
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol. , 13 March 2020
Sec. Thyroid Endocrinology
Volume 11 - 2020 | https://doi.org/10.3389/fendo.2020.00102
This article is part of the Research Topic An Update on Thyroid Diseases at the 1st Italian Thyroid Preceptorship View all 8 articles
 Alessandro Prete1*
Alessandro Prete1* Patricia Borges de Souza2
Patricia Borges de Souza2 Simona Censi3
Simona Censi3 Marina Muzza4
Marina Muzza4 Nicole Nucci5
Nicole Nucci5 Marialuisa Sponziello6
Marialuisa Sponziello6The incidence of thyroid cancer (TC) has increased worldwide over the past four decades. TC is divided into three main histological types: differentiated (papillary and follicular TC), undifferentiated (poorly differentiated and anaplastic TC), and medullary TC, arising from TC cells. This review discusses the molecular mechanisms associated to the pathogenesis of different types of TC and their clinical relevance. In the last years, progresses in the genetic characterization of TC have provided molecular markers for diagnosis, risk stratification, and treatment targets. Recently, papillary TC, the most frequent form of TC, has been reclassified into two molecular subtypes, named BRAF-like and RAS-like, associated to a different range of cancer risks. Similarly, the genetic characterization of follicular TC has been proposed to complement the new histopathological classification in order to estimate the prognosis. New analyses characterized a comprehensive molecular profile of medullary TC, raising the role of RET mutations. More recent evidences suggested that immune microenvironment associated to TC may play a critical role in tumor invasion, with potential immunotherapeutic implications in advanced and metastatic TC. Several types of ancillary approaches have been developed to improve the diagnostic value of fine needle aspiration biopsies in indeterminate thyroid nodules. Finally, liquid biopsy, as a non-invasive diagnostic tool for body fluid genotyping, brings a new prospective of disease and therapy monitoring. Despite all these novelties, much work remains to be done to fully understand the pathogenesis and biological behaviors of the different types of TC and to transfer this knowledge in clinical practice.
Thyroid cancer (TC) represents the most common endocrine malignancy, accounting for 3.4% of all cancers diagnosed annually (1). The transformation of thyroid follicular cells may result in differentiated or undifferentiated TC, through a multistep process that is the most accepted theory of follicular cell carcinogenesis (2). In this model, distinct molecular alterations have been associated with specific stages, driving progression from well-differentiated to undifferentiated follicular-derived thyroid carcinomas. More recently, the cancer stem-like cells theory has been proposed, according to which phenotypically different cancer cells could be generated by a small subpopulation of stem cells after genetic and epigenetic transformations (3). Differentiated TC, accounting for more than 90% of thyroid malignancies, comprises papillary thyroid carcinoma (PTC) and follicular thyroid carcinoma (FTC). Poorly differentiated thyroid carcinoma (PDTC) and anaplastic thyroid carcinoma (ATC) are rare tumors (5 and 1%, respectively) associated with aggressive behavior and short median time of survival (5 years and 6 months, respectively). Differently, medullary thyroid carcinoma (MTC), representing 5% of TC, arises from parafollicular C cells.
In the last 30 years, the availability of the genome sequence has produced much progress in elucidating the molecular mechanisms underlying TC (4). TC is a genetically simple disease with a relatively low somatic mutation burden in each tumor. Driver mutations, i.e., mutations that provide a selective growth advantage thus promoting cancer development, are identified in more than 90% of TC (4). The molecular pathogenesis of the majority of TC involves dysregulation of the mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3 kinase (PI3K)/AKT signaling pathways. MAPK activation is considered to be crucial for PTC initiation, through point mutations of the BRAF and RAS genes or gene fusions of RET/PTC and TRK. On the other hand, PI3K/AKT activation is thought to be critical in FTC initiation and can be triggered by activating mutations in RAS, PIK3CA, and AKT1 as well as by inactivation of PTEN, which negatively regulates this pathway. TC progression and dedifferentiation to PDTC and ATC involves a number of additional mutations affecting other cell signaling pathways, such as p53 and Wnt/β-catenin. More recently, TERT promoter mutations have been described in all the histological TC type, with a significantly higher prevalence in aggressive and undifferentiated tumors, indicating their role in TC progression (Figure 1). Mutations in the RET (Rearranged during transfection) proto-oncogene account for most MTC cases and can occur sporadically or as inherited germline events in the multiple endocrine neoplasia type 2A (MEN2A) and 2B (MEN2B) syndromes. A minority of sporadic MTC are caused by H-, K-, and N-RAS mutations (Table 1).
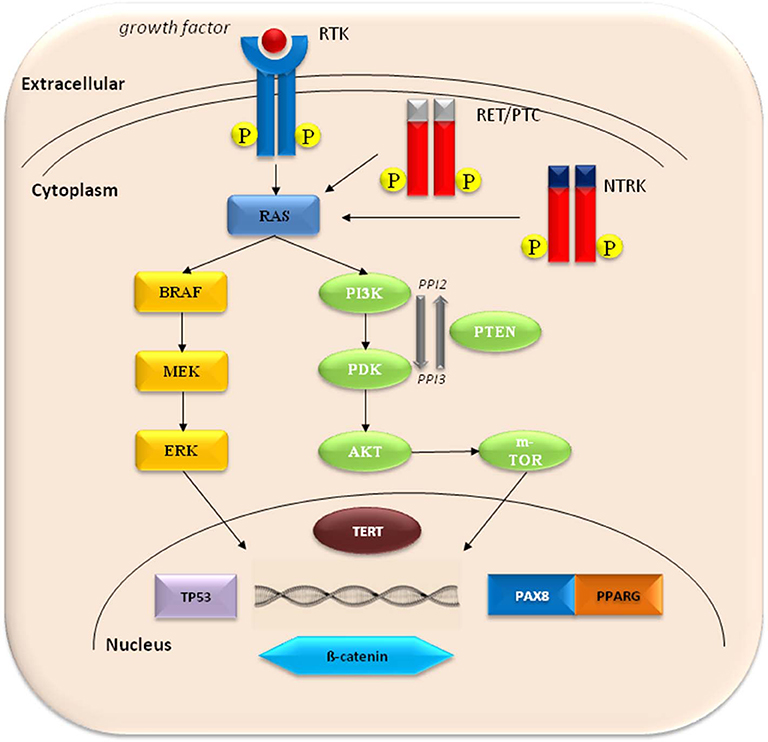
Figure 1. The molecular pathogenesis of thyroid cancer involves dysregulation of the mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3 kinase (PI3K)/AKT pathways. Common activating mutations in the MAPK pathway include RET-PTC and NTRK rearrangements, and RAS and BRAF mutations. Common genetic alterations in the PI3K pathway include RAS mutations, PTEN mutations or deletions, PIK3K mutations or amplifications, and AKT1 mutations. PAX8-PPARG fusions are common in FTC. Activation of Wnt/b-catenin pathway, inactivating mutations in TP53, and activating mutations in TERT promoter are frequent in undifferentiated thyroid cancer.
