 He Zhou
He Zhou Lin Sun
Lin Sun Siwen Zhang
Siwen Zhang Xue Zhao
Xue Zhao Xiaokun Gang
Xiaokun Gang Guixia Wang
Guixia Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 24 March 2020
Sec. Clinical Diabetes
Volume 11 - 2020 | https://doi.org/10.3389/fendo.2020.00125
Type 1 diabetes (T1D) is a multifactorial autoimmune disease mediated by genetic, epigenetic, and environmental factors. In recent years, the emergence of high-throughput sequencing has allowed us to investigate the role of gut microbiota in the development of T1D. Significant changes in the composition of gut microbiome, also termed dysbiosis, have been found in subjects with clinical or preclinical T1D. However, whether the dysbiosis is a cause or an effect of the disease remains unclear. Currently, increasing evidence has supported a causal link between intestine microflora and T1D development. The current review will focus on recent research regarding the associations between intestine microbiome and T1D progression with an intention to evaluate the causality. We will also discuss the possible mechanisms by which imbalanced gut microbiota leads to the development of T1D.
Type 1 diabetes (T1D) is an autoimmune disease characterized by insufficient insulin production, which is caused by autoreactive T-cell-mediated partial or complete destruction of islet beta cells in patients (1, 2), with a high incidence in children and young adults (3). Genetic factors play important roles in T1D etiology, and a number of genetic loci associated with T1D have already been identified (4). Environmental factors are also a pivotal contributor to the disease (5, 6), which is evidenced in these observations that fewer than 10% of genetically predisposed individuals develop the disease (7), the increasing frequency of lower-risk genotypes in diagnosed patients (8, 9), the disparate incidence of diabetes in monozygotic twins (10), and the substantially risen prevalence in recent decades (11).
In recent years, gastrointestinal microbiota has been recognized as one of the key environmental factors associated with the development of T1D. Microbes in the human gut make up to 100 trillion cells, 10 times the number of human cells (12). More than 95% species of gut microbiota can be classified into four major microbial phyla: Firmicutes, Bacteroidetes, Actinobacteria, and Protecteobacteria (13, 14). The intestinal microbiota is sometimes described as a “hidden organ” (15) based on their capacity to perform diverse physiological functions comprising fighting against pathogens (16), producing energy (17), maintaining intestinal epithelial integrity (18, 19), and regulating immunological activities (20). There is a mutualistic relationship between the human host and the intestinal flora to maintain homeostasis (21).
A perturbation to the normal composition of commensal communities, also termed dysbiosis, may break the homeostasis and lead to various autoimmune diseases (15, 22). With the introduction of high-throughput sequencing technologies, accumulating evidence has shown that there are remarkable differences in the intestinal microbial profile between T1D patients and healthy controls, indicating a close interplay between diabetes development and gut microbiota (23). In addition, a growing number of evidence suggests that compositional changes in gut microbiota may be involved in the pathophysiology of T1D, and gut dysbiosis-mediated immunological deregulation and gut leakiness are the possible pathogenic mechanisms. However, whether these microbial changes are causal, responsive, or both so far has not been completely elucidated. Once the causal role of gut community in the onset of T1D is determined, it will provide a new opportunity for preventative or therapeutic strategies for T1D on account of the modifiable nature of gut microbiota. In this review, we will elaborate on common compositional changes of intestinal microbiome associated with T1D patients and review the evidence supporting a causal role of intestinal microbiome in the onset and progression of the disease. Furthermore, we will discuss the possible mechanisms whereby intestinal microbiome influences the T1D progression, which holds the key to unravel the complex interaction between microbiota community and host.
To determine whether there is objective evidence supporting a causal role of gut community in the onset of T1D, we reviewed literatures from different databases including MEDLINE, EMBASE, Web of Science, and the Cochrane Library. The search strategy used a combination of MeSH terms and keywords pertaining to gut microbiota and T1D. The inclusion criteria were as follows: (1) case–control or cohort studies comparing gut microbiota in patients with T1D or islet autoimmunity and healthy controls and (2) well-controlled intervention studies in human and murine models detecting the bacterial changes in fecal or mucosal samples. Studies that did not assign a control group were excluded. Two investigators independently performed the literature search and assessed the eligibility of selected studies based on the established inclusion criteria. Any discrepancies between investigators were resolved by discussion until consensus was reached. Then, we proposed the possible mechanisms whereby aberrant microbiota composition influences the T1D development.
Previous studies have found large significant differences in the microbial composition between subjects with T1D or islet autoimmunity and healthy controls (Table 1). Most of the findings originated from cross-sectional studies, which cannot directly determinize causality, but still an important initial step toward evaluating the causal role of gut microbiota in T1D pathogenesis.

Table 1. Gut microbiota changes in preclinical and clinical type 1 diabetes (T1D).
One case–control study analyzed the gut microbiota in 16 children with T1D and 16 unaffected children. Compared with healthy control group, the Firmicutes/Bacteroides (F/B) ratio in T1D patients was significantly reduced (24). A reduction in F/B ratio was also seen in other studies. Recent studies have identified that the intestinal community profile of T1D patients in China and Turkey is also characterized by a decreased F/B ratio in comparison to healthy subjects (25, 41). Additionally, a cohort study revealed that the F/B ratio declined over time as children developed islet autoimmunity and autoimmune diabetes ultimately (26). However, conflicting data with regard to F/B ratio in individuals diagnosed with T1D have been published too (28, 29), which may be attributed to different sample sizes, data analysis approaches, and geographical location. Another common gut microbiome shift associated with the T1D development is the decreased microbial diversity, which has been reported both in T1D children (30, 31) and in autoantibody-positive children (26, 27, 32). It was further found that the decline in bacterial diversity was specific to seroconverters that eventually developed into T1D but not in undeveloped seroconverters (33). Moreover, metagenomic data from several studies showed a significant reduction in the abundance of butyrate producers such as Clostridium clusters IV and XIVa and mucin-degrading bacteria such as Prevotella and Akkermansia in T1D patients. A study comparing gut microbiota composition between 28 children with new-onset T1D and 27 age-matched unaffected children revealed that control children exhibited a higher number of butyrate-producing species within Clostridium clusters IV and XIVa compared to corresponding diabetic children (34). Children with new-onset T1D in another study also showed a similar trend regarding Clostridium clusters IV and XIVa (35). In addition, the decreased butyrate-producing species have been documented in adults with longstanding T1D (36) and children with pancreas autoimmunity (27, 32). In line with the reduction in butyrate-producing species that can induce mucin synthesis, Brown and colleagues have identified that Prevotella and Akkermansia, as perspective signatures of elevated mucin synthesis, were substantially lower in seropositive individuals than in healthy children (27). A decrease in Bifidobacterium was also reported in T1D subjects by several studies. In a study investigating the bacterial compositional differences among children with T1D and maturity-onset diabetes of the young 2 (MODY2) and healthy control subjects by 16S ribosomal RNA (rRNA) gene sequencing, the authors concluded that T1D children presented with a reduced level of Bifidobacterium, and perhaps even more interestingly, it was observed that the intestinal microbiota profile of T1D patients was different from not only healthy subjects but also subjects with MODY2 (30). Another study also using 16S rRNA gene sequencing concluded the similar trends regarding Bifidobacterium (37). Additionally, a lower proportion of Bifidobacterium in T1D patients was also detected by other techniques including analysis of the microbial proteome (38) and stool cultures (39). Additionally, the colonization of intestinal Candida albicans was also reported to be positively linked to T1D development (39, 40).
In the past decade, a growing number of animal studies have suggested a causal link between intestine microflora and T1D development. This may be due to the fact that, in contrast to human studies, the gut microbiota in murine models can be disturbed under strictly controlled conditions to minimize confounding factors (42). Non-obese diabetic (NOD) mice (43) and bio-breeding diabetes-prone (BB-DP) rats (44) are two widely studied animal models of autoimmune diabetes. Both models carry the risk genes of T1D and develop T1D spontaneously (43, 45, 46).
Antibiotic intervention modulates the bacterial composition by selective removal of certain microbial lineages. It was reported that the alterations of bacterial profile in the gut induced by vancomycin remarkably increased the incidence of T1D in NOD mice (47, 48), while in another study, vancomycin displayed T1D-protecting effect in NOD mice (49). Other specific antibiotics or combinations of antibiotics have also been found to cause distinct alterations in gut microbial composition compared to untreated control NOD mice and subsequently delayed or accelerated disease progression (50–52). The types of antibiotics or the time of administration were different in each study, which may account for their disparate outcomes.
Probiotic supplement is another important intervention to change the composition of intestinal flora in NOD mice or BB-DP rats. Probiotics are defined as specific live microorganisms that can create a favorable gut environment when administered in sufficient amounts (53). Probiotics were proved to prevent diabetes development in animal models, indicating that altered intestinal microbiome has a role in manipulating the onset of T1D. A published study showed that oral administration of VSL#3, a combination of several probiotics, could prevent diabetes in NOD mice when administrated from 4 weeks of age (54). The protecting effect of VSL#3 in NOD mice was further confirmed by another study performed by Dolpady et al. (55). Probiotics Clostridium butyricum CGMCC0313.1 were also demonstrated to suppress the onset of T1D in NOD mice by selectively modulating the structure of gut microbiota, including increasing the F/B ratio, Clostridium, and butyrate-producing bacteria (56). Conferred protection against diabetes by probiotics was also shown in BB-DP rats (57).
