 Pieter Vancamp
Pieter Vancamp Barbara A. Demeneix
Barbara A. Demeneix Sylvie Remaud
Sylvie Remaud- UMR 7221 Molecular Physiology and Adaptation, Centre National de le Recherche Scientifique-Muséum National d'Histoire Naturelle, Paris, France
Monocarboxylate transporter 8 (MCT8) deficiency or the Allan-Herndon-Dudley Syndrome (AHDS) is an X-linked psychomotor disability syndrome with around 320 clinical cases described worldwide. SLC16A2 gene mutations, encoding the thyroid hormone (TH) transporter MCT8, result in intellectual disability due to impaired TH uptake in the developing brain. MCT8 deficiency is a multi-organ affecting disease with a predominant neuronal cell-based pathology, with the glial component inadequately investigated. However, deficiency in myelin, a key component of white matter (WM) enabling fast nerve conduction, is a TH-dependent hallmark of the disease. Nevertheless, analysis of the myelin status in AHDS patients has led to conflicting interpretations. The majority of individual case studies reported delayed myelination, that was restored later in life. In contrast, post-mortem studies and high-resolution MRIs detected WM (micro-) abnormalities throughout adolescence, suggesting permanent hypomyelination. Thus, interpretations vary depending on methodology to investigate WM microstructure. Further, it is unknown whether the mutation within the MCT8 is linked to the severity of the myelin deficiency. Consequently, terminology is inconsistent among reports, and AHDS is occasionally misdiagnosed as another WM disorder. The evolutionary conserved TH signaling pathway that promotes the generation of myelinating oligodendrocytes enabled deciphering how the lack of MCT8 might affect myelinogenesis. Linking patient findings on myelination to those obtained from models of MCT8 deficiency revealed underlying pathophysiological mechanisms, but knowledge gaps remain, notably how myelination progresses both spatially and temporally in MCT8 deficiency. This limits predicting how myelin integrity might benefit therapeutically, and when to initiate. A recurrent observation in clinical trials is the absence of neurological improvement. Testing MCT8-independent thyromimetics in models, and evaluating treatments used in other demyelinating diseases, despite different etiologies, is crucial to propose new therapeutic strategies combatting this devastating disease.
Introduction
The Allan-Herndon-Dudley Syndrome (AHDS, OMIM #300523) is a very rare, X-linked psychomotor disability syndrome first described in 1944, almost exclusively affecting boys (1). The clinical picture of AHDS is diverse, including muscle wasting, hypotonia, spastic paraplegia, and severe intellectual disability with an IQ generally lower than 30 (2). Patients often develop complications and are prone to infections, and consequently have a decreased life expectancy, although some have lived beyond the age of 60. A hallmark of the disease are abnormal thyroid hormone (TH) serum levels. AHDS patients have low thyroxine (3,5,3′,5′-tetraiodothyronine, or T4), high 3,5′,3′-triiodothyronine (T3) and borderline-increased thyroid stimulating hormone levels (3, 4).
Soon after the identification of monocarboxylate transporter 8 (MCT8) as a highly-specific transmembrane TH transporter (5), inactivating mutations in the human SLC16A2 gene encoding MCT8 were linked to the altered TH levels detected in AHDS patients (3, 4), now often referred to as the MCT8 deficiency syndrome. Since then, several point mutations, translocations and deletions have been described in more than 320 diagnosed AHDS patients worldwide (6, 7), though many more are to be expected. It is estimated that up to 4% of the X-linked intellectual disability syndromes involve SLC16A2 mutations (8).
MCT8 facilitates cellular influx and efflux of the mainly inactive (pro)hormone T4 and the bioactive T3. The uptake of lipophilic THs is a prerequisite for T3 to bind to nuclear TH receptors and act on gene transcriptional activity. The amount of available T3 to bind receptors is additionally controlled by deiodinases (DIOs), either activating T4 into T3 (DIO2), or inactivating T4 into reverse T3, or T3 into T2 (DIO3) (9). TH action is of vital importance for the development of many organs, including the central nervous system (CNS). Genome-wide studies in rodent brains revealed hundreds of TH target genes whose expression is directly and indirectly regulated (10, 11) through dynamic interactions of (un)liganded TH receptors with chromatin and DNA (12), as well as through epigenetic modifications (13). A strict spatiotemporal regulation of gene expression underlies the highly orchestrated development of the human CNS that occurs over a period of many years up to adolescence, and continues throughout life in some brain regions (14–17). Animal studies showed that THs orchestrate many cellular processes underlying the foundation of the CNS cyto-architecture (18, 19), including cell proliferation, cell fate decisions, migration and differentiation, but also maturation including axogenesis, synaptogenesis and myelinogenesis (20).
A key developmental process regulated by TH is myelination (21). Neuronal axons in the vertebrate CNS with a diameter > 0.4 μm are encapsulated by myelin enabling faster electrical conduction over relatively long distances (22). Any damage to the white matter (WM) has devastating consequences for neural connectivity, impairing neurological performance and motor-coordination (23). Myelin is a lipid-rich (fatty) substance, giving it a white appearance in post-mortem tissue, hence the name WM for myelinated CNS regions. In humans, the peak of myelination occurs primarily during the first 2 years after birth, and is quasi complete at 5 years of age, but continues until early adulthood in some regions like the frontal cortex (24). Myelination follows a fixed temporal-spatial pattern, with axons in the brain stem being the first to be myelinated, followed by those in the cerebellum, and finally the cerebral cortex (25, 26). Compact myelin is a hallmark of the CNS of craniates that allowed the expansion and diversification of the vertebrate subphylum (27, 28). Furthermore, it is a well-known TH-dependent process, and associates with a neonatal peak in TH levels detected in all vertebrates (29). Accordingly, congenital hypothyroidism, resulting from an ineffective thyroid gland at birth, is characterized by delayed myelination. Treating new-borns with levothyroxine can restore myelination progress (30), showing that TH is a key player modulating myelin integrity.
Impaired myelination is also a common trait in AHDS patients, the majority of them having an abnormal WM content during the 1st years after birth (31, 32). However, as discussed herein, conflicting interpretations among case reports and brain imaging analyzes, reporting the myelin deficiency as either delayed or permanent, impede the unequivocal classification of AHDS within the broad family of demyelinating disorders. Consequently, there is sometimes an inconsistent use of terminology in different studies. A detailed description of the myelin phenotype is still missing in patient's later life, but also in animal models that replicate various aspects of the human disease. This complicates correct diagnosis of new AHDS patients and hampers prognosis of the extent to which the deficit in myelin might benefit from a potential treatment. Several MCT8-independent thyromimetics, currently tested to combat the peripheral thyrotoxicosis due to the excess of circulating T3 levels, might also improve myelination if their cellular mode of action is thoroughly understood, and if they can be administered early enough to obtain a maximal response.
Here, we review recent literature data to clarify the myelin deficit associated with AHDS and models of MCT8 deficiency. We also discuss potential mechanisms to investigate how the lack of MCT8 could hamper myelination, and we provide insights on MCT8-independent TH analogs and other drugs as promising candidates for myelin repair therapies.
Clinical Data From Allan-Herndon-Dudley Syndrome Patients: Permanent or Delayed Myelination?
To classify AHDS correctly within the broad group of demyelinating disorders, it is imperative to use correct terminology. Clinical myelin disorders have been subdivided according to WM appearance and developmental progression, mostly assessed using brain magnetic resonance imaging (MRI) (26, 31). A leukoencephalopathy is any clinical neurological disorder characterized by abnormal development or degeneration of WM in the CNS. In particular, a leukodystrophy is a congenital leukoencephalopathy, i.e., with a genetic etiology (25). The fact that myelination primarily takes place during the first 2 years of postnatal life means it is important to determine whether a congenital myelination deficit is permanent or delayed. If the same pattern of myelination is observed in at least two MRIs, taken > 6 months apart in a child older than 1 year, the paucity is considered permanent and referred to as “permanent hypomyelination” (26). Whenever progress in myelination or WM content can be observed within this time interval, it is defined as “delayed myelination” (26).
During the Delphi convention in 2015, leukodystrophy experts redefined and subdivided the WM disorders into two new groups (31, 33). The first group comprises the “leukodystrophies.” These are defined as WM disorders, with glial cells being the main cause of the pathology and myelin sheath abnormalities the most apparent feature. The second group are the “genetic leukoencephalopathies” (gLEs), characterized by neuronal cell pathologies and/or by other systemic disease manifestations, whereby WM abnormalities are subordinate to the neurological component of the disease (31, 33).
To determine whether myelination is delayed or permanent in AHDS patients, data on WM status and progression is needed. Azzolini et al. performed a meta-analysis of the myelin status in 32 diagnosed AHDS patients from January 2004 until December 2012, and also reported a new case (34). We build on this list, citing cases found up until December 2019 and taking case reports into account that described the WM phenotype. We employed a similar search strategy to (34) using the following search terms: “MCT8” and “mutation,” “SLC16A2” and “mutation,” “Allan–Herndon–Dudley syndrome,” “AHDS” and “case report,” “AHDS” and “MRI” and “myelination.” We found an additional 18 peer-reviewed articles reporting on 61 patients for whom MRI was used to assess myelin status in the brain (Table 1). Hence, we had MRI data on 94 AHDS patients in total, representing roughly a quarter of all known cases. We only included data on a patient's last MRI to simplify the analysis (Figure 1). Four patients from Remerand et al. (32) were excluded, since it was unclear whether the WM profile was normal or abnormal for their last MRI. We divided the remaining 90 patients in groups younger than 2 years old (2 year-olds included), between 2 and 6 years old (6 year-olds included), and those older than 6 years. Two years was chosen as a first cut-off point based on (26), stating that it is difficult to evaluate a myelination deficit before the age of 2 years when the myelination process is still ongoing. The age of 6 years was chosen as an arbitrary cut-off given that myelination is virtually complete by 5 years (24).

Table 1. MRI data on case-reports of AHDS patients from October 2004 to December 2019.
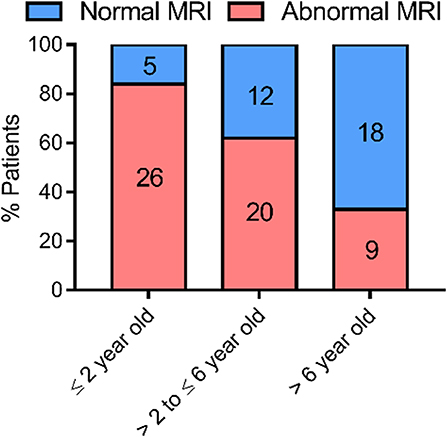
Figure 1. Evolution of myelination based on MRI data obtained from case-reports of 90 AHDS patients. The WM status as observed in the last MRI of each patient was used for the analysis. Sequential MRIs were not taken up in the analysis, so that each data point represents one patient. We ascribed a patient to the group “normal MRI” whenever the report specifically mentioned a normal WM status or the complete absence or disappearance of WM abnormalities in the last exam. In case a delay of any degree was reported or WM status showed abnormalities, we ascribed the patient to the group “abnormal MRI.” The references for the patient reports used for this analysis can be found in Table 1. This graph was made using GraphPad Prism v7.00.
Abnormal or delayed myelination was evident in 84% of the patients ≤ 2 years of age. Of the AHDS patients with MRI data that were older than 2 and ≤ 6 years of age 63% displayed some form of myelination delay. The percentage was reduced to 33% in patients older than 6 years (Figure 1), although the latter group consisted of only 27 patients. Thus, there tends to be a very slow but gradual improvement in myelin status in the patient cohort during this developmental period. A gradual progression in WM status was also observed in several of the patients that underwent sequential MRIs (Table 1). This finding defines MCT8 deficiency as a special case. Pouwels et al., with their long-time experience of demyelination studies, suggested that whenever deficient myelination is still observable in 2 year olds, it seems unlikely a child will ever display a normal WM content (26). The observed progression is developmental in nature and not a case of spontaneous regeneration of myelin, since no partially remyelinating sites, also called shadow plaques, were observed that are otherwise typical in myelinating disorders such as Multiple Sclerosis (MS) (66).
