 Kaspars Megnis1
Kaspars Megnis1 Raitis Peculis1
Raitis Peculis1 Vita Rovite1Pola Laksa1Helvijs Niedra1Inga Balcere2
Vita Rovite1Pola Laksa1Helvijs Niedra1Inga Balcere2 Olivija Caune2Austra Breiksa3Jurijs Nazarovs3Janis Stukens4
Olivija Caune2Austra Breiksa3Jurijs Nazarovs3Janis Stukens4 Ilze Konrade1,2Valdis Pirags1,5,6Janis Klovins1*
Ilze Konrade1,2Valdis Pirags1,5,6Janis Klovins1*- 1Human Genetics and Molecular Medicine, Latvian Biomedical Research and Study Centre, Riga, Latvia
- 2Department of Internal Medicine, Riga Stradins University, Riga, Latvia
- 3Institute of Pathology, Pauls Stradins Clinical University Hospital, Riga, Latvia
- 4Department of Neurosurgery, Pauls Stradins Clinical University Hospital, Riga, Latvia
- 5Department of Endocrinology, Pauls Stradins Clinical University Hospital, Riga, Latvia
- 6Faculty of Medicine, University of Latvia, Riga, Latvia
Objective: Circulating free DNA (cfDNA) in general and circulating tumor DNA (ctDNA) in particular is becoming an increasingly used form of liquid biopsy biomarkers. In this study, we are investigating the ability to detect ctDNA from the plasma of pituitary adenoma (PA) patients.
Design: Tumor tissue samples were obtained from planed PA resections, before which blood plasma samples were taken. Somatic variants found in PA tissue samples were evaluated in related cfDNA, isolated from plasma samples.
Methods: Sanger sequencing, as well as previously obtained whole-exome sequencing data, were used to evaluate somatic variants composition in tumor tissue samples. cfDNA was isolated from the same PA patients and competitive allele-specific TaqMan PCR and amplicon-based next-generation sequencing (NGS) approach were used for targeted detection of variants found in corresponding tumor tissue samples.
Results: Using NGS-based analysis, we detected five out of 17 somatic variants in 40 to 60% of total reads, three variants in 0.50–5.00% of total read count, including GNAS c.601C>T, which was detected using ultra-deep NGS (1.78 million X) in 0.77% of amplicons reads. Nine variants were not detected. We also detected We were not able to detect variant found in PA tissue in cfDNA using cast-PCR, indicating that the portion of variant-containing ctDNA in total isolated cfDNA is too small to be detected with this method.
Conclusions: For the first time, we demonstrate the possibility to detect somatic variants of PA in cfDNA isolated from patients' blood plasma. Whether the source of variant detected in cfDNA is PA should be further tested.
Introduction
Pituitary adenomas (PAs) are a group of diverse benign pituitary tumors associated with increased morbidity and mortality attributed to their hormonal hypersecretion, mass effects and therapy-related adverse sequelae (1). Information about PAs' prevalence varies between different studies and populations. For example, the reported prevalence of symptomatic PAs varies from 3.9 cases per 100,000 people in western Sweden (2) to 77.6 cases per 100,000 people in Banbury (Oxfordshire, UK) (1) or 115.6 cases per 100,000 people in Iceland (3). Several studies estimating the PAs' prevalence in general population disregarding their clinical significance (using radiologic or post mortem studies) have found that PAs are present in 10.4% (1) to 22.5% (4) of people.
The pathogenesis of sporadic PAs remains unclear. These adenomas have lower somatic variant rate than malignant tumors and variants in classic oncogenes and tumor-suppressor genes are relatively rare (5–7). Despite that, as mentioned by Caimari and Korbonits (5) and supported by Song et al. (8), somatic variants in specific genes, like, GNAS, USP8, or PIK3CA, can be found in ~40% of sporadic PAs, and in some cases, knowing the status of these somatic variants could be useful while choosing treatment options (5, 8).
