 James E. Foley
James E. Foley
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 08 November 2019
Sec. Molecular and Structural Endocrinology
Volume 10 - 2019 | https://doi.org/10.3389/fendo.2019.00780
This article is part of the Research Topic GLP-1 analogues and DPP-4 inhibitors in type 2 diabetes therapy: from structural biology to clinics View all 8 articles
Vildagliptin blocks glucagon like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) inactivation of the meal induced increases in GLP-1 and GIP so that elevated GLP-1 and GIP levels are maintained over 24 h. The primary insulin secretion effect of vildagliptin is to improve the impaired sensitivity of the β-cells to glucose in subjects with impaired fasting glucose (IFG) and impaired glucose tolerance (IGT) and in patients with type 2 diabetes mellitus (T2DM); this effect was seen acutely and maintained over at least 2 years in patients with T2DM. Vildagliptin was also associated with improved β-cell function that is likely secondary to the improved metabolic state. Although there was no evidence of restoration of β-cell mass, the preponderance of the vildagliptin data does indicate that for at least 2 years β-cell function was maintained in vildagliptin treated patients but not in the untreated patients. Vildagliptin suppressed an inappropriate glucagon response to an oral glucose challenge in patients with T2DM, to a mixed meal challenge in patients with T2DM and type 1 diabetes mellitus, and to a mixed meal challenge in subjects with IGT and IFG. The improved glucagon response was maintained for at least 2 years in patients with T2DM and there was no change in the glucagon response in normoglycemic individuals. Vildagliptin lowered glucose levels into the normal range without increasing hypoglycemia. These hypoglycemic benefits appear to be secondary in large part to the improved sensitivity of both the β and α-cell to glucose. In the case of the α-cell, if glucose levels are high, GLP-1 attenuates the glucagon levels and if glucose levels are low, GIP increases glucagon levels. Vildagliptin reduces fatty acid flux from the adipocyte leading to reduced liver fat which in turn leads to increased glucose utilization. The reduced glycosuria and reduced lipo-toxicity associated with vildagliptin therapy does not lead to weight gain presumably due to increased fat mobilization and oxidation during meals and to reduced fat extraction from the gut.
The loss of sensitivity of the islets to glucose characterizes type 2 diabetes mellitus (T2DM). Insulin levels are higher than normal, but inadequate to overcome insulin resistance due initially to inappropriate glucagon secretion and to the lipo-toxicity that is characterized by greater triacylglycerol storage in non-fat tissues. Increasing hyperglycemia in T2DM is associated with increasing glucose toxicity and diminished maximum capacity of the β-cells to secrete insulin. The loss of sensitivity of the islets to glucose not only leads to hyperglycemia, but also to increased hypoglycemia (1). The Sandoz (which became Novartis in 1997) interest in glucagon like peptide-1 (GLP-1) followed directly from this problem statement.
At the 1990 EASD it was first reported that GLP-1 was a useful tool in the treatment of T2DM in humans and in 1992 the data was published (2). Since that first report at the EASD Sandoz then Novartis has played a pioneering role in GLP-1 based therapies (3). In 1993 after an aggressive but failed attempt over 2 years to make a non-peptide GLP-1 peptide mimetic, attention switched to inactivating dipeptidyl peptidase-4 (DPP-4) (3) when it was reported that GLP-1 was inactivated solely by DPP-4 (4). The same is true for the inactivation of the other incretin hormone glucose-dependent insulinotropic polypeptide (GIP), but this was not a focus at that time (3). The DPP-4 program was accelerated in 1995 after it was reported that DPP-4 inhibition raised GLP-1 in vitro (3, 5). By the end of 1995 using valine pyrrolidide, a known orally active DPP-4 inhibitor from the Sandoz library, it was shown that a DPP-4 inhibitor could lower blood glucose levels in rodents and non-human primates (3). In 1996, using combinatorial chemistry and valine pyrrolidide as a starting point DPP-728 was discovered (3); only 3 years later DPP-728 provided the first proof of concept that a DPP-4 inhibitor improves glycemia in patients with T2DM (6). Kinetic studies of DPP-728 determined that it was a slow substrate for the catalytic site of the DPP-4 enzyme, rather than a simple competitive inhibitor, thereby blocking GLP-1 and GIP inactivation rather than simply slowing these rates of inactivation (3, 7).
Engineering the kinetic properties of DPP-728 led to the discovery of vildagliptin in 1998 (3, 8). Vildagliptin was first evaluated in man in 2002 (3, 9). Early clinical mechanism studies with vildagliptin demonstrated that by blocking GLP-1 and GIP inactivation the meal induced increases in GLP-1 and GIP were maintained over 24 h (1, 3, 10).
The purpose of this article is to integrate the expected as well as many unexpected GLP-1 and GIP actions emerging from the vildagliptin studies in man. A systematic review of the differences and similarities among DPP-4 inhibitors has been published previously (1). The therapeutic utility of DPP-4 inhibitors including vildagliptin is discussed in an accompanying article (11). The safety of vildagliptin has been reviewed previously (12); briefly the profile includes rare cases of mild to moderate elevations in hepatic enzymes, rare cases of angioedema (mostly in patients taking a concomitant ACE inhibitor) that resolved with ongoing treatment, and rare adverse drug reactions, including pancreatitis, bullous or exfoliative skin lesions, and arthralgia.