Fecal microbiota transplantation (FMT) also can change the risk of progression to T1D by altering the gut microbiome profile in NOD mice. A study demonstrated that intestinal microflora transferred from diabetes protective NOD mice, which are genetically deficient in myeloid differentiation primary response gene 88 (MyD88), could stably alter the gut bacterial composition and eventually reduced insulitis significantly and delayed the onset of T1D in wild-type NOD mice (58). It is well-established that NOD mice harbor more diabetogenic microbes in the gut compared with non-obese diabetes-resistant (NOR) mice. A study found that the FMT from NOD mice to NOR mice elicited insulitis in NOR mice (59). Nevertheless, a recent study found that the transfer of the whole microbiota from the low- to the high-incidence colony of NOD mice did not reduce diabetes incidence. Intriguingly, single symbionts transfer of Akkermansia muciniphila could delay diabetes development in the high-incidence NOD colony, suggesting that individual microbiota members might have potential significance in the pathogenesis of T1D (60).
Animal studies have indicated that dietary factors such as gluten and fiber may change the incidence of T1D by altering the composition of gut microbiota. A gluten-free diet has been shown to reduce diabetes incidence in NOD mice along with elevated Akkermansia and reduced Bifidobacterium, Tannerella, and Barnesiella species compared to gluten-containing diet. Notably, adding gluten to the gluten-free diet reversed the antidiabetogenic effect as a result of reversing the gut bacterial composition (61). A study conducted by Toivonen et al. noted that the two fermentable fibers, pectin and xylan, exerted diabetes-promoting effect in NOD mice by significantly enhancing the level of Bacteroides and depleting the mucin-degrading bacteria such as Verrucomicrobiales and Prevotellaceae (62). Moreover, a recent study showed that the low esterified pectin, a novel dietary fiber, could decrease the diabetes incidence in NOD mice by selectively enriching specific microbial species that produce short-chain fatty acids (SCFAs) (63).
This chicken–egg situation (causal or consequence) can be partially clarified in several cohort studies, which have identified that the changes in gut microbiota composition occurred before the T1D development (26, 27, 32, 33). Although these prospective studies will provide a timeline of disease progression associated with microbial dysbiosis, a causal contribution can only be derived from intervention studies.
It was reported that breast milk, as an independent protective factor, could lead to a drop in the incidence of T1D (64), and the microbial composition of breast-fed infants was characterized by a more stable gut microbiome dominated by Bifidobacteria compared to infants who were not breastfed (65). Studies showing the protective effects of probiotics on T1D in humans are limited. One well-known study is that of Uusitalo, who previously reported that early supplementation of probiotics during the first four postnatal weeks reduced the risk of beta cell autoimmunity in infants genetically susceptible to T1D compared to those with no supplementation (66). Recently, a single-center, randomized, double-blind, placebo-controlled pilot study in children with T1D for at least 1 year showed that consumption of prebiotics could alter the gut microbiota composition and decrease the intestinal permeability, leading to improved beta cell function. However, there was no improvement in glycemic control in the prebiotic group, possibly due to the small sample size and relatively short intervention time (67). In addition, Groele et al. are conducting a double-blind, randomized, placebo-controlled study to discuss whether probiotics will improve beta-cell function by modulating the immune system in children with newly diagnosed T1D (68). Nevertheless, such randomized controlled clinical trials are still rare, and to date, there are no studies in humans supporting the link between FMT or antibiotics-induced microbial changes and T1D risk. Overall, although interventional human studies show a hint of promise, a causal relationship still cannot be concluded due to the lack of large-scale prospective studies demonstrating that long-term changes in the bacterial composition alter T1D risk. Further randomized controlled studies in large human cohorts will need to be undertaken.
Collectively, the evidence generated from well-controlled intervention studies in murine models is promising for establishing a causal relationship. However, conclusions from animal studies must be interpreted with caution, as animal models have a number of limitations, making it difficult to translate to humans. In addition, human studies showing a clear association between the long-term changes of gut microbiota and altered T1D risk are still lacking. Thus, the evidence supporting causative relationship summarized above is inconclusive and needs to be further confirmed.
There was a proposed complicated interaction between intestinal microbiota, immune system, and gut permeability (69). A large body of evidence suggested that defective profile of intestinal microbiome may influence the pathogenesis of T1D by affecting immune homeostasis and/or gut permeability (Figure 1) (70).
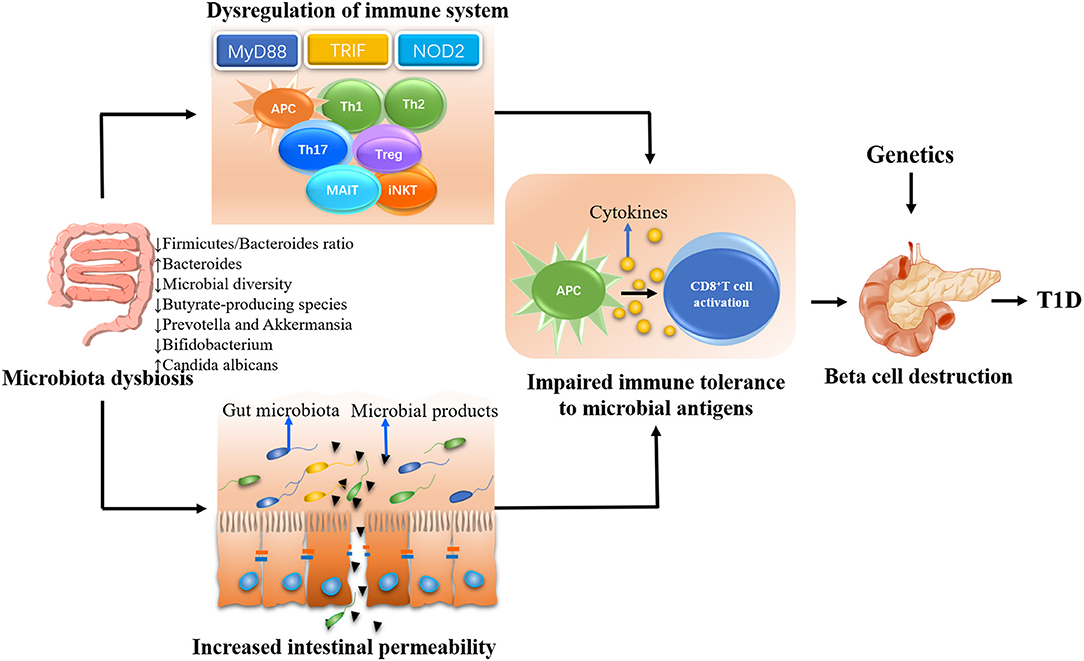
Figure 1. The possible mechanisms whereby gut microbiota influences the type 1 diabetes (T1D) development. The gut microbiota plays a decisive role in the maturation of immune system in early life. Gut dysbiosis will lead to the dysregulation of immune response including both innate and adaptive immune system, eventually resulting in beta cell destruction and the onset of T1D in genetically susceptible individuals. On the other hand, the gut dysbiosis can lead to the disassembly of tight junctions, thereby disrupting the integrity of intestinal barrier. The enhanced intestinal permeability will allow unregulated passage of microbial antigens such as microbiota and their products. These antigens escaping from intestinal tract could be untaken by antigen-presenting cells (APCs), which can process and present antigens to autoreactive T cells and subsequently promote the destruction of pancreatic beta cells in genetically predisposed individuals.
Gastrointestinal microbiota has been considered as the largest organ of the immune system (71) owing to their ability to constantly interact with immunological cells (20). It was demonstrated that there are disorders of the mucosal immune system in NOD mice before diabetes onset (72). Gastrointestinal microflora plays an essential role in the development and maturation of the immune system, and early stages of life are the critical time window for the establishment of immune tolerance (13, 73). According to the Hygiene Hypothesis, it is the lack of microbial stimulation in early childhood resulting from advances in medicine and improved sanitation that leads to the rise in the prevalence of immune-related disorders (74). Thus, the infants, who can benefit from early exposure to specific microorganisms, may be much less likely to develop autoimmune diseases later in life (75, 76). The Hygiene Hypothesis has been supported by murine studies. For example, NOD mice were more likely to develop diabetes when they lived in a clean environment, and it was reported that infection of NOD mice with various bacteria in early life could prevent T1D development (77, 78). In general, hence, it seems that gut microbiome-mediated appropriate immune maturation during early life is critical to prevent T1D development (79). Conversely, alterations of gut microbiota composition will lead to a poorly educated immunity and eventually result in insulin-secreting beta cell damage and the onset of T1D in genetically predisposed subjects (80, 81).
Another proposed T1D pathogenesis associated with microbial dysbiosis is the increased intestinal permeability, often referred to as a “leaky gut” (69), which may act either independently or coincidently with the immunological deregulation (82). A “leaky gut” in T1D has been observed from both human studies and animal research (83–86), indicating that the barrier dysfunction is a primary feature of T1D. Some researchers argued that hyperglycemia and insulitis may contribute to the enhanced leakiness of the gut epithelial barrier in T1D (87). Nevertheless, other researchers favor the notion that the “leaky gut” may be more of a cause than an effect, as a highly permeable gut has been observed before the development of both insulitis and clinical onset of T1D (88, 89). One of the most cited studies is that of Bosi et al., who measured intestinal permeability of 81 subjects at different stages of T1D as well as 40 healthy controls. They found that the increased gut permeability occurred before the manifestation of the disease (90). Similarly, a recent study performed by Harbison and others observed that children at risk for T1D had higher intestinal permeability and were associated with gut microbiota dysbiosis (91). Furthermore, administration of hydrolyzed casein diet has been reported to be able to protect from T1D by improving the gut integrity in BB-DP rats (92), indicating that the melioration of increased gut permeability can protect against autoimmune diabetes development. In line with this notion, infection with barrier-disrupting C. rodentium accelerated insulitis in NOD mice, while NOD mice failed to develop insulitis when infected with the DeltaespF strain, which cannot disrupt gut integrity (93). These studies support the concept that such a “leaky gut” could be a component within the natural history of T1D progression and perhaps is the initial steps of evolution to the disease. It was suggested that the enhanced intestinal permeability, as a consequence of impaired integrity of the intestinal barrier (94), might allow unregulated passage of exogenous antigens especially the microbial components (95). The translocation of these microbial components to systemic compartment could trigger systemic inflammation and autoimmune progression by directly damaging pancreatic beta cells (96). Alternately, these microbial components could be untaken by antigen-presenting cells (APCs), which can process and present the antigen to autoreactive T cells (87, 97), leading to the destruction of islet beta cells (57, 89). Another possible mechanism by which these translocated microbial antigens initiate the diabetes onset is molecular mimicry. Some microbial antigens have homology with islet self-antigen and may result in the destruction of islet beta cells by T-cell cross-reactivity (98). A recent study reported that oral administration of Bacteroides fragilis under loss of gut barrier integrity condition induced by chemical approach could lead to rapid disease progression in NOD mice, further highlighting the role of microbial translocation in contributing to T1D (99).