Due to the relatively high prevalence of PAs and the inaccessibility of a tumor for tissue biopsies, it is useful to evaluate the circulating biomarkers in blood for PAs' diagnostics and prognostics. Circulating free DNA (cfDNA) in general and circulating tumor DNA (ctDNA) in particular is becoming a more and more popular class of liquid biopsy biomarkers (9). One of the biggest advantages of its uses as a biomarker is that cfDNA can be extracted from blood plasma, which makes sampling a minimally invasive procedure, compared to tissue biopsies (10).
ctDNA has proven itself as a useful marker in several cancer types. For instance, detection of estrogen receptor 1 (ESR1) variants in ctDNA can be used for assessing possible response to endocrine therapies or resistance to aromatase inhibitor therapy in breast cancer (11). In the case of lung cancer, variants in the epidermal growth factor receptor gene (EGFR) can be used to predict the epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs) therapy effectivity (12). Whereas, in the case of colorectal cancer, ctDNA quantity can be used to detect tumor relapse in post-surgery surveillance months in advance compared to conventional follow-up (13).
However, so far, there have been conflicting results regarding the detection of ctDNA of benign neoplasms in blood plasma. There are cases where ctDNA from aldosterone-producing adenoma and benign colorectal adenomas reportedly have been detected in patients' plasma (14–16). However, other studies have failed to detect ctDNA in patients with colorectal adenomas or other neoplasms like thyroid nodules (17–19). Most of these studies have evaluated differences in the total cfDNA quantity or tried to detect tumor-specific variants in cfDNA using the PCR based methods, which does not always have the highest sensitivity comparing to next-generation sequencing (NGS) (20, 21). As benign neoplasms have a potentially lower fraction of ctDNA in plasma compared to cancers, methods that provide higher sensitivity might be needed for comprehensive analysis in benign tumors. Recently, it has been reported that even 0.3% variant allele fractions of ctDNA can be detected by specifically designed targeted NGS on Illumina NextSeq 500 (22).
The variants in the guanine nucleotide-binding protein, alpha stimulating complex locus (GNAS) gene are frequently encountered in PA tissue material studies. It is reported that about 40% of somatotropinomas and with lower frequency also other types of PAs have GNAS variants and these alterations affect clinical characteristics of the disease (23–25). Therefore, the ability to detect the GNAS variants in the blood of PA patients can serve as a tool for the detection of prognostic markers.
No studies on the analysis of cfDNA in case of PA have been published so far. This study is the first to investigate the ability to detect ctDNA from the plasma of PA patients. Our strategy includes the use of competitive allele-specific TaqMan PCR (cast-PCR) and NGS for targeted detection of GNAS c.601C>T variant. In addition, we employed NGS for a targeted search of other adenoma specific variants identified from whole-exome sequencing (WES) of germline—tumor DNA pairs from the corresponding patients.
Materials and Methods
Patients, Tissue, and Blood Samples
All samples and clinical information were obtained from patients who underwent planned PA resections at Pauls Stradins Clinical University Hospital and Riga East University Hospital. All patients were recruited to the Genome Database of the Latvian population (LGDB), a government-funded national biobank (26). Two informed consents were obtained from each patient after full explanation of the purpose and nature of all procedures used, broad consent for LGDB for use of biological material and medical data for human health and hereditary research, and project-specific consent to the research of pituitary tumors. Both biobank and PA research study have been approved by the Central Medical Ethics Committee of Latvia (protocol No. 22.03.07/A7 and 01.29.1/28, respectively). Sample obtainment and research process comply with the Declaration of Helsinki.
All patients (Table 1) had macroadenomas, eight of them had confirmed extrasellar adenomas, but two patients had endosellar adenomas. Three of them had clinical manifestations characteristic to somatotropinomas and seven had clinical manifestations characteristic to non-functioning pituitary adenomas. According to IHC(P) analysis of PIT1, SF1, and TPIT transcription profiles, HA074, HA090, and HA091 were predominantly of PIT1 lineage, HA065, and HA067 were predominantly of SF1 lineage, HA066 was predominantly of TPIT lineage but HA068, HA069, HA070, and HA073 had no data about expression of above-mentioned transcription factors. Four of them were females, and six were males. Their age distribution varied from 40 to 74 years. For eight patients this was their first-time diagnosis of pituitary adenoma, but for two patients this was relapse from previously operated PA. One patient had somatostatin and dopamine analog therapy before resection, but the rest of the patients had no medicamentous therapy before surgery.
