 Livia Lenke
Livia Lenke Gonzalo Martínez de la Escalera
Gonzalo Martínez de la Escalera Carmen Clapp
Carmen Clapp Thomas Bertsch
Thomas Bertsch Jakob Triebel
Jakob Triebel- 1Institute for Clinical Chemistry, Laboratory Medicine and Transfusion Medicine, Nuremberg General Hospital & Paracelsus Medical University, Nuremberg, Germany
- 2Instituto de Neurobiología, Universidad Nacional Autónoma de México (UNAM), Campus UNAM-Juriquilla, Querétaro, Mexico
Preeclampsia is a hypertensive disorder affecting 3–5% of all pregnancies. The only curative treatment is delivery of the placenta and the pathophysiology is poorly understood. Studies have demonstrated altered levels of antiangiogenic factors in patients with preeclampsia. One such factor is the antiangiogenic and antivasodilatatory peptide hormone vasoinhibin, which is higher in the circulation, urine, and amniotic fluid of women with preeclampsia. Normal pregnancy is characterized by elevated circulating prolactin and placental lactogen levels, both of which can serve as vasoinhibin precursors when they are enzymatically cleaved. A dysregulation of vasoinhibin generation during preeclampsia is indicated by higher vasoinhibin, prolactin, placental lactogen, and vasoinhibin-generating enzymes levels and activity. The present article integrates known vasoinhibin levels, effects, and signaling mechanisms to the clinical characteristics of preeclampsia to substantiate the notion that vasoinhibin dysregulation can be causally linked to the development of preeclampsia. If this view is demonstrated, assessment of vasoinhibin levels and regulation of its activity could help estimate the risk of preeclampsia and improve its treatment.
Introduction
Preeclampsia (PE) is a severe complication of pregnancy, defined by gestational hypertension and proteinuria after the 20th week of pregnancy (1). Hypertension is defined as blood pressure >140/90 mmHg in mild PE and >160/110 in severe presentations. Proteinuria is defined as more than 0.3 or 5 g protein in urine collected for 24 h in mild or severe states, respectively (2, 3). In about one of five cases, PE is associated with small-for-gestational-age (SGA) neonates (birth weight below the 10th percentile of the gestational age) (4). The worldwide incidence of PE is described as 3–5% of all pregnancies (5). Beside hypertension and proteinuria, patients may present edema, headache, right upper quadrant or epigastric pain, blurred vision, nausea, and shortness of breath due to pulmonary edema (6). Treatment options are limited since there is no curative therapy except delivery of the placenta. Complications of PE can lead to more severe diseases like eclampsia, characterized by generalized seizure attacks not attributed to other causes (3, 7), or HELLP syndrome (hemolysis, elevated liver enzymes, and low platelets) (8). Besides hypertension and microalbuminuria, long term complications include the development of ischemic heart disease risk elevation, higher stroke rate, venous thromboembolisms and cardiovascular disease risk elevation (9). Numerous studies have investigated anti-angiogenic and angiogenic factors in the circulation of patients with PE, and it is generally accepted that a dysbalance of these factors may not merely represent an epiphenomenon, but rather have a causal role in the pathophysiology of PE. While the exact pathomechanism for preeclampsia is still unclear, it is known that various pro- and anti-angiogenic factors as well as their receptors, play a key role in the development of preeclampsia, since they regulate placental blood flow and vascular development. One of the most widely studied pro-angiogenic factors is vascular endothelial growth factor (VEGF) and the anti-angiogenic soluble form of the VEGF receptor 1 or soluble fms-like tyrosine kinase 1 (sFlt1 or sVEGFR1) (10). A comparatively less studied factor with vascular effects is the pituitary hormone prolactin (PRL), which is the focus of the present article.