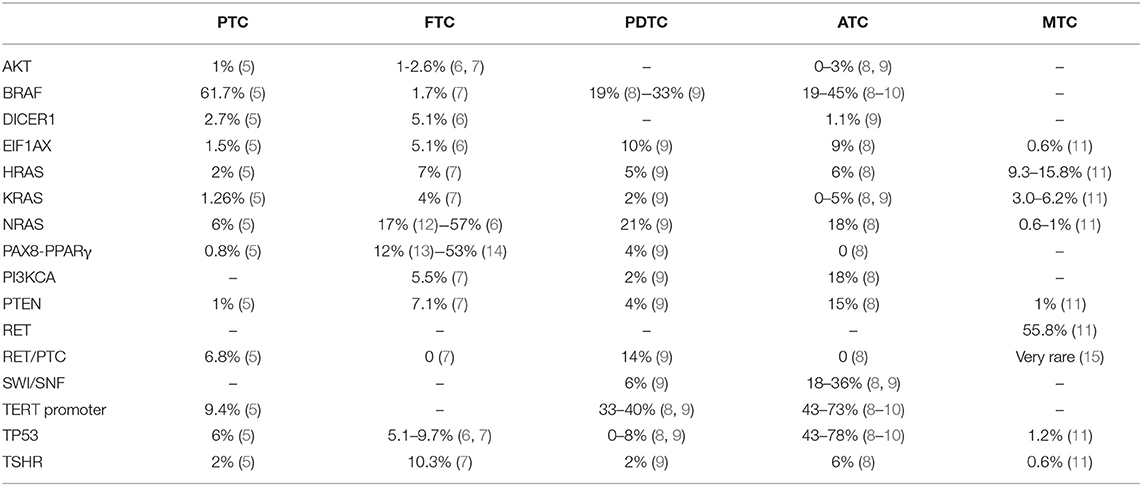
Table 1. Distribution and frequency of known somatic mutations in different histotypes of thyroid cancer.
The majority of PTC has an excellent prognosis in terms of long-term survival (>90%) (4, 16), although recurrent disease rates reported are rather high, occurring in 25–35% of patients (4, 17, 18). The clinical challenge relies in the early identification of those patients who need aggressive treatment from the beginning from those who will have an indolent course.
At the molecular level, several driver mutations have been associated with PTC malignancy and clinicopathological features, but none has proven useful in directing treatment and determining clinical outcome. Among these, RET rearrangements or point mutations of RAS or BRAF proto-oncogenes have been described and are found in an almost mutually exclusive modality in nearly 70% of PTC (Table 1). These genetic alterations are common in PTC, leading to (constitutively) activation of MAPK or PI3K signaling pathways (19).
RET proto-oncogene encodes for a tyrosine kinase receptor and its activation invokes intracellular signaling cascades, leading to gene expression modulation and biological responses. RET/PTC fusion protein maintains the tyrosine kinase domain intact and enables uncontrolled activation of the MAPK signaling cascade (20). RET rearrangement was first reported by Fusco et al. (21), and in the following years, different types of RET/PTC rearrangements have been identified (15). RET/PTC1 and RET/PTC3 are the most common (5), the latter being frequent in post-Chernobyl children due to radiation exposure. The prevalence of RET rearrangements in PTC has varied deeply among studies (2.5–73%) (22, 23) probably due to ethnical and geographical variations as well as to the method used for their identification and genetic heterogeneity, as demonstrated by Zhu et al. (24); recent reports, however, from the Tumor Cancer Genome Atlas (TCGA) in a series of 484 PTC belonging to different ethnic groups, only 6.8% presented RET rearrangements (5). Some reports have indicated RET/PTC1 as being associated with a more favorable prognosis, while RET/PTC3 was associated with a more aggressive and malignant phenotype (25, 26). However, patients harboring these rearrangements usually follow a favorable course, owing to their ability to respond well to radioactive iodine (RAI) therapy (27). It is of interest that in post-Chernobyl TC, other rearrangements have been found: in particular, TRK gene and BRAF gene fusions (28). Recently, NTRK fusions have been reported in some series of advanced cancers and have been proposed as novel targets of cancer therapy (29).
BRAF, a member of the raf family of serine/threonine protein kinases, has been shown to be mutated and constitutively activated in ~7% of all cancers. Prevalence of BRAF mutation in PTC varies among different series ranging from 29 to 83% (30–37). Most recently, the TCGA reported 74.6% of BRAF mutations in PTC, of which 61.7% were V600E substitutions. Different genetic alterations have been identified in this gene; however, the majority of classic PTC (cPTC) harbor the BRAFV600E variant (32). The mutation of BRAF promotes the activation of downstream transcription factors, leading to cell differentiation, proliferation, growth, and apoptosis. Several studies reported an association between the V600E variant and aggressive disease features, including lymph node metastases, invasion, and recurrence (38, 39). Intratumor genetic heterogeneity involving BRAF mutation has been demonstrated and the clonal/subclonal status of BRAFV600E could account for the conflicting results on the prognostic value of this variant (5, 40, 41), as well as may explain the lack of complete response to targeted therapies (42).
RAS is a family of GTP-binding proteins, upstream of BRAF, that acts through the MAPK and PI3K-AKT signaling pathways. HRAS, KRAS, and NRAS encode for four different but related proteins (H-Ras, N-Ras, K-Ras4A, and K-Ras4B) that are cardinal in controlling cell growth, differentiation, and survival. Missense mutations at codons 12, 13, and 61 lead to constitutive activation of RAS signaling, which is found mutated in >30% of all tumors, including thyroid lesions both benign and malignant (43). In fact, RAS mutations may be found in FTC (28–68%), in follicular-variant PTC [FVPTC; up to 43% (31)], and in its non-invasive FVPTC [NIFTP; up to 47% (44)], as well as in follicular adenomas (20–25%) (45), showing the limited role for RAS mutations alone in the clinical outcomes of TC (5, 46).
TERT encodes for the telomerase reverse transcriptase, and two hotspot genetic alterations have been reported (C228T and C250T). These mutations promote telomerase activity and telomere length maintenance in cancer cells and are present in nearly 10% of PTC (Table 1). There are consistent data linking them to PTC aggressiveness when in co-presence of a driver mutation, indicating a possible role for TERT mutations in PTC progression and prognosis (47).
Among PTC variants, NIFTP represents a novel entity with an almost negligible risk of negative outcome (46). Many efforts have been produced in order to identify a unique NIFTP genomic profile that helps to diagnose these tumors by FNAB. A recent article showed that the NIFTP genomic profile is more similar to FTC than PTC. Interestingly, 67% of NIFTP harbored RAS mutations alone or in tandem with other mutations (p53 and PTEN mutations), whereas BRAFV600E mutation was not described. Furthermore, 22% of NIFTP presented PAX8/PPARG and THADA/IGF2BP3 gene fusion mutations (48). However, although NIFTP genomic profile seems to be different from those of other PTC variants, the degree of overlap makes it difficult to identify NIFTP with FNAB (49) and molecular analysis.