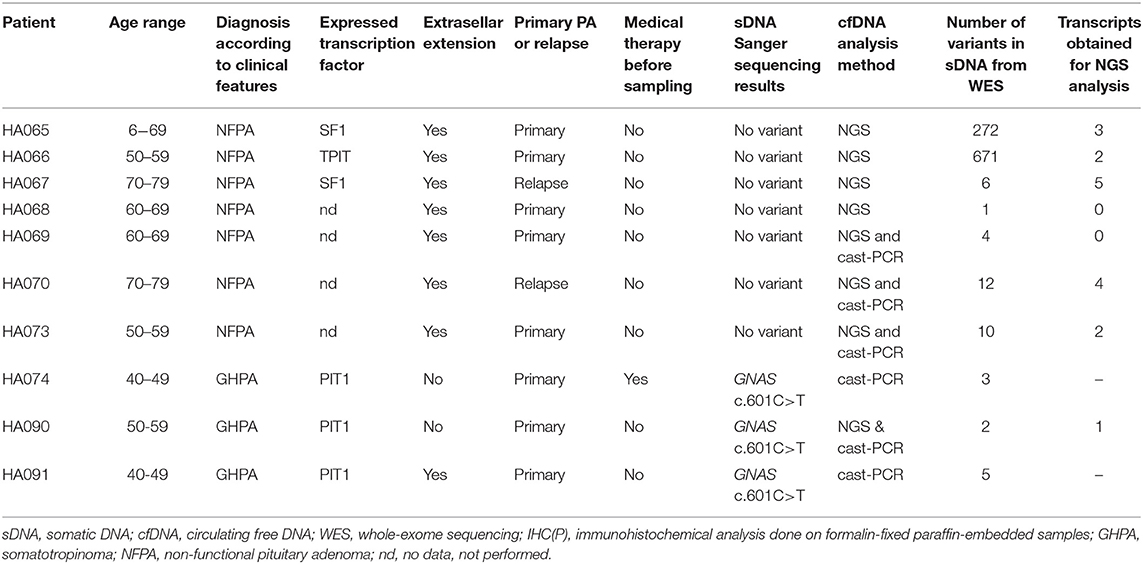
Table 1. Study sample description.
Whole blood samples were collected in EDTA vacutainers from patients before resection and in multiple cases 24 h after resection as well. Plasma samples were obtained by centrifuging whole blood for 10 min, at 2,000 g, in room temperature. After that, upper plasma layers were transferred to new 15 ml tubes and centrifuged for 10 min, at 4,000 g, in room temperature in order to remove remaining blood cells and prevent future cfDNA contamination with germline DNA (gDNA). Obtained plasma samples were aliquoted per 1 ml and frozen in −80°C for later use within 2 h after whole blood sampling.
After resection, adenoma tissue samples were carefully separated with a scalpel by the surgeon from any accompanying tissue lesions or particles that have originated from adenoma or neighboring tissue during surgery and adhered to the tumor sample. The sample was submerged in RNAlater® Solution (Thermo Fisher Scientific, USA) for later nucleic acid extraction.
DNA Extraction
cfDNA was extracted from 2 ml of plasma using QIAamp Circulating Nucleic Acid Kit (Qiagen, Germany) following the manufacturer's instructions. Extraction outcome was evaluated and cfDNA concentration measured using Agilent High Sensitivity DNA kit and Bioanalyzer 2100 (Agilent Technologies, USA).
gDNA was acquired from LGDB [details on the sample preparation and DNA extraction are described by Rovite et al. (26)]. Somatic DNA (sDNA) was extracted from tissue samples using AllPrep DNA/RNA Mini Kit (Qiagen, Germany), following the manufacturer's instructions. The concentration of extracted gDNA and sDNA was measured using Qubit™ dsDNA HS Assay Kit and Qubit 2.0 Fluorometer (Thermo Fisher Scientific, USA).
Sanger Sequencing and Cast-PCR
Sanger sequencing was applied to patients' sDNA and gDNA to estimate the presence of GNAS c.601C>T (NC_000020.11:g.58909365C>T) and c.680A>T (NC_000020.11:g.58909541A>T) variants. DNA was amplified using GNAS_Fw, GNAS_Rs, GNAS227_Fw and GNAS227_Rs primers (Supplementary Table 1) and sequenced using BigDye chemistry and ABI PRISM 3130xl Genetic Analyzer (Thermo Fisher Scientific, USA). All chromatograms were manually inspected using Finch TV software (Geospiza Inc., USA).