A role of PRL in preeclampsia has first suggested around 1975 by Redman, Horrobin (11, 12), and others, but at the time, the possible contribution of antiangiogenic PRL-fragments was not considered as their vascular actions were discovered later, in 1991 (13) (Table 1). Since then, antiangiogenic fragments of PRL, but also of placental lactogen (PL) and growth hormone (GH) (22, 23), evolved as a the vasoinhibin family, which shares specific structural features, biological effects, and signaling mechanisms (24, 25). Vasoinhibin features a variety of endocrine, paracrine, and autocrine effects (25, 26). It is generated by matrix metalloproteinases (MMP), cathepsin-D, and bone morphogenic protein 1 (BMP-1) which cleave PRL in the region of the long loop connecting the third and fourth alpha helix (27–29). The vascular system is one of its major target tissues and its effects include the inhibition of vasopermeability, angiogenesis, and vasodilation (25). All three vasoinhibin-precursor molecules- PRL, GH, and hPL—show major changes in their circulating levels during normal pregnancy (30). In PE, the changes in circulating hormonal levels are altered compared to normal pregnancy, as are the level and activity of the cleaving enzymes determining the generation. However, the studies addressing vasoinhibin levels in PE, the changing values of their precursors, and the altered levels and activity of the cleaving enzymes have not been analyzed in conjunction with each other and with the altered physiology of the prolactin/vasoinhibin axis, the endocrine system that controls the generation and action of vasoinhibin (31). The present article is an attempt to integrate the various findings pointing to dysregulation of vasoinhibin generation and action as contributors to the development of PE (19).
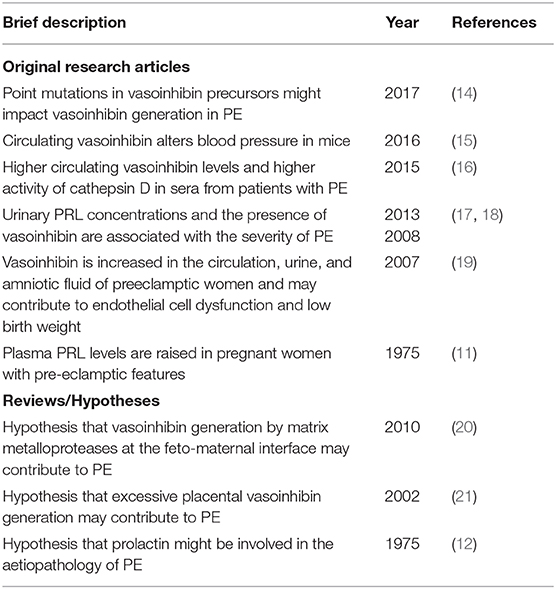
Table 1. Selected original research articles and reviews supporting the involvement of the prolactin/vasoinhibin axis in preeclampsia.
The Prolactin/Vasoinhibin Axis in Pregnancy and Preeclampsia
The Levels of the Vasoinhibin Precursors, PRL, hPL and GH, Are Elevated in PE
Due to the rise of vasoinhibin precursor levels, the state of pregnancy as such is a predisposition for a dysregulation of the prolactin/vasoinhibin axis. Vasoinhibin-generating enzymes may utilize a manifold higher supply of substrates, and their activity and regulators in the tissue microenvironment would control both, their normal pregnancy-adapted vasoinhibin levels, as well as the pathological (suppressed or excessive) vasoinhibin levels. Circulating PRL levels start to rise in pregnant women at week 10 and reach up to ≅ 200 ng/ml at week 40 of pregnancy (32). At 1 week postpartum, PRL levels in serum are only about 50 ng/ml and return to baseline levels 4–6 weeks after birth. The pregnancy rise in PRL levels is physiological and occurs to prepare for lactation. Remarkably, women with PE demonstrate higher serum and urine levels of PRL, compared to women with normal pregnancy. Compared to normal PRL levels (about 139 ng/ml), mild PE (162.6 ng/ml), and severe PE patients (190.5 ng/ml) show higher PRL levels in serum before induction of labor or cesarean delivery (17), although other studies did not report such findings (19, 33). Similarly, PRL median levels in urine of women with PE are elevated (195.3, 1,342, and 9,830 pg/ml for normal pregnancy, mild, and severe PE, respectively) (17). Of note, the synciotrophoblast secretes hPL [3] and the concentration of PL in serum of pregnant women is detectable at week 5 and rises continuously to very high levels (≅ 4 μg/ml) at the end of pregnancy (34). One week after term, PL level is very low or already undetectable (34). The circulating levels of pituitary growth hormone decline during pregnancy in response to the rise of placental GH (PGH, also named GH-2), which demonstrates agonism at the GH-receptor. PGH levels can be detected after the week 5 of gestation and continually rise until it becomes dominant over the pituitary isoform during the second half of pregnancy (35–37). PGH levels are about 12 ng/ml in normal and 23 ng/ml in preeclamptic pregnancies (38). Hence, all three hormone vasoinhibin pecursors are higher in PE.