Understanding such mechanisms through which gut microbiota influences the T1D development is of great importance for developing novel prevention and treatment strategies of autoimmune diabetes. However, before making much research effort on microbiota-based therapies, it would seem to be pivotal to uncover first whether and how dysbiotic gut microbiota contributes to immunological aberrancies and gut leakiness.
Alterations of the intestinal microbiota during early life have been hypothesized to impact T1D pathogenesis by disturbing the normal pattern of immunological maturation (81), and various bacterial taxa and bacterial metabolites as well as bacteria-derived components, which were able to affect immune responses, have been identified (100, 101).
Innate immunity plays an essential role in the etiology of T1D, and investigations showed that interaction of commensal bacteria with the innate immune system was involved in the onset and progression of T1D (3). Toll-like receptors (TLRs), known as important players in the innate immunity, are critical for intestinal homeostasis. TLRs as one of pathogen recognition receptors (PRRs) expressed on immune and non-immune cells can recognize pathogen-associated molecular patterns (PAMPs) derived from microbiota (102) and enable the initiation of the innate immune system (103). There are many different microbes that can facilitate or inhibit autoimmunity of T1D by signaling through different receptors of the TLR family (104–108). The first attempt to investigate innate immune pathway associated with microbial exposure in T1D was conducted in MyD88-deficient NOD mice. MyD88 is an adaptor protein of multiple TLRs that can recognize microbial stimuli and contribute to downstream signaling pathways of TLRs (102, 109). NOD mice deficient in molecule MyD88 were completely protected from T1D under conventional conditions, and the protective effect was derived from beneficial microbial composition, which differed from that of wild-type controls, indicating that the composition of microbiota was changed by host MyD88 deficiency (3). Conversely, MyD88-deficient mice had an increased risk of developing T1D under germ-free (GF) conditions, while the incidence of diabetes was reduced in these mice when exposed to a defined microbial mixture, which further supports the intimate interaction between microbial community and host innate immune system (3). A recent study conducted by Gulden and colleagues unraveled a novel innate immune pathway influenced by gastrointestinal microbiota in T1D development. They found that the deletion of the Toll/interleukin-1 receptor (TIR)-domain-containing adapter-inducing interferon-β (TRIF), another critical adaptor protein downstream of TLRs, could protect NOD mice from diabetes. Importantly, a different microbiota profile was found in TRIF-deficient NOD mice compared to wild-type NOD mice, suggesting that the protective effect of TRIF deficiency is through changing microbial composition (110). In addition, a recent study performed by Simon et al. found that NOD TLR4−/− animals had an increased risk of progression to diabetes along with higher abundance of Bacteroidetes and lower Firmicutes in the large intestine before the onset of T1D when compared to NOD TLR4+/+ mice, indicating that TLR4 expression status determined early alterations of gut microbial composition (111). NOD-like receptors (NLRs), another important PRR, have also been reported to be involved in T1D development through the recognition of bacterial products. Costa et al. found that NOD2 receptor could be activated by translocated gut bacteria in pancreatic lymph nodes (PLNs) of streptozotocin (STZ)-treated mice and contribute to the pathogenesis of T1D, while broad-spectrum antibiotics treatment and NOD2 receptor deletion could protect STZ-treated mice from T1D (112). Additionally, it has been documented that early-life gut microbiota is critical to the development and function of APCs (113). Studies conducted by Hu et al. found that antibiotic-treated NOD mice displayed protective effect for T1D primarily achieved by the generation of immune-tolerogenic APCs, which possessed the impaired self-antigen presentation function and eventually led to reduced activation of cytotoxic CD8+ T cells. Interestingly, the tolerogenic APC-mediated diabetes protection was able to be transferred to the second generation (48, 50). This mechanism of tolerogenic APCs induced by changes in the intestinal microbiome is reinforced by a study investigating macrophages, a potent subset of APCs, in which these cells showed hyporesponsiveness to lipopolysaccharide (LPS) stimulation in streptomycin and cefotaxime-treated mice, whose gut microorganisms were nearly completely eliminated after 3 weeks treatment (114). Dendritic cells (DCs), another major subset of APCs, have also been demonstrated to become tolerogenic when exposed to specific microbial stimuli including Lactobacillus reuteri (115) and a mixture of Bifidobacteriaceae, Lactobacil-laceae, and Streptococcus (55).
Intestinal immune health is achieved, in part, through the development of adaptive immune response (116), while perturbations in the composition of gut microbiome can affect the adaptive immune system development at multiple levels including CD4+ T cells, CD8+ T cells, mucosal-associated invariant T (MAIT) cells, and invariant natural killer T (iNKT) cells.
T helper 1 (Th1) and T helper 2 (Th2) cells as major components of the adaptive immune response are vital for controlling the autoimmune reactions (117). An imbalanced Th1/Th2 response has been reported to be involved in the damage of islet beta cells in T1D (117). It has been demonstrated that the commensal community has a decisive role in establishing this equilibrium (118, 119). For instance, GF mice present with a highly Th2-skewed cytokine profile (120, 121), and a shift from Th2 to Th1 immune response has been identified to be promoted by exposures to microorganisms early in life (118). The maturation of regulatory T cells (Tregs) expressing Foxp3 transcription factor is crucial for immune homeostasis and induction of tolerance (122). Numerous studies have revealed that reduced frequency or function of Foxp3+ Tregs in NOD mice is a major susceptibility factor for T1D (123–125). Notably, the Foxp3+ Tregs in mesenteric lymph nodes of GF mice showed a significant reduction in relative and total numbers as well as an impaired regulatory function, indicating a critical role of the gut microbiota in regulating Tregs development (126). Interestingly, monocolonization of GF mice with B. fragilis, a member of the Bacteroidetes phyla, could restore the differentiation of Foxp3+ Tregs (121, 127). Other defined bacterial strains that have the capacity to increase Foxp3+ Tregs number and function in GF mice are Roseburia faecis (a member of Clostridium cluster XIVa) and Faecalibacterium prausnitzii (a member of Clostridium cluster IV) (128). It has been postulated that these microbes induce Tregs by producing butyrate (129). Intestinal commensal microorganisms can ferment the dietary fibers and produce SCFAs such as acetate, propionate, and butyrate (130). Butyrate can not only provide energy for colonic epithelial cells (131) but also enhance the abundance and function of splenic and colonic Foxp3+ Tregs via histone modification (15, 132). Th17 cells have also been considered crucially involved in the etiology of T1D (133, 134). The main function of Th17 cells is producing interleukin (IL)-17 and clearing the extracellular pathogens during infection, while an excessive inflammatory response triggered by Th17 cells may promote autoimmunity (135). However, much uncertainty still exists about its role in autoimmune diabetes because a diabetes-protective effect of Th17 immunity has also been reported in T1D (136). Alam et al. found that the level of IL-17 expression was decreased in the small intestinal lamina propria of GF mice (137). Of note, the reduced frequencies of Th17 cells in GF mice could be restored upon colonization with segmented filamentous bacteria (SFB) (138). At the same time, it was demonstrated that SFB colonization could protect male GF NOD mice from diabetes development and conferred a protective effect in female NOD mice when there were other intestinal microbes (139). Moreover, there are other Th17 cells inducers such as altered Schaedler flora (ASF). ASF, a mixture of eight intestinal bacterial species, also has the capacity of inducing the Th17 responses in GF mice (140). CD8+ T cells are essential for beta cell destruction (141). A recent study revealed that insulin-reactive pathogenic CD8+ T cells were activated as a result of bacterial composition change caused by antibiotic treatment and thus led to the acceleration of diabetes progression in a NOD transgenic mouse model (142), suggesting that compositional change are likely responsible for CD8+ T cells regulation. Recently, Rouxel et al. suggested that MAIT cells known as innate-like lymphocytes might exert protective impact on T1D by maintaining gut integrity and controlling anti-islet autoimmune responses (143). It has been reported that the number of MAIT cells in GF mice was lower than that of specific pathogen-free mice, indicating a close interplay between MAIT cells and gut commensals (144). The iNKT cells have also been recently considered to play a key pathogenic role in the development of T1D since the cytokine-secretion phenotype of iNKT17 cells, mainly IL-17-producing phenotype, could directly trigger autoimmune diabetes (145). It was identified that the gastrointestinal microbiota could promote iNKT17 cell differentiation and acquire a specific cytokine-secretion phenotype (146). One piece of evidence supporting this notion is that GF mice displayed less mature and hyporesponsive iNKT cells (147, 148). In addition, De Giorgi et al. found that the increase in iNKT17 cell differentiation in NOD mice correlated with specific intestine bacterial composition, which was characterized by increased microbial richness, elevated frequency of Bacteroidales, and reduced relative abundance of Clostridiales strains (149).
There is emerging evidence suggesting that the permeability of the intestinal barrier can be regulated by microbial community (72, 150–153). Several commensals have been demonstrated to have a beneficial impact on mucosal barrier integrity, while some other bacteria exert adverse effects (154).