Cast-PCR was performed on sDNA and cfDNA from selected samples, using TaqMan® Mutation Detection Mutant Allele Assay GNAS_27887_mu /c.601C>T/ (Thermo Fisher Scientific, USA) and ViiA7 Real-Time PCR System (Thermo Fisher Scientific, USA). Twenty nanogram of sDNA samples were used for the reaction, as recommended by the manufacturer. Since cfDNA amount that could be extracted from blood plasma was too low to meet the manufacturers' recommendations for cast-PCR reaction input, 2 μl of cfDNA samples were used for the reaction, regardless of cfDNAs' concentration. All reactions were done in triplicates.
cfDNA Amplification and NGS
For this study, we used data from WES study (to be published separately), analyzing DNA samples from peripheral blood and pituitary tumor obtained from 10 PA patients. DNA libraries were prepared with Illumina Nextera TruSeq Exome kit (Illumina, USA) following manufacturer's instructions and sequencing was performed with Illumina NextSeq 500/550 High Output v2 kit (150 cycles) (Illumina, USA). Paired-end sequencing with read length 75 bp was carried out on Illumina NexSeq 500 sequencer (Illumina, USA). Illumina exome target manifest TruSeq Rapid Exome TargetedRegions v1.2 (Illumina, USA) was used to define exome regions and Illumina Basespace Enrichment App (v3.0.0) was used for data analysis. Variant analysis revealed a median of 10 somatic variants per tumor. All variants selected for this study were found in sDNA, but not in gDNA, as well as they were validated with Sanger sequencing (Supplementary Figure 2). Only those somatic variants that were detected in sDNA of each individual patient were tested in their corresponding cfDNA. Insertions and deletions were not considered. Initially, we selected all validated variants with the exception for HA065 and HA066, where due to the high number of somatic variants three variants from each sample were selected. In addition, variant in GNAS and VPS13D genes that have been previously reported in relation to PA were included. This resulted in total of 24 variants. We were able to obtain good quality PCR fragments from cfDNA for 17 of these variants that were further used in the study.
Primers (Supplementary Table 1) were designed according to the variant positions found in whole-exome NGS using Primer3plus (available at http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi). Primer sequences were checked for unique human genome binding with UCSC In-Silico PCR (available at http://rohsdb.cmb.usc.edu/GBshape/cgi-bin/hgPcr). Amplicons were designed not to surpass the average length of cfDNA fragment (165 bp).
Amplification of cfDNA regions, containing studied variants, was done using HOT FIREPol® (Solis BioDyne, Estonia). PCR products were visualized in 1.2% agarose gel.
Due to different amplicon sizes (105, 111, and 119 bp), they were divided into three libraries for cfDNAs' NGS, which were prepared with Ion Plus Fragment Library Kit (Thermo Fisher Scientific, USA). Size selection and clean-up between library preparation in all stages, except during end repair, was performed with NucleoMag® NGS Clean-up and Size beads (Macherey-Nagel, Germany). Since NucleoMag® beads guaranteed size selection is between 150 and 800 bp, during end repair stage NucleoMag® beads were replaced with Sephadex™ G-50 Fine (GE Healthcare, USA) in order to avoid excessive fragment loss as our fragments were between 105 and 136 bp long. After ligation of barcode and adapters, size selection was done with BluePippin Automated DNA Size Selection System (Sage Science, USA) to decrease the risk of contamination by adapter dimers. Quality and quantity of prepared libraries were verified with High Sensitivity DNA Reagents kit, High Sensitivity DNA Chips and Agilent 2100 Bioanalyzer (Agilent Technologies, USA). Preparation of template-positive ion PI™ Ion Sphere™ Particles was done on Ion OneTouch™ 2 instrument and Ion PI™ Hi-Q™ OT2 Solutions 200 kit (Thermo Fisher Scientific, USA). Prepared libraries were sequenced using Ion PI™ Hi-Q™ Sequencing 200 Solution kit, Ion PI™ Chip Kit V3 and Ion Proton System (Thermo Fisher Scientific, USA). 7 pMol of each library were used for sequencing, generating from 73,356 to 1,856,095 reads per library.
Sequences from Ion Proton system were exported in fastq format. Sequences were aligned to GRCh37—hg19 using BWA (v 0.7.17-r1188) (27). Position statistics were calculated with Samtools (v 0.1.19) (28). Alignments were reviewed with IGV (v 2.3.14) (Broad Institute, USA) (29).