The Levels of Vasoinhibin and Vasoinhibin Generating Enzymes Are Elevated in PE
Vasoinhibin derived from PRL is elevated in serum, urine, and amniotic fluid of women with PE (18, 19). It appears to be generated locally in utero placental tissue through the action of various cleaving enzymes such as cathepsin-D. The incubation of PRL with lysates from placental trophoblasts results in its conversion to vasoinhibin, and addition of pepstatin A, a cathepsin D inhibitor, abolishes such conversion. The generation of vasoinhibin is significantly higher when lysates from women with PE are used, compared to lysates from normal pregnant women, indicating a higher activity of cathepsin D in the placenta during PE (19). The detection rate of vasoinhibin in urine occurs in proportional manner to the severity of preeclampsia and can also be demonstrated in other pregnancy complications such as placental abruption, pulmonary edema, and renal failure (17). About 21% of urine samples of women with severe PE demonstrate the presence of vasoinhibin (14 and 16 kDa isoforms) (17). In patients with HELLP syndrome, vasoinhibin appears in about 60% of urine samples (18). The activity of the vasoinhibin-generating enzyme cathepsin D is elevated in serum of patients with PE compared to normal pregnancy (16). During pregnancy, CD activity is about 156%, compared to non-pregnant women, whereas preeclamptic values are around 183%. Soon after delivery this difference is even more pronounced (129 vs. 217%) and still maintained 1 month after (102 vs. 197%) (16). Similar to this observation, MMP serum levels change during pregnancy. MMP-2 and MMP 3 levels are elevated in sera of pregnant women, although circulating MMP-9 level is lower than in the absence of pregnancy (39, 40). Also, MMP-2 levels increase in PE amniotic fluid (40, 41). These data show that both, substrates and enzymes needed for vasoinhibin generation, are higher in pregnancy compared to the normal non-pregnant state. Comparisons between normal pregnancies and PE reveal an even more pronounced elevation of substrate/enzyme levels in patients with PE.
Mechanistic Implications of Vasoinhibin Actions in PE
VEGF is a major promoter of angiogenesis and vasodilation in the placenta, and its actions are partially mediated by the production of endothelium-derived nitric oxide (NO). Decreased levels of VEGF and NO occur in PE and inhibition of placental VEGF action and NO production causes reduced perfusion of the fetoplacental unit, hypertension, proteinuria, and fetal growth restriction, indicating that blockage of VEGF-induced NO production is causal in preeclampsia (42). Vasoinhibin suppress VEGF-induced endothelial NO synthase (eNOS) activity in endothelial cells and an NO donor reverses the vasoinhibin-mediated inhibition of VEGF-induced endothelial cell proliferation and acetylcholine-induced vasodilation (43). Genetically modified mice, which overexpress hepatic PRL and develop an increase in systemic PRL and vasoinhibin levels, show an increase in systemic blood pressure, a reduced ejection fraction of the left ventricle, and a reduction of the thickness of the intraventricular septum and the left ventricular posterior wall. In mice deficient in eNOS no variation in systemic blood pressure is found when PRL and vasoinhibin levels are elevated, indicating that eNOS plays a critical role in vasoinhibin-mediated blood pressure regulation (15).
A Dysregulation of the Prolactin/Vasoinhibin Axis Appears to Contribute to Preeclampsia
Altogether, a sequence of events throughout pregnancy, from the rise of vasoinhibin-precursors, an upregulation of vasoinhibin-generating enzymes, an excessive vasoinhibin generation in the placenta or elsewhere, high vasoinhibin levels in the maternal circulation and other compartments, and the consequence of PE manifestation, points toward a dysregulation of the prolactin/vasoinhibin axis in PE (Table 2). A causal inference of the role of vasoinhibin in PE is further consolidated when the various reports are evaluated for their agreement with the 2015 Bradford Hill Criteria (48), whereby any criteria is supported by one or more studies (Table 3).

Table 2. Proposed mechanistic pathways involved in preeclampsia.

Table 3. Causal inference between vasoinhibin dysregulation and preeclampsia: application of the 2015 Bradford Hill criteria (48).