Remarkably, some patients never acquire a completely normal WM content, even after several years. Even though older patients more likely have normal myelination, some MRI data show no normalization in patients above the age of 10 [e.g., (32, 51)] (Figure 2). Clear signs of hypomyelination were also found during a post-mortem histological exam of the brain of a deceased 11-years-old AHDS boy (52). Inadequate myelination also coincides with several functional outcomes observed in AHDS patients, such as muscle dystonia, spastic paraplegia with poor head and limb control (32). A previous comparison of MRI data even identified three patients with a worsening myelin phenotype (34), and Remerand et al. also described three deteriorating cases out of their 24 patient cohort (32). The two patients examined in Anik et al. clearly illustrate the discrepancy among patients as a function of age (50): the two boys of the second investigated family are 6 months and 6 years old, but only the older boy displays hypomyelination.
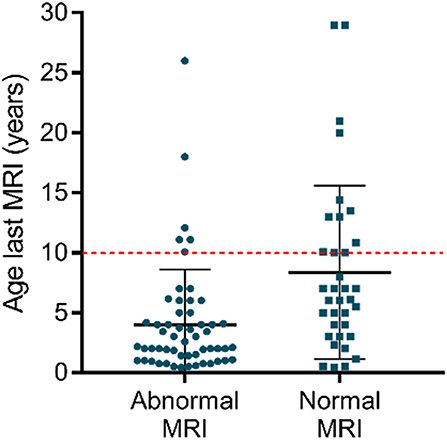
Figure 2. Myelin appearance (abnormal MRI vs. normal MRI) in AHDS patients in function of the age at the last MRI. Each point represents one patient, and the scatter plots also depict mean ± standard deviation. Finding normal WM status on MRI is more likely in older patients. Notice that above the age of 10 (red dotted line), there are still several patients with an abnormal WM status as observed on MRI. The references for the patient reports used for this graph can be found in Table 1. This graph was made using GraphPad Prism v7.00.
In this last case, the myelin status differs extensively despite the brothers sharing the same 2.8 kb deletion in the SLC16A2 gene that results in a complete loss of MCT8 uptake activity due to the lack of exons 3 and 4 (50). This case prompts the question whether or not a genotype-phenotype relationship exists between the type of gene mutation, its impact on MCT8 protein structure and function, and the severity of the myelin deficiency. In vitro cultures using human-derived fibroblasts, or JEG3- and COS9-cells previously identified several point mutations that resulted in residual TH uptake activity. These mutations are often associated with milder neurocognitive phenotypes in patients [e.g., (37, 65, 67)]. Other point mutations affecting proper substrate binding, trafficking or folding [e.g., (68, 69)], as well as large deletions, substitutions or premature stop-codons, render the MCT8 protein completely inactive, and result in severe mental and motor disabilities [e.g., (50, 70)].
Remerand and co-workers determined that for a group of AHDS patients for which radio- and neurological features were available in literature, 27% of them presented with moderate-to-mild intellectual disability, irrespective of age (32). However, how the type of mutation in each of these patients differentially affects MCT8 uptake activity is unknown. A genotype-phenotype relationship is expected for MCT8 mutations and the neurocognitive outcome (71), but a general trend has yet to be uncovered (6). Data on WM status in particular are currently too fragmentary to identify a potential link with the kind of mutation and the severity of the myelin deficiency.
More detailed MRI examinations also revealed that while the general WM structure seems intact as a whole, subtle localized myelin abnormalities might go unnoticed. Matheus and colleagues found a normally myelinated corpus callosum in three 6-years-old AHDS patients, but distinct tracts remained inadequately myelinated (59). The hypomyelination of subcortical U-fibers and periventricular tracts even became more discernible (59). This is corroborated by several other reports mentioning local, more subtle WM abnormalities too, as for instance in the periventricular WM tracts (31, 32, 42) and indicates regionally hypomyelinated areas may still be present in later life. Similar microstructural WM abnormalities were also observed in children with severe congenital hypothyroidism (72). This indicates that a global brain MRI is insufficient to discern between “normal” vs. “abnormal” myelination, and advocates a high-resolution and regional examination of the myelin status of the entire brain, and by extension the spinal cord.
Future studies employing a more accurate exploration of the WM phenotype by MRI imaging at regular time intervals would reveal if, and how fast exactly myelination improves in AHDS patients. Whenever possible, additional post-mortem analyses should include accurate WM examinations. Clinical data from newly discovered patients extend the database, and could unmask subgroups responding differently to MCT8 deficiency, or could uncover mutations harboring individual-specific traits related to myelination, or even other clinical features. An important observation is that in the patients that do exhibit a gradual increase in myelination, neurological functions do not improve. This suggests persistent neuronal problems throughout the CNS as being the main cause of abnormal sensorimotor functioning (58), although one can also not rule out a significant impact of poor feeding and extensive muscle wasting (37). Here too, additional MRI imaging examining the spinal cord WM should be performed to rule out any problems in electrical conductance along downstream axonal tracts. Moreover, novel brain imaging techniques such as positron emission tomography (PET) used to image progression of WM lesions in MS (73) could provide new ways to dynamically follow up myelination.
The enigmatic myelin phenotype accompanying MCT8 deficiency impedes the unequivocal classification of AHDS into a distinct group of WM disorders (Table 2), and it leads to unintentional inconsistencies in literature. For instance, Charzewska et al. proposed to classify it as a leukodystrophy (31), whereas Barkovich et al. did not include it in their list of “hypomyelinating disorders” (74), while Remerand et al. described the myelin deficit as hypomyelination (32). Furthermore, the unawareness of changing clinical terminology has led to occasionally mixed use of the terms “delayed” and “hypomyelination” in animal research too (see section myelin deficiency in animal models of MCT8 deficiency: permanent or delayed?). At the Delphi convention, AHDS was categorized as a gLE (31, 33). This classification is justified if delayed myelination is indeed a typical hallmark during the 1st years of development, but eventually becomes a non-pathognomonic trait if myelin formation recovers.
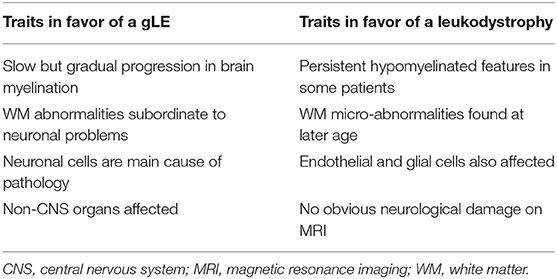
Table 2. Characteristics of AHDS to classify it as a genetic leukoencephalopathy (gLE) vs. a leukodystrophy.
That would also mean that AHDS differs from the group of leukodystrophies displaying permanent hypomyelination without any progress noticeable on sequential MRIs (31, 75). An example is Pelizaeus-Merzbacher Disease (PMD) most often caused by duplications in the PLP1 gene that encodes two primary components of myelin. In PMD, brain regions are permanently hypomyelinated and patients suffer from muscle hypotonia, ataxia, and speech problems (76).
However, 11% of patients originally diagnosed with PMD had mutations in the SLC16A2 gene (42), indicating WM status itself is insufficient as a diagnostic marker. In addition, while AHDS is considered primarily as a neuronal cell-based pathology, a criterion for classifying it as a gLE, the glial component remains poorly investigated. It is however almost certain that glial cell types are affected in MCT8 deficiency (see section discovering underlying mechanisms: the oligodendroglial cell and the lack of MCT8), which is a typical hallmark of a leukodystrophy (Table 2). It might therefore be diagnostically more interesting to characterize AHDS as a hypomyelinating disorder (31), allowing it's integration into a new proposed classification system for leukodystrophies (77). As such, severe cases of myelin deficiency or ambiguous MRIs will require additional genetic and serum TH level testing as conclusive diagnostic criteria to discriminate between AHDS and other WM disorders such as PMD (78). That accelerates finding a diagnosis for many undiagnosed patients (78), but is also crucial to anticipate on how and when to therapeutically intervene. An earlier intervention might increase chances of a maximal response (26, 79).
Discovering Underlying Mechanisms: The Oligodendroglial Cell and the Lack of MCT8
A crucial aspect of vertebrate brain evolution was the appearance of myelin, enabling fast electrical conduction across axons spanning increased distances with CNS expansion (28). TH levels peak during neonatal stages (29) during which myelination processes occurs (21, 27). The evolutionary conserved TH signaling pathways are thus an asset for studying how MCT8 deficiency might hamper TH-dependent myelination in animal models and AHDS patients.
Thyroid Hormone Action in Oligodendroglial Cells
In the CNS, myelin is produced by oligodendrocytes, a glial cell type that also provides trophic support for neurons (80). While myelination occurs mainly postnatally in vertebrates, the pool of oligodendrocyte precursor cells (OPCs) is established during pre- and perinatal development. The premature CNS consists of multipotent neural stem cells that generate a pool of OPCs (gliogenic wave) after the neurogenic wave (29). These OPCs can further differentiate into oligodendrocytes, which can mature into fully functional, myelinating oligodendrocytes.
Decades of research revealed that TH action on OPCs and oligodendrocytes is strongly conserved [for a comprehensive review, see (80)]. Classic experiments in the 90s using in vitro OPC cultures derived from optic nerves collected from neonatal rats showed that T3 inhibits OPC proliferation in the presence of mitogens, induces differentiation into mature oligodendrocytes, and facilitates the formation of myelin extensions (81–85). Subsequent experiments showed that T3 stimulates OPC cell cycle exit by binding to the increasingly expressed TH receptor α1, while its interaction with TH receptor β induces timely expression of genes crucial for normal oligodendrogenesis and myelinogenesis (86–88). Important direct target genes include myelin basic protein (MBP), proteolipid protein (PLP) and Krüppel-like factor 9 (KLF9) (89, 90).
Temporal Aspects of Thyroid Dependent Myelination in vivo
The crucial role of TH in promoting oligodendrocyte differentiation in vitro echoes the in vivo need of adequate TH levels for proper oligodendrogenesis in the CNS. Induction of hypothyroidism in postnatal rodents reduced oligodendrocyte numbers and delayed oligodendrocyte maturation, leading to insufficient myelination in several brain regions such as the anterior commissure and the corpus callosum (82, 91, 92). A recent study showed that both transient postnatal (from postnatal day 0 – 21) and permanent hypothyroidism in methimazole-treated rats decreased the number of myelinated axons in the anterior commissure at postnatal day 150, while the number of unmyelinated axons slightly increased (93). Since prenatal hypothyroidism in rats did not alter the total number of axons in the anterior commissure (94), it can be concluded that the ratio of unmyelinated vs. myelinated axons increased. The axons that were myelinated had similar g-ratios (reflecting the ratio of the myelin sheath thickness relative to the axon diameter) (93). This suggests that in TH-depleted conditions, some axons might fail to reach a threshold diameter required to induce myelination (95, 96). Importantly, a short T4 pulse from postnatal day 15–21 during the period of postnatal hypothyroidism did not restore the myelin deficiency at postnatal day 150 (93), emphasizing the vulnerability of myelin integrity to endocrine changes during this crucial developmental window.
The key role of THs in myelination is also evident in human cases of maternal hypothyroxinemia and congenital hypothyroidism. Children born to hypothyroxinemic mothers have significantly smaller corpus callosums (97), demonstrating that maternal THs during the first half of pregnancy, when the fetal thyroid gland is not yet functional, already promote cellular events that later drive myelination. Children with severe congenital hypothyroidism displayed poorer hearing and communication skills that could be directly correlated to disrupted WM microstructural integrity (72). WM abnormalities were present despite early postnatal treatment with levothyroxine, again underlining the importance of the developmental period for normal myelination (72). Lastly, maternal T4 levels during early pregnancy correlate with a higher gray to WM ratio in childhood (98), indicating that reaching optimal WM content requires strictly controlled TH levels. While these studies collectively illustrate that TH signaling during the fetal period is essential for later WM status, myelination is a protracted process particularly in primates (99), expanding the period to which WM development might be susceptible to altered TH levels, or responsive to treatment, up to adolescence.
