 Shan Wan1†
Shan Wan1† Xijie Yu
Xijie Yu- 1Laboratory of Endocrinology and Metabolism, Department of Endocrinology, National Clinical Research Center for Geriatrics, West China Hospital, Sichuan University, Chengdu, China
- 2Histology and Imaging Platform, Core Facility of West China Hospital, Sichuan University, Chengdu, China
- 3Department of Orthopedics, West China Hospital, Sichuan University, Chengdu, China
Objective: To clarify the role and mechanism of miR-17-92 cluster in islet beta-cell repair after streptozotocin intervention.
Methods: Genetically engineered mice (miR-17-92βKO) and control RIP-Cre mice were intraperitoneally injected with multiple low dose streptozotocin. Body weight, random blood glucose (RBG), fasting blood glucose, and intraperitoneal glucose tolerance test (IPGTT) were monitored regularly. Mice were sacrificed for histological analysis 8 weeks later. Morphological changes of pancreas islets, quantity, quality, apoptosis, and proliferation of beta-cells were measured. Islets from four groups were isolated. MiRNA and mRNA were extracted and quantified.
Results: MiR-17-92βKO mice showed dramatically elevated fasting blood glucose and impaired glucose tolerance after streptozotocin treatment in contrast to control mice, the reason of which is reduced beta-cell number and total mass resulting from reduced proliferation, enhanced apoptosis of beta-cells. Genes related to cell proliferation and insulin transcription repression were significantly elevated in miR-17-92βKO mice treated with streptozotocin. Furthermore, genes involved in DNA biosynthesis and damage repair were dramatically increased in miR-17-92βKO mice with streptozotocin treatment.
Conclusion: Collectively, our results demonstrate that homozygous deletion of miR-17-92 cluster in mouse pancreatic beta-cells promotes the development of experimental diabetes, indicating that miR-17-92 cluster may be positively related to beta-cells restoration and adaptation after streptozotocin-induced damage.
Introduction
Defective beta-cell function is one of the key reasons underlying the pathological process of both type 1 and 2 diabetes mellitus. Normal insulin-producing pancreatic beta-cells possess the powerful ability of adaptation and proliferation in response to chronic metabolic challenges such as obesity and gestation. For example, at the end of pregnancy, the beta-cell mass in normal rodents is increased by about 50% compared with non-pregnant female rodents (1, 2). Long-term high-fat diet feeding for 4 months also leads to a threefold increase in beta-cell mass and more insulin secretion in response to glucose stimulation (3). Individuals with the failed beta-cell function will gradually develop diabetes. Therefore, exploring the molecular and cellular mechanisms underlying the beta-cell adaptation and proliferation is critical for the intervention of diabetes.
Several studies have reported that microRNAs (miRNAs) were involved in the regulation of pancreatic beta-cell development, differentiation and insulin secretion (4–7). For example, Dicer1 governs the maturation of miRNAs, conditional deletion of which in mouse pancreas leads to abnormal development and differentiation of pancreatic cell lineages (8), indicating miRNAs are important for mouse pancreatic organogenesis. MiR-21, miR-29, miR-34a, miR-146, and miR-200a have been shown to be related to beta-cell apoptosis, whereas miR-7, miR-124a, miR-375, and miR-184 control insulin secretion (9–11). Furthermore, researchers have unveiled that the promoter region of miR-375 contains the binding domain of Pdx-1, Ngn3, and NeuroD1 which are pivotal transcriptional factors involved in beta-cell differentiation (12–15). Downregulation of miR-375 results in dedifferentiation of insulin secreting cells (16), whereas overexpression of miR-375 promotes the differentiation of pluripotent stem cells into beta-cell-like clusters (17). Intriguingly, overexpression of miR-375 together with downregulation of miR-9 show synergistic effects on the differentiation of human mesenchymal stem cells (hMSCs) into functional insulin-producing cells (18). Besides, miR-338-3p plays a key role in beta-cell adaptation to pregnancy and obesity, which is dramatically downregulated in rats islets at the 14th day of gestation (19). MiR-15a/b, miR-16, and miR-195 also show important roles in beta-cell development and specification (20). Moreover, overexpression of miR-124a2 leads to a decrease in several target genes such as Pdx-1, Kir6.2, and Sur-1, which are involved in glucose metabolism and insulin secretion (21).
Recent studies suggest that miR-17-92 cluster is also involved in islet beta-cell differentiation and development. Patients with gestational diabetes mellitus showed a high level of miR-17-92 cluster in plasma (22). In addition, the miR-17-92 cluster especially miR-17 was dramatically up-regulated in MIN6 cells and mouse islets after high glucose stimulation (23). When the nutrient substance was transited from fatty milk to full of carbohydrate diet at weaning, miR-17-92 cluster was down-regulated in rodent islets (24). MiR-18a inhibits pancreatic progenitor proliferation by suppressing the activation of proliferation-related signaling pathways phosphatidylinositol 3-kinase-protein kinase B (PI3K/AKT) and extracellular signal-regulated kinase (ERK) (25). MiR-19a promotes beta-cell proliferation and insulin secretion, while suppresses pancreatic beta-cell apoptosis through suppressor of cytokine signaling 3 (SOCS3), a direct target gene of miR-19a (26). Furthermore, the expression of miR-19b-1 in pancreatic progenitor cells is high, miR-19b-1 directly binds to the 3′UTR of NeuroD1 mRNA to downregulate its transcription, thus reduces the expression of insulin 1 gene, and consequently alters beta-cell differentiation and function (27). MiR-20a is upregulated in the peripheral blood mononuclear cells from type 1 diabetes patients (28). Overexpression of miR-92a-1 reduced insulin expression, in contrast, down-regulation of miR-92a-1 promoted insulin expression and ultimately enhanced glucose-induced insulin secretion (29). In addition, our previous studies have demonstrated the highest expression levels of miR-17 and miR-92a-1 in mouse pancreatic beta-cells, followed by miR-19b-1, miR-19a, miR-18a, and miR-20a (30). Furthermore, our studies suggested conditional knockout of miR-17-92 cluster in mouse pancreatic beta-cells impaired glucose tolerance and the first phase insulin secretion during intraperitoneal glucose tolerance test (IPGTT), which was further deteriorated by chronic high-fat diet feeding (30), suggesting that miR-17-92 cluster may be involved in the adaptation and proliferation of pancreatic beta-cells in response to chronic metabolic challenges.
Previous researches have shown that miR-17-92 was involved in regulation of development of multiple organs including heart, lung, lymphatic system, and reproductive system through targeting transcription factors that regulate cell proliferation and cell cycle and inhibiting the expression of apoptosis-related proteins (31, 32). However, little research has been done on its modulation of pancreatic beta-cell function. Because the function of beta-cell determines the development of diabetes, it is important to determine whether homozygous deletion of miR-17-92 cluster aggravates beta-cell dysfunction after streptozotocin treatment. In the current study, we used mice with conditional deletion of miR-17-92 cluster in islet beta-cells to investigate the role of miR-17-92 cluster in beta-cell adaptation as well as regeneration.
Materials and Methods
Experimental Animals
All animal procedures were approved by the Institute's Animal Care and Use Committee of the West China Hospital and followed the Guide for the care and use of laboratory animals. The rat insulin promoter Cre mice (RIP-Cre) (33) and the miR-17-92flox/flox mice (34) both derived from C57BL/6J backgrounds, were acquired from the Jackson Laboratory (Bar Harbor, Maine, USA). RIP-Cre mice were mated with miR-17-92flox/flox mice to generate experimental male mice with miR-17-92 cluster conditional deletion in pancreatic beta-cells (miR-17-92βKO mice). The RIP-Cre male mice were used as the control. All mice were maintained in a standard light-dark cycle and provided with free access to water and a standard diet.
Streptozotocin Treatment
Male miR-17-92βKO and RIP-Cre mice at 12–16-week-old were injected with STZ (Sigma, Lot# WXBC2544V, P-Code: 101809717, USA) intraperitoneally for 5 consecutive days. The control mice of both genotypes were injected with citrate buffer. The STZ was dissolved in citrate buffer (PH = 4.5) at a dose of 50 mg/kg body weight. Random blood glucose (RBG) was measured at the third, seventh, and ninth day after the last STZ intervention. The mouse with RBG exceeding 16.7 mmol/l was recognized as a successful diabetic model, or the mouse was excluded from the diabetic groups. Body weight was measured once per week. The blood glucose levels after 16 h of fasting were recognized as fast blood glucose (FBG) and were tested once per week. RBG was monitored twice per week at 9 a.m. or 2 p.m.
