 Roberto Coccurello
Roberto Coccurello Cinzia Volonté
Cinzia Volonté- 1Institute for Complex System (ISC), National Research Council (CNR), Rome, Italy
- 2Preclinical Neuroscience, European Center for Brain Research (CERC)/IRCCS Santa Lucia Foundation, Rome, Italy
- 3Institute for Systems Analysis and Computer Science, National Research Council (CNR), Rome, Italy
Whole-body energy metabolism entails the highly regulated balance between food intake, nutrient breakdown, energy generation (ATP), and energy storage for the preservation of vital functions and body mass. Purinergic signaling has attracted increasing attention in the regulatory mechanisms not only for the reverse processes of white adipose tissue lipogenesis and lipolysis, but also for brown adipocyte-dependent thermogenesis and leptin production. This regulatory role has remarkable implications in the handling of body's energy expenditure and energy reservoir. Hence, selected purinergic receptors can play a relevant function in lipid metabolism, endocrine activity, glucose uptake, ATP-dependent increased expression of uncoupling protein 1, and browning of adipose tissue. Indeed, purinergic P2 receptors regulate adipogenesis and lipid metabolism and are involved in adipogenic differentiation. In particular, the ionotropic ATP-activated P2X7 subtype is involved in fat distribution, as well as in the modulation of inflammatory pathways in white adipose tissue. Within this context, very recent evidence has established a direct function of P2X7 in energy metabolism. Specifically, either genetic deletion (P2X7 knockout mice) or subchronic pharmacological inhibition of the receptor produces a decrease of whole-body energy expenditure and, concurrently, an increase of carbohydrate oxidation. As further evidence, lipid accumulation, increased fat mass distribution, and weight gain are reported in P2X7-depleted mice. Conversely, the stimulation of P2X7 enhances energy expenditure. Altogether, this knowledge supports the role of P2X7 signaling in the fight against obesity and insulin resistance, as well as in the promotion of adaptive thermogenesis.
Introduction
At difference with the primary function of intracellular nucleotides that provides the energy supply for cell viability and survival, extracellular nucleotides such as ATP can act as signaling molecules when released into the extracellular compartment. By means of their capacity to use extracellular ATP, adenosine, and other nucleotides and nucleosides for triggering intracellular signal transduction mechanisms, the purinergic receptors have a distinctive role also in the regulation of intracellular metabolism and energy homeostasis (1–3). Basically, there are two wide families of purinergic receptors, the adenosine-binding (P1) and the ATP-binding (P2) subtypes that are, in turn, categorized in P2X ion channels and P2Y G protein–coupled receptors (4). Respectively seven and eight different subtypes have been identified for P2Xs (P2X1–7) and P2Ys (P2Y1, 2, 4, 6, 11–14) receptors, all possessing different binding affinities depending on the various nucleotides (4).
P2x Receptors, Latent Inflammation, And Obesity-Associated Insulin Resistance
After the discovery by Geoffrey Burnstock of ATP as a noncanonical extracellular mediator (5), accumulating evidence corroborated the idea that ATP being constitutively released by many different cell types allows for cell-to-cell communication. Due to the pleiotropic functional role of ATP, P2X, and P2Y receptors are involved also in a great number of disease conditions. For instance, intense investigation is focused on the therapeutic potential of P2X in cardiovascular diseases (e.g., heart failure, ischemia) (6), cancer (7), neuropathic and inflammatory pain (8), neurodegenerative diseases (9), and muscle and bone disorders (10, 11). Within the context of these diseases, it is also relevant that purinergic receptors are distributed not only in neurons, but also in astrocytes, oligodendrocytes, and immunocompetent microglia (12–14).
In addition to trophic support (15), ATP can also act as a danger signal upon tissue injury or cell damage, by triggering the consecutive phases of inflammatory insurgence and resolution, as well as immune system activation by regulation of T lymphocyte proliferation, T-helper 1 production, and macrophage chemotaxis (16–19). A major common feature underlying systemic inflammation in which P2X receptors are known to play a pathogenic role is altered regulation of energy homeostasis (20, 21). Moreover, there is now large consensus about the role of chronic “low-grade systemic inflammation” as a common factor connecting aging, neurodegenerative diseases, diabetes, and metabolic syndrome (22–24). By definition, obesity is a low-grade systemic inflammatory disease where excessive/uncontrolled energy intake coexists with insufficient energy expenditure (EE) causing abnormal fat accumulation with expansion of adipose tissue (AT) and energy stores (25). It is now accepted that the study of excessive white AT storage (i.e., adiposity) is of major importance for the comprehension of maladaptive chronic inflammatory response and the generation of low-grade systemic inflammation and obesity comorbidities, such as type 2 diabetes, dyslipidemia, cardiovascular disease, and neurodegenerative diseases (26–28). Indeed, over the last decade, the concept of AT as inert energy reservoir has been finally replaced by the notion of endocrine organ, with the identification of a growing number of AT-secreted hormonal factors or adipokines involved in the control of energy homeostasis (29). In parallel, mounting evidence has helped to clarify that adipocytes hypertrophy and aberrant secretory activity involving inflammatory adipokines, such as tumor necrosis factor α (TNF-α) and interleukin 6 (IL-6), are significantly associated with recruitment, infiltration, and accumulation of B and T lymphocytes and macrophages into the white AT and development of insulin resistance (30, 31). On the other hand, before the occurrence of insulin resistance, several mechanisms, such as early activation of proinflammatory and anti-inflammatory immune cells, release of adipocyte-derived cytokines, and secretion of lipolytic hormones (e.g., leptin), can activate the sympathetic nervous system and trigger brown AT-mediated thermogenic responses (32, 33), thus limiting white AT accumulation. However, in the case of protracted positive energy balance, adaptive inflammation and acute immune activation become inadequate responses, and white AT enlargement will produce the alteration of fat distribution, ectopic lipid (i.e., triglycerides) deposition, dyslipidemia, and insulin resistance (34).
Both P2X and P2Y receptors have been found to be regulators of adipogenesis and adipocyte differentiation from bone marrow—and AT-derived mesenchymal stromal cells. In particular, P2Y1, P2Y4, P2Y14, and P2X6 receptors are involved in adipogenic differentiation (35, 36), whereas P2Y2 and P2Y13 receptors have been described in bone marrow–derived adipocyte differentiation (37, 38) (Table 1). The present survey on the role of purinergic signaling in obesity and insulin resistance will be focused on the pleiotropic P2X7 receptor subtype (41) for its major involvement not only in the modulation of fat distribution and inflammation in white AT (42), but also in energy metabolism and nonshivering thermogenesis (39, 40).
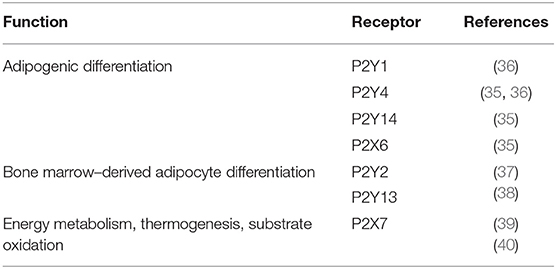
Table 1. Role of P2 receptors in adipogenic functions and energy metabolism.
P2x7: From Inflammation Gatekeeping To Energy Metabolism
As other components of the P2X receptor family, also the P2X7 member is largely expressed on immune cells such as B and T lymphocytes, macrophages/microglia, mast cells, and natural killer cells (43), thus contributing to the orchestration of innate and adaptive immune responses (44). Because ATP is also constitutively and/or passively released upon tissue injury or cell damage, the ATP-mediated signaling downstream from activation of P2X7 receptor (possessing a higher Kb for ATP with respect to the other P2 receptors) is often related to the damage- or pathogen-associated molecular pattern molecules and inflammasome formation [e.g., nucleotide-binding oligomerization domain, leucine rich repeat and pyrin domain containing protein 3 (NLRP3)] (43, 45). For this reason, the P2X7 is of major importance for host defense, being considered a sensor of danger signals (46, 47). Indeed, the activation of P2X7 is associated with a remarkable number of inflammatory clinical conditions, including systemic lupus erythematosus and rheumatoid arthritis (48), cancer (49), cardiovascular diseases (50), liver disease (51), colitis and inflammatory bowel disease (52, 53), pain development (54), epilepsy (55), and depression (56).
