 Morgane Sébert
Morgane Sébert Nuria Sola-Tapias
Nuria Sola-Tapias Emmanuel Mas
Emmanuel Mas Frédérick Barreau
Frédérick Barreau Audrey Ferrand
Audrey Ferrand
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 24 October 2019
Sec. Neuroendocrine Science
Volume 10 - 2019 | https://doi.org/10.3389/fendo.2019.00717
This article is part of the Research Topic GPCR in Inflammatory and Cancer Diseases View all 7 articles
Protease-activated receptors (PARs) belong to the G protein-coupled receptor (GPCR) family. Compared to other GPCRs, the specificity of the four PARs is the lack of physiologically soluble ligands able to induce their activation. Indeed, PARs are physiologically activated after proteolytic cleavage of their N-terminal domain by proteases. The resulting N-terminal end becomes a tethered activation ligand that interact with the extracellular loop 2 domain and thus induce PAR signal. PARs expression is ubiquitous and these receptors have been largely described in chronic inflammatory diseases and cancer. In this review, after describing their discovery, structure, mechanisms of activation, we then focus on the roles of PARs in the intestine and the two main diseases affecting the organ, namely inflammatory bowel diseases and cancer.
Protease-activated receptors (PARs) belong to the family of G-protein coupled receptors (GPCRs). Their activation results from the specific cleavage, by proteases, of the amino terminal sequence that exposes a new N-terminal sequence as a tethered ligand, which then binds intramolecularly to activate the receptor. PARs are ubiquitous throughout the organism, although predominantly expressed in vascular, immune, intestinal epithelial cells and the nervous system. Thus, their activations regulate a set of crucial biological processes involved in physiology and diseases (1, 2).
In the intestine, cleavage and activation of PARs have been largely described in the modulation of pain (3), but are also linked to inflammation (3) and cancer (4–6). Indeed, the gastrointestinal tract being an important source of proteases, PARs might play crucial roles in multiple pathophysiological processes.
In polarized intestinal epithelial cells, these receptors are expressed at both apical and basolateral sides, suggesting that luminal, circulating and secreted proteases can reach and activate them (7). As a consequence, proteases coming from either coagulation cascade, inflammatory cells, microbiota, or intestinal epithelial cells are able to cleave and trigger PAR signaling to maintain gut homeostasis, regulate ion exchange, motility, permeability and healing mechanisms, but also lead to visceral hypersensitivity, inflammation, or cancer when upregulated (8–10).
Four PARs have been identified, PAR1, PAR2, PAR3, and PAR4, according to their cloning order (11). Surprisingly, the genes coding for PAR1, PAR2, and PAR3 are located on chromosome 5q, whereas PAR4 gene is located on chromosome 19p. These PARs are ubiquitous, with variable expression depending on the tissues and physiopathological context (12).
The first receptor of this family to be cloned was PAR1, in 1991. Originally, the authors wanted to identify the receptors involved in the mechanisms of action of thrombin in inflammation and haemostasis (13, 14). They ended discovering a new type of receptor activated, after proteolytic cleavage, by the protease.
The PARs cloning followed in 1994 with PAR2. After the isolation of a DNA sequence coding for a G-protein-coupled receptor from a mouse genomic library, the predicted protein displayed a structure similar to PAR1 and activated by a similar mechanism. This receptor was first described activated by trypsin, but not by thrombin. In addition, the authors described that an exogenous agonist peptide allowed the receptor activation, suggesting the importance of the proteolytic cleavage in this process. This receptor was indeed named PAR2 (15). Nowadays, it is established that thrombin can actually indirectly activate PAR2 by transactivation (16). More recently, in a murine PAR1 KO model, thrombin-induced PAR2 activation was described to trigger aortic vasodilatation and MAPK signaling (17).
A similar approach allowed PAR3 discovery in 1997. This thrombin-activated receptor seems to be a PAR4 cofactor (18). To date, no exogenous and specific agonist peptide has been shown able to induce its activation.
Finally PAR4 was discovered shortly afterwards. Both trypsin and thrombin can activate this receptor (19).
The genes encoding the PARs consist in two exons. The exon 1 generates a larger N-terminal sequence than most GPCRs that includes the site of cleavage. The exon 2 codes for the receptor itself (20). PARs have seven transmembrane domains (TM), with an N-terminal domain of 17–26 amino acids, a pro-domain of 11–30 amino acids, 3 intracellular loops (ICL) and 3 extracellular loops (ECL), and a C-terminal domain of 13–51 amino acids. PAR1 and PAR3 display a “hirudin-like” domain, allowing a more specific binding for thrombin (13) (Figure 1). These receptors may also undergo post-transcriptional modifications (phosphorylation, ubiquitination, etc.). Each receptor can be stimulated by an activator ligand—TFLLR for PAR1, SLIGKV for PAR2, TFRGAP for PAR3, and GYPGQV for PAR4—followed by an amino acid sequence involved in the inhibition of its self-activation (21).
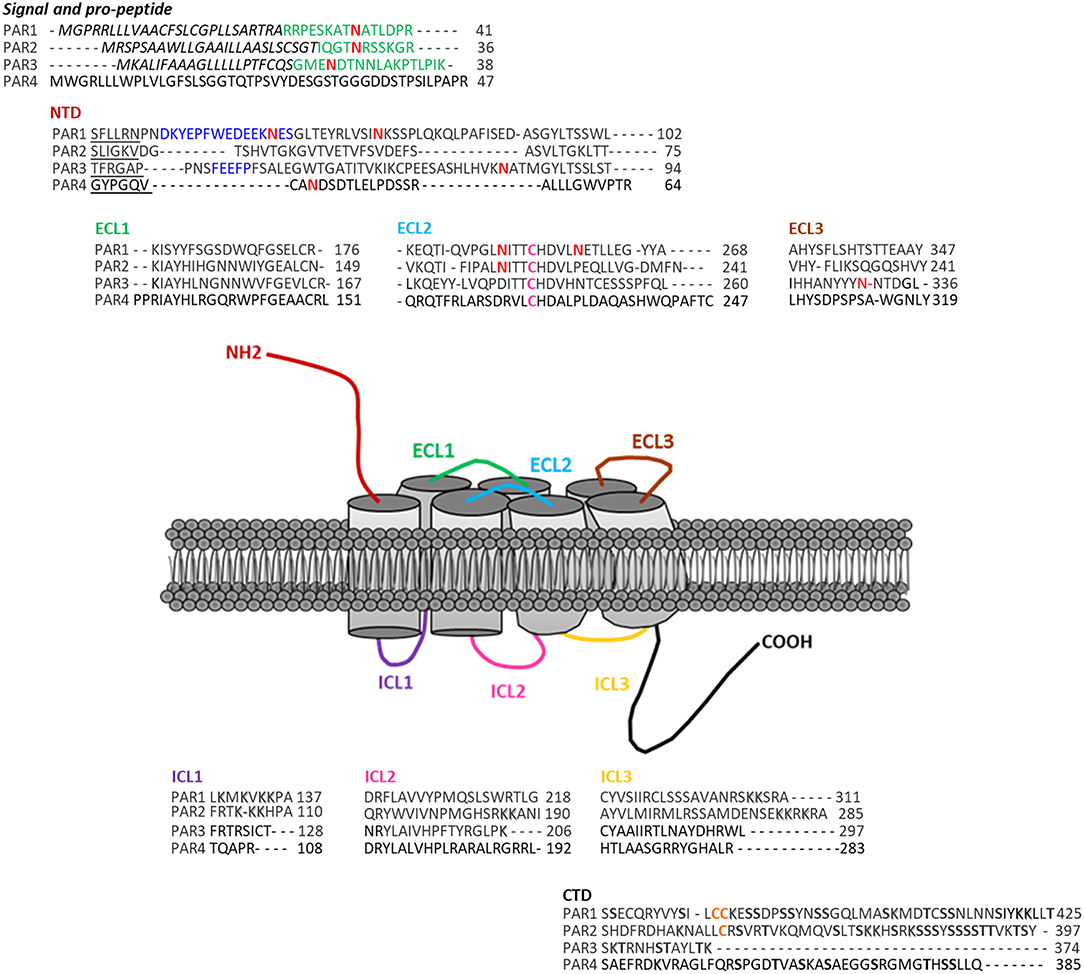
Figure 1. Protease-activated receptors structure. These receptors present several domains within their structures: the signal (italics lettering) and pro-peptide (green lettering) domains, a NH2-terminal domain (NTD), three extracellular loops (ECL1-3), three intracellular loops (ICL1-3), and a COOH-terminal domain (CTD). Within each receptor, the sequence of their specific tethered ligand are underlined. The blue lettering represents PAR1 and PAR3 Hirudin-like domains. Pink Cysteines are the ones forming a disulfide linkage between the transmembrane domain 3 and ECL2. PARs also present several post-translational modifications sites (N-glycosylation-red N, PAR1 and PAR2 putative palmitoylation sites–Orange lettering, Shadowed and bold lettering, respectively, represent ubiquitination and phosphorylation sites). Finally within PAR1 CTD, the YKKL motif is involved in the regulation of its trafficking.
Under the action of a protease, the proteolytic cleavage of the receptor activator ligand within the canonical N-terminal domain site is irreversible (Figure 2A). This ligand will then bind to the second extracellular loop of the receptor, resulting in its conformational change that will induce signaling cascades (22).