In recent years, a considerable amount of studies have recognized a negative correlation between butyrate-producing species and the risk of T1D, as already mentioned above (27, 32, 34, 36). These butyrate-producing species have been reported to have a pivotal role in the maintenance of intestinal barrier integrity. It was identified that butyrate could reduce gut permeability by promoting the assembly of tight junctions (TJ) (155) as well as inducing mucin synthesis (131), and the role of butyrate in restoring TJ barrier was achieved by affecting the expression of TJ proteins comprising claudin-2, occludin, cingulin, and zonula occludens (ZO) proteins (156). Despite the great variability of intestinal microbiota associated with T1D, most published studies have found that Bacteroides were positively associated with T1D development (24, 27, 28, 30, 32, 34, 38). These bacteria are able to ferment glucose and lactate to propionate, acetate, and succinate (157), which cannot induce the biosynthesis of mucin-like butyrate (158). Conversely, these bacteria would reduce the assembly of TJ and generate an increase in gut permeability, eventually promoting the T1D-associated autoimmunity (27, 150). Collectively, the bacteria capable of converting lactate to butyrate contribute to increased mucin synthesis and TJ formation, thereby facilitating gut health. In contrast, bacteria metabolizing lactate to other SCFAs are related to impaired TJ with a consequential increase in gut permeability and result in the onset of T1D (27). Certain producers of lactate may produce net butyrate (159); thus, these bacteria may also be of great significance for the intestinal barrier function. For instance, Bifidobacterium longum subspecies infantis, lactate-producing bacteria, were demonstrated to have the capacity to protect intestinal permeability (160). Several bacteria with probiotic effect including Lactobacillus johnsonii N6.2 (57), B. lactis (161), Lactobacillus rhamnosus and Lactobacillus reuter (162), and Lactobacillus plantarum (163) were also reported to be able to decrease intestinal permeability (164).
However, as yet, the mechanisms underlying the modulation of epithelial barrier function by gut microbiota are complex and remain unclear (152). One of the potential pathways through which gut microbiota affects intestinal permeability appears to be dependent on high levels of zonulin, whose production can be regulated by bacterial colonization (165, 166). Zonulin has been discovered to reversibly regulate intestinal permeability by modulating TJ (95, 167, 168). It was reported that there was increased zonulin release coincident with an increased permeability before the onset of clinically evident T1D (5). It was supposed that imbalanced microflora colonization could induce the upregulation of zonulin into gut lumen (7). The released zonulin was then recognized by receptors on the surface of intestinal epithelial cells and elicited changes in TJ dynamics including the remodeling of cytoskeleton and the phosphorylation of ZO-1 and occludin (169). In the end, gut permeability was enhanced as a consequence of the disassembly of TJ (169, 170).
A large number of studies have demonstrated that the altered abundance of specific members or reduced diversity of gut microbiota was associated with the progression of T1D. However, the exact role of gut microbiota in the pathogenesis of T1D remains controversial. Up to now, the most convincing evidence for a causal link between intestinal microbiome and the disease comes from well-controlled intervention studies in murine models. These studies illustrated the efficacy of probiotic supplement, antibiotic use, FMT, and diet intervention in modifying the risk of T1D via changing the gut colonization patterns. Furthermore, increasing evidence has indicated that the involvement of intestinal microflora in T1D pathogenesis may be through exerting impact on immune homeostasis and/or gut permeability. Taken together, these findings reviewed here underscore the importance of maintaining healthy microbial composition and provide a new insight into T1D prevention or treatment measures. However, translating these insights into feasible therapeutic measures might be challenging. Further randomized controlled studies in large human cohorts are needed to help answer this chicken–egg question and confirm the efficacy of microbiota-based therapeutic approaches.
HZ designed this review. LS, SZ, and XZ performed the paper selection. HZ, GW, and XG wrote the manuscript. All authors approved it for publication.
This work was supported by National Natural Science Foundation of China (81670732) and Engineering Research Center of Individualized Accurate Diagnosis and Treatment of Jilin Province Science and Technology Department Project (20170623005TC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. Lancet. (2014) 383:69–82. doi: 10.1016/S0140-6736(13)60591-7
2. Abdellatif AM, Sarvetnick NE. Current understanding of the role of gut dysbiosis in type 1 diabetes. J Diabetes. (2019) 11:632–44. doi: 10.1111/1753-0407.12915
3. Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, et al. Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature. (2008) 455:1109–13. doi: 10.1038/nature07336
4. Bakay M, Pandey R, Grant SFA, Hakonarson H. The genetic contribution to type 1 diabetes. Curr Diab Rep. (2019) 19:116. doi: 10.1007/s11892-019-1235-1
5. Bibbo S, Dore MP, Pes GM, Delitala G, Delitala AP. Is there a role for gut microbiota in type 1 diabetes pathogenesis? Ann Med. (2017) 49:11–22. doi: 10.1080/07853890.2016.1222449
6. Xu J, Huang G, Nagy T, Guo TL. Bisphenol A alteration of type 1 diabetes in non-obese diabetic (NOD) female mice is dependent on window of exposure. Arch Toxicol. (2019) 93:1083–93. doi: 10.1007/s00204-019-02419-4
7. Visser J, Rozing J, Sapone A, Lammers K, Fasano A. Tight junctions, intestinal permeability, and autoimmunity: celiac disease and type 1 diabetes paradigms. Ann N Y Acad Sci. (2009) 1165:195–205. doi: 10.1111/j.1749-6632.2009.04037.x
8. Gillespie KM, Bain SC, Barnett AH, Bingley PJ, Christie MR, Gill GV, et al. The rising incidence of childhood type 1 diabetes and reduced contribution of high-risk HLA haplotypes. Lancet. (2004) 364:1699–700. doi: 10.1016/S0140-6736(04)17357-1
9. Fourlanos S, Varney MD, Tait BD, Morahan G, Honeyman MC, Colman PG, et al. The rising incidence of type 1 diabetes is accounted for by cases with lower-risk human leukocyte antigen genotypes. Diabetes Care. (2008) 31:1546–9. doi: 10.2337/dc08-0239
10. Tai N, Wong FS, Wen L. The role of gut microbiota in the development of type 1, type 2 diabetes mellitus and obesity. Rev Endocr Metab Disord. (2015) 16:55–65. doi: 10.1007/s11154-015-9309-0
11. Tuomilehto J. The emerging global epidemic of type 1 diabetes. Curr Diab Rep. (2013) 13:795–804. doi: 10.1007/s11892-013-0433-5
12. Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. (2010) 464:59–65. doi: 10.1038/nature08821
13. Sommer F, Backhed F. The gut microbiota–masters of host development and physiology. Nat Rev Microbiol. (2013) 11:227–38. doi: 10.1038/nrmicro2974
14. Landman C, Quevrain E. Gut microbiota: description, role and pathophysiologic implications. Rev Med Interne. (2016) 37:418–23. doi: 10.1016/j.revmed.2015.12.012
15. Harsch IA, Konturek PC. The role of gut microbiota in obesity and type 2 and type 1 diabetes mellitus: new insights into “old” diseases. Med Sci. (2018) 6:32. doi: 10.3390/medsci6020032
16. Stecher B, Hardt WD. Mechanisms controlling pathogen colonization of the gut. Curr Opin Microbiol. (2011) 14:82–91. doi: 10.1016/j.mib.2010.10.003
17. Pociot F, McDermott MF. Genetics of type 1 diabetes mellitus. Genes Immun. (2002) 3:235–49. doi: 10.1038/sj.gene.6363875
18. Kim YS, Ho SB. Intestinal goblet cells and mucins in health and disease: recent insights and progress. Curr Gastroenterol Rep. (2010) 12:319–30. doi: 10.1007/s11894-010-0131-2
19. Natividad JM, Petit V, Huang X, de Palma G, Jury J, Sanz Y, et al. Commensal and probiotic bacteria influence intestinal barrier function and susceptibility to colitis in Nod1-/-; Nod2-/- mice. Inflamm Bowel Dis. (2012) 18:1434–46. doi: 10.1002/ibd.22848
20. McDermott AJ, Huffnagle GB. The microbiome and regulation of mucosal immunity. Immunology. (2014) 142:24–31. doi: 10.1111/imm.12231
