release of immune cells in ob/ob mice and humans with leptin deficiency (40, 56, 58, 59). In B cells, leptin induces a pro-inflammatory cytokine response through stimulation of TNF-α, IL-6, and IL-10 secretion (60). Similar to T cells, leptin limits apoptosis of B cells by enhancing expression of blc-2 and by promoting cell cycle entry via cyclin D1 (61). Reduced number of total B cells, including pre-B cells and immature B cells, and defective lymphopoiesis in ob/ob mice could be restored by administration of recombinant leptin (62). Taken together, it is evident that leptin has pro-inflammatory effects and exerts a crucial function in modulating immune response of both the innate and the adaptive immune system.
In the cardiovascular system, the role of leptin still remains controversial. Many studies suggest that leptin is involved in the pathogenesis of chronic inflammation. In this regard, elevated leptin levels in obese patients are believed to contribute to the low-grade systemic inflammation, which makes obese individuals more susceptible to develop cardiovascular diseases (Figure 2). In addition, elevated leptin levels have been reported in patients with dilated cardiomyopathy, which are used as a biomarker for the progression of heart failure independent of immune responses (63). In a prospective study of 4,080 men, aged 60–79 years, with no diagnosed heart failure followed for 9 years, increased BMI and circulating leptin levels were used as independent predictors for the incidence of heart failure. Elevated leptin levels were associated with an increased risk of heart failure in men without pre-existing coronary heart disease, independent of BMI and potential mediators (64). In the Framingham study with 818 participants (mean age 79 years, 62% women), leptin levels were strongly associated with incidence of congestive heart failure and cardiovascular disease. After adjustment for BMI, the association of leptin with congestive heart failure was nullified, but the relation to an enhanced incidence for development of cardiovascular disease was only modestly attenuated (65). In contrast, the Multi-Ethnic Study of Atherosclerosis with randomly selected 1,905 participants (mean age 64.5 years, 50% women) without underlying cardiovascular disease revealed that leptin levels were not associated with incidence of cardiovascular disease events after adjustment for cardiovascular risk factors, BMI or waist circumference (66). Recently, the use of BMI for the risk assessment of cardiovascular disease is on a core controversy because BMI does not discriminate between body fat mass and muscle mass. It has been suggested that visceral adiposity, rather than a high BMI, is correlated with increased risk of cardiovascular diseases and diabetes (67). Based on these epidemiologic data, it remains unclear whether leptin is associated with the development of heart failure. It is possible that chronic effects of leptin may have adverse consequences on myocardial function.
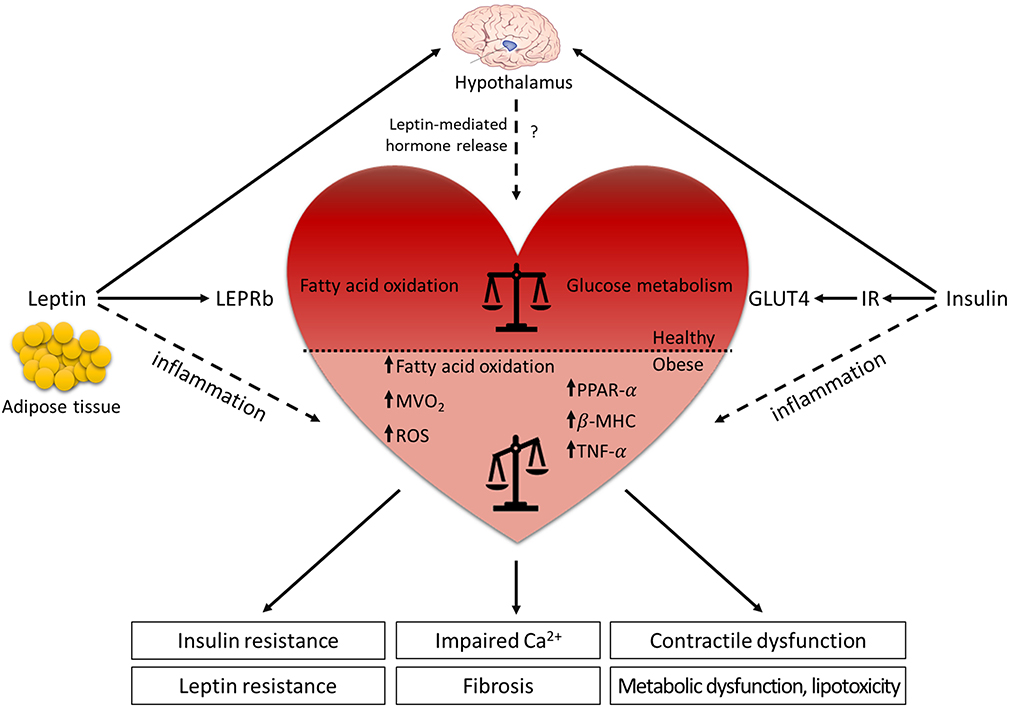
Figure 2. Leptin and LEPR signaling in the (dys-)regulation of the cardiovascular system. Leptin is mainly secreted from the adipose tissue and binds to the LEPR in the arcuate nucleus of the hypothalamus where it regulates food intake, energy expenditure and hormone release. The actions of leptin and insulin are interconnected and contribute to optimal metabolic control. Although leptin role in the cardiovascular system is still controversial, diabetic and obese animal models demonstrate that leptin has beneficial effects on cardiac metabolism. Under physiological condition, leptin signaling supports the balance between glucose metabolism and fatty acid oxidation in the heart, while the absence of leptin or LEPR results in the lack of the metabolic flexibility. The reduced dynamic between the energy substrates results in systemic metabolic disorders (insulin resistance, leptin resistance, metabolic dysfunction, and lipotoxicity), leading to decreased cardiac efficiency (impaired Ca2+ handling, contractile dysfunction, and fibrosis). In contrast, elevated leptin levels in obese patients contribute to the low-grade systemic inflammation, which increases the risk to develop cardiovascular diseases. Horizontal black line demarcates differences between the healthy heart and the heart in obesity/diabetes. LEPRb, leptin receptor b; MVO2, myocardial oxygen consumption; ROS, reactive oxygen species; PPAR-α, peroxisome proliferator-activated receptor α; β-MHC, myosin heavy chain β; TNF-α, tumor necrosis factor-α; GLUT4, glucose transporter 4; IR, insulin receptor.
Nevertheless, many studies using rodent obese and diabetic models demonstrate that leptin has beneficial effects on cardiac metabolism by supporting glucose metabolism and fatty acid oxidation. Therefore, acute effects of leptin may provide a compensatory response to cardiac insults, such as ischemia or heart failure (68).