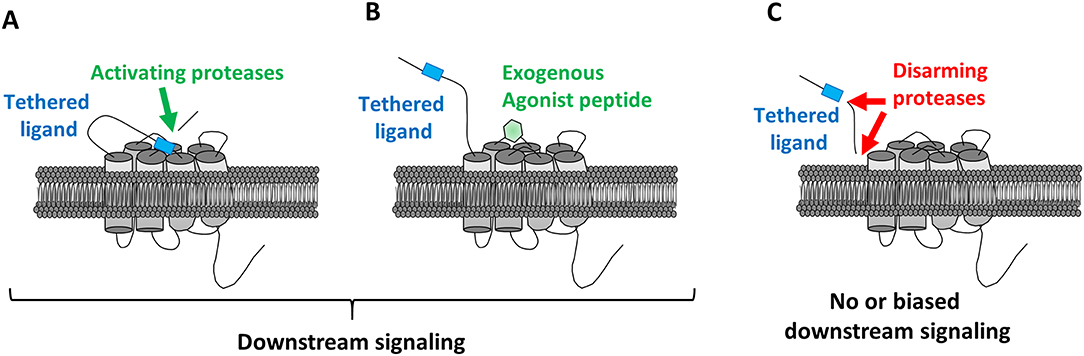
Figure 2. Mechanisms regulating protease-activated receptors activation. (A) Proteolytic cleavage by an activating proteases resulting in the binding of the tethered ligand to the ECL2, and downstream signaling activation. (B) Binding of an exogenous synthetic agonist peptide on ECL2 without proteolytic cleavage inducing downstream signaling activation. (C) No or Biased activation by disarming proteases cleaving the N-terminal domain after the tethered ligand either inhibiting the signal transduction or inducing biased downstream signaling compared to the one induced par the activating proteases. Moreover, disarmed receptors can be retained at the cell membrane making them available for a future activation by synthetic agonist peptides.
PAR1 is cleaved (↓) at its canonical site, LDPR41 ↓ S42FLLRN, by thrombin (23).
PAR2 is activated at its canonical cleavage site, SKGR34 ↓ S35LIGKV, by trypsin (15).
PAR3 presents a putative cleavage site for thrombin, LPIK38 ↓ T39FRGAP (18). However, no signal transduction seems to result from this putative cleavage, and compared to other receptors, no protease able to activate PAR3 has been yet identified.
PAR4 can be cleaved by thrombin and trypsin, at similar doses, at this canonical site: PAPR47 ↓ G48YPGQV (19). PAR4 can also be activated by cathepsin G, plasmin, factor X and kallikreins (24–27).
Table 1 gives examples of activating proteases as well as synthetic peptides and tethered ligand sequences, but also the sites of expression and the induced effects for each of the PARs.
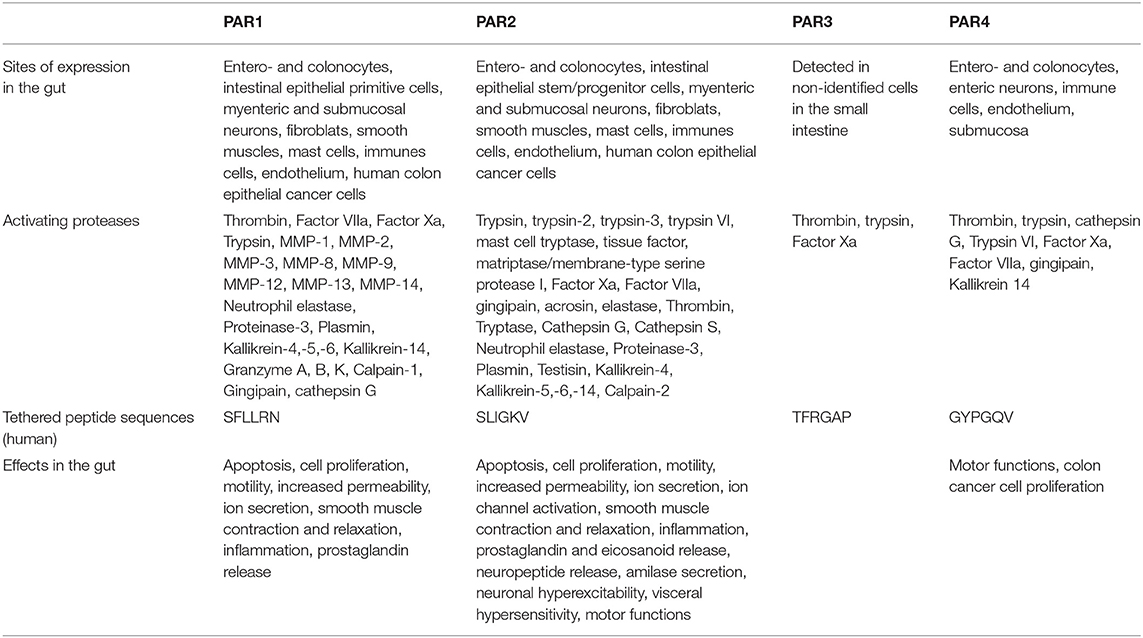
Table 1. PARs expression sites, activating proteases, tethered peptides, and main effects in the intestine.
Then, the notion of biased activation, characterized by a stimulation of the receptor on different sites than the canonical cleavage site, called “non-canonical sites,” appeared. This biased activation causes incomplete or different signaling compared to the ones observed after canonical activation. Proteases can activate PARs in a biased way (Figure 2C).
Biased activation was first described for PAR1 signaling (28). In this study, the authors demonstrated that MMP1 activates PAR1 via an activator ligand located two amino acids upstream of the one generated by thrombin (PRSFLLR ligand), but resulting in the same signaling as the one induced by thrombin. Another team showed a biased PAR1 activation leading to a different signaling. Indeed, PAR1 activation by activated protein C via a non-canonical site favors opposite effects than thrombin, i.e., anti-inflammatory effect and endothelial barrier protection (29). PAR1 can also be activated in a biased manner by other proteases and coagulation cascade actors, such as plasmin, factor X, granzymes A, trypsins, kallikreins, and cathepsin G (13, 25, 30–35).
Regarding PAR2, a study demonstrates the role of neutrophil elastase in MAPK signaling through biased activation of PAR2 (36). PAR2 can also be activated by other serine proteases, such as tryptase, granzymes, and kallikreins (23, 27, 37). To date, no studies demonstrating the biased activation of PAR3 and PAR4 have been reported.
Thus, considering the diversity of elements able to cleave and activate the PARs, it has not been easy to decipher for each individual receptor its own mechanisms of activation. For example, thrombin can activate PAR1, PAR3, and PAR4. Deciphering the specific signaling triggered by PAR1 via thrombin is in consequence difficult. In that context, using synthetic peptide sequences or agonist peptides of 5–6 amino acids is paramount (12, 13, 38) (Figure 2B).
Several peptide sequences, with a different number of amino acids, additional hydrophilic residues or amino acid substitutions relative to the PAR1 activator ligand sequence, have been developed to activate PAR1. The most efficient one is in fact similar to PAR1 activator ligand sequence, TFLLR (39). Another point is the signaling induced by the agonist peptides. Indeed, it has been observed that the signaling generated via an agonist peptide is not identical in all respects to the one induced by proteolytic cleavage, confirming the biased activation. For example, several agonist peptides for PAR1 have shown various effects on signaling triggering platelet activation: no activation, little activation or complete activation (40). In addition, the MAPK pathway generated by the activation of PAR1 via thrombin is not triggered by the SFLLRN-NH2 agonist peptide (41) unless the doses of agonist peptides used are significantly higher (100-fold) than the commonly used doses (42).
Regarding PAR2, here again, depending on the peptide tested, the results are not identical. Indeed, PAR2 activation via the SLAAAA agonist peptide results in intracellular calcium release, MAPK pathway signaling and receptor internalization (43), whereas the SLAAAA-NH2 agonist peptide only induces intracellular calcium release (44). An activator sequence, SLIGKV, resulting in intracellular calcium release in rat and human cell lines was then validated (45–47). Next, further studies have allowed to design a more potent PAR2 agonist peptide by adding a seventh or eighth amino acid, leucine type (48). However, although these agonist peptides are stable, they display low bioavailability and low solubility.
No PAR3 specific agonist peptides have been generated. Indeed, the peptides designed with that aim, such as TFRGAP-NH2, seem actually to activate PAR4. An explanation could be a PAR3 and PAR4 dimerization as described in response to thrombin (49, 50).
Regarding PAR4, the agonist peptide GYPGQV-NH2 specifically activates the receptor, causing contractility of the aorta and longitudinal gastric muscles in the rat (51).
PARs activation can be inhibited by disarming the receptor. Indeed, some proteases can prevent the canonical proteolytic cleavage by a proteolytic cleavage upstream of the activator ligand sequence of the receptor (Figure 2C). A second mechanism involves proteolytic cleavage within the receptor sequence to prevent signaling induction (52–54). For example, kallikrein 14 (KLK14), trypsin, cathepsin G, elastase, and plasmin disarm PAR1 (27, 31, 52, 55, 56). The disruption of PAR2 can be achieved by plasmin, PR3, elastase, and cathepsin G (57, 58).
PARs can also be activated through co-activation or transactivation. Indeed, the hirudin-like domain present on the PAR1 and PAR3 sequences allows increasing the affinity of these receptors for thrombin, helping in turns to activate PAR4, which does not have such a domain (Figure 3). One study reports that the only expression of PAR3 in COS7 cells does not induce any signaling in response to thrombin, while co-transfecting PAR4, allowed an inositol triphosphate signaling as if they only expressed PAR4, but at a lower dose of thrombin. PAR4 co-activation with PAR3 was then confirmed (49). PAR3 binds thrombin through its exosite I, allowing the active site of thrombin to remain free and to activate other PARs. PAR3 then changes the conformation of thrombin and increases its affinity for PAR4. This mechanism has been described by crystallography (59). Activation of PAR4 via PAR1 has also been confirmed. By FRET, a study evidenced the heterodimerization of PAR1 and PAR4 in response to thrombin, this heterodimerization leading to an increased platelet aggregation compared to the one induced by PAR4 alone (60).