Results
Sanger Sequencing and Cast-PCR
Both gDNA and sDNA obtained from 10 PA patients samples were analyzed for the presence of GNAS c.601C>T and c.680A>T somatic variants in tumor tissue by direct PCR amplification of corresponding DNA fragment and subsequent Sanger sequencing. The GNAS c.601C>T variant was present in three tumor samples while none of the gDNA samples contained this variant. None of the analyzed tumor tissue samples contained GNAS c.680A>T somatic variant.
After determining the presence of tumor-specific c.601C>T variant in tissue samples, we examined the ability of cast-PCR to detect this variant in the cfDNA obtained from the plasma of the corresponding patients. Amount of the cfDNA that we were able to extract from 2 ml blood plasma varied from 279.20 pg to 4.49 ng (13.96–224.40 pg/μl) (Supplementary Figure 1). None of the samples met the manufacturer's requirements for cfDNA input amount for the cast-PCR (20 ng). Therefore, we used the maximum amount (2 μl) of cfDNA per reaction, regardless of its concentration. That constitutes a 44–716 times lower amount of input DNA compared to the recommended one. sDNA from variant-positive tissue samples were used as positive control while sDNA from variant-negative tissue samples and related cfDNAs were used as a negative control. As a result, we were able to obtain a positive signal only from positive controls (Ct = 28.21 ± 0.90). No signal was obtained from neither negative controls nor cfDNA from GNAS c.601C>T positive samples, indicating that the portion of GNAS c.601C>T variant-containing ctDNA in total isolated cfDNA is too small to be detected with cast-PCR or cfDNA does not contain ctDNA of PA origin.
NGS Based Analysis
For this study, we used WES data to identify cfDNA amplicon targets. A number of identified somatic variants per patient, in their sDNA but not gDNA, as well as the number of tumor variants selected for this study, are shown in Table 1. Of the 24 somatic variants, 17 amplicons encompassing their location were obtained from cfDNA while amplification of seven cfDNA regions with somatic variants was not successful. All 17 obtained amplicons were successfully sequenced with NGS. 82.35% of NGS reads mapped to the target region with an average mapping quality 57.0. The obtained depth of coverage at the variant sites ranged from 444 X to 13,048 X (median 4,833 X).
In a result, eight out of 17 analyzed variants were convincingly detected in cfDNA amplicons. Results of the cfDNA sequencing can be divided into three categories with respect to the variant frequency in the reads (Table 2). The first group includes five variants in genes VPS13D, LDLRAD2, SPEN, GPATCH4, and G6PC2 that were detected between 40 and 60% of the total reads obtained from corresponding amplicons. Interestingly, all these five variants were identified in two individuals that also harbored the highest number of total somatic variants (272 and 671) in tumor tissue as identified by WES. The second group includes three variants from three individuals in genes—MPRIP and RYR (frequencies of 3.65 and 2.27%, respectively) who were identified in sDNA, but not in gDNA in previously mentioned WES study, and GNAS (0.77% frequency) who were identified in sDNA, but not in gDNA in Sanger sequencing, mentioned in section Sanger sequencing and cast-PCR (Supplementary Figure 2). The third group includes identified somatic variants in genes CLEC1B, ATF4, CLCNKA, SMARCAD1, PDE3A, MTFMT, CCDC138, FXR1, and PRPF8 that were either not detected in any of the NGS reads or detected in a small fraction of reads, ranging from 0.02to 0.15%. It should be noted that the substitution of the different nucleotides can be found across all positions examined or any other position of the alignment that corresponds to the rate of the technical error of the used sequencing platform which at this study were 0.22%.