Conclusion
Several publications reported a possible involvement of vasoinhibin to the pathophysiology of preeclampsia (Table 1). The majority of studies are associational, however, they include strong mechanistic implications. The present theory is an attempt to integrate the findings reported in these studies addressing measurements of vasoinhibin in biological samples from patients, vascular effects of vasoinhibin, and clinical characteristics of preeclampsia (Table 2). Both, substrate levels and enzyme activity required for vasoinhibin generation, are higher in pregnancy compared to the non-pregnant state. In PE, there is an even more pronounced elevation of vasoinhibin-precursor levels, cleaving enzymes, and vasoinhibin. Increased vasoinhibin production and its antiangiogenic activity imply that elevated levels may be causally involved in PE, for example by contributing to the development of hypertension (Table 2).
The present analysis reinforces the recommendation that a detection of vasoinhibin levels in blood, urine and amniotic fluid can be useful for screening of PE onset, and as a prognostic marker when PE is manifested (19). Also, placental tissue from women with and without PE should be investigated for vasoinhibin, its precursors and vasoinhibin-generating enzymes to validate that the placenta is origin of vasoinhibin which may not only act locally, but also enter the maternal circulation. These implications underscore the need to develop a quantitative assay for vasoinhibin in body fluids that is suitable for routine clinical use, and to improve detection methods to assess vasoinhibin in human tissues. The question of which vasoinhibin isoforms are generated in the placenta, and whether they are generated by cleavage of PRL, PL, PGH, or all three of them, including their overall quantitative composition, requires particular attention.
Further consolidation and clinical evaluation will demonstrate whether the assessment of vasoinhibin levels, its precursors and regulatory enzymes can contribute to estimate the risk of preeclampsia or to improve its treatment.
Author Contributions
LL and JT wrote the draft of the manuscript. GM, CC, and TB contributed critical revisions and intellectual content. All authors approved the final version of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Steegers EA, von Dadelszen P, Duvekot JJ, Pijnenborg R. Pre-eclampsia. Lancet. (2010) 376:631–44. doi: 10.1016/S0140-6736(10)60279-6
2. Schroeder BM, American College of O, Gynecologists. ACOG practice bulletin on diagnosing and managing preeclampsia and eclampsia. American College of Obstetricians and Gynecologists. Am Fam Phys. (2002) 66:330–1.
3. American College of O, Gynecologists, Task Force on Hypertension in P. Hypertension in pregnancy. Report of the American College of Obstetricians and Gynecologists' Task Force on Hypertension in Pregnancy. Obstetr Gynecol. (2013) 122:1122–31. doi: 10.1097/01.AOG.0000437382.03963.88
4. Villar J, Carroli G, Wojdyla D, Abalos E, Giordano D, Ba'aqeel H, et al. Preeclampsia, gestational hypertension and intrauterine growth restriction, related or independent conditions? Am J Obstetr Gynecol. (2006) 194:921–31. doi: 10.1016/j.ajog.2005.10.813
5. Abalos E, Cuesta C, Grosso AL, Chou D, Say L. Global and regional estimates of preeclampsia and eclampsia: a systematic review. Eur J Obstetr Gynecol Reprod Biol. (2013) 170:1–7. doi: 10.1016/j.ejogrb.2013.05.005
6. Uzan J, Carbonnel M, Piconne O, Asmar R, Ayoubi JM. Pre-eclampsia: pathophysiology, diagnosis, and management. Vasc Health Risk Manag. (2011) 7:467–74. doi: 10.2147/VHRM.S20181
7. Lambert G, Brichant JF, Hartstein G, Bonhomme V, Dewandre PY. Preeclampsia: an update. Acta Anaesthesiol Belg. (2014) 65:137–49.