The main liability of P2X7 in AT inflammation and obesity was at first suggested by the fact that visceral and subcutaneous AT expresses functional P2X7 receptors (both mRNA and protein) and by the notion that such expression is higher in subjects with metabolic syndrome (57). Remarkably, stimulation of isolated adipocytes with the prototypic P2X7 agonist 3′-O-(4-benzoyl)benzoyl ATP (BzATP) triggers the production of IL-6, TNF-α, and plasminogen activator inhibitor 1 (57). Moreover, the exacerbation of the inflammatory status and formation of the P2X7-NLRP3 inflammasome complex have been reported in perivascular AT of subjects with heavy smoking habits (58), in which the overactivation of P2X7 was also associated with higher IL-1β and IL-18 plasma levels. Indeed, P2X7 activation is a potent trigger of IL-1β release and a major cause of innate immune cells activation, proinflammatory activity, and chronic inflammatory diseases (59). However, the same activation of P2X7 is also involved in the release of the anti-inflammatory IL-1 receptor antagonist (IL-1Ra), for instance, in macrophages (60). Of note, it has been shown that an increase of IL-1Ra may provide a protective potential against pancreatic beta cell damage and improve glucose homeostasis in patients with type 2 diabetes, whereas IL-1Ra pancreatic deletion can impair glucose tolerance in mice (61). In agreement with the role of IL-1Ra in glucose homeostasis and beta cell function, the targeted deletion of pancreatic IL-1Ra in mice has been reported to decrease glucose-stimulated insulin secretion and induce glucose intolerance (62). In particular, the study of P2X7-mediated control over IL-1Ra secretion in both lean and obese diabetic patients has helped to disclose a potential regulatory function exerted by PX7 activation on pancreatic beta cell function (63). Moreover, pancreatic IL-1Ra resulted downregulated in beta cell islets of diabetic patients, and P2X7 knockout mice show reduced IL-1Ra secretory capacity, hyperglycemia, glucose intolerance, and impaired beta cell compensation in response to high-sucrose diet (63). Recently, higher IL-1Ra serum levels and improved beta cell function have been found in diabetic patients bearing a polymorphism of the P2X7 gene [i.e., 1,068 G>A, single nucleotide polymorphism (SNP)], although in the lack of significant improvement of glycemic control (64). Together, while the functional association between P2X7-mediated regulation of IL-1Ra secretion and disruption of beta cell function and glucose homeostasis awaits further experimental confirmation, the interplay between IL-1β and the P2X7R is “solid as rock”(65).
Persistent exposure to high-fat diet (HFD) is a well-known experimental model of metabolic syndrome, obesity, and type 2 diabetes, which are relevant risk factors for the increasing incidence of chronic kidney disease (66). High-fat diet–induced renal inflammation involves the P2X7 receptor via the activation of NLRP3 inflammasome, whereas reduced kidney damage, inflammation, and decreased NLRP3 upregulation are observed in mice genetically depleted of the receptor (67). Moreover, while renal P2X7 expression is enhanced in diabetic patients and is associated with damage of glomerular filtration and increased fibrosis, the pharmacological blockade of P2X7 receptor reduces renal macrophage accumulation in experimental diabetic nephropathy (68). It should be noted that P2X7 receptors are expressed by different types of pancreatic cells such as alpha and beta cells, and multiple evidence corroborates the regulatory action exerted by the P2X7 on pancreatic stellate cell proliferation, insulin secretion, and involvement in type 2 diabetes pathogenesis (69–71). Also, the study of P2X7 polymorphisms further demonstrates that P2X7 hypofunction dysregulates glucose homeostasis with reduced insulin sensitivity and compromised glucose tolerance (72). Accordingly, as recently shown (71), P2X7 receptors are activated by glucose elevation, and through their expression on beta cells, they are able to regulate Ca2+ signaling, cell proliferation, and insulin secretion. As reported recently, the P2X7 receptor of the beta cells line INS-1E plays a functional role in the regulation of glucose-dependent ATP release. The results prompt for a model according to which glucose is metabolized to ATP after its entry in beta cells, and ATP is also released via pannexin 1 channels under the cooperative regulation of P2X7-dependent modulation of Ca2+ influx and potentiation of insulin secretion (71). Within the same line of investigation, it has been observed that P2X7 stimulation induces internalization of GLUT2 in intestinal epithelial cell line (73), and mice lacking P2X7 show upregulation of GLUT2 expression at enterocyte level (74). As a consequence, the authors have found that blood glucose is increased in P2X7 knockout together with hypercholesterolemia, hypertriglyceridemia, and insulin resistance (74).
As for HFD-induced renal inflammation and diabetes-associated renal failure, diabetic retinopathy is exacerbated by sustained P2X7 signaling causing proinflammatory TNF-α and IL-1β release and apoptosis of retinal microvessels (75), which can be limited by P2X7 pharmacological blockade (76). In the context of type 2 diabetes and pathogenesis of insulin resistance, P2X7 activation behaves like a double-edged sword, which may have beneficial effects on glucose homeostasis and pancreatic islet function (71), while in conditions of overnutrition (77, 78) and exposure to obesogenic environments (79), a sustained P2X7 activation concurs to weight gain, hyperglycemia, AT inflammation, and excess of circulating free fatty acids (FFAs).
Indeed, changes of dietary patterns involving the overconsumption of high-energy-dense diets and low-quality foods represent the current nutritional scenario, at least for Western people and developing countries. In such a scenario, it becomes of great relevance to understand the role of P2X7 not only on the impact of nutritional overload, AT enlargement, ectopic fat deposition, and adipose inflammation for the development of insulin resistance, but also its role on defective thermogenesis in obesity.
Control of Energy Expenditure And P2x7 Signaling
Among the several different hypotheses that have been proposed to explain body mass regulation (e.g., the dual intervention point model) (80), there is the idea that body weight is regularly adjusted and preserved according to a given set point, which results from the balance between energy intake and EE. Because the active defense of body weight set point is constantly threatened by both living in an obesogenic environment and tendency of human genome to store energy as fat, the restriction of energy intake and the increase of EE are the only options to fight body weight gain, adiposity accrual, and obesity. As three major components, EE includes the basal metabolic rate that keeps internal temperature and vital functions, physical activity, and the dissipation of cell energy via heat production (i.e., thermogenesis). It is known that, in addition to white adipocytes where extra calories are stored as triglycerides and released as FFAs, there are also brown and beige/brite adipocytes that are required for adaptive thermogenesis (81–83). The brown AT is highly vascularized and rich in mitochondria and is able to burn energy and generate heat by the activation of the uncoupling protein 1 (UCP1), which “uncouple” mitochondrial respiration and electron transport chain from ATP generation. Because the energy produced by the uncoupling of respiration from ATP production is dissipated as heat into a futile cycle (i.e., proton leak), the brown AT is the major responsible of adaptive or nonshivering thermogenesis. Although UCP1 is mostly brown AT-restricted, low levels of UCP1 are also present in beige/brite adipocytes that are localized within the white AT and exhibit, upon stimulation, inducible thermogenic capacity. Notably, cold exposure, sympathetic nervous system activation, noradrenaline release, binding to β3-adrenergic receptors, prostaglandins, fibroblast growth factor 21, as well as drugs such as thiazolidinediones, may trigger the differentiation from white to brown-like beige adipocytes (83–85). Of importance for thermogenesis in humans, in large adult mammals and not only in rodents, a scattered accumulation of brown adipocytes can still be identified in the neck, in the supraclavicular area, and in paravertebral and perirenal AT (86–88).
