 Kelli A. Duncan
Kelli A. Duncan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Endocrinol. , 29 May 2020
Sec. Neuroendocrine Science
Volume 11 - 2020 | https://doi.org/10.3389/fendo.2020.00345
This article is part of the Research Topic Recent Progress and Perspectives in Neurosteroid Research View all 20 articles
Traumatic brain injury (TBI) is responsible for various neuronal and cognitive deficits as well as psychosocial dysfunction. Characterized by damage inducing neuroinflammation, this response can cause an acute secondary injury that leads to widespread neurodegeneration and loss of neurological function. Estrogens decrease injury induced neuroinflammation and increase cell survival and neuroprotection and thus are a potential target for use following TBI. While much is known about the role of estrogens as a neuroprotective agent following TBI, less is known regarding their formation and inactivation following damage to the brain. Specifically, very little is known surrounding the majority of enzymes responsible for the production of estrogens. These estrogen metabolizing enzymes (EME) include aromatase, steroid sulfatase (STS), estrogen sulfotransferase (EST/SULT1E1), and some forms of 17β-hydroxysteroid dehydrogenase (HSD17B) and are involved in both the initial conversion and interconversion of estrogens from precursors. This article will review and offer new prospective and ideas on the expression of EMEs following TBI.
Traumatic brain injury (TBI) is a leading cause of human death and morbidity worldwide. TBI is a broad term to explain any injury or damage to the central nervous system, ranging from concussive, penetrating, to ischemic stroke. Over 70,000 cases are reported a year and it affects millions of people in the United States alone (1, 2). At-risk groups range from children, who frequently participate in contact sports to military personnel, and to an increasingly active elderly population (3, 4). Disturbingly high injury rates have made TBI not only an issue of social and economic concern, but also the target of nearly 50 years of dedicated clinical research (5–7). Despite research and improvements in effective patient care, there still remain very few recommended treatments which leads to poor patient outcomes (8).
Injury to the brain occurs in two phases, regardless of the cause. The irreversible primary phase is the injury itself. The majority of research around the primary phase focuses on decreasing the chance of injury (e.g., helmet design). The secondary and potentially reversible phase, which begins after the initial injury and continues for days to weeks afterword, is characterized by an induction of a neuroinflammatory response. This response is characterized by a permeation of inflammatory cells around the injury, followed by endothelial activation, and an accumulation of inflammatory cytokines (9–11). This secondary phase has some beneficial effects, but commonly results in an exacerbation of deleterious inflammatory effects, decreased cognitive ability, motor loss, and increased risk of neurodegenerative diseases, such as multiple sclerosis (12–17). This secondary response to injury is well-characterized across vertebrates (rodents, birds, fish) (18–20). The majority of research has focused on preventing this secondary response (21, 22). One pharmacotherapeutic strategy for treatment of secondary injury has revolved around the role of sex steroids, specifically estrogens on the attenuation of the immune response (22–25). Briefly, we will review the effects of estrogens on cell survival and neuroinflammation following TBI.
Following damage to the brain, estrogens can promote neurogenesis and neural recovery by attenuating neural outgrowth and glial activity (25–29). Estrogens are also indicated to serve as an antioxidant against intrinsic free radical production following TBI (30, 31). The majority of these effects are through estrogen receptor (ER) signaling pathways. The genomic signal transduction pathway of estrogens involves the dimerization of ERs and transcriptional regulation of estrogen mediated target genes which in turn can provide neuroprotective effects, evidenced by decreased inflammation, reactive gliosis, and edema (30, 32–34). Furthermore, activation of ER alpha, the primary mediator of steroid induced neuroprotection, can also alter neurovascular function and promote myelin repair (24, 35). However, rapid non-genomic signaling has been shown to upregulate estrogen mediated neuroprotection via the pro-survival signaling pathway, phosphoinositide 3-kinase/Akt. Activation of this pathway results in increased cell survival, differentiation, and growth (36). Estrogens can also mediate cell death and apoptosis by inhibiting subcellular trafficking of p38α. Jun N-terminal kinase (JNK) and p38 are necessary for the activation of pro-apoptotic signaling pathways (34, 36, 37). Moreover, when estrogens are given or produced immediately after injury, they can decrease pro-inflammatory and increase anti-inflammatory cytokines and thus have strong antioxidant and anti-inflammatory effects. In terms of behavior and cognitive ability following TBI, very little is known about the role of estrogens in ameliorating these symptoms. However, estrogens can also enhance memory function and neurological outcome in male rodents following TBI (38–47). In addition to these effects on cell/neuronal survival, estrogens can also regulate neuroinflammation following injury.
Estrogen synthesis following TBI reduces the concentration of pro-inflammatory cytokines and thus decreases the secondary wave of degeneration observed following damage to the brain (30, 48–50). Through regulation of pro-inflammatory cytokines, estrogens subsequently reduce leukocyte recruitment, cerebral edema, apoptosis, and reactive astrogliosis, thusly improving outcomes following injury (51). Thus, estrogens are neuroprotective on many fronts following TBI (52, 53). While, I've only touched on a few of the pre-clinical studies, the majority of these studies suggest that estrogens are neuroprotective following TBI. However, clinical trials in humans involving estrogen administration have not shown as robust of a beneficial result, thus identifying how the natural/systemic productions of estrogens differs from that of exogenous/synthetic production is key. Aside from the neuroprotective effects of estrogens, they exert many biological effects as well. Estrogens are mostly widely known for their sex-specific effects related to sexual determination and differentiation (54). Both genomic and non-genomic signal transduction pathways of estrogens alter the regulation of the cell cycle, this includes, but is not limited to proliferation and differentiation, but specifically cyclines and cycline-2dependent kinases (55–57). As a result of this effect on cell growth and cell progression, estrogens have direct cancerogenic effects and damage DNA and cellular proteins via highly reactive oxygen reactivity (55, 58). Thus, understanding and parsing out the hormonal effects from neuroprotective actions of estrogens are key to their use as therapeutics.
Four estrogens are produced naturally: estrone (E1), 17β-estradiol (estradiol or E2), estriol (E3), and estetrol (E4; Figure 1) The weakest form estrone is primarily found following menopause while estriol and estetrol are the predominant estrogens produced during pregnancy (30). Weak estrogens can bind to estrogen receptors, but generally lack a dramatic effect within tissues or cells. However, some weak estrogens such as estriol exhibit greater protection than estradiol in autoimmune disorders, such as multiple sclerosis (59). Estradiol is the most common and strongest of the estrogens and is thought to mediate the neuroprotection following various damage to the nervous system (30). Estradiol is produced during the menstrual cycle and also de novo in the brain (60–62).