A Cellular Lack of MCT8 Could Disrupt Oligodendrogenesis
Knowing how TH promotes the generation of myelinating oligodendrocytes allows understanding how a lack of MCT8 could interfere with this process. Several rodent studies showed how MCT8 expression changes in oligodendroglial lineage cells during development. RNA-Seq analysis on the postnatal mouse brain showed enrichment of MCT8 mRNA transcripts primarily in OPCs, but also in newly differentiated oligodendrocytes. In more mature myelinating oligodendrocytes, OATP3A1 was the only potential TH transporter expressed (100). Another transcriptome analysis revealed MCT10 transcripts in mature oligodendrocytes of the postnatal day 21 mouse brain (101). In the developing rat cerebellum, the myelination markers OLIG1 and PDGFRα were expressed strongly at perinatal stages, while MBP mRNA expression and protein levels increased at postnatal day 14 when high levels of circulating THs are detected (21). These temporal expression patterns of important myelin markers coincided with moderately decreased MCT8 mRNA expression and strongly elevated expression levels of DIO2 vs. DIO3 during the first two postnatal weeks, all strictly regulating TH availability (21).
Turning to human evidence, MCT8 was also enriched in immature OPCs and pre-myelinating oligodendrocytes derived from the NKX2.1-GFP human embryonic stem cell (ESC) line, suggesting MCT8 could enable T3 uptake during the maturation phase (90). A transcriptome analysis on human brain cell types also demonstrated moderate MCT8 expression in mature oligodendrocytes (102). Using immunohistochemistry, one paper suggested MCT8 protein expression in oligodendrocytes of healthy human brain tissue (80). Furthermore, inadequate TH uptake due to the lack of a functional MCT8 protein in cultured human ESCs prohibited OPC cell cycle exit and maturation into oligodendrocytes (80). Hence, it is likely that a lack of MCT8 at the OPC level strongly reduces intracellular T3 action, blocking the switch from OPC to mature and myelinating oligodendrocytes (81).
Of note, impaired TH uptake might even occur before TH reaches the oligodendroglial cell membrane. Animal and in vitro models of MCT8 deficiency (see section myelin deficiency in animal models of MCT8 deficiency: permanent or delayed?) indicate a crucial role for MCT8 in TH uptake at the endothelial cells of the blood-brain barrier (BBB), and probably also the blood-cerebrospinal fluid barrier (103–106). In addition, one cannot exclude that a lack of MCT8 also affects OPC generation during embryonic development. Unfortunately, no immunohistochemical data were provided for the presence of MCT8 protein in OPCs in a set of prenatal post mortem human brains, although MCT8 was widely expressed in several other cell types including radial glia progenitors (107). The latter finding was recently also observed in neural progenitors in human brain organoid cultures (106).
The observation that myelination still progresses in most AHDS patients, albeit at a slow rate, implies that intracellular TH levels might however not be completely depleted in the MCT8-deficient brain, in contrast to deeply hypothyroid brains, which probably never reach a normal WM content (72). Since in vitro cellular assays demonstrate that most mutations render the MCT8 protein completely inactive (6), OPCs could be supplied with THs via alternative routes. One possibility is via the T4-selective organic anion transporting polypeptide 1C1 (OATP1C1), expressed in radial glial cells (107) for THs supplied by the cerebrospinal fluid during early development (108). OATP1C1 was not found to be expressed in relevant amounts elsewhere in the human pre- and perinatal brain except for the hypothalamic region (105, 107, 109, 110). Therefore, other transporters, such as MCT10 and the large amino acid transporter 1 (LAT1) and LAT2, for which expression patterns in the developing human brain are still largely unexplored, might play a role in TH uptake at the OPC membrane itself. If such an alternative oligodendroglial TH transporter(s) exists, it should probably be able to take up T3, since the majority of T3 for neurons and other cell types is generated by DIO2 in astrocytes (111). Mouse brain OPCs express some DIO2 (100), but mature oligodendrocytes in the postnatal brain do not (112). So far, there is no data on DIO2 expression in human brain oligodendrocyte lineage cells. Sufficient T3 uptake could induce cell-cycle exit and differentiation of some OPCs promoting slow but gradual myelination. An emerging aspect herein is that the oligodendrocyte differentiation potential changes as function of age (113), questioning whether OPCs still respond the same to cues in later life.
Together, these data suggest that MCT8 is a key cellular component driving TH-dependent oligodendrocyte differentiation and myelination. Impaired oligodendrogenesis caused by MCT8 deficiency could therefore functionally impair nerve conduction and thus, contributing to at least some psychomotor problems observed in AHDS.
Myelin Deficiency in Animal Models of MCT8 Deficiency: Permanent or Delayed?
So far, several genetically altered vertebrates mimic at least some of the characteristics of MCT8 deficiency [for a review, see (114, 115)]. The relevance of mammalian and non-mammalian vertebrate models relies on the fact that TH action during neurodevelopment is highly conserved amongst the vertebrate subphylum (116, 117). The involvement of THs in regulation of the oligodendroglial pathway is universal and conserved throughout evolution (80, 90, 96). Even though some differences might occur in vivo due to imperfect sequence homology, and slight alterations in protein stability and substrate selection, MCT8 orthologues of zebrafish, chicken, mice, and humans possess similar transport characteristics (114, 118). The myelin deficit associated with AHDS was explored in some animal models of MCT8 deficiency.
The Myelin Status in Mouse Models of MCT8 Deficiency
The Mct8 knockout (KO) mouse was the first available model to study the pathophysiological mechanisms underlying AHDS (119). It reproduces the altered TH serum levels found in AHDS patients, making it an excellent model for the peripheral component of the disease (120). However, Mct8 KO mice have a normal brain morphology, a normal myelin content as evaluated on frontal brain sections (121), and only mildly altered neurological and locomotor behavior (119, 122). The current consensus is that other TH transporters, notably the highly enriched OATP1C1 at the murine BBB (105), are able to ensure T4 uptake that via astrocyte-dependent DIO2 provides sufficient T3 to neural cells (119, 120, 123). In contrast, OATP1C1 is only very faintly expressed at the human BBB (105). The generation of Mct8/Oatp1c1 double KO mice was thus essential to replicate the neurological component of AHDS.
Genetic deletion of both Mct8 and Oatp1c1 blocked TH uptake by 90%, rendering mice strongly hypothyroid in the CNS, allowing studying the impact on brain structure and function (121). These Mct8/Oatp1c1 double KO mice displayed pronounced hypomyelination, notably in the corpus callosum as corroborated by the observations of thinner WM tracts (121), consistent with findings in AHDS patients (59). A reduction in FluoroMyelin-stained axons and a lower MBP fluorescent signal intensity on frontal brain sections reflected a hypomyelinated status, suggesting impaired oligodendrocyte differentiation and maturation.
Electron-microscope observations revealed many unmyelinated axons, hampering their conductance, thereby contributing to the neurological deficits and locomotor behavioral impairments (121). The study provided evidence that the myelination deficit is probably permanent given the absence of a normal WM structure at postnatal day 180. Remarkably, the few axons that were myelinated had a normal myelin thickness (121), but whether or not this was directly related to axonal diameter was not investigated. This was also observed in postnatal hypothyroid mice (93), and again indicates that the axonal diameter surpassing a certain threshold is a crucial prerequisite for myelination. This observation suggests that MCT8 deficiency increases the ratio of unmyelinated vs. myelinated axons, but might not affect the myelin sheath itself, i.e., how much myelin per axon is produced. Since ~10% of the normal TH levels were preserved in the brain (121), some oligodendrocytes could thus still take up TH in absence of both transporters, and myelinate axons appropriately. This implies that OPCs express other secondary TH transporters although RNA-Seq analysis of rodent brain cells only showed low expression of several uncharacterized TH transporters (100). Above all, these findings question whether the myelin deficit is a specific problem of a too small axon diameter, or whether is it caused by blocked OPC differentiation, and requires further investigation. Another possibility is that the fraction of oligodendrocytes still able to myelinate under MCT8-deficient conditions represents a unique subpopulation with a specific (epi)genetic signature, or one that functions more independently of MCT8. Purifying these subpopulations and performing high-throughput analysis should provide more answers.
The Dio2/Mct8 double KO mouse represents another interesting model (124, 125). Following T4 uptake by OATP1C1 at the BBB, increased DIO2 activity in astrocytes provides sufficient T3 for transport to neurons and oligodendrocytes (126). Therefore, eliminating both genes depletes T3 levels in various brain regions, in a slightly different manner than the Mct8/Oatp1c1 double KO mouse. If OPCs express TH transporters other than MCT8 and OATP1C1, loss of DIO2 might prevent astrocyte-dependent T3 supply to cells, resulting in more profound intracellular hypothyroidism. In that case, one can expect a more severe myelin deficiency in Dio2/Mct8 double KO mice.
The Myelin Status in the MCT8-deficient Zebrafish
Several zebrafish models were established to investigate the role of TH regulators in development thanks to the zebrafish genetic (127) and physiological similarities with mammalian counterparts, amongst which the thyroid axis [for a review, see (128)]. The myelination process is also strikingly similar to that of mammals. OPCs are generated at day 1 of the 3-day long embryonic development, and following migration form myelinating oligodendrocytes at hatching [i.e., 3 days post-fertilization (3 dpf)]. During this period, a first TH peak occurs (29). Then, myelination starts and myelinated axons are clearly distinguishable in 7 dpf larvae (129, 130). Mct8 is expressed in most oligodendrocytes throughout the zebrafish CNS from 3 dpf onwards (129, 131, 132). The Mct8-deficient zebrafish, first generated by knocking down (131) and then by knocking out mct8 (129), was the first alternative non-mammalian model for AHDS.
The expression of several genes related to oligodendrogenesis was significantly affected in mct8−/− zebrafish. Increased olig2 expression together with decreased mbp, p0, and plpb1 expression during post-hatch development suggested a block in OPC differentiation to mature oligodendrocytes, corroborated by reduced oligodendrocyte numbers in the CNS, including the spinal cord (129, 132). The oligodendrocytes that still developed in a Mct8-absent environment generated fewer extensions (132), suggesting that associated physiological functions could be impaired. Locomotor activity, sleep-awake cycles and responses to light/dark stimuli were abnormal (129), but could have also been caused by microscopic structural alterations of sensory and motor neurons including reduced axon branching and synaptic density. Thus, it remains unknown to what extent behavioral deficits in these mct8−/− fish are a direct consequence of the myelination impairment. An increased number of Schwann cells could indicate that myelination of the peripheral nervous system is hampered too (129). However, the myelin status of nerve axons of the peripheral nervous system has never been explored neither in AHDS patients, nor in any animal model of MCT8 deficiency. Another striking observation was the persistent dysregulated expression of TH-responsive genes related to myelination in the adult mct8−/− zebrafish brain (132), though no data is available on oligodendrocyte differentiation and maturation. It is therefore uncertain whether myelination is delayed or persistent in these mutants, although the term hypomyelination is commonly used.
Future Aspects of Investigating the Myelin Status in Models of MCT8 Deficiency
It is clear that data on myelination phenotypes in animal models of MCT8 deficiency gave preliminary insights into the pathophysiological mechanisms, but so far remain incomplete. We do not know if myelination is delayed or permanent in adult Mct8-deficient zebrafish or 1-year-old Mct8/Oatp1c1 double KO mice, and the myelin status in other models of MCT8 deficiency still requires further examination. A more thorough investigation of myelination in different CNS regions such as the cerebral cortex and spinal cord are necessary to obtain a more detailed picture of myelin progression. The corpus callosum is often investigated as a reference region for brain WM status, because this large structure is easy discernible and primarily composed of interhemispheric WM tracts. However, it is known that under transient hypothyroid conditions, neural stem cells in the adjacent dorsal subventricular zone generate new OPCs that can populate the corpus callosum and provide functional remyelination in 3-months-old mice (133, 134). It therefore remains uncertain to what extent the myelin status of the corpus callosum reflects the WM status of the entire CNS, especially under MCT8-deficient conditions.