Adaptive thermogenesis is described by the capacity of UCP1 inside the inner mitochondrial membrane to dissipate the electrochemical proton (H+) gradient generated by the electron transport chain and to release energy as heat, a mechanism identified 25 years ago (89) and now widely accepted. Uncoupling protein 1 can be activated by the long-chain fatty acids released in brown adipocytes via adipose triglyceride lipase-dependent hydrolysis of triglycerides (90, 91). Recently, UCP1 currents in the inner mitochondrial membrane have been studied by a patch-clamp technique demonstrating that long-chain fatty acids are not only required for UCP1-mediated H+ transport activity, but they are also “anchored” in a substrate-like fashion to UCP1 substrates, thus allowing to carry H+ across UCP1 (92). Of note, while UCP1 is activated by FFAs, there is also evidence that UCP1 is inhibited by purine nucleotides and, mostly, by ATP-mediated binding on the cytosolic portion of UCP1 that hampers the kinetics of UCP1 translocation (93, 94). Considering the role of P2X7 as sensor of the extracellular ATP environment (being activated by higher ATP concentrations with respect to other P2X receptors) and that loss of P2X7 function in mice disrupts adipocyte distribution inducing adipocyte hyperplasia and lipid ectopic depots (42), a possible opposite impact of loss- or gain-of-function of P2X7 in whole-body energy metabolism has been hypothesized. Indeed, a recent work has demonstrated that genetic depletion and, to a lesser extent, also pharmacological inhibition of P2X7 elicit a pronounced decrease of the whole-body EE and metabolic rate in mice, with a significant increase of the respiratory exchange ratio, thus indicating a prevalent increase of carbohydrate oxidation (39). The relative sparing of fatty acid storage and concomitant defective energy homeostasis were also associated to body weight gain (39), thus generating lipid accumulation, increased fat mass distribution, and, ultimately, weight gain that is also reported in P2X7-depleted mice (42). In line with these results, under standard nutritional conditions, both metabolic rate as measured in terms of O2 intake and heat production were significantly increased by subchronic administration of BzATP for seven consecutive days (40). Notably, the increase of metabolic rate/EE was generated also in the lack of motor activity, as detected during the overall resting period recorded across the entire light–dark cycle. Moreover, because the enhancement of EE was not attributable to changes of food intake, it was possible to rule out the potential contribution of diet-induced thermogenesis. On the other hand, because respiratory exchange ratio was decreased by subchronic BzATP administration, the activation of P2X7 produced a significant shift in the level of nutrient substrate utilization, and in particular of lipid consumption over other food energy sources (i.e., carbohydrates and proteins). Being simultaneously consumed more O2 without evident alteration of motor behavior, an increase of fatty acid oxidation and a prevalent use of lipids as energy source after P2X7 receptor stimulation, was demonstrated. Providing further confirmation, the preadministration of the specific P2X7 antagonist A804589 delayed and attenuated the increase in O2 consumption and EE induced by BzATP-induced potentiation of P2X7 activity (40) (Figure 1, Table 1). Thus, these data support the notion that an opposite regulation of P2X7 function (namely, suppression vs potentiation) is able to produce antithetical effects on whole-body energy metabolism, being the EE reduced by P2X7 suppression and boosted by P2X7 potentiation. Accordingly, while lipid oxidation is decreased by suppression of P2X7 function, fatty acids are more selectively oxidized by the stimulation of P2X7 function (39, 40).
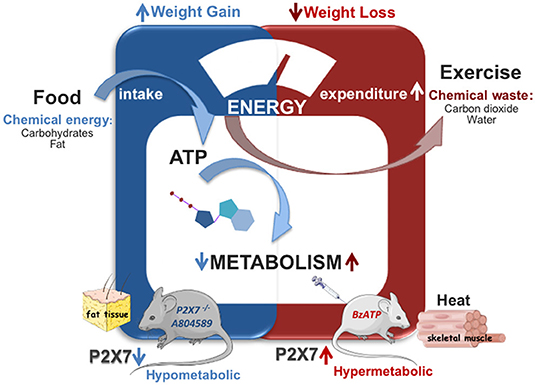
Figure 1. Role of P2X7 in energy metabolism. Genetic deletion or subchronic pharamacological inhibition of P2X7 (A804589) decreases the whole body energy expenditure causing weight gain without affecting food intake (in blue). Stimulation of P2X7 by BzATP enhances energy expenditure and fatty acid oxidation causing weight loss and heat production in mice (in red).
Muscle Machinery, Fatty Acids Oxidation, And P2x7 Signaling
As one of the largest tissue accounting for ~50% of body mass, the skeletal muscle is a major determinant of the whole-body metabolic rate (95), possessing a remarkable capacity to rapidly shift from carbohydrates to fatty acids utilization in response to increasing energy demand and intensity of physical exercise (96–98). As a consequence, generation of reactive oxygen species (ROS) is augmented in skeletal muscles as part of a physiological response to exercise, adaptation to increased workload, and optimization of the contractile capacity (99). In particular, in the muscle gastrocnemius the physical exercise specifically stimulates the mRNA levels of NADPH oxidase 2 (NOX2) (100), a superoxide-generating enzyme and major source of ROS under resting and contractile muscle conditions (101). Importantly, BzATP-induced activation of P2X7 increases the content of the glycoprotein gp91phox (a component of the membrane bound NADPH oxidase complex) in the gastrocnemius (possessing both fast- and slow-twitch fibers) and tibialis anterior (with 95% fast-twitch fibers) muscles, thus indicating that P2X7 activation might have a role in the contraction-induced intracellular signaling mediated by nonmitochondrial ROS sources such as NOX2 (40). We do not exclude that the BzATP-induced activation of P2X7 might mimic the increase of oxygen consumption (102, 103) and the NOX2-dependent redox adaptation that occur in mixed and fast-twitch glycolytic fiber types (104) after high-intensity training and endurance exercise. Under this perspective, the sustained activation of P2X7 might cause a remodeling of fatigue-susceptible muscles (i.e., type II fibers) and reprogramming of the global muscle gene expression toward a slower, more oxidative transcription program.
Skeletal muscle exhibits shivering and adaptive thermogenic capacity, as well as the ability to increase EE as a function of different types of physical exercise, adjusting the intensity of contractile activity and metabolic pathways to the changing energy requests (i.e., by shifting from carbohydrates to prevalent fatty acids utilization). These remarkable features, combined with the well-known possibility to breakdown almost 80% of the insulin-stimulated glucose uptake, make the skeletal muscle a specialized tissue to manage insulin sensitivity and, whenever defective, a primary player in type 2 diabetes pathogenesis. It is known that P2X7 activation triggers the opening of cations permeable channels allowing a considerable mobilization of intracellular Ca2+ (105) and the increase of mitochondrial Ca2+ and cellular ATP levels (106). Interestingly, within the context of Ca2+ handling and preservation of muscle physiology, the activity of sarco(endo)plasmic reticulum Ca2+ATPase (SERCA) provides a key contribution not only to the maintenance of intracellular Ca2+ homeostasis, but also to the nonshivering thermogenesis. By subserving the removal of cytosolic Ca2+ and its reuptake into the sarcoplasmic reticulum lumen (107), the SERCA pump contributes to Ca2+ handling and muscle contraction–relaxation cycle (108). As a result, a protracted inhibition of SERCA activity may lead to cytosolic Ca2+ overload, as observed in defective muscle regeneration and muscular dystrophies (109, 110). Indeed, excessive increase of intracellular Ca2+ is an early pathogenetic event in the progression of Duchenne muscular dystrophy (109–112), and P2X7 receptor seems to have a role in muscular dystrophies (113, 114). Thus, the excessive levels of extracellular ATP observed in dystrophic muscle might contribute to the overactivation of the P2X7 receptor and then to abnormal intracellular Ca2+ homeostasis and chronic inflammation (113–115). Because sarcolipin binding to SERCA is facilitated by high cytosolic Ca2+ (116), there is the possibility that P2X7 overactivation and increase of intracellular Ca2+ may promote sarcolipin binding to SERCA and thus increase nonshivering muscle thermogenesis.