As expected vildagliptin increased the insulin secretion rate (corrected for the concentration of the glucose stimulus {abbreviated ISR/G}) in patients with T2DM (1). In contrast, 24-h insulin exposure was not increased with vildagliptin treatment indicating that insulin secretion was increased when needed without increasing total insulin exposure (13). There was a residual effect when GLP-1 action was blocked after 10 days of vildagliptin treatment suggesting that at least part of vildagliptin's effect on insulin secretion was due to GIP (14), which is consistent with the incretin hormone concept.
During a study where vildagliptin was given acutely before an evening meal both GLP-1 and GIP levels rose at the beginning of the meal as expected, but unexpectedly the levels were maintained above placebo during the entire overnight period (1, 10). Furthermore, vildagliptin's acute effect to increase ISR/G was the same from early in the evening meal until breakfast the next morning (Figure 1), suggesting that enhanced ISR/G was not due to the pharmacological rise in GLP-1 at the beginning of the meal, but rather to the more physiological rise in GLP-1 maintained during the entire overnight period (1, 10). Although the GIP profile is similar to the GLP-1 profile, GIP is not predicted to be insulinotropic when given acutely (15).
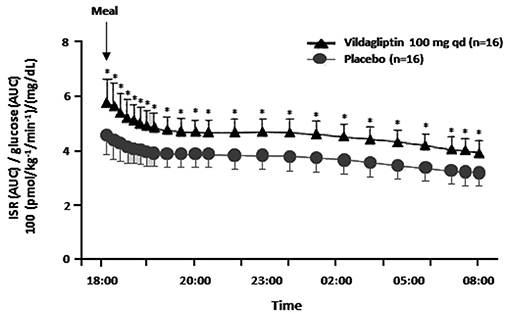
Figure 1. The insulin secretion rate (ISR) divided by the glucose area under the curve (AUC) (reflecting the relative glucose stimulus) from dinner until before breakfast in placebo patients vs. patients receiving an acute dose of vildagliptin before dinner. *P < 0.05; qd, once daily; ISR/glucose AUC was calculated from data in Balas et al. (10).
Vildagliptin increased ISR/G in both impaired fasting glucose (IFG) (16) and impaired glucose tolerance (IGT) (17) but had no effect in healthy volunteers (18). These results are consistent with extensive studies with vildagliptin confirming the primary insulin secretion effect of vildagliptin was to improve the impaired sensitivity of the β-cells to glucose (1, 19, 20). Interestingly this improved sensitivity to glucose extends into the hypoglycemic range such that there was even less insulin secretion in the hypoglycemic range with vildagliptin than in the placebo group (21). Vildagliptin's effect to improve the sensitivity of the β-cells to glucose [ISR/G] was seen acutely and maintained over at least 2 years in patients with T2DM (22).
Interestingly HOMA-B was increased, but paradoxically this was due to the reduction in the fasting glucose levels and not to an increase in fasting insulin levels (23, 24). Vildagliptin's effect to increase insulin sensitivity to glucose was seen with glucose given orally or intravenously (25); this lack of the classic incretin effect was unexpected. This unexpected lack of the classic incretin effect has more recently been explained by the incretin effect being due to GIP and the GIP effect being overwhelmed by the GLP-1 effect secondary to DPP-4 inhibition (15). Thus, the vildagliptin effect to improve insulin secretion acutely was likely due to GLP-1 (15), but over days the GIP effect to stimulate insulin may have been restored (14). Vildagliptin was also associated with improved β-cell function that is likely secondary to the improved metabolic state such as improved proinsulin processing (a reduction in the proinsulin to insulin ratio) (24) and the acute insulin response to IV glucose after 6 weeks treatment with vildagliptin in subjects with IFG (16), and after 12 weeks treatment with vildagliptin in patients with T2DM (26).
T2DM has been associated with diminished maximum capacity of the β-cell to secrete insulin which has often been ascribed to a reduction in β-cell mass (27). The greatest promise of GLP-1 based therapies was that they would restore β-cell mass and thus the maximum capacity of the β-cell to secrete insulin. In neonatal rodents vildagliptin increased β-cell mass via increased β-cell neogenesis as well by a decrease in β-cell apoptosis (28). In a 1-year study with vildagliptin treatment in patients with T2DM that was followed by a three-month washout and where the maximum insulin secretory rate was assessed it was clear that vildagliptin had no disease modifying effect to increase β-cell mass (27). It was later demonstrated in mature rodents that there was also no disease modifying effect of vildagliptin to increase β-cell mass (28) suggesting that GLP-1 based therapy is only effective to increase β-cell mass in developing β-cells. Although there was no evidence of restoration of β-cell mass, the preponderance of the vildagliptin data does indicate that for at least 2 years β-cell function was maintained in vildagliptin treated patients but not in the untreated patients (20, 22, 27).