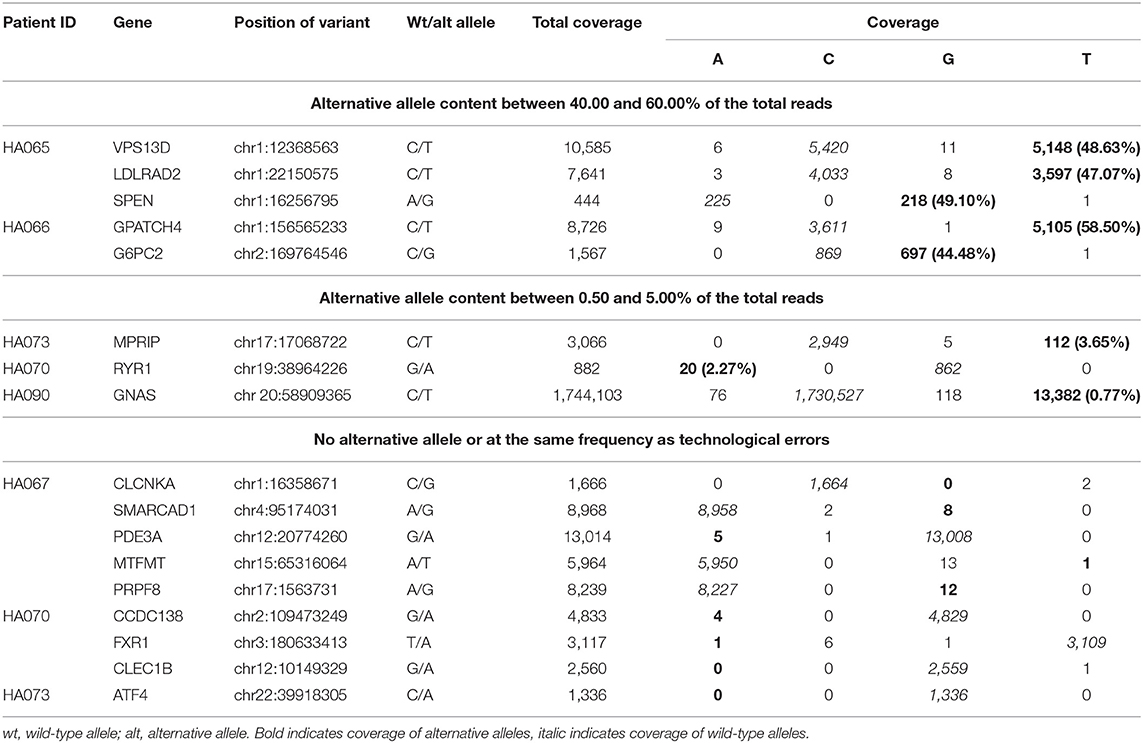
Table 2. Summary of NGS results.
Discussion
During recent years, liquid biopsies and cfDNA has been studied in relation to different tumors and other diseases (30, 31). Despite the theoretically high prevalence of PAs and the broad effect on the organism caused by these tumors (32, 33), there are no studies that would examine the usability of ctDNA in PA diagnostics and prognostics. In our knowledge, this is the first study that attempts to detect ctDNA of PA origin in the patient's bloodstream.
cfDNA has been mostly studied as a prognostic tumor marker in malignant tumor types. It is widely reported that cfDNA amount in patients' bloodstream is significantly higher in cancer patients than a healthy subject, with higher amounts indicated in latter tumor stages compared to early (34–36). In other studies, researchers have applied WES to cfDNA and matching tumor samples, and they had found that cfDNA represents the genomic characteristics of a tumor (37, 38). In benign tumors, ctDNA detection has proven to be more challenging as in some cases this has been successful (14–16), but in others not (17–19).
Apoptosis, necrosis and possibly active secretion have been thought to be the main mechanisms of cfDNA release in the blood (39). However, a recent study has highlighted a stronger correlation of ctDNA detection with cellular proliferation than with cell death (40). It can be further theoreticized, that in the case of fast-growing cancers, rapid cell proliferation could result in relatively higher amounts of ctDNA released in the blood, which can be further isolated and analyzed. However, since the benign tumors generally have slower growth and consequently the proliferation rate, ctDNA could be released in lower amounts, resulting in a lower concentration in blood. For this reason, in case of the benign tumors, ctDNA could be much harder to detect and analyse. On the other hand, many pituitary cells being secretory by nature may contribute to the appearance of cfDNA from these cells into plasma. So far, several attempts have been published to analyse variants in ctDNA from benign tumors of different origin. Lupo et al. measured ctDNA yield in blood plasma from patients with thyroid nodules and analyzed variant content, using commercial assay and NGS. Although they managed to detect ctDNA in two patients, authors concluded that neither cfDNA measurements nor ctDNA variants detection were sensitive or specific enough to be used in clinical practice instead of analysis of tissue biopsy (19).