The most part of whole-body adaptive thermogenesis that occurs in skeletal muscle cannot rely on UCP1 (not expressed in muscle), but on the analog UCP3 that is prevalently expressed in both skeletal muscle and brown AT (117). Uncoupling protein 3 is activated by FFAs and can be inhibited by the direct competition between FFAs and purine nucleotides (118). The expression of UCP3 in muscle appears regulated by the levels of fatty acid oxidation occurring during prolonged physical activity (119), fasting (120), or under HFD regimen (121), supporting the concept of UCP3 as functionally involved in muscle fatty acid transport, beta-oxidation (122), and protection against HFD-induced insulin resistance (123). Although muscle UCP3 does not contribute to brown AT-mediated thermogenesis (124, 125), there is nevertheless evidence that in the lack of UCP3 both the thermogenic response to sympathomimetic drugs and lipopolysaccharide challenge are blunted (126, 127), thus supporting the notion that UCP3 may be important in amplifying brown AT thermogenesis and promoting skeletal muscle thermogenic activity and/or fatty acid oxidation (127, 128). Most importantly, it should be underlined the protective mechanism exerted by UCP3 against mitochondrial fatty acid accumulation and fatty acid–induced damage of mitochondrial oxidative capacity (129–131). Further support to this aspect comes from experiments on UCP3 knockout mice in which an increase in mitochondrial ROS production is observed together with reduced uncoupling activity but in the absence of body weight alterations or cold-induced thermogenesis (132, 133). Under this regard, the result that direct P2X7 activation increases fatty acid oxidation and reduces body weight might also suggest a potential UCP3-mediated facilitatory action of the receptor.
Conclusive Discussion
In the present survey, we have highlighted the major involvement of P2X7 in determining and regulating fat distribution, white AT inflammation, energy metabolism, and nonshivering thermogenesis. Indeed, functional P2X7 receptors are found expressed in visceral and subcutaneous AT, and the alteration of P2X7 function greatly contributes to AT inflammation and adiposity.
Next, we reported recent evidence that modulation of P2X7 can change whole-body EE, also determining the rate of macronutrient oxidation and the adjustment of fuel selection (i.e., carbohydrate vs lipid) to achieve body weight regulation. In particular, we have reported that BzATP-induced activation of P2X7 increases EE and induces NOX2 expression in gastrocnemius and in fast-twitch tibialis anterior. Because balanced levels of NOX2 and ROS are viewed as mediators of glucose transport in muscle (134), we suggest that activation of P2X7 function may not only increase fatty acid oxidation and EE and decrease body weight, but also possibly reduce the susceptibility to insulin resistance under high-fat feeding and/or during the dysregulation of the muscle P2X7/Ca2+/ UCP3/sarcolipin-SERCA axis.
Because selective overexpression of UCP3 in muscle has been found to act as exercise mimetic not only by increasing fatty acid oxidation and EE (135), but also by modulating the mutual interplay between ROS generation and fatty acid oxidation (133), it might become important to next improve our knowledge about the ability of P2X7 to shape the metabolic signature of muscle fibers mimicking high-intensity training and endurance exercise (40, 102). However, given the “double-edged sword” nature of P2X7 (136), caution must still be recommended because continuous or excessive stimulation of P2X7 could exacerbate endoplasmic reticulum stress that has been recently recognized to be mechanistically involved in the decrease of brown AT-dependent EE and obesity progression (137).
Author Contributions
RC conceived the study. RC and CV wrote the manuscript.
Funding
The Italian Ministry of Health have supported the current research (Ricerca Corrente to RC and CV).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr. Maria Cristina Marrone for her critical reading of the manuscript.
Abbreviations
AT, Adipose tissue; BzATP, 3′-O-(4-Benzoyl)benzoyl ATP; EE, energy expenditure; FFAs, free fatty acids; HFD, high-fat diet; IL, interleukin; IL-1Ra, interleukin-1 receptor antagonist; NLRP3, nucleotide-binding oligomerization domain, leucine rich repeat and pyrin domain containing protein 3; ROS, reactive oxygen species; SERCA, sarco(endo)plasmic reticulum Ca2+ATPase; TNF-α, tumor necrosis factor α; UCP1, uncoupling protein 1; UCP3, uncoupling protein 3.
References
1. la Sala A, Ferrari D, Di Virgilio F, Idzko M, Norgauer J, Girolomoni G. Alerting and tuning the immune response by extracellular nucleotides. J Leukoc Biol. (2003) 73:339–43. doi: 10.1189/jlb.0802418
2. Burnstock G, Gentile D. The involvement of purinergic signalling in obesity. Purinergic Signal. (2018) 14:97–08. doi: 10.1007/s11302-018-9605-8
3. Tang Z, Ye W, Chen H, Kuang X, Guo J, Xiang M, et al. Role of purines in regulation of metabolic reprogramming. Purinergic Signal. (2019) 15:423–38. doi: 10.1007/s11302-019-09676-z
4. Burnstock G. Purine and purinergic receptors. Brain Neurosci Adv. (2018) 2:2398212818817494. doi: 10.1177/2398212818817494
6. Burnstock G. Purinergic Signaling in the Cardiovascular System. Circ Res. (2017) 120:207–28. doi: 10.1161/CIRCRESAHA.116.309726
7. Burnstock G, Di Virgilio F. Purinergic signalling and cancer. Purinergic Signal. (2013) 9:491–40. doi: 10.1007/s11302-013-9372-5
8. Tsuda M, Tozaki-Saitoh H, Inoue K. Pain and purinergic signaling. Brain Res Rev. (2010) 63:222–32. doi: 10.1016/j.brainresrev.2009.11.003
9. Volonté C, Amadio S, Cavaliere F, D'Ambrosi N, Vacca F, Bernardi G. Extracellular ATP and neurodegeneration. Curr Drug Targets CNS Neurol Disord. (2003) 2:403–12. doi: 10.2174/1568007033482643
10. Ryten M, Yang SY, Dunn PM, Goldspink G, Burnstock G. Purinoceptor expression in regenerating skeletal muscle in the mdx mouse model of muscular dystrophy and in satellite cell cultures. FASEB J. (2004) 18:1404–06. doi: 10.1096/fj.03-1175fje
11. Orriss IR, Burnstock G, Arnett TR. Purinergic signalling and bone remodelling. Curr Opin Pharmacol. (2010) 10:322–30. doi: 10.1016/j.coph.2010.01.003
12. del Puerto A, Wandosell F, Garrido JJ. Neuronal and glial purinergic receptors functions in neuron development and brain disease. Front Cell Neurosci. (2013) 7:197. doi: 10.3389/fncel.2013.00197
13. Burnstock G, Krügel U, Abbracchio MP, Illes P. Purinergic signalling: From normal behaviour to pathological brain function. Prog Neurobiol. (2011) 95:229–74. doi: 10.1016/j.pneurobio.2011.08.006