21. Yap YA, Marino E. An insight into the intestinal web of mucosal immunity, microbiota, and diet in inflammation. Front Immunol. (2018) 9:2617. doi: 10.3389/fimmu.2018.02617
22. Petersen C, Round JL. Defining dysbiosis and its influence on host immunity and disease. Cell Microbiol. (2014) 16:1024–33. doi: 10.1111/cmi.12308
23. Gavin PG, Hamilton-Williams EE. The gut microbiota in type 1 diabetes: friend or foe? Curr Opin Endocrinol Diabetes Obes. (2019) 26:207–12. doi: 10.1097/MED.0000000000000483
24. Murri M, Leiva I, Gomez-Zumaquero JM, Tinahones FJ, Cardona F, Soriguer F, et al. Gut microbiota in children with type 1 diabetes differs from that in healthy children: a case-control study. BMC Med. (2013) 11:46. doi: 10.1186/1741-7015-11-46
25. Huang Y, Li SC, Hu J, Ruan HB, Guo HM, Zhang HH, et al. Gut microbiota profiling in Han Chinese with type 1 diabetes. Diabetes Res Clin Pract. (2018) 141:256–63. doi: 10.1016/j.diabres.2018.04.032
26. Giongo A, Gano KA, Crabb DB, Mukherjee N, Novelo LL, Casella G, et al. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. (2011) 5:82–91. doi: 10.1038/ismej.2010.92
27. Brown CT, Davis-Richardson AG, Giongo A, Gano KA, Crabb DB, Mukherjee N, et al. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS ONE. (2011) 6:e25792. doi: 10.1371/journal.pone.0025792
28. Mejia-Leon ME, Petrosino JF, Ajami NJ, Dominguez-Bello MG, de la Barca AM. Fecal microbiota imbalance in Mexican children with type 1 diabetes. Sci Rep. (2014) 4:3814. doi: 10.1038/srep03814
29. Pellegrini S, Sordi V, Bolla AM, Saita D, Ferrarese R, Canducci F, et al. Duodenal mucosa of patients with type 1 diabetes shows distinctive inflammatory profile and microbiota. J Clin Endocrinol Metab. (2017) 102:1468–77. doi: 10.1210/jc.2016-3222
30. Leiva-Gea I, Sanchez-Alcoholado L, Martin-Tejedor B, Castellano-Castillo D, Moreno-Indias I, Urda-Cardona A, et al. Gut microbiota differs in composition and functionality between children with type 1 diabetes and MODY2 and healthy control subjects: a case-control study. Diabetes Care. (2018) 41:2385–95. doi: 10.2337/dc18-0253
31. Traversi D, Rabbone I, Ignaccolo MG, Carletto G, Racca I, Vallini C, et al. Gut microbiota diversity and T1DM onset: preliminary data of a case-control study. Human Microb J. (2017) 5–6:11–3. doi: 10.1016/j.humic.2017.11.002
32. de Goffau MC, Luopajarvi K, Knip M, Ilonen J, Ruohtula T, Harkonen T, et al. Fecal microbiota composition differs between children with beta-cell autoimmunity and those without. Diabetes. (2013) 62:1238–44. doi: 10.2337/db12-0526
33. Kostic AD, Gevers D, Siljander H, Vatanen T, Hyotylainen T, Hamalainen AM, et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe. (2015) 17:260–73. doi: 10.1016/j.chom.2015.01.001
34. de Goffau MC, Fuentes S, van den Bogert B, Honkanen H, de Vos WM, Welling GW, et al. Aberrant gut microbiota composition at the onset of type 1 diabetes in young children. Diabetologia. (2014) 57:1569–77. doi: 10.1007/s00125-014-3274-0
35. Cinek O, Kramna L, Mazankova K, Odeh R, Alassaf A, Ibekwe MU, et al. The bacteriome at the onset of type 1 diabetes: a study from four geographically distant African and Asian countries. Diabetes Res Clin Pract. (2018) 144:51–62. doi: 10.1016/j.diabres.2018.08.010
36. de Groot PF, Belzer C, Aydin O, Levin E, Levels JH, Aalvink S, et al. Distinct fecal and oral microbiota composition in human type 1 diabetes, an observational study. PLoS ONE. (2017) 12:e0188475. doi: 10.1371/journal.pone.0188475
37. Higuchi BS, Rodrigues N, Gonzaga MI, Cicogna Paiolo JC, Stefanutto N, Omori WP, et al. Intestinal dysbiosis in autoimmune diabetes is correlated with poor glycemic control and increased interleukin-6: a pilot study. Front Immunol. (2018) 9:1689. doi: 10.3389/fimmu.2018.01689
38. Pinto E, Anselmo M, Calha M, Bottrill A, Duarte I, Andrew PW, et al. The intestinal proteome of diabetic and control children is enriched with different microbial and host proteins. Microbiology. (2017) 163:161–74. doi: 10.1099/mic.0.000412
39. Soyucen E, Gulcan A, Aktuglu-Zeybek AC, Onal H, Kiykim E, Aydin A. Differences in the gut microbiota of healthy children and those with type 1 diabetes. Pediatr Int. (2014) 56:336–43. doi: 10.1111/ped.12243
40. Gursoy S, Kockar T, Atik SU, Onal Z, Onal H, Adal E. Autoimmunity and intestinal colonization by Candida albicans in patients with type 1 diabetes at the time of the diagnosis. Korean J Pediatr. (2018) 61:217–20. doi: 10.3345/kjp.2018.61.7.217
41. Demirci M, Bahar Tokman H, Taner Z, Keskin FE, Cagatay P, Ozturk Bakar Y, et al. Bacteroidetes and Firmicutes levels in gut microbiota and effects of hosts TLR2/TLR4 gene expression levels in adult type 1 diabetes patients in Istanbul, Turkey. J Diabetes Complications. (2019) 34:107449. doi: 10.1016/j.jdiacomp.2019.107449
42. Meijnikman AS, Gerdes VE, Nieuwdorp M, Herrema H. Evaluating causality of gut microbiota in obesity and diabetes in humans. Endocr Rev. (2018) 39:133–53. doi: 10.1210/er.2017-00192
43. Pearson JA, Wong FS, Wen L. The importance of the non obese diabetic (NOD) mouse model in autoimmune diabetes. J Autoimmun. (2016) 66:76–88. doi: 10.1016/j.jaut.2015.08.019
44. Nakhooda AF, Like AA, Chappel CI, Murray FT, Marliss EB. The spontaneously diabetic Wistar rat. Metab Morphologic Stud Diabetes. (1977) 26:100–12. doi: 10.2337/diab.26.2.100
45. Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature. (2010) 464:1293–300. doi: 10.1038/nature08933
46. Pearson JA, Agriantonis A, Wong FS, Wen L. Modulation of the immune system by the gut microbiota in the development of type 1 diabetes. Hum Vaccin Immunother. (2018) 14:1–17. doi: 10.1080/21645515.2018.1514354
47. Candon S, Perez-Arroyo A, Marquet C, Valette F, Foray A-P, Pelletier B, et al. Antibiotics in early life alter the gut microbiome and increase disease incidence in a spontaneous mouse model of autoimmune insulin-dependent diabetes. PLoS ONE. (2015) 10:e0125448. doi: 10.1371/journal.pone.0125448
48. Hu Y, Jin P, Peng J, Zhang X, Wong FS, Wen L. Different immunological responses to early-life antibiotic exposure affecting autoimmune diabetes development in NOD mice. J Autoimmun. (2016) 72:47–56. doi: 10.1016/j.jaut.2016.05.001
49. Hansen CHF, Krych L, Nielsen DS, Vogensen FK, Hansen LH, Sørensen SJ, et al. Early life treatment with vancomycin propagates Akkermansia muciniphila and reduces diabetes incidence in the NOD mouse. Diabetologia. (2012) 55:2285–94. doi: 10.1007/s00125-012-2564-7
50. Hu Y, Peng J, Tai N, Hu C, Zhang X, Wong FS, et al. Maternal antibiotic treatment protects offspring from diabetes development in nonobese diabetic mice by generation of tolerogenic APCs. J Immunol. (2015) 195:4176–84. doi: 10.4049/jimmunol.1500884
51. Livanos AE, Greiner TU, Vangay P, Pathmasiri W, Stewart D, McRitchie S, et al. Antibiotic-mediated gut microbiome perturbation accelerates development of type 1 diabetes in mice. Nat Microbiol. (2016) 1:16140. doi: 10.1038/nmicrobiol.2016.140
52. Zhang XS, Li J, Krautkramer KA, Badri M, Battaglia T, Borbet TC, et al. Antibiotic-induced acceleration of type 1 diabetes alters maturation of innate intestinal immunity. Elife. (2018) 7:e37816. doi: 10.7554/eLife.37816.059
53. Hill C, Guarner F, Reid G, Gibson GR, Merenstein DJ, Pot B, et al. Expert consensus document. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat Rev Gastroenterol Hepatol. (2014) 11:506–14. doi: 10.1038/nrgastro.2014.66
54. Calcinaro F, Dionisi S, Marinaro M, Candeloro P, Bonato V, Marzotti S, et al. Oral probiotic administration induces interleukin-10 production and prevents spontaneous autoimmune diabetes in the non-obese diabetic mouse. Diabetologia. (2005) 48:1565–75. doi: 10.1007/s00125-005-1831-2
55. Dolpady J, Sorini C, Di Pietro C, Cosorich I, Ferrarese R, Saita D, et al. Oral probiotic VSL#3 prevents autoimmune diabetes by modulating microbiota and promoting indoleamine 2,3-dioxygenase-enriched tolerogenic intestinal environment. J Diabetes Res. (2016) 2016:7569431. doi: 10.1155/2016/7569431
56. Jia L, Shan K, Pan LL, Feng N, Lv Z, Sun Y, et al. Clostridium butyricum CGMCC0313.1 protects against autoimmune diabetes by modulating intestinal immune homeostasis and inducing pancreatic regulatory T cells. Front Immunol. (2017) 8:1345. doi: 10.3389/fimmu.2017.01345
57. Valladares R, Sankar D, Li N, Williams E, Lai K-K, Abdelgeliel AS, et al. Lactobacillus johnsonii N6.2 mitigates the development of type 1 diabetes in BB-DP rats. PLoS ONE. (2010) 5:e10507. doi: 10.1371/journal.pone.0010507