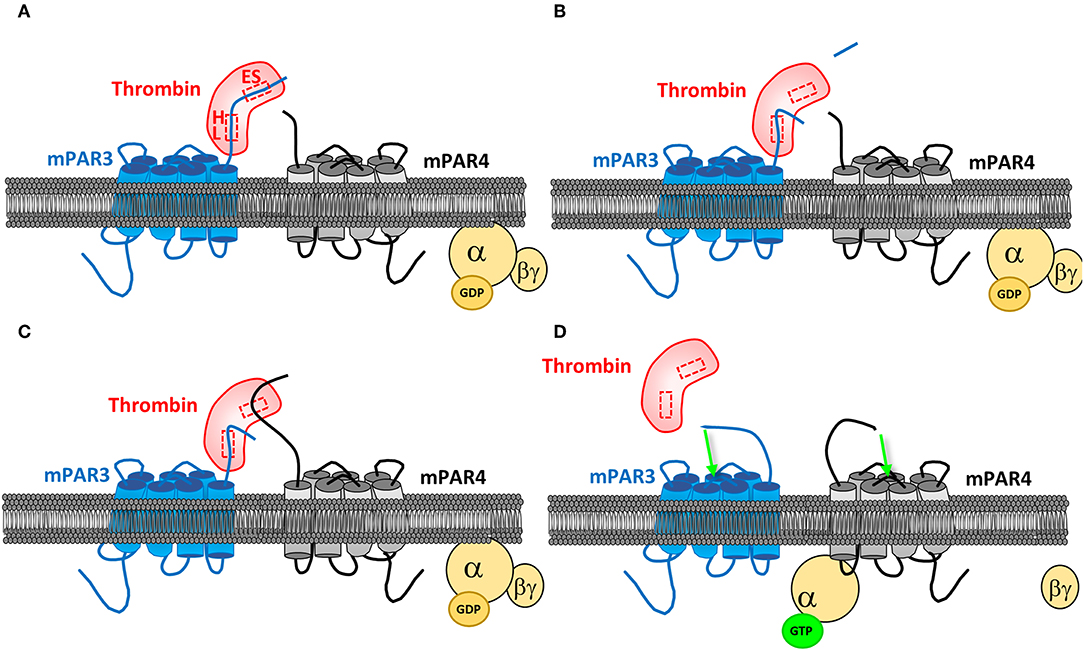
Figure 3. PAR4 co-activation by PAR3. (A) PAR3 binds thrombin active site via its cleavage site. PAR3 hirudin-like domain (HL) allows a more specific binding to the protease, on its exosite I (ES). (B) After PAR3 proteolytic cleavage by thrombin, which releases PAR3 N-terminal peptide, the receptor remains linked to the protease via the HL domain. (C) The active site of thrombin being free, it can bind to PAR4. (D) The PAR3:PAR4 heterodimerization results in a conformational change of both receptors and their activation, allowing them to couple to the G proteins and transduce signaling.
PAR1 can also be activated by a mechanism called transactivation (Figure 4). The first study to highlight this phenomenon dates back to 1996 (61). Blackhart et al. wanted to characterize the specificity of PAR1 and PAR2 ligands (SFLLRN and SLIGKV) by analyzing the cross-reactivity of these receptors. In view of their sequence similarities, the authors looked at whether the PAR2 agonist peptide could activate PAR1. The results confirmed their hypothesis. The second step was to see whether thrombin could activate PAR2 through PAR1, as it is the case with the PAR1 agonist peptide. In this aim, researchers have mutated PAR1 making sure that it is still able to bind to thrombin via the exosite I, but without the thrombin being able to cleave PAR1 and cause signaling. The mutated PAR1 was then co-expressed with PAR2 in human endothelial cells. This coexpression caused signaling, similar to that caused by the individual expression of PAR1 and PAR2 in endothelial cells, after stimulation with thrombin (16). In order to confirm the activation of PAR2 via PAR1, a PAR1 antagonist was used, resulting in the inhibition of 75% of PAR1-induced signaling. The fact that the signal could be completely blocked with the addition of a PAR2 antagonist further supported this mechanism of activation between the two receptors.
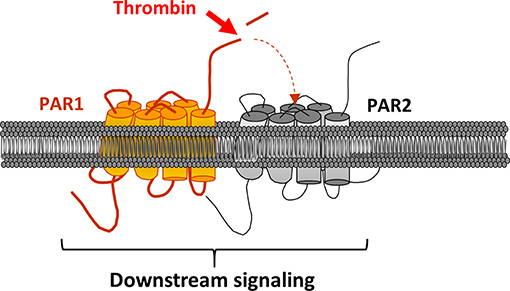
Figure 4. PAR2 transactivation via PAR1. PAR1 is clived and activated by thrombin. In turns, its activating ligand links PAR2 ECL2 leading to the receptor and downstream signaling activation.
In addition, this transactivation leads to ERK1/2 signaling (Extracellular signal-Regulated Kinases 1/2) and appears to be more prevalent during pathological events, such as sepsis in endothelial cells, chronic inflammation and carcinogenesis (62–64). FRET and co-immunoprecipitation approaches allowed to confirmed this mechanism of transactivation by heterodimerization (63).
Proteases, also known as proteinases and peptidases, are degradative enzymes for protein catabolism that hydrolyse a peptide bond to generate amino acids (65). The genes coding for proteases represent 2% of the mammalian genome and can be distributed in five groups depending on their mechanism of hydrolytic cleavage: serine-, metallo-, cysteine-, aspartate-, and threonine proteases. Serine-, Metallo-, and Cysteine-proteases are able to cleave and activate PARs, triggering different signaling pathways, although PAR4 is mainly activated by thrombin, while PAR2 is cleaved and activated by trypsin.
MMPs are a group of zinc- dependent endopeptidases known to degrade and remodel the components of the extracellular matrix. Depending on their substrate specificities, MMPs are subdivided into six groups: Collagenases, gelatinases, stromelysins, matrilysins, membrane-types MMPs, and non-classified MMPs. Besides the extracellular matrix turn over, MMPs are involved in other tissue maintenance functions, such as wound healing, and regulation of a broad range of molecules, such as chemokines, cytokines, growth factors, cytoskeleton, and junctional proteins (66). Dysregulation of MMP activity leads to the development of several pathologies including chronic inflammatory diseases as inflammatory Bowel Diseases (IBD) and cancer (67).
Theses proteases are enzymes that hydrolyze peptide bonds within the protein sequence, in which serine serves as nucleophilic amino acid at the active site. Serine proteases, the most abundant group of proteases, are widely distributed in nature and present in the three domains of life (archaea, bacteria, and eukaryotes) as well as in viral genomes (68). Widespread throughout the human body, serine protease are usually endopeptidases that hydrolyse the peptide bond in the middle of a polypeptide chain. However, some are exopeptidases cleaving only terminal amino acid residue. Mammalian serine proteases comprise matriptase, plasminogen activators, chymotrypsin, trypsin, and proteolytic enzymes produced by polymorphonuclear cells, such as cathepsin G and neutrophil elastase. Bacteria from commensal microbiota are also an important source of proteases present in the GI tract.
Most are found intracellularly. Besides their fundamental functions of catabolism and protein processing, cysteine proteases mediate other signaling pathways involved in programmed cell death, inflammation and intestinal mucosa integrity (epithelium turnover and homeostasis) (69, 70). Cysteine group comprises caspases, autophagins, calpains, and deubiquitinases intracellularly, and cathepsins B, K, and L extracellularly (9) Altered activity of cysteine proteases is associated with IBD (71).
As mentioned above, the colon is highly exposed to proteases, whether pancreatic, bacterial proteases from resident colon cells or proteases produced and secreted by epithelial cells (72). These proteases intervene in a multitude of physiological and pathological processes, through activation of PARs (72). Although the roles of proteases in the colonic epithelium are not fully understood, PARs have been extensively studied with agonist peptides, even if these peptides do not always reflect in a precise way the functions of the PARs in the physiology and the pathology.
In the colon, smooth muscle cells, endothelial cells, enteric neurons, fibroblasts and immune cells, such as neutrophils, lymphocytes, macrophages, express PAR1. Originally, PAR1 expression was only reported in cancerous colon epithelial cells, but not in normal epithelial cells (4). An explanation is that, at that time, no anti-PAR1 antibody was effective. Since then, our team has described its, and PAR2, expression in normal human and murine colonic epithelium by immunostaining (73). The expression of PAR3 is poorly studied and has never been reported in the colon. PAR4 is expressed in colonic epithelial cells, at the submucosa level (10).
One study demonstrated that PAR1 activation on intact cultured monolayers of intestinal epithelial cells in Using chambers resulted in chloride ion release, whereas in intact tissues, PAR1 activation would not result in this release (74). This difference between cultured cell monolayer and tissue would emphasize the importance of the location of the receptor on the cell. Indeed, it is thought that the activation of PAR1 on the apical side (toward the intestinal lumen), would have a protective role, generating a release of chloride ions, allowing a faster transit and the elimination of pathogens or at least of their toxins. Activation of PAR2 also promotes the secretion of chloride ions and is correlated with diarrhea in inflammatory conditions, as found in the tissues of IBD patients (75). In addition, other elements, such as amylase and mucin, are secreted in response to PAR2 activation (52).
Another colon function is to allow the transit of chime. This involves a regulation of the intestinal motility. The role of PAR1 and PAR2 in this process was demonstrated by the stimulation of circular and longitudinal rat colonic muscle layers with either thrombin, trypsin or agonist peptides. This study showed that both PAR1 and PAR2 activations result in muscle contraction and relaxation (76). The same authors demonstrated that the nervous system is involved in this regulation by secreting tachykinins (77).
PAR1 and PAR2 play a role in nociception. Thus, several studies have reported that colorectal distension performed by intracolonic administration of trypsin and PAR2 agonist peptide in rats caused visceral pain (78). PAR2 would have a pro-nociceptive role. Regarding PAR1, its role seems to be anti-nociceptive. Indeed, a study showed that the injection of PAR1 agonist into the mouse paw does not induce a response to a mechanical or thermal stimulus, but still increases the sensitivity threshold of the perception of pain (79). In addition, PAR1 would inhibit the transmission of nociceptive signals (80). Intracolonic administration of PAR4 agonist peptide decreased visceral motility, after colorectal distention and decreased pain induced by PAR2 activation (81).