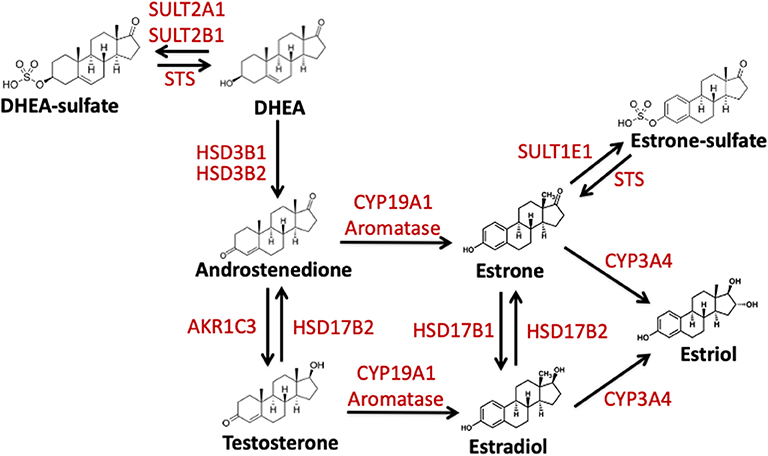
Figure 1. Schematic representation of the enzymatic conversion and synthesis of biologically active estrogens. Estrogens are produced from C19 steroid precursors through several enzymatic conversions. DHEA, dehydroepiandrosterone. DHEA-S, dehydroepiandrosterone-sulfate; Aromatase/CYP19A1, estrogen synthase; HSD3B1, hydroxysteroid 3 beta-1; HSD3B2, hydroxysteroid 3 beta-2; HSD17B1, hydroxysteroid 17-beta dehydrogenase; HSD17B2, hydroxysteroid 17-beta dehydrogenase 2; STS, steroid sulfatase; SULT2A1, Sulfotransferase Family 2A Member 1; SULT2B1, Sulfotransferase Family 2B Member 1; SULT1E1, estrogen sulfotransferase; AKR1C3, Aldo-Keto Reductase Family 1 Member C3; CYP3A4, Cytochrome P450 3A4.
Estrogens are formed following the enzymatic conversion and interconversion from cholesterol-based precursors via a subset of enzymes termed estrogen-metabolizing enzymes (EME). The most prevalent of these enzymes is aromatase or CYP19A1. The aromatase pathway forms estrone and estradiol from androgenic precursors androstenedione and testosterone, respectively (Figure 1) (63). In addition to this estrogen synthase activity, aromatase has been proposed to regulate estrogen-2-hydroxylase activity in placental tissue and in Japanese quail brains (64–66). This activity also paired with aromatase's interaction with TH and DA signaling suggest that aromatase plays a role in catecholaminergic transmission (67, 68). Thus, aromatase may be involved in both the production and inactivation of estrogens (68). Another EME, 17β-hydroxysteroid dehydrogenases 1 and 2 (HSD17B1, HSD17B2) is also necessary for the conversion of estrone to estradiol (61, 69, 70). Finally, estrogens can be made inactive by both degradation and sulfonation. In the sulfatase pathway, inactive estrogen sulfate is the source or precursor for the active estradiol and estrone. This is mediated via the enzymes steroid sulfatase (STS) and estrogen sulfotransferase (SULT1E1) (Figure 1)(71, 72). Below I will review what is known about these EMEs and their role following TBI.
Among the EMEs, aromatase is the most prominent and widely studied. Across vertebrates aromatase expression is found in gonads, placenta, adipose tissue, bone, and other tissues including both male and female brains (73–75). Within the vertebrate brain, high concentrations of aromatase are expressed within the hypothalamus, amygdala, hippocampus, and cerebral cortex (76, 77). Aromatase is broadly expressed within neurons and not glial cells in the above listed brain areas of the uninjured brain (78–80). Aromatase is also present in pre-synaptic boutons, suggesting direct perisynaptic actions (81). Following injury or neuroinflammation, aromatase is also found in glial cells, specifically astrocytes (80, 82). In the songbird brain, females exhibit higher expression immediately after injury, but these differences disappear by 24 h post injury (83). This upregulated glial aromatase appears to affect neurodegenerative pathways by decreasing apoptosis (84, 85). In songbirds, as in the mammals (86), administration of fadrozole (aromatase inhibitor) dramatically increases the volume of damage induced by penetrating mechanical injury (84), sometimes in a sexually dimorphic manner (87). Replacing estradiol at the time of injury prevents this fadrozole-induced damage (88).
Cytokines increase aromatase expression without concurrent cell death or damage to neuronal tissues (25, 26, 89). Using TNF-α and IL-1β KO mice, we were able to determine that TNF-α, but not IL-1β signaling is necessary for the induction of aromatase following brain injury (25). Interestingly, while inflammation appears to regulate aromatase expression, increasing aromatase decreases expression of TNF-α and IL-1β following injury furthermore aromatase inhibition results in prolonged elevation of TNF-α and IL-1β (29, 89). Another mechanism by which estrogens may become inactive following TBI is through aromatase's estrogen-2-hydroxylase activity, that converts estrogens to catechol-estrogens (64–66). The role of this method of estrogen inactivation following TBI remains unknown. This cycle of both upregulation and inhibition of neuronal aromatase and cytokine expression may suggest a broadly conserved mechanism for protecting the CNS following detection of a threat (25).
In addition to the aromatase pathway described previously, estrogens can also be formed from inactive precursors by the removal of sulfate groups (90–93). When sulfated, estrogens are unable to bind and dimerize to estrogen receptors. This protects cells and tissues from excess estrogen activity (55). Thus, sulfonation can potentially regulate active estrogen signaling and serve as a hormone “reservoir” for future use (91, 94, 95). Steroid sulfatase (STS) hydrolyzes the removal of sulfate groups from estrone sulfate (E1-S) to E1 and dehydroepiandrosterone sulfate (DHEA-S) to dehydroepiandrosterone (DHEA), also known as androstenolone (Figure 1) (96). STS is expressed broadly across vertebrates in both males and females with highest levels being found in the placenta, but low levels found across the majority of steroid sensitive tissues. STS expression and the mechanism that control its expression remain poorly understood (97). However, estrogen signaling pathways regulate expression of STS, and thus potentially create a positive feedback loop to increase estrogen production and signaling (71).
The majority of studies examining STS expression and TBI have focused more on the beneficial effects DHEA vs. E2. As STS alters both estrogens and DHEA at this time we are unable to separate out the neuroprotective effects of estrogens vs. those of DHEA. Both DHEA and DHEA-S are neuroprotective following damage to the CNS in rodents and birds. Specifically, DHEA and DHEA-S protect rodent neurons against various forms of toxicity including overactivation of N-methyl-D-aspartate (NMDA) glutamate receptors (98, 99), amyloid beta expression (100), and oxygen-glucose deprivation (101). Importantly, DHEA-S prevents against rodent hippocampal neuronal cell loss and damage observed following ischemia and stroke (102). Additionally, the progesterone precursor pregnenolone and pregnenolone-sulfate also exhibit neuroprotective benefits (103–105). More work is necessary to discern estrogen mediated STS effects vs. DHEA mediated ones following TBI.
Like aromatase, we see a connection between STS expression and inflammatory signals. Interleukin (IL)-1β suppresses STS expression in endometrial stromal cells (106). However, in breast cancer cells, various other cytokines IL-1α, IL-6, TNFα, and enhanced or increased activity of STS (107). For both IL-6 and TNFα, STS mRNA expression was unchanged and the difference in activity was due to posttranslational modifications via STS glycosylation (72). Our studies examining the effects of a single penetrating injury in the adult zebra finch (Taeniopygia guttata) brain showed that STS expression was unchanged following TBI (Figures 2A,B). The zebra finch serves as a model organism for the study of steroid induced neuroprotection, because they rapidly and robustly respond to injury and express the full suite of steroidogenic enzymes (53, 82, 108). Furthermore, increased expression of pro-inflammatory cytokines is observed anywhere from 2 to 4 h following injury (83). Thus, the lack of a change in STS mRNA expression was surprising. As STS mRNA levels are unaffected in zebra finches, more research is necessary to determine if STS glycosylation or estrone bioavailability is increased following injury.