As expected vildagliptin suppressed an inappropriate glucagon response to an oral glucose challenge in patients with T2DM (29) and to a mixed meal challenge in patients with T2DM (9, 10, 23), but unexpectedly to a mixed meal challenge in subjects with IGT (17) and IFG (16). The improved glucagon response was maintained for at least 2 years in patients with T2DM (30). Importantly, vildagliptin had no effect on the glucagon response in normoglycemic individuals (18). Since GLP-1 levels were elevated in all cases, it appears that the glucagon response to GLP-1 was glucose dependent (1). The lack of adequate GLP-1 receptors on the α-cells suggested that the GLP-1 α-cell response was secondary to a paracrine effect by insulin. However, vildagliptin suppressed an inappropriate glucagon response to a mixed meal challenge in patients with type 1 diabetes mellitus (31, 32) indicating that this effect is not secondary to a paracrine effect by insulin; there is evidence that it may be mediated by a local paracrine effect of somatostatin (33).
Vildagliptin treatment increased insulin secretion and decreased glucagon secretion in hyperglycemia, providing a clear explanation for the improvement in glucose tolerance that persist for at least 2 years that has been discussed above. An important added benefit to this mechanism is that as the patients are treated to the normal glycemic range, they do so without increasing hypoglycemia (1, 3). This is presumably due to the vildagliptin mechanism leading to improved sensitivity of the β and α-cells to glucose. As discussed previously the insulin secretion response to glucose was attenuated in the hypoglycemic range (21) and it was expected that the decrease in glucagon due to GLP-1 seen in hyperglycemia would disappear. Unexpectedly, in T2DM patents with mild hyperglycemia the α-cell response to hypoglycemia was enhanced (21). However, the α-cell response to hypoglycemia was not enhanced in insulin-treated, metformin-treated and in type 1 diabetes and thus the α-cell response to hypoglycemia may be unaltered or enhanced depending on the study population (34).
Interestingly, GIP is known to stimulate glucagon secretion in hypoglycemia and not influence glucagon secretion in hyperglycemia (35). The dual role of GLP-1 and GIP is visualized in Figure 2. Thus, the physiological consequences of an increase in the sensitivity of the β and α-cells to glucose following a carbohydrate rich meal such as fruit is that hepatic glucose production is rapidly inhibited while glucose utilization is enhanced so that glucose excursions are minimized with the least amount of insulin; and following a low or no carbohydrate meal such as meat that hepatic glucose production is increased to maintain euglycemia in order to compensate for the increased insulin required to store fat and protein.
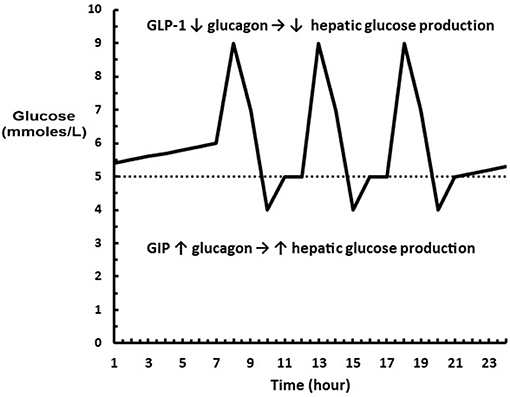
Figure 2. Diagram showing where GLP-1 and GIP are acting on glucagon relative to a glucose profile. (…..) represents euglycemia where neither GLP-1 or GIP are acting on the α-cell. The excursions above euglycemia cause GLP-1 to decrease glucagon levels leading to a decrease in hepatic glucose production. The excursions below euglycemia cause GIP to increase glucagon levels leading to an increase hepatic glucose production.
Very early in the vildagliptin clinical program it was surprising that there was a decrease in fasting palmitate flux (Figure 3A) without any change in fasting insulin levels or fasting glucagon levels (23). This suggested an extra-pancreatic effect on the fat cell to either inhibit lipolysis or to stimulate esterification of FA. Rodent studies have shown that GIP improves the sensitivity of the adipocyte to insulin leading to increased esterification of FA relative to lipolysis resulting in reduced FFA release (36). The rate limiting step in esterification of fatty acids is glucose transport and the sensitivity to insulin of glucose transport was also increased by GIP in rodents (37). Thus, extending the physiological surge in GIP beyond the beginning of the evening meal into the fasting overnight period with vildagliptin presumably leads to reduced fatty acid flux from the adipocyte. This mechanism can be visualized in Figure 4. This could correct the underlying increased abdominal adipose cell size risk of developing T2DM where larger fat cells are associated with increased lipolysis and circulating FFA levels (38). This finding begs the question of the physiological relevance of GIP increasing esterification of FA. Under physiological conditions GIP is only increased at the beginning of meals. It is known that protein can markedly stimulate GIP release (39). As discussed above, when eating only meat, GIP presumably allows the insulin levels to rise enough to store the fat and protein and the glucagon levels to rise enough to prevent hypoglycemia. By increasing glucose transport in adipocytes in the presence of the low insulin levels associated with low carbohydrate meals, GIP may play an important role to further enhance fat storage.