Animal models have helped to understand leptin signaling in the heart (69) and both ob/ob and db/db mice show an age-dependent progression of hypertrophy, mostly effecting left ventricular (LV) mass and LV wall thickness (70, 71). In particular, db/db mice develop both systolic and diastolic dysfunctions and contractile abnormalities, but with maintenance of cardiac output by favorable loading conditions (increased preload and decreased afterload) at 12 weeks of age (72, 73). Cardiomyocyte-specific deletion of Lepr (Lepr−/−) in mice, in the presence of increased Cre recombinase expression, caused significant thinning of the ventricular wall and reduced ejection fraction, which was associated with impaired energy production via AMPK- and mTOR signaling. After myocardial infarction, cardiomyocyte-specific Lepr−/− mice revealed greater cardiac dysfunction and increased morbidity compared to wild-type mice, which were associated with attenuated cardiac STAT3, PI3K, and AKT activity and mitochondrial function (74, 75). Furthermore, these studies demonstrated that impaired cardiac leptin signaling in Lepr−/− mice resulted in metabolic inflexibility for glucose utilization in the face of cardiac stress (75). Similar to mouse models, impaired leptin signaling also contributes to the cardiac-specific phenotype in fa/fa and ZDF rats, as illustrated by LV hypertrophy and defects in cardiac contractility, such as reduced fractional shortening, yet these effects are less pronounced in fa/fa rats (76, 77). Moreover, db/db mice and ZDF rats are more susceptible to ischemic injury and show impaired recovery of cardiac function after myocardial infarction induced by coronary artery ligation at stages when animals have developed diabetes (69). Interestingly, while either leptin administration or caloric restriction resulted in weight loss in ob/ob mice, leptin infusion but not caloric restriction reversed the obesity-related cardiac remodeling process, indicating that leptin has anti-hypertrophic effects not attributable to weight loss alone (70). However, it has not been completely understood whether leptin directly affects cardiac function or acts through a leptin-regulated neurohumoral pathway (Figure 2).
Under physiological conditions, the heart uses both glucose and free fatty acids as energy substrates. In leptin- or Lepr-deficient rodent models, impaired cardiac performance is linked to altered myocardial substrate uptake and metabolic inflexibility due to increased vesicular fatty acid uptake and fatty acid oxidation with reduced glucose uptake and carbohydrate oxidation (Figure 2). As glucose is a more efficient energy substrate for cardiomyocytes than free fatty acids, a metabolic switch from glucose metabolism to free fatty acid oxidation results in increased myocardial oxygen consumption (MVO2) and decreased cardiac efficiency (ratio of cardiac work to energy input), which leads to systemic metabolic disorders (76–78). Leptin- or Lepr-deficient animals show elevated triglyceride levels and lipid accumulation in the myocardium that may promote lipotoxicity and directly impede cardiac contractility (79–82). The prominent effects of leptin in reducing high triglyceride levels have been confirmed in an acyl CoA synthase transgenic mouse model showing severe lipotoxic cardiomyopathy (83). Liver-specific overexpression of wild-type Lepr in fa/fa rats emphasized the prominent effects of leptin in reducing high triglyceride levels only in the liver but nowhere else (80). Cardiomyocyte-restricted re-expression of Lepr in db/db mice reduced cardiac triglyceride levels, without changes in the levels of plasma triglycerides, and these mice displayed lower heart weight and reduced LV wall thickness in comparison to db/db mice (84). These studies suggest that leptin might act as an anti-lipotoxic adipokine that protects the heart from harmful lipid accumulation and progression of cardiac steatosis, especially under cardiac stress conditions.
Notably, metabolic modulation to sustain glucose utilization by perinatal overexpression of the glucose transporter GLUT4 prevented cardiac dysfunction in db/db mice (72, 85). High levels of insulin and glucose normalized cardiac metabolism, restored cardiac efficiency, and improved post-ischemic recovery in db/db mice (86). Administration of a peroxisome proliferator-activated receptor γ (PPARγ) agonist to ZDF rats resulted in an increase in glucose metabolism, reduced accumulation of triacylglycerol in the myocardium, and restored cardiac function (76). Although chronic administration of db/db mice with a PPARγ agonist induced insulin sensitization, enhanced glucose oxidation, and decreased palmitate oxidation, it failed to restore cardiac function (87). These studies indicate that alterations in glucose uptake, which are caused by cardiac insulin resistance, facilitate the cardiac metabolic switch from glucose metabolism to free fatty acid oxidation, leading to increased MVO2 and reduced cardiac efficiency and performance. Upon ischemic injury, energy shortage in the myocardium is exacerbated, which reduces ischemic tolerance of the diabetic heart. Restoring the balance of glucose and fatty acid metabolism in cardiomyocytes following injury is of fundamental importance for functional recovery of the heart.
Increased MVO2 can also be a result of increased production of reactive oxygen species (ROS), which is associated with augmented fatty acid oxidation, mitochondrial uncoupling, and oxidative stress in hearts of db/db mice (88). Comparable to db/db mice, fa/fa rats display an increased oxidative stress response, as indicated by high levels of lipid peroxide and elevated activity of the superoxide dismutase (89). Another factor contributing to cardiac dysfunction in rodent models is perturbed intracellular Ca2+ handling. In db/db mice, systolic and diastolic Ca2+ levels, sarcoplasmic reticulum Ca2+ load, Ca2+ transient decay, and L-type Ca2+ current are all reduced while Ca2+ leakage from the sarcoplasmic reticulum is increased. These data demonstrate that perturbations in cardiac Ca2+ handling are a major cause of contractile dysfunction (90, 91). In conclusion, studies using leptin- or Lepr-deficient rodent models indicate that leptin acts as a metabolic, cardio-protective adipokine, particularly under ischemic conditions.
Abnormal myocardial structure and impaired cardiac performance in the absence of overt cardiovascular risk factors, such as coronary artery disease, valvular heart disease, hypertension, or dyslipidemia in patients with T2DM, are described as diabetic cardiomyopathy (92). Although diabetic cardiomyopathy is usually asymptomatic in very early stages, its progression results in LV diastolic dysfunction and/or reduced ejection fraction, LV hypertrophy, and increased interstitial fibrosis (92–94). Patients with diabetic cardiomyopathy display a diastolic, but not systolic dysfunction at early stages, which gradually progress to severe diastolic heart failure with preserved ejection fraction, known as HFpEF. At later stages, these patients develop systolic dysfunction and heart failure with reduced ejection fraction (HFrEF). The prevalence of diabetic cardiomyopathy is increasing in parallel with an increase in T2DM. With an incidence of 19–26% to develop heart failure, cardiovascular remodeling processes resulting in diabetic cardiomyopathy are a major cause of disease-related deaths in patients with T2DM (92).