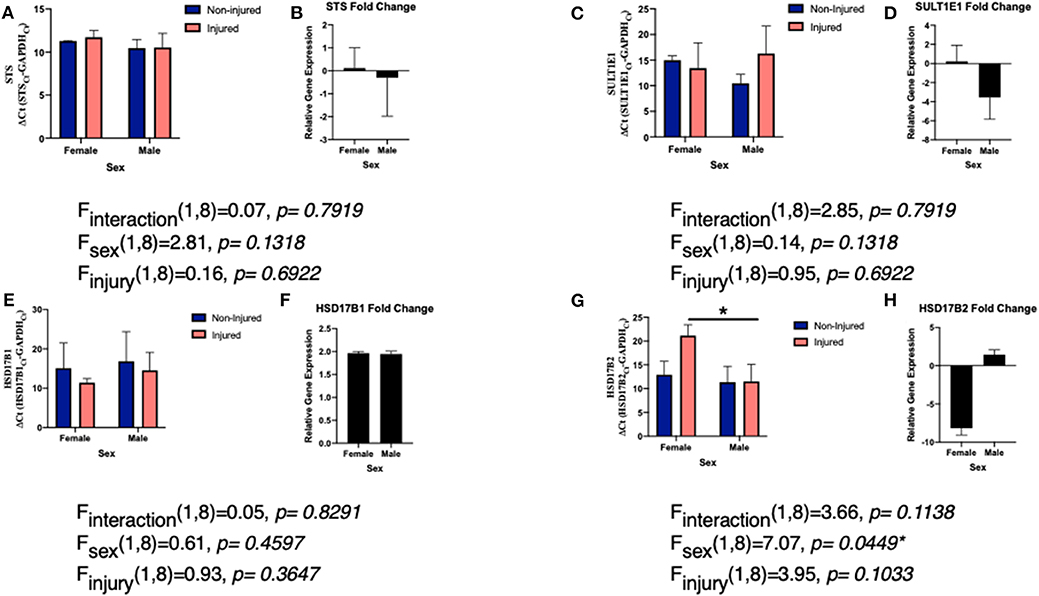
Figure 2. Levels of EMEs mRNA relative to glyceraldehyde-3-phosphate dehydrogenase (GAPDH; delta CT value and fold change) in both male and female zebra finches 24 h post-surgery. Adult male and female zebra finches were subjected to an unilateral penetrating injury directed toward the entopallium and collected 24 h later. Finches were sacrificed and total RNA was extracted from microdissections immediately adjacent to the cortical needle and the corresponding location on uninjured hemisphere. Expression of STS (A), SULT1E1 (C), HSD17B1 (E), and HSD17B2 (G) relative to GAPDH was measured using quantitative PCR using primers specific for the ZF mRNA sequence. Mean ΔCT values were compared with a two-way ANOVA. Fold change in gene expression was also calculated STS (B), SULT1E1 (D), HSD17B1 (F), and HSD17B2 (H). Neither expression of STS, SULT1E1, or HSD17B1 were significantly changed following injury. However, HSD17B2 was significantly downregulated following injury. *denotes a significant difference at P < 0.05. All protocols were approved by the Vassar College Institutional Animal Care and Use Committee following National Institutes of Health Guidelines.
SULT1E1 (Figure 1) when compared to other members of the steroid sulfotransferase (SULT) family has greater affinity for estrogens than other steroids, and thus readily inactivates estrone and estradiol. SULT1E1 is expressed in various tissues in both sexes (109) including the brain (110) and its expression is regulated by steroid hormones, specifically progesterone (96). Estrone-sulfate and its naturally occurring counterpart estradiol-sulfate (E2-SO4) are biologically important as they regulate neuronal network formation, activity, and synaptogenesis (111–114) in vertebrates.
Very little is known about the expression of SULT1E1 following TBI, however there is some data on the role of estradiol-sulfate (E2-SO4) in rodent models. In terms of expression, like STS, we did not see a significant increase or decrease in expression of SULT1E1 following TBI (Figures 2C,D), however the expression was far more variable than that of STS. Studies are on-going to determine if protein expression matched the mRNA expression. Sulfated estrogens reduce neurodegeneration by altering cell and tissue damage and oxidative stress in rodents (115, 116). This is accomplished by improving blood and water flow to damaged tissue and thus increasing cerebral perfusion pressure and decreasing cell loss (22). Furthermore, the solubility of E2-SO4 as opposed to estradiol enables intravenous delivery of supraphysiological quantities and thus one could give much higher doses and have it be eliminated from the body much more quickly (117, 118). Sulfated estrogens provide a noteworthy avenue for both future research and future treatment options following TBI (118).
With respect to SULT1E1 and TBI induced inflammation, very little is known, but there appear to be a strong connection between SULT1E1 expression and release of cytokines. Knockdown of SULT1E1 increases pro-inflammatory cytokines and decreases anti-inflammatory signals (119). Furthermore, both edema and intercranial pressure are altered by SULT1E1 expression (115, 118). Understanding SULT1E1 expression and its role following TBI is key to understanding how sulfate estrogens can be used as a treatment. The time of SULT1E1 expression may also change the results. In the finch brain, we examined expression at 24 h, however the knockdown expression studies were 72 h. It would be interesting to extend the timepoints at which expression was examined.
17β-hydroxysteroid dehydrogenases (HSD17B) are enzymes responsible for the synthesis and deactivation of estrogens and androgens, specifically the formation of testosterone and estradiol from precursors (61, 76, 108, 120, 121) (Figure 1). HSD17B1 primarily catalyzes the conversion of estrone to estradiol in various tissues including the brain. In addition to its role in estrogen synthesis, HSD17B1 facilitates the formation of various androgens as well (76, 122–124). Conversely, the enzyme HSD17B2 mediates the oxidation of estradiol back to estrone, and testosterone and androstendiol back to androstenedione dehydroepiandrosterone. Furthermore, HSD17B2, is responsible for the production of the active progestin, progesterone (76, 125). The expression of HSD17B2 has previously only been identified in non-neuronal tissues, however a few reports have identified transcript and protein in the brain (126).
Surprisingly, there remains a dearth of information and studies on the role or expression of either HSD17B1 or HSD17B2 following TBI. The majority of past studies have examined the role of HSD17Bs in steroid sensitive cancers (69). Specifically, in terms of brain damage, there has been some work showing a connection between HSD17B1 and risk of Alzheimer's disease in Down Syndrome patients (127). In our work, we did not find a significant change in expression of HSD17B1 following TBI, suggesting that conversion to estrone is not a probable path to increasing estrogen signaling following injury (Figures 2E,F). While estradiol is the predominate estrogen mediating neuroprotection, estrone, has been shown to be neuroprotective following various damage or insults to the brain (128–130). Estrone increases the signaling of neuroprotective pathways (ERK1/2 and BDNF) and decreases cell death and thus ischemic injury size (38). These results suggest that despite the lack of change in HSD17B1, vertebrates may still be getting the protection from higher levels of estrone (38). As estrone is the most abundant estrogen in menopausal women (131, 132), it is hypothesized to be extremely important in mediating neuroprotection in this population (38). Much more work is necessary to understand the levels of estrone relative to estradiol following TBI.