Intraperitoneal Glucose Tolerance Test
IPGTT was performed every week after STZ intervention, and total eight times were performed for each group. Glucose (2.0 g/kg body weight) was intraperitoneally injected after 16 h fasting. Blood was collected from the tail vein and blood glucose was measured with the Accu-Check glucometer (Roche, Indianapolis, IN) at 0, 15, 30, 60, and 120 min post-injection.
Islets Isolation and Beta-Cell Sorting
The mice were sacrificed for histologic analysis 8 weeks after STZ intervention. The pancreatic islets isolation was performed as previously reported (35, 36). In short, collagenase P (Roche, Lot# 11036922, Germany) was dissolved in pre-cooling Hank's balanced salt solution without magnesium and calcium (Solarbio, Cat. No. H1045-500, Beijing, China) at a concentration of 1 mg/ml, and then the solution was retrogradely poured into the common bile duct. Subsequently, the pancreas was excised and digested in the 37°C water bath for 15 min. Finally, the islets were purified by density gradient along with hand sorting using an islet-specific coloring agent-dithizone (DTZ) (Shanghai Ryon Biological Technology CO., Ltd, Lot. RS31081B032, Shanghai, China).
RNA Extraction and Quantitative RT-PCR Analysis
Total RNA was extracted from mice islets using Trizol reagent (Invitrogen, Bleiswijk, Netherlands), and both miRNA and mRNA were reversely transcribed into cDNA with PrimeScriptTM RT reagent kit (TaKaRa Biotechnology Co., Ltd., Dalian, China). Following reverse transcription, the cDNA was amplified and quantified with SYBR Green premix kit (TaKaRa Biotechnology Co., Ltd., Dalian, China) on Light Cycler 96 system (Roche, Basel, Switzerland). GAPDH and U6 were used as endogenous control for mRNA and miRNA, respectively. Each sample was conducted in triplicate and was analyzed with the 2−ΔΔCT method.
Pancreatic Histology and Immunofluorescent Staining
Mice pancreatic tissues were fixed in 10% paraformaldehyde solution and then embedded in paraffin. Pancreatic tissue blocks were further sectioned into 5 mm slices as previously reported (37). H&E (hematoxylin-eosin) staining was performed for histomorphological analysis.
Immunofluorescent staining was performed to evaluate the expression and location of insulin and glucagon using anti-insulin (cat# 8138, Cell Signaling Technology) and anti-glucagon (cat# sc-13091, Santa Cruz) antibodies. For BrdU labeling, mice were subcutaneously injected with BrdU (cat# B8010, Solarbo) dissolved in saline at a dose of 50 mg/kg body weight once a day for 4 days before sacrifice. The slices were successively incubated with anti-insulin and anti-BrdU (cat# sc-32323, Santa Cruz) antibodies then counterstained with DAPI (cat# 4083, Cell Signaling Technology) (38). The TUNEL assay kit (cat# G3250, Promega) was used to detect the apoptosis of pancreatic beta-cells. The LCA software was used to calculate pancreatic alpha-cell and beta-cell fraction automatically. The beta-cell mass per pancreas was calculated by multiplying the beta-cell fraction by the initial wet pancreatic weight. All data were obtained from at least three mice in each genotype as previously reported (15, 39).
Statistical Analysis
SPSS 22.0 was used for data analysis. All data were shown as the mean ± standard error of the mean (SEM). The mRNA data were normalized to control conditions and presented as the relative expression. Multiple groups' comparison was achieved by one-way ANOVA. The Fisher's PLSD post hoc test was further conducted if there was a significant difference. Two-way ANOVA was used to detect the interactions between genotype and STZ treatment. The significant difference was set at P < 0.05 (two tails).
Results
The miR-17-92 Cluster Is Induced by Streptozotocin
As reported before, all members of the miR-17-92 cluster especially miR-17 and miR-92a-1 were highly expressed in mouse pancreatic islets and beta-cell line (30). We further studied the effect of STZ intervention on the expression of the miR-17-92 cluster in mouse islets. When compared to control mice treated with citrate buffer, control mice treated with STZ exhibited an increase in miR-17 expression by 116%, in miR-18a by 81%, in miR-19a by 89%, in miR-19b-1 by 76%, in miR-20a by 85%, in miR-92a-1 by 99% in pancreatic islets, respectively (Figure 1), suggesting that miR-17-92 cluster might play a key role in STZ-induced beta-cell damage and repair. However, miR-17-92βKO mice given either intervention (citrate buffer or STZ) showed similar few expression levels of miR-17-92 cluster in islets (Figure 1), indicating successful miR-17-92 cluster deletion in mouse pancreatic beta-cells.
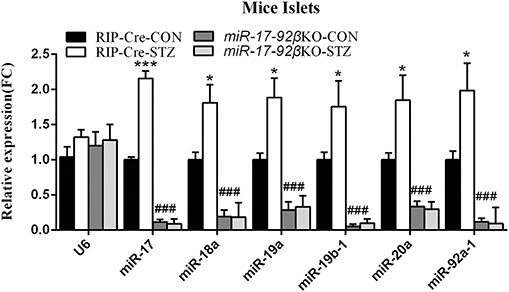
Figure 1. MiR-17-92 changes after streptozotocin treatment. Quantitative RT-PCR revealed that expression levels of miR-17-92 cluster significantly up-regulated in the isolated pancreatic islets from control mice but not miR-17-92βKO mice after STZ intervention. Statistical significance was shown as *P < 0.05, ***P < 0.001 compared to the same genotype, ###P < 0.001 compared to the same treatment. N = 16–20 mice/group.
MiR-17-92 Homozygous Deletion in Mouse Pancreatic Beta-Cells Promotes Streptozotocin-Induced Metabolic Abnormities
To investigate the pathophysiologic roles of the miR-17-92 cluster during type 1 diabetes development, we treated RIP-Cre and miR-17-92βKO mice with MLD-STZ. The MLD-STZ is proved to induce hyperglycemia through direct beta-cell DNA damage or indirect inflammatory response that leads to beta-cell dysfunction even death (40). In the present study, we treated 12 to 16-week-old male RIP-Cre and miR-17-92βKO mice with MLD-STZ and then performed metabolic and histological analysis on various time points after STZ intervention. Data showed that intraperitoneal injection of STZ for 5 consecutive days resulted in gradually body weight reduction in both RIP-Cre and miR-17-92βKO mice with the progress of the study. Especially, miR-17-92βKO mice injected with STZ exhibited more serious body weight loss than counterpart RIP-Cre-STZ mice, and at the end of the observation, the body weight of mice in RIP-Cre-STZ and miR-17-92βKO-STZ group reduced by 3.6 ± 0.3 g (13%) and 5.7 ± 0.5 g (22%) compared to RIP-Cre-CON and miR-17-92βKO-CON group, respectively (Figure 2A). The epididymal fat of miR-17-92βKO-CON group mice was higher than that of RIP-Cre-CON group. However, the epididymal fat pad of both genotypes was significantly reduced by 0.05 ± 0.02 g (17%) and 0.216 ± 0.03 g (38%), respectively, after STZ intervention (Figure 2B).