Activation of PARs present in epithelial cells leads to changes in paracellular permeability. Activation of PAR1, in response an agonist peptide, increases the intestinal permeability via the apoptotic process through caspase 3, and an alteration of ZO1 expression (82). Moreover, neutrophil proteases, such as elastase and PR3, via the basolateral activation of PAR1 and PAR2, can also modulate the intestinal permeability (83). Cenac et al. have shown that in mouse model, PAR2 activation by trypsin, tryptase, and chymase (all from serine proteases family) promotes an increase in colonic permeability displaying inflammation and disruption on the intestinal barrier integrity (84). These results have been supported by other studies, which have shown an alteration on the intestinal permeability using PAR2 agonists (78, 85). The mechanism of action through PAR2-mediated modification of intestinal permeability involved the calmodulin and MLCK. PAR2 agonist, via the calmodulin, increases MLCK phosphorylation, which leads to epithelial cell cytoskeleton contraction and the enhancement of the mucosal permeability. ML-7, an MLCK inhibitor, abolished the disruption of the tight junctions composition and function (86). Another study revealed that activation of ERK1/2 by tryptase, in cultured colonocytes, also phosphorylates MLCK, leading to epithelial cells disruption (87). Trypsin-3, released by intestinal epithelial cells in response to LPS, is able to cleave and activate PAR2 to increase the intestinal permeability (88). Thus, these results also highlight the involvement of PARs in intestinal inflammation.
PAR1 and PAR2 have been both described involved in the stimulation of colorectal cancer cell proliferation (4, 89). In addition, activation of PAR2 leads to reduced apoptosis of colonic epithelial cells by activating MEK1/2 and PI3K (90). PAR2 activation also regulates the survival of colonic stem cells in the organoid model via the GSK3β pathway (73). More recently, in human colon organoid cultures, we reported that thrombin significantly reduces the size of budding structures, metabolic activity and proliferation, while increasing apoptosis. In the same study, we reported that both PAR1 and PAR4 antagonists inhibited apoptosis regardless of thrombin doses (91).
The IBD, Crohn's disease (CD) and ulcerative colitis (UC), are chronic diseases causing inflammation of the gut (92). Since the second part of the last century, IBD has emerged as a public health challenge worldwide. In North America and Europe, more than 1.5 million people exhibit these pathologies, respectively (93). Outside the industrialized countries, the number of people affected by IBD remains unclear. Recently, the highest reported prevalence values for UC and CD were reported in Europe and North America (94). Although the molecular mechanisms of IBD are poorly understood, recent data suggest that IBD occurs in genetically predisposed individuals developing an abnormal immune response to intestinal microbes after being exposed to specific environmental triggers (95). Moreover, stools from IBD patients showed increased levels of active proteases, which are secreted meanly by infiltrated and resident cells, intestinal epithelial cells or smooth muscle (7). In addition, increased fecal proteases in IBD might result from both commensal and pathogenic gut bacteria, which can secrete serine proteases, cysteine proteases and MMPs (96–98).
In the gut, PARs are stimulated by endogenous proteases, such as pancreas trypsin, cells of the intestinal mucosa (immune cells including mast cells, epithelial cells including goblet, neuroendocrine, and enterocyte cells), or gut microbiota. Moreover, PARs expressions in the intestinal epithelium are different between IBD patients and healthy individuals. Colonic biopsies from UC and CD patients exhibited increased expression PAR1, while PAR2 and PAR4 are just upregulated in UC conditions. Enhanced levels of these receptors is linked to its activation-internalization-degradation signaling induced by proteases released from eukaryotic host cells but also from gut micro-organisms. Indeed, on neutrophil cells, Candida albicans induces a TLR2-dependent PAR1 stimulation and expression, while Aspergillus fumigatus inhibits a TLR4-dependent PAR2 activation and expression (99).
PAR1 stimulation triggers apoptosis of the epithelial cells within the gut mucosa through a mechanism involving caspase-3 activation. This excessive apoptosis is associated to a disruption of the intestinal barrier function, promoting then the development and/or severity of the colitis (82). PAR1 is expressed by intestinal epithelial cells but also by endothelial cells, enteric neurons, myocytes, and immune cells (52). PAR1 expression by intestinal epithelium is linked to the presence of microbiota (100), and its colonic stimulation leads to colitis (10, 101). Moreover, deletion or blockage of PAR1 reduce inflammatory signs and mortality in a murine model of IBD (102). In addition to an increased PAR1 expression in IBD patient colons, it has been recently shown that thrombin level is increased in CD colonic biopsies (103). Moreover, an increased incidence of thrombosis has been observed in IBD patients (104). Altogether, these studies evidence a potential role for PAR1 in IBD pathophysiology, however it is still not clear whether the intestinal bacteria directly activate PAR1 through the release of proteases. Nevertheless, this notion has been evidenced by a study showing that a cysteine protease released by Porphyromonas gingivalis increases the expression of pro-inflammatory cytokines through PAR1 activation (105).
PAR2 receptor is localized at the apical and basolateral membranes (84, 106, 107) of the gut epithelium cells and can be stimulated by trypsin, tryptase, and bacterial proteases (108). PAR2 is also present in the cells membrane of numerous immune cells, stromal cells or endothelial cells. Thus, PAR2-associated inflammation coming from numerous pathways from either systemic or local locations. Systemically, PAR2 stimulation promotes the rolling, adhesion and extravasation of leukocytes (109). Locally at colonic level, activation of this receptor triggers colitis (7). Additionally, blockage of PAR2 activation reduces the severity of the colitis induced by either TNBS or a PAR2 agonist (110). All together, the majority of the studies show that stimulation of PAR2 triggers an inflammatory response. Nevertheless, one study has evidenced a protective effect of chronic stimulation of PAR2 in a model of colitis (111). This protective effect might be the result of a local desensitization, or anti-inflammatory effects on macrophages (112). Additionally, although we do not know which of the numerous mechanisms described for PAR2 stimulation in the intestine triggers colitis in rodent models, excessive PAR2 stimulation by trypsin and tryptase has been speculated to mediate colitis in IBD patients. Moreover, in IBD, PAR2 expression is increased at the membrane of mast cells, then participating in PAR2-induced colitis (113). Thus, although, these studies have highlighted the major role played by PAR-2 in the colitis mediated by mast cells, recent papers have also reported that proteases coming from gut micro-organisms could participate to PAR2 activation altering then colonic homeostasis (96). PAR2 can be also stimulated by gut micro-organisms either directly by bacterial proteases, as demonstrated for P. gingivalis (108) or C. difficile (114), or indirectly by the release of host cells proteases triggered by bacteria stimulation (37). Moreover, as antibiotic treatment diminished intestinal PAR2 expression, this suggests that, in addition to its activation, PAR2 expression can also be regulated by the gut microbiota (115).
Altogether, these studies report the role for PAR2 in IBD pathophysiology. However, the most important source of the proteases activating PAR2 to promote IBD is largely unclear.
The biological importance of PAR3 is not fully demonstrated. Thus, PAR3 does not exhibit, as the other PARs, a C-terminal intracytoplasmic tail. However, as described previously, PAR3 could play a role as co-factor or co-receptor for PARs and/or other receptors. Although PAR3 mRNA has been evidenced in the gut, no study has reported its involvement in intestinal inflammation (18).
PAR4 expression has been detected in the gut (19) and on colonocytes (116). It can be cleaved and then activated by several proteases including thrombin, trypsin and by the neutrophil granule protease cathepsin G (24). PAR4 stimulation on leukocyte has been reported to promote their rolling and adherence, then suggesting a pro-inflammatory role (79). Moreover, colonic exposure to PAR4 agonists increases the paracellular permeability of the colic epithelium, suggesting that PAR4 could favor the genesis of IBD (117). In human colon, PAR4 expression is very weak in non-IBD patients, while its expression is drastically increased in UC patients. Interestingly, cathepsin G activity is enhanced in fecal supernatant from UC individuals compared to controls (117). Moreover, inhibition of this cathepsin G activity resulted in a restored gut paracellular permeability (117). Thus, cathepsin G, via PAR4 receptor, could participate in the increase of the gut permeability in UC patients.
Worldwide, colorectal cancer is the second and the third most commonly occurring cancer in women and men, respectively. There were over 1.8 million new cases detected in 2018 (GLOBOCAN database 2018, http://gco.iarc.fr/). CRC is the fourth most common cause of death from cancer in the industrialized world. The survival depends on the stage of the pathology at the time of the diagnosis. Indeed, patients with the later-stage diagnosis have the poorer survival. Indeed, the 5-years survival rate is 90% for colorectal cancers diagnosed at an early stage but drops down to 13% for those diagnosed at a late stage.
Since a long time now, proteases have been associated with tumor progression mainly due to their ability to degrade the extracellular matrix (ECM), favoring thus tumor cell invasion and metastasis process (118). However, it is now well-established that their roles in cancer is not only restricted to their ECM degradation capacities, these enzymes acting directly on the cancer cells via the PARs.
Evidence that thrombin potentiated the tumor growth and the metastatic process in vivo was obtained in 1991. Nierodzik et al. treated cancer cell lines, including the murine carcinoma cell line CT26, with thrombin (0.5–1 U/mL). They then injected these cells intravenously in the mouse and observed an increase in the metastatic power of these cells (119). Nevertheless, a study shows that thrombin concentration may have opposite effects on tumor cells (120). Indeed, at a low dose of thrombin, 0.1–0.5 U/mL, PAR1 activation resulted in the growth of tumor cells. Whereas, at a higher dose of thrombin, 0.5–1 U/ml, growth was decreased in favor of cell apoptosis. Since, it has been demonstrated that PAR1 plays a direct role in the progression of epithelial colon tumors, regulating both cell proliferation and migration (4, 121, 122) have pro- and anti-apoptotic effects, depending on the dose of thrombin or agonist peptides used (123).