While HSD17B1 expression was not altered by TBI, HSD17B2 expression was downregulated in a sex specific manner following TBI in the finch brain (Figures 2G,H). Females decreased expression of HSD17B2 following injury while males did not. Again, much remains unknown surrounding the function of HSD17B2 in the brain, specifically because it is noticeably absent from human and rodent brains (133). Notably other HSDs are found throughout the avian brain and regulate conversion from cholesterol to the sex steroids (108, 120, 134). Further research is needed in order to conclude if the expression of HSD17B2 following injury is unique to songbird brains, or if this represents a more evolutionarily conserved pathway. Despite the abundant questions remaining, the results are promising. One could hypothesize that females are decreasing expression of HSD17B2 in order to keep more estradiol available for neuroprotection and repair, while not having to produce more estrogens. As males use estrogen to undergo sexual differentiation, having a local upregulation of estrogens in the female brain may not be beneficial long term. Furthermore, while HSD17B2 is the key HSD17B isozyme in androgen and estrogen inactivation, it also activates 20α-hydroxyprogesterone into progesterone (70, 135). Females may be using progesterone in combination with estradiol as neuroprotective steroids following injury. These results are compelling as we have previously found a role for progesterone in cell survival following TBI in the brain (105) and this supports a large body of research examining progesterone treatment following TBI (136, 137). Interestingly, like with estradiol despite positive pre-clinical studies, larger clinical, and human trials of progesterone use following TBI have been unsuccessful (138). Together, these data suggest that much more research is needed in order to understand the very complex steroidal milieu following TBI.
As has been discussed in the preceding sections, there is ample evidence to support that estrogens are neuroprotective following injury. Yet, when estrogen use has been used in clinical trials of TBI, the majority of evidence has not been positive and has led many to question its use as a treatment option (139, 140). Thus, more research on the formation of these estrogens and how they are inactivated is necessary in order to better develop treatment plans and options for mirroring estrogen's endogenous neuroprotective effects found preclinically without the undesirable hormonal ones observed.
The animal study was reviewed and approved by Vassar College IACUC.
The author confirms being the sole contributor of this work and has approved it for publication.
This work was supported by Vassar College Salmon Fund to KD.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author wishes to acknowledge Dr. Colin Saldanha, and the many undergraduates that contributed to the work presented here.
1. Lenzlinger PM, Morganti-Kossmann MC, Laurer HL, McIntosh TK. The duality of the inflammatory response to traumatic brain injury. Mol Neurobiol. (2001) 24:169–81. doi: 10.1385/MN:24:1-3:169
2. Maas AI, Stocchetti N, Bullock R. Moderate and severe traumatic brain injury in adults. Lancet Neurology. (2008) 7:728–41. doi: 10.1016/S1474-4422(08)70164-9
3. Stein DG. Is progesterone a worthy candidate as a novel therapy for traumatic brain injury? Dialogues Clin Neurosci. (2011) 13:352–9.
4. Griesbach GS, Kreber LA, Harrington D, Ashley MJ. Post-acute traumatic brain injury rehabilitation: effects on outcome measures and life care costs. J Neurotrauma. (2015) 32:704–11. doi: 10.1089/neu.2014.3754
5. Bruns J, Hauser WA. The epidemiology of traumatic brain injury: a review. Epilepsia. (2003) 44:2–10. doi: 10.1046/j.1528-1157.44.s10.3.x
6. Corrigan JD, Selassie AW, Orman JA. The epidemiologyo of traumatic brain injury. J Head Trauma Rehabil. (2010) 25:72–80. doi: 10.1097/HTR.0b013e3181ccc8b4
7. Langlois JA, Rutland-Brown W, Wald MM. The epidemiology and impact of traumatic brain injury: a brief overview. J Head Trauma Rehabil. (2006) 21:375–8. doi: 10.1097/00001199-200609000-00001
8. Abou-Abbass H, Bahmad H, Ghandour H, Fares J, Wazzi-Mkahal R, Yacoub B, et al. Epidemiology and clinical characteristics of traumatic brain injury in Lebanon: a systematic review. Medicine. 95:e5342. doi: 10.1097/MD.0000000000005342
9. Morganti-Kossmann MC, Satgunaseelan L, Bye N, Kossmann T. Modulation of immune response by head injury. Injury. (2007) 38:1392–400. doi: 10.1016/j.injury.2007.10.005
10. Xu J, Fan G, Chen S, Wu Y, Xu XM, Hsu CY. Methylprednisolone inhibition of TNF-alpha expression and NF-kB activation after spinal cord injury in rats. Brain Res Mol Brain Res. (1998) 59:135–42. doi: 10.1016/S0169-328X(98)00142-9
11. Ziebell JM, Morganti-Kossmann MC. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics. (2010) 7:22–30. doi: 10.1016/j.nurt.2009.10.016
12. Kang JH, Lin HC. Increased risk of multiple sclerosis after traumatic brain injury: a nationwide population-based study. J Neurotrauma. (2012) 29:90–5. doi: 10.1089/neu.2011.1936
13. Prins M, Greco T, Alexander D, Giza CC. The pathophysiology of traumatic brain injury at a glance. DMM Dis Model Mech. (2013) 6:1307–15. doi: 10.1242/dmm.011585
14. Lye TC, Shores EA. Traumatic brain injury as a risk factor for Alzheimer's disease: a review. Neuropsychol Rev. (2000) 10:115–29. doi: 10.1023/A:1009068804787
15. Sorby-Adams AJ, Marcoionni AM, Dempsey ER, Woenig JA, Turner RJ. The role of neurogenic inflammation in blood-brain barrier disruption and development of cerebral oedema following acute central nervous system (CNS) injury. Int J Mol Sci. (2017) 18:1788. doi: 10.3390/ijms18081788
16. Fitch MT, Doller C, Combs CK, Landreth GE, Silver J. Cellular and molecular mechanisms of glial scarring and progressive cavitation: in vivo and in vitro analysis of inflammation-induced secondary injury after CNS trauma. J Neurosci. (1999) 19:8182–98. doi: 10.1523/JNEUROSCI.19-19-08182.1999
17. Thelin EP, Hall CE, Gupta K, Carpenter KLH, Chandran S, Hutchinson PJ, et al. Elucidating pro-inflammatory cytokine responses after traumatic brain injury in a human stem cell model. J Neurotrauma. (2018) 35:341–52. doi: 10.1089/neu.2017.5155
18. Rothwell NJ, Strijbos PJ. Cytokines in neurodegeneration and repair. Int J Dev Neurosci. (1995) 13:179–85. doi: 10.1016/0736-5748(95)00018-C