Recently, the TCGA has unfolded the genomic landscape of TC, reducing to <4% the unknown genomics of PTC (5). Based on a BRAFV600E-RAS gene expression score, PTCs may be grouped according to their molecular differences as BRAFV600E-like and RAS-like PTC. In fact, BRAFV600E mutation is more frequent in cPTC and tall-cell variant PTC, showing increased MAPK activation, whereas RAS mutations occur mostly in FVPTC and NIFTP, having a genomic profile more similar to FTC. The genomic landscape described by these studies reveals that PTC bears a relatively stable genome, which could explain the usually indolent course of this disease. Nonetheless, aggressive PTC may occur and, therefore, additional investigation is necessary in order to early identify those PTCs that will dedifferentiate and become life-threatening.
Hyalinizing trabecular tumor (HTT) is a rare benign follicular neoplasm characterized by thick trabeculae and cells with nuclear elements shared with PTC, producing false-positive cytology (50). However, using whole-exome and RNA-Seq analyses, Nikiforova et al. presented a unique genomic signature of HTT and showed that GLIS fusions, especially PAX8-GLIS3, are highly prevalent in HTT but not in PTC. These fusions were related to overexpression of GLIS, inducing upregulation of extracellular deposition of collagen IV (51).
In 2017, the World Health Organization guidelines proposed to re-classify the FTC into minimally invasive (miFTC), encapsulated angioinvasive (eaFTC), and widely invasive (wiFTC) subtypes, according to their different clinical and biological behaviors (52). Although the genomic landscape of PTC is nearly complete, the molecular characterization of FTC and its progression from miFTC to wiFTC are still not totally clear.
In FTC, the most common mutations are in the RAS gene family (HRAS, KRAS, and NRAS), and NRAS gene was found mutated in 17% (12) to 57% (6) of cases. Although a previous study had demonstrated that RAS mutations are negative prognostic markers (53), recent evidences did not describe RAS mutations as predictors of disease-specific mortality (54). Intriguingly, RAS mutations appear to be mutually exclusive with TSH receptor mutations, which were found in 10.3% of FTC cases (54) (Table 1).
The fusion gene PAX8-PPARγ was identified in one-third of FTC cases, ranging from 12% (13) to 53% (14) (Table 1). PAX8 is a member of paired box family of transcription factors, and it is necessary for the physiological thyroid development, promoting thyroid progenitor survival and driving the expression of thyroid-specific genes (55). Conversely, PPARγ is a member of the nuclear receptor family of transcription factors, and, besides its role of master of adipogenesis, it seems to be a tumor suppressor gene (56). The fusion protein PAX8-PPARγ can act as a negative inhibitor of oncosuppressor PPARγ activity or as a novel transcriptional factor with protooncogene activity (57). However, it seems to not affect FTC prognosis (13).
TERT promoter mutations have been described in about 15% of FTCs (Table 1) and associated with worst clinical and prognostic features (58). Furthermore, many groups (54, 59, 60) described point mutations of driver genes EIF1AX and DICER1 and somatic arm-level copy changes (e.g., loss of 22q), the significance of which needs to be clarified.
In this complex scenario, the total mutational burden seems to be a prognostic factor: the bigger is the number of mutations, the worse is the prognosis. Furthermore, since multivariate analysis describes the total mutational burden as an independent indicator of histopathology, the genetic analysis may be used to predict survival as a complement to the histological informations (54).
The frequency of molecular mutations in PDTC is not the same in all studies, and this can be related not only to the sensitivity of the molecular technique used to ascertain the molecular pattern (Next-generation sequencing vs. Sanger sequencing) but also to the histological criteria used to define the PDTC. Indeed, two main classifications for PDTC exist: Turin (the presence of a solid/trabecular/insular pattern of growth, in the absence of the conventional nuclear features of PTC and at least one of the following: convoluted nuclei, high mitotic rate, or tumor necrosis) and MSKCC criteria (high mitotic rate and necrosis independently from the growth pattern) (8, 61–63). BRAF mutations are found in 19% (8)−33% (61) of PDTC, while H-, K-, and N-RAS mutations were found in 5% (61)−28% (8) of cases (Table 1). BRAFV600E was found to be more frequent in PDTC when defined following the MSKCC criteria, while RAS mutations are more common in PDTC fulfilling Turin definition (8). Moreover, BRAF and RAS are mutually exclusive and correlate with a different clinical behavior: BRAF-mutated PDTCs were found to have a higher rate of nodal metastases vs. a higher rate of distant metastases found in RAS-mutated PDTCs. Furthermore, the expression of thyroid-specific genes related to radioiodine avidity was found to be lowered in BRAF-mutated PDTCs, but not in their RAS mutated counterparts (8).
TERT promoter mutation can co-occur with BRAF and RAS mutations (8)and is particularly frequent in advanced tumors: 33% (61)−40% (8) of PDTCs are found to carry a TERT promoter mutation (Table 1), inducing a higher risk of distant metastases and mortality (8). Intriguingly, TERT promoter mutation in PTC is subclonal, while it is clonal in advanced cancers (PDTC and ATC), suggesting an advancement in TC due to a selected immortalized TERT-positive clone (8). By contrast, p53 was rarely found in PDTC, even using sensible techniques, having a frequency of 8% in the Landa et al. analysis, with no patients carrying p53 mutation in PDTC in the Elisei et al. series (8, 61). EIF1AX mutations are present in 1% of PTC and in 10% of PDTC, inducing a worse survival (8) (Table 1). Interestingly, in advanced cancers, it has a strong association with RAS mutations, while in PTC, these two mutations are mutually exclusive. The significance of this observation is yet to be clarified. EIF1AX mutation does not overlap with PI3K/AKT/mTOR pathway mutations, suggesting similar functions in thyroid progression. Also, PTEN/PI3KCA is uncommon (8) or even absent in PDTC (61).
Another substantial difference in thyroid advanced tumors compared to PTC is in the chromosome number variation. The genome of PTC is largely diploid, while in PDTC and ATC, chromosome copy number alterations are widespread and more frequent in those tumors lacking a driver gene mutation (8). Gene rearrangements common in PTC (RET/PTC, PAX8-PPARγ, ALK fusions) may be found in 14% of PDTC (specially in younger patients), but are absent in ATC (8).
In ATC, BRAF and H-, K-, and N-RAS mutations have a frequency of 19–45% and 9.5–27%, respectively (8, 9, 61), lower than that of DTC. Conversely, the two most frequent mutations occurring in ATC are TERT promoter mutations, occurring in 43–73% of cases and TP53 mutations that have a frequency ranging from 48 to 73% of cases (8, 9, 61). Interestingly, while TERT promoter mutations are quite common also in PDTC, TP53 is highly frequent only in ATC and thus it may be considered pathognomonic for this tumor and its severe aggressiveness (8). Also, mutations in PTEN and PI3KCA are rather frequent in ATC, 15 and 18%, respectively, in comparison with well-differentiated cancers and PDTC (8). Moreover, in ATC, other mutations less typical for thyroid tumors are also found: 18–36% of ATC carry mutations in SWI/SNF chromatin remodeling complex (8, 9) and in genes associated with histone modifications (8) (Table 1); mutations in genes involved in cell-cycle regulation (CDKN2A, CDKN2B, and CCNE1) are present in 29% of ATC (8), and finally, few ATCs were also found to be mutated in tumor immune regulation genes (PDL1, PDL2, and JAK2) (8). The real pathogenic role of these alterations is unknown and could be related to the genomic instability of these tumors.