Another major unresolved issue is to what extent the lack of MCT8 in both OPCs and oligodendrocytes and the resulting myelin deficit explain motor and behavioral defects. Lack of TH uptake in global knockouts such as the Mct8/Oatp1c1 double KO mouse affects nearly all CNS cell types controlling sensorimotor functions, and therefore do not allow addressing how deficient myelination contributes to altered behavior. Use of single cell type genetic technologies such as conditional KO mice or zebrafish lacking MCT8 or both MCT8 and OATP1C1 (135) in OPCs/oligodendrocytes could enable isolating neuronal or neuro-muscular defects caused by pathologies linked to other cell types. Measuring electric potentials across major axonal tracts might also help to identify impaired neuromuscular communication. Models, such as in vitro induced pluripotent stem cell-based OPC cultures (136, 137) comparing healthy and MCT8-deficient patients (103), or human ESCs (90) have also been used. To these approaches, one could add Crispr-Cas9 techniques to induce specific gene mutations thereby delivering important insights on oligodendrogenesis and myelination, as well as to identify genotype-phenotype relationships. Another powerful in vitro model includes investigating the myelination process in co-cultures of neurons and oligodendrocytes (138).
Therapeutic Strategies to Ameliorate the Myelin Deficit in MCT8 Deficiency
The neurological symptoms associated with MCT8 deficiency severely affect quality of life and decrease life expectancy. Enhancing myelination is a promising endpoint to alleviate many of the neurological symptoms (80, 90). The therapeutic aim is to restore intracellular TH action in TH-deprived cells, while avoiding side effects in other euthyroid or non-target cells. For OPCs and pre-myelinating oligodendrocytes, this strategy aims to normalize expression of key TH-target genes such as KLF9, MPB, PLP, and other candidates to promote proliferation, differentiation and maturation of oligodendroglial cells. Using animal models, different TH analogs could potentially improve or even reverse certain symptoms (104, 129, 139–142). Several of them have also gathered interest as potential therapeutic agents to alleviate the symptoms of AHDS patients. These compounds must meet several a priori requirements. They should be taken up independently of MCT8, have thyromimetic actions by binding to nuclear TH receptors α and β, and be metabolized and excreted in a similar fashion to THs (143). However, the transporter(s) responsible for TH analogue in- and efflux remain(s) to be fully identified.
Table 3 summarizes all available data on myelination following the use of TH analogs and other therapeutic strategies in models of MCT8 deficiency and human patients. Some general trends arise from these pilot experiments. First, physiological doses of T3 alone were not able to mediate any measurable effects on myelination in in vivo models, but remain necessary to confirm the lack of uptake by MCT8 or another secondary transporter. An exception was a high dose of T3 that could rescue oligodendrocyte numbers during zebrafish embryonic development (132), probably because a functional BBB was not yet developed at this stage (145). Irrespectively, T3 will never provide a therapeutic option due to its severe side effects in off target organs, such as the heart and liver. Second, the dosage of TH analogue(s) is critical. Administration of a high (400 nM) but not a low dose (50 nM) of 3,5,3′-triiodothyroacetic acid (Triac) from postnatal day 1 to 12 to Mct8/Oatp1c1 double KO mice improved myelination in the cerebral cortex, as analyzed at postnatal day 12 (139). The dose eventually applied in human trials will be a delicate trade-off between beneficial and adverse effects, and also depends on the half-life of a given compound. In the case of Triac, the half-life is ~6 h in humans (142, 146), which limits its therapeutic use, even though it was already successfully used on a long-term in patients with TH receptor β mutations that suffer from the resistance to TH syndrome (147, 148). Three,5,3′,5′-tetraiodothyroacetic acid, or Tetrac, has been proposed as an alternative TH analogue with a half-life in the order of several days (149), but there is far less experience with this drug in animal research and clinical settings. Thirdly, zebrafish experiments showed that the developmental period during which a drug is administered could differentiate between improved oligodendrogenesis or no effect, as already shown above for T3. For instance, an Mct8-expressing construct in the endothelial cells of the BBB successfully restored oligodendrocyte numbers, but only when it was transfected in later post-hatch development (129, 132).
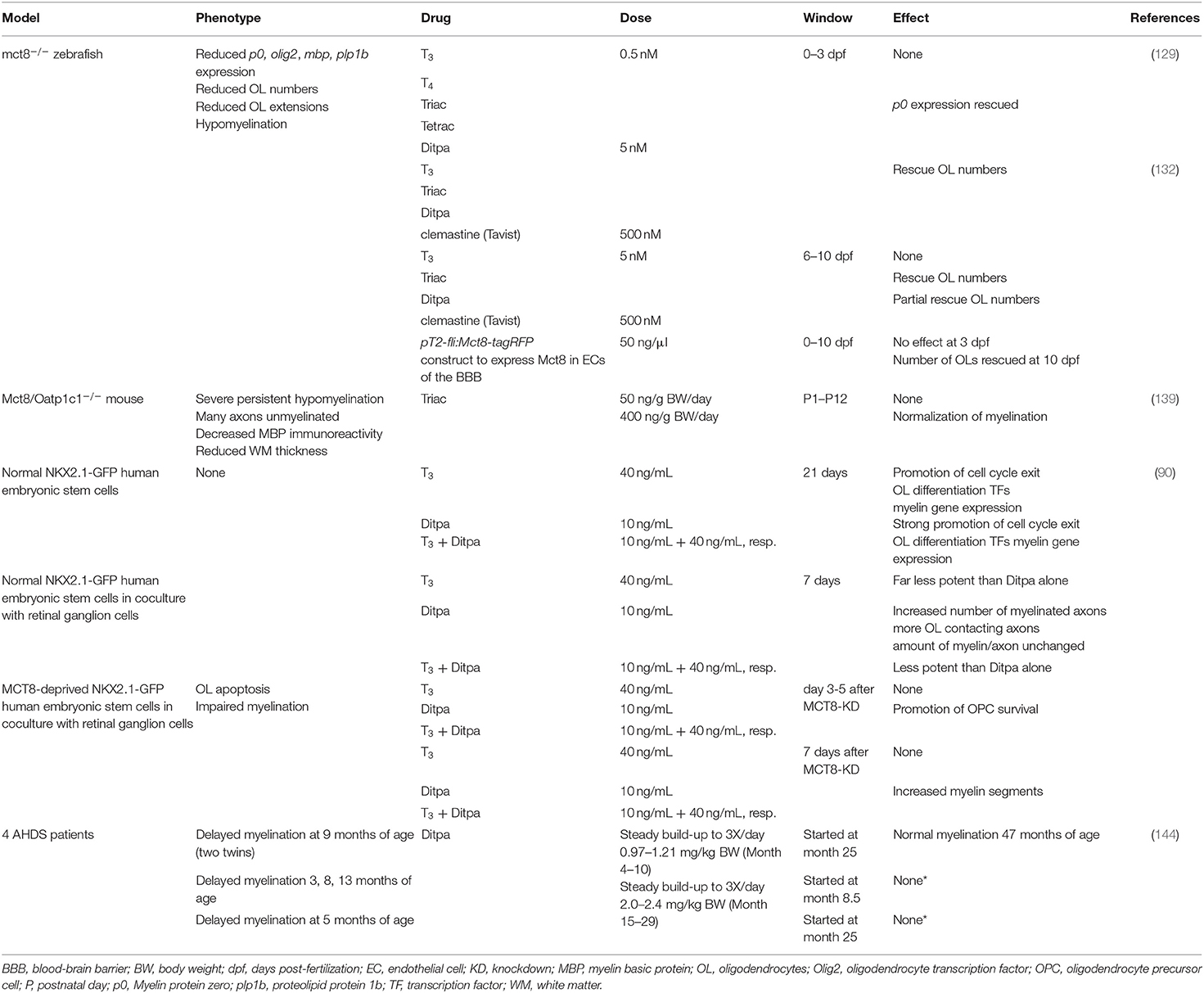
Table 3. Current studies investigating effects of THs, TH analogs and other therapies on the myelination phenotype in models of MCT8 deficiency and human AHDS patients.
Currently, in animal models there are no evaluations of long-term effects following administration of a TH analogue, nor whether rescuing effects at the cellular level translate into improved functional and behavioral outcomes directly related to improved myelination. Here too, evaluating the effects of TH analogue treatments might be more straightforward in conditional KO models. Another promising approach comprised in vitro administration of the TH analogue 3,5-diiodothyropropionic acid or Ditpa, to cultured human NKX2.1-GFP ESCs reprogrammed to become oligodendrocytes (Table 3). Ditpa rescued T3-dependent OPC cell-cycle exit and differentiation into oligodendrocytes that were both absent in MCT8-deprived cultures (90). This indicates that T3 uptake in human OPCs is strongly MCT8-dependent, and that Ditpa is taken up via another TH transporter. Furthermore, Ditpa also inhibited apoptosis of oligodendrocytes and stimulated myelination (90). Importantly, Ditpa crosses the placental barrier in mice (150) showing its potential for prenatal treatment.
The beneficial effects on oligodendrocyte differentiation and myelination of some other treatments that have already been evaluated in other demyelinating diseases [for excellent reviews, see (80, 151)] might hold potential for MCT8 deficiency too, despite different aetiologies. In Mct8 KO zebrafish, early administration of the antihistamine drug clemastine also partially rescued oligodendrocyte numbers and myelin-related gene expression (132) (Table 3). Similarly, clemastine stimulated OPC proliferation, differentiation and oligodendrocyte maturation in the prefrontal cortex of socially isolated mice that suffer from demyelination, and it also successfully promoted remyelination in animal models of the demyelinating disease MS (152) that is characterized by a loss of myelin-forming oligodendrocytes and locomotor deficits (153).
Another promising drug is sobetirome, also called GC-1, and its CNS-selective prodrug Sob-AM2, a methyl amide derivative (154). The latter crosses the BBB and is converted into the active compound by the CNS-specific enzyme fatty acid amide hydrolase (154), limiting side-effects in peripheral organs. An additional advantage of the prodrug is that it produces increased brain sobetirome levels as compared to equimolar doses of sobetirome (155). Sobetirome is a TH receptor β agonist, previously tested as a drug to combat hypercholesterolemia (156). Sobetirome had myelin-promoting effects by stimulating myelin-associated gene expression and oligodendrocyte differentiation in mouse and human in vitro cultures, but also in several mouse brain regions during development (157). The drug was successful in partially remyelinating brain areas in mouse models of MS (158).
Thus, clemastine and sobetirome are two interesting candidates for testing their effects on myelination in MCT8 deficiency models. Sob-AM2 showed already promising thyromimetic effects on gene expression in the brain of Mct8 KO and Mct8/Dio2 double KO mice (159). Other drugs already commercially available or in preparation for testing in MS models (160), or stem cell-based treatments proposed for PMD patients (161) might extend to MCT8 deficiency models too. A last promising therapy would only apply to patients carrying SLC16A2 mutations (e.g., ΔPhe501) that lead to specific MCT8 protein folding defects and reduced uptake activity. In vitro experiments showed that the drug phenylbutyrate can stabilize the misfolded MCT8 protein and restore TH uptake (71). It will be interesting to see whether this compound can rescue the myelin deficit and neurological manifestations in mouse models carrying these specific mutations.
Turning to clinical trials, TH analogs have shown potential in AHDS patients to combat the peripheral effects of thyrotoxicosis [e.g., (144, 162) and Triac trial I: NCT02060474], but have so far proven largely inadequate in improving patients' mental status. However, the youngest patients might still benefit from treatment: a clinical trial using Triac in the youngest new-borns is currently underway (Triac trial II: NCT02396459). Other TH analogs were tested in small cohorts of AHDS patients, but only one study included an examination of the myelin status (144). Two out of 4 patients that received increasing doses of Ditpa from 2 years on, showed improved myelination at 47 months of age (144) (Table 3). However, given that myelination improves in many patients during the early years of childhood (Figures 1, 2), the authors could not conclude whether Ditpa was responsible for this progress. Nevertheless, restoring TH action could relieve the quasi-blocked oligodendroglial maturation and myelination at the cellular level, and thus it seems interesting to incorporate this parameter into a more extensive study like the Triac trial that includes more detailed MRI examinations in a larger cohort.