Clinical studies reported an association of plasma leptin levels with LV hypertrophy. In hypertensive insulin-resistant men, fasting plasma leptin levels were higher than in controls and correlated with increased myocardial wall thickness. This correlation persisted after adjustment for BMI and was independent of blood pressure (95). Moreover, Perego et al. showed that plasma leptin levels were higher in morbidly obese patients than in lean controls and were positively correlated to LV mass after adjustment for BMI. One year after bariatric surgery, obese patients showed profound weight loss and significant reductions in BMI and insulin resistance. Nevertheless, the decrease in LV mass was correlated only with a decrease in leptin levels on multivariable analyses (96). These clinical findings propose that leptin participates in the progression and development of LV hypertrophy in obese patients. Studies have also shown that leptin accounts for the increase in blood pressure associated with obesity, as obese patients with a loss-of-function mutation in leptin or LEPR displayed low blood pressure (97), and obese patients are frequently characterized by sympathetic hyperactivity and higher plasma levels of adrenaline and noradrenaline (98). Chronic leptin infusion in male rats has been shown to elevate arterial blood pressure and heart rate through the stimulation of the sympathetic nervous system activity (99). Therefore, leptin-mediated increase in heart rate and blood pressure may provoke an increase in myocardial workload, which contributes to cardiac hypertrophy over the long term.
It is important to emphasize that human patients with obesity-associated impairment of cardiac function commonly suffer from HFpEF, in combination with sodium retention, plasma volume expansion, microvascular inflammation, and atypical deposition of fibrotic tissue (100, 101). Most of these symptoms are correlated with elevated plasma leptin levels (102). Intriguingly, patients with advanced stages of chronic heart failure are hyperleptinemic, as serum levels of leptin and its soluble receptor LEPRe are increased after the adjustment for BMI (103).
The underlying mechanisms of diabetic cardiomyopathy, particularly in obesity-related T2DM, have mainly been investigated in ob/ob and db/db mice or fa/fa and ZDF rats (93). As already mentioned earlier, various metabolic disturbances are involved in the pathogenesis of diabetic cardiomyopathy, such as systemic tissue inflammation, insulin resistance, altered myocardial substrate utilization, elevated fatty acid levels, oxidative stress, and activation of the renin-angiotensin-aldosterone system. Animal models with a genetic defect in leptin signaling have helped to increase our knowledge about mechanisms contributing to the cardiac remodeling process in the diabetic, leptin-deficient heart. However, it is hard to draw any conclusions from the observations in rodent models for patients with diet-related obesity in the clinical setting, as obese patients normally display high circulating levels of leptin and are leptin resistant.
Obese patients with insulin resistance display high levels of myocardial triglycerides, which are linked to transcriptional upregulation of PPARα-regulated genes, myosin heavy chain β (β-MHC), and TNF-α, in addition to cardiac contractile dysfunction (82, 104). A study by Nyman et al. uncovered that cardiac steatosis caused by lipid oversupply in patients with metabolic syndrome was associated with LV diastolic function. Although the amount of epicardial and pericardial fat correlated with the severity of LV diastolic dysfunction, increase in myocardial triglyceride levels did not act as a mere fat deposit compared to epicardial and pericardial fat (105). In vitro experiments using cardiomyocytes from either patients with dilated cardiomyopathy or controls indicate that leptin acts in concert with resistin causing a TNF-α- and IL6-dependent cardiac redox stress response (63). Human and neonatal rat cardiomyocytes treated with recombinant leptin were more susceptible to progression of cardiac hypertrophy, as indicated by increased cell size, elevated matrix metalloproteinase-2 (MMP-2) activity, and endothelin-1-induced rise in ROS levels (106, 107).
Taken together, leptin exerts physiological effects that may be detrimental in states of cardiac dysfunction or heart failure. Leptin's hemodynamic effects, such as elevation of resting heart rate and blood pressure, generally increase myocardial workload via activation of the sympathetic nervous system. Leptin may act synergistically with other factors that are associated with obesity, such as hyperglycemia, inflammation, and oxidative stress, to accelerate development and progression of cardiovascular diseases (Figure 2). Recently, studies have investigated the role of sodium glucose transporter 2 (SGLT2) inhibitors, such as empagliflozin, on modulating cardiac alterations in obesity-related T2DM. As leptin is implemented to regulate the expression of SGLT2 in renal tubules through activation of the sympathetic nervous system and the renin-angiotensin system, SGLT2 inhibitors have been shown to reverse leptin-induced renal sodium and glucose retention and inflammatory response in adipocytes by reducing secretion of leptin and limiting its paracrine effects on cardiac and renal metabolism (108). SGLT2 inhibitors are known not only to improve cardiac function and metabolism, but also to promote weight loss by reducing visceral fat depots and to attenuate the obesity-induced inflammatory response and insulin resistance by modulating the activity of M2 macrophages (109, 110). Most importantly, SGLT2 inhibitors significantly reduced the number of deaths arising from T2DM-associated complications on the cardiovascular system (111).
Leptin-mediated aldosterone production is considered a novel mechanism of obesity-associated endothelial dysfunction and cardiac fibrosis, which impair myocardial relaxation and thereby contribute to cardiovascular disease (112). This explains why the aldosterone antagonist spironolactone is particularly effective in reducing morbidity and mortality in patients with obesity-related HFpEF (113). Moreover, spironolactone improved LV function and reduced levels of circulating procollagen in obese patients without any other comorbidities (114). The correlation between leptin and aldosterone levels was also observed in cardiac myofibroblasts of high-fat diet-fed rats. Here, the aldosterone antagonist eplerenone reduced the leptin-induced increase in protein levels of pro-fibrotic factors collagen I, TGFβ, connective tissue growth factor, and galectin-3 as well as the levels of both total and mitochondrial ROS (115).
In a study of 294 healthy adolescents (aged 13–16 years) with a broad ranged BMI, an elevation in leptin levels was associated with impaired arterial distensibility, independent of metabolic and inflammatory disturbances associated with obesity (116). Moreover, increased plasma leptin levels were positively correlated with the total number of stenotic coronary arteries in patients with coronary artery disease (117). These data indicate that leptin plays a key role in the development of vascular dysfunction. This is supported by studies showing that ob/ob mice did not develop any atherosclerotic lesions or plaques despite being fed with a chow diet containing 2% cholesterol over 2 months, which is contrary to expectations in obese, diabetic individuals with hypercholesterolemia (41, 118). In vitro studies using human umbilical vein endothelial cells demonstrate that leptin induces chronic oxidative stress in endothelial cells, which may promote atherogenic processes and contribute to the development of vascular pathology (119). Leptin may stimulate proliferation and migration of vascular smooth muscle cells (120) and induce the calcification of vascular cells (121), thereby contributing to the formation and development of vascular lesions. In addition, it is generally accepted that leptin-mediated release of endothelial nitric oxide (NO) causes vasodilatation, which opposes the pressor effect of sympathoexcitatory activity induced by leptin (122, 123). However, in patients with obesity and T2DM, hyperleptinemia caused a reduction of NO bioavailability and attenuated NO-dependent vasodilation, which contributes to vascular dysfunction (124).