8. Sibai BM. Diagnosis, controversies, and management of the syndrome of hemolysis, elevated liver enzymes, and low platelet count. Obstetr Gynecol. (2004) 103(5 Pt 1):981–91. doi: 10.1097/01.AOG.0000126245.35811.2a
9. Bellamy L, Casas JP, Hingorani AD, Williams DJ. Pre-eclampsia and risk of cardiovascular disease and cancer in later life: systematic review and meta-analysis. BMJ. (2007) 335:974. doi: 10.1136/bmj.39335.385301.BE
10. Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Investig. (2003) 111:649–58. doi: 10.1172/JCI17189
11. Redman CW, Bonnar J, Beilin LJ, McNeilly AS. Prolactin in hypertensive pregnancy. Br Med J. (1975) 1:304–6. doi: 10.1136/bmj.1.5953.304
12. Horrobin DF. The possible role of prolactin in pre-eclampsia. Med Hypoth. (1975) 1:159–64. doi: 10.1016/0306-9877(75)90013-4
13. Ferrara N, Clapp C, Weiner R. The 16K fragment of prolactin specifically inhibits basal or fibroblast growth factor stimulated growth of capillary endothelial cells. Endocrinology. (1991) 129:896–900. doi: 10.1210/endo-129-2-896
14. Triebel J, Friedrich CJ, Leuchs A, Martinez de la Escalera G, Clapp C, Bertsch T. Human prolactin point mutations and their projected effect on vasoinhibin generation and vasoinhibin-related Diseases. Front Endocrinol. (2017) 8:294. doi: 10.3389/fendo.2017.00294
15. Chang AS, Grant R, Tomita H, Kim HS, Smithies O, Kakoki M. Prolactin alters blood pressure by modulating the activity of endothelial nitric oxide synthase. Proc Natl Acad Sci USA. (2016) 113:12538–43. doi: 10.1073/pnas.1615051113
16. Nakajima R, Ishida M, Kamiya CA, Yoshimatsu J, Suzuki M, Hirota A, et al. Elevated vasoinhibin derived from prolactin and cathepsin D activities in sera of patients with preeclampsia. Hypertens Res. (2015) 38:899–901. doi: 10.1038/hr.2015.99
17. Leanos-Miranda A, Marquez-Acosta J, Cardenas-Mondragon GM, Chinolla-Arellano ZL, Rivera-Leanos R, Bermejo-Huerta S, et al. Urinary prolactin as a reliable marker for preeclampsia, its severity, and the occurrence of adverse pregnancy outcomes. J Clin Endocrinol Metab. (2008) 93:2492–9. doi: 10.1210/jc.2008-0305
18. Leanos-Miranda A, Campos-Galicia I, Ramirez-Valenzuela KL, Chinolla-Arellano ZL, Isordia-Salas I. Circulating angiogenic factors and urinary prolactin as predictors of adverse outcomes in women with preeclampsia. Hypertension. (2013) 61:1118–25. doi: 10.1161/HYPERTENSIONAHA.111.00754
19. Gonzalez C, Parra A, Ramirez-Peredo J, Garcia C, Rivera JC, Macotela Y, et al. Elevated vasoinhibins may contribute to endothelial cell dysfunction and low birth weight in preeclampsia. Lab Investig. (2007) 87:1009–17. doi: 10.1038/labinvest.3700662
20. Reuwer AQ, Reuwer PJ, van der Post JA, Cramer MJ, Kastelein JJ, Twickler MT. Prolactin fragmentation by trophoblastic matrix metalloproteinases as a possible contributor to peripartum cardiomyopathy and pre-eclampsia. Med Hypoth. (2010) 74:348–52. doi: 10.1016/j.mehy.2009.08.029
21. Parra A, Ramirez-Peredo J. The possible role of prolactin in preeclampsia: 2001, a hypothesis revisited a quarter of century later. Med Hypoth. (2002) 59:378–84. doi: 10.1016/S0306-9877(02)00124-X