Interestingly, in this panorama of apparently heterogeneous molecular scenario, four distinct subtypes of molecular pattern of ATC have been proposed: (1) type 1 ATC, BRAF-positive ATC, with a genetic landscape similar to PTC (it is likely to evolve from PTC); (2) type 2 ATC, NRAS-positive ATC, which may originate from FTC; (3) type 3 ATC, which carries RAS mutations or more atypical ones (e.g., PTEN, NF1 and RB1) and is likely to originate from FTC or from Hürthle cell carcinoma; and (4) mixed ATC, which harbor loss-of-function genetic alterations and mutations in the genes of cell-cycle regulations (CDKN2A and CDKN2B) (9) and do not seem to derive from a pre-existing DTC.
Intriguingly, ATC presents a deeper status of dedifferentiation than DTC and PDTC: in ATC, mRNA levels for TG, TSHR, TPO, PAX8, SLC26A4, DIO1, and DUOX2 genes are profoundly supressed (8). In BRAF-positive ATC, TP53 or PIK3CA mutations are frequently found and may drive the dedifferentiation process. Among ATC tumors carrying RAS mutations, the mechanism of dedifferentiation is less clear, although EIF1AX is a good candidate, given its frequent co-occurrence with RAS mutations in advanced cancers (10). Among ATC tumors not carrying BRAF or RAS mutations, the atypical mutations of NF1, ERBB2, mTOR, and MHL genes may enhance the dedifferentiation process (10).
MTC can be either familial (25%) or sporadic (75%), and in both cases, proto-oncogene RET exerts a crucial role in its oncogenesis. Virtually, all familial cases (>98%) present germline RET mutations (64). However, two cases of familial MTC without any RET germline mutations have been recently described with one case carrying a germline mutation of ESR2 gene (65) and another one of MET gene (66). In sporadic cases, RET is mutated in 44% and RAS genes (mainly HRAS and KRAS) are mutated in 13% of cases, according to COSMIC (catalog of somatic mutations in cancer) database (7). Intriguingly, oncogenesis of a relevant group of sporadic (more than 40%) and some rare familial MTCs is still unclear.
In MTC, RET gene is typically harboring point mutations, while its deletions or insertions are rare. Activating point mutations of RET may affect both extracellular and intracellular domains, inducing different effects: intracellular mutations domain induce a ligand-independent constitutive dimerization, promoting the activation of the tyrosine kinase receptor; otherwise, extracellular mutation domains induce a ret activation, which is ligand and dimerization independent (15). Mutations of different ret domains induce different clinical features in familial MTC cases. They are grouped into MEN2A and MEN2B syndromes, and the MEN2A syndrome is subdivided by clinical characteristics into MEN2A associated with cutaneous lichen amyloidosis (CLA), MEN2A with Hirschsprung Disease (HD), and familial MTC (FMTC) (67). There is an evident genotype–phenotype correlation with patients affected by classical MEN2A harboring almost exclusively RET codon 634 mutations (68), patients affected by MEN2A and HD carrying RET germline “Janus” mutations in exon 10 (codons 609, 611, 618 and 620) (69), and patients with MEN2B harboring almost exclusively RET germline mutations in exon 16 (codon M918T) (70).
The genetic profile in sporadic MTCs is more heterogeneous than in familial MTCs. Recently, the genetic landscape of 208 cases of sporadic MTCs, identified by using a deep sequencing technique, has been published (11). In this large series, the number of RET or RAS negative cases was highly reduced (18.3%), and the crucial pathogenic role of RET and RAS gene, which are affected by mutually exclusive alterations, has been confirmed. According to these data, RET mutations remain the most common genetic variant in sporadic MTCs (55.8%) followed by RAS mutations (24.3%) (Table 1). Interestingly, the study also demonstrated that patients with RET-positive MTCs have a lower survival than those with RAS mutations. Moreover, the variant allele frequency represents an additional prognostic marker in RET-positive MTCs (11).
Increasing evidences confirm that solid tumors are composed by different clusters of cells including cancer cells, cancer stem cells, fibroblasts, and stroma cells, and also a variety of cells belonging to the innate and adaptive immune system (71). Nowadays, the evidence that tumor cells and immune cells have an important relationship inside tumor microenvironment is recognized worldwide. Furthermore, the presence in some solid tumors of an intensive infiltration and the evidence of a contemporary anti-tumor response and a tolerant microenvironment, essential for tumor growth and progression, enforce the role of the immune system inside cancer (72). In this context, a comprehensive study of the immune profile of TC to clarify the mechanisms involved in immune escape and characterize the tumor microenvironment results is pivotal.
Over the last years, many studies have focused on cancer gene expression profiling, outlying a detailed immune profile also for TC. In 2018, a classification arisen from a huge research on the TCGA databases detected six immune subtypes of cancers (73): C1—Wound healing; C2—IFN-γ dominant; C3—Inflammatory; C4—Lymphocyte depleted; C5—Immunologically quiet; C6—TGF-β dominant. Following this classification, the majority of PTCs were classified as C3 tumors, with a balance in T helper1:T helper2 presence, elevated T helper 17 genes, low tumor cell proliferation, and lower levels of aneuploidy and somatic copy number alterations. This was in agreement with the former observation of a complex immune network inside PTCs consisting of a rich infiltration by tumor-associated macrophages, myeloid-derived suppressor cells, and T helper 17 cells (74).
More recently, the development of an immunoscore stratification, based on immune contexture within the tumor, has allowed the classification of cancers by their immune phenotype. Indeed, thanks to the distribution of T CD3+ and T CD8+ lymphocytes in the center of the cancer or at the invasive margin, it could be possible to distinguish four different phenotypes of cancers: (1) the hot ones, with a high infiltration of cells all over the tumor; (2) altered–excluded with the presence of cells only at the invasive margin; (3) altered–immunosuppressed with sparse immune cells within all the tumor; and (4) cold tumors, without infiltration (75). A study published in 2019 analyzed a cluster of about 730 immune-related genes in the three major histotypes of TC (i.e., PTC, PDTC, and ATC) with the aim to investigate and clarify the immune profiling of advanced TCs (76). The histotypes are segregated into two different clusters of expression: a first group, including PDTCs, part of PTCs, and normal thyroid, and a second group including ATCs and part of PTCs. Interestingly, the regulation of gene expression was different between ATCs and PDTCs: the first ones had a marked overexpression of about all immune genes analyzed, compared to normal tissue; the second ones had expression levels that were very similar to normal thyroid. The results obtained indicated the existence of two major immune phenotypes in TCs: an ATC-like one, including hot and altered–immunosuppressed tumors, and a PDTC-like one, including altered–excluded and cold tumors. Moreover, TCs, mostly the anaplastic ones, showed an increased overexpression of immune checkpoints, including PDL1, PDL2, PD1, LAG-3, TIM-3, PVR, and TIGIT. These data confirm a strong activation of adaptive immune escape strategies for blocking tumor-infiltrating leucocytes, especially in ATC (77).