Overall, leptin has emerged as an adipocyte-derived factor with pleiotropic effects in the nutritional and metabolic state, in modulating immune responses and inflammation, and in controlling cardiovascular functions (Figure 2). Animal studies using leptin- or Lepr-deficient rodents demonstrate that the cardiac dysfunction is associated with (i) a metabolic switch from glucose metabolism to fatty acid oxidation, promoting lipotoxicity, (ii) systemic inflammation, (iii) insulin resistance, and (iv) activation of the renin-angiotensin-aldosterone system. However, the alteration of lipid profile in leptin- or Lepr-deficient animal models cannot fully recapitulate the phenotypes of patients with T2DM. Leptin resistance in obese or T2DM patients adversely affects the heart's response to stress conditions and promotes cardiac remodeling as a result of impaired cardiac metabolism, increased fibrosis, vascular dysfunction, and enhanced inflammation, all contributing to impairment of cardiac function. Even after decades of research on rodents and clinical studies, existing differences in the underlying mechanisms and pathways between animal models and patients with obesity and T2DM make data interpretation challenging. This hampers the development of new strategies to treat patients with diabetic cardiomyopathy (or HFpEF).
Since leptin is a critical regulator of the cardiovascular system, new strategies targeting leptin's action precisely in the cardiovascular system in normal and diseased states might be important. Future research is required to understand the precise role of leptin directly in different cardiac cell types (e.g., cardiomyocytes, endothelial cells or fibroblasts), which may open up new avenues for the treatment of obesity- and diabetes-associated cardiomyopathy. Disease modeling using human induced pluripotent stem cells combined with the CRISPR/Cas9 technique may allow us to study and better understand abnormal leptin signaling in cardiomyocytes as well as in endothelial cells in vitro, which might cause obesity- and T2DM-associated cardio-metabolic remodeling processes. Consequently, a shift from leptin- and Lepr-deficient rodent models to human-based models will be of great importance to overcome the translational barrier and to develop innovative therapeutic approaches.
MP, AS, and KG conducted a review of the literature and wrote the first draft of the review. KG contributed to conception and design of the article and finalized the review. All authors read and approved the final manuscript.
This study was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Project Number 288034826–IRTG 2251: Immunological and Cellular Strategies in Metabolic Disease. We acknowledge the support of the Open Access Funding by the Publication Fund of the TU Dresden.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
2. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart disease and stroke statistics−2015 update: a report from the American Heart Association. Circulation. (2015) 131:e29–322. doi: 10.1161/CIR.0000000000000152
3. Scherer PE, Hill JA. Obesity, diabetes, and cardiovascular diseases: a compendium. Circ Res. (2016) 118:1703–5. doi: 10.1161/CIRCRESAHA.116.308999
4. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. (1994) 372:425–32. doi: 10.1038/372425a0
5. Timper K, Bruning JC. Hypothalamic circuits regulating appetite and energy homeostasis: pathways to obesity. Dis Model Mech. (2017) 10:679–89. doi: 10.1242/dmm.026609
6. Williams KW, Margatho LO, Lee CE, Choi M, Lee S, Scott MM, et al. Segregation of acute leptin and insulin effects in distinct populations of arcuate proopiomelanocortin neurons. J Neurosci. (2010) 30:2472–9. doi: 10.1523/JNEUROSCI.3118-09.2010
7. Balland E, Chen W, Dodd GT, Conductier G, Coppari R, Tiganis T, et al. Leptin signaling in the arcuate nucleus reduces insulin's capacity to suppress hepatic glucose production in obese mice. Cell Rep. (2019) 26:346–55 e343. doi: 10.1016/j.celrep.2018.12.061
8. Zhao S, Zhu Y, Schultz RD, Li N, He Z, Zhang Z, et al. Partial leptin reduction as an insulin sensitization and weight loss strategy. Cell Metab. (2019) 30:706–19 e706. doi: 10.1016/j.cmet.2019.08.005
9. Sinha MK, Sturis J, Ohannesian J, Magosin S, Stephens T, Heiman ML, et al. Ultradian oscillations of leptin secretion in humans. Biochem Biophys Res Commun. (1996) 228:733–8. doi: 10.1006/bbrc.1996.1724
10. Ye F, Than A, Zhao Y, Goh KH, Chen P. Vesicular storage, vesicle trafficking, and secretion of leptin and resistin: the similarities, differences, and interplays. J Endocrinol. (2010) 206:27–36. doi: 10.1677/JOE-10-0090
11. Mercer JG, Moar KM, Hoggard N, Strosberg AD, Froguel P, Bailleul B. B219/OB-R 5'-UTR and leptin receptor gene-related protein gene expression in mouse brain and placenta: tissue-specific leptin receptor promoter activity. J Neuroendocrinol. (2000) 12:649–55. doi: 10.1046/j.1365-2826.2000.00501.x
12. Belouzard S, Delcroix D, Rouille Y. Low levels of expression of leptin receptor at the cell surface result from constitutive endocytosis and intracellular retention in the biosynthetic pathway. J Biol Chem. (2004) 279:28499–508. doi: 10.1074/jbc.M400508200
13. Seron K, Couturier C, Belouzard S, Bacart J, Monte D, Corset L, et al. Endospanins regulate a postinternalization step of the leptin receptor endocytic pathway. J Biol Chem. (2011) 286:17968–81. doi: 10.1074/jbc.M111.224857
14. Lee GH, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, Lee JI, et al. Abnormal splicing of the leptin receptor in diabetic mice. Nature. (1996) 379:632–5. doi: 10.1038/379632a0
15. Ge H, Huang L, Pourbahrami T, Li C. Generation of soluble leptin receptor by ectodomain shedding of membrane-spanning receptors in vitro and in vivo. J Biol Chem. (2002) 277:45898–903. doi: 10.1074/jbc.M205825200
16. Uotani S, Bjorbaek C, Tornoe J, Flier JS. Functional properties of leptin receptor isoforms: internalization and degradation of leptin and ligand-induced receptor downregulation. Diabetes. (1999) 48:279–86. doi: 10.2337/diabetes.48.2.279
18. Nakashima K, Narazaki M, Taga T. Leptin receptor (OB-R) oligomerizes with itself but not with its closely related cytokine signal transducer gp130. FEBS Lett. (1997) 403:79–82. doi: 10.1016/S0014-5793(97)00013-6
19. Dunn SL, Bjornholm M, Bates SH, Chen Z, Seifert M, Myers MG Jr. Feedback inhibition of leptin receptor/Jak2 signaling via Tyr1138 of the leptin receptor and suppressor of cytokine signaling 3. Mol Endocrinol. (2005) 19:925–38. doi: 10.1210/me.2004-0353