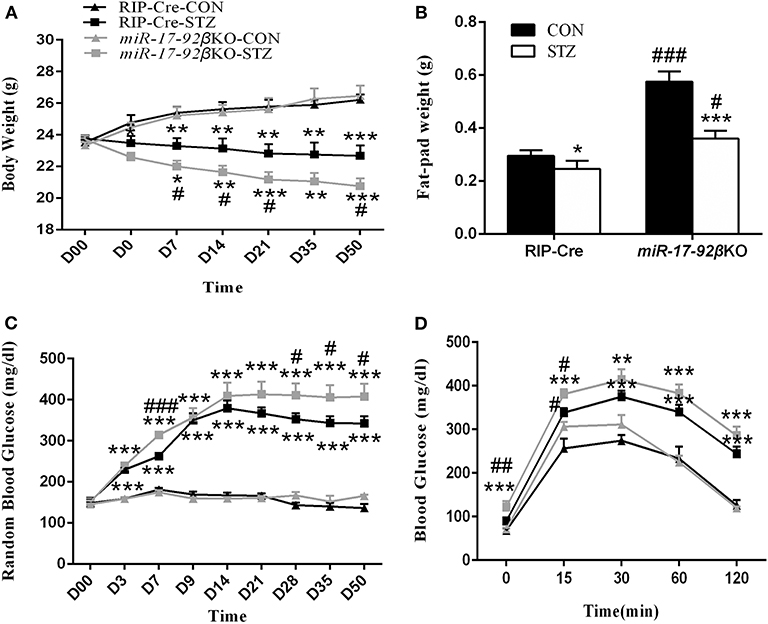
Figure 2. MiR-17-92 homozygous deletion in mouse pancreatic beta-cells promotes streptozotocin-induced metabolic abnormities. (A–D) Metabolic profiles of RIP-Cre and miR-17-92βKO mice treated with citrate buffer or STZ. Differences between control and miR-17-92βKO mice, including body weight (A), fat-pad weight (B), RBG (C), and blood glucose levels of IPGTT (D). Significant differences were shown as *P < 0.05, **P < 0.01, ***P < 0.001 compared to the same genotype, #P < 0.05, ##P < 0.01, ###P < 0.001 compared to the same treatment. N = 6–8 mice/group. “D00” and “D0” in (A) indicated the initial body weight of all mice and the body weight of mice after 5 consecutive day intraperitoneal STZ injection, respectively. The “D3” indicated the body weight of the third day of mice after 5 consecutive day intraperitoneal STZ intervention, so did the other related labels.
Additionally, before STZ intervention, the levels of RBG in four groups were similar. Whereas, the levels of RBG in both genotypes began to increase significantly since the third day after STZ injection. At the end of the experimental observation, the levels of RBG in RIP-Cre-STZ and miR-17-92βKO-STZ group increased by 206 ± 9.6 mg/dl (121%) and 242 ± 2.4 mg/dl (146%), respectively (Figure 2C). Meanwhile, miR-17-92βKO-STZ mice exhibited fasting hyperglycemia in contrast to RIP-Cre-STZ mice (121 ± 14.4 mg/dl vs. 90 ± 2.7 mg/dl; P < 0.05). When challenged with IPGTT, the changes in blood glucose in the miR-17-92βKO-CON group was similar as those in the RIP-Cre-CON group, which increased at 15, 30, and 60 min, but recovered to the fasting level at 120 min (Figure 2D, result at the sixth week; similar to other weeks, data not shown), indicating the glucose tolerance in the miR-17-92βKO-CON group was still in the compensatory state. Nevertheless, after STZ treatment, both genotypes of mice showed remarkable glucose intolerance. At the first IPGTT test, RIP-Cre-STZ group and miR-17-92βKO-STZ group began to exhibit elevated blood glucose levels at all time points (data not shown), the most obvious of which were in miR-17-92βKO-STZ group at the sixth IPGTT test with an 35, 13, 10, 13, and 17% elevation in 0, 15, 30, 60, and 120 min in contrast to the RIP-Cre-STZ group (Figure 2D).
Along with the research, the blood glucose levels of two genotypes of mice with STZ treatment restored slightly, but at the end of the experiment, the blood glucose levels were still higher than that of the CON mice, and the highest was in the miR-17-92βKO-STZ group. Taken together, these results suggest that mice with conditional deletion of the miR-17-92 cluster in islet beta-cells can physiologically maintain normal glucose metabolism homeostasis through the compensatory mechanism. However, when given exogenous stimulation (such as IPGTT or STZ-induced pancreatic beta-cell damage), the mice will lose its compensation, displaying beta-cell dysfunction, which indicates that the miR-17-92 cluster is required for the beta-cell function to STZ-induced damage.
Reduced Proliferation and Elevated Apoptosis of Beta-Cells in miR-17-92βKO Mice Treated With Streptozotocin
In order to further confirm whether the miR-17-92 cluster modulates beta-cell regeneration after MLD-STZ intervention, we performed histomorphological analyses of pancreatic islets from the four groups. The pancreatic islets from four groups were first analyzed by H&E staining. Compared with RIP-Cre-CON mice, the islets of miR-17-92βKO-CON mice showed normal morphology, complete structure, and distinct division of cytoplasm and nucleus (Figure 3A), while the islets of both genotypes were significantly impaired after STZ intervention, manifested by abnormal islet morphology, scattered structure, and increased peri-islet neovascularization, which was more serious in miR-17-92βKO-STZ mice (Figure 3A).
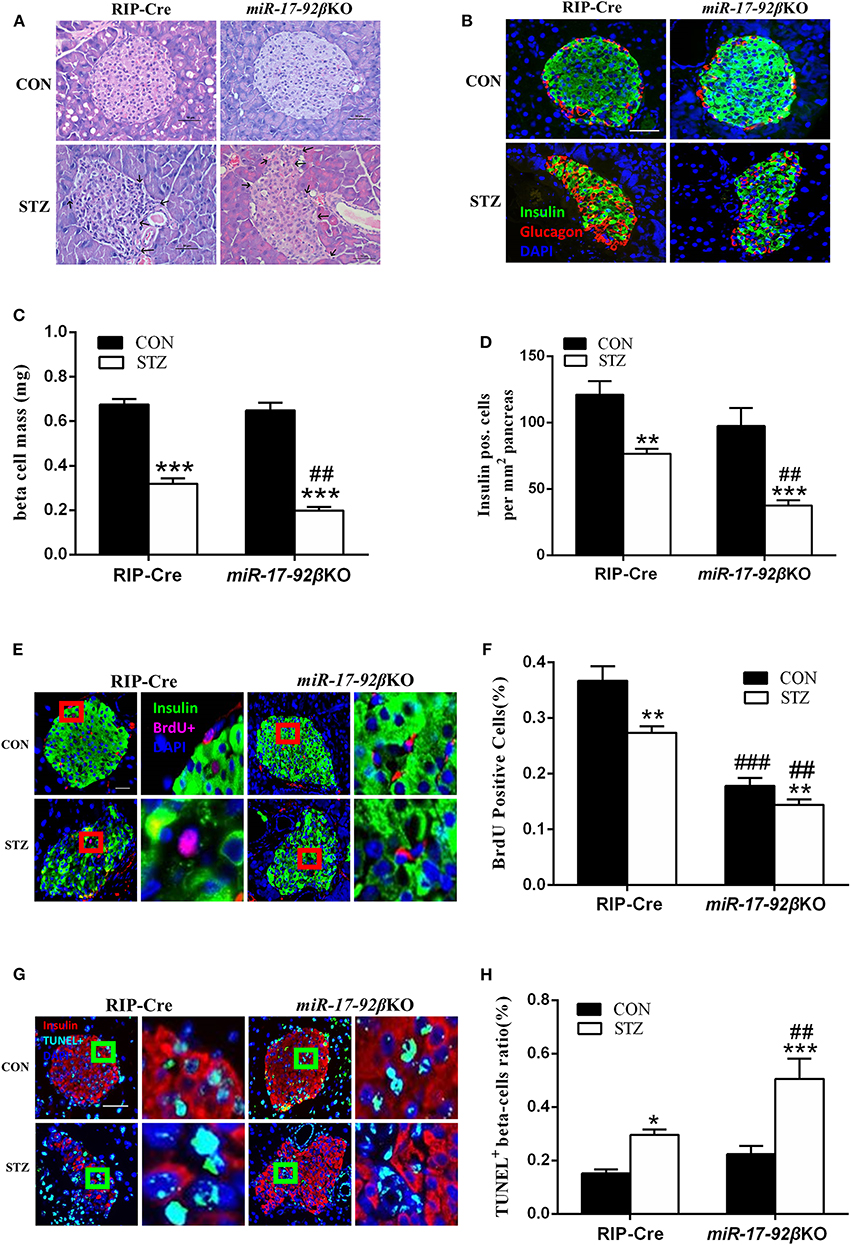
Figure 3. Reduced proliferation and elevated apoptosis of beta-cells in miR-17-92βKO mice treated with streptozotocin. (A) Microscopic photographs of islets from control and miR-17-92βKO mice after citrate buffer or STZ intervention, H&E staining, and original magnification 200×. Black arrows represent the abnormal islet morphology in mice treated with STZ. (B) Immunofluorescence staining for insulin (green), glucagon (red), and DNA (DAPI-blue) of islets from two genotypes, and original magnification 200×. (C,D) Quantitation of pancreatic islets beta-cell mass (C) and insulin positive cells per mm2 pancreas (D) of control and miR-17-92βKO mice after different treatments. (E–H) Analysis of BrdU positive cells (% of insulin-positive beta-cells) (E,F) and TUNEL positive beta-cells ratio (%) (G,H) by immunofluorescence staining of insulin-positive beta-cells in pancreatic islets from both genotypes. The pictures on the right are the corresponding enlarged pictures of the red/green box in the left pictures. Significant differences were shown as *P < 0.05, **P < 0.01, ***P < 0.001 compared to the same genotype, ##P < 0.01, ###P < 0.001 compared to the same treatment. N = 16–20 mice/group. Scale Bar = 50 μm.