Patients with IBD are 10–20 times more likely to develop CRC (124). One study revealed the role of thrombin in the development of colon cancer from an inflammatory context. Using the murine DSS-induced colitis model and the colitis-associated cancer model treating the mice with DSS and AOM, the authors observed the formation of adenomas. The result obtained was a uniform development of adenomas from aberrant crypts in control mice (Factor II+/+ mice) and, in Factor II +/− mice, a decrease in these precancerous lesions as well as the number of adenomas (125). The involvement of thrombin in cancer development from a previous inflammatory state has been proven by treating the mice with hirudin, an inhibitor of thrombin, these mice displaying then a slower adenoma development. However, a recent study reports that PAR1-deficient APCMin/+ mice display an increased number of adenomas and larger adenomas than PAR1-expressing mice suggesting a protecting role of PAR1 in some CRC favoring context (126). Actually, the same authors already described earlier that the growth of colonic adenocarcinoma in PAR-1-deficient mice was decreased compared to control animals, and proposed an implication of the stromal cell-associated PAR-1 as target important for tumor development (127).
Regarding the role of PAR2, several human colon cancer cell lines, namely T84, Caco-2, HT-29, and C1.19A, produce and secrete trypsin at concentrations compatible with PAR2 activation, supporting the idea of a possible autocrine/paracrine regulation of PAR2 activity by trypsin in colon cancer cells (128). Trypsin is produced by human cancer colon cells and activates protease-activated receptor-2 within these cells, depicting an autocrine loop. Interestingly, EGF-R transactivation by PAR2 results in the growth of colon cancer cells after the activation of a Src/ERK1/2 pathway (85).
More recently, we reported in three-dimensional cultures of murine colorectal crypt and in Caco-2 cells, that PAR2 activation decreases the numbers and the size of normal or cancerous spheroids. Spheroids deficient for display an increased proliferation, suggesting that cell proliferation is repressed by PAR2. However, in the same study, PAR2-stimulated normal cells are more resistant to stress, suggesting PAR2 pro-survival roles. Indeed, PAR2-deficient normal spheroids display an increase of active caspase-3. Moreover, we showed that PAR2, but not PAR1, was able to trigger GSK3β activation in normal and tumor cells. The PAR2-triggered GSK3β activation involves an arrestin/PP2A/GSK3β complex that is dependent on the activity of the Rho kinase. Finally, the survival of PAR2-stimulated cultures can be pharmacologically inhibited using a GSK3 inhibitor. This study highlighted the PAR2/GSK3β pathway as a novel critical player in the regulation of stem/progenitor cell survival and proliferation in normal colon crypts and colon cancer (73).
To date, no clear role of PAR3 in CRC has been reported.
Regarding PAR4, its expression is increased in colorectal cancer tissues compared to the associated normal tissues. This overexpression seems to promote colorectal cancer cell proliferation, survival and metastasis, making PAR4 a potential therapeutic target in CRC (129).
PAR4 mediates thrombin effects on human colon cancer cells. AP4, a specific PAR4 agonist, mimics the effects of thrombin on cell proliferation. Its effects on calcium mobilization in CHO-PAR4-expressing cells are similar to the one observed in HT-29 cells, while HT-29 cells treatment with a reverse peptide has no effect on calcium mobilization. Finally, AP4 promotes colon cancer cell proliferation. More recently, PAR4 overexpression in LoVo cells has been shown, via activation of the ERK1/2 pathway, to increase their proliferation and migration and tumorigenesis capacities, while its knock-down in HT29 results in opposite effect (129). Thus, PAR4 should be regarded as a crucial receptor by which thrombin modulates colon carcinogenesis (130).
Since their discovery in the early nineties, PARs have been shown to be largely involved in the regulation of the intestine physiological processes, but also in the two main diseases affecting the organ, namely inflammatory bowel diseases and colorectal cancer. Consequently, these G-protein coupled receptors represent attractive targets for therapeutic drug development. More than aiming to target their ligands, efforts to develop specific receptor inhibitors are currently regarded as a priority although to date, the development of effective PAR antagonist yet remains in its early stages.
MS, NS-T, FB, and AF wrote the manuscript. EM did a careful reading and review of the manuscript before its submission.
INSERM, Direction Générale des Offres de Soins (DGSO), Association François Aupetit, Rotary France.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Hyun E, Andrade-Gordon P, Steinhoff M, Vergnolle N. Protease-activated receptor-2 activation: a major actor in intestinal inflammation. Gut. (2008) 57:1222–9. doi: 10.1136/gut.2008.150722
2. Vergnolle N, Bunnett NW, Sharkey KA, Brussee V, Compton SJ, Grady EF, et al. Proteinase-activated receptor-2 hyperalgesia: a novel pain pathway. Nat Med. (2001) 7:821–6. doi: 10.1038/89945
3. Dale C. Vergnolle N. Protease signaling to G protein-coupled receptors: implications for inflammation and pain. J Recept Signal Transduct Res. (2008) 28:29–37. doi: 10.1080/10799890801941913
4. Darmoul D, Gratio V, Devaud H, Lehy T, Laburthe M. Aberrant expression and activation of the thrombin receptor protease-activated receptor-1 induces cell proliferation and motility in human colon cancer cells. Am J Pathol. (2003) 162:1503–13. doi: 10.1016/S0002-9440(10)64283-6
5. Gratio V, Walker F, Lehy T, Laburthe M, Darmoul D. Aberrant expression of proteinase-activated receptor 4 promotes colon cancer cell proliferation through a persistent signaling that involves Src ErbB-2 kinase. Int J Cancer. (2009) 124:1517–25. doi: 10.1002/ijc.24070
6. Trusevych EH. MacNaughton WK. Proteases and their receptors as mediators of inflammation-associated colon cancer. Curr Pharm Des. (2015) 21:2983–92. doi: 10.2174/1381612821666150514104800
7. Vergnolle N. Proteinase-activated receptors (PARs) in infection and inflammation in the gut. Int J Biochem Cell Biol. (2008) 40:1219–27. doi: 10.1016/j.biocel.2008.01.016
8. Amadesi S, Bunnett N. Protease-activated receptors: protease signaling in the gastrointestinal tract. Curr Opin Pharmacol. (2004) 4:551–6. doi: 10.1016/j.coph.2004.08.004
9. Vergnolle N. Protease inhibition as new therapeutic strategy for GI diseases. Gut. (2016) 65:1215–24. doi: 10.1136/gutjnl-2015-309147
10. Vergnolle N. Clinical relevance of proteinase activated receptors (PARs) in the gut. Gut. (2005) 54:867–74. doi: 10.1136/gut.2004.048876
11. Hollenberg MD, Compton SJ. International Union of Pharmacology. XXVIII. Proteinase-activated receptors. Pharmacol Rev. (2002) 54:203–17. doi: 10.1124/pr.54.2.203
12. Ossovskaya VS, Bunnett NW. Protease-activated receptors: contribution to physiology and disease. Physiol Rev. (2004) 84:579–621. doi: 10.1152/physrev.00028.2003
13. Vu TK, Hung DT, Wheaton VI, Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. (1991) 64:1057–68. doi: 10.1016/0092-8674(91)90261-V
14. Vu TK, Wheaton VI, Hung DT, Charo I, Coughlin SR. Domains specifying thrombin-receptor interaction. Nature. (1991) 353:674–7. doi: 10.1038/353674a0
15. Nystedt S, Emilsson K, Wahlestedt C, Sundelin J. Molecular cloning of a potential proteinase activated receptor. Proc Natl Acad Sci USA. (1994) 91:9208–12. doi: 10.1073/pnas.91.20.9208
16. O'Brien PJ, Prevost N, Molino M, Hollinger MK, Woolkalis MJ, Woulfe DS, et al. Thrombin responses in human endothelial cells. Contributions from receptors other than PAR1 include the transactivation of PAR2 by thrombin-cleaved PAR1. J Biol Chem. (2000) 275:13502–9. doi: 10.1074/jbc.275.18.13502
17. Mihara K, Ramachandran R, Saifeddine M, Hansen KK, Renaux B, Polley D, et al. Thrombin-mediated direct activation of proteinase-activated receptor-2: another target for thrombin signaling. Mol Pharmacol. (2016) 89:606–14. doi: 10.1124/mol.115.102723
18. Ishihara H, Connolly AJ, Zeng D, Kahn ML, Zheng YW, Timmons C, et al. Protease-activated receptor 3 is a second thrombin receptor in humans. Nature. (1997) 386:502–6. doi: 10.1038/386502a0
19. Xu WF, Andersen H, Whitmore TE, Presnell SR, Yee DP, Ching A, et al. Cloning and characterization of human protease-activated receptor 4. Proc Natl Acad Sci USA. (1998) 95:6642–6. doi: 10.1073/pnas.95.12.6642
20. Kahn ML, Hammes SR, Botka C, Coughlin SR. Gene and locus structure and chromosomal localization of the protease-activated receptor gene family. J Biol Chem. (1998) 273:23290–6. doi: 10.1074/jbc.273.36.23290
21. Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. (2000) 407:258–64. doi: 10.1038/35025229
22. Macfarlane SR, Seatter MJ, Kanke T, Hunter GD, Plevin R. Proteinase-activated receptors. Pharmacol Rev. (2001) 53:245–82.