22. Struman I, Bentzien F, Lee H, Mainfroid V, D'Angelo G, Goffin V, et al. Opposing actions of intact and N-terminal fragments of the human prolactin/growth hormone family members on angiogenesis: an efficient mechanism for the regulation of angiogenesis. Proc Nat Acad Sci USA. (1999) 96:1246–51. doi: 10.1073/pnas.96.4.1246
23. Corbacho AM, Martinez De La Escalera G, Clapp C. Roles of prolactin and related members of the prolactin/growth hormone/placental lactogen family in angiogenesis. J Endocrinol. (2002) 173:219–38. doi: 10.1677/joe.0.1730219
24. Triebel J, Robles JP, Zamora M, Martinez de la Escalera G, Bertsch T, Clapp C. Regulator of angiogenesis and vascular function: a 2019 update of the vasoinhibin nomenclature. Front Endocrinol. (2019) 10:214. doi: 10.3389/fendo.2019.00214
25. Clapp C, Thebault S, Macotela Y, Moreno-Carranza B, Triebel J, Martinez de la Escalera G. Regulation of blood vessels by prolactin and vasoinhibins. Adv Exp Med Biol. (2015) 846:83–95. doi: 10.1007/978-3-319-12114-7_4
26. Clapp C, Thebault S, Jeziorski MC, Martinez De La Escalera G. Peptide hormone regulation of angiogenesis. Physiol Rev. (2009) 89:1177–215. doi: 10.1152/physrev.00024.2009
27. Ge G, Fernandez CA, Moses MA, Greenspan DS. Bone morphogenetic protein 1 processes prolactin to a 17-kDa antiangiogenic factor. Proc Natl Acad Sci USA. (2007) 104:10010–5. doi: 10.1073/pnas.0704179104
28. Baldocchi RA, Tan L, King DS, Nicoll CS. Mass spectrometric analysis of the fragments produced by cleavage and reduction of rat prolactin: evidence that the cleaving enzyme is cathepsin D. Endocrinology. (1993) 133:935–8. doi: 10.1210/endo.133.2.8344226
29. Macotela Y, Aguilar MB, Guzman-Morales J, Rivera JC, Zermeno C, Lopez-Barrera F, et al. Matrix metalloproteases from chondrocytes generate an antiangiogenic 16 kDa prolactin. J Cell Sci. (2006) 119(Pt 9):1790–800. doi: 10.1242/jcs.02887
30. Tyson JE, Hwang P, Guyda H, Friesen HG. Studies of prolactin secretion in human pregnancy. Am J Obstetr Gynecol. (1972) 113:14–20. doi: 10.1016/0002-9378(72)90446-2
31. Triebel J, Bertsch T, Bollheimer C, Rios-Barrera D, Pearce CF, Hufner M, et al. Principles of the prolactin/vasoinhibin axis. Am J Physiol Regul Integr Comp Physiol. (2015) 309:R1193–203. doi: 10.1152/ajpregu.00256.2015
32. Hwang P, Guyda H, Friesen H. A radioimmunoassay for human prolactin. Proc Natl Acad Sci USA. (1971) 68:1902–6. doi: 10.1073/pnas.68.8.1902
33. Ranta T, Stenman UH, Unnerus HA, Rossi J, Seppala M. Maternal plasma prolactin levels in preeclampsia. Obstetr Gynecol. (1980) 55:428–30.
34. Rastogi GK, Sinha MK, Jayaraman S, Devi PK. Serum human placental lactogen (HPL) levels in normal pregnancy. J Obstetr Gynaecol India. (1976) 26:65–9.
35. Schiessl B, Strasburger CJ, Bidlingmaier M, Gutt B, Kirk SE, Oberhoffer R, et al. Role of placental growth hormone in the alteration of maternal arterial resistance in pregnancy. J Reprod Med. (2007) 52:313–6.