19. Duncan KA, Walters BJ, Saldanha CJ. Traumatized and inflamed - but resilient: glial aromatization and the avian brain. Horm Behav. (2013) 63:208–15. doi: 10.1016/j.yhbeh.2012.02.026
20. McCutcheon V, Park E, Liu E, Sobhebidari P, Tavakkoli J, Wen XY, et al. A novel model of traumatic brain injury in adult zebrafish demonstrates response to injury and treatment comparable with mammalian models. J Neurotrauma. (2017) 34:1382–93. doi: 10.1089/neu.2016.4497
21. Bramlett HM, Dietrich WD. Progressive damage after brain and spinal cord injury: pathomechanisms and treatment strategies. Prog Brain Res. (2007) 161:125–41. doi: 10.1016/S0079-6123(06)61009-1
22. Day NL, Floyd CL, D'Alessandro TL, Hubbard WJ, Chaudry IH. 17β-estradiol confers protection after traumatic brain injury in the rat and involves activation of G protein-coupled estrogen receptor 1. J Neurotrauma. (2013) 30:1531–41. doi: 10.1089/neu.2013.2854
23. Herson PS, Koerner IP, Hurn PD. Sex, sex steroids, and brain injury. Semin Reprod Med. (2009) 27:229–39. doi: 10.1055/s-0029-1216276
24. Saldanha CJ, Duncan KA, Walters BJ. Neuroprotective actions of brain aromatase. Front Neuroendocrinol. (2009) 30:106–18. doi: 10.1016/j.yfrne.2009.04.016
25. Duncan KA, Saldanha CJ. Central aromatization: a dramatic and responsive defense against threat and trauma to the vertebrate brain. Front Neuroendocrinol. (2020) 56:100816. doi: 10.1016/j.yfrne.2019.100816
26. Duncan KA, Saldanha CJ. Neuroinflammation induces glial aromatase expression in the uninjured songbird brain. J Neuroinflammation. (2011) 8:81. doi: 10.1186/1742-2094-8-81
27. Stein DG. Brain damage, sex hormones and recovery: a new role for progesterone and estrogen? Trends Neurosci. (2001) 24:386–91. doi: 10.1016/S0166-2236(00)01821-X
28. Acaz-Fonseca E, Avila-Rodriguez M, Garcia-Segura LM, Barreto GE. Regulation of astroglia by gonadal steroid hormones under physiological and pathological conditions. Prog Neurobiol. (2016) 144:5–26. doi: 10.1016/j.pneurobio.2016.06.002
29. Pedersen AL, Nelson LH, Saldanha CJ. Centrally synthesized estradiol is a potent anti-inflammatory in the injured zebra finch brain. Endocrinology. (2016) 157:2041–51. doi: 10.1210/en.2015-1991
30. Raghava N, Das BC, Ray SK. Neuroprotective effects of estrogen in CNS injuries: insights from animal models. Neurosci Neuroeconomics. (2017) 6:15–29. doi: 10.2147/NAN.S105134
31. Culmsee C, Vedder H, Ravati A, Junker V, Otto D, Ahlemeyer B, et al. Neuroprotection by estrogens in a mouse model of focal cerebral ischemia and in cultured neurons: evidence for a receptor-independent antioxidative mechanism. J Cereb Blood Flow Metab. (1999) 19:1263–9. doi: 10.1097/00004647-199911000-00011
32. Lee JY, Choi HY, Na WH, Ju BG, Yune TY. 17β-estradiol inhibits MMP-9 and SUR1/TrpM4 expression and activation and thereby attenuates BSCB disruption/hemorrhage after spinal cord injury in male rats. Endocrinology. (2015) 156:1838–50. doi: 10.1210/en.2014-1832
33. Cox A, Varma A, Barry J, Vertegel A, Banik N. Nanoparticle estrogen in rat spinal cord injury elicits rapid anti-inflammatory effects in plasma, cerebrospinal fluid, and tissue. J Neurotrauma. (2015) 32:1413–21. doi: 10.1089/neu.2014.3730
34. Han D, Scott EL, Dong Y, Raz L, Wang R, Zhang Q. Attenuation of mitochondrial and nuclear p38α signaling: a novel mechanism of estrogen neuroprotection in cerebral ischemia. Mol Cell Endocrinol. (2015) 400:21–31. doi: 10.1016/j.mce.2014.11.010
35. Dubal DB, Zhu H, Yu J, Rau SW, Shughrue PJ, Merchenthaler I, et al. Estrogen receptor alpha, not beta, is a critical link in estradiol-mediated protection against brain injury. Proc Natl Acad Sci USA. (2001) 98:1952–7. doi: 10.1073/pnas.98.4.1952
36. Zhu C, Wang S, Wang B, Du F, Hu C, Li H, et al. 17β-estradiol up-regulates Nrf2 via PI3K/AKT and estrogen receptor signaling pathways to suppress light-induced degeneration in rat retina. Neuroscience. (2015) 304:328–39. doi: 10.1016/j.neuroscience.2015.07.057
37. Hill RA, Chua HK, Jones ME, Simpson ER, Boon WC. Estrogen deficiency results in apoptosis in the frontal cortex of adult female aromatase knockout mice. Mol Cell Neurosci. (2009) 41:1–7. doi: 10.1016/j.mcn.2008.12.009
38. Gatson JW, Liu MM, Abdelfattah K, Wigginton JG, Smith S, Wolf S, et al. Estrone is neuroprotective in rats after traumatic brain injury. J Neurotrauma. (2012) 29:2209–19. doi: 10.1089/neu.2011.2274
39. Simpkins JW, Rajakumar G, Zhang YQ, Simpkins CE, Greenwald D, Yu CJ, et al. Estrogens may reduce mortality and ischemic damage caused by middle cerebral artery occlusion in the female rat. J Neurosurg. (1997) 87:724–30. doi: 10.3171/jns.1997.87.5.0724
40. Kondo Y, Suzuki K, Sakuma Y. Estrogen alleviates cognitive dysfunction following transient brain ischemia in ovariectomized gerbils. Neurosci Lett. (1997) 238:45–8. doi: 10.1016/S0304-3940(97)00847-1
41. McCullough LD, Blizzard K, Simpson ER, Oz OK, Hurn PD. Aromatase cytochrome P450 and extragonadal estrogen play a role in ischemic neuroprotection. J Neurosci. (2003) 23:8701–5. doi: 10.1523/JNEUROSCI.23-25-08701.2003
42. Merchenthaler I, Dellovade TL, Shughrue PJ. Neuroprotection by estrogen in animal models of global and focal ischemia. Ann. N. Y. Acad. Sci. 1007:89–100. doi: 10.1196/annals.1286.009
43. Sudo S, Wen TC, Desaki J, Matsuda S, Tanaka J, Arai T, et al. Beta-estradiol protects hippocampal CA1 neurons against transient forebrain ischemia in gerbil. Neurosci Res. (1997) 29:345–54. doi: 10.1016/S0168-0102(97)00106-5
44. Suzuki S, Gerhold LM, Böttner M, Rau SW, Dela Cruz C, Yang E, et al. Estradiol enhances neurogenesis following ischemic stroke through estrogen receptors α and β. J Comp Neurol. (2007) 500:1064–75. doi: 10.1002/cne.21240
45. Yang SH, Shi J, Day AL, Simpkins JW. Estradiol exerts neuroprotective effects when administered after ischemic insult. Stroke. (2000) 31:745–50. doi: 10.1161/01.STR.31.3.745
46. Brotfain EE, Gruenbaum S, Boyko M, Kutz R, Zlotnik A, Klein M. Neuroprotection by estrogen and progesterone in traumatic brain injury and spinal cord injury. Curr Neuropharmacol. (2016) 14:641–53. doi: 10.2174/1570159X14666160309123554
47. Emerson CS, Headrick JP, Vink R. Estrogen improves biochemical and neurologic outcome following traumatic brain injury in male rats, but not in females. Brain Res. (1993) 608:95–100. doi: 10.1016/0006-8993(93)90778-L