20. Hekerman P, Zeidler J, Bamberg-Lemper S, Knobelspies H, Lavens D, Tavernier J, et al. Pleiotropy of leptin receptor signalling is defined by distinct roles of the intracellular tyrosines. FEBS J. (2005) 272:109–19. doi: 10.1111/j.1432-1033.2004.04391.x
21. Gong Y, Ishida-Takahashi R, Villanueva EC, Fingar DC, Munzberg H, Myers MG Jr. The long form of the leptin receptor regulates STAT5 and ribosomal protein S6 via alternate mechanisms. J Biol Chem. (2007) 282:31019–27. doi: 10.1074/jbc.M702838200
22. Ren D, Li M, Duan C, Rui L. Identification of SH2-B as a key regulator of leptin sensitivity, energy balance, and body weight in mice. Cell Metab. (2005) 2:95–104. doi: 10.1016/j.cmet.2005.07.004
23. Kastin AJ, Pan W, Maness LM, Koletsky RJ, Ernsberger P. Decreased transport of leptin across the blood-brain barrier in rats lacking the short form of the leptin receptor. Peptides. (1999) 20:1449–53. doi: 10.1016/S0196-9781(99)00156-4
24. Huang L, Wang Z, Li C. Modulation of circulating leptin levels by its soluble receptor. J Biol Chem. (2001) 276:6343–9. doi: 10.1074/jbc.M009795200
25. Paz-Filho G, Mastronardi CA, Licinio J. Leptin treatment: facts and expectations. Metab Clin Exp. (2015) 64:146–56. doi: 10.1016/j.metabol.2014.07.014
26. Lindström P. The physiology of obese-hyperglycemic mice [ob/ob mice]. Sci World J. (2007) 7:666–85. doi: 10.1100/tsw.2007.117
27. Wang B, Chandrasekera PC, Pippin JJ. Leptin- and leptin receptor-deficient rodent models: relevance for human type 2 diabetes. Curr Diabetes Rev. (2014) 10:131–45. doi: 10.2174/1573399810666140508121012
28. Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. (1996) 334:292–5. doi: 10.1056/NEJM199602013340503
29. Fried SK, Ricci MR, Russell CD, Laferrere B. Regulation of leptin production in humans. J Nutr. (2000) 130:3127S−3131S. doi: 10.1093/jn/130.12.3127S
30. Papaspyrou-Rao S, Schneider SH, Petersen RN, Fried SK. Dexamethasone increases leptin expression in humans in vivo. J Clin Endocrinol Metab. (1997) 82:1635–7. doi: 10.1210/jcem.82.5.3928
31. Ricci MR, Fried SK, Mittleman KD. Acute cold exposure decreases plasma leptin in women. Metab Clin Exp. (2000) 49:421–3. doi: 10.1016/S0026-0495(00)80001-3
32. Gettys TW, Harkness PJ, Watson PM. The beta 3-adrenergic receptor inhibits insulin-stimulated leptin secretion from isolated rat adipocytes. Endocrinology. (1996) 137:4054–7. doi: 10.1210/endo.137.9.8756584
33. Slieker LJ, Sloop KW, Surface PL, Kriauciunas A, LaQuier F, Manetta J, et al. Regulation of expression of ob mRNA and protein by glucocorticoids and cAMP. J Biol Chem. (1996) 271:5301–4. doi: 10.1074/jbc.271.10.5301
34. Wannamethee SG, Tchernova J, Whincup P, Lowe GD, Kelly A, Rumley A, et al. Plasma leptin: associations with metabolic, inflammatory and haemostatic risk factors for cardiovascular disease. Atherosclerosis. (2007) 191:418–26. doi: 10.1016/j.atherosclerosis.2006.04.012
35. Myers MG Jr, Heymsfield SB, Haft C, Kahn BB, Laughlin M, Leibel RL, et al. Challenges and opportunities of defining clinical leptin resistance. Cell Metab. (2012) 15:150–6. doi: 10.1016/j.cmet.2012.01.002
36. Santoro A, Mattace Raso G, Meli R. Drug targeting of leptin resistance. Life Sci. (2015) 140:64–74. doi: 10.1016/j.lfs.2015.05.012
37. Gruzdeva O, Borodkina D, Uchasova E, Dyleva Y, Barbarash O. Leptin resistance: underlying mechanisms and diagnosis. Diabetes Metab Syndr Obes. (2019) 12:191–8. doi: 10.2147/DMSO.S182406
38. Nunziata A, Funcke JB, Borck G, von Schnurbein J, Brandt S, Lennerz B, et al. Functional and phenotypic characteristics of human leptin receptor mutations. J Endocr Soc. (2019) 3:27–41. doi: 10.1210/js.2018-00123
39. Clement K, Vaisse C, Lahlou N, Cabrol S, Pelloux V, Cassuto D, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. (1998) 392:398–401. doi: 10.1038/32911
40. Farooqi IS, Wangensteen T, Collins S, Kimber W, Matarese G, Keogh JM, et al. Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor. N Engl J Med. (2007) 356:237–47. doi: 10.1056/NEJMoa063988
41. Nishina PM, Naggert JK, Verstuyft J, Paigen B. Atherosclerosis in genetically obese mice: the mutants obese, diabetes, fat, tubby, and lethal yellow. Metab Clin Exp. (1994) 43:554–8. doi: 10.1016/0026-0495(94)90195-3
42. Griffen SC, Wang J, German MS. A genetic defect in beta-cell gene expression segregates independently from the fa locus in the ZDF rat. Diabetes. (2001) 50:63–8. doi: 10.2337/diabetes.50.1.63
43. de Heredia FP, Gomez-Martinez S, Marcos A. Obesity, inflammation and the immune system. Proc Nutr Soc. (2012) 71:332–8. doi: 10.1017/S0029665112000092
44. Procaccini C, La Rocca C, Carbone F, De Rosa V, Galgani M, Matarese G. Leptin as immune mediator: Interaction between neuroendocrine and immune system. Dev Comp Immunol. (2017) 66:120–9. doi: 10.1016/j.dci.2016.06.006
45. Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol. (2006) 6:772–83. doi: 10.1038/nri1937
46. La Cava A, Matarese G. The weight of leptin in immunity. Nat Rev Immunol. (2004) 4:371–9. doi: 10.1038/nri1350
47. Abella V, Scotece M, Conde J, Pino J, Gonzalez-Gay MA, Gomez-Reino JJ, et al. Leptin in the interplay of inflammation, metabolism and immune system disorders. Nat Rev Rheumatol. (2017) 13:100–9. doi: 10.1038/nrrheum.2016.209
48. Caldefie-Chezet F, Poulin A, Vasson MP. Leptin regulates functional capacities of polymorphonuclear neutrophils. Free Radic Res. (2003) 37:809–14. doi: 10.1080/1071576031000097526
49. Bruno A, Conus S, Schmid I, Simon HU. Apoptotic pathways are inhibited by leptin receptor activation in neutrophils. J Immunol. (2005) 174:8090–6. doi: 10.4049/jimmunol.174.12.8090
50. Zarkesh-Esfahani H, Pockley AG, Wu Z, Hellewell PG, Weetman AP, Ross RJ. Leptin indirectly activates human neutrophils via induction of TNF-alpha. J Immunol. (2004) 172:1809–14. doi: 10.4049/jimmunol.172.3.1809
51. Mattioli B, Giordani L, Quaranta MG, Viora M. Leptin exerts an anti-apoptotic effect on human dendritic cells via the PI3K-Akt signaling pathway. FEBS Lett. (2009) 583:1102–6. doi: 10.1016/j.febslet.2009.02.029
52. Zhao Y, Sun R, You L, Gao C, Tian Z. Expression of leptin receptors and response to leptin stimulation of human natural killer cell lines. Biochem Biophys Res Commun. (2003) 300:247–52. doi: 10.1016/S0006-291X(02)02838-3
53. Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature. (1998) 394:897–901. doi: 10.1038/29795
54. Takeda K, Kaisho T, Yoshida N, Takeda J, Kishimoto T, Akira S. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J Immunol. (1998) 161:4652–60.