Then, the pancreatic islets were analyzed by immunofluorescent staining. Compared to mice treated with citrate buffer, mice treated with STZ showed dramatically reduced insulin-positive and total mass of pancreatic beta-cells, and the distribution of alpha-cells changing from peripheral to scattered, among which the total mass of beta-cells declined by 38%, and the insulin-positive beta-cells decreased by 51% in islets from miR-17-92βKO-STZ mice in contrast to the RIP-Cre-STZ mice, suggesting damaged beta-cell regeneration in mice with miR-17-92 homozygous deletion in beta-cells after MLD-STZ treatment (Figures 3B–D). To clarify whether the damaged regeneration resulted from impaired beta-cell proliferation, we further conducted BrdU-insulin immunofluorescent staining to access beta-cell mitotic rate. Data showed that the proliferation of beta-cells diminished in miR-17-92βKO-CON mice compared to RIP-Cre-CON mice (Figures 3E,F). In addition, the proliferation of islet beta-cells further decreased by 47% in miR-17-92βKO-STZ group compared with RIP-Cre-STZ group (Figure 3F). Interestingly, we carried out TUNEL-insulin immunofluorescent staining to evaluate the apoptosis of the beta-cells and found that the apoptosis increased by 71% in the miR-17-92βKO-STZ group on contrast to the RIP-Cre-STZ mice (Figures 3G,H). Collectively, the decreased proliferation and elevated apoptosis reflect declining regeneration and conversion capacity of islet beta-cells in response to STZ-induced damage.
The miR-17-92 Cluster Regulates Beta-Cell Number and Function
In order to further clarify the molecular mechanisms, we focused on the validated target genes of the miR-17-92 cluster involved in beta-cell proliferation and apoptosis. Compared with RIP-Cre-CON mice, mRNA expression of Pten (phosphatase and tensin homolog deleted on chromosome ten) in islets from miR-17-92βKO-CON mice was up-regulated by 44% (Figure 4A). After STZ intervention, mRNA expression of Pten was up-regulated by 36% in RIP-Cre-STZ group and 70% in the miR-17-92βKO-STZ group, respectively (Figure 4A), which may be the reason for the declining number and total mass of islet beta-cells in miR-17-92βKO-STZ mice.
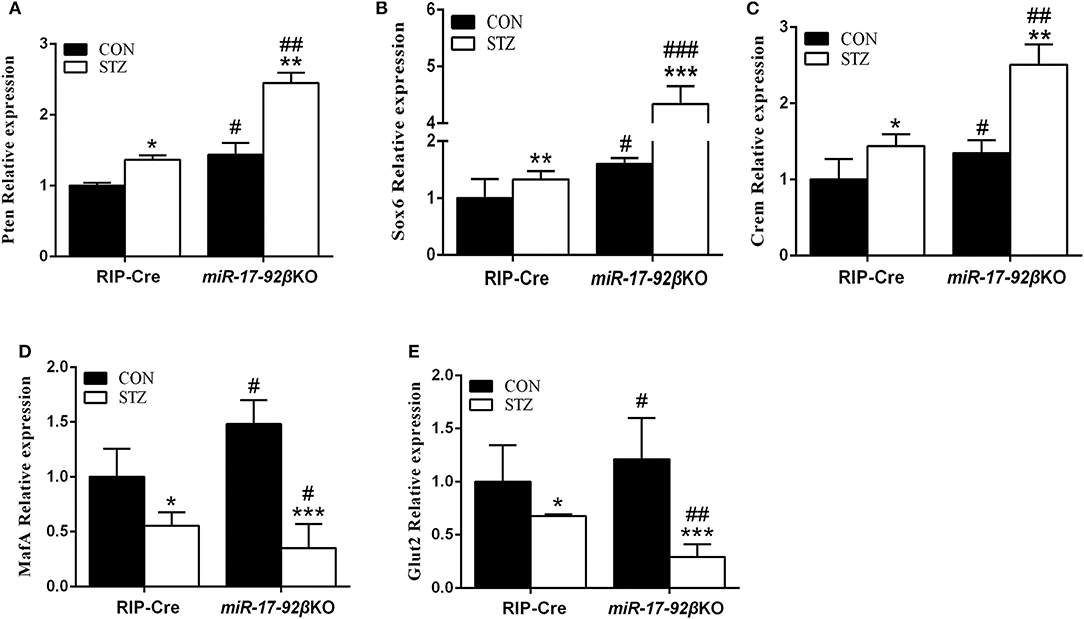
Figure 4. Impaired signaling pathways of pancreatic beta-cells' proliferation and insulin gene transcription. (A) Quantitative RT-PCR demonstrated that expression level of Pten significantly up-regulated in isolated islets from miR-17-92βKO mice treated with STZ. (B,C) Expression levels of Sox6 and Crem dramatically elevated in islets from miR-17-92βKO-STZ mice. (D,E) Expression levels of MafA and Glut2 remarkably decreased in islets from miR-17-92βKO mice given STZ treatment. Significant differences were shown as *P < 0.05, **P < 0.01, ***P < 0.001 compared to the same genotype, #P < 0.05, ##P < 0.01, ###P < 0.001 compared to the same treatment. N = 16–20 mice/group.
Furthermore, the expression of genes related to insulin biosynthesis and secretion was further studied in islets from four groups of mice. Compared to RIP-Cre-CON mice, mRNA expressions of Sox6 (Sex-determination region Y-box 6) and Crem (cAMP response element modulator), genes related to insulin synthesis inhibition, up-regulated by 60 and 35% in islets of miR-17-92βKO-CON mice. The mRNA expressions of MafA (Mus musculus v-maf musculoapontic fibrocoma oncogene protein A) and Glut2 (Glucose transporter 2), genes related to insulin synthesis activation, in islets of miR-17-92βKO-CON mice were also up-regulated by 48 and 21% (Figures 4B–E). The mRNA expressions of Sox6 and Crem up-regulated by 32 and 44%, while mRNA expressions of MafA and Glut2 down-regulated by 45 and 32%, respectively in RIP-Cre-STZ mice (Figures 4B–E). At the same time, mRNA expression levels of Sox6 and Crem dramatically elevated by 171 and 86%, while mRNA expressions of MafA and Glut2 down-regulated by 76 and 75% separately in islets from miR-17-92βKO mice treated with STZ, resulting in inhibition of insulin transcription pathway in miR-17-92βKO-STZ mice (Figures 4B–E). Collectively, these results suggest that the miR-17-92 cluster is crucial for the quantity maintenance and insulin-producing function of the beta-cells during STZ treatment.