48. Cook S, Hung V, Duncan KA. Crosstalk between estrogen withdrawal and NFκB signaling following penetrating brain injury. Neuroimmunomodulation. (2018) 25:193–200. doi: 10.1159/000493506
49. Pozzi S, Benedusi V, Maggi A, Vegeto E. Estrogen action in neuroprotection and brain inflammation. Ann N Y Acad Sci. (2006) 1089:302–23. doi: 10.1196/annals.1386.035
50. Wen Y, Yang S, Liu R, Perez E, Yi KD, Koulen P, et al. Estrogen attenuates nuclear factor-kappa B activation induced by transient cerebral ischemia. Brain Res. (2004) 1008:147–54. doi: 10.1016/j.brainres.2004.02.019
51. Pedersen AL, Saldanha CJ. Reciprocal interactions between prostaglandin E2- and estradiol-dependent signaling pathways in the injured zebra finch brain. J Neuroinflammation. (2017) 14:262. doi: 10.1186/s12974-017-1040-1
52. Walters BJ, Alexiades NG, Saldanha CJ. Intracerebral estrogen provision increases cytogenesis and neurogenesis in the injured zebra finch brain. Dev Neurobiol. (2011) 71:170–81. doi: 10.1002/dneu.20839
53. Duncan KA, Saldanha CJ. Inducible Aromatase in Astroglia: Protection and Recovery from Neural Perturbation in Birds. New York, NY: Oxford University Press (2013). doi: 10.1093/acprof:oso/9780199841196.003.0020
54. MacLusky N, Naftolin F. Sexual differentiation of the central nervous system. Science. (1981) 211:1294–302. doi: 10.1126/science.6163211
55. Secky L, Svoboda M, Klameth L, Bajna E, Hamilton G, Zeillinger R, et al. The sulfatase pathway for estrogen formation: targets for the treatment and diagnosis of hormone-associated tumors. J Drug Deliv. (2013) 2013:957605. doi: 10.1155/2013/957605
56. Leitman DC, Paruthiyil S, Vivar OI, Saunier EF, Herber CB, Cohen I, et al. Regulation of specific target genes and biological responses by estrogen receptor subtype agonists. Curr Opin Pharmacol. (2010) 10:629–36. doi: 10.1016/j.coph.2010.09.009
57. Koos RD. Minireview: putting physiology back into estrogens' mechanism of action. Endocrinology. (2011) 152:4481–8. doi: 10.1210/en.2011-1449
58. Bolton JL, Thatcher GRJ. Potential mechanisms of estrogen quinone carcinogenesis. Chem Res Toxicol. (2008) 21:93–101. doi: 10.1021/tx700191p
59. Voskuhl RR, Gold SM. Sex-related factors in multiple sclerosis susceptibility and progression. Nat Rev Neurol. (2012) 8:255–63. doi: 10.1038/nrneurol.2012.43
60. Arnold AP, Schlinger BA. Sexual differentiation of brain and behavior: the zebra finch is not just a flying rat. Brain Behav Evol. (1993) 42:231–41. doi: 10.1159/000114157
61. Tsutsui K. Neurosteroid biosynthesis and function in the brain of domestic birds. Front Endocrinol. (2011) 2:37. doi: 10.3389/fendo.2011.00037
63. Labrie F. Intracrinology. Mol Cell Endocrinol. (1991) 78:C113–8. doi: 10.1016/0303-7207(91)90116-A
64. Balthazart J, Stoop R, Foidart A, Granneman JCM, Lambert JGD. Distribution and regulation of estrogen-2-hydroxylase in the quail brain. Brain Res Bull. (1994) 35:339–45. doi: 10.1016/0361-9230(94)90111-2
65. Almadhidi J, Moslemi S, Drosdowsky MA, Séralini GE. Equine cytochrome p450 aromatase exhibits an estrogen 2-hydroxylase activity in vitro. J Steroid Biochem Mol Biol. (1996) 59:55–61. doi: 10.1016/S0960-0760(96)00085-4
66. Osawa Y, Higashiyama T, Shimizu Y, Yarborough C. Multiple functions of aromatase and the active site structure; aromatase is the placental estrogen 2-hydroxylase. J Steroid Biochem Mol Biol. (1993) 44:469–80. doi: 10.1016/0960-0760(93)90252-R
67. Balthazart J, Ball GF. New insights into the regulation and function of brain estrogen synthase (aromatase). Trends Neurosci. (1998) 21:243–9. doi: 10.1016/S0166-2236(97)01221-6
68. Charlier TD, Cornil CA, Patte-Mensah C, Meyer L, Mensah-Nyagan AG, Balthazart J. Local modulation of steroid action: rapid control of enzymatic activity. Front Neurosci. (2015) 9:83. doi: 10.3389/fnins.2015.00083
69. Hilborn E, Stål O, Jansson A. Estrogen and androgen-converting enzymes 17β-hydroxysteroid dehydrogenase and their involvement in cancer: with a special focus on 17β-hydroxysteroid dehydrogenase type 1, 2, and breast cancer. Oncotarget. (2017) 8:30552–62. doi: 10.18632/oncotarget.15547
70. Moeller G, Adamski J. Multifunctionality of human 17β-hydroxysteroid dehydrogenases. Mol. Cell. Endocrinol. (2006) 248:47–55. doi: 10.1016/j.mce.2005.11.031
71. Zaichuk T, Ivancic D, Scholtens D, Schiller C, Khan SA. Tissue-specific transcripts of human steroid sulfatase are under control of estrogen signaling pathways in breast carcinoma. J Steroid Biochem Mol Biol. (2007) 105:76–84. doi: 10.1016/j.jsbmb.2006.12.101
72. Newman SP, Purohit A, Ghilchik MW, Potter B V, Reed MJ. Regulation of steroid sulphatase expression and activity in breast cancer. J Steroid Biochem Mol Biol. (2000) 75:259–64. doi: 10.1016/S0960-0760(00)00177-1
73. Stocco C. Tissue physiology and pathology of aromatase. Steroids. (2012) 77:27–35. doi: 10.1016/j.steroids.2011.10.013
74. Stocco C. Aromatase expression in the ovary: hormonal and molecular regulation. Steroids. (2008) 73:473–87. doi: 10.1016/j.steroids.2008.01.017
75. Naftolin F, Horvath TL, Jakab RL, Leranth C, Harada N, Balthazart J. Aromatase immunoreactivity in axon terminals of the vertebrate brain. an immunocytochemical study on quail, rat, monkey and human tissues. Neuroendocrinology. (1996) 63:149–55. doi: 10.1159/000126951
76. Stoffel-Wagner B, Watzka M, Schramm J, Bidlingmaier F, Klingmüller D. Expression of CYP19 (aromatase) mRNA in different areas of the human brain. J Steroid Biochem Mol Biol. 70:237–41. doi: 10.1016/S0960-0760(99)00114-4
77. Hojo Y, Hattori TA, Enami T, Furukawa A, Suzuki K, Ishii HT, et al. Adult male rat hippocampus synthesizes estradiol from pregnenolone by cytochromes P45017α and P450 aromatase localized in neurons. Proc Natl Acad Sci USA. (2004) 101:865–70. doi: 10.1073/pnas.2630225100
78. Saldanha CJ, Remage-Healey L, Schlinger BA. Synaptocrine signaling: steroid synthesis and action at the synapse. Endocr Rev. (2011) 32:532–49. doi: 10.1210/er.2011-0004
79. Yague JG, Wang AC, Janssen WG, Hof PR, Garcia-Segura LM, Azcoitia I, et al. Aromatase distribution in the monkey temporal neocortex and hippocampus. Brain Res. (2008) 1209:115–27. doi: 10.1016/j.brainres.2008.02.061
80. Zhang QG, Wang R, Tang H, Dong Y, Chan A, Sareddy GR, et al. Brain-derived estrogen exerts anti-inflammatory and neuroprotective actions in the rat hippocampus. Mol Cell Endocrinol. (2014) 389:84–91. doi: 10.1016/j.mce.2013.12.019