Significant advances in the understanding of TC biology, coupled with advances in high-throughput technologies, are contributing to the development of novel diagnostic, prognostic, predictive, and therapeutic tools for TC patients.
Most efforts have been made in the development of molecular tests for cancer diagnosis in thyroid nodules. Panels of gene expression markers [e.g., Afirma Genomic Sequencing Classifier (78)] or somatic mutation panels [e.g., ThyroSeq Genomic Classifier (79)] have improved the pre-operative diagnostic accuracy for patients with indeterminate cytology by addressing the problem of unnecessary surgery for benign thyroid nodules. Much effort should be done in order to pre-operatively identify a subset of aggressive cancers or to increase positive predictive value in some tumor subtypes (i.e., in RAS-mutated cases).
An alternative approach for early diagnosis and prompt detection of disease persistence or relapse is liquid biopsy (i.e., the sampling and non-invasive analysis of circulating tumor-derived material, the tumor circulome) (80), and its details are reported elsewhere (81). Although the development of circulating biomarkers for TC is still in its infancy and at present liquid biopsy does not find any application, it presents several advantages, such as the rapid, low-cost, non-invasive nature of sample collection and the capture of intratumoral and intermetastatic genetic heterogeneity. The diagnostic application of circulating tumor DNA (ctDNA) in follicular cell-derived TC is still questioned. In contrast with other advanced cancers, only 25% of metastatic TCs have detectable ctDNA (82). These data are confirmed by multiple studies focusing on detection of BRAFV600E mutation in PTCs, which showed low or no concordance between plasma and tissue samples (83), also when sensitive techniques were employed (84). Conversely, higher concordance was found in ATC (85) and MTC (86) patients with important implications in guiding treatment selection and clinical trial enrollment.
Circulating miRNAs represent an alternative and valuable source for real-time thyroid tumor monitoring, due to their high stability in biological fluids (87) and tissue specificity (88). Most studies published thus far have been conducted on PTC patients, and in this setting, unlike ctDNAs, circulating miRNAs show undeniable promise as novel diagnostic and predictive biomarkers. Higher circulating levels of miR-221-3p, miR-222-3p, and miR-146b-5p were detected in PTC patients than in healthy controls, while miR-222 and miR-146b levels also discriminate between PTCs and benign nodules. Moreover, circulating levels of miR-146b-5p, miR-221-3p, miR-222-3p, and miR-146a-5p have been shown to decline after tumor excision (89). Recently, miRNAs of tumor tissue have been proposed to face the challenge of indeterminate FNAB category. Stokowy et al. showed that none of the miRNAs could be used as an alone malignancy marker but the classifier made by miR-484/miR-148b-3p identified TC with a sensitivity of 89% and a specificity of 87% (90).
Furthermore, miR-221-3p and miR-146a-5p blood levels in PTC patients have been shown to predict clinical responses, with significantly increased levels observed at the 2 year follow-up in patients with structural evidence of disease, including some in which serum thyroglobulin assays remained persistently negative (91).
Realization of this enormous potential will depend on our ability to develop standardized methods for detection of circulating biomarkers and to validate their performance in clinical setting.
We discussed the molecular mechanisms involved into the pathogenesis of the different types of TC and their clinical relevance. In the last years, many steps forward have been made in the genetic characterization of TC, providing molecular markers for diagnosis, risk stratification, and treatment targets. However, many other steps need to be done in order to diagnose TCs with aggressive behavior, to tailor the most appropriate target therapy, and to monitor the response to the therapies using new molecular approaches.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with the author MM.
We thank Prof. Francesco Frasca from the Endocrine Unit of the Department of Clinical and Experimental Medicine of Catania University, Italy; Prof. Efisio Puxeddu from the Internal Medicine and Endocrine and Metabolic Science Section of the Department of Medicine, University of Perugia, Italy; Prof. Mario Vitale from the Endocrine Unit, University of Salerno, Italy; and Prof. Rossella Elisei from the Endocrine Unit of the Department of Clinical and Experimental Medicine of Pisa University, Italy, for the invaluable contribution in preparing this manuscript.
1. Seib CD, Sosa JA. Evolving understanding of the epidemiology of thyroid cancer. Endocrinol Metab Clin N Am. (2019) 48:23–35. doi: 10.1016/j.ecl.2018.10.002
2. Nowell PC. The clonal evolution of tumor cell populations. Science. (1976) 194:23–8. doi: 10.1126/science.959840
3. Takano T. Fetal cell carcinogenesis of the thyroid: a modified theory based on recent evidence. Endocr J. (2014) 61:311–20. doi: 10.1507/endocrj.EJ13-0517
4. Mazzaferri EL. An overview of the management of papillary and follicular thyroid carcinoma. Thyroid. (1999) 9:421–7. doi: 10.1089/thy.1999.9.421
5. The Cancer Genome Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell. (2014) 159:676–90. doi: 10.1016/j.cell.2014.09.050
6. Fukahori M, Yoshida A, Hayashi H, Yoshihara M, Matsukuma S, Sakuma Y, et al. The associations between ras mutations and clinical characteristics in follicular thyroid tumors: new insights from a single center and a large patient cohort. Thyroid. (2012) 22:683–9. doi: 10.1089/thy.2011.0261
7. Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. (2019) 47:D941–7. doi: 10.1093/nar/gky1015
8. Landa I, Ibrahimpasic T, Boucai L, Sinha R, Knauf JA, Shah RH, et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J Clin Invest. (2016) 126:1052–66. doi: 10.1172/JCI85271
9. Pozdeyev N, Gay LM, Sokol ES, Hartmaier R, Deaver KE, Davis S, et al. Genetic analysis of 779 advanced differentiated and anaplastic thyroid cancers. Clin Cancer Res. (2018) 24:3059–68. doi: 10.1158/1078-0432.CCR-18-0373