23. Molino M, Barnathan ES, Numerof R, Clark J, Dreyer M, Cumashi A, et al. Interactions of mast cell tryptase with thrombin receptors and PAR-2. J Biol Chem. (1997) 272:4043–9. doi: 10.1074/jbc.272.7.4043
24. Sambrano GR, Huang W, Faruqi T, Mahrus S, Craik C, Coughlin SR. Cathepsin G activates protease-activated receptor-4 in human platelets. J Biol Chem. (2000) 275:6819–23. doi: 10.1074/jbc.275.10.6819
25. Camerer E, Kataoka H, Kahn M, Lease K, Coughlin SR. Genetic evidence that protease-activated receptors mediate factor Xa signaling in endothelial cells. J Biol Chem. (2002) 277:16081–7. doi: 10.1074/jbc.M108555200
26. Quinton TM, Kim S, Derian CK, Jin J, Kunapuli SP. Plasmin-mediated activation of platelets occurs by cleavage of protease-activated receptor 4. J Biol Chem. (2004) 279:18434–9. doi: 10.1074/jbc.M401431200
27. Oikonomopoulou K, Hansen KK, Saifeddine M, Vergnolle N, Tea I, Blaber M, et al. Kallikrein-mediated cell signalling: targeting proteinase-activated receptors (PARs). Biol Chem. (2006) 387:817–24. doi: 10.1515/BC.2006.104
28. Boire A, Covic L, Agarwal A, Jacques S, Sherifi S, Kuliopulos A. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell. (2005) 120:303–13. doi: 10.1016/j.cell.2004.12.018
29. Russo A, Soh UJ, Paing MM, Arora P, Trejo J. Caveolae are required for protease-selective signaling by protease-activated receptor-1. Proc Natl Acad Sci USA. (2009) 106:6393–7. doi: 10.1073/pnas.0810687106
30. Suidan HS, Bouvier J, Schaerer E, Stone SR, Monard D, Tschopp J. Granzyme A released upon stimulation of cytotoxic T lymphocytes activates the thrombin receptor on neuronal cells and astrocytes. Proc Natl Acad Sci USA. (1994) 91:8112–6. doi: 10.1073/pnas.91.17.8112
31. Kuliopulos A, Covic L, Seeley SK, Sheridan PJ, Helin J, Costello CE. Plasmin desensitization of the PAR1 thrombin receptor: kinetics, sites of truncation, and implications for thrombolytic therapy. Biochemistry. (1999) 38:4572–85. doi: 10.1021/bi9824792
32. Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science. (2002) 296:1880–2. doi: 10.1126/science.1071699
33. Knecht W, Cottrell GS, Amadesi S, Mohlin J, Skaregarde A, Gedda K, et al. Trypsin IV or mesotrypsin and p23 cleave protease-activated receptors 1 and 2 to induce inflammation and hyperalgesia. J Biol Chem. (2007) 282:26089–100. doi: 10.1074/jbc.M703840200
34. Wilson TJ, Nannuru KC, Futakuchi M, Singh RK. Cathepsin G-mediated enhanced TGF-beta signaling promotes angiogenesis via upregulation of VEGF and MCP-1. Cancer Lett. (2010) 288:162–9. doi: 10.1016/j.canlet.2009.06.035
35. Gao L, Chao L, Chao J. A novel signaling pathway of tissue kallikrein in promoting keratinocyte migration: activation of proteinase-activated receptor 1 and epidermal growth factor receptor. Exp Cell Res. (2010) 316:376–89. doi: 10.1016/j.yexcr.2009.10.022
36. Ramachandran R, Mihara K, Chung H, Renaux B, Lau CS, Muruve DA, et al. Neutrophil elastase acts as a biased agonist for proteinase-activated receptor-2 (PAR2). J Biol Chem. (2011) 286:24638–48. doi: 10.1074/jbc.M110.201988
37. Hansen KK, Sherman PM, Cellars L, Andrade-Gordon P, Pan Z, Baruch A, et al. A major role for proteolytic activity and proteinase-activated receptor-2 in the pathogenesis of infectious colitis. Proc Natl Acad Sci USA. (2005) 102:8363–8. doi: 10.1073/pnas.0409535102
38. Scarborough RM, Naughton MA, Teng W, Hung DT, Rose J, Vu TK, et al. Tethered ligand agonist peptides. Structural requirements for thrombin receptor activation reveal mechanism of proteolytic unmasking of agonist function. J Biol Chem. (1992) 267:13146–9.
39. Feng DM, Veber DF, Connolly TM, Condra C, Tang MJ, Nutt RF. Development of a potent thrombin receptor ligand. J Med Chem. (1995) 38:4125–30. doi: 10.1021/jm00020a029
40. Mao Y, Jin J, Kunapuli SP. Characterization of a new peptide agonist of the protease-activated receptor-1. Biochem Pharmacol. (2008) 75:438–47. doi: 10.1016/j.bcp.2007.09.002
41. Vouret-Craviari V, Van Obberghen-Schilling E, Rasmussen UB, Pavirani A, Lecocq JP, Pouyssegur J. Synthetic alpha-thrombin receptor peptides activate G protein-coupled signaling pathways but are unable to induce mitogenesis. Mol Biol Cell. (1992) 3:95–102. doi: 10.1091/mbc.3.1.95
42. Blackhart BD, Ruslim-Litrus L, Lu CC, Alves VL, Teng W, Scarborough RM, et al. Extracellular mutations of protease-activated receptor-1 result in differential activation by thrombin and thrombin receptor agonist peptide. Mol Pharmacol. (2000) 58:1178–87. doi: 10.1124/mol.58.6.1178
43. Al-Ani B, Hansen KK, Hollenberg MD. Proteinase-activated receptor-2: key role of amino-terminal dipeptide residues of the tethered ligand for receptor activation. Mol Pharmacol. (2004) 65:149–56. doi: 10.1124/mol.65.1.149
44. Ramachandran R, Mihara K, Mathur M, Rochdi MD, Bouvier M, Defea K, et al. Agonist-biased signaling via proteinase activated receptor-2: differential activation of calcium and mitogen-activated protein kinase pathways. Mol Pharmacol. (2009) 76:791–801. doi: 10.1124/mol.109.055509
45. Maryanoff BE, Santulli RJ, McComsey DF, Hoekstra WJ, Hoey K, Smith CE, et al. Protease-activated receptor-2 (PAR-2): structure-function study of receptor activation by diverse peptides related to tethered-ligand epitopes. Arch Biochem Biophys. (2001) 386:195–204. doi: 10.1006/abbi.2000.2207
46. Kawabata A, Kanke T, Yonezawa D, Ishiki T, Saka M, Kabeya M, et al. Potent and metabolically stable agonists for protease-activated receptor-2: evaluation of activity in multiple assay systems in vitro and in vivo. J Pharmacol Exp Ther. (2004) 309:1098–107. doi: 10.1124/jpet.103.061010
47. McGuire JJ Proteinase-activated Receptor 2 (PAR2): a challenging new target for treatment of vascular diseases. Curr Pharm Des. (2004) 10:2769–78. doi: 10.2174/1381612043383656
48. Barry GD, Suen JY, Low HB, Pfeiffer B, Flanagan B, Halili M, et al. A refined agonist pharmacophore for protease activated receptor 2. Bioorg Med Chem Lett. (2007) 17:5552–7. doi: 10.1016/j.bmcl.2007.08.026
49. Nakanishi-Matsui M, Zheng YW, Sulciner DJ, Weiss EJ, Ludeman MJ, Coughlin SR. PAR3 is a cofactor for PAR4 activation by thrombin. Nature. (2000) 404:609–13. doi: 10.1038/35007085
50. McLaughlin JN, Patterson MM, Malik AB. Protease-activated receptor-3 (PAR3) regulates PAR1 signaling by receptor dimerization. Proc Natl Acad Sci USA. (2007) 104:5662–7. doi: 10.1073/pnas.0700763104
51. Hollenberg MD, Saifeddine M, Al-Ani B, Gui Y. Proteinase-activated receptor 4 (PAR4): action of PAR4-activating peptides in vascular and gastric tissue and lack of cross-reactivity with PAR1 and PAR2. Can J Physiol Pharmacol. (1999) 77:458–64. doi: 10.1139/y99-090
52. Kawabata A, Kuroda R, Nishikawa H, Kawai K. Modulation by protease-activated receptors of the rat duodenal motility in vitro: possible mechanisms underlying the evoked contraction and relaxation. Br J Pharmacol. (1999) 128:865–72. doi: 10.1038/sj.bjp.0702755
53. Hansen KK, Oikonomopoulou K, Baruch A, Ramachandran R, Beck P, Diamandis EP, et al. Proteinases as hormones: targets and mechanisms for proteolytic signaling. Biol Chem. (2008) 389:971–82. doi: 10.1515/BC.2008.120
54. Ramachandran R, Hollenberg MD. Proteinases and signalling: pathophysiological and therapeutic implications via PARs and more. Br J Pharmacol. (2008) 153(Suppl. 1):S263–82. doi: 10.1038/sj.bjp.0707507
55. Molino M, Blanchard N, Belmonte E, Tarver AP, Abrams C, Hoxie JA, et al. Proteolysis of the human platelet and endothelial cell thrombin receptor by neutrophil-derived cathepsin G. J Biol Chem. (1995) 270:11168–75. doi: 10.1074/jbc.270.19.11168
56. Renesto P, Si-Tahar M, Moniatte M, Balloy V, Van Dorsselaer A, Pidard D, et al. Specific inhibition of thrombin-induced cell activation by the neutrophil proteinases elastase, cathepsin G, and proteinase 3: evidence for distinct cleavage sites within the aminoterminal domain of the thrombin receptor. Blood. (1997) 89:1944–53.
57. Loew D, Perrault C, Morales M, Moog S, Ravanat C, Schuhler S, et al. Proteolysis of the exodomain of recombinant protease-activated receptors: prediction of receptor activation or inactivation by MALDI mass spectrometry. Biochemistry. (2000) 39:10812–22. doi: 10.1021/bi0003341
58. Dulon S, Cande C, Bunnett NW, Hollenberg MD, Chignard M, Pidard D. Proteinase-activated receptor-2 and human lung epithelial cells: disarming by neutrophil serine proteinases. Am J Respir Cell Mol Biol. (2003) 28:339–46. doi: 10.1165/rcmb.4908
59. Bah A, Chen Z, Bush-Pelc LA, Mathews FS, Di Cera E. Crystal structures of murine thrombin in complex with the extracellular fragments of murine protease-activated receptors PAR3 and PAR4. Proc Natl Acad Sci USA. (2007) 104:11603–8. doi: 10.1073/pnas.0704409104
60. Leger AJ, Jacques SL, Badar J, Kaneider NC, Derian CK, Andrade-Gordon P, et al. Blocking the protease-activated receptor 1-4 heterodimer in platelet-mediated thrombosis. Circulation. (2006) 113:1244–54. doi: 10.1161/CIRCULATIONAHA.105.587758
61. Blackhart BD, Emilsson K, Nguyen D, Teng W, Martelli AJ, Nystedt S, et al. Ligand cross-reactivity within the protease-activated receptor family. J Biol Chem. (1996) 271:16466–71. doi: 10.1074/jbc.271.28.16466
62. Shi X, Gangadharan B, Brass LF, Ruf W, Mueller BM. Protease-activated receptors (PAR1 and PAR2) contribute to tumor cell motility and metastasis. Mol Cancer Res. (2004) 2:395–402.