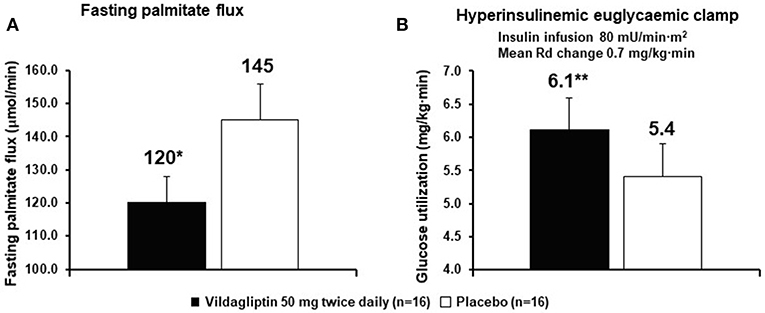
Figure 3. Vildagliptin decreases fasting fatty acid (palmitate dilution) flux from adipocytes (A) and increases glucose utilization (B). The rates of glucose utilization are calculated from the last 30 min a long duration clamp (the second step of a two-step clamp) at a very high insulin concentration so the capacity of cells to oxidize glucose and capacity of the muscle to store glycogen are exceeded. Under such conditions the reduction of glucose to fatty acids by the liver becomes the rate limiting step (1, 3, 7). *P < 0.01; **P < 0.05; bid, twice daily; Rd, rate of glucose utilization. Data extracted from Azuma et al. (23).
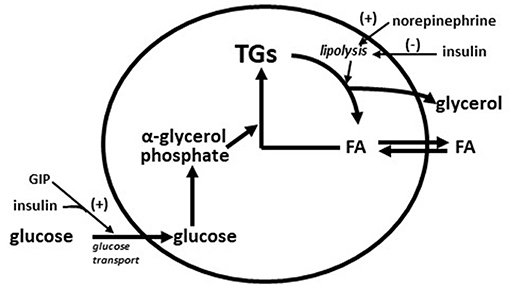
Figure 4. Diagram of storage and mobilization of fatty acids (FA) from adipocytes. GIP enhances insulin stimulated glucose transport (37) leading to increased α-glycerol phosphate which in turn increases rate of esterification of fatty acids into triglyceride (TG). Norepinephrine from nerve ending in adipose tissue stimulates lipolysis (breakdown of triglycerides to fatty acids and glycerol) and insulin inhibits lipolysis. Increased adipose cells size increases the lipolysis rate (38). A GIP effect on glucose transport during fasting is predicted to increase the rate of esterification of fatty acids into triglyceride to compensate for the increased rate of lipolysis from larger adipose cells.
It was also shown that after only 6 weeks vildagliptin treatment was associated with improved glucose disposal (Figure 3B) under conditions where the rate limiting step exceeded the capacity of the muscle to store glycogen and thus is presumed to be lipogenesis in the liver (1, 3, 7, 23). Thiazolidinediones are known to reduce fasting fatty acid flux from adipocytes (40) leading to a reduction in liver fat (41, 42) and improved glucose utilization under conditions like those shown with vildagliptin (43). Vildagliptin was also shown to decrease liver fat [Figure 5, (44)]. As can be seen in Figure 6 a reduction in FA flux to the liver is predicted to decrease liver fat in the absence of increased hepatic export of lipid. Hepatic export of lipid is unlikely to have increased given the 13% fall in plasma triglyceride concentration (44). Furthermore, there was no vildagliptin treatment effect on postprandial triglyceride levels secondary to reduced flux of lipoproteins from the liver (45). Lipogenesis is diminished by increased liver fat via FA feedback inhibition and reducing liver fat is predicted to reduce FA feedback inhibition of lipogenesis (Figure 6). Since there was no decrease in fasting hepatic glucose production and a decrease in fasting glucose levels (44) it follows that fasting glucose utilization increased and that this increase was mostly likely due to increased lipogenesis. This is important since at the end of each day any glucose not oxidized or stored as glycogen must be converted to fat in the liver to avoid a rise in fasting glucose levels. This is also consistent with the observation that HOMA-B increased due to a reduction fasting glucose levels with no change in the fasting insulin levels that was discussed earlier.
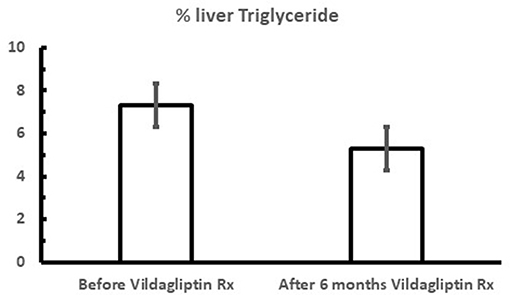
Figure 5. Mean fasting % liver triglyceride content during vildagliptin therapy (the Redirect Study) where metformin treated patients were additionally treated with and without 50 mg bid vildagliptin for 6 months. P < 0.001; Data extracted from Macauley et al. (44).