14. Lindberg D, Shan D, Ayers-Ringler J, Oliveros A, Benitez J, Prieto M, et al. Purinergic Signaling and Energy Homeostasis in Psychiatric Disorders. Curr Mol Med. (2015) 15:275–95. doi: 10.2174/1566524015666150330163724
15. Burnstock G. Short-and long-term (Trophic) purinergic signalling. Philos Trans R Soc B Biol Sci. (2016) 371:1700. doi: 10.1098/rstb.2015.0422
16. Verkhratsky A, Burnstock G. Biology of purinergic signalling: Its ancient evolutionary roots, its omnipresence and its multiple functional significance. BioEssays. (2014) 36:697–05. doi: 10.1002/bies.201400024
17. Junger WG. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol. (2011) 11:201–12. doi: 10.1038/nri2938
18. Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. (2009) 461:282–86. doi: 10.1038/nature08296
19. Trautmann A. Extracellular ATP in the immune system: More than just a “danger signal.” Sci Signal. (2009) 2:pe6. doi: 10.1126/scisignal.256pe6
20. Kotas ME, Medzhitov R. Homeostasis, Inflammation, and Disease Susceptibility. Cell. (2015) 160:816–27. doi: 10.1016/j.cell.2015.02.010
21. Wang H, Ye J. Regulation of energy balance by inflammation: Common theme in physiology and pathology. Rev Endocr Metab Disord. (2015) 16:47–4. doi: 10.1007/s11154-014-9306-8
22. Holmes C. Review: Systemic inflammation and Alzheimer's disease. Neuropathol Appl Neurobiol. (2013) 39:51–8. doi: 10.1111/j.1365-2990.2012.01307.x
23. Yin F, Sancheti H, Patil I, Cadenas E. Energy metabolism and inflammation in brain aging and Alzheimer's disease. Free Radic Biol Med. (2016) 100:108–22. doi: 10.1016/j.freeradbiomed.2016.04.200
24. Reddy P, Lent-Schochet D, Ramakrishnan N, McLaughlin M, Jialal I. Metabolic syndrome is an inflammatory disorder: A conspiracy between adipose tissue and phagocytes. Clin Chim Acta. (2019) 496:35–44. doi: 10.1016/j.cca.2019.06.019
25. Gregor MF, Hotamisligil GS. Inflammatory Mechanisms in Obesity. Annu Rev Immunol. (2011) 29:415–45. doi: 10.1146/annurev-immunol-031210-101322
26. Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. (2011) 121:2111–17. doi: 10.1172/JCI57132
27. Parimisetty A, Dorsemans AC, Awada R, Ravanan P, Diotel N, Lefebvre d'Hellencourt C. Secret talk between adipose tissue and central nervous system via secreted factors-an emerging frontier in the neurodegenerative research. J Neuroinflammation. (2016) 13:67. doi: 10.1186/s12974-016-0530-x
28. Nakamura K, Fuster JJ, Walsh K. Adipokines: A link between obesity and cardiovascular disease. J Cardiol. (2014) 63:250–9. doi: 10.1016/j.jjcc.2013.11.006
29. Ahima RS, Lazar MA. Adipokines and the peripheral and neural control of energy balance. Mol Endocrinol. (2008) 22:1023–31. doi: 10.1210/me.2007-0529
30. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. (2003) 112:1796–08. doi: 10.1172/JCI200319246
31. Sell H, Habich C, Eckel J. Adaptive immunity in obesity and insulin resistance. Nat Rev Endocrinol. (2012) 8:709–16. doi: 10.1038/nrendo.2012.114
32. Lee BC, Lee J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim Biophys Acta. (2014) 1842:446–62. doi: 10.1016/j.bbadis.2013.05.017
33. Reilly SM, Saltiel AR. Adapting to obesity with adipose tissue inflammation. Nat Rev Endocrinol. (2017) 13:633–43. doi: 10.1038/nrendo.2017.90
34. Liu L, Mei M, Yang S, Li Q. Roles of chronic low-grade inflammation in the development of ectopic fat deposition. Mediators Inflamm. (2014) 2014:418185. doi: 10.1155/2014/418185
35. Zippel N, Limbach CA, Ratajski N, Urban C, Luparello C, Pansky A, et al. Purinergic receptors influence the differentiation of human mesenchymal stem cells. Stem Cells Dev. (2012) 21:884–00. doi: 10.1089/scd.2010.0576
36. Ciciarello M, Zini R, Rossi L, Salvestrini V, Ferrari D, Manfredini R, et al. Extracellular purines promote the differentiation of human bone marrow-derived mesenchymal stem cells to the osteogenic and adipogenic lineages. Stem Cells Dev. (2013) 22:1097–11. doi: 10.1089/scd.2012.0432
37. Biver G, Wang N, Gartland A, Orriss I, Arnett TR, Boeynaems JM, et al. Role of the P2Y13 receptor in the differentiation of bone marrow stromal cells into osteoblasts and adipocytes. Stem Cells. (2013) 31:2747–58. doi: 10.1002/stem.1411
38. Li W, Wei S, Liu C, Song M, Wu H, Yang Y. Regulation of the osteogenic and adipogenic differentiation of bone marrow-derived stromal cells by extracellular uridine triphosphate: The role of P2Y2 receptor and ERK1/2 signaling. Int J Mol Med. (2016) 37:63–3. doi: 10.3892/ijmm.2015.2400
39. Giacovazzo G, Apolloni S, Coccurello R. Loss of P2X7 receptor function dampens whole body energy expenditure and fatty acid oxidation. Purinergic Signal. (2018) 14:299–05. doi: 10.1007/s11302-018-9610-y
40. Giacovazzo G, Fabbrizio P, Apolloni S, Coccurello R, Volonté C. Stimulation of P2X7 Enhances Whole Body Energy Metabolism in Mice. Front Cell Neurosci. (2019) 13:390. doi: 10.3389/fncel.2019.00390
41. Volonté C, Apolloni S D., Skaper S, Burnstock G. P2X7 Receptors: Channels, Pores and More. CNS Neurol Disord - Drug Targets. (2012) 11:705–21. doi: 10.2174/187152712803581137
42. Beaucage KL, Xiao A, Pollmann SI, Grol MW, Beach RJ, Holdsworth DW, et al. Loss of P2X7 nucleotide receptor function leads to abnormal fat distribution in mice. Purinergic Signal. (2014) 10:291–04. doi: 10.1007/s11302-013-9388-x
43. Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 Receptor in Infection and Inflammation. Immunity. (2017) 47:15–1. doi: 10.1016/j.immuni.2017.06.020
44. Di Virgilio F, Sarti AC, Grassi F. Modulation of innate and adaptive immunity by P2X ion channels. Curr Opin Immunol. (2018) 52:51–9. doi: 10.1016/j.coi.2018.03.026
45. Idzko M, Ferrari D, Eltzschig HK. Nucleotide signalling during inflammation. Nature. (2014) 509:310–7. doi: 10.1038/nature13085
46. Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A, Idzko M, et al. The P2X 7 receptor: a key player in IL-1 processing and release. J Immunol. (2007) 176:3877–83. doi: 10.4049/jimmunol.179.12.8569-b
47. Okumura H, Shiba D, Kubo T, Yokoyama T. P2X7 receptor as sensitive flow sensor for ERK activation in osteoblasts. Biochem Biophys Res Commun. (2008) 3:486–90. doi: 10.1016/j.bbrc.2008.05.066
48. Portales-Cervantes L, Niño-Moreno P, Doníz-Padilla L, Baranda-Candido L, García-Hernández M, Salgado-Bustamante M, et al. Expression and function of the P2X7 purinergic receptor in patients with systemic lupus erythematosus and rheumatoid arthritis. Hum Immunol. (2010) 71:818–25. doi: 10.1016/j.humimm.2010.05.008