63. Kaneider NC, Leger AJ, Agarwal A, Nguyen N, Perides G, Derian C, et al. 'Role reversal' for the receptor PAR1 in sepsis-induced vascular damage. Nat Immunol. (2007) 8:1303–12. doi: 10.1038/ni1525
64. Lin H, Trejo J. Transactivation of the PAR1-PAR2 heterodimer by thrombin elicits beta-arrestin-mediated endosomal signaling. J Biol Chem. (2013) 288:11203–15. doi: 10.1074/jbc.M112.439950
65. Lopez-Otin C, Bond JS. Proteases: multifunctional enzymes in life and disease. J Biol Chem. (2008) 283:30433–7. doi: 10.1074/jbc.R800035200
66. Rodriguez D, Morrison CJ, Overall CM. Matrix metalloproteinases: what do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim Biophys Acta. (2010) 1803:39–54. doi: 10.1016/j.bbamcr.2009.09.015
67. Louis E, Ribbens C, Godon A, Franchimont D, De Groote D, Hardy N, et al. Increased production of matrix metalloproteinase-3 and tissue inhibitor of metalloproteinase-1 by inflamed mucosa in inflammatory bowel disease. Clin Exp Immunol. (2000) 120:241–6. doi: 10.1046/j.1365-2249.2000.01227.x
68. Page MJ, Di Cera E. Serine peptidases: classification, structure and function. Cell Mol Life Sci. (2008) 65:1220–36. doi: 10.1007/s00018-008-7565-9
69. Ruemmele FM, Seidman EG, Lentze MJ. Regulation of intestinal epithelial cell apoptosis and the pathogenesis of inflammatory bowel disorders. J Pediatr Gastroenterol Nutr. (2002) 34:254–60. doi: 10.1097/00005176-200203000-00005
70. Tamhane T, Lllukkumbura R, Lu S, Maelandsmo GM, Haugen MH, Brix K. Nuclear cathepsin L activity is required for cell cycle progression of colorectal carcinoma cells. Biochimie. (2016) 122:208–18. doi: 10.1016/j.biochi.2015.09.003
71. Menzel K, Hausmann M, Obermeier F, Schreiter K, Dunger N, Bataille F, et al. Cathepsins B, L and D in inflammatory bowel disease macrophages and potential therapeutic effects of cathepsin inhibition in vivo. Clin Exp Immunol. (2006) 146:169–80. doi: 10.1111/j.1365-2249.2006.03188.x
72. Bustos D, Negri G, De Paula JA, Di Carlo M, Yapur V, Facente A, et al. Colonic proteinases: increased activity in patients with ulcerative colitis. Medicina (B Aires). (1998) 58:262–4.
73. Nasri I, Bonnet D, Zwarycz B, d'Aldebert E, Khou S, Mezghani-Jarraya R, et al. PAR2-dependent activation of GSK3beta regulates the survival of colon stem/progenitor cells. Am J Physiol Gastrointest Liver Physiol. (2016) 311:G221–36. doi: 10.1152/ajpgi.00328.2015
74. Buresi MC, Schleihauf E, Vergnolle N, Buret A, Wallace JL, Hollenberg MD, et al. Protease-activated receptor-1 stimulates Ca2+-dependent Cl− secretion in human intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. (2001) 281:G323–32. doi: 10.1152/ajpgi.2001.281.2.G323
75. Lau C, Lytle C, Straus DS, DeFea KA. Apical and basolateral pools of proteinase-activated receptor-2 direct distinct signaling events in the intestinal epithelium. Am J Physiol Cell Physiol. (2011) 300:C113–23. doi: 10.1152/ajpcell.00162.2010
76. Mule F, Baffi MC, Cerra MC. Dual effect mediated by protease-activated receptors on the mechanical activity of rat colon. Br J Pharmacol. (2002) 136:367–74. doi: 10.1038/sj.bjp.0704746
77. Mule F, Baffi MC, Capparelli A, Pizzuti R. Involvement of nitric oxide and tachykinins in the effects induced by protease-activated receptors in rat colon longitudinal muscle. Br J Pharmacol. (2003) 139:598–604. doi: 10.1038/sj.bjp.0705273
78. Coelho AM, Vergnolle N, Guiard B, Fioramonti J, Bueno L. Proteinases and proteinase-activated receptor 2: a possible role to promote visceral hyperalgesia in rats. Gastroenterology. (2002) 122:1035–47. doi: 10.1053/gast.2002.32387
79. Asfaha S, Brussee V, Chapman K, Zochodne DW, Vergnolle N. Proteinase-activated receptor-1 agonists attenuate nociception in response to noxious stimuli. Br J Pharmacol. (2002) 135:1101–6. doi: 10.1038/sj.bjp.0704568
80. Fang M, Kovacs KJ, Fisher LL, Larson AA. Thrombin inhibits NMDA-mediated nociceptive activity in the mouse: possible mediation by endothelin. J Physiol. (2003) 549:903–17. doi: 10.1113/jphysiol.2002.036384
81. Auge C, Balz-Hara D, Steinhoff M, Vergnolle N, Cenac N. Protease-activated receptor-4 (PAR 4): a role as inhibitor of visceral pain and hypersensitivity. Neurogastroenterol Motil. (2009) 21:1189–e107. doi: 10.1111/j.1365-2982.2009.01310.x
82. Chin AC, Vergnolle N, MacNaughton WK, Wallace JL, Hollenberg MD, Buret AG. Proteinase-activated receptor 1 activation induces epithelial apoptosis and increases intestinal permeability. Proc Natl Acad Sci USA. (2003) 100:11104–9. doi: 10.1073/pnas.1831452100
83. Chin AC, Lee WY, Nusrat A, Vergnolle N, Parkos CA. Neutrophil-mediated activation of epithelial protease-activated receptors-1 and−2 regulates barrier function and transepithelial migration. J Immunol. (2008) 181:5702–10. doi: 10.4049/jimmunol.181.8.5702
84. Cenac N, Coelho AM, Nguyen C, Compton S, Andrade-Gordon P, MacNaughton WK, et al. Induction of intestinal inflammation in mouse by activation of proteinase-activated receptor-2. Am J Pathol. (2002) 161:1903–15. doi: 10.1016/S0002-9440(10)64466-5
85. Darmoul D, Gratio V, Devaud H, Laburthe M. Protease-activated receptor 2 in colon cancer: trypsin-induced MAPK phosphorylation and cell proliferation are mediated by epidermal growth factor receptor transactivation. J Biol Chem. (2004) 279:20927–34. doi: 10.1074/jbc.M401430200
86. Cunningham KE, Turner JR. Myosin light chain kinase: pulling the strings of epithelial tight junction function. Ann NY Acad Sci. (2012) 1258:34–42. doi: 10.1111/j.1749-6632.2012.06526.x
87. Cenac N, Chin AC, Garcia-Villar R, Salvador-Cartier C, Ferrier L, Vergnolle N, et al. PAR2 activation alters colonic paracellular permeability in mice via IFN-gamma-dependent and -independent pathways. J Physiol. (2004) 558:913–25. doi: 10.1113/jphysiol.2004.061721
88. Rolland-Fourcade C, Denadai-Souza A, Cirillo C, Lopez C, Jaramillo JO, Desormeaux C, et al. Epithelial expression and function of trypsin-3 in irritable bowel syndrome. Gut. (2017) 66:1767–78. doi: 10.1136/gutjnl-2016-312094
89. Darmoul D, Marie JC, Devaud H, Gratio V, Laburthe M. Initiation of human colon cancer cell proliferation by trypsin acting at protease-activated receptor-2. Br J Cancer. (2001) 85:772–9. doi: 10.1054/bjoc.2001.1976
90. Iablokov V, Hirota CL, Peplowski MA, Ramachandran R, Mihara K, Hollenberg MD, et al. Proteinase-activated receptor 2 (PAR2) decreases apoptosis in colonic epithelial cells. J Biol Chem. (2014) 289:34366–77. doi: 10.1074/jbc.M114.610485
91. Sebert M, Denadai-Souza A, Quaranta M, Racaud-Sultan C, Chabot S, Lluel P, et al. Thrombin modifies growth, proliferation and apoptosis of human colon organoids: a protease-activated receptor 1- and protease-activated receptor 4-dependent mechanism. Br J Pharmacol. (2018) 175:3656–68. doi: 10.1111/bph.14430
92. Baumgart DC, Carding SR. Inflammatory bowel disease: cause and immunobiology. Lancet. (2007) 369:1627–40. doi: 10.1016/S0140-6736(07)60750-8
93. Burisch J, Jess T, Martinato M, Lakatos PL, EpiCom E. The burden of inflammatory bowel disease in Europe. J Crohns Colitis. (2013) 7:322–37. doi: 10.1016/j.crohns.2013.01.010
94. Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. (2018) 390:2769–78. doi: 10.1016/S0140-6736(17)32448-0
95. Ferrand A, Al Nabhani Z, Tapias NS, Mas E, Hugot JP, Barreau F. NOD2 expression in intestinal epithelial cells protects toward the development of inflammation and associated carcinogenesis. Cell Mol Gastroenterol Hepatol. (2019) 7:357–69. doi: 10.1016/j.jcmgh.2018.10.009
96. Macfarlane GT, Cummings JH, Allison C. Protein degradation by human intestinal bacteria. J Gen Microbiol. (1986) 132:1647–56. doi: 10.1099/00221287-132-6-1647
97. Carroll IM, Ringel-Kulka T, Ferrier L, Wu MC, Siddle JP, Bueno L, et al. Fecal protease activity is associated with compositional alterations in the intestinal microbiota. PLoS ONE. (2013) 8:e78017. doi: 10.1371/journal.pone.0078017
98. Steck N, Mueller K, Schemann M, Haller D. Bacterial proteases in IBD and IBS. Gut. (2012) 61:1610–8. doi: 10.1136/gutjnl-2011-300775