It is also unknown to what extent the embryonic neural stem cell and OPC populations are affected by the central hypothyroidism caused by the mutated MCT8 protein. In mice, the pre- and perinatal period includes the generation of the OPC pool that will persist throughout life within the parenchyma and occurs before and during a postnatal TH peak (29). In the human brain, many cell types express regulators of cellular TH availability from embryonic stages onwards (107). Thus, shifting the treatment window toward a period before birth could increase the efficacy of TH-replacement therapies. Triac has been shown to cross the placental barrier in pregnant women (13), but there are scarce data regarding safety and dose range. In one case, prenatal Triac therapy successfully treated fetal hypothyroidism due to resistance to TH syndrome, although some complications occurred at birth (163). Apart from the risk of major side effects, ethical and methodological issues are raised for human use. Treatments should target and therefore ameliorate the CNS phenotype to avoid worsening the condition of other body organs that suffer from thyrotoxicosis. Furthermore, epidemiological data indicate that subtle hypo- and hyperthyroidism during CNS development can lead to adverse effects on gray matter and WM content (98). Further research along the entire developmental period is necessary to understand the interplay between THs and MCT8 in the oligodendroglial lineage and how that mediates myelination.
Conclusions
This review underlines the degree of inconsistency existing in the literature on how to classify AHDS amongst the diverse group of WM disorders. One the one hand, this is a consequence of variation amongst patient cases, most showing a gradual improvement of myelination, some still reporting WM abnormalities after several years. On the other hand, MRI studies are not always performed in detail, and follow-up data is often missing. The increasing numbers of case reports in which high resolution, sequential MRIs are performed will progressively give a clearer view on myelin deficits in the disease and whether subtle WM micro-abnormalities are still present later in life. The growing case numbers of AHDS patients identified will also provide more information on patient heterogeneity. Longitudinal assessment of the myelination process in animal models of MCT8 deficiency can further clarify the underlying cellular and molecular mechanisms. Models are also needed to evaluate the translational potential of therapeutic drugs, such as the TH analogs. Here, reciprocal exchanges of new therapeutic evolvements used in other demyelinating diseases sharing some of the hallmarks of AHDS, such as MS or PMD will be particularly edifying. Lastly, comparing data from basic and clinical research assist this same goal, thereby paving the way to improve the health and life span of AHDS patients. Such input will also help those involved in their daily care.
Author Contributions
PV worked out the concept of the paper and made the figures and tables. PV, BD, and SR wrote the paper.
Funding
PV was supported by a basic science grant from the European Thyroid Association and a grant (SPF201909009111) from the Fondation pour la Recherche Médicale. This work was also supported by the Centre national de la recherche scientifique (CNRS) and the EU H2020 contract Thyrage (Grant n°666869).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AHDS, Allan-Herndon-Dudley Syndrome; BBB, blood-brain barrier; CNS, central nervous system; DIO, deiodinase; Ditpa, 3,5-diiodothyropropionic acid; dpf, days post-fertilization; ESC, embryonic stem cell; gLE, genetic leukoencephalopathy; KLF9, Krüppel-like factor 9; KO, knockout; LAT, large amino acid transporter; MBP, myelin basic protein; MCT, monocarboxylate transporter; MRI, magnetic resonance imaging; MS, multiple sclerosis; OATP1C1, organic anion transporting polypeptide 1C1; OPC, oligodendrocyte precursor cell; PET, positron emission tomography; PLP, proteolipid protein; PMD, Pelizaeus-Merzbacher Disease; SLC16A2, solute carrier 16A2; T3, 3,5,3′-triiodothyronine; T4, 3,5,3′,5′-tetraiodothyronine; TH, thyroid hormone; Triac, 3,5,3′-triiodothyroacetic acid; Tetrac, 3,5,3′,5′-tetraiodothyroacetic acid; WM, white matter.
References
1. Allan W, Herndon CN, Dudley FC. Some examples of the inheritance of mental deficiency: apparently sex-linked iodicy and microcephaly. Am J Ment Defic. (1944) 48:325–34.
2. Schwartz CE, Stevenson RE. The MCT8 thyroid hormone transporter and Allan–Herndon–Dudley syndrome. Best Pract Res Clin Endocrinol Metab. (2007) 21:307–21. doi: 10.1016/j.beem.2007.03.009
3. Dumitrescu AM, Liao X-H, Best TB, Brockmann K, Refetoff S. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet. (2004) 74:168–75. doi: 10.1086/380999
4. Friesema EC, Grueters A, Biebermann H, Krude H, von Moers A, Reeser M, et al. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet. (2004) 364:1435–7. doi: 10.1016/S0140-6736(04)17226-7
5. Friesema ECH, Ganguly S, Abdalla A, Fox JEM, Halestrap AP, Visser TJ. Identification of monocarboxylate transporter 8 as a specific thyroid hormone transporter. J Biol Chem. (2003) 278:40128–35. doi: 10.1074/jbc.M300909200
6. Groeneweg S, van Geest FS, Peeters RP, Heuer H, Visser WE. Thyroid hormone transporters. Endocr Rev. (2020) 41:146–201. doi: 10.1210/endrev/bnz008
7. Groeneweg S, Visser WE, Visser TJ. Disorder of thyroid hormone transport into the tissues. Best Pract Res Clin Endocrinol Metab. (2017) 31:241–53. doi: 10.1016/j.beem.2017.05.001
8. Visser WE, Vrijmoeth P, Visser FE, Arts WFM, van Toor H, Visser TJ. Identification, functional analysis, prevalence and treatment of monocarboxylate transporter 8 (MCT8) mutations in a cohort of adult patients with mental retardation. Clin Endocrinol. (2013) 78:310–5. doi: 10.1111/cen.12023
9. Gereben B, Zeöld A, Dentice M, Salvatore D, Bianco AC. Activation and inactivation of thyroid hormone by deiodinases: local action with general consequences. Cell Mol Life Sci. (2008) 65:570–90. doi: 10.1007/s00018-007-7396-0
10. Bernal J. Thyroid hormone regulated genes in cerebral cortex development. J Endocrinol. (2017) 232:R83–97. doi: 10.1530/JOE-16-0424
11. Chatonnet F, Flamant F, Morte B. A temporary compendium of thyroid hormone target genes in brain. Biochim Biophys Acta–Gene Regul Mech. (2015) 1849:122–9. doi: 10.1016/j.bbagrm.2014.05.023
12. Grøntved L, Waterfall JJ, Kim DW, Baek S, Sung MH, Zhao L, et al. Transcriptional activation by the thyroid hormone receptor through ligand-dependent receptor recruitment and chromatin remodelling. Nat Commun. (2015) 6:7048. doi: 10.1038/ncomms8048
13. Martinez ME, Duarte CW, Stohn JP, Karaczyn A, Wu Z, DeMambro VE, et al. Thyroid hormone influences brain gene expression programs and behaviors in later generations by altering germ line epigenetic information. Mol Psychiatry. (2018) 25:939–50. doi: 10.1038/s41380-018-0281-4
14. Spalding KL, Bergmann O, Alkass K, Bernard S, Salehpour M, Huttner HB, et al. Dynamics of hippocampal neurogenesis in adult humans. Cell. (2013) 153:1219–27. doi: 10.1016/j.cell.2013.05.002
15. Ernst A, Alkass K, Bernard S, Salehpour M, Perl S, Tisdale J, et al. Neurogenesis in the striatum of the adult human brain. Cell. (2014) 156:1072–83. doi: 10.1016/j.cell.2014.01.044
16. Moreno-Jiménez EP, Flor-García M, Terreros-Roncal J, Rábano A, Cafini F, Pallas-Bazarra N, et al. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer's disease. Nat Med. (2019) 25:554–60. doi: 10.1038/s41591-019-0375-9
17. Sanai N, Tramontin AD, Quiñones-Hinojosa A, Barbaro NM, Gupta N, Kunwar S, et al. Unique astrocyte ribbon in adult human brain contains neural stem cells but lacks chain migration. Nature. (2004) 427:740–4. doi: 10.1038/nature02301
18. Zoeller RT, Rovet J. Timing of thyroid hormone action in the developing brain: clinical observations and experimental findings. J Neuroendocrinol. (2004) 16:809–18. doi: 10.1111/j.1365-2826.2004.01243.x
19. Bernal J. Thyroid hormone receptors in brain development and function. Nat Clin Pract Endocrinol Metab. (2007) 3:249–59.
20. Moog NK, Entringer S, Heim C, Wadhwa PD, Kathmann N, Buss C. Influence of maternal thyroid hormones during gestation on fetal brain development. Neuroscience. (2017) 342:68–100. doi: 10.1016/j.neuroscience.2015.09.070
21. Calzà L, Fernández M, Giardino L. Role of the thyroid system in myelination and neural connectivity. Compr. Physiol. (2015) 5:1405–21. doi: 10.1002/cphy.c140035
22. Lee S, Leach MK, Redmond SA, Chong SYC, Mellon SH, Tuck SJ, et al. A culture system to study oligodendrocyte myelination processes using engineered nanofibers. Nat Methods. (2012) 9:917–22. doi: 10.1038/nmeth.2105
23. Alizadeh A, Dyck SM, Karimi-Abdolrezaee S. Myelin damage and repair in pathologic CNS: challenges and prospects. Front Mol Neurosci. (2015) 8:35. doi: 10.3389/fnmol.2015.00035
24. van Tilborg E, de Theije CGM, van Hal M, Wagenaar N, de Vries LS, Benders MJ, et al. Origin and dynamics of oligodendrocytes in the developing brain: implications for perinatal white matter injury. Glia. (2018) 66:221–38. doi: 10.1002/glia.23256
25. Schiffmann R, Van Der Knaap MS. Invited Article: an MRI-based approach to the diagnosis of white matter disorders. Neurology. (2009) 72: 750–9. doi: 10.1212/01.wnl.0000343049.00540.c8
26. Pouwels PJW, Vanderver A, Bernard G, Wolf NI, Dreha-Kulczewksi SF, Deoni SCL, et al. Hypomyelinating leukodystrophies: translational research progress and prospects. Ann Neurol. (2014) 76:5–19. doi: 10.1002/ana.24194
27. Zalc B, Goujet D, Colman D. The origin of the myelination program in vertebrates. Curr Biol. (2008) 18:R511–2. doi: 10.1016/j.cub.2008.04.010
28. Stassart RM, Möbius W, Nave K-A, Edgar JM. The axon-myelin unit in development and degenerative disease. Front Neurosci. (2018) 12:467. doi: 10.3389/fnins.2018.00467
29. Gothié J, Vancamp P, Demeneix B, Remaud S. Thyroid hormone regulation of neural stem cell fate: from development to ageing. Acta Physiol. (2020) 228:e13316. doi: 10.1111/apha.13316
30. Jagannathan NR, Tandon N, Raghunathan P, Kochupillai N. Reversal of abnormalities of myelination by thyroxine therapy in congenital hypothyroidism: localized in vivo proton magnetic resonance spectroscopy (MRS) study. Dev Brain Res. (1998) 109:179–86. doi: 10.1016/S0165-3806(98)00081-9
31. Charzewska A, Wierzba J, Izycka-Swieszewska E, Bekiesinska-Figatowska M, Jurek M, Gintowt A, et al. Hypomyelinating leukodystrophies—a molecular insight into the white matter pathology. Clin Genet. (2016) 90:293–304. doi: 10.1111/cge.12811
32. Remerand G, Boespflug-Tanguy O, Tonduti D, Touraine R, Rodriguez D, Curie A, et al. Expanding the phenotypic spectrum of Allan–Herndon–Dudley syndrome in patients with SLC 16A2 mutations. Dev Med Child Neurol. (2019) 61:1439–47. doi: 10.1111/dmcn.14332