Homozygous Deletion of the miR-17-92 Cluster in Beta-Cells Suppresses DNA Biosynthesis but Promotes DNA Damage Repair
As mentioned above, MLD-STZ intervention leads to both impaired glucose tolerance and hyperglycemia and simulates human type 1 diabetes partially through direct beta-cell DNA damage. To determine whether miR-17-92 is involved in beta-cell DNA damage repair, we explored the expression profiles of some identified target genes of STZ and related to DNA synthesis and damage repair in islets from four groups of mice. Compared with RIP-Cre-CON mice, the expression of Cdkn1a which inhibit DNA synthesis while promoting DNA damage repair and ATM (ataxia telangiectasia mutated) kinase that is the key enzyme of DNA damage repair up-regulated by 29 and 46%, respectively, in miR-17-92βKO-CON mice (Figures 5A,B). Furthermore, the expression levels of Cdkn1a and ATM kinase upregulated by 24 and 59% in RIP-Cre-STZ mice, the same as previous studies (41–43), and 2,144 and 631% in islets of miR-17-92βKO-STZ mice (Figures 5A,B), leading to cell cycle arrest and DNA synthesis inhibition, but at the same time promotes the repair of damaged DNA, improves the effectiveness of homologous recombinant DNA repair, renovates STZ-induced beta-cell damage, restores the function of damaged beta-cells, and regulates the regeneration and compensation of islet beta-cells for STZ-induced damage. Taken together, these results imply that the miR-17-92 cluster is pivotal for beta-cell adaptation to STZ treatment.
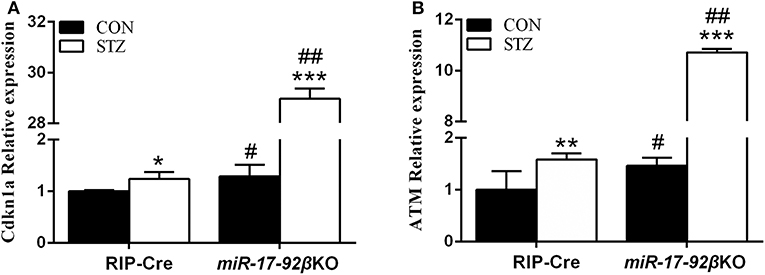
Figure 5. Different expression profiles of genes-related to DNA biosynthesis and damage repair in islets from two genotypes of mice after STZ intervention. (A,B) Quantitative RT-PCR illustrated high expression levels of Cdkn1a and ATM kinase in isolated islets of miR-17-92βKO mice treated with STZ. Significant differences were shown as *P < 0.05, **P < 0.01, ***P < 0.001 compared to the same genotype, #P < 0.05, ##P < 0.01 compared to the same treatment. N = 16–20 mice/group.
Discussion
In the present study, the RBG levels of the two genotypes were higher than 300 mg/dl (16.7 mmol/l) after STZ intervention, indicating the diabetic model was successful. Our previous study has revealed high expression levels of miR-17-92 cluster in mouse islets and beta-cell line (30), indicating a significant role of miR-17-92 cluster in normal beta-cell function. In the current study, we found the expression levels of miR-17-92 cluster were elevated to different extent in islets from RIP-Cre-STZ mice, suggesting that the miR-17-92 cluster may be involved in the adaptive response of islet beta-cells to STZ-induced injury.
Consistent with our previous study, the body weight, RBG, and fasting blood glucose of miR-17-92βKO-CON mice were similar to those in the RIP-Cre-CON mice (30), implying that there might be other mechanisms in islet beta-cells that coordinated with the miR-17-92 signaling pathway to regulate glucose metabolism, which could partially rectify the abnormal glucose metabolism induced by the homozygous deletion of miR-17-92 cluster. However, after STZ treatment, the miR-17-92βKO mice showed more pronounced metabolic disturbance characterized with more obvious body weight loss, higher random and fasting blood glucose levels and more serious glucose intolerance. Histomorphological analysis showed that STZ intervention impaired the morphology and structure of islets in two genotypes of mice. Meanwhile, insulin-glucagon immunofluorescent staining revealed that both RIP-Cre-STZ and miR-17-92βKO-STZ mice showed reduced number and total mass of islet beta-cells, which is more severe in islets from the latter group, suggesting the islet beta-cells in two genotypes of mice were in decompensation stage. Taken together, the above results suggest that conditional deletion of miR-17-92 cluster in islet beta-cells reduces their adaptation ability to stress stimulation, and MLD-STZ intervention further deteriorates the function of islet beta-cells, so as to result in disturbance of glucose homeostasis. Our data indicate that miR-17-92 cluster is involved in the adaptive response of islet beta-cells to STZ treatment.
Additionally, previous studies have shown that the number of islet beta-cell fluctuates. Physiologically, the body can adjust the number of islet beta-cells according to the changes of the internal and external environment along with the functional status of beta-cells, so as to maintain a relatively stable blood glucose levels to adapt to the environmental variation (44). The pancreatic insulin-producing beta-cells are derived from the differentiation process during embryo development or from replication which appears postnatally (45, 46), and it is essential to maintain sufficient beta-cell number to respond to aging and metabolic stresses. Limited beta-cell proliferation is extensive in human type 1 and type 2 diabetes. Studies have shown that miR-17-92 targets a series of cell proliferation-related genes, such as Cdkn1a, p57 (47), and apoptosis-related genes including Pten, Bcl2L11 (48) to modulate cell proliferation and apoptosis. Nevertheless, the regulation of proliferation and apoptosis of islet beta-cells by miR-17-92 cluster remains largely unclear. Recent studies have found that lipid phosphatase encoded by Pten, is a potent negative regulator of phosphoinositide 3-kinase (PI3K)-Akt signaling pathway, which plays an important role in cell proliferation and insulin biosynthesis and secretion (49). Importantly, Pten is known to be the target gene of miR-19a and miR-19b-1 (50). Pten is also a critical determinant of body size and glucose metabolism in mice (51). Studies have demonstrated that conditional deletion of Pten in insulin-producing cells during mouse pancreatic embryonic development (E17.5) or in adult beta-cells significantly increased islet mass and beta-cell proliferation (49), and exerted protective effects against high-fat diet feeding and STZ-induced diabetes (52, 53). In short, Pten is a critical negative effector of both beta-cell mass and function, and its expression was up-regulated in diabetic animal models (53). Previous studies have suggested that deletion of miR-17-92 cluster in beta-cells led to impaired glucose tolerance and reduced first-phase insulin secretion, which may be partly mediated by Pten-Akt signaling pathway (24, 30). Moreover, in the present study we found higher expression level of Pten in beta-cells from both genotypes of mice given STZ intervention, which was obvious in the miR-17-92βKO-STZ mice as the synergistic effect of conditional deletion of miR-17-92 cluster and the STZ treatment. Further studies showed that STZ treatment reduced proliferation and increased apoptosis in islet beta-cells in two genotypes of mice, which may be achieved by regulating the expression of Pten. It also indicates that STZ intervention decompensated islet beta-cells of two genotypes of mice and resulted in a low transformation status of the beta-cells particularly in the miR-17-92βKO-STZ group.
Diabetes mellitus shows features of hyperglycemia because of absolutely or relatively insufficient serum insulin secretion. Pancreatic beta-cells have fundamental functions to modulate insulin synthesis and secretion, which is regulated by various transcriptional activators such as Pdx-1 (pancreas/duodenum homeobox factor-1) (54), MafA (55, 56), and Glut2 (57–59) and transcriptional repressors including Sox6 (60) and Crem (61). Pdx-1 plays pivotal roles in the early differentiation, maturation and regeneration of pancreatic islet cells (57). MafA is highly expressed in pancreatic beta-cells, which activates the transcription of the insulin gene by targeting RIPE3b1, a cis-regulatory element (62–64). The expression of MafA gene is down-regulated, thus inhibiting the transcription of insulin in diabetes mellitus (55, 56). MafA can regulate the expression of insulin gene as well as other related factors including Pdx-1 and Glut2 (57). Glut2 is a glucose transporter in pancreatic beta-cells, which can transport glucose and regulate ion channels on cell membrane, thus promoting insulin secretion (57). Sox6, a member of the high mobility group box superfamily, acts as a co-repressor with Pdx-1 and plays critical roles in pancreatic beta-cell replication and insulin gene transcription (65). Over-expression of Sox6 is involved in decreased insulin gene transcription through inhibition of the activation of insulin gene promoter by Pdx-1 (60). In addition, Crem has also been reported to directly repress the insulin gene promoter activation in beta-cell (66). In the present study, the expression of MafA and Glut2 in islets of RIP-Cre-STZ and miR-17-92βKO-STZ group was down-regulated, which reduced insulin gene transcription as previous studies (57–59, 67). Meanwhile, the upregulation of Sox6 and Crem resulted in the decrease of insulin transcription. In summary, the downregulation of MafA and Glut2 along with the upregulation of Sox6 and Crem may be related to the decrease of the transcription of insulin gene and islet beta-cell proliferation in the STZ group. Nevertheless, there is no literature on the relationship between the miR-17-92 cluster and the above factors, which needs further studies.