10. Kunstman JW, Juhlin CC, Goh G, Brown TC, Stenman A, Healy JM, et al. Characterization of the mutational landscape of anaplastic thyroid cancer via whole-exome sequencing. Hum Mol Genet. (2015) 24:2318–29. doi: 10.1093/hmg/ddu749
11. Ciampi R, Romei C, Ramone T, Prete A, Tacito A, Cappagli V, et al. Genetic landscape of somatic mutations in a large cohort of sporadic medullary thyroid carcinomas studied by next-generation targeted sequencing. iScience. (2019) 20:324–36. doi: 10.1016/j.isci.2019.09.030
12. Vuong HG, Kondo T, Oishi N, Nakazawa T, Mochizuki K, Inoue T, et al. Genetic alterations of differentiated thyroid carcinoma in iodine-rich and iodine-deficient countries. Cancer Med. (2016) 5:1883–9. doi: 10.1002/cam4.781
13. Boos LA, Dettmer M, Schmitt A, Rudolph T, Steinert H, Moch H, et al. Diagnostic and prognostic implications of the PAX8-PPARγ translocation in thyroid carcinomas-a TMA-based study of 226 cases. Histopathology. (2013) 63:234–41. doi: 10.1111/his.12150
14. Nikiforova MN, Biddinger PW, Caudill CM, Kroll TG, Nikiforov YE. PAX8-PPARγ rearrangement in thyroid tumors: RT-PCR and immunohistochemical analyses. Am J Surg Pathol. (2002) 26:1016–23. doi: 10.1097/00000478-200208000-00006
15. Romei C, Ciampi Elisei RR. A comprehensive overview of the role of the RET proto-oncogene in thyroid carcinoma. Nat Rev Endocrinol. (2016) 12:192–202. doi: 10.1038/nrendo.2016.11
16. Ito Y, Miyauchi A, Kihara M, Fukushima M, Higashiyama T, Miya A. Overall survival of papillary thyroid carcinoma patients: a single-institution long-term follow-up of 5897 patients. World J Surg. (2018) 42:615–22. doi: 10.1007/s00268-018-4479-z
17. Shah MD, Hall FT, Eski SJ, Witterick IJ, Walfish PG, Freeman JL. Clinical course of thyroid carcinoma after neck dissection. Laryngoscope. (2003) 12:2102–7. doi: 10.1097/00005537-200312000-00008
18. Grogan RH, Kaplan SP, Cao H, Weiss RE, Degroot LJ, Simon CA, et al. A study of recurrence and death from papillary thyroid cancer with 27 years of median follow-up. Surgery. (2013) 154:1436–47. doi: 10.1016/j.surg.2013.07.008
19. Zou M, Baitei EY, Alzahrani AS, BinHumaid FS, Alkhafaji D, Al-Rijjal RA, et al. Concomitant RAS. RET/PTC, or BRAF mutations in advanced stage of papillary thyroid carcinoma. Thyroid. (2014) 24:1256–66. doi: 10.1089/thy.2013.0610
20. Knauf JA, Kuroda H, Basu S, Fagin JA. RET/PTC-induced dedifferentiation of thyroid cells is mediated through Y1062 signaling through SHC-RAS-MAP kinase. Oncogene. (2003) 22:4406–12. doi: 10.1038/sj.onc.1206602
21. Fusco A, Grieco M, Santoro M, Berlingieri MT, Pilotti S, Pierotti MA, et al. A new oncogene in human thyroid papillary carcinomas and their lymph-nodal metastases. Nature. (2003) 328:170–2. doi: 10.1038/328170a0
22. Zou M, Shi Y, Farid NR. Low rate of ret proto-oncogene activation (PTC/retTPC) in papillary thyroid carcinomas from Saudi Arabia. Cancer. (1994) 73:176–803
23. Chua EL, Wu WM, Tran KT, McCarthy SW, Lauer CS, Dubourdieu D, et al. Prevalence and distribution of ret/ptc 1, 2, and 3 in papillary thyroid carcinoma in New Caledonia and Australia 1. J Clin Endocrinol Metab. (2000) 85:2733–9. doi: 10.1210/jc.85.8.2733
24. Zhu Z, Ciampi R, Nikiforova MN, Gandhi M, Nikiforov YE. Prevalence of RET/PTC rearrangements in thyroid papillary carcinomas: effects of the detection methods and genetic heterogeneity. J Clin Endocrinol Metab. (2006) 91:3603–10. doi: 10.1210/jc.2006-1006
25. Adeniran AJ, Zhu Z, Gandhi M, Steward DL, Fidler JP, Giordano TJ, et al. Correlation between genetic alterations and microscopic features, clinical manifestations, and prognostic characteristics of thyroid papillary carcinomas. Am J Surg Pathol. (2006) 30:216–22. doi: 10.1097/01.pas.0000176432.73455.1b
26. Khan MS, Qadri Q, Makhdoomi MJ, Wani MA, Malik AA, Niyaz M, et al. RET/PTC gene rearrangements in thyroid carcinogenesis: assessment and clinico-pathological correlations. Pathol Oncol Res. (2018). doi: 10.1007/s12253-018-0540-3. [Epub ahead of print].
27. Paulson VA, Rudzinski ER, Hawkins DS. Thyroid cancer in the pediatric population. Genes. (2019) 10:723. doi: 10.3390/genes10090723
28. Ciampi R, Knauf JA, Kerler R, Gandhi M, Zhu Z, Nikiforova MN, et al. Oncogenic AKAP9-BRAF fusion is a novel mechanism of MAPK pathway activation in thyroid cancer. J Clin Invest. (2005) 115:94–101. doi: 10.1172/JCI23237
29. Solomon JP, Benayed R, Hechtman JF, Ladanyi M. Identifying patients with NTRK fusion cancer. Ann Oncol. (2019) 30:viii16–22. doi: 10.1093/annonc/mdz384
30. Kim KH, Kang DW, Kim SH, Seong IO, Kang DY. Mutations of the BRAF gene in papillary thyroid carcinoma in a Korean population. Yonsei Med J. (2004) 45:818–21. doi: 10.3349/ymj.2004.45.5.818
31. Kimura ET, Nikiforova MN, Zhu Z, Knauf JA, Nikiforov YE, Fagin JA. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. (2003) 63:1454–7.
32. Trovisco V, Vieira de Castro I, Soares P, Máximo V, Silva P, Magalhães J, et al. BRAF mutations are associated with some histological types of papillary thyroid carcinoma. J Pathol. (2004) 202:247–51. doi: 10.1002/path.1511
33. Soares P, Trovisco V, Rocha AS, Lima J, Castro P, Preto A, et al. BRAF mutations and RET/PTC rearrangements are alternative events in the etiopathogenesis of PTC. Oncogene. (2003) 22:4578–80. doi: 10.1038/sj.onc.1206706
34. Nikiforova MN, Kimura ET, Gandhi M, Biddinger PW, Knauf JA, Basolo F, et al. BRAF mutations in thyroid tumors are restricted to papillary carcinomas and anaplastic or poorly differentiated carcinomas arising from papillary carcinomas. J Clin Endocrinol Metab. (2003) 88:5399–404. doi: 10.1210/jc.2003-030838
35. Cohen Y, Rosenbaum E, Clark DP, Zeiger MA, Umbricht CB, Tufano RP, et al. Mutational analysis of BRAF in fine needle aspiration biopsies of the thyroid: a potential application for the preoperative assessment of thyroid nodules. Clin Cancer Res. (2004) 10:2761–5. doi: 10.1158/1078-0432.CCR-03-0273
36. Fugazzola L, Mannavola D, Cirello V, Vannucchi G, Muzza M, Vicentini L, et al. BRAF mutations in an Italian cohort of thyroid cancers. Clin Endocrinol. (2004) 61:239–43. doi: 10.1111/j.1365-2265.2004.02089.x