81. Remage-Healey L, Saldanha CJ, Schlinger BA. Estradiol synthesis and action at the synapse: evidence for “synaptocrine” signaling. Front Endocrinol. (2011) 2:28. doi: 10.3389/fendo.2011.00028
82. Peterson RS, Saldanha CJ, Schlinger BA. Rapid upregulation of aromatase mRNA and protein following neural injury in the zebra finch (taeniopygia guttata). J Neuroendocrinol. (2001) 13:317–23. doi: 10.1046/j.1365-2826.2001.00647.x
83. Saldanha CJ, Burstein SR, Duncan KA. Induced synthesis of oestrogens by glia in the songbird brain. J Neuroendocrinol. (2013) 25:1032–8. doi: 10.1111/jne.12067
84. Wynne RD, Saldanha CJ. Glial aromatization decreases neural injury in the zebra finch (taeniopygia guttata): influence on apoptosis. J Neuroendocrinol. (2004) 16:676–83. doi: 10.1111/j.1365-2826.2004.01217.x
85. Wynne RD, Walters BJ, Bailey DJ, Saldanha CJ. Inhibition of injury-induced glial aromatase reveals a wave of secondary degeneration in the songbird brain. Glia. (2008) 56:97–105. doi: 10.1002/glia.20594
86. Azcoitia I, Sierra A, Veiga S, Honda S, Harada N, Garcia-Segura LM. Brain aromatase is neuroprotective. J Neurobiol. (2001) 47:318–29. doi: 10.1002/neu.1038
87. Gölz C, Kirchhoff FP, Westerhorstmann J, Schmidt M, Hirnet T, Rune GM, et al. Sex hormones modulate pathogenic processes in experimental traumatic brain injury. J Neurochem. (2019) 150:173–87. doi: 10.1111/jnc.14678
88. Saldanha CJ, Rohmann KN, Coomaralingam L, Wynne RD. Estrogen provision by reactive glia decreases apoptosis in the zebra finch (taeniopygia guttata). J Neurobiol. (2005) 64:192–201. doi: 10.1002/neu.20147
89. Pedersen AL, Brownrout JL, Saldanha CJ. Neuroinflammation and neurosteroidogenesis: reciprocal modulation during injury to the adult zebra finch brain. Physiol Behav. (2018) 187:51–6. doi: 10.1016/j.physbeh.2017.10.013
90. Purohit A, Potter B V, Parker MG, Reed MJ. Steroid sulphatase: expression, isolation and inhibition for active-site identification studies. Chem Biol Interact. (1998) 109:183–93. doi: 10.1016/S0009-2797(97)00132-4
91. Hobkirk R. Steroid sulfation current concepts. Trends Endocrinol Metab. (1993) 4:69–74. doi: 10.1016/S1043-2760(05)80018-9
92. Aksoy IA, Wood TC, Weinshilboum R. Human liver estrogen sulfotransferase: identification by cDNA cloning and expression. Biochem Biophys Res Commun. (1994) 200:1621–29. doi: 10.1006/bbrc.1994.1637
93. Falany CN, Vazquez ME, Kalb JM. Purification and characterization of human liver dehydroepiandrosterone sulphotransferase. Biochem J. (1989) 260:641–6. doi: 10.1042/bj2600641
94. Pasqualini JR, Chetrite G, Blacker C, Feinstein MC, Delalonde L, Talbi M, et al. Concentrations of estrone, estradiol, and estrone sulfate and evaluation of sulfatase and aromatase activities in pre- and postmenopausal breast cancer patients. J Clin Endocrinol Metab. (1996) 81:1460–4. doi: 10.1210/jcem.81.4.8636351
95. Adjei AA, Thomae BA, Prondzinski JL, Eckloff BW, Wieben ED, Weinshilboum RM. Human estrogen sulfotransferase (SULT1E1) pharmacogenomics: gene resequencing and functional genomics. Br J Pharmacol. (2003) 139:1373–82. doi: 10.1038/sj.bjp.0705369
96. RiŽner TL. The important roles of steroid sulfatase and sulfotransferases in gynecological diseases. Front Pharmacol. (2016) 7:30. doi: 10.3389/fphar.2016.00030
97. Nardi A, Pomari E, Zambon D, Belvedere P, Colombo L, Dalla Valle L. Transcriptional control of human steroid sulfatase. J Steroid Biochem Mol Biol. (2009) 115:68–74. doi: 10.1016/j.jsbmb.2009.02.017
98. Mao X, Barger SW. Neuroprotection by dehydroepiandrosteronesulfate: role of an NF?B-like factor. Neuroreport. (1998) 9:759–63. doi: 10.1097/00001756-199803090-00036
99. Kimonides VG, Khatibi NH, Svendsen CN, Sofroniew M V, Herbert J. Dehydroepiandrosterone (DHEA) and DHEA-sulfate (DHEAS) protect hippocampal neurons against excitatory amino acid-induced neurotoxicity. Proc Natl Acad Sci USA. (1998) 95:1852–7. doi: 10.1073/pnas.95.4.1852
100. Cardounel A, Regelson W, Kalimi M. Dehydroepiandrosterone protects hippocampal neurons against neurotoxin-induced cell death: mechanism of action. Proc Soc Exp Biol Med. (1999) 222:145–9. doi: 10.1046/j.1525-1373.1999.d01-124.x
101. Kaasik A, Kalda A, Jaako K, Zharkovsky A. Dehydroepiandrosterone sulphate prevents oxygen-glucose deprivation-induced injury in cerebellar granule cell culture. Neuroscience. (2001) 102:427–32. doi: 10.1016/S0306-4522(00)00489-9
102. Li ZK, Feng JX, Zhao CY, Ke H, Shen L. Protection of androgen against hypoxic-ischemic brain damage in neonatal rats and possible mechanisms. Zhongguo Dang Dai Er Ke Za Zhi. (2006) 8:441–6.
103. Weaver CE, Marek P, Park-Chung M, Tam SW, Farb DH. Neuroprotective activity of a new class of steroidal inhibitors of the N-methyl-D-aspartate receptor. Proc Natl Acad Sci USA. (1997) 94:10450–4. doi: 10.1073/pnas.94.19.10450
104. Gursoy E, Cardounel A, Kalimi M. Pregnenolone protects mouse hippocampal (HT-22) cells against glutamate and amyloid beta protein toxicity. Neurochem Res. (2001) 26:15–21. doi: 10.1023/a:1007668213330
105. Blackshear K, Giessner S, Hayden JP, Duncan KA. Exogenous progesterone is neuroprotective following injury to the male zebra finch brain. J Neurosci Res. (2018) 96:545–555. doi: 10.1002/jnr.24060
106. Matsuoka R, Yanaihara A, Saito H, Furusawa Y, Toma Y, Shimizu Y, et al. Regulation of estrogen activity in human endometrium: effect of IL-1beta on steroid sulfatase activity in human endometrial stromal cells. Steroids. (2002) 67:655–9. doi: 10.1016/S0039-128X(02)00016-8
107. Ren X, Wu X, Hillier SG, Fegan KS, Critchley HOD, Mason JI, et al. Local estrogen metabolism in epithelial ovarian cancer suggests novel targets for therapy. J Steroid Biochem Mol Biol. (2015) 150:54–63. doi: 10.1016/j.jsbmb.2015.03.010
108. Mirzatoni A, Spence RD, Naranjo KC, Saldanha CJ, Schlinger BA. Injury-induced regulation of steroidogenic gene expression in the cerebellum. J Neurotrauma. (2010) 27:1875–82. doi: 10.1089/neu.2010.1330
109. Coughtrie MWH. Sulfation through the looking glass - recent advances in sulfotransferase research for the curious. Pharmacogenomics J. (2002) 2:297–308. doi: 10.1038/sj.tpj.6500117
110. Uhlén M, Björling E, Agaton C, Szigyarto CAK, Amini B, Andersen E, et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol Cell Proteomics. (2005) 4:1920–32. doi: 10.1074/mcp.M500279-MCP200