STZ is a kind of natural compounds produced by streptococcus that specifically damages pancreatic islet beta-cells in mammals. The underlying mechanism is to form isocyanate compounds in vivo, which bind to nucleic acid proteins to inhibit the activity of DNA polymerase, and interfere with the synthesis and damage repair of DNA, thus to regulate pancreatic beta-cell proliferation and apoptosis (68). Cdkn1a encodes a potential cyclin-dependent kinase inhibitor which represses the activity of cyclin-cyclin-dependent kinase 2/4, then modulates G1 cell cycle progression. The encoded protein also can interact with PCNA (proliferating cell nuclear antigen), a kind of cofactor of DNA polymerase, and regulate DNA replication and damage repair in S cell cycle. Additionally, mice lack of Cdkn1a have the ability to regenerate damaged tissue. Besides, ATM kinase increases the phosphorylation levels of H2AX and 53BP1, thus promoting DNA damage repair, improving the effectiveness of homologous recombinant DNA repair to inhibit beta-cell proliferation and reduce their apoptosis. Conditional deletion of the master DNA repair gene-ATM kinase in mouse pancreatic beta-cells protects mice against STZ-induced diabetes (69). Importantly, it has been reported that Cdkn1a and ATM kinase are involved in DNA damage repair process in islet beta-cells induced by STZ, and the expression of Cdkn1a and ATM kinase were up-regulated after STZ intervention (42, 43, 70). Moreover, studies have found that miR-17 and miR-92a-1 could inhibit Cdkn1a, and miR-18a targets ATM kinase thus promoting DNA synthesis and inhibiting DNA damage repair (71, 72). In the current study, the expression of Cdkn1a and ATM kinase was up-regulated in miR-17-92βKO-CON mice, which was similar as previous studies (71, 72). Interestingly, the expression of Cdkn1a and ATM kinase were further up-regulated after STZ intervention in two genotypes of mice (42, 43, 70), which resulted in cell cycle arrest and DNA synthesis repression. At the same time, it promotes the repairment of damaged DNA induced by STZ, restores the function of damaged beta-cells thereby modulating the adaptation of islet beta-cells after STZ-induced injury.
Last but not least, our studies found high epididymal fat pad content in miR-17-92βKO-CON mice compared with control mice (30). However, STZ treatment led to the low epididymal fat content in both genotypes of mice, possibly due to the more deteriorated metabolic disturbance. Therefore, miR-17-92βKO mice showed lower body weight, higher glucose levels, and more epididymal fat content after STZ treatment, indicating miR-17-92βKO mice may be a more suitable animal model to develop diabetes induced by STZ treatment compared with control mice.
In conclusion, our results showed that mice with conditional deletion of the miR-17-92 cluster in pancreatic beta-cells exerted profound metabolic abnormalities due to beta-cell dysfunction and reduced beta-cell proliferation after STZ treatment, indicating that the miR-17-92 cluster is essential for pancreatic beta-cell restoration and adaptation in a chemical-induced diabetes animal model.
Data Availability Statement
All datasets generated for this study are included in the article/supplementary material.
Ethics Statement
The animal study was reviewed and approved by the Institute's Animal Care and Use Committee of the West China Hospital.
Author Contributions
XY designed this research. SW, JL, YM, LL, FC, and JZ were responsible for the experiments. Among them, LL, FC, and JZ were mainly responsible for the histopathological part. The other three were in charge of the animal experiments and the next molecular experiments after the animal were sacrificed. XC, LT, and XY are responsible for the revision of the whole article.
Funding
This work was supported by grants from the National Natural Science Foundation of China (Nos. 81770875, 81572639, and 81702156), the Science and Technology Department of Sichuan Province (2018SZ0142), the Sichuan University (2018SCUH0093), and the National Clinical Research Center for Geriatrics of West China Hospital (No. Z2018B05), 1.3.5 project for disciplines of excellence, West China Hospital, Sichuan University (ZYGD18022).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Y. Liang, Z. Yang, and L. Bai (Histology and Imaging platform, Core Facility of West China Hospital) for their contribution to this study.
References
1. Scaglia L, Smith FE, Bonner-Weir S. Apoptosis contributes to the involution of beta cell mass in the post partum rat pancreas. Endocrinology. (1995) 136:5461–8. doi: 10.1210/endo.136.12.7588296
2. Blondeau B, Garofano A, Czernichow P, Breant B. Age-dependent inability of the endocrine pancreas to adapt to pregnancy: a long-term consequence of perinatal malnutrition in the rat. Endocrinology. (1999) 140:4208–13. doi: 10.1210/endo.140.9.6960
3. Sone H, Kagawa Y. Pancreatic beta cell senescence contributes to the pathogenesis of type 2 diabetes in high-fat diet-induced diabetic mice. Diabetologia. (2005) 48:58–67. doi: 10.1007/s00125-004-1605-2
4. Filios SR, Shalev A. beta-cell microRNAs: small but powerful. Diabetes. (2015) 64:3631–44. doi: 10.2337/db15-0831
5. Dumortier O, Fabris G, Van Obberghen E. Shaping and preserving beta-cell identity with microRNAs. Diabetes Obes Metab. (2016) 18 (Suppl. 1):51–7. doi: 10.1111/dom.12722
6. LaPierre MP, Stoffel M. MicroRNAs as stress regulators in pancreatic beta cells and diabetes. Mol Metab. (2017) 6:1010–23. doi: 10.1016/j.molmet.2017.06.020
7. Esguerra JLS, Nagao M, Ofori JK, Wendt A, Eliasson L. MicroRNAs in islet hormone secretion. Diabetes Obes Metab. (2018) 20 (Suppl. 2):11–9. doi: 10.1111/dom.13382
8. Lynn FC, Skewes-Cox P, Kosaka Y, McManus MT, Harfe BD, German MS. MicroRNA expression is required for pancreatic islet cell genesis in the mouse. Diabetes. (2007) 56:2938–45. doi: 10.2337/db07-0175
9. Nesca V, Guay C, Jacovetti C, Menoud V, Peyot ML, Laybutt DR, et al. Identification of particular groups of microRNAs that positively or negatively impact on beta cell function in obese models of type 2 diabetes. Diabetologia. (2013) 56:2203–12. doi: 10.1007/s00125-013-2993-y
10. Tattikota SG, Sury MD, Rathjen T, Wessels HH, Pandey AK, You X, et al. Argonaute2 regulates the pancreatic beta-cell secretome. Mol Cell Proteomics. (2013) 12:1214–25. doi: 10.1074/mcp.M112.024786
11. Belgardt BF, Ahmed K, Spranger M, Latreille M, Denzler R, Kondratiuk N, et al. The microRNA-200 family regulates pancreatic beta cell survival in type 2 diabetes. Nat Med. (2015) 21:619–27. doi: 10.1038/nm.3862
12. Keller DM, McWeeney S, Arsenlis A, Drouin J, Wright CV, Wang H, et al. Characterization of pancreatic transcription factor Pdx-1 binding sites using promoter microarray and serial analysis of chromatin occupancy. J Biol Chem. (2007) 282:32084–92. doi: 10.1074/jbc.M700899200
13. El Ouaamari A, Baroukh N, Martens GA, Lebrun P, Pipeleers D, van Obberghen E. miR-375 targets 3'-phosphoinositide-dependent protein kinase-1 and regulates glucose-induced biological responses in pancreatic beta-cells. Diabetes. (2008) 57:2708–17. doi: 10.2337/db07-1614
14. Avnit-Sagi T, Kantorovich L, Kredo-Russo S, Hornstein E, Walker MD. The promoter of the pri-miR-375 gene directs expression selectively to the endocrine pancreas. PLoS ONE. (2009) 4:e5033. doi: 10.1371/journal.pone.0005033
15. Poy MN, Hausser J, Trajkovski M, Braun M, Collins S, Rorsman P, et al. miR-375 maintains normal pancreatic alpha- and beta-cell mass. Proc Natl Acad Sci USA. (2009) 106:5813–8. doi: 10.1073/pnas.0810550106
16. Nathan G, Kredo-Russo S, Geiger T, Lenz A, Kaspi H, Hornstein E, et al. MiR-375 promotes redifferentiation of adult human beta cells expanded in vitro. PLoS ONE. (2015) 10:e0122108. doi: 10.1371/journal.pone.0122108
17. Shaer A, Azarpira N, Karimi MH. Differentiation of human induced pluripotent stem cells into insulin-like cell clusters with miR-186 and miR-375 by using chemical transfection. Appl Biochem Biotechnol. (2014) 174:242–58. doi: 10.1007/s12010-014-1045-5
18. Plaisance V, Abderrahmani A, Perret-Menoud V, Jacquemin P, Lemaigre F, Regazzi R. MicroRNA-9 controls the expression of Granuphilin/Slp4 and the secretory response of insulin-producing cells. J Biol Chem. (2006) 281:26932–42. doi: 10.1074/jbc.M601225200
19. Jacovetti C, Abderrahmani A, Parnaud G, Jonas JC, Peyot ML, Cornu M, et al. MicroRNAs contribute to compensatory beta cell expansion during pregnancy and obesity. J Clin Invest. (2012) 122:3541–51. doi: 10.1172/JCI64151
20. Joglekar MV, Parekh VS, Mehta S, Bhonde RR, Hardikar AA. MicroRNA profiling of developing and regenerating pancreas reveal post-transcriptional regulation of neurogenin3. Dev Biol. (2007) 311:603–12. doi: 10.1016/j.ydbio.2007.09.008
21. Baroukh N, Ravier MA, Loder MK, Hill EV, Bounacer A, Scharfmann R, et al. MicroRNA-124a regulates Foxa2 expression and intracellular signaling in pancreatic beta-cell lines. J Biol Chem. (2007) 282:19575–88. doi: 10.1074/jbc.M611841200
22. Zhu Y, Tian F, Li H, Zhou Y, Lu J, Ge Q. Profiling maternal plasma microRNA expression in early pregnancy to predict gestational diabetes mellitus. Int J Gynaecol Obstet. (2015) 130:49–53. doi: 10.1016/j.ijgo.2015.01.010
23. Lu Y, Fei XQ, Yang SF, Xu BK, Li YY. Glucose-induced microRNA-17 promotes pancreatic beta cell proliferation through down-regulation of Menin. Eur Rev Med Pharmacol Sci. (2015) 19:624–9.