58. Peng J, Narasimhan S, Marchesi JR, Benson A, Wong FS, Wen L. Long term effect of gut microbiota transfer on diabetes development. J Autoimmunity. (2014) 53:85–94. doi: 10.1016/j.jaut.2014.03.005
59. Brown K, Godovannyi A, Ma C, Zhang Y, Ahmadi-Vand Z, Dai C, et al. Prolonged antibiotic treatment induces a diabetogenic intestinal microbiome that accelerates diabetes in NOD mice. ISME J. (2015) 10:321. doi: 10.1038/ismej.2015.114
60. Hanninen A, Toivonen R, Poysti S, Belzer C, Plovier H, Ouwerkerk JP, et al. Akkermansia muciniphila induces gut microbiota remodelling and controls islet autoimmunity in NOD mice. Gut. (2018) 67:1445–53. doi: 10.1136/gutjnl-2017-314508
61. Marietta EV, Gomez AM, Yeoman C, Tilahun AY, Clark CR, Luckey DH, et al. Low incidence of spontaneous type 1 diabetes in non-obese diabetic mice raised on gluten-free diets is associated with changes in the intestinal microbiome. PLoS ONE. (2013) 8:e78687. doi: 10.1371/journal.pone.0078687
62. Toivonen RK, Emani R, Munukka E, Rintala A, Laiho A, Pietila S, et al. Fermentable fibres condition colon microbiota and promote diabetogenesis in NOD mice. Diabetologia. (2014) 57:2183–92. doi: 10.1007/s00125-014-3325-6
63. Wu C, Pan LL, Niu W, Fang X, Liang W, Li J, et al. Modulation of gut microbiota by low methoxyl pectin attenuates type 1 diabetes in non-obese diabetic mice. Front Immunol. (2019) 10:1733. doi: 10.3389/fimmu.2019.01733
64. Sadauskaite-Kuehne V, Ludvigsson J, Padaiga Ž, Jašinskiene E, Samuelsson U. Longer breastfeeding is an independent protective factor against development of type 1 diabetes mellitus in childhood. Diabetes Metab Res Rev. (2004) 20:150–7. doi: 10.1002/dmrr.425
65. Ho NT, Li F, Lee-Sarwar KA, Tun HM, Brown BP, Pannaraj PS, et al. Meta-analysis of effects of exclusive breastfeeding on infant gut microbiota across populations. Nat Commun. (2018) 9:4169. doi: 10.1038/s41467-018-06473-x
66. Uusitalo U, Liu X, Yang J, Aronsson CA, Hummel S, Butterworth M, et al. Association of early exposure of probiotics and islet autoimmunity in the TEDDY study. JAMA Pediatr. (2016) 170:20–8. doi: 10.1001/jamapediatrics.2015.2757
67. Ho J, Nicolucci AC, Virtanen H, Schick A, Meddings J, Reimer RA, et al. Effect of prebiotic on microbiota, intestinal permeability and glycemic control in children with type 1 diabetes. J Clin Endocrinol Metab. (2019) 104:4427–40. doi: 10.1210/jc.2019-00481
68. Groele L, Szajewska H, Szypowska A. Effects of Lactobacillus rhamnosus GG and Bifidobacterium lactis Bb12 on beta-cell function in children with newly diagnosed type 1 diabetes: protocol of a randomised controlled trial. BMJ Open. (2017) 7:e017178. doi: 10.1136/bmjopen-2017-017178
69. Vaarala O, Atkinson MA, Neu J. The: perfect storm for type 1 diabetes: the complex interplay between intestinal microbiota, gut permeability, and mucosal immunity. Diabetes. (2008) 57:2555–62. doi: 10.2337/db08-0331
70. Davis-Richardson AG, Triplett EW. A model for the role of gut bacteria in the development of autoimmunity for type 1 diabetes. Diabetologia. (2015) 58:1386–93. doi: 10.1007/s00125-015-3614-8
71. Fasano A. Leaky gut and autoimmune diseases. Clin Rev Allergy Immunol. (2012) 42:71–8. doi: 10.1007/s12016-011-8291-x
72. Miranda MCG, Oliveira RP, Torres L, Aguiar SLF, Pinheiro-Rosa N, Lemos L, et al. Frontline Science: Abnormalities in the gut mucosa of non-obese diabetic mice precede the onset of type 1 diabetes. J Leukoc Biol. (2019) 106:513–29. doi: 10.1002/JLB.3HI0119-024RR
73. Siljander H, Honkanen J, Knip M. Microbiome and type 1 diabetes. EBioMedicine. (2019) 46:512–21. doi: 10.1016/j.ebiom.2019.06.031
74. Ownby DR, Johnson CC, Peterson EL. Exposure to dogs and cats in the first year of life and risk of allergic sensitization at 6 to 7 years of age. JAMA. (2002) 288:963–72. doi: 10.1001/jama.288.8.963
75. Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med. (2002) 347:911–20. doi: 10.1056/NEJMra020100
76. Bach JF, Chatenoud L. The hygiene hypothesis: an explanation for the increased frequency of insulin-dependent diabetes. Cold Spring Harb Perspect Med. (2012) 2:a007799. doi: 10.1101/cshperspect.a007799
77. Martins TC, Aguas AP. Mechanisms of Mycobacterium avium-induced resistance against insulin-dependent diabetes mellitus (IDDM) in non-obese diabetic (NOD) mice: role of Fas and Th1 cells. Clin Exp Immunol. (1999) 115:248–54. doi: 10.1046/j.1365-2249.1999.00781.x
78. Zaccone P, Raine T, Sidobre S, Kronenberg M, Mastroeni P, Cooke A. Salmonella typhimurium infection halts development of type 1 diabetes in NOD mice. Eur J Immunol. (2004) 34:3246–56. doi: 10.1002/eji.200425285
79. Belkaid Y, Hand Timothy W. Role of the microbiota in immunity and inflammation. Cell. (2014) 157:121–41. doi: 10.1016/j.cell.2014.03.011
80. Sudo N, Yu XN, Aiba Y, Oyama N, Sonoda J, Koga Y, et al. An oral introduction of intestinal bacteria prevents the development of a long-term Th2-skewed immunological memory induced by neonatal antibiotic treatment in mice. Clin Exp Allergy. (2002) 32:1112–6. doi: 10.1046/j.1365-2222.2002.01430.x
81. Bendtsen KM, Fisker L, Hansen AK, Hansen CHF, Nielsen DS. The influence of the young microbiome on inflammatory diseases—lessons from animal studies. Birth Defects Res. (2015) 105:278–95. doi: 10.1002/bdrc.21116
82. Dunne JL, Triplett EW, Gevers D, Xavier R, Insel R, Danska J, et al. The intestinal microbiome in type 1 diabetes. Clin Exp Immunol. (2014) 177:30–7. doi: 10.1111/cei.12321
83. Carratu R, Secondulfo M, de Magistris L, Iafusco D, Urio A, Carbone MG, et al. Altered intestinal permeability to mannitol in diabetes mellitus type I. J Pediatr Gastroenterol Nutr. (1999) 28:264–9. doi: 10.1097/00005176-199903000-00010
84. Kuitunen M, Saukkonen T, Ilonen J, Akerblom HK, Savilahti E. Intestinal permeability to mannitol and lactulose in children with type 1 diabetes with the HLA-DQB1*02 allele. Autoimmunity. (2002) 35:365–8. doi: 10.1080/0891693021000008526
85. Secondulfo M, Iafusco D, Carratu R, deMagistris L, Sapone A, Generoso M, et al. Ultrastructural mucosal alterations and increased intestinal permeability in non-celiac, type I diabetic patients. Dig Liver Dis. (2004) 36:35–45. doi: 10.1016/j.dld.2003.09.016
86. Sapone A, de Magistris L, Pietzak M, Clemente MG, Tripathi A, Cucca F, et al. Zonulin upregulation is associated with increased gut permeability in subjects with type 1 diabetes and their relatives. Diabetes. (2006) 55:1443–9. doi: 10.2337/db05-1593
87. Li X, Atkinson MA. The role for gut permeability in the pathogenesis of type 1 diabetes–a solid or leaky concept? Pediatr Diabetes. (2015) 16:485–92. doi: 10.1111/pedi.12305
88. Meddings JB, Jarand J, Urbanski SJ, Hardin J, Gall DG. Increased gastrointestinal permeability is an early lesion in the spontaneously diabetic BB rat. Am J Physiol. (1999) 276:G951–7. doi: 10.1152/ajpgi.1999.276.4.G951
89. Neu J, Reverte CM, Mackey AD, Liboni K, Tuhacek-Tenace LM, Hatch M, et al. Changes in intestinal morphology and permeability in the biobreeding rat before the onset of type 1 diabetes. J Pediatr Gastroenterol Nutr. (2005) 40:589–95. doi: 10.1097/01.MPG.0000159636.19346.C1
90. Bosi E, Molteni L, Radaelli MG, Folini L, Fermo I, Bazzigaluppi E, et al. Increased intestinal permeability precedes clinical onset of type 1 diabetes. Diabetologia. (2006) 49:2824–7. doi: 10.1007/s00125-006-0465-3
91. Harbison JE, Roth-Schulze AJ, Giles LC, Tran CD, Ngui KM, Penno MA, et al. Gut microbiome dysbiosis and increased intestinal permeability in children with islet autoimmunity and type 1 diabetes: a prospective cohort study. Pediatr Diabetes. (2019) 20:574–83. doi: 10.1111/pedi.12865
92. Visser JT, Lammers K, Hoogendijk A, Boer MW, Brugman S, Beijer-Liefers S, et al. Restoration of impaired intestinal barrier function by the hydrolysed casein diet contributes to the prevention of type 1 diabetes in the diabetes-prone BioBreeding rat. Diabetologia. (2010) 53:2621–8. doi: 10.1007/s00125-010-1903-9
93. Lee AS, Gibson DL, Zhang Y, Sham HP, Vallance BA, Dutz JP. Gut barrier disruption by an enteric bacterial pathogen accelerates insulitis in NOD mice. Diabetologia. (2010) 53:741–8. doi: 10.1007/s00125-009-1626-y
94. Viggiano D, Ianiro G, Vanella G, Bibbo S, Bruno G, Simeone G, et al. Gut barrier in health and disease: focus on childhood. Eur Rev Med Pharmacol Sci. (2015) 19:1077–85.