55. Fujita Y, Murakami M, Ogawa Y, Masuzaki H, Tanaka M, Ozaki S, et al. Leptin inhibits stress-induced apoptosis of T lymphocytes. Clin Exp Immunol. (2002) 128:21–6. doi: 10.1046/j.1365-2249.2002.01797.x
56. Lord GM, Matarese G, Howard JK, Bloom SR, Lechler RI. Leptin inhibits the anti-CD3-driven proliferation of peripheral blood T cells but enhances the production of proinflammatory cytokines. J Leukoc Biol. (2002) 72:330–8.
57. Savino W. The thymus gland is a target in malnutrition. Eur J Clin Nutr. (2002) 56 Suppl 3:S46–49. doi: 10.1038/sj.ejcn.1601485
58. Fantuzzi G, Faggioni R. Leptin in the regulation of immunity, inflammation, and hematopoiesis. J Leukoc Biol. (2000) 68:437–46. doi: 10.1189/jlb.68.4.437
59. Farooqi IS, Matarese G, Lord GM, Keogh JM, Lawrence E, Agwu C, et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest. (2002) 110:1093–103. doi: 10.1172/JCI0215693
60. Agrawal S, Gollapudi S, Su H, Gupta S. Leptin activates human B cells to secrete TNF-alpha, IL-6, and IL-10 via JAK2/STAT3 and p38MAPK/ERK1/2 signaling pathway. J Clin Immunol. (2011) 31:472–8. doi: 10.1007/s10875-010-9507-1
61. Lam QL, Wang S, Ko OK, Kincade PW, Lu L. Leptin signaling maintains B-cell homeostasis via induction of Bcl-2 and Cyclin D1. Proc Natl Acad Sci USA. (2010) 107:13812–7. doi: 10.1073/pnas.1004185107
62. Claycombe K, King LE, Fraker PJ. A role for leptin in sustaining lymphopoiesis and myelopoiesis. Proc Natl Acad Sci USA. (2008) 105:2017–21. doi: 10.1073/pnas.0712053105
63. Bobbert P, Jenke A, Bobbert T, Kuhl U, Rauch U, Lassner D, et al. High leptin and resistin expression in chronic heart failure: adverse outcome in patients with dilated and inflammatory cardiomyopathy. Eur J Heart Fail. (2012) 14:1265–75. doi: 10.1093/eurjhf/hfs111
64. Wannamethee SG, Shaper AG, Whincup PH, Lennon L, Sattar N. Obesity and risk of incident heart failure in older men with and without pre-existing coronary heart disease: does leptin have a role? J Am Coll Cardiol. (2011) 58:1870–7. doi: 10.1016/j.jacc.2011.06.057
65. Lieb W, Sullivan LM, Harris TB, Roubenoff R, Benjamin EJ, Levy D, et al. Plasma leptin levels and incidence of heart failure, cardiovascular disease, and total mortality in elderly individuals. Diabetes Care. (2009) 32:612–6. doi: 10.2337/dc08-1596
66. Martin SS, Blaha MJ, Muse ED, Qasim AN, Reilly MP, Blumenthal RS, et al. Leptin and incident cardiovascular disease: the Multi-ethnic Study of Atherosclerosis (MESA). Atherosclerosis. (2015) 239:67–72. doi: 10.1016/j.atherosclerosis.2014.12.033
67. Kim SH, Despres JP, Koh KK. Obesity and cardiovascular disease: friend or foe? Eur Heart J. (2016) 37:3560–8. doi: 10.1093/eurheartj/ehv509
68. Hall ME, Harmancey R, Stec DE. Lean heart: role of leptin in cardiac hypertrophy and metabolism. World J Cardiol. (2015) 7:511–24. doi: 10.4330/wjc.v7.i9.511
69. Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. (2007) 115:3213–23. doi: 10.1161/CIRCULATIONAHA.106.679597
70. Barouch LA, Berkowitz DE, Harrison RW, O'Donnell CP, Hare JM. Disruption of leptin signaling contributes to cardiac hypertrophy independently of body weight in mice. Circulation. (2003) 108:754–9. doi: 10.1161/01.CIR.0000083716.82622.FD
71. Yue P, Arai T, Terashima M, Sheikh AY, Cao F, Charo D, et al. Magnetic resonance imaging of progressive cardiomyopathic changes in the db/db mouse. Am J Physiol Heart Circ Physiol. (2007) 292:H2106–2118. doi: 10.1152/ajpheart.00856.2006
72. Semeniuk LM, Kryski AJ, Severson DL. Echocardiographic assessment of cardiac function in diabetic db/db and transgenic db/db-hGLUT4 mice. Am J Physiol Heart Circ Physiol. (2002) 283:H976–82. doi: 10.1152/ajpheart.00088.2002
73. Van den Bergh A, Flameng W, Herijgers P. Type II diabetic mice exhibit contractile dysfunction but maintain cardiac output by favourable loading conditions. Eur. J. Heart Fail. (2006) 8:777–83. doi: 10.1016/j.ejheart.2006.03.001
74. Hall ME, Smith G, Hall JE, Stec DE. Cardiomyocyte-specific deletion of leptin receptors causes lethal heart failure in Cre-recombinase-mediated cardiotoxicity. Am J Physiol Regul Integr Comp Physiol. (2012) 303:R1241–1250. doi: 10.1152/ajpregu.00292.2012
75. Witham W, Yester K, O'Donnell CP, McGaffin KR. Restoration of glucose metabolism in leptin-resistant mouse hearts after acute myocardial infarction through the activation of survival kinase pathways. J Mol Cell Cardiol. (2012) 53:91–100. doi: 10.1016/j.yjmcc.2012.03.016
76. Golfman LS, Wilson CR, Sharma S, Burgmaier M, Young ME, Guthrie PH, et al. Activation of PPARgamma enhances myocardial glucose oxidation and improves contractile function in isolated working hearts of ZDF rats. Am J Physiol Endocrinol Metab. (2005) 289:E328–36. doi: 10.1152/ajpendo.00055.2005
77. Wang P, Lloyd SG, Zeng H, Bonen A, Chatham JC. Impact of altered substrate utilization on cardiac function in isolated hearts from Zucker diabetic fatty rats. Am J Physiol Heart Circ Physiol. (2005) 288:H2102–2110. doi: 10.1152/ajpheart.00935.2004
78. Buchanan J, Mazumder PK, Hu P, Chakrabarti G, Roberts MW, Yun UJ, et al. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology. (2005) 146:5341–9. doi: 10.1210/en.2005-0938
79. Zhou YT, Grayburn P, Karim A, Shimabukuro M, Higa M, Baetens D, et al. Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci USA. (2000) 97:1784–9. doi: 10.1073/pnas.97.4.1784