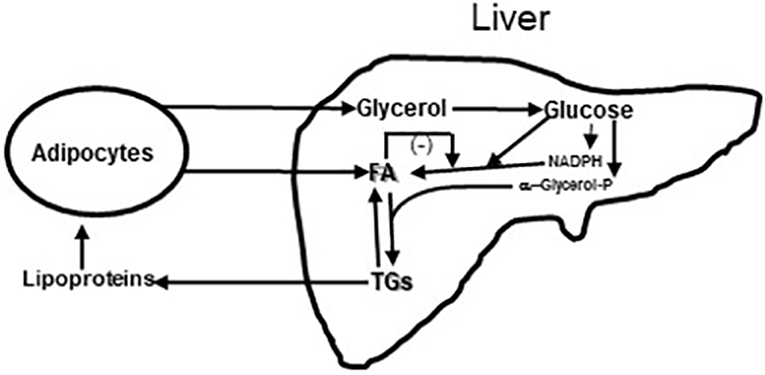
Figure 6. Diagram of Glycerol and Fatty acids (FA) coming to liver from fat cells and lipoproteins coming from liver to fat cells. In liver glycerol is metabolized to glucose. Some of the glucose is metabolized to provide reducing equivalents in the form of NADPH so that a second part of the glucose can undergo reduction to fatty acids (FA); this process of de novo fatty acid synthesis can be restrained by fatty acid feedback inhibition (44). A third part of the glucose is metabolized to α-glycerol phosphate to esterify fatty acids to triglyceride (TG). Triglycerides can be exported from the liver to fat and muscle or undergo lipolysis in the liver. Increased lipolysis in large adipocytes increases the flux of fatty acids and glycerol to the liver leading increased liver fatty acid and triglyceride levels. Under these conditions the capacity of the liver to metabolize glucose to fat is impaired and fasting glucose levels rise. This figure is a simplification of Figure 3 from Rui (46).
Overall vildagliptin was associated with weight neutrality. The low probability of hypoglycemia which precludes the weight gain associated with defensive eating to avoid hypoglycemia is consistent with such weight neutrally. However, the caloric penalty associated with a reduction of glucose levels from above to below the renal threshold predicts weight gain. When the weight changes from pooled monotherapy studies after 24 weeks of therapy with vildagliptin were assessed vs. the fasting plasma glucose (FPG) levels at baseline it was clear that no weight change was observed at the renal threshold in T2DM (FPG of 14.6 mmol/L or 263 mg/dL). Baseline FPG values below and above this threshold were associated with weight loss and weight gain, respectively; a baseline FPG of 8 mmol/L (144 mg/dL) predicted a weight loss of 1 kg (47). There was no evidence of a satiety effect with vildagliptin that could explain this apparent weight loss when glucosuria was considered (48). However, two other potential weight mitigating mechanisms were identified in the vildagliptin studies. In the first there was a reduction in Apo B-48 secretion which presumably decreases fat extraction from the gut (45). In the second there was an increase postprandial lipolysis from adipose tissue and increased postprandial fat oxidation in the muscle so that during the fed state fat acids are mobilized and burned (49). Although such a mechanism is predicted to cause glucose intolerance, the pancreatic mechanisms to improve glucose tolerance clearly prevails. Thus, as discussed above, in the fasted state vildagliptin treatment results in greater fat storage while in the fed state it mobilizes and burns fat. This has the effect of reducing lipo-toxicity without the weight gain associated with thiazolidinediones (50).
The clinical studies with vildagliptin in T2DM have shown that DPP-4 inhibition with vildagliptin attenuated the diminished sensitivity of the islets to glucose, reduced the insulin resistance due to inappropriate glucagon secretion and lipo-toxicity characterized by greater triacylglycerol storage in non-fat tissues, decreased the hyperglycemia that is associated with glucose toxicity, and maintained but did not restore the diminished maximum capacity of the β-cells to secrete insulin. The improved sensitivity of the islets to glucose not only led to a reduction in hyperglycemia but did so without increased hypoglycemia. The VERIFY study just reported that an early metformin and vildagliptin combination was more durable than adding vildagliptin to patients failing metformin and that over 5 years vildagliptin plus metformin resulted in a small weight loss in patients that were clearly below the renal threshold during this 5-year period (51). The contributions of reduced lipo-toxicity and improved islet sensitivity to glucose in the face of no weight gain to vildagliptin's durability remains to be determined.
The author confirms being the sole contributor of this work and has approved it for publication.
Prior to Nov 2017, the author was an employee of Novartis Pharmaceuticals.