24. Jacovetti C, Matkovich SJ, Rodriguez-Trejo A, Guay C, Regazzi R. Postnatal beta-cell maturation is associated with islet-specific microRNA changes induced by nutrient shifts at weaning. Nat Commun. (2015) 6:8084. doi: 10.1038/ncomms9084
25. Li X, Zhang Z, Li Y, Zhao Y, Zhai W, Yang L, et al. miR-18a counteracts AKT and ERK activation to inhibit the proliferation of pancreatic progenitor cells. Sci Rep. (2017) 7:45002. doi: 10.1038/srep45002
26. Li Y, Luo T, Wang L, Wu J, Guo S. MicroRNA-19a-3p enhances the proliferation and insulin secretion, while it inhibits the apoptosis of pancreatic β cells via the inhibition of SOCS3. Int J Mol Med. (2016) 38:1515–24. doi: 10.3892/ijmm.2016.2748
27. Zhang ZW, Zhang LQ, Ding L, Wang F, Sun YJ, An Y, et al. MicroRNA-19b downregulates insulin 1 through targeting transcription factor NeuroD1. FEBS Lett. (2011) 585:2592–8. doi: 10.1016/j.febslet.2011.06.039
28. Azhir Z, Dehghanian F, Hojati Z. Increased expression of microRNAs, miR-20a and miR-326 in PBMCs of patients with type 1 diabetes. Mol Biol Rep. (2018) 45:1973–80. doi: 10.1007/s11033-018-4352-z
29. Setyowati Karolina D, Sepramaniam S, Tan HZ, Armugam A, Jeyaseelan K. miR-25 and miR-92a regulate insulin I biosynthesis in rats. RNA Biol. (2013) 10:1365–78. doi: 10.4161/rna.25557
30. Chen Y, Tian L, Wan S, Xie Y, Chen X, Ji X, et al. MicroRNA-17-92 cluster regulates pancreatic beta-cell proliferation and adaptation. Mol Cell Endocrinol. (2016) 437:213–23. doi: 10.1016/j.mce.2016.08.037
31. Mendell JT. miRiad roles for the miR-17-92 cluster in development and disease. Cell. (2008) 133:217–22. doi: 10.1016/j.cell.2008.04.001
32. Carraro G, El-Hashash A, Guidolin D, Tiozzo C, Turcatel G, Young BM, et al. miR-17 family of microRNAs controls FGF10-mediated embryonic lung epithelial branching morphogenesis through MAPK14 and STAT3 regulation of E-Cadherin distribution. Dev Biol. (2009) 333:238–50. doi: 10.1016/j.ydbio.2009.06.020
33. Herrera PL. Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development. (2000) 127:2317–22.
34. Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell. (2008) 132:875–86. doi: 10.1016/j.cell.2008.02.019
35. Zmuda EJ, Powell CA, Hai T. A method for murine islet isolation and subcapsular kidney transplantation. J Vis Exp. (2011) 50:2096. doi: 10.3791/2096
36. Colvin ES, Ma HY, Chen YC, Hernandez AM, Fueger PT. Glucocorticoid-induced suppression of beta-cell proliferation is mediated by Mig6. Endocrinology. (2013) 154:1039–46. doi: 10.1210/en.2012-1923
37. Bock T, Pakkenberg B, Buschard K. Increased islet volume but unchanged islet number in ob/ob mice. Diabetes. (2003) 52:1716–22. doi: 10.2337/diabetes.52.7.1716
38. Mason DY, Micklem K, Jones M. Double immunofluorescence labelling of routinely processed paraffin sections. J Pathol. (2000) 191:452–61. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH665>3.0.CO;2-O
39. Ardestani A, Paroni F, Azizi Z, Kaur S, Khobragade V, Yuan T, et al. MST1 is a key regulator of beta cell apoptosis and dysfunction in diabetes. Nat Med. (2014) 20:385–97. doi: 10.1038/nm.3482
40. Lenzen S. The mechanisms of alloxan- and streptozotocin-induced diabetes. Diabetologia. (2008) 51:216–26. doi: 10.1007/s00125-007-0886-7
41. Agudo J, Ayuso E, Jimenez V, Salavert A, Casellas A, Tafuro S, et al. IGF-I mediates regeneration of endocrine pancreas by increasing beta cell replication through cell cycle protein modulation in mice. Diabetologia. (2008) 51:1862–72. doi: 10.1007/s00125-008-1087-8
42. Tian LM, Liu J, Sun XL, Gao CX, Fan Y, Guo Q. A protective effect of endomorphins on the oxidative injury of islet. Exp Clin Endocrinol Diabetes. (2010) 118:467–72. doi: 10.1055/s-0030-1252068
43. Tonne JM, Sakuma T, Deeds MC, Munoz-Gomez M, Barry MA, Kudva YC, et al. Global gene expression profiling of pancreatic islets in mice during streptozotocin-induced beta-cell damage and pancreatic Glp-1 gene therapy. Dis Model Mech. (2013) 6:1236–45. doi: 10.1242/dmm.012591
44. Bonner-Weir S. Life and death of the pancreatic beta cells. Trends Endocrinol Metab. (2000) 11:375–8. doi: 10.1016/S1043-2760(00)00305-2
45. Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. (2004) 429:41–6. doi: 10.1038/nature02520
46. Teta M, Rankin MM, Long SY, Stein GM, Kushner JA. Growth and regeneration of adult beta cells does not involve specialized progenitors. Dev Cell. (2007) 12:817–26. doi: 10.1016/j.devcel.2007.04.011
47. Ivanovska I, Ball AS, Diaz RL, Magnus JF, Kibukawa M, Schelter JM, et al. MicroRNAs in the miR-106b family regulate p21/CDKN1A and promote cell cycle progression. Mol Cell Biol. (2008) 28:2167–74. doi: 10.1128/MCB.01977-07
48. Woods K, Thomson JM, Hammond SM. Direct regulation of an oncogenic micro-RNA cluster by E2F transcription factors. J Biol Chem. (2007) 282:2130–4. doi: 10.1074/jbc.C600252200
49. Yang KT, Bayan JA, Zeng N, Aggarwal R, He L, Peng Z, et al. Adult-onset deletion of Pten increases islet mass and beta cell proliferation in mice. Diabetologia. (2014) 57:352–61. doi: 10.1007/s00125-013-3085-8
50. Chen J, Huang ZP, Seok HY, Ding J, Kataoka M, Zhang Z, et al. mir-17-92 cluster is required for and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts. Circ Res. (2013) 112:1557–66. doi: 10.1161/CIRCRESAHA.112.300658
51. Nguyen KT, Tajmir P, Lin CH, Liadis N, Zhu XD, Eweida M, et al. Essential role of Pten in body size determination and pancreatic beta-cell homeostasis in vivo. Mol Cell Biol. (2006) 26:4511–8. doi: 10.1128/MCB.00238-06