33. Vanderver A, Prust M, Tonduti D, Mochel F, Hussey HM, Helman G, et al. Case definition and classification of leukodystrophies and leukoencephalopathies. Mol Genet Metab. (2015) 114:494–500. doi: 10.1016/j.ymgme.2015.01.006
34. Azzolini S, Nosadini M, Balzarin M, Sartori S, Suppiej A, Mardari R, et al. Delayed myelination is not a constant feature of Allan–Herndon–Dudley syndrome: report of a new case and review of the literature. Brain Dev. (2014) 36:716–20. doi: 10.1016/j.braindev.2013.10.009
35. Holden KR, Zuñiga OF, May MM, Su H, Molinero MR, Rogers RC, et al. X-linked MCT8 gene mutations: characterization of the pediatric neurologic phenotype. J Child Neurol. (2005) 20:852–7. doi: 10.1177/08830738050200101601
36. Kakinuma H, Itoh M, Takahashi H. A novel mutation in the monocarboxylate transporter 8 gene in a boy with putamen lesions and low free T4 levels in cerebrospinal fluid. J Pediatr. (2005) 147:552–4. doi: 10.1016/j.jpeds.2005.05.012
37. Schwartz CE, May MM, Carpenter NJ, Rogers RC, Martin J, Bialer MG, et al. Allan-Herndon-Dudley syndrome and the monocarboxylate transporter 8 (MCT8) gene. Am J Hum Genet. (2005) 77:41–53. doi: 10.1086/431313
38. Namba N, Etani Y, Kitaoka T, Nakamoto Y, Nakacho M, Bessho K, et al. Clinical phenotype and endocrinological investigations in a patient with a mutation in the MCT8 thyroid hormone transporter. Eur J Pediatr. (2008) 167:785–91. doi: 10.1007/s00431-007-0589-6
39. Papadimitriou A, Dumitrescu AM, Papavasiliou A, Fretzayas A, Nicolaidou P, Refetoff S. A novel monocarboxylate transporter 8 gene mutation as a cause of severe neonatal hypotonia and developmental delay. Pediatrics. (2008) 121:e199–202. doi: 10.1542/peds.2007-1247
40. Sijens PE, Rödiger LA, Meiners LC, Lunsing RJ. 1H magnetic resonance spectroscopy in monocarboxylate transporter 8 gene deficiency. J Clin Endocrinol Metab. (2008) 93:1854–9. doi: 10.1210/jc.2007-2441
41. Fuchs O, Pfarr N, Pohlenz J SH. Elevated serum triiodothyronine and intellectual and motor disability with paroxysmal dyskinesia caused by a monocarboxylate transporter 8 gene mutation. Dev Med Child Neurol. (2009) 51:240–4. doi: 10.1111/j.1469-8749.2008.03125.x
42. Vaurs-Barrière C, Deville M, Sarret C, Giraud G, Des Portes V, Prats-Viñas J-M, et al. Pelizaeus-Merzbacher-Like disease presentation of MCT8 mutated male subjects. Ann Neurol. (2009) 65:114–8. doi: 10.1002/ana.21579
43. Boccone L, Mariotti S, Dessì V, Pruna D, Meloni A, Loudianos G. Allan–Herndon–Dudley syndrome (AHDS) caused by a novel SLC16A2 gene mutation showing severe neurologic features and unexpectedly low TRH-stimulated serum TSH. Eur J Med Genet. (2010) 53:392–5. doi: 10.1016/j.ejmg.2010.08.001
44. Gika AD, Siddiqui A, Hulse AJ, Edward S, Fallon P, McEntagart ME, et al. White matter abnormalities and dystonic motor disorder associated with mutations in the SLC16A2 gene. Dev Med Child Neurol. (2009) 52:475–82. doi: 10.1111/j.1469-8749.2009.03471.x
45. Crushell E, Reardon W. Elevated TSH levels in a mentally retarded boy. Eur J Pediatr. (2010) 169:573–5. doi: 10.1007/s00431-009-1075-0
46. Tsurusaki Y, Osaka H, Hamanoue H, Shimbo H, Tsuji M, Doi H, et al. Rapid detection of a mutation causing X-linked leucoencephalopathy by exome sequencing. J Med Genet. (2011) 48:606–9. doi: 10.1136/jmg.2010.083535
47. Zung A, Visser TJ, Uitterlinden AG, Rivadeneira F, Friesema ECH. A child with a deletion in the monocarboxylate transporter 8 gene: 7-years follow-up and effects of thyroid hormone treatment. Eur J Endocrinol. (2011) 165:823–30. doi: 10.1530/EJE-11-0358
48. Tonduti D, Vanderver A, Berardinelli A, Schmidt JL, Collins CD, Novara F, et al. MCT8 deficiency. J Child Neurol. (2013) 28:795–800. doi: 10.1177/0883073812450944
49. Boccone L, Dessì V, Meloni A, Loudianos G. Allan–Herndon–Dudley syndrome (AHDS) in two consecutive generations caused by a missense MCT8 gene mutation. Phenotypic variability with the presence of normal serum T3 levels. Eur J Med Genet. (2013) 56:207–10. doi: 10.1016/j.ejmg.2013.02.001
50. Anik A, Kersseboom S, Demir K, Çatli G, Yiş U, Böber E, et al. Psychomotor retardation caused by a defective thyroid hormone transporter: report of two families with different MCT8 mutations. Horm Res Paediatr. (2014) 82:261–71. doi: 10.1159/000365191
51. Kobayashi S, Onuma A, Inui T, Wakusawa K, Tanaka S, Shimojima K, et al. Clinical course and images of four familial cases of Allan-Herndon-Dudley syndrome with a novel monocarboxylate transporter 8 gene mutation. Pediatr Neurol. (2014) 51:414–6. doi: 10.1016/j.pediatrneurol.2014.05.004
52. López-Espíndola D, Morales-Bastos C, Grijota-Martínez C, Liao XH, Lev D, Sugo E, et al. Mutations of the thyroid hormone transporter MCT8 cause prenatal brain damage and persistent hypomyelination. J Clin Endocrinol Metab. (2014) 99:E2799–804. doi: 10.1210/jc.2014-2162
53. Rodrigues F, Grenha J, Ortez C, Nascimento A, Morte B, M-Belinchón M, et al. Hypotonic male infant and MCT8 deficiency–a diagnosis to think about. BMC Pediatr. (2014) 14:252. doi: 10.1186/1471-2431-14-252
54. Yamamoto T, Shimojima K, Umemura A, Uematsu M, Nakayama T, Inoue K. SLC16A2 mutations in two Japanese patients with Allan–Herndon–Dudley syndrome. Hum Genome Var. (2014) 1:14010. doi: 10.1038/hgv.2014.10
55. Armour CM, Kersseboom S, Yoon G, Visser TJ. Further insights into the Allan-Herndon-Dudley syndrome: clinical and functional characterization of a novel MCT8 mutation. PLoS ONE. (2015) 10:e0139343. doi: 10.1371/journal.pone.0139343
56. Gagliardi L, Nataren N, Feng J, Schreiber AW, Hahn CN, Conwell LS, et al. Allan-Herndon-Dudley syndrome with unusual profound sensorineural hearing loss. Am J Med Genet A. (2015) 167A:1872–6. doi: 10.1002/ajmg.a.37075
57. Kim JH, Kim YM, Yum MS, Choi JH, Lee BH, Kim GH, et al. Clinical and endocrine features of two allan-herndon-dudley syndrome patients with monocarboxylate transporter 8 mutations. Horm Res Paediatr. (2015) 83:288–92. doi: 10.1159/000371466
58. La Piana R, Vanasse M, Brais B, Bernard G. Myelination delay and Allan-Herndon-Dudley syndrome caused by a novel mutation in the SLC16A2 gene. J Child Neurol. (2015) 30:1371–4. doi: 10.1177/0883073814555189
59. Matheus MG, Lehman RK, Bonilha L, Holden KR. Redefining the pediatric phenotype of X-linked monocarboxylate transporter 8 (MCT8) deficiency. J Child Neurol. (2015) 30:1664–8. doi: 10.1177/0883073815578524
60. Bedoyan JK, Tim-aroon T, Deeb KK, Ganganna ST, Bass NE. The value of comprehensive thyroid function testing and family history for early diagnosis of MCT8 deficiency. Clin Pediatr. (2016) 55:286–9. doi: 10.1177/0009922815584219
61. Ono E, Ariga M, Oshima S, Hayakawa M, Imai M, Ochiai Y, et al. Three novel mutations of the MCT8 (SLC16A2) gene: individual and temporal variations of endocrinological and radiological features. Clin Pediatr Endocrinol. (2016) 25:23–35. doi: 10.1297/cpe.25.23
62. Shimojima K, Maruyama K, Kikuchi M, Imai A, Inoue K, Yamamoto T. Novel SLC16A2 mutations in patients with Allan-Herndon-Dudley syndrome. Intractable Rare Dis Res. (2016) 5:214–7. doi: 10.5582/irdr.2016.01051
63. Novara F, Groeneweg S, Freri E, Estienne M, Reho P, Matricardi S, et al. Clinical and molecular characteristics of SLC16A2 (MCT8) mutations in three families with the Allan-Herndon-Dudley syndrome. Hum Mutat. (2017) 38:260–4. doi: 10.1002/humu.23140
64. Rego T, Lado CG, Rodríguez PC, Santos FS, Angueira FB, Castro-Feijóo L, et al. Severe neurological abnormalities in a young boy with impaired thyroid hormone sensitivity due to a novel mutation in the MCT8 gene. Hormones. (2017) 16:194–9. doi: 10.14310/horm.2002.1733
65. Masnada S, Groenweg S, Saletti V, Chiapparini L, Castellotti B, Salsano E, et al. Novel mutations in SLC16A2 associated with a less severe phenotype of MCT8 deficiency. Metab Brain Dis. (2019) 34:1565–75. doi: 10.1007/s11011-019-00464-7
66. Frohman EM, Racke MK, Raine CS. Multiple sclerosis—the plaque and its pathogenesis. N Engl J Med. (2006) 354:942–55. doi: 10.1056/NEJMra052130
67. Braun D, Schweizer U. Efficient activation of pathogenic ΔPhe501 mutation in monocarboxylate transporter 8 by chemical and pharmacological chaperones. Endocrinology. (2015) 156:4720–30. doi: 10.1210/en.2015-1393
68. Jansen J, Friesema ECH, Kester MHA, Schwartz CE, Visser TJ. Genotype-phenotype relationship in patients with mutations in thyroid hormone transporter MCT8. Endocrinology. (2008) 149:2184–90. doi: 10.1210/en.2007-1475
69. Groeneweg S, Friesema ECH, Kersseboom S, Klootwijk W, Visser WE, Peeters RP, et al. The role of Arg445 and Asp498 in the human thyroid hormone transporter MCT8. Endocrinology. (2014) 155:618–26. doi: 10.1210/en.2013-1521
70. Wang J, Zhang Q, Bao X, Chen Y, Yu S. Clinical and genetic features of five patients with Allan-Herndon-Dudley syndrome. Chinese J Med Genet. (2018) 35:484–8. doi: 10.3760/cma.j.issn.1003-9406.2018.04.005
71. Braun D, Schweizer U. The chemical chaperone phenylbutyrate rescues MCT8 mutations associated with milder phenotypes in patients with AHDS. Endocrinology. (2016) 158:678–91. doi: 10.1210/en.2016-1530
72. Cooper HE, Kaden E, Halliday LF, Bamiou D-E, Mankad K, Peters C, et al. White matter microstructural abnormalities in children with severe congenital hypothyroidism. NeuroImage Clin. (2019) 24:101980. doi: 10.1016/j.nicl.2019.101980
73. Bodini B, Veronese M, García-Lorenzo D, Battaglini M, Poirion E, Chardain A, et al. Dynamic imaging of individual remyelination profiles in multiple sclerosis. Ann Neurol. (2016) 79:726–38. doi: 10.1002/ana.24620
74. Barkovich AJ, Deon S. Hypomyelinating disorders: an MRI approach. Neurobiol Dis. (2016) 87:50–8. doi: 10.1016/j.nbd.2015.10.015
75. van der Knaap MS, Wolf NI. Hypomyelination versus delayed myelination. Ann Neurol. (2009) 68:115. doi: 10.1002/ana.21751
76. Hobson G, Garbern J. Pelizaeus-merzbacher disease, pelizaeus-merzbacher-like disease 1, and related hypomyelinating disorders. Semin Neurol. (2012) 32:62–7. doi: 10.1055/s-0032-1306388
77. van der Knaap MS, Bugiani M. Leukodystrophies: a proposed classification system based on pathological changes and pathogenetic mechanisms. Acta Neuropathol. (2017) 134:351–82. doi: 10.1007/s00401-017-1739-1
78. Parikh S, Bernard G, Leventer RJ, van der Knaap MS, van Hove J, Pizzino A, et al. A clinical approach to the diagnosis of patients with leukodystrophies and genetic leukoencephelopathies. Mol Genet Metab. (2015) 114:501–15. doi: 10.1016/j.ymgme.2014.12.434
79. van der Knaap MS, Bugiani M. Leukodystrophies—much more than just diseases of myelin. Nat Rev Neurol. (2018) 14:747–8 doi: 10.1038/s41582-018-0093-9
80. Kim MJ, Petratos S. Oligodendroglial lineage cells in thyroid hormone-deprived conditions. Stem Cells Int. (2019) 2019:1–13. doi: 10.1155/2019/5496891
81. Barres BA, Lazar MA, Raff MC. A novel role for thyroid hormone, glucocorticoids and retinoic acid in timing oligodendrocyte development. Development. (1994) 120:1097–108.