The author declares that this review was written in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Ahrén B, Foley JE. Improved glucose regulation in type 2 diabetic patients with DPP-4 inhibitors: focus on alpha and beta cell function and lipid metabolism. Diabetologia. (2016) 59:907–17. doi: 10.1007/s00125-016-3899-2
2. Gutnaik M, Ørskov C, Holst JJ, Ahrén B, Efendić S. Antidiabetic effect of glucagon-like peptide-1 (7-36) amide in normal subjects and patients with diabetes mellitus. N Engl J Med. (1992) 326:1316–22. doi: 10.1056/NEJM199205143262003
3. Foley JE, Ahrén B. The vildagliptin experience – 25 years since the initiation of the novartis glucagon-like peptide-1 based therapy programme and 10 years since the first vildagliptin registration. Eur Endocrinol. (2017) 13:56–61. doi: 10.17925/EE.2017.13.02.56
4. Mentlein R, Gallwitz B, Schmidt WE Dipeptidyl-peptidase IV hydrolyses gastric inhibitory polypeptide glucagon-like peptide-1(7-36) amide peptide histidine methionine and is responsible for their degradation in human serum. Eur J Biochem. (1993) 214:829–35. doi: 10.1111/j.1432-1033.1993.tb17986.x
5. Deacon CF, Johnsen AH, Holst JJ. Degradation of glucagon-like peptide-1 by human plasma in vitro yields an N-terminally truncated peptide that is a major endogenous metabolite in vivo. J Clin Endocrinol Metab. (1995) 80:952–7. doi: 10.1210/jcem.80.3.7883856
6. Ahrén B, Simonsson E, Larsson H, Landin-Olsson M, Torgeirsson H, Jansson P, et al. Inhibition of dipeptidyl peptidase IV improves metabolic control over a 4-week study period in type 2 diabetes. Diabetes Care. (2002) 25:869–75. doi: 10.2337/diacare.25.5.869
7. Ahrén B, Schweizer A, Dejager S, Villhauer EB, Dunning BE, Foley JE. Mechanisms of action of the dipeptidyl peptidase-4 inhibitor vildagliptin in humans. Diabetes Obes Metab. (2011) 13:775–83. doi: 10.1111/j.1463-1326.2011.01414.x
8. Villhauer EB, Brinkman JA, Naderi GB, Burkey BF, Dunning BE, Prasad K, et al. 1-[[(3-hydroxy1-adamantyl)amino]acetyl]-2-cyano-(S)-pyrrolidine: a potent, selective, and orally bioavailable dipeptidyl peptidase IV inhibitor with antihyperglycemic properties. J Med Chem. (2003) 46:2774–89. doi: 10.1021/jm030091l
9. Ahrén B, Landin-Olsson M, Jansson PA, Jansson P, Svensson M, Holmes D, et al. Inhibition of dipeptidyl peptidase-4 reduces glycemia, sustains insulin levels, and reduces glucagon levels in type 2 diabetes. J Clin Endocrinol Metab. (2004) 89:2078–84. doi: 10.1210/jc.2003-031907
10. Balas B, Baig MR, Watson C, Dunning BE, Ligueros-Saylan M, Wang Y, et al. The dipeptidyl peptidase IV inhibitor vildagliptin suppresses endogenous glucose production and enhances islet function after single dose administration in type 2 diabetic patients. J Clin Endocrinol Metab. (2007) 92:1249–55. doi: 10.1210/jc.2006-1882
11. Ahrén B. DPP-4 inhibition and the path to clinical proof. Front Endocrinol. (2019) 19:376. doi: 10.3389/fendo.2019.00376
12. Mathieu C, Kozlovski P, Paldánius PM, Foley JE, Modgill V, Evans M, et al. Clinical safety and tolerability of vildagliptin – insights from randomised trials, observational studies and postmarketing surveillance. Eur Endocrinol. (2017) 13:68–72. doi: 10.17925/EE.2017.13.02.68
13. Ahren B, Gomis R, Standl E, Mills D, Schweizer A. Twelve– and fifty-two–week efficacy of the dipeptidyl peptidase iv inhibitor laf237 in metformin-treated patients with type 2 diabetes. Diabetes Care. (2004) 27:2874–880. doi: 10.2337/diacare.27.12.2874
14. Nauck MA, Kind J, Köthe LD, Holst JJ, Deacon CF, Broschag M, He YL, et al. Quantification of the contribution of GLP-1 to mediating insulinotropic effects of DPP-4 inhibition with vildagliptin in healthy subjects and type 2-diabetic patients using exendin [9-39] as a GLP-1 receptor antagonist. Diabetes. (2016) 65:2440–7. doi: 10.2337/db16-0107
15. Nauck NA, Meier JJ. The incretin effect in healthy individuals and those with type 2 diabetes: physiology, pathophysiology, and response to therapeutic interventions. Lancet Diabetes Endocrinol. (2016) 4:525–36. doi: 10.1016/S2213-8587(15)00482-9
16. Utzschneider KM, Tong J, Montgomery B, Udayasankar J, Gerchman F, Marcovina SM, et al. The dipeptidylpeptidase-4 inhibitor vildagliptin improves β-cell function and insulin sensitivity in subjects with impaired fasting glucose. Diabetes Care. (2008) 31:108–13. doi: 10.2337/dc07-1441
17. Rosenstock J, Foley JE, Rendell M, Landin-Olsson M, Holst JJ, Deacon CF, et al. Effects of the dipeptidyl peptidase-IV inhibitor vildagliptin on incretin hormones, islet function, and postprandial glycemia in subjects with impaired glucose tolerance. Diabetes Care. (2008) 31:30–5. doi: 10.2337/dc07-1616