37. Zatelli MC, Trasforini G, Leoni S, Frigato G, Buratto M, Tagliati F, et al. BRAF V600E mutation analysis increases diagnostic accuracy for papillary thyroid carcinoma in fine-needle aspiration biopsies. Eur J Endocrinol. (2009) 161:467–73. doi: 10.1530/EJE-09-0353
38. Cappola AR, Mandel SJ. Molecular testing in thyroid cancer: BRAF mutation status and mortality. JAMA. (2013) 309:1529–30. doi: 10.1001/jama.2013.3620
39. Rossi M, Buratto M, Bruni S, Filieri C, Tagliati F, Trasforini G, et al. Role of ultrasonographic/clinical profile, cytology, and BRAF V600E mutation evaluation in thyroid nodule screening for malignancy: a prospective study. J Clin Endocrinol Metab. (2012) 97:2354–61. doi: 10.1210/jc.2011-3494
40. Guerra A, Sapio MR, Marotta V, Campanile E, Rossi S, Forno I, et al. The primary occurrence of BRAFV600E is a rare clonal event in papillary thyroid carcinoma. J Clin Endocrinol Metab. (2012) 7:517–24. doi: 10.1210/jc.2011-0618
41. Finkel A, Liba L, Simon E, Bick T, Prinz E, Sabo E, et al. Subclonality for BRAF mutation in papillary thyroid carcinoma is associated with earlier disease stage. J Clin Endocrinol Metab. (2016) 101:1407–13. doi: 10.1210/jc.2015-4031
42. Antonello ZA, Hsu N, Bhasin M, Roti G, Joshi M, Van Hummelen P, et al. Vemurafenib-resistance via de novo RBM genes mutations and chromosome 5 aberrations is overcome by combined therapy with palbociclib in thyroid carcinoma with BRAFV600E. Oncotarget. (2017) 8:84743–60. doi: 10.18632/oncotarget.21262
43. Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. (2011) 11:761–74. doi: 10.1038/nrc3106
44. Kim M, Jeon MJ, Oh HS, Park S, Kim TY, Shong YK, et al. BRAF and RAS mutational status in noninvasive follicular thyroid neoplasm with papillary-like nuclear features and invasive subtype of encapsulated follicular variant of papillary thyroid carcinoma in Korea. Thyroid. (2018) 28:504–10. doi: 10.1089/thy.2017.0382
45. Xing M. Clinical utility of RAS mutations in thyroid cancer: a blurred picture now emerging clearer. BMC Med. (2016) 14:12 doi: 10.1186/s12916-016-0559-9
46. Nikiforov YE, Seethala RR, Tallini G, Baloch ZW, Basolo F, Thompson LD, et al. Nomenclature revision for encapsulated follicular variant of papillary thyroid carcinoma. JAMA Oncol. (2016) 2:1023–9. doi: 10.1001/jamaoncol.2016.2205
47. Vuong HG, Altibi AMA, Duong UNP, Hassell L. Prognostic implication of BRAF and TERT promoter mutation combination in papillary thyroid carcinoma—a meta-analysis. Clin Endocrinol. (2017) 87:411–17. doi: 10.1111/cen.13413
48. Brandler TC, Liu CZ, Cho M, Zhou F, Cangiarella J, Yee-Chang M, et al. Does noninvasive follicular thyroid neoplasm with papillary-like nuclear features (NIFTP) have a unique molecular profile? Am J Clin Pathol. (2018) 150:451–60. doi: 10.1093/ajcp/aqy075
49. Zhao L, Dias-Santagata D, Sadow PM, Faquin WC. Cytological molecular, and clinical features of noninvasive follicular thyroid neoplasm with papillary-like nuclear features versus invasive forms of follicular variant of papillary thyroid carcinoma. Cancer Cytopathol. (2017) 125:323–31. doi: 10.1002/cncy.21839
50. Rossi ED, Papotti M, Faquin W, Larocca LM, Pantanowitz L. The diagnosis of hyalinizing trabecular tumor: a difficult and controversial thyroid entity. Head Neck Pathol. (2019). doi: 10.1007/s12105-019-01083-5
51. Nikiforova MN, Nikitski AV, Panebianco F, Kaya C, Yip L, Williams M, et al. GLIS rearrangement is a genomic hallmark of hyalinizing trabecular tumor of the thyroid gland. Thyroid. (2019) 29:161–73. doi: 10.1089/thy.2018.0791
52. Lloyd RV, Osamura YR, Kloppel G, Rosai J. WHO Classification of Tumours of Endocrine Organs. WHO Press (2017).
53. Garcia-Rostan G, Zhao H, Camp RL, Pollan M, Herrero A, Pardo J, et al. ras Mutations are associated with aggressive tumor phenotypes and poor prognosis in thyroid cancer. J Clin Oncol. (2003) 21:3226–35. doi: 10.1200/JCO.2003.10.130
54. Nicolson NG, Murtha TD, Dong W, Paulsson JO, Choi J, Barbieri AL, et al. Comprehensive genetic analysis of follicular thyroid carcinoma predicts prognosis independent of histology. J Clin Endocrinol Metab. (2018) 103:2640–50. doi: 10.1210/jc.2018-00277
55. Nilsson M, Fagman H. Development of the thyroid gland. Development. (2017) 144:2123–40. doi: 10.1242/dev.145615
56. Patel L, Pass I, Coxon P, Downes CP, Smith SA, Macphee CH. Tumor suppressor and anti-inflammatory actions of PPARγ agonists are mediated via upregulation of PTEN. Curr Biol. (2001) 11:764–8. doi: 10.1016/S0960-9822(01)00225-1
57. Raman P, Koenig RJ. Pax-8-PPAR-γ 3 fusion protein in thyroid carcinoma. Nat Rev Endocrinol. (2014) 10:616–23. doi: 10.1038/nrendo.2014.115
58. Yang J, Gong Y, Yan S, Chen H, Qin S, Gong R. Association between TERT promoter mutations and clinical behaviors in differentiated thyroid carcinoma: a systematic review and meta-analysis. Endocrine. (2019) 67:44–57. doi: 10.1007/s12020-019-02117-2
59. Jung SH, Kim MS, Jung CK, Park HC, Kim SY, Liu J, et al. Mutational burdens and evolutionary ages of thyroid follicular adenoma are comparable to those of follicular carcinoma. Oncotarget. (2016)7:69638–48. doi: 10.18632/oncotarget.11922
60. Yoo SK, Lee S, Kim SJ, Jee HG, Kim BA, Cho H, et al. Comprehensive analysis of the transcriptional and mutational landscape of follicular and papillary thyroid cancers. PLoS Genet. (2016) 12:1–23. doi: 10.1371/journal.pgen.1006239
61. Romei C, Tacito A, Molinaro E, Piaggi P, Cappagli V, Pieruzzi L, et al. Clinical, pathological and genetic features of anaplastic and poorly differentiated thyroid cancer: a single institute experience. Oncol Lett. (2018) 15:9174–82. doi: 10.3892/ol.2018.8470
62. Volante M, Collini P, Nikiforov YE, Sakamoto A, Kakudo K, Katoh R, et al. Poorly differentiated thyroid carcinoma: the turin proposal for the use of uniform diagnostic criteria and an algorithmic diagnostic approach. Am J Surg Pathol. (2007) 31:1256–64. doi: 10.1097/PAS.0b013e3180309e6a
63. Hiltzik D, Carlson DL, Tuttle RM, Chuai S, Ishill N, Shaha A, et al. Poorly differentiated thyroid carcinomas defined on the basis of mitosis and necrosis. Cancer. (2006) 106:1286–95. doi: 10.1002/cncr.21739