80. Lee Y, Wang MY, Kakuma T, Wang ZW, Babcock E, McCorkle K, et al. Liporegulation in diet-induced obesity. The antisteatotic role of hyperleptinemia. J Biol Chem. (2001) 276:5629–35. doi: 10.1074/jbc.M008553200
81. Christoffersen C, Bollano E, Lindegaard ML, Bartels ED, Goetze JP, Andersen CB, et al. Cardiac lipid accumulation associated with diastolic dysfunction in obese mice. Endocrinology. (2003) 144:3483–90. doi: 10.1210/en.2003-0242
82. Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, et al. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. (2004) 18:1692–700. doi: 10.1096/fj.04-2263com
83. Lee Y, Naseem RH, Duplomb L, Park BH, Garry DJ, Richardson JA, et al. Hyperleptinemia prevents lipotoxic cardiomyopathy in acyl CoA synthase transgenic mice. Proc Natl Acad Sci USA. (2004) 101:13624–9. doi: 10.1073/pnas.0405499101
84. Hall ME, Maready MW, Hall JE, Stec DE. Rescue of cardiac leptin receptors in db/db mice prevents myocardial triglyceride accumulation. Am J Physiol Endocrinol Metab. (2014) 307:E316–325. doi: 10.1152/ajpendo.00005.2014
85. Belke DD, Larsen TS, Gibbs EM, Severson DL. Altered metabolism causes cardiac dysfunction in perfused hearts from diabetic (db/db) mice. Am J Physiol Endocrinol Metab. (2000) 279:E1104–1113. doi: 10.1152/ajpendo.2000.279.5.E1104
86. Hafstad AD, Khalid AM, How OJ, Larsen TS, Aasum E. Glucose and insulin improve cardiac efficiency and postischemic functional recovery in perfused hearts from type 2 diabetic (db/db) mice. Am J Physiol Endocrinol Metab. (2007) 292:E1288–1294. doi: 10.1152/ajpendo.00504.2006
87. Carley AN, Semeniuk LM, Shimoni Y, Aasum E, Larsen TS, Berger JP, et al. Treatment of type 2 diabetic db/db mice with a novel PPARgamma agonist improves cardiac metabolism but not contractile function. Am J Physiol Endocrinol Metab. (2004) 286:E449–455. doi: 10.1152/ajpendo.00329.2003
88. Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, et al. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. (2007) 56:2457–66. doi: 10.2337/db07-0481
89. Vincent HK, Powers SK, Dirks AJ, Scarpace PJ. Mechanism for obesity-induced increase in myocardial lipid peroxidation. Int J Obes Relat Metab Disord. (2001) 25:378–88. doi: 10.1038/sj.ijo.0801536
90. Belke DD, Swanson EA, Dillmann WH. Decreased sarcoplasmic reticulum activity and contractility in diabetic db/db mouse heart. Diabetes. (2004) 53:3201–8. doi: 10.2337/diabetes.53.12.3201
91. Pereira L, Matthes J, Schuster I, Valdivia HH, Herzig S, Richard S, et al. Mechanisms of [Ca2+]i transient decrease in cardiomyopathy of db/db type 2 diabetic mice. Diabetes. (2006) 55:608–15. doi: 10.2337/diabetes.55.03.06.db05-1284
92. Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res. (2018) 122:624–38. doi: 10.1161/CIRCRESAHA.117.311586
93. Jia G, DeMarco VG, Sowers JR. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol. (2016) 12:144–53. doi: 10.1038/nrendo.2015.216
94. Lourenco AP, Leite-Moreira AF, Balligand JL, Bauersachs J, Dawson D, de Boer RA, et al. An integrative translational approach to study heart failure with preserved ejection fraction: a position paper from the working group on myocardial function of the european society of cardiology. Eur J Heart Fail. (2018) 20:216–27. doi: 10.1002/ejhf.1059
95. Paolisso G, Tagliamonte MR, Galderisi M, Zito GA, D'Errico A, Marfella R, et al. Plasma leptin concentration, insulin sensitivity, and 24-hour ambulatory blood pressure and left ventricular geometry. Am J Hypertens. (2001) 14:114–20. doi: 10.1016/S0895-7061(00)01241-3
96. Perego L, Pizzocri P, Corradi D, Maisano F, Paganelli M, Fiorina P, et al. Circulating leptin correlates with left ventricular mass in morbid (grade III) obesity before and after weight loss induced by bariatric surgery: a potential role for leptin in mediating human left ventricular hypertrophy. J Clin Endocrinol Metab. (2005) 90:4087–93. doi: 10.1210/jc.2004-1963
97. Simonds SE, Pryor JT, Ravussin E, Greenway FL, Dileone R, Allen AM, et al. Leptin mediates the increase in blood pressure associated with obesity. Cell. (2014) 159:1404–16. doi: 10.1016/j.cell.2014.10.058
98. Uckaya G, Ozata M, Sonmez A, Kinalp C, Eyileten T, Bingol N, et al. Plasma leptin levels strongly correlate with plasma renin activity in patients with essential hypertension. Horm Metab Res. (1999) 31:435–8. doi: 10.1055/s-2007-978769
99. Carlyle M, Jones OB, Kuo JJ, Hall JE. Chronic cardiovascular and renal actions of leptin: role of adrenergic activity. Hypertension. (2002) 39 (2 Pt 2), 496–501. doi: 10.1161/hy0202.104398
100. Altara R, Giordano M, Norden ES, Cataliotti A, Kurdi M, Bajestani SN, et al. Targeting obesity and diabetes to treat heart failure with preserved ejection fraction. Front Endocrinol (Lausanne). (2017) 8:160. doi: 10.3389/fendo.2017.00160
101. Obokata M, Reddy YNV, Pislaru SV, Melenovsky V, Borlaug BA. Evidence Supporting the Existence of a Distinct Obese Phenotype of Heart Failure With Preserved Ejection Fraction. Circulation. (2017) 136:6–19. doi: 10.1161/CIRCULATIONAHA.116.026807
102. Faxen UL, Hage C, Andreasson A, Donal E, Daubert JC, Linde C, et al. HFpEF and HFrEF exhibit different phenotypes as assessed by leptin and adiponectin. Int J Cardiol. (2017) 228:709–16. doi: 10.1016/j.ijcard.2016.11.194