52. Stiles BL, Kuralwalla-Martinez C, Guo W, Gregorian C, Wang Y, Tian J, et al. Selective deletion of Pten in pancreatic beta cells leads to increased islet mass and resistance to STZ-induced diabetes. Mol Cell Biol. (2006) 26:2772–81. doi: 10.1128/MCB.26.7.2772-2781.2006
53. Wang L, Liu Y, Yan Lu S, Nguyen KT, Schroer SA, Suzuki A, et al. Deletion of Pten in pancreatic ss-cells protects against deficient ss-cell mass and function in mouse models of type 2 diabetes. Diabetes. (2010) 59:3117–26. doi: 10.2337/db09-1805
54. Petersen HV, Serup P, Leonard J, Michelsen BK, Madsen OD. Transcriptional regulation of the human insulin gene is dependent on the homeodomain protein STF1/IPF1 acting through the CT boxes. Proc Natl Acad Sci USA. (1994) 91:10465–9. doi: 10.1073/pnas.91.22.10465
55. Zhao L, Guo M, Matsuoka TA, Hagman DK, Parazzoli SD, Poitout V, et al. The islet beta cell-enriched MafA activator is a key regulator of insulin gene transcription. J Biol Chem. (2005) 280:11887–94. doi: 10.1074/jbc.M409475200
56. Kaneto H, Miyatsuka T, Kawamori D, Yamamoto K, Kato K, Shiraiwa T, et al. PDX-1 and MafA play a crucial role in pancreatic beta-cell differentiation and maintenance of mature beta-cell function. Endocr J. (2008) 55:235–52. doi: 10.1507/endocrj.K07E-041
57. Yin XL, Liu HY, Zhang YQ. Mulberry branch bark powder significantly improves hyperglycemia and regulates insulin secretion in type II diabetic mice. Food Nutr Res. (2017) 61:1368847. doi: 10.1080/16546628.2017.1368847
58. Srivastava A, Dadheech N, Vakani M, Gupta S. Swertisin ameliorates diabetes by triggering pancreatic progenitors for islet neogenesis in Streptozotocin treated BALB/c mice. Biomed Pharmacother. (2018) 100:221–5. doi: 10.1016/j.biopha.2018.01.096
59. Zhuo J, Zeng Q, Cai D, Zeng X, Chen Y, Gan H, et al. Evaluation of type 2 diabetic mellitus animal models via interactions between insulin and mitogenactivated protein kinase signaling pathways induced by a high fat and sugar diet and streptozotocin. Mol Med Rep. (2018) 17:5132–42. doi: 10.3892/mmr.2018.8504
60. Iguchi H, Ikeda Y, Okamura M, Tanaka T, Urashima Y, Ohguchi H, et al. SOX6 attenuates glucose-stimulated insulin secretion by repressing PDX1 transcriptional activity and is down-regulated in hyperinsulinemic obese mice. J Biol Chem. (2005) 280:37669–80. doi: 10.1074/jbc.M505392200
61. Andrali SS, Sampley ML, Vanderford NL, Ozcan S. Glucose regulation of insulin gene expression in pancreatic beta-cells. Biochem J. (2008) 415:1–10. doi: 10.1042/BJ20081029
62. Matsuoka TA, Zhao L, Artner I, Jarrett HW, Friedman D, Means A, et al. Members of the large Maf transcription family regulate insulin gene transcription in islet beta cells. Mol Cell Biol. (2003) 23:6049–62. doi: 10.1128/MCB.23.17.6049-6062.2003
63. Zhang C, Moriguchi T, Kajihara M, Esaki R, Harada A, Shimohata H, et al. MafA is a key regulator of glucose-stimulated insulin secretion. Mol Cell Biol. (2005) 25:4969–76. doi: 10.1128/MCB.25.12.4969-4976.2005
64. Wang H, Brun T, Kataoka K, Sharma AJ, Wollheim CB. MAFA controls genes implicated in insulin biosynthesis and secretion. Diabetologia. (2007) 50:348–58. doi: 10.1007/s00125-006-0490-2
65. Kulkarni RN, Jhala US, Winnay JN, Krajewski S, Montminy M, Kahn CR. PDX-1 haploinsufficiency limits the compensatory islet hyperplasia that occurs in response to insulin resistance. J Clin Invest. (2004) 114:828–36. doi: 10.1172/JCI21845
66. Zhou YP, Marlen K, Palma JF, Schweitzer A, Reilly L, Gregoire FM, et al. Overexpression of repressive cAMP response element modulators in high glucose and fatty acid-treated rat islets. A common mechanism for glucose toxicity and lipotoxicity? J Biol Chem. (2003) 278:51316–23. doi: 10.1074/jbc.M307972200
67. Kang HS, Yang H, Ahn C, Kang HY, Hong EJ, Jaung EB. Effects of xenoestrogens on streptozotocin-induced diabetic mice. J Physiol Pharmacol. (2014) 65:273–82.
68. Kanasaki K, Shi S, Kanasaki M, He J, Nagai T, Nakamura Y, et al. Linagliptin-mediated DPP-4 inhibition ameliorates kidney fibrosis in streptozotocin-induced diabetic mice by inhibiting endothelial-to-mesenchymal transition in a therapeutic regimen. Diabetes. (2014) 63:2120–31. doi: 10.2337/db13-1029
69. Horwitz E, Krogvold L, Zhitomirsky S, Swisa A, Fischman M, Lax T, et al. beta-Cell DNA Damage Response Promotes Islet Inflammation in Type 1 Diabetes. Diabetes. (2018) 67:2305–18. doi: 10.2337/db17-1006
70. Zeng L, Ding QF, Xu TY, Luo F, Ge N, Li ST. The role of DNA double-strain damage repairing mechanisms in diabetic atheroscolersis. Sichuan Da Xue Xue Bao Yi Xue Ban. (2017) 48:191–6.
71. Song L, Lin C, Wu Z, Gong H, Zeng Y, Wu J, et al. miR-18a impairs DNA damage response through downregulation of ataxia telangiectasia mutated (ATM) kinase. PLoS ONE. (2011) 6:e25454. doi: 10.1371/journal.pone.0025454
Keywords: miR-17-92 cluster, pancreatic beta-cells, streptozotocin, restoration, Cdkn1a, ATM kinase
Citation: Wan S, Zhang J, Chen X, Lang J, Li L, Chen F, Tian L, Meng Y and Yu X (2020) MicroRNA-17-92 Regulates Beta-Cell Restoration After Streptozotocin Treatment. Front. Endocrinol. 11:9. doi: 10.3389/fendo.2020.00009
Received: 05 November 2019; Accepted: 07 January 2020;
Published: 23 January 2020.
Edited by:
Åke Sjöholm, Gävle Hospital, SwedenReviewed by:
Gordon C. Weir, Joslin Diabetes Center, Harvard Medical School, United StatesBashoo Naziruddin, Baylor Simmons Transplant Institute, United States
Copyright © 2020 Wan, Zhang, Chen, Lang, Li, Chen, Tian, Meng and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xijie Yu, eGlqaWV5dSYjeDAwMDQwO3NjdS5lZHUuY24=; eGlqaWV5dSYjeDAwMDQwO2hvdG1haWwuY29t
†These authors have contributed equally to this work