64. Elisei R, Tacito A, Ramone T, Ciampi R, Bottici V, Cappagli V, et al. Twenty-five years experience on RET genetic screening on hereditary MTC: an update on the prevalence of germline RET mutations. (2019) 10:E698. doi: 10.3390/genes10090698
65. Smith J, Read ML, Hoffman J, Brown R, Bradshaw B, Campbell C, et al. Germline ESR2 mutation predisposes to medullary thyroid carcinoma and causes up-regulation of RET expression. Hum Mol Genet. (2016) 25:1836–45. doi: 10.1093/hmg/ddw057
66. Sponziello M, Benvenuti S, Gentile A, Pecce V, Rosignolo F, Virzì AR, et al. Whole exome sequencing identifies a germline MET mutation in two siblings with hereditary wild-type RET medullary thyroid cancer. Hum Mutat. (2018) 39:371–7. doi: 10.1002/humu.23378
67. Wells SA Jr, Asa SL, Dralle H, Elisei R, Evans DB, Gagel RF, et al. Revised american thyroid association guidelines for the management of medullary thyroid carcinoma. Thyroid. (2015) 25:567–610. doi: 10.1089/thy.2014.0335
68. Verga U, Fugazzola L, Cambiaghi S, Pritelli C, Alessi E, Cortelazzi D, et al. Frequent association between MEN 2A and cutaneous lichen amyloidosis. Clin Endocrinol. (2003) 59:156–61. doi: 10.1046/j.1365-2265.2003.01782.x
69. Moore S, Zaahl M. The Hirschsprung's–multiple endocrine neoplasia connection. Clinics. (2012) 67(Suppl. 1):63–7. doi: 10.6061/clinics/2012(Sup01)12
70. Castinetti F, Waguespack SG, Machens A, Uchino S, Hasse-Lazar K, Sanso G, et al. Natural history, treatment, and long-term follow up of patients with multiple endocrine neoplasia type 2B: an international, multicentre, retrospective study. Lancet Diabetes Endocrinol. (2019) 7:213–20. doi: 10.1016/S2213-8587(18)30336-X
71. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
72. Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. (1986) 315:1650–9. doi: 10.1056/NEJM198612253152606
73. Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The immune landscape of cancer. Immunity. (2018) 48:812–30. doi: 10.1016/j.immuni.2018.03.023
74. Cunha LL, Morari EC, Guihen AC, Razolli D, Gerhard R, Nonogaki S, et al. Infiltration of a mixture of different immune cells may be related to molecular profile of differentiated thyroid cancer. Endocr Relat Cancer. (2012) 19:31–6. doi: 10.1530/ERC-11-0285
75. Galon J, Mlecnik B, Bindea G, Angell HK, Berger A, Lagorce C, et al. Towards the introduction of the ‘Immunoscore' in the classification of malignant tumours. J. Pathol. (2014) 232:199–209. doi: 10.1002/path.4287
76. Giannini R, Moretti S, Ugolini C, Macerola E, Menicali E, Nucci N, et al. Immune profiling of thyroid carcinomas suggests the existence of two major phenotypes: an ATC-like and a PDTC-like. J Clin Endocrinol Metab. (2019) 104:3557–75. doi: 10.1210/jc.2018-01167
77. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. (2012) 12:252–64. doi: 10.1038/nrc3239
78. Patel KN, Angell TE, Babiarz J, Barth NM, Blevins T, Duh QY, et al. Performance of a genomic sequencing classifier for the preoperative diagnosis of cytologically indeterminate thyroid nodules. JAMA Surg. (2018) 153:817–24. doi: 10.1001/jamasurg.2018.1153
79. Steward DL, Carty SE, Sippel RS, Yang SP, Sosa JA, Sipos JA, et al. Performance of a multigene genomic classifier in thyroid nodules with indeterminate cytology. JAMA Oncol. (2019) 5:204–12. doi: 10.1001/jamaoncol.2018.4616
80. De Rubis G, Rajeev Krishnan S, Bebawy M. Liquid biopsies in cancer diagnosis, monitoring, and prognosis. Trends Pharmacol Sci. (2019) 40:172–86. doi: 10.1016/j.tips.2019.01.006
81. Tumino D, Grani G, Di Stefano M, Di Mauro M, Scutari M, Rago T, et al. Nodular thyroid disease in the era of precision medicine. Front Endocrinol. (2020) 10:907. doi: 10.3389/fendo.2019.00907
82. Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. (2014) 6:224ra24. doi: 10.1158/1538-7445.AM2014-5606
83. Khatami F, Tavangar SM. Liquid biopsy in thyroid cancer: new insight. Int J Hematol Oncol Stem Cell Res. (2018) 12:235–48.
84. Condello V, Macerola E, Ugolini C, De Napoli L, Romei C, Materazzi G, et al. Analysis of circulating tumor DNA does not improve the clinical management of patients with locally advanced and metastatic papillary thyroid carcinoma. Head Neck. (2018) 40:1752–8. doi: 10.1002/hed.25155
85. Sandulache VC, Williams MD, Lai SY, Lu C, William WN, Busaidy NL, et al. Real-time genomic characterization utilizing circulating cell-free DNA in patients with anaplastic thyroid carcinoma. Thyroid. (2017) 27:81–7. doi: 10.1089/thy.2016.0076
86. Cote GJ, Evers C, Hu MI, Grubbs EG, Williams MD, Hai T, et al. Prognostic significance of circulating RET M918T mutated tumor DNA in patients with advanced medullary thyroid carcinoma. J Clin Endocrinol Metab. (2017) 102:3591–9. doi: 10.1210/jc.2017-01039
87. Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan E, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci USA. (2008) 105:10513–8. doi: 10.1073/pnas.0804549105
88. Rosignolo F, Memeo L, Monzani F, Colarossi C, Pecce V, Verrienti A, et al. MicroRNA-based molecular classification of papillary thyroid carcinoma. Int J Oncol. (2017) 50:1767–77. doi: 10.3892/ijo.2017.3960
89. Celano M, Rosignolo F, Maggisano V, Pecce V, Iannone M, Russo D, et al. MicroRNAs as biomarkers in thyroid carcinoma. Int J Genomics. (2017) 2017:6496570. doi: 10.1155/2017/6496570
90. Stokowy T, Wojtas B, Jarzab B, Krohn K, Fredman D, Dralle H, et al. Two-miRNA classifiers differentiate mutation-negative follicular thyroid carcinomas and follicular thyroid adenomas in fine needle aspirations with high specificity. Endocrine. (2016) 54:440–7. doi: 10.1007/s12020-016-1021-7
Keywords: thyroid cancer, oncogenes, RET, RAS, BRAF
Citation: Prete A, Borges de Souza P, Censi S, Muzza M, Nucci N and Sponziello M (2020) Update on Fundamental Mechanisms of Thyroid Cancer. Front. Endocrinol. 11:102. doi: 10.3389/fendo.2020.00102
Received: 03 December 2019; Accepted: 17 February 2020;
Published: 13 March 2020.
Edited by:
Luca Persani, University of Milan, ItalyReviewed by:
Massimo Bongiovanni, Synlab Pathology SA, SwitzerlandCopyright © 2020 Prete, Borges de Souza, Censi, Muzza, Nucci and Sponziello. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessandro Prete, YWxlc3NhbmRyby5wcmV0ZTIyQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.