82. Rodriguez-Peña A, Ibarrola N, Iñiguez MA, Muñoz A, Bernal J. Neonatal hypothyroidism affects the timely expression of myelin-associated glycoprotein in the rat brain. J Clin Invest. (1993) 91:812–8. doi: 10.1172/JCI116301
83. Johe KK, Hazel TG, Muller T, Dugich-Djordjevic MM, McKay RDG. Single factors direct the differentiation of stem cells from the fetal and adult central nervous system. Genes Dev. (1996) 10:3129–40. doi: 10.1101/gad.10.24.3129
84. Ahlgren SC, Wallace H, Bishop J, Neophytou C, Raff MC. Effects of thyroid hormone on embryonic oligodendrocyte precursor cell development in vivo and in vitro. Mol Cell Neurosci. (1997) 9:420–32. doi: 10.1006/mcne.1997.0631
85. Baas D, Bourbeau D, Sarliève LL, Ittel ME, Dussault JH, Puymirat J. Oligodendrocyte maturation and progenitor cell proliferation are independently regulated by thyroid hormone. Glia. (1997) 19:324–32. doi: 10.1002/(SICI)1098-1136(199704)19:4<324::AID-GLIA5>3.0.CO;2-X
86. Billon N, Tokumoto Y, Forrest D, Raff M. Role of thyroid hormone receptors in timing oligodendrocyte differentiation. Dev Biol. (2001) 235:110–20. doi: 10.1006/dbio.2001.0293
87. Billon N. Normal timing of oligodendrocyte development depends on thyroid hormone receptor alpha 1 (TRalpha1). EMBO J. (2002) 21:6452–60. doi: 10.1093/emboj/cdf662
88. Younes-Rapozo V, Berendonk J, Savignon T, Manhães AC, Barradas PC. Thyroid hormone deficiency changes the distribution of oligodendrocyte/myelin markers during oligodendroglial differentiation in vitro. Int J Dev Neurosci. (2006) 24:445–53. doi: 10.1016/j.ijdevneu.2006.08.004
89. Dugas JC, Ibrahim A, Barres BA. The T3-induced gene KLF9 regulates oligodendrocyte differentiation and myelin regeneration. Mol Cell Neurosci. (2012) 50:45–57. doi: 10.1016/j.mcn.2012.03.007
90. Lee JY, Petratos S. Thyroid hormone signaling in oligodendrocytes: from extracellular transport to intracellular signal. Mol Neurobiol. (2016) 53:6568–83. doi: 10.1007/s12035-016-0013-1
91. Berbel P, Guadan~o-Ferraz A, Angulo A, Cerezo J. Role of thyroid hormones in the maturation of interhemispheric connections in rats. Behav Brain Res. (1994) 64:9–14. doi: 10.1016/0166-4328(94)90114-7
92. Schoonover CM, Seibel MM, Jolson DM, Stack MJ, Rahman RJ, Jones SA, et al. Thyroid hormone regulates oligodendrocyte accumulation in developing rat brain white matter tracts. Endocrinology. (2004) 145:5013–20. doi: 10.1210/en.2004-0065
93. Lucia FS, Pacheco-Torres J, González-Granero S, Canals S, Obregón M-J, García-Verdugo JM, et al. Transient hypothyroidism during lactation arrests myelination in the anterior commissure of rats. A magnetic resonance image and electron microscope study. Front Neuroanat. (2018) 12:31. doi: 10.3389/fnana.2018.00031
94. Ferraz AG, del Rey FE, de Escobar GM, Innocenti GM, Berbel P. The development of the anterior commissure in normal and hypothyroid rats. Dev Brain Res. (1994) 81:293–308. doi: 10.1016/0165-3806(94)90315-8
95. Waxman SG, Bennett MVL. Relative conduction velocities of small myelinated and non-myelinated fibres in the central nervous system. Nat New Biol. (1972) 238:217–9. doi: 10.1038/newbio238217a0
97. Samadi A, Skocic J, Rovet JF. Children born to women treated for hypothyroidism during pregnancy show abnormal corpus callosum development. Thyroid. (2015) 25:494–502. doi: 10.1089/thy.2014.0548
98. Korevaar TIM, Muetzel R, Medici M, Chaker L, Jaddoe VWV, de Rijke YB, et al. Association of maternal thyroid function during early pregnancy with offspring IQ and brain morphology in childhood: a population-based prospective cohort study. Lancet Diabetes Endocrinol. (2016) 4:35–43. doi: 10.1016/S2213-8587(15)00327-7
99. Miller DJ, Duka T, Stimpson CD, Schapiro SJ, Baze WB, McArthur MJ, et al. Prolonged myelination in human neocortical evolution. Proc Natl Acad Sci USA. (2012) 109:16480–5. doi: 10.1073/pnas.1117943109
100. Bernal J, Guadaño-Ferraz A, Morte B. Thyroid hormone transporters-functions and clinical implications. Nat Rev Endocrinol. (2015) 11:406–17. doi: 10.1038/nrendo.2015.66
101. Müller J, Heuer H. Expression pattern of thyroid hormone transporters in the postnatal mouse brain. Front Endocrinol. (2014) 5:92. doi: 10.3389/fendo.2014.00092
102. Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron. (2016) 89:37–53. doi: 10.1016/j.neuron.2015.11.013
103. Vatine GD, Al-Ahmad A, Barriga BK, Svendsen S, Salim A, Garcia L, et al. Modeling psychomotor retardation using iPSCs from MCT8-deficient patients indicates a prominent role for the blood-brain barrier. Cell Stem Cell. (2017) 20:831–43.e5. doi: 10.1016/j.stem.2017.04.002
104. Ceballos A, Belinchon MM, Sanchez-Mendoza E, Grijota-Martinez C, Dumitrescu AM, Refetoff S, et al. Importance of monocarboxylate transporter 8 for the blood-brain barrier-dependent availability of 3,5,3′ -triiodo-L-thyronine. Endocrinology. (2009) 150:2491–6. doi: 10.1210/en.2008-1616
105. Roberts LM, Woodford K, Zhou M, Black DS, Haggerty JE, Tate EH, et al. Expression of the thyroid hormone transporters monocarboxylate transporter-8 (SLC16A2) and organic ion transporter-14 (SLCO1C1) at the blood-brain barrier. Endocrinology. (2008) 149:6251–61. doi: 10.1210/en.2008-0378
106. Wilpert N-M, Krueger M, Opitz R, Sebinger D, Paisdzior S, Mages B, et al. Spatiotemporal changes of cerebral monocarboxylate transporter 8 expression. Thyroid. (2020). doi: 10.1089/thy.2019.0544. [Epub ahead of print].
107. López-Espíndola D, García-Aldea Á, Gómez de la Riva I, Rodríguez-García AM, Salvatore D, Visser TJ, et al. Thyroid hormone availability in the human fetal brain: novel entry pathways and role of radial glia. Brain Struct Funct. (2019) 224:2103–19. doi: 10.1007/s00429-019-01896-8
108. Quignodon L, Legrand C, Allioli N, Guadaño-Ferraz A, Bernal J, Samarut J, et al. Thyroid hormone signaling is highly heterogeneous during pre- and postnatal brain development. J Mol Endocrinol. (2004) 33:467–76. doi: 10.1677/jme.1.01570
109. Chan S-Y, Martín-Santos A, Loubière LS, González AM, Stieger B, Logan A, et al. The expression of thyroid hormone transporters in the human fetal cerebral cortex during early development and in N-Tera-2 neurodifferentiation. J Physiol. (2011) 589:2827–45. doi: 10.1113/jphysiol.2011.207290
110. Alkemade A, Friesema ECH, Kalsbeek A, Swaab DF, Visser TJ, Fliers E. Expression of thyroid hormone transporters in the human hypothalamus. J Clin Endocrinol Metab. (2011) 96:E967–71. doi: 10.1210/jc.2010-2750
111. Guadano-Ferraz A, Obregon MJ, Germain DLS, Bernal J. The type 2 iodothyronine deiodinase is expressed primarily in glial cells in the neonatal rat brain. Proc Natl Acad Sci USA. (1997) 94:10391–6. doi: 10.1073/pnas.94.19.10391
112. Guadaño-Ferraz A, Escámez MJ, Rausell E, Bernal J. Expression of type 2 iodothyronine deiodinase in hypothyroid rat brain indicates an important role of thyroid hormone in the development of specific primary sensory systems. J Neurosci. (1999) 19:3430–9. doi: 10.1523/JNEUROSCI.19-09-03430.1999
113. Spitzer SO, Sitnikov S, Kamen Y, Evans KA, Kronenberg-Versteeg D, Dietmann S, et al. Oligodendrocyte progenitor cells become regionally diverse and heterogeneous with age. Neuron. (2019) 101:459–71.e5. doi: 10.1016/j.neuron.2018.12.020
114. Vancamp P, Darras VM. From zebrafish to human: a comparative approach to elucidate the role of the thyroid hormone transporter MCT8 during brain development. Gen Comp Endocrinol. (2018) 265:219–29. doi: 10.1016/j.ygcen.2017.11.023
115. Richard S, Flamant F. Regulation of T3 availability in the developing brain: the mouse genetics contribution. Front Endocrinol. (2018) 9:265. doi: 10.3389/fendo.2018.00265
116. Darras VM, Houbrechts AM, Van Herck SLJ. Intracellular thyroid hormone metabolism as a local regulator of nuclear thyroid hormone receptor-mediated impact on vertebrate development. Biochim Biophys Acta–Gene Regul Mech. (2015) 1849:130–41. doi: 10.1016/j.bbagrm.2014.05.004
117. Walter KM, Dach K, Hayakawa K, Giersiefer S, Heuer H, Lein PJ, et al. Ontogenetic expression of thyroid hormone signaling genes: an in vitro and in vivo species comparison. PLoS ONE. (2019) 14:e0221230. doi: 10.1371/journal.pone.0221230
118. Groeneweg S, Kersseboom S, van den Berge A, Dolcetta-Capuzzo A, van Geest FS, van Heerebeek REA, et al. In vitro characterization of human, mouse, and zebrafish MCT8 orthologues. Thyroid. (2019) 29:1499–510. doi: 10.1089/thy.2019.0009
119. Dumitrescu AM, Liao X-H, Weiss RE, Millen K, Refetoff S. Tissue-specific thyroid hormone deprivation and excess in monocarboxylate transporter (Mct) 8-deficient mice. Endocrinology. (2006) 147:4036–43. doi: 10.1210/en.2006-0390
120. Trajkovic M, Visser TJ, Mittag J, Horn S, Lukas J, Darras VM, et al. Abnormal thyroid hormone metabolism in mice lacking the monocarboxylate transporter 8. J Clin Invest. (2007) 117:627–35. doi: 10.1172/JCI28253
121. Mayerl S, Müller J, Bauer R, Richert S, Kassmann CM, Darras VM, et al. Transporters MCT8 and OATP1C1 maintain murine brain thyroid hormone homeostasis. J Clin Invest. (2014) 124:1987–99. doi: 10.1172/JCI70324
122. Wirth EK, Roth S, Blechschmidt C, Hölter SM, Becker L, Racz I, et al. Neuronal 3′,3,5-triiodothyronine (T3) uptake and behavioral phenotype of mice deficient in Mct8, the neuronal T3 transporter mutated in Allan-Herndon-Dudley syndrome. J Neurosci. (2009) 29:9439–49. doi: 10.1523/JNEUROSCI.6055-08.2009
123. Mayerl S, Visser TJ, Darras VM, Horn S, Heuer H. Impact of Oatp1c1 deficiency on thyroid hormone metabolism and action in the mouse brain. Endocrinology. (2012) 153:1528–37. doi: 10.1210/en.2011-1633
124. Morte B, Ceballos A, Diez D, Grijota-Martínez C, Dumitrescu AM, Di Cosmo C, et al. Thyroid hormone-regulated mouse cerebral cortex genes are differentially dependent on the source of the hormone: a study in monocarboxylate transporter-8- and deiodinase-2-deficient mice. Endocrinology. (2010) 151:2381–7. doi: 10.1210/en.2009-0944
125. Bárez-López S, Grijota-Martínez C, Ausó E, Fernández-de Frutos M, Montero-Pedrazuela A, Guadaño-Ferraz A. Adult mice lacking Mct8 and Dio2 proteins present alterations in peripheral thyroid hormone levels and severe brain and motor skill impairments. Thyroid. (2019) 29:1669–82. doi: 10.1089/thy.2019.0068
126. Morte B, Bernal J. Thyroid hormone action: astrocyte-neuron communication. Front Endocrinol. (2014) 5:82. doi: 10.3389/fendo.2014.00082
127. Howe K, Clark MD, Torroja CF, Torrance J, Berthelot C, Muffato M, et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. (2013) 496:498–503. doi: 10.1038/nature12111
128. Vancamp P, Houbrechts AM, Darras VM. Insights from zebrafish deficiency models to understand the impact of local thyroid hormone regulator action on early development. Gen Comp Endocrinol. (2019) 279:45–52. doi: 10.1016/j.ygcen.2018.09.011
129. Zada D, Tovin A, Lerer-Goldshtein T, Vatine GD, Appelbaum L. Altered behavioral performance and live imaging of circuit-specific neural deficiencies in a zebrafish model for psychomotor retardation. PLoS Genet. (2014) 10:e1004615. doi: 10.1371/journal.pgen.1004615
130. Preston MA, Macklin WB. Zebrafish as a model to investigate CNS myelination. Glia. (2015) 63:177–93. doi: 10.1002/glia.22755
131. Vatine GD, Zada D, Lerer-Goldshtein T, Tovin A, Malkinson G, Yaniv K, et al. Zebrafish as a model for monocarboxyl transporter 8-deficiency. J Biol Chem. (2013) 288:169–80. doi: 10.1074/jbc.M112.413831
132. Zada D, Tovin A, Lerer-Goldshtein T, Appelbaum L. Pharmacological treatment and BBB-targeted genetic therapy for MCT8-dependent hypomyelination in zebrafish. DMM Dis Model Mech. (2016) 9:1339–48. doi: 10.1242/dmm.027227
133. Remaud S, Gothié JD, Morvan-Dubois G, Demeneix BA. Thyroid hormone signaling and adult neurogenesis in mammals. Front Endocrinol. (2014) 5:62. doi: 10.3389/fendo.2014.00062
134. Remaud S, Ortiz FC, Perret-Jeanneret M, Aigrot M-S, Gothié J-D, Fekete C, et al. Transient hypothyroidism favors oligodendrocyte generation providing functional remyelination in the adult mouse brain. Elife. (2017) 6:6568–83. doi: 10.7554/eLife.29996
135. Salveridou E, Mayerl S, Sundaram SM, Markova B, Heuer H. Tissue-specific function of thyroid hormone transporters: new insights from mouse models. Exp Clin Endocrinol Diabetes. (2019). doi: 10.1055/a-1032-8328. [Epub ahead of print].