111. Garcia-Segura LM, Luquin S, Parducz A, Naftolin F. Gonadal hormone regulation of glial fibrillary acidic protein immunoreactivity and glial ultrastructure in the rat neuroendocrine hypothalamus. Glia. (1994) 10:59–69. doi: 10.1002/glia.440100108
112. Roof RL, Hall ED. Gender differences in acute CNS trauma and stroke: neuroprotective effects of estrogen and progesterone. J Neurotrauma. (2000) 17:367–88. doi: 10.1089/neu.2000.17.367
113. McEwen B. Estrogen actions throughout the brain. Recent Prog Horm Res. (2002) 57:357–84. doi: 10.1210/rp.57.1.357
114. Beyer C. Estrogen and the developing mammalian brain. Anat Embryol. (1999) 199:379–90. doi: 10.1007/s004290050236
115. Liu X, Huang J, Sun Y, Zhan K, Zhang Z, Hong M. Identification of multiple binding sites for substrate transport in bovine organic anion transporting polypeptide 1a2. Drug Metab Dispos. (2013) 41:602–7. doi: 10.1124/dmd.112.047910
116. Stein DG, Hoffman SW. Estrogen and progesterone as neuroprotective agents in the treatment of acute brain injuries. Pediatr Rehabil. (2003) 6:13–22. doi: 10.1080/1363849031000095279
117. Kim H, Yu T, Cam-Etoz B, Van Groen T, Hubbard WJ, Chaudry IH. Treatment of traumatic brain injury with 17α-ethinylestradiol-3-sulfate in a rat model. J Neurosurg. (2017) 127:23–31. doi: 10.3171/2016.7.JNS161263
118. Kim H, Cam-Etoz B, Zhai G, Hubbard WJ, Zinn KR, Chaudry IH. Salutary effects of estrogen sulfate for traumatic brain injury. J Neurotrauma. (2015) 32:1210–6. doi: 10.1089/neu.2014.3771
119. Xu Y, Yang X, Wang Z, Li M, Ning Y, Chen S, et al. Estrogen sulfotransferase (SULT1E1) regulates inflammatory response and lipid metabolism of human endothelial cells via PPARγ. Mol Cell Endocrinol. (2013) 369:140–9. doi: 10.1016/j.mce.2013.01.020
120. London SE, Itoh Y, Lance VA, Wise PM, Ekanayake PS, Oyama RK, et al. Neural expression and post-transcriptional dosage compensation of the steroid metabolic enzyme 17β-HSD type 4. BMC Neurosci. (2010) 11:47. doi: 10.1186/1471-2202-11-47
121. Tomaszycki ML, Peabody C, Replogle K, Clayton DF, Tempelman RJ, Wade J. Sexual differentiation of the zebra finch song system: potential roles for sex chromosome genes. BMC Neurosci. (2009) 10:24. doi: 10.1186/1471-2202-10-24
122. Peltoketo H, Isomaa V, Mäentausta O, Vihko R. Complete amino acid sequence of human placental 17 beta-hydroxysteroid dehydrogenase deduced from cDNA. FEBS Lett. (1988) 239:73–7. doi: 10.1016/0014-5793(88)80548-9
123. The VL, Labrie C, Zhao HF, Couët J, Lachance Y, Simard J, et al. Characterization of cdnas for human estradiol 17β-dehydrogenase and assignment of the gene to chromosome 17: evidence of two mrna species with distinct 5'-termini in human placenta. Mol Endocrinol. (1989) 3:1301–9. doi: 10.1210/mend-3-8-1301
124. Dumont M, Luu-The V, de Launoit Y, Labrie F. Expression of human 17 beta-hydroxysteroid dehydrogenase in mammalian cells. J Steroid Biochem Mol Biol. (1992) 41:605–8. doi: 10.1016/0960-0760(92)90391-U
125. Wu L, Einstein M, Geissler WM, Chan HK, Elliston KO, Andersson S. Expression cloning and characterization of human 17 beta-hydroxysteroid dehydrogenase type 2, a microsomal enzyme possessing 20 alpha-hydroxysteroid dehydrogenase activity. J Biol Chem. (1993) 268:12964–9.
126. Wang CL, Ying SJ, Wang ZY, Xing HJ, Wang LZ, He DY, et al. Molecular cloning and expression of 17β-hydroxysteroid dehydrogenase type 2 gene in Hu sheep. Mol Biol Rep. (2013) 40:1073–80. doi: 10.1007/s11033-012-2149-z
127. Lee JH, Gurney S, Pang D, Temkin A, Park N, Janicki SC, et al. Polymorphisms in HSD17B1: early onset and increased risk of Alzheimer's disease in women with down syndrome. Curr Gerontol Geriatr Res. (2012) 2012:361218. doi: 10.1155/2012/361218
128. Bae YH, Hwang JY, Kim YH, Koh JY. Anti-oxidative neuroprotection by estrogens in mouse cortical cultures. J Korean Med Sci. (2000) 15:327–36. doi: 10.3346/jkms.2000.15.3.327
129. Perez E, Liu R, Yang S-H, Cai ZY, Covey DF, Simpkins JW. Neuroprotective effects of an estratriene analog are estrogen receptor independent in vitro and in vivo. Brain Res. (2005) 1038:216–22. doi: 10.1016/j.brainres.2005.01.026
130. Regan RF, Guo Y. Estrogens attenuate neuronal injury due to hemoglobin, chemical hypoxia, and excitatory amino acids in murine cortical cultures. Brain Res. (1997) 764:133–40. doi: 10.1016/S0006-8993(97)00437-X
131. Blair IA. Analysis of estrogens in serum and plasma from postmenopausal women: past present, and future. Steroids. (2010) 75:297–306. doi: 10.1016/j.steroids.2010.01.012
132. Cirigliano M. Bioidentical hormone therapy: a review of the evidence. J Women's Heal. (2007) 16:600–31. doi: 10.1089/jwh.2006.0311
133. Stoffel-Wagner B, Watzka M, Steckelbroeck S, Schramm J, Bidlingmaier F, Klingmüller D. Expression of 17β-hydroxysteroid dehydrogenase types 1, 2, 3 and 4 in the human temporal lobe. J Endocrinol. (1999) 160:119–26. doi: 10.1677/joe.0.1600119
134. Tsutsui KSBA. Steroidogenesis in the avian brain. In: Dawson ACCM, editor. Avian Endocrinology (New Delhi: Narosa Publishing House), 59–77.
135. Mindnich R, Möller G, Adamski J. The role of 17 beta-hydroxysteroid dehydrogenases. Mol Cell Endocrinol. (2004) 218:7–20. doi: 10.1016/j.mce.2003.12.006
136. Stein DG. Progesterone exerts neuroprotective effects after brain injury. Brain Res Rev. (2008) 57:386–97. doi: 10.1016/j.brainresrev.2007.06.012
137. Stein DG, Wright DW, Kellermann AL. Does progesterone have neuroprotective properties? Ann Emerg Med. (2008) 51:164–72. doi: 10.1016/j.annemergmed.2007.05.001
138. Stein DG. Embracing failure: what the phase III progesterone studies can teach about TBI clinical trials. Brain Inj. (2015) 29:1259–72. doi: 10.3109/02699052.2015.1065344
139. Engler-Chiurazzi EB, Covey DF, Simpkins JW. A novel mechanism of non-feminizing estrogens in neuroprotection. Exp Gerontol. (2017) 94:99–102. doi: 10.1016/j.exger.2016.10.013
Keywords: TBI, estrogen, aromatase, androgen, sulfatase, HSD17B
Citation: Duncan KA (2020) Estrogen Formation and Inactivation Following TBI: What we Know and Where we Could go. Front. Endocrinol. 11:345. doi: 10.3389/fendo.2020.00345
Received: 01 March 2020; Accepted: 04 May 2020;
Published: 29 May 2020.
Edited by:
Hubert Vaudry, Université de Rouen, FranceReviewed by:
Barney A. Schlinger, University of California, Los Angeles, United StatesCopyright © 2020 Duncan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kelli A. Duncan, a2VkdW5jYW5AdmFzc2FyLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.