99. Moretti S, Bellocchio S, Bonifazi P, Bozza S, Zelante T, Bistoni F, et al. The contribution of PARs to inflammation and immunity to fungi. Mucosal Immunol. (2008) 1:156–68. doi: 10.1038/mi.2007.13
100. Reinhardt C, Bergentall M, Greiner TU, Schaffner F, Ostergren-Lunden G, Petersen LC, et al. Tissue factor and PAR1 promote microbiota-induced intestinal vascular remodelling. Nature. (2012) 483:627–31. doi: 10.1038/nature10893
101. Cenac N, Cellars L, Steinhoff M, Andrade-Gordon P, Hollenberg MD, Wallace JL, et al. Proteinase-activated receptor-1 is an anti-inflammatory signal for colitis mediated by a type 2 immune response. Inflamm Bowel Dis. (2005) 11:792–8. doi: 10.1097/01.mib.0000177506.71784.bd
102. Van Spaendonk H, Ceuleers H, Witters L, Patteet E, Joossens J, Augustyns K, et al. Regulation of intestinal permeability: the role of proteases. World J Gastroenterol. (2017) 23:2106–23. doi: 10.3748/wjg.v23.i12.2106
103. Denadai-Souza A, Bonnart C, Tapias NS, Marcellin M, Gilmore B, Alric L, et al. Functional proteomic profiling of secreted serine proteases in health and inflammatory Bowel disease. Sci Rep. (2018) 8:7834. doi: 10.1038/s41598-018-26282-y
104. Thompson NP, Wakefield AJ, Pounder RE. Inherited disorders of coagulation appear to protect against inflammatory bowel disease. Gastroenterology. (1995) 108:1011–5. doi: 10.1016/0016-5085(95)90197-3
105. Giacaman RA, Asrani AC, Ross KF, Herzberg MC. Cleavage of protease-activated receptors on an immortalized oral epithelial cell line by Porphyromonas gingivalis gingipains. Microbiology. (2009) 155:3238–46. doi: 10.1099/mic.0.029132-0
106. Cuffe JE, Bertog M, Velazquez-Rocha S, Dery O, Bunnett N, Korbmacher C. Basolateral PAR-2 receptors mediate KCl secretion and inhibition of Na+ absorption in the mouse distal colon. J Physiol. (2002) 539:209–22. doi: 10.1113/jphysiol.2001.013159
107. Kong W, McConalogue K, Khitin LM, Hollenberg MD, Payan DG, Bohm SK, et al. Luminal trypsin may regulate enterocytes through proteinase-activated receptor 2. Proc Natl Acad Sci USA. (1997) 94:8884–9. doi: 10.1073/pnas.94.16.8884
108. Holzhausen M, Spolidorio LC, Ellen RP, Jobin MC, Steinhoff M, Andrade-Gordon P, et al. Protease-activated receptor-2 activation: a major role in the pathogenesis of Porphyromonas gingivalis infection. Am J Pathol. (2006) 168:1189–99. doi: 10.2353/ajpath.2006.050658
109. Vergnolle N. Proteinase-activated receptor-2-activating peptides induce leukocyte rolling, adhesion, and extravasation in vivo. J Immunol. (1999) 163:5064–9.
110. Lohman RJ, Cotterell AJ, Barry GD, Liu L, Suen JY, Vesey DA, et al. An antagonist of human protease activated receptor-2 attenuates PAR2 signaling, macrophage activation, mast cell degranulation, and collagen-induced arthritis in rats. FASEB J. (2012) 26:2877–87. doi: 10.1096/fj.11-201004
111. Fiorucci S, Mencarelli A, Palazzetti B, Distrutti E, Vergnolle N, Hollenberg MD, et al. Proteinase-activated receptor 2 is an anti-inflammatory signal for colonic lamina propria lymphocytes in a mouse model of colitis. Proc Natl Acad Sci USA. (2001) 98:13936–41. doi: 10.1073/pnas.241377298
112. Nhu QM, Shirey KA, Pennini ME, Stiltz J, Vogel SN. Proteinase-activated receptor 2 activation promotes an anti-inflammatory and alternatively activated phenotype in LPS-stimulated murine macrophages. Innate Immun. (2012) 18:193–203. doi: 10.1177/1753425910395044
113. Kim JA, Choi SC, Yun KJ, Kim DK, Han MK, Seo GS, et al. Expression of protease-activated receptor 2 in ulcerative colitis. Inflamm Bowel Dis. (2003) 9:224–9. doi: 10.1097/00054725-200307000-00002
114. Cottrell GS, Amadesi S, Pikios S, Camerer E, Willardsen JA, Murphy BR, et al. Protease-activated receptor 2, dipeptidyl peptidase I, and proteases mediate Clostridium difficile toxin A enteritis. Gastroenterology. (2007) 132:2422–37. doi: 10.1053/j.gastro.2007.03.101
115. Roka R, Ait-Belgnaoui A, Salvador-Cartier C, Garcia-Villar R, Fioramonti J, et al. Dexamethasone prevents visceral hyperalgesia but not colonic permeability increase induced by luminal protease-activated receptor-2 agonist in rats. Gut. (2007) 56:1072–8. doi: 10.1136/gut.2006.115352
116. Mule F, Pizzuti R, Capparelli A, Vergnolle N. Evidence for the presence of functional protease activated receptor 4 (PAR4) in the rat colon. Gut. (2004) 53:229–34. doi: 10.1136/gut.2003.021899
117. Dabek M, Ferrier L, Annahazi A, Bezirard V, Polizzi A, Cartier C, et al. Intracolonic infusion of fecal supernatants from ulcerative colitis patients triggers altered permeability and inflammation in mice: role of cathepsin G and protease-activated receptor-4. Inflamm Bowel Dis. (2011) 17:1409–14. doi: 10.1002/ibd.21454
118. Mook OR, Frederiks WM, Van Noorden CJ. The role of gelatinases in colorectal cancer progression and metastasis. Biochim Biophys Acta. (2004) 1705:69–89. doi: 10.1016/j.bbcan.2004.09.006
119. Nierodzik ML, Kajumo F, Karpatkin S. Effect of thrombin treatment of tumor cells on adhesion of tumor cells to platelets in vitro and tumor metastasis in vivo. Cancer Res. (1992) 52:3267–72.
120. Zain J, Huang YQ, Feng X, Nierodzik ML, Li JJ, Karpatkin S. Concentration-dependent dual effect of thrombin on impaired growth/apoptosis or mitogenesis in tumor cells. Blood. (2000) 95:3133–8.
121. Chiang HS, Yang RS, Huang TF. Thrombin enhances the adhesion and migration of human colon adenocarcinoma cells via increased beta 3-integrin expression on the tumour cell surface and their inhibition by the snake venom peptide, rhodostomin. Br J Cancer. (1996) 73:902–8. doi: 10.1038/bjc.1996.161
122. Darmoul D, Gratio V, Devaud H, Peiretti F, Laburthe M. Activation of proteinase-activated receptor 1 promotes human colon cancer cell proliferation through epidermal growth factor receptor transactivation. Mol Cancer Res. (2004) 2:514–22.
123. Flynn AN, Buret AG. Proteinase-activated receptor 1 (PAR-1) and cell apoptosis. Apoptosis. (2004) 9:729–37. doi: 10.1023/B:APPT.0000045784.49886.96
124. Dyson JK, Rutter MD. Colorectal cancer in inflammatory bowel disease: what is the real magnitude of the risk? World J Gastroenterol. (2012) 18:3839–48. doi: 10.3748/wjg.v18.i29.3839
125. Turpin B, Miller W, Rosenfeldt L, Kombrinck K, Flick MJ, Steinbrecher KA, et al. Thrombin drives tumorigenesis in colitis-associated colon cancer. Cancer Res. (2014) 74:3020–30. doi: 10.1158/0008-5472.CAN-13-3276
126. Adams GN, Sharma BK, Rosenfeldt L, Frederick M, Flick MJ, Witte DP, et al. Protease-activated receptor-1 impedes prostate and intestinal tumor progression in mice. J Thromb Haemost. (2018) 16:2258–69. doi: 10.1111/jth.14277
127. Adams GN, Rosenfeldt L, Frederick M, Miller W, Waltz D, Kombrinck K, et al. Colon cancer growth and dissemination relies upon thrombin, stromal PAR-1, and fibrinogen. Cancer Res. (2015) 75:4235–43. doi: 10.1158/0008-5472.CAN-15-0964
128. Ducroc R, Bontemps C, Marazova K, Devaud H, Darmoul D, Laburthe M. Trypsin is produced by and activates protease-activated receptor-2 in human cancer colon cells: evidence for new autocrine loop. Life Sci. (2002) 70:1359–67. doi: 10.1016/S0024-3205(01)01519-3
129. Zhang H, Jiang P, Zhang C, Lee S, Wang W, Zou H. PAR4 overexpression promotes colorectal cancer cell proliferation and migration. Oncol Lett. (2018) 16:5745–52. doi: 10.3892/ol.2018.9407
Keywords: protease-activated receptors (PARs), small intestine, colon, gut, inflammation, cancer
Citation: Sébert M, Sola-Tapias N, Mas E, Barreau F and Ferrand A (2019) Protease-Activated Receptors in the Intestine: Focus on Inflammation and Cancer. Front. Endocrinol. 10:717. doi: 10.3389/fendo.2019.00717
Received: 09 July 2019; Accepted: 04 October 2019;
Published: 24 October 2019.
Edited by:
Alain Couvineau, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
David Poyner, Aston University, United KingdomCopyright © 2019 Sébert, Sola-Tapias, Mas, Barreau and Ferrand. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Audrey Ferrand, YXVkcmV5LmZlcnJhbmRAaW5zZXJtLmZy
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.