49. Adinolfi E, De Marchi E, Orioli E, Pegoraro A, Di Virgilio F. Role of the P2X7 receptor in tumor-associated inflammation. Curr Opin Pharmacol. (2019) 47:59–4. doi: 10.1016/J.COPH.2019.02.012
50. Guerra Martinez C. P2X7 receptor in cardiovascular disease: The heart side. Clin Exp Pharmacol Physiol. (2019) 46:513–26. doi: 10.1111/1440-1681.13079
51. Savio LEB, de Andrade Mello P, Figliuolo VR, de Avelar Almeida TF, Santana PT, Oliveira SDS, et al. CD39 limits P2X7 receptor inflammatory signaling and attenuates sepsis-induced liver injury. J Hepatol. (2017) 67:716–26. doi: 10.1016/j.jhep.2017.05.021
52. Antonioli L, Giron MC, Colucci R, Pellegrini C, Sacco D, Caputi V, et al. Involvement of the P2X7 purinergic receptor in colonic motor dysfunction associated with bowel inflammation in rats. PLoS One. (2014) 9:e116253 doi: 10.1371/journal.pone.0116253
53. Wan P, Liu X, Xiong Y, Ren Y, Chen J, Lu N, et al. Extracellular ATP mediates inflammatory responses in colitis via P2 × 7 receptor signaling. Sci Rep. (2016) 6:19108. doi: 10.1038/srep19108
54. Alves LA, Soares Bezerra RJ, Faria RX, Ferreira LGB, Frutuoso VDS. Physiological roles and potential therapeutic applications of the P2X7 receptor in inflammation and pain. Molecules. (2013) 18:10953–72. doi: 10.3390/molecules180910953
55. Beamer E, Fischer W, Engel T. The ATP-gated P2X7 receptor as a target for the treatment of drug-resistant epilepsy. Front Neurosci. (2017) 11:21. doi: 10.3389/fnins.2017.00021
56. Ribeiro DE, Roncalho AL, Glaser T, Ulrich H, Wegener G, Joca S. P2X7 Receptor Signaling in Stress and Depression. Int J Mol Sci. (2019) 20:E2778. doi: 10.3390/ijms20112778
57. Madec S, Rossi C, Chiarugi M, Santini E, Salvati A, Ferrannini E, et al. Adipocyte P2X7 receptors expression: A role in modulating inflammatory response in subjects with metabolic syndrome? Atherosclerosis. (2011) 219:552–58. doi: 10.1016/j.atherosclerosis.2011.09.012
58. Rossi C, Santini E, Chiarugi M, Salvati A, Comassi M, Vitolo E, et al. The complex P2X7receptor/inflammasome in perivascular fat tissue of heavy smokers. Eur J Clin Invest. (2014) 44:295–02. doi: 10.1111/eci.12232
59. Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. (2011) 117:3720–32. doi: 10.1182/blood-2010-07-273417
60. Wilson HL, Francis SE, Dower SK, Crossman DC. Secretion of Intracellular IL-1 Receptor Antagonist (Type 1) Is Dependent on P2X 7 Receptor Activation. J Immunol. (2004) 173:1202–08. doi: 10.4049/jimmunol.173.2.1202
61. Larsen CM, Faulenbach M, Vaag A, Vølund A, Ehses JA, Seifert B, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med. (2007) 356:1517–26. doi: 10.1056/NEJMoa065213
62. Burke SJ, Batdorf HM, Burk DH, Martin TM, Mendoza T, Stadler K, et al. Pancreatic deletion of the interleukin-1 receptor disrupts whole body glucose homeostasis and promotes islet β-cell de-differentiation. Mol Metab. (2018) 14:95–07. doi: 10.1016/j.molmet.2018.06.003
63. Glas R, Sauter NS, Schulthess FT, Shu L, Oberholzer J, et al. Purinergic P2X7 receptors regulate secretion of interleukin-1 receptor antagonist and beta cell function and survival. Diabetologia. (2009) 52:1579–88. doi: 10.1007/s00125-009-1349-0
64. Uresti-Rivera EE, García-Jacobo RE, Méndez-Cabañas JA, Gaytan-Medina LE, Cortez-Espinosa N, Portales-Pérez DP, et al. The presence of the 1068 G>A variant of P2X7 receptors is associated to an increase in IL-1Ra levels, insulin secretion and pancreatic β-cell function but not with glycemic control in type 2 diabetes patients. Gene. (2018) 652:1–6. doi: 10.1016/j.gene.2018.01.084
65. Giuliani AL, Sarti AC, Falzoni S, Di Virgilio F. The P2X7 receptor-interleukin-1 liaison. Front Pharmacol. (2017) 8:123. doi: 10.3389/fphar.2017.00123
66. Prasad GR. Metabolic syndrome and chronic kidney disease: Current status and future directions. World J Nephrol. (2014) 3:210–9. doi: 10.5527/wjn.v3.i4.210
67. Solini A, Menini S, Rossi C, Ricci C, Santini E, Blasetti Fantauzzi C, et al. The purinergic 2X7 receptor participates in renal inflammation and injury induced by high-fat diet: Possible role of NLRP3 inflammasome activation. J Pathol. (2013) 231:342–53. doi: 10.1002/path.4237
68. Menzies RI, Booth JWR, Mullins JJ, Bailey MA, Tam FWK, Norman JT, et al. Hyperglycemia-induced Renal P2X7 Receptor Activation Enhances Diabetes-related Injury. EBioMedicine. (2017) 19:73–3. doi: 10.1016/j.ebiom.2017.04.011
69. Haanes KA, Schwab A, Novak I. The P2X7 Receptor Supports Both Life and Death in Fibrogenic Pancreatic Stellate Cells. PLoS One. (2012) 7:e51164. doi: 10.1371/journal.pone.0051164
70. Coutinho-Silva R, Parsons M, Robson T, Lincoln J, Burnstock G. P2X and P2Y purinoceptor expression in pancreas from streptozotocin-diabetic rats. Mol Cell Endocrinol. (2003) 204:141–54. doi: 10.1016/S0303-7207(03)00003-0
71. Tozzi M, Larsen AT, Lange SC, Giannuzzo A, Andersen MN, Novak I. The P2X7 receptor and pannexin-1 are involved in glucose-induced autocrine regulation in β-cells. Sci Rep. (2018) 8:8926. doi: 10.1038/s41598-018-27281-9
72. Todd JN, Poon W, Lyssenko V, Groop L, Nichols B, Wilmot M, et al. Variation in glucose homeostasis traits associated with P2RX7 polymorphisms in mice and humans. J Clin Endocrinol Metab. (2015) 100:e688–96. doi: 10.1210/jc.2014-4160
73. Bourzac JF, L'Ériger K, Larrivée JF, Arguin G, Bilodeau MS, Stankova J, et al. Glucose transporter 2 expression is down regulated following P2X7 activation in enterocytes. J Cell Physiol. (2013) 228:120–9. doi: 10.1002/jcp.24111
74. Arguin G, Bourzac JF, Placet M, Molle CM, Paquette M, Beaudoin JF, et al. The loss of P2X7 receptor expression leads to increase intestinal glucose transit and hepatic steatosis. Sci Rep. (2017) 7:12917. doi: 10.1038/s41598-017-13300-8
75. Sugiyama T, Kobayashi M, Kawamura H, Li Q, Puro DG. Enhancement of P2X 7 -Induced Pore Formation and Apoptosis: An Early Effect of Diabetes on the Retinal Microvasculature. Investig Opthalmology Vis Sci. (2004) 45:1026–32. doi: 10.1167/iovs.03-1062
76. Platania CBM, Giurdanella G, Di Paola L, Leggio GM, Drago F, Salomone S, et al. P2X7 receptor antagonism: Implications in diabetic retinopathy. Biochem Pharmacol. (2017) 138:130–9. doi: 10.1016/j.bcp.2017.05.001
77. Novak I, Solini A. P2X receptor-ion channels in the inflammatory response in adipose tissue and pancreas - potential triggers in onset of type 2 diabetes? Curr Opin Immunol. (2018) 52:1–7. doi: 10.1016/j.coi.2018.02.002
78. Solini A, Novak I. Role of the P2X7 receptor in the pathogenesis of type 2 diabetes and its microvascular complications. Curr Opin Pharmacol. (2019) 47:75–81. doi: 10.1016/j.coph.2019.02.009