95. Watts T, Berti I, Sapone A, Gerarduzzi T, Not T, Zielke R, et al. Role of the intestinal tight junction modulator zonulin in the pathogenesis of type I diabetes in BB diabetic-prone rats. Proc Natl Acad Sci USA. (2005) 102:2916–21. doi: 10.1073/pnas.0500178102
96. Myers MA, Hettiarachchi KD, Ludeman JP, Wilson AJ, Wilson CR, Zimmet PZ. Dietary microbial toxins and type 1 diabetes. Ann NY Acad Sci. (2003) 1005:418–22. doi: 10.1196/annals.1288.071
97. Sorini C, Cosorich I, Lo Conte M, De Giorgi L, Facciotti F, Luciano R, et al. Loss of gut barrier integrity triggers activation of islet-reactive T cells and autoimmune diabetes. Proc Natl Acad Sci USA. (2019) 116:15140–9. doi: 10.1073/pnas.1814558116
98. Tai N, Peng J, Liu F, Gulden E, Hu Y, Zhang X, et al. Microbial antigen mimics activate diabetogenic CD8 T cells in NOD mice. J Exp Med. (2016) 213:2129–46. doi: 10.1084/jem.20160526
99. Sofi MH, Johnson BM, Gudi RR, Jolly A, Gaudreau MC, Vasu C. Polysaccharide A-dependent opposing effects of mucosal and systemic exposures to human gut commensal Bacteroides fragilis in type 1 diabetes. Diabetes. (2019) 68:1975–89. doi: 10.2337/db19-0211
100. Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. (2012) 336:1268–73. doi: 10.1126/science.1223490
101. Brown EM, Kenny DJ, Xavier RJ. Gut microbiota regulation of T cells during inflammation and autoimmunity. Annu Rev Immunol. (2019) 37:599–624. doi: 10.1146/annurev-immunol-042718-041841
102. Li J, Wang X, Zhang F, Yin H. Toll-like receptors as therapeutic targets for autoimmune connective tissue diseases. Pharmacol Ther. (2013) 138:441–51. doi: 10.1016/j.pharmthera.2013.03.003
103. Beutler BA. TLRs and innate immunity. Blood. (2009) 113:1399–407. doi: 10.1182/blood-2008-07-019307
104. Wong FS, Hu C, Zhang L, Du W, Alexopoulou L, Flavell RA, et al. The role of Toll-like receptors 3 and 9 in the development of autoimmune diabetes in NOD mice. Ann NY Acad Sci. (2008) 1150:146–8. doi: 10.1196/annals.1447.039
105. Clanchy FIL, Sacre SM. Modulation of toll-like receptor function has therapeutic potential in autoimmune disease. Expert Opin Biol Therapy. (2010) 10:1703–16. doi: 10.1517/14712598.2010.534080
106. Zhang Y, Lee AS, Shameli A, Geng X, Finegood D, Santamaria P, et al. TLR9 blockade inhibits activation of diabetogenic CD8+ T cells and delays autoimmune diabetes. J Immunol. (2010) 184:5645. doi: 10.4049/jimmunol.0901814
107. Gülden E, Ihira M, Ohashi A, Reinbeck AL, Freudenberg MA, Kolb H, et al. Toll-like receptor 4 deficiency accelerates the development of insulin-deficient diabetes in non-obese diabetic mice. PLoS ONE. (2013) 8:e75385. doi: 10.1371/journal.pone.0075385
108. Alkanani AK, Hara N, Lien E, Ir D, Kotter CV, Robertson CE, et al. Induction of diabetes in the RIP-B7.1 mouse model is critically dependent on TLR3 and MyD88 pathways and is associated with alterations in the intestinal microbiome. Diabetes. (2014) 63:619–31. doi: 10.2337/db13-1007
109. Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. (2011) 34:637–50. doi: 10.1016/j.immuni.2011.05.006
110. Gulden E, Chao C, Tai N, Pearson JA, Peng J, Majewska-Szczepanik M, et al. TRIF deficiency protects non-obese diabetic mice from type 1 diabetes by modulating the gut microbiota and dendritic cells. J Autoimmun. (2018) 93:57–65. doi: 10.1016/j.jaut.2018.06.003
111. Simon MC, Reinbeck AL, Wessel C, Heindirk J, Jelenik T, Kaul K, et al. Distinct alterations of gut morphology and microbiota characterize accelerated diabetes onset in non-obese diabetic mice. J Biol Chem. (2020) 295:969–80. doi: 10.1074/jbc.RA119.010816
112. Costa FR, Francozo MC, de Oliveira GG, Ignacio A, Castoldi A, Zamboni DS, et al. Gut microbiota translocation to the pancreatic lymph nodes triggers NOD2 activation and contributes to T1D onset. J Exp Med. (2016) 213:1223–39. doi: 10.1084/jem.20150744
113. Katz JD, Ondr JK, Opoka RJ, Garcia Z, Janssen EM. Cutting edge: merocytic dendritic cells break T cell tolerance to beta cell antigens in nonobese diabetic mouse diabetes. J Immunol. (2010) 185:1999–2003. doi: 10.4049/jimmunol.1001398
114. Umenai T, Hirai H, Shime N, Nakaya T, Asahara T, Nomoto K, et al. Eradication of the commensal intestinal microflora by oral antimicrobials interferes with the host response to lipopolysaccharide. Eur J Clin Microbiol Infect Dis. (2010) 29:633–41. doi: 10.1007/s10096-010-0905-3
115. Haileselassie Y, Navis M, Vu N, Qazi KR, Rethi B, Sverremark-Ekström E. Lactobacillus reuteri and Staphylococcus aureus differentially influence the generation of monocyte-derived dendritic cells and subsequent autologous T cell responses. Immunity Inflamm Dis. (2016) 4:315–26. doi: 10.1002/iid3.115
116. Dong C. Diversification of T-helper-cell lineages: finding the family root of IL-17-producing cells. Nat Rev Immunol. (2006) 6:329. doi: 10.1038/nri1807
117. Neurath MF, Finotto S, Glimcher LH. The role of Th1/Th2 polarization in mucosal immunity. Nat Med. (2002) 8:567–73. doi: 10.1038/nm0602-567
118. Bowman LM, Holt PG. Selective enhancement of systemic Th1 immunity in immunologically immature rats with an orally administered bacterial extract. Infect Immun. (2001) 69:3719–27. doi: 10.1128/IAI.69.6.3719-3727.2001
120. Shi HN, Walker A. Bacterial colonization and the development of intestinal defences. Can J Gastroenterol. (2004) 18:493–500. doi: 10.1155/2004/690421
121. Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. (2005) 122:107–18. doi: 10.1016/j.cell.2005.05.007
122. Campbell DJ, Koch MA. Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nat Rev Immunol. (2011) 11:119–30. doi: 10.1038/nri2916
123. You S, Leforban B, Garcia C, Bach JF, Bluestone JA, Chatenoud L. Adaptive TGF-beta-dependent regulatory T cells control autoimmune diabetes and are a privileged target of anti-CD3 antibody treatment. Proc Natl Acad Sci USA. (2007) 104:6335–40. doi: 10.1073/pnas.0701171104
124. Endesfelder D, Castell W, Ardissone A, Davis-Richardson AG, Achenbach P, Hagen M, et al. Compromised gut microbiota networks in children with anti-islet cell autoimmunity. Diabetes. (2014) 63:2006–14. doi: 10.2337/db13-1676
125. Zoka A, Barna G, Somogyi A, Muzes G, Olah A, Al-Aissa Z, et al. Extension of the CD4(+)Foxp3(+)CD25(-/low) regulatory T-cell subpopulation in type 1 diabetes mellitus. Autoimmunity. (2015) 48:289–97. doi: 10.3109/08916934.2014.992518
126. Ostman S, Rask C, Wold AE, Hultkrantz S, Telemo E. Impaired regulatory T cell function in germ-free mice. Eur J Immunol. (2006) 36:2336–46. doi: 10.1002/eji.200535244
127. Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci USA. (2010) 107:12204. doi: 10.1073/pnas.0909122107
128. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. (2011) 331:337–41. doi: 10.1126/science.1198469
129. Longman RS, Yang Y, Diehl GE, Kim SV, Littman DR. Microbiota: host interactions in mucosal homeostasis and systemic autoimmunity. Cold Spring Harb Symp Quant Biol. (2013) 78:193–201. doi: 10.1101/sqb.2013.78.020081
130. Rescigno M. Intestinal microbiota and its effects on the immune system. Cell Microbiol. (2014) 16:1004–13. doi: 10.1111/cmi.12301
131. Hague A, Butt AJ, Paraskeva C. The role of butyrate in human colonic epithelial cells: an energy source or inducer of differentiation and apoptosis? Proc Nutr Soc. (1996) 55:937–43. doi: 10.1079/PNS19960090
132. Tanca A, Palomba A, Fraumene C, Manghina V, Silverman M, Uzzau S. Clostridial butyrate biosynthesis enzymes are significantly depleted in the gut microbiota of nonobese diabetic mice. mSphere. (2018) 3:e00492–18. doi: 10.1128/mSphere.00492-18
133. Emamaullee JA, Davis J, Merani S, Toso C, Elliott JF, Thiesen A, et al. Inhibition of Th17 cells regulates autoimmune diabetes in NOD mice. Diabetes. (2009) 58:1302–11. doi: 10.2337/db08-1113
134. Honkanen J, Nieminen JK, Gao R, Luopajarvi K, Salo HM, Ilonen J, et al. IL-17 immunity in human type 1 diabetes. J Immunol. (2010) 185:1959–67. doi: 10.4049/jimmunol.1000788
135. Atkinson MA, Chervonsky A. Does the gut microbiota have a role in type 1 diabetes? Early evidence from humans and animal models of the disease. Diabetologia. (2012) 55:2868–77. doi: 10.1007/s00125-012-2672-4
136. Lau K, Benitez P, Ardissone A, Wilson TD, Collins EL, Lorca G, et al. Inhibition of type 1 diabetes correlated to a Lactobacillus johnsonii N6.2-mediated Th17 bias. J Immunol. (2011) 186:3538. doi: 10.4049/jimmunol.1001864