36. Eriksson L, Frankenne F, Eden S, Hennen G, Von Schoultz B. Growth hormone 24-h serum profiles during pregnancy–lack of pulsatility for the secretion of the placental variant. Br J Obstet Gynaecol. (1989) 96:949–53. doi: 10.1111/j.1471-0528.1989.tb03352.x
37. Frankenne F, Closset J, Gomez F, Scippo ML, Smal J, Hennen G. The physiology of growth hormones (GHs) in pregnant women and partial characterization of the placental GH variant. J Clin Endocrinol Metab. (1988) 66:1171–80. doi: 10.1210/jcem-66-6-1171
38. Mittal P, Espinoza J, Hassan S, Kusanovic JP, Edwin SS, Nien JK, et al. Placental growth hormone is increased in the maternal and fetal serum of patients with preeclampsia. J Mater fetal Neonatal Med. (2007) 20:651–9. doi: 10.1080/14767050701463571
39. Montagnana M, Lippi G, Albiero A, Scevarolli S, Salvagno GL, Franchi M, et al. Evaluation of metalloproteinases 2 and 9 and their inhibitors in physiologic and pre-eclamptic pregnancy. J Clin Lab Analysis. (2009) 23:88–92. doi: 10.1002/jcla.20295
40. Laskowska M. Altered Maternal Serum Matrix Metalloproteinases MMP-2, MMP-3, MMP-9, and MMP-13 in Severe Early- and Late-Onset Preeclampsia. BioMed Res Int. (2017) 2017:6432426. doi: 10.1155/2017/6432426
41. Lavee M, Goldman S, Daniel-Spiegel E, Shalev E. Matrix metalloproteinase-2 is elevated in midtrimester amniotic fluid prior to the development of preeclampsia. Reprod Biol Endocrinol. (2009) 7:85. doi: 10.1186/1477-7827-7-85
42. Tranquilli AL, Giannubilo SR, Bezzeccheri V, Ciavattini A, Scagnoli C, Mazzanti L. Amniotic levels of nitric oxide and vascular endothelial growth factor in pregnancy with subsequent intrauterine fetal death. Eur J Obstetr Gynecol Reprod Biol. (2004) 114:162–5. doi: 10.1016/j.ejogrb.2003.10.016
43. Gonzalez C, Corbacho AM, Eiserich JP, Garcia C, Lopez-Barrera F, Morales-Tlalpan V, et al. 16K-prolactin inhibits activation of endothelial nitric oxide synthase, intracellular calcium mobilization, and endothelium-dependent vasorelaxation. Endocrinology. (2004) 145:5714–22. doi: 10.1210/en.2004-0647
44. Sinha YN, Gilligan TA, Lee DW, Hollingsworth D, Markoff E. Cleaved prolactin: evidence for its occurrence in human pituitary gland and plasma. J Clin Endocrinol Metab. (1985) 60:239–43. doi: 10.1210/jcem-60-2-239
45. Narumiya H, Zhang Y, Fernandez-Patron C, Guilbert LJ, Davidge ST. Matrix metalloproteinase-2 is elevated in the plasma of women with preeclampsia. Hypertens Preg. (2001) 20:185–94. doi: 10.1081/PRG-100106968
46. Triebel J, Silawal S, Willauschus M, Schulze-Tanzil G, Bertsch T. Analysing point mutations in protein cleavage sites by using enzyme specificity matrices. Front Endocrinol. (2019) 10:267. doi: 10.3389/fendo.2019.00267
47. Perimenis P, Bouckenooghe T, Delplanque J, Moitrot E, Eury E, Lobbens S, et al. Placental antiangiogenic prolactin fragments are increased in human and rat maternal diabetes. Biochim Biophys Acta. (2014) 1842:1783–93. doi: 10.1016/j.bbadis.2014.06.026
48. Fedak KM, Bernal A, Capshaw ZA, Gross S. Applying the Bradford Hill criteria in the 21st century: how data integration has changed causal inference in molecular epidemiology. Emerg Themes Epidemiol. (2015) 12:14. doi: 10.1186/s12982-015-0037-4
49. Khan F, Belch JJ, MacLeod M, Mires G. Changes in endothelial function precede the clinical disease in women in whom preeclampsia develops. Hypertension. (2005) 46:1123–8. doi: 10.1161/01.HYP.0000186328.90667.95
50. Rodgers GM, Taylor RN, Roberts JM. Preeclampsia is associated with a serum factor cytotoxic to human endothelial cells. Am J Obstetr Gynecol. (1988) 159:908–14. doi: 10.1016/S0002-9378(88)80169-8
Keywords: vasoinhibin, prolactin, preeclampsia, 16K PRL, PRL, prolactin/vasoinhibin axis, hypertensive pregnancy disorders
Citation: Lenke L, Martínez de la Escalera G, Clapp C, Bertsch T and Triebel J (2020) A Dysregulation of the Prolactin/Vasoinhibin Axis Appears to Contribute to Preeclampsia. Front. Endocrinol. 10:893. doi: 10.3389/fendo.2019.00893
Received: 03 May 2019; Accepted: 05 December 2019;
Published: 09 January 2020.
Edited by:
GianLuca Colussi, University of Udine, ItalyReviewed by:
Frank Spradley, University of Mississippi Medical Center, United StatesDerek Boeldt, University of Wisconsin-Madison, United States
Copyright © 2020 Lenke, Martínez de la Escalera, Clapp, Bertsch and Triebel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jakob Triebel, amFrb2IudHJpZWJlbEBnbXguZGU=