18. El-Ouaghlidi A, Rehring E, Holst JJ, El-Ouaghlidi A, Rehring E, Holst JJ, et al. The dipeptidyl peptidase 4 inhibitor vildagliptin does not accentuate glibenclamide-induced hypoglycemia but reduces glucoseinducedglucagon-likepeptide1andgastricinhibitorypolypeptide secretion. J Clin Endocrinol Metab. (2007) 92:4165–71. doi: 10.1210/jc.2006-1932
19. Mari A, Sallas WM, He YL, Watson C, Ligueros-Saylan M, Dunning BE, et al. Vildagliptin, a dipeptidyl peptidase-IV inhibitor, improves model-assessed β-cell function in patients with type 2 diabetes. J Clin Endocrinol Metab. (2005) 90:4888–94. doi: 10.1210/jc.2004-2460
20. Mari A, Scherbaum WA, Nilsson PM, Lalanne G, Schweizer A, Dunning BE, et al. Characterization of the influence of vildagliptin on model-assessed-cell function in patients with type 2 diabetes and mild hyperglycemia. J Clin Endocrinol Metab. (2008) 93:103–9. doi: 10.1210/jc.2007-1639
21. Ahrén B, Schweizer A, Dejager S, Dunning BE, Nilsson PM, Persson M, et al. Vildagliptin enhances islet responsiveness to both hyper- and hypoglycemia in patients with type 2 diabetes. J Clin Endocrinol Metab. (2009) 94:1236–43. doi: 10.1210/jc.2008-2152
22. Scherbaum WA, Schweizer A, Mari A, Nilsson M, Lalanne G, Wang Y, et al. Evidence that vildagliptin attenuates deterioration of glycaemic control during 2 year treatment of patients with type 2 diabetes and mild hyperglycaemia. Diabetes Obes Metab. (2008) 10:1114–24. doi: 10.1111/j.1463-1326.2008.00875.x
23. Azuma K, Rádiková Z, Mancino J, Toledo FGS, Thomas E, Kangani C, et al. Measurements of islet function and glucose metabolism with the dipeptidyl peptidase 4 inhibitor vildagliptin inpatients with type 2 diabetes. J Clin Endocrinol Metab. (2008) 93:459–64. doi: 10.1210/jc.2007-1369
24. Pratley RE, Schweizer A, Rosenstock J, Foley JE., Banerji MA, Pi-Sunyer FX, et al. Robust improvements in fasting and prandial measures of beta-cell function with vildagliptin in drug-naive patients: analysis of pooled vildagliptin monotherapy database. Diabetes Obes Metab. (2008) 10:931–38. doi: 10.1111/j.1463-1326.2007.00835.x
25. Vardarli I, Nauck MA, Köthe LD, Deacon CF, Holst JJ, Schweizer A, et al. Inhibition of DPP-4 with vildagliptin improved insulin secretion in response to oral as well as “isoglycemic” intravenous glucose without numerically changing the incretin effect in patients with type 2 diabetes. J Clin Endocrinol Metab. (2011) 96:945–54. doi: 10.1210/jc.2010-2178
26. D'Alessio DA, Denney AM, Hermiller LM, Prigeon RL, Martin JM, Tharp WG, et al. Treatment with the DPP-4 inhibitor vildagliptin improves fasting islet-cell function in subjects with type 2 diabetes. J Clin Endocrinol Metab. (2008) 94:81–8. doi: 10.1210/jc.2008-1135
27. Foley JE, Bunck MC, Möller-Goede DL, Poelma M, Nijpels G, Eekhoff EM, et al. Beta cell function following 1year vildagliptin or placebo treatment and after 12week washout in drug-naive patients with type 2 diabetes and mild hyperglycaemia: a randomised controlled trial. Diabetologia. (2011) 54:1985–91. doi: 10.1007/s00125-011-2167-8
28. Omar BA, Vikman J, Sörhede Winzell M, Voss BAU, Ekblad E, Foley JE, et al. Enhanced beta cell function and anti-inflammatory effect after chronic treatment with the dipeptidyl peptidase 4 inhibitor vildagliptin in an advanced age diet induced obesity mouse model. Diabetologia. (2013) 56:1752–60. doi: 10.1007/s00125-013-2927-8
29. He YL, Wang Y, Bullock JM, Deacon CF, Holst JJ, Dunning BE, et al. Pharmacodynamics of vildagliptin in patients with type 2 diabetes during OGTT. J Clin Pharmacol. (2007) 47:633–41. doi: 10.1177/0091270006299137
30. Ahrén B, Foley JE, Ferrannini E, Matthews DR, Zinman B, Dejager S, et al. Changes in prandial glucagon levels after 2-year treatment with vildagliptin or glimepiride inpatients with type 2 diabetes mellitus inadequately controlled with metformin monotherapy. Diabetes Care. (2010) 33:730–2. doi: 10.2337/dc09-1867
31. Foley JE, Ligueros-Saylan M, He YL. Effect of vildagliptin on glucagon concentration during meals in patients with type 1 diabetes. Horm Metab Res. (2008) 40:727–30. doi: 10.1055/s-2008-1078754
32. Farngren J, Persson M, Schweizer A, Foley JE, Ahrén B. Vildagliptin reduces glucagon during hyperglycemia and sustains glucagon counter regulation during hypoglycemia in type 1 diabetes. J Clin Endocrinol Metab. (2012) 97:3799–806. doi: 10.1210/jc.2012-2332
33. de Heer J, Rasmussen C, Coy DH, Holst JJ. Glucagon-like peptide-1, but not glucose-dependent insulinotropic peptide, inhibits glucagon secretion via somatostatin (receptor subtype 2) in the perfused rat pancreas. Diabetologia. (2008) 51:2263–70. doi: 10.1007/s00125-008-1149-y