79. Coccurello R, Maccarrone M. Hedonic eating and the “delicious circle”: From lipid-derived mediators to brain dopamine and back. Front Neurosci. (2018) 12:271. doi: 10.3389/fnins.2018.00271
80. Speakman JR, Levitsky DA, Allison DB, Bray MS, De Castro JM, Clegg DJ, et al. Set points, settling points and some alternative models: Theoretical options to understand how genes and environments combine to regulate body adiposity. DMM Dis Model Mech. (2011) 4:733–45. doi: 10.1242/dmm.008698
81. Lowell BB, Spiegelman BM. Towards a molecular understanding of adaptive thermogenesis. Nature. (2000) 404:652–60. doi: 10.1038/35007527
82. Oelkrug R, Polymeropoulos ET, Jastroch M. Brown adipose tissue: physiological function and evolutionary significance. J Comp Physiol B Biochem Syst Environ Physiol. (2015) 185:587–06. doi: 10.1007/s00360-015-0907-7
83. Harms M, Seale P. Brown and beige fat: Development, function and therapeutic potential. Nat Med. (2013) 19:1252–63. doi: 10.1038/nm.3361
84. Sidossis L, Kajimura S. Brown and beige fat in humans: Thermogenic adipocytes that control energy and glucose homeostasis. J Clin Invest. (2015) 125:478–86. doi: 10.1172/JCI78362
85. Bonet ML, Oliver P, Palou A. Pharmacological and nutritional agents promoting browning of white adipose tissue. Biochim Biophys Acta - Mol Cell Biol Lipids. (2013) 1831:969–85. doi: 10.1016/j.bbalip.2012.12.002
86. Van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JMAFL, Kemerink GJ, et al. Cold-activated brown adipose tissue in healthy men. N Engl J Med. (2009) 360:1500–8. doi: 10.1056/NEJMoa0808718
87. Jespersen NZ, Feizi A, Andersen ES, Heywood S, Hattel HB, Daugaard S, et al. Heterogeneity in the perirenal region of humans suggests presence of dormant brown adipose tissue that contains brown fat precursor cells. Mol Metab. (2019) 24:30–43. doi: 10.1016/j.molmet.2019.03.005
88. Sacks H, Symonds ME. Anatomical locations of human brown adipose tissue: Functional relevance and implications in obesity and type 2 diabetes. Diabetes. (2013) 62:1783–90. doi: 10.2337/db12-1430
89. Nicholls DG, Rial E. A history of the first uncoupling protein, UCP1. J Bioenerg Biomembr. (1999) 31:399–06. doi: 10.1023/A:1005436121005
90. Haemmerle G, Lass A, Zimmermann R, Gorkiewicz G, Meyer C, Rozman J, et al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science. (2006) 312:734–7. doi: 10.1126/science.1123965
91. Cannon B, Nedergaard J. Brown Adipose Tissue: Function and Physiological Significance. Physiol Rev. (2004) 84:277–59. doi: 10.1152/physrev.00015.2003
92. Fedorenko A, Lishko P V., Kirichok Y. Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell. (2012) 151:400–13. doi: 10.1016/j.cell.2012.09.010
93. Klingenberg M. Wanderings in bioenergetics and biomembranes. Biochim Biophys Acta - Bioenerg. (2010) 1797:579–94. doi: 10.1016/j.bbabio.2010.02.012
94. Shabalina IG, Ost M, Petrovic N, Vrbacky M, Nedergaard J, Cannon B. Uncoupling protein-1 is not leaky. Biochim Biophys Acta. (2010) 1797:773–84. doi: 10.1016/j.bbabio.2010.04.007
95. Zurlo F, Larson K, Bogardus C, Ravussin E. Skeletal muscle metabolism is a major determinant of resting energy expenditure. J Clin Invest. (1990) 86:1423–27. doi: 10.1172/JCI114857
96. Kiens B, Essen-Gustavsson B, Christensen NJ, Saltin B. Skeletal muscle substrate utilization during submaximal exercise in man: effect of endurance training. J Physiol. (1993) 469:459–78. doi: 10.1113/jphysiol.1993.sp019823
97. Romijn JA, Coyle EF, Sidossis LS, Gastaldelli A, Horowitz JF, Endert E, et al. Regulation of endogenous fat and carbohydrate metabolism in relation to exercise intensity and duration. Am J Physiol. (1993) 265:E380–91. doi: 10.1152/ajpendo.1993.265.3.E380
98. Van Loon LJC, Greenhaff PL, Constantin-Teodosiu D, Saris WHM, Wagenmakers AJM. The effects of increasing exercise intensity on muscle fuel utilisation in humans. J Physiol. (2001) 536:295–04. doi: 10.1111/j.1469-7793.2001.00295.x
99. Forman HJ, Ursini F, Maiorino M. An overview of mechanisms of redox signaling. J Mol Cell Cardiol. (2014) 73:2–9. doi: 10.1016/j.yjmcc.2014.01.018
100. Loureiro ACC, Rêgo-Monteiro IC do, Louzada RA, Ortenzi VH, Aguiar AP de, Abreu ES de, et al. Differential Expression of NADPH Oxidases Depends on Skeletal Muscle Fiber Type in Rats. Oxid Med Cell Longev. (2016) 2016:6738701. doi: 10.1155/2016/6738701
101. Pearson T, Kabayo T, Ng R, Chamberlain J, McArdle A, Jackson MJ. Skeletal muscle contractions induce acute changes in cytosolic superoxide, but slower responses in mitochondrial superoxide and cellular hydrogen peroxide. PLoS One. (2014) 9:e96378. doi: 10.1371/journal.pone.0096378
102. Trapp EG, Chisholm DJ, Freund J, Boutcher SH. The effects of high-intensity intermittent exercise training on fat loss and fasting insulin levels of young women. Int J Obes. (2008) 32:684–91. doi: 10.1038/sj.ijo.0803781
103. Hazell TJ, Olver TD, Hamilton CD, Lemon PWR. Two minutes of sprint-interval exercise elicits 24-hr oxygen consumption similar to that of 30 min of continuous endurance exercise. Int J Sport Nutr Exerc Metab. (2012) 22:276–83. doi: 10.1123/ijsnem.22.4.276
104. Henríquez-Olguín C, Renani LB, Arab-Ceschia L, Raun SH, Bhatia A, Li Z, et al. Adaptations to high-intensity interval training in skeletal muscle require NADPH oxidase 2. Redox Biol. (2019) 24:101188. doi: 10.1016/j.redox.2019.101188
105. Di Virgilio F. The P2Z purinoceptor: an intriguing role in immunity, inflammation and cell death. Immunol Today. (1995) 16:524–8. doi: 10.1016/0167-5699(95)80045-X
106. Adinolfi E, Callegari MG, Ferrari D, Bolognesi C, Minelli M, Wieckowski MR, et al. Basal Activation of the P2X7 ATP Receptor Elevates Mitochondrial Calcium and Potential, Increases Cellular ATP Levels, and Promotes Serum-independent Growth. Mol Biol Cell. (2005) 16:3260–72. doi: 10.1091/mbc.e04-11-1025
107. Primeau JO, Armanious GP, Fisher ME, Young HS. The sarcoendoplasmic reticulum calcium ATPase. In: Membrane Protein Complexes: Structure and Function Singapore: Springer (2018). p. 229–58. doi: 10.1007/978-981-10-7757-9_8
108. Periasamy M, Herrera JL, Reis FCG. Skeletal muscle thermogenesis and its role in whole body energy metabolism. Diabetes Metab J. (2017) 41:327–36. doi: 10.4093/dmj.2017.41.5.327
109. Divet A, Lompré AM, Huchet-Cadiou C. Effect of cyclopiazonic acid, an inhibitor of the sarcoplasmic reticulum Ca-ATPase, on skeletal muscles from normal and mdx mice. Acta Physiol Scand. (2005) 184:173–86. doi: 10.1111/j.1365-201X.2005.01450.x