103. Schulze PC, Kratzsch J, Linke A, Schoene N, Adams V, Gielen S, et al. Elevated serum levels of leptin and soluble leptin receptor in patients with advanced chronic heart failure. Eur J Heart Fail. (2003) 5:33–40. doi: 10.1016/S1388-9842(02)00177-0
104. Utz W, Engeli S, Haufe S, Kast P, Hermsdorf M, Wiesner S, et al. Myocardial steatosis, cardiac remodelling and fitness in insulin-sensitive and insulin-resistant obese women. Heart. (2011) 97:1585–9. doi: 10.1136/hrt.2011.224451
105. Nyman K, Graner M, Pentikainen MO, Lundbom J, Hakkarainen A, Siren R, et al. Cardiac steatosis and left ventricular function in men with metabolic syndrome. J Cardiovasc Magn Reson. (2013) 15:103. doi: 10.1186/1532-429X-15-103
106. Xu FP, Chen MS, Wang YZ, Yi Q, Lin SB, Chen AF, et al. Leptin induces hypertrophy via endothelin-1-reactive oxygen species pathway in cultured neonatal rat cardiomyocytes. Circulation. (2004) 110:1269–75. doi: 10.1161/01.CIR.0000140766.52771.6D
107. Madani S, De Girolamo S, Munoz DM, Li RK, Sweeney G. Direct effects of leptin on size and extracellular matrix components of human pediatric ventricular myocytes. Cardiovasc Res. (2006) 69:716–25. doi: 10.1016/j.cardiores.2005.11.022
108. Packer M. Do sodium-glucose co-transporter-2 inhibitors prevent heart failure with a preserved ejection fraction by counterbalancing the effects of leptin? A novel hypothesis Diabetes Obes Metab. (2018) 20:1361–6. doi: 10.1111/dom.13229
109. Martens P, Mathieu C, Verbrugge FH. Promise of SGLT2 Inhibitors in Heart Failure: Diabetes and Beyond. Curr Treat Options Cardiovasc Med. (2017) 19:23. doi: 10.1007/s11936-017-0522-x
110. Xu L, Ota T. Emerging roles of SGLT2 inhibitors in obesity and insulin resistance: focus on fat browning and macrophage polarization. Adipocyte. (2018) 7:121–8.
111. Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. (2015) 373:2117–28. doi: 10.1056/NEJMoa1504720
112. Huby AC, Antonova G, Groenendyk J, Gomez-Sanchez CE, Bollag WB, Filosa JA, et al. Adipocyte-derived hormone leptin is a direct regulator of aldosterone secretion, which promotes endothelial dysfunction and cardiac fibrosis. Circulation. (2015) 132:2134–45. doi: 10.1161/CIRCULATIONAHA.115.018226
113. Pitt B, Pfeffer MA, Assmann SF, Boineau R, Anand IS, Claggett B, et al. Spironolactone for heart failure with preserved ejection fraction. N Engl J Med. (2014) 370:1383–92. doi: 10.1056/NEJMoa1313731
114. Kosmala W, Przewlocka-Kosmala M, Szczepanik-Osadnik H, Mysiak A, Marwick TH. Fibrosis and cardiac function in obesity: a randomised controlled trial of aldosterone blockade. Heart. (2013) 99:320–6. doi: 10.1136/heartjnl-2012-303329
115. Gutierrez-Tenorio J, Marin-Royo G, Martinez-Martinez E, Martin R, Miana M, Lopez-Andres N, et al. The role of oxidative stress in the crosstalk between leptin and mineralocorticoid receptor in the cardiac fibrosis associated with obesity. Sci Rep. (2017) 7:16802. doi: 10.1038/s41598-017-17103-9
116. Singhal A, Farooqi IS, Cole TJ, O'Rahilly S, Fewtrell M, Kattenhorn M, et al. Influence of leptin on arterial distensibility: a novel link between obesity and cardiovascular disease? Circulation. (2002) 106:1919–24. doi: 10.1161/01.CIR.0000033219.24717.52
117. Tsai JP, Wang JH, Chen ML, Yang CF, Chen YC, Hsu BG. Association of serum leptin levels with central arterial stiffness in coronary artery disease patients. BMC Cardiovasc Disord. (2016) 16:80. doi: 10.1186/s12872-016-0268-5
118. Yen TT, Allan JA, Pearson DV, Schinitsky MR. Dissociation of obesity, hypercholesterolemia and diabetes from atherosclerosis in ob/ob mice. Experientia. (1977) 33:995–6. doi: 10.1007/BF01945927
119. Bouloumie A, Marumo T, Lafontan M, Busse R. Leptin induces oxidative stress in human endothelial cells. FASEB J. (1999) 13:1231–8. doi: 10.1096/fasebj.13.10.1231
120. Oda A, Taniguchi T, Yokoyama M. Leptin stimulates rat aortic smooth muscle cell proliferation and migration. Kobe J Med Sci. (2001) 47:141–50. doi: 10.24546/00082677
121. Parhami F, Tintut Y, Ballard A, Fogelman AM, Demer LL. Leptin enhances the calcification of vascular cells: artery wall as a target of leptin. Circ Res. (2001) 88:954–60. doi: 10.1161/hh0901.090975
122. Fruhbeck G. Pivotal role of nitric oxide in the control of blood pressure after leptin administration. Diabetes. (1999) 48:903–8. doi: 10.2337/diabetes.48.4.903
123. Kimura K, Tsuda K, Baba A, Kawabe T, Boh-oka S, Ibata M, et al. Involvement of nitric oxide in endothelium-dependent arterial relaxation by leptin. Biochem. Biophys. Res. Commun. (2000) 273:745–49. doi: 10.1006/bbrc.2000.3005
Keywords: leptin receptor, leptin resistance, metabolism, cardiovascular disease, obesity, type 2 diabetes mellitus
Citation: Poetsch MS, Strano A and Guan K (2020) Role of Leptin in Cardiovascular Diseases. Front. Endocrinol. 11:354. doi: 10.3389/fendo.2020.00354
Received: 26 March 2020; Accepted: 06 May 2020;
Published: 16 June 2020.
Edited by:
Daryl Owen Schwenke, University of Otago, New ZealandReviewed by:
Evgenya Uchasova, Research Institute for Complex Issues of Cardiovascular Diseases, RussiaCopyright © 2020 Poetsch, Strano and Guan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kaomei Guan, a2FvbWVpLmd1YW5AdHUtZHJlc2Rlbi5kZQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.