137. Alam C, Bittoun E, Bhagwat D, Valkonen S, Saari A, Jaakkola U, et al. Effects of a germ-free environment on gut immune regulation and diabetes progression in non-obese diabetic (NOD) mice. Diabetologia. (2011) 54:1398–406. doi: 10.1007/s00125-011-2097-5
138. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. (2009) 139:485–98. doi: 10.1016/j.cell.2009.09.033
139. Yurkovetskiy L, Burrows M, Khan Aly A, Graham L, Volchkov P, Becker L, et al. Gender bias in autoimmunity is influenced by microbiota. Immunity. (2013) 39:400–12. doi: 10.1016/j.immuni.2013.08.013
140. Geuking MB, Cahenzli J, Lawson MA, Ng DC, Slack E, Hapfelmeier S, et al. Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity. (2011) 34:794–806. doi: 10.1016/j.immuni.2011.03.021
141. Lieberman SM, Evans AM, Han B, Takaki T, Vinnitskaya Y, Caldwell JA, et al. Identification of the beta cell antigen targeted by a prevalent population of pathogenic CD8+ T cells in autoimmune diabetes. Proc Natl Acad Sci USA. (2003) 100:8384–8. doi: 10.1073/pnas.0932778100
142. Pearson JA, Kakabadse D, Davies J, Peng J, Warden-Smith J, Cuff S, et al. Altered gut microbiota activate and expand insulin B15-23-reactive CD8+ T cells. Diabetes. (2019) 68:1002–13. doi: 10.2337/db18-0487
143. Rouxel O, Da Silva J, Beaudoin L, Nel I, Tard C, Cagninacci L, et al. Cytotoxic and regulatory roles of mucosal-associated invariant T cells in type 1 diabetes. Nat Immunol. (2017) 18:1321–31. doi: 10.1038/ni.3854
144. Koay HF, Gherardin NA, Enders A, Loh L, Mackay LK, Almeida CF, et al. A three-stage intrathymic development pathway for the mucosal-associated invariant T cell lineage. Nat Immunol. (2016) 17:1300–11. doi: 10.1038/ni.3565
145. Simoni Y, Gautron AS, Beaudoin L, Bui LC, Michel ML, Coumoul X, et al. NOD mice contain an elevated frequency of iNKT17 cells that exacerbate diabetes. Eur J Immunol. (2011) 41:3574–85. doi: 10.1002/eji.201141751
146. Zeissig S, Blumberg RS. Commensal microbiota and NKT cells in the control of inflammatory diseases at mucosal surfaces. Curr Opin Immunol. (2013) 25:690–6. doi: 10.1016/j.coi.2013.09.012
147. Olszak T, An D, Zeissig S, Vera MP, Richter J, Franke A, et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science. (2012) 336:489–93. doi: 10.1126/science.1219328
148. Wingender G, Stepniak D, Krebs P, Lin L, McBride S, Wei B, et al. Intestinal microbes affect phenotypes and functions of invariant natural killer T cells in mice. Gastroenterology. (2012) 143:418–28. doi: 10.1053/j.gastro.2012.04.017
149. De Giorgi L, Sorini C, Cosorich I, Ferrarese R, Canducci F, Falcone M. Increased iNKT17 cell frequency in the intestine of non-obese diabetic mice correlates with high bacterioidales and low clostridiales abundance. Front Immunol. (2018) 9:1752. doi: 10.3389/fimmu.2018.01752
150. Tlaskalova-Hogenova H, Stepankova R, Kozakova H, Hudcovic T, Vannucci L, Tuckova L, et al. The role of gut microbiota (commensal bacteria) and the mucosal barrier in the pathogenesis of inflammatory and autoimmune diseases and cancer: contribution of germ-free and gnotobiotic animal models of human diseases. Cell Mol Immunol. (2011) 8:110–20. doi: 10.1038/cmi.2010.67
151. Jakobsson HE, Rodriguez-Pineiro AM, Schutte A, Ermund A, Boysen P, Bemark M, et al. The composition of the gut microbiota shapes the colon mucus barrier. EMBO Rep. (2015) 16:164–77. doi: 10.15252/embr.201439263
152. Kelly JR, Kennedy PJ, Cryan JF, Dinan TG, Clarke G, Hyland NP. Breaking down the barriers: the gut microbiome, intestinal permeability and stress-related psychiatric disorders. Front Cell Neurosci. (2015) 9:392. doi: 10.3389/fncel.2015.00392
153. Michielan A, D'Inca R. Intestinal permeability in inflammatory bowel disease: pathogenesis, clinical evaluation, and therapy of leaky gut. Mediators Inflamm. (2015) 2015:628157. doi: 10.1155/2015/628157
154. Ulluwishewa D, Anderson RC, McNabb WC, Moughan PJ, Wells JM, Roy NC. Regulation of tight junction permeability by intestinal bacteria and dietary components. J Nutr. (2011) 141:769–76. doi: 10.3945/jn.110.135657
155. Holzman IR, Green RS, Lin J, Peng L, Li Z-R. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of amp-activated protein kinase in Caco-2 Cell monolayers. J Nutr. (2009) 139:1619–25. doi: 10.3945/jn.109.104638
156. Ploger S, Stumpff F, Penner GB, Schulzke JD, Gabel G, Martens H, et al. Microbial butyrate and its role for barrier function in the gastrointestinal tract. Ann NY Acad Sci. (2012) 1258:52–9. doi: 10.1111/j.1749-6632.2012.06553.x
157. Burger-van Paassen N, Vincent A, Puiman PJ, van der Sluis M, Bouma J, Boehm G, et al. The regulation of intestinal mucin MUC2 expression by short-chain fatty acids: implications for epithelial protection. Biochem J. (2009) 420:211–9. doi: 10.1042/BJ20082222
158. Flint HJ, Scott KP, Duncan SH, Louis P, Forano E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes. (2012) 3:289–306. doi: 10.4161/gmic.19897
159. Duncan SH, Louis P, Flint HJ. Lactate-utilizing bacteria, isolated from human feces, that produce butyrate as a major fermentation product. Appl Environ Microbiol. (2004) 70:5810–7. doi: 10.1128/AEM.70.10.5810-5817.2004
160. Underwood MA, German JB, Lebrilla CB, Mills DA. Bifidobacterium longum subspecies infantis: champion colonizer of the infant gut. Pediatr Res. (2015) 77:229–35. doi: 10.1038/pr.2014.156
161. Stratiki Z, Costalos C, Sevastiadou S, Kastanidou O, Skouroliakou M, Giakoumatou A, et al. The effect of a bifidobacter supplemented bovine milk on intestinal permeability of preterm infants. Early Hum Dev. (2007) 83:575–9. doi: 10.1016/j.earlhumdev.2006.12.002
162. Rosenfeldt V, Benfeldt E, Valerius NH, Paerregaard A, Michaelsen KF. Effect of probiotics on gastrointestinal symptoms and small intestinal permeability in children with atopic dermatitis. J Pediatr. (2004) 145:612–6. doi: 10.1016/j.jpeds.2004.06.068
163. Karczewski J, Troost FJ, Konings I, Dekker J, Kleerebezem M, Brummer RJ, et al. Regulation of human epithelial tight junction proteins by Lactobacillus plantarum in vivo and protective effects on the epithelial barrier. Am J Physiol Gastrointest Liver Physiol. (2010) 298:G851–9. doi: 10.1152/ajpgi.00327.2009
164. Krumbeck JA, Rasmussen HE, Hutkins RW, Clarke J, Shawron K, Keshavarzian A, et al. Probiotic Bifidobacterium strains and galactooligosaccharides improve intestinal barrier function in obese adults but show no synergism when used together as synbiotics. Microbiome. (2018) 6:121. doi: 10.1186/s40168-018-0494-4
165. Wang W, Uzzau S, Goldblum SE, Fasano A. Human zonulin, a potential modulator of intestinal tight junctions. J Cell Sci. (2000) 113(Pt 24):4435–40.
166. El Asmar R, Panigrahi P, Bamford P, Berti I, Not T, Coppa GV, et al. Host-dependent zonulin secretion causes the impairment of the small intestine barrier function after bacterial exposure. Gastroenterology. (2002) 123:1607–15. doi: 10.1053/gast.2002.36578
167. Fasano A. Intestinal permeability and its regulation by zonulin: diagnostic and therapeutic implications. Clin Gastroenterol Hepatol. (2012) 10:1096–100. doi: 10.1016/j.cgh.2012.08.012
168. Kelly CP, Green PH, Murray JA, Dimarino A, Colatrella A, Leffler DA, et al. Larazotide acetate in patients with coeliac disease undergoing a gluten challenge: a randomised placebo-controlled study. Aliment Pharmacol Ther. (2013) 37:252–62. doi: 10.1111/apt.12147
169. Groschwitz KR, Hogan SP. Intestinal barrier function: molecular regulation and disease pathogenesis. J Allergy Clin Immunol. (2009) 124:3–20; quiz 21–2. doi: 10.1016/j.jaci.2009.05.038
Keywords: gut microbiota, dysbiosis, type 1 diabetes, causality, mechanisms
Citation: Zhou H, Sun L, Zhang S, Zhao X, Gang X and Wang G (2020) Evaluating the Causal Role of Gut Microbiota in Type 1 Diabetes and Its Possible Pathogenic Mechanisms. Front. Endocrinol. 11:125. doi: 10.3389/fendo.2020.00125
Received: 13 September 2019; Accepted: 25 February 2020;
Published: 24 March 2020.
Edited by:
Jan Polák, Charles University, CzechiaCopyright © 2020 Zhou, Sun, Zhang, Zhao, Gang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guixia Wang, Z3dhbmcxNjhAamx1LmVkdS5jbg==; Xiaokun Gang, aW5zaWdodGZ1bEAxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.