110. Allen DG, Gervasio OL, Yeung EW, Whitehead NP. Calcium and the damage pathways in muscular dystrophy. Canadian J Physiol Pharmacol. (2010) 88:83–1. doi: 10.1139/Y09-058
111. Franco A, Lansman JB. Calcium entry through stretch-inactivated ion channels in mdx myotubes. Nature. (1990) 344:670–3. doi: 10.1038/344670a0
112. Whitehead NP, Yeung EW, Allen DG. Muscle damage in mdx (dystrophic) mice: Role of calcium and reactive oxygen species. Clin Exp Pharmacol Physiol. (2006) 33:657–62. doi: 10.1111/j.1440-1681.2006.04394.x
113. Young CNJ, Sinadinos A, Gorecki DC. P2X receptor signaling in skeletal muscle health and disease. Wiley Interdiscip Rev Membr Transp Signal. (2013) 2:265–74. doi: 10.1002/wmts.96
114. Yeung D, Zabłocki K, Lien C-F, Jiang T, Arkle S, Brutkowski W, et al. Increased susceptibility to ATP via alteration of P2X receptor function in dystrophic mdx mouse muscle cells. FASEB J. (2006) 20:610–20. doi: 10.1096/fj.05-4022com
115. Young CNJ, Brutkowski W, Lien CF, Arkle S, Lochmüller H, Zabłocki K, et al. P2X7 purinoceptor alterations in dystrophic mdx mouse muscles: Relationship to pathology and potential target for treatment. J Cell Mol Med. (2012) 16:1026–37. doi: 10.1111/j.1582-4934.2011.01397.x
116. Sahoo SK, Shaikh SA, Sopariwala DH, Bal NC, Periasamy M. Sarcolipin protein interaction with Sarco(endo)plasmic reticulum CA 2+ ATPase (SERCA) Is distinct from phospholamban protein,and only sarcolipin can promote uncoupling of the serca pump. J Biol Chem. (2013) 288:6881–9. doi: 10.1074/jbc.M112.436915
117. Vidal-Puig A, Solanes G, Grujic D, Flier JS, Lowell BB. UCP3: An uncoupling protein homologue expressed preferentially and abundantly in skeletal muscle and brown adipose tissue. Biochem Biophys Res Commun. (1997) 235:79–2. doi: 10.1006/bbrc.1997.6740
118. Macher G, Koehler M, Rupprecht A, Kreiter J, Hinterdorfer P, Pohl EE. Inhibition of mitochondrial UCP1 and UCP3 by purine nucleotides and phosphate. Biochim Biophys Acta Biomembr. (2018) 1860:664–72. doi: 10.1016/j.bbamem.2017.12.001
119. Zhou M, Lin BZ, Coughlin S, Vallega G, Pilch PF. UCP-3 expression in skeletal muscle: Effects of exercise, hypoxia, and AMP-activated protein kinase. Am J Physiol - Endocrinol Metab. (2000) 279:E622–9. doi: 10.1152/ajpendo.2000.279.3.e622
120. Millet L, Vidal H, Andreelli F, Larrouy D, Riou JP, Ricquier D, et al. Increased uncoupling protein-2 and−3 mRNA expression during fasting in obese and lean humans. J Clin Invest. (1997) 100:2265–70. doi: 10.1172/JCI119811
121. Matsuda J, Hosoda K, Itoh H, Son C, Doi K, Tanaka T, et al. Cloning of rat uncoupling protein-3 and uncoupling protein-2 cDNAs: Their gene expression in rats fed high-fat diet. FEBS Lett. (1997) 418:200–4. doi: 10.1016/S0014-5793(97)01381-1
122. Bezaire V, Spriet LL, Campbell S, Sabet N, Gerrits M, Bonen A, et al. Constitutive UCP3 overexpression at physiological levels increases mouse skeletal muscle capacity for fatty acid transport and oxidation. FASEB J. (2005) 19:977–9. doi: 10.1096/fj.04-2765fje
123. Cheol SC, Fillmore JJ, Kim JK, Liu ZX, Kim S, Collier EF, et al. Overexpression of uncoupling protein 3 in skeletal muscle protects against fat-induced insulin resistance. J Clin Invest. (2007) 117:1995–03. doi: 10.1172/JCI13579
124. Schrauwen P, Hesselink M. UCP2 and UCP3 in muscle controlling body metabolism. J Exp Biol. (2002) 205:2275–85.
125. Brand MD, Esteves TC. Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab. (2005) 2:85–93. doi: 10.1016/j.cmet.2005.06.002
126. Mills EM, Rusyniak DE, Sprague JE. The role of the sympathetic nervous system and uncoupling proteins in the thermogenesis induced by 3,4-methylenedioxymethamphetamine. J Mol Med. (2004) 82:787–99. doi: 10.1007/s00109-004-0591-7
127. Riley CL, Dao C, Kenaston MA, Muto L, Kohno S, Nowinski SM, et al. The complementary and divergent roles of uncoupling proteins 1 and 3 in thermoregulation. J Physiol. (2016) 594:7455–64. doi: 10.1113/JP272971
128. Samec S, Seydoux J, Dulloo AG. Role of UCP homologues in skeletal muscles and brown adipose tissue: mediators of thermogenesis or regulators of lipids as fuel substrate? FASEB J. (1998) 12:715–24. doi: 10.1096/fasebj.12.9.715
129. Schrauwen P, Saris WHM, Hesselink MKC. An alternative function for human uncoupling protein 3: protection of mitochondria against accumulation of nonesterified fatty acids inside the mitochondrial matrix. FASEB J. (2001) 15:2497–502. doi: 10.1096/fj.01-0400hyp
130. Goglia F, Skulachev VP. A function for novel uncoupling proteins: antioxidant defense of mitochondrial matrix by translocating fatty acid peroxides from the inner to the outer membrane leaflet. FASEB J. (2003) 17:1585–91. doi: 10.1096/fj.03-0159hyp
131. Brand MD, Buckingham JA, Esteves TC, Green K, Lambert AJ, Miwa S, et al. Mitochondrial superoxide and aging: Uncoupling-protein activity and superoxide production. Biochem Soc Symp. (2004) 71:203–13. doi: 10.1042/bss0710203
132. Vidal-Puig AJ, Grujic D, Zhang CY, Hagen T, Boss O, Ido Y, et al. Energy metabolism in uncoupling protein 3 gene knockout mice. J Biol Chem. (2000) 275:16258–66. doi: 10.1074/jbc.M910179199
133. Pohl EE, Rupprecht A, Macher G, Hilse KE. Important trends in UCP3 investigation. Front Physiol. (2019) 10:470 doi: 10.3389/fphys.2019.00470
134. Ferreira LF, Laitano O. Regulation of NADPH oxidases in skeletal muscle. Free Radic Biol Med. (2016) 98:18–28. doi: 10.1016/j.freeradbiomed.2016.05.011
135. Aguer C, Fiehn O, Seifert EL, Bézaire V, Meissen JK, Daniels A, et al. Muscle uncoupling protein 3 overexpression mimics endurance training and reduces circulating biomarkers of incomplete β-oxidation. FASEB J. (2013) 27:4213–25. doi: 10.1096/fj.13-234302
136. Leung YM. P2X7 receptor as a double-edged sword: Neurotrophic and neurotoxic effects. Biomed. (2011) 1:16–20. doi: 10.1016/j.biomed.2011.10.003
Keywords: P2X7 receptor, energy metabolism, lipid oxidation, adipose tissue, thermogenesis, skeletal muscle
Citation: Coccurello R and Volonté C (2020) P2X7 Receptor in the Management of Energy Homeostasis: Implications for Obesity, Dyslipidemia, and Insulin Resistance. Front. Endocrinol. 11:199. doi: 10.3389/fendo.2020.00199
Received: 03 December 2019; Accepted: 19 March 2020;
Published: 12 May 2020.
Edited by:
Carolyn Mary Ecelbarger, Georgetown University, United StatesReviewed by:
Tobias Fromme, Technical University of Munich, GermanyHayley Maree O'Neill, Bond University, Australia
Copyright © 2020 Coccurello and Volonté. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roberto Coccurello, cm9iZXJ0by5jb2NjdXJlbGxvQGNuci5pdA==