136. Wang S, Bates J, Li X, Schanz S, Chandler-Militello D, Levine C, et al. Human iPSC-derived oligodendrocyte progenitor cells can myelinate and rescue a mouse model of congenital hypomyelination. Cell Stem Cell. (2013) 12:252–64. doi: 10.1016/j.stem.2012.12.002
137. Yamashita T, Miyamoto Y, Bando Y, Ono T, Kobayashi S, Doi A, et al. Differentiation of oligodendrocyte progenitor cells from dissociated monolayer and feeder-free cultured pluripotent stem cells. PLoS ONE. (2017) 12:e0171947. doi: 10.1371/journal.pone.0171947
138. Lubetzki C, Demerens C, Anglade P, Villarroya H, Frankfurter A, Lee VMY, et al. Even in culture, oligodendrocytes myelinate solely axons. Proc Natl Acad Sci USA. (1993) 90:6820–4. doi: 10.1073/pnas.90.14.6820
139. Kersseboom S, Horn S, Edward Visser W, Chen J, Friesema ECH, Vaurs-Barrière C, et al. In vitro and mouse studies supporting therapeutic utility of triiodothyroacetic acid in MCT8 deficiency. Mol Endocrinol. (2014) 28:1961–70. doi: 10.1210/me.2014-1135
140. Di Cosmo C, Liao X-H, Dumitrescu AM, Weiss RE, Refetoff S. A thyroid hormone analog with reduced dependence on the monocarboxylate transporter 8 for tissue transport. Endocrinology. (2009) 150:4450–8. doi: 10.1210/en.2009-0209
141. Ferrara AM, Liao X-H, Ye H, Weiss RE, Dumitrescu AM, Refetoff S. The thyroid hormone analog DITPA ameliorates metabolic parameters of male mice with Mct8 deficiency. Endocrinology. (2015) 156:3889–94. doi: 10.1210/en.2015-1234
142. Groeneweg S, Peeters RP, Visser TJ, Visser WE. Triiodothyroacetic acid in health and disease. J Endocrinol. (2017) 234:R99–121. doi: 10.1530/JOE-17-0113
143. Visser WE, Visser TJ. Finding the Way into the Brain without MCT8. J Clin Endocrinol Metab. (2012) 97:4362–5. doi: 10.1210/jc.2012-3759
144. Verge CF, Konrad D, Cohen M, Di Cosmo C, Dumitrescu AM, Marcinkowski T, et al. Diiodothyropropionic Acid (DITPA) in the treatment of MCT8 deficiency. J Clin Endocrinol Metab. (2012) 97:4515–23. doi: 10.1210/jc.2012-2556
145. Fleming A, Diekmann H, Goldsmith P. Functional characterisation of the maturation of the blood-brain barrier in larval zebrafish. PLoS ONE. (2013) 8:e77548. doi: 10.1371/journal.pone.0077548
146. Anzai R, Adachi M, Sho N, Muroya K, Asakura Y, Onigata K. Long-term 3,5,3′-triiodothyroacetic acid therapy in a child with hyperthyroidism caused by thyroid hormone resistance: pharmacological study and therapeutic recommendations. Thyroid. (2012) 22:1069–75. doi: 10.1089/thy.2011.0450
147. Radetti G, Persani L, Molinaro G, Mannavola D, Cortelazzi D, Chatterjee VKB-PP. Clinical and hormonal outcome after 2 years of triiodothyroacetic acid treatment in a child with thyroid hormone resistance. Thyroid. (1997) 7:775–8. doi: 10.1089/thy.1997.7.775
148. Torre P, Bertoli M, Di Giovanni S, Scommegna S, Conte C, Novelli G, et al. Endocrine and neuropsychological assessment in a child with a novel mutation of thyroid hormone receptor: response to 12- months triiodothyroacetic acid (TRIAC) therapy. J Endocrinol Invest. (2005) 28:657–62. doi: 10.1007/BF03347267
149. Horn S, Kersseboom S, Mayerl S, Müller J, Groba C, Trajkovic-Arsic M, et al. Tetrac can replace thyroid hormone during brain development in mouse mutants deficient in the thyroid hormone transporter Mct8. Endocrinology. (2013) 154:968–79. doi: 10.1210/en.2012-1628
150. Ferrara AM, Liao X-H, Gil-Ibáñez P, Bernal J, Weiss RE, Dumitrescu AM, et al. Placenta passage of the thyroid hormone analog DITPA to male wild-type and Mct8-deficient mice. Endocrinology. (2014) 155:4088–93. doi: 10.1210/en.2014-1085
151. Hartley MD, Kirkemo L, Galipeau D, Calkins E, Chaudhary P, Pocius E, et al. 86 th annual meeting of the american thyroid association. Thyroid. (2016) 26:P-1-A-129. doi: 10.1089/thy.2016.29022.abstracts
152. Bove RM, Green AJ. Remyelinating pharmacotherapies in multiple sclerosis. Neurotherapeutics. (2017) 14:894–904. doi: 10.1007/s13311-017-0577-0
153. Hartley MD, Altowaijri G, Bourdette D. Remyelination and multiple sclerosis: therapeutic approaches and challenges. Curr Neurol Neurosci Rep. (2014) 14:485. doi: 10.1007/s11910-014-0485-1
154. Meinig JM, Ferrara SJ, Banerji T, Banerji T, Sanford-Crane HS, Bourdette D, et al. Structure–activity relationships of central nervous system penetration by Fatty Acid Amide Hydrolase (FAAH)-targeted thyromimetic prodrugs. ACS Med Chem Lett. (2019) 10:111–6. doi: 10.1021/acsmedchemlett.8b00501
155. Ferrara SJ, Meinig JM, Placzek AT, Banerji T, McTigue P, Hartley MD, et al. Ester-to-amide rearrangement of ethanolamine-derived prodrugs of sobetirome with increased blood-brain barrier penetration. Bioorganic Med Chem. (2017) 25:2743–53. doi: 10.1016/j.bmc.2017.03.047
156. Lammel Lindemann J, Webb P. Sobetirome: the past, present and questions about the future. Expert Opin Ther Targets. (2016) 20:145–9. doi: 10.1517/14728222.2016.1090429
157. Baxi EG, Schott JT, Fairchild AN, Kirby LA, Karani R, Uapinyoying P, et al. A selective thyroid hormone β receptor agonist enhances human and rodent oligodendrocyte differentiation. Glia. (2014) 62:1513–29. doi: 10.1002/glia.22697
158. Hartley MD, Banerji T, Tagge IJ, Kirkemo LL, Chaudhary P, Calkins E, et al. Myelin repair stimulated by CNS-selective thyroid hormone action. JCI Insight. (2019) 4:e126329. doi: 10.1172/jci.insight.126329
159. Bárez-López S, Hartley MD, Grijota-Martínez C, Scanlan TS, Guadaño-Ferraz A. Sobetirome and its amide prodrug Sob-AM2 exert thyromimetic actions in Mct8-deficient brain. Thyroid. (2018) 28:1211–20. doi: 10.1089/thy.2018.0008
160. Kremer D, Küry P, Dutta R. Promoting remyelination in multiple sclerosis: current drugs and future prospects. Mult Scler J. (2015) 21:541–9. doi: 10.1177/1352458514566419
161. Osorio MJ, Rowitch DH, Tesar P, Wernig M, Windrem MS, Goldman SA. Concise review: stem cell-based treatment of pelizaeus-merzbacher disease. Stem Cells. (2017) 35:311–5. doi: 10.1002/stem.2530
162. Groeneweg S, Peeters RP, Moran C, Stoupa A, Auriol F, Tonduti D, et al. Effectiveness and safety of the tri-iodothyronine analogue Triac in children and adults with MCT8 deficiency: an international, single-arm, open-label, phase 2 trial. Lancet Diabetes Endocrinol. (2019) 7:695–706. doi: 10.1016/S2213-8587(19)30155-X
Keywords: Allan-Herndon-Dudley Syndrome, SLC16A2 gene, monocarboxylate transporter 8, oligodendrocyte, myelination and myelin repair, thyroid hormones, thyroid hormone analog, neurodevelopment
Citation: Vancamp P, Demeneix BA and Remaud S (2020) Monocarboxylate Transporter 8 Deficiency: Delayed or Permanent Hypomyelination? Front. Endocrinol. 11:283. doi: 10.3389/fendo.2020.00283
Received: 14 February 2020; Accepted: 15 April 2020;
Published: 13 May 2020.
Edited by:
Frédéric Flamant, Université de Lyon, FranceReviewed by:
Ana Guadaño-Ferraz, Consejo Superior de Investigaciones Científicas (CSIC), SpainBijay Vaidya, University of Exeter, United Kingdom
Copyright © 2020 Vancamp, Demeneix and Remaud. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pieter Vancamp, cGlldGVyLnZhbmNhbXBAbW5obi5mcg==