34. Farngren J, Persson M, Schweizer A., Foley JE, Ahrén B. Glucagon dynamics during hypoglycemia and food-re-challenge following treatment with vildagliptin in insulin-treated patients with type 2 diabetes. Diabetes Obes Metab. (2014) 16:812–4. doi: 10.1111/dom.12284
35. Christensen M, Vedtofte L, Hot JJ, Vilsbøll T, Knop FK. Glucose-dependent insulinotropic polypeptide: a bifunctional glucose-dependent regulator of glucagon and insulin secretion in humans. Diabetes. (2011) 60:3103–9. doi: 10.2337/db11-0979
36. Getty-Kaushik L, Song DH, Boylan MO, Corkey BE, Wolfe MM. GIP effect on basal and isoproterenol-stimulated rat adipocyte FFA release. Obesity. (2006) 14:1124–31. doi: 10.1038/oby.2006.129
37. Starich GH, Bar RS, Mazzaferri E. GIP increases insulin receptor affinity and cellular sensitivity in adipocytes. Am J Physiol. (1985) 249:E603–7. doi: 10.1152/ajpendo.1985.249.6.E603
38. Weyer C, Foley JE, Bogardus C, Tataranni PA, Pratley RE. Enlarged subcutaneous abdominal adipocyte size, but not obesity itself, predicts type II diabetes independent of insulin resistance Diabetologia. (2000) 43:1498–506. doi: 10.1007/s001250051560
39. Carr R, Larsen MO, Winzell MS, Jelic K, Lindgren O, Deacon CF, et al. Incretin and islet hormonal responses to fat and protein ingestion in healthy men. Am J Physiol. (2008) 295:E779–84. doi: 10.1152/ajpendo.90233.2008
40. Cheng AYY, Fantus G. Oral antihyperglycemic therapy for type 2 diabetes mellitus. Can Med Assoc J. (2005) 172:213–6. doi: 10.1503/cmaj.1031414
41. Ravikumar B, Gerrard J, Dalla Man C, Firbank MJ, Lane A, English PT, et al. Pioglitazone decreases fasting and postprandial endogenous glucose production in proportion to decrease in hepatic triglyceride content. Diabetes. (2008) 57:2288–95. doi: 10.2337/db07-1828
42. Ferre P, Foufelle F. Hepatic steatosis: a role for de novo lipogenesis and the transcription factor SREBP-1c. Diabetes Obes Metab. (2010) 12(Suppl 2):82–93. doi: 10.1111/j.1463-1326.2010.01275.x
43. Miyazaki Y, Mahankali A, Matsuda M, Glass L, Mahankali S, Ferrannini E, et al. Improved glycemic control and enhanced insulin sensitivity in type 2 diabetic subjects treated with pioglitazone. Diabetes Care. (2001) 24:710–9. doi: 10.2337/diacare.24.4.710
44. Macauley M, Hollingsworth KG, Smith FE, Thelwall PE, Al-Mrabeh A, Schweizer A, et al. Effect of vildagliptin on hepatic steatosis. J Clin Endocrinol Metab. (2015) 100:1578–85. doi: 10.1210/jc.2014-3794
45. Matikainen N, Mänttäri S, Schweizer A, Ulvestad A, Mills D, Dunning BE, et al. Vildagliptin therapy reduces postprandial intestinal triglyceride-rich lipoprotein particles in patients with type 2 diabetes. Diabetologia. (2006) 49:2049–57. doi: 10.1007/s00125-006-0340-2
47. Blüher M, Schweizer A, Bader G, Foley JE. Changes in body weight after 24 weeks of vildagliptin therapy as a function of fasting glucose levels in patients with type 2 diabetes. Vasc Health Risk Manage. (2014) 10:661–4. doi: 10.2147/VHRM.S73608
48. Vella A, Bock G, Giesler PD, Burton DB, Serra DB, Ligueros Saylan M, et al. The effect of dipeptidyl peptidase-4 inhibition on gastric volume, satiation and enteroendocrine secretion in type 2 diabetes: a double-blind, placebo-controlled crossover study. Clin Endocrinol. (2008) 69:737–44. doi: 10.1111/j.1365-2265.2008.03235.x
49. Boschmann M, Engeli S, Dobberstein K, Budziarek P, Strauss A, Boehnke J, et al. Dipeptidylpeptidase-IV inhibition augments postprandial lipid mobilization andoxidation in type 2 diabetic patients. J Clin Endocrinol Metab. (2009) 94:846–52. doi: 10.1210/jc.2008-1400
50. Foley JE, Jordan J. Weight neutrality with the DPP-4 inhibitor, vildagliptin: mechanistic basis and clinical experience. Vasc Health Risk Manage. (2010) 6:541–8. doi: 10.2147/VHRM.S10952
51. Matthews DR, Paldanius PM, Proot P, Chiang YT, Strumvoll M, Del Prato S. Glycaemic durability of an early combination therapy with vildagliptin and metformin versus sequential metformin monotherapy in newly diagnosed type 2 diabetes (VERIFY): a 5-year, multicentre, randomised, double-blind trial. Lancet. (2019) 394:1519–29. doi: 10.1016/S0140-6736(19)32131-2
Keywords: β-cell, α-cell, hypoglycemia, lipo-toxicity, weight, type 2 diabetes
Citation: Foley JE (2019) Insights Into GLP-1 and GIP Actions Emerging From Vildagliptin Mechanism Studies in Man. Front. Endocrinol. 10:780. doi: 10.3389/fendo.2019.00780
Received: 28 June 2019; Accepted: 25 October 2019;
Published: 08 November 2019.
Edited by:
Jens Juul Holst, University of Copenhagen, DenmarkReviewed by:
Alain Couvineau, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceCopyright © 2019 Foley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James E. Foley, U2VhbXVzZm9sZXkxMDBAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.