In our first attempt to analyse ctDNA from PAs, we have chosen well-known recurring somatic variant (c.601C>T) in the GNAS gene and succeeded to identify three tumors from PA patients with this variant. We used cast-PCR and allele-specific TaqMan assay to detect this variant in the cfDNA obtained from the plasma of the corresponding patients. Although cast-PCR is reported to have 0.10–1.00% variant detection limit in cfDNA (21), we were not able to find the presence of c.601C>T variant in any of these samples. Cast-PCR sensitivity can be affected by the fact, that amount of cfDNA input that we used was lower than manufacturer's recommended, due to a limited amount of cfDNA that we were able to extract from blood plasma. It is, of course, plausible that the amount of ctDNA that comes from PA in the bloodstream could be too low to be detected with this method because PAs are generally slow-growing benign tumors.
The method described above, however, has significant limitations. First, only patients with specific GNAS variant are investigated. Secondly, also the number of genomic sites per tumor/patient is limited to one. We, therefore, used NGS based sequencing of amplicons from WES determined genomic regions to estimate and quantify the possible presence of ctDNA in eight patients. This approach requires the prior knowledge of patient specific somatic variants found in tumor tissue. In our case, we selected variants identified in WES and Sanger sequencing study analyzing gDNA obtained from peripheral blood cells and sDNA from a pituitary tumor obtained from the same PA patients. In total 17 somatic variants, that were found in sDNA, but not in gDNA, were analyzed in six patients from corresponding cfDNA amplicons. From all analyzed variants the GNAS gene variant has been reported in several other studies, as mentioned above (7, 23–25). Notably, two genes VPS13D and RYR1 have also been reported to contain somatic variants in GH secreting PA tissue (41), but the localization of the variants was different than we found in our study. Further investigation is needed to explore the potential functional role of these genes in PA development.
In three patients majority of somatic variants were not detectable at the convincing level and only three variants were detected between 0.5 and 5.0% of the total reads that in general corresponds to the levels of a tumor-specific DNA found in the cfDNA shown in the studies exploring different cancers. One of these variants was c.601C>T that could not be identified with cast-PCR and allele-specific TaqMan assay. From these results, we conclude that the concentration of ctDNA in case of PAs are at the boundary of detection with semiconductor sequencing technologies and below the boundary for detection with cast-PCR and TaqMan technologies. Although the results of this study indicate successful detection of the variants in plasma-derived cfDNA in some cases, the read count obtained from the sequencing is on the borderline of sensitivity and cannot be used for tumor variant monitoring in a clinical setting with the presented technology. Potentially, other molecular techniques, for example, digital droplet PCR could be further studied to develop more sensitive monitoring methods.
The results obtained from two patients included in the study are unexpected. First of all, tumors from these patients are characterized with a higher number of somatic variants (272 and 671) than usually observed in benign PAs, but no clinical manifestations indicating increased aggressiveness or recurrent pathological tumors were identified in these patients. All five variants from these patients that we were able to detect had between 40 and 60% of the total reads that is unusually high for a ctDNA. One can hypothesize that high amount of genetic changes in these tumors could contribute to mechanisms that are related with ctDNA release into the bloodstream or these variants could be present in other tissues in patients bodies with high activity of mechanisms that promote cfDNA release.
In summary, for the first time, we demonstrate the possibility to detect somatic variants of PA in cfDNA isolated from patients' blood plasma. Beside others, we were able to detect variant in the GNAS gene, whose impact on PA has been widely described in the literature. Whether the source of variant detected in cfDNA is PA should be further tested.
Data Availability
Circulating free DNA amplicon sequencing data can be found in public repository under the following links: http://www.ncbi.nlm.nih.gov/bioproject/546469; https://www.ncbi.nlm.nih.gov/biosample/11959605; https://www.ncbi.nlm.nih.gov/biosample/11959606; https://www.ncbi.nlm.nih.gov/biosample/11959607.
Ethics Statement
The studies involving human participants were reviewed and approved by Central Medical Ethics Committee of Latvia (protocol No. 22.03.07/A7 and 01.29.1/28). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
KM participated in study design and organization of experiments and performed manuscript writing. RP participated in study design and data analysis. VR, IK, and VP participated in study design and manuscript editing. PL performed DNA isolation, Sanger sequencing, and cast-PCR. HN performed DNA isolation, amplification and next-generation sequencing. IB performed patient enrolment in the study. OC performed clinical data obtainment and description. AB and JN performed tumor tissue immunohistochemical characterization. JS performed tumor resection and tumor material obtainment. JK was the project leader, performed study design, and manuscript editing.
Funding
This research was supported by the European Regional Development Fund (ERDF), Measure 1.1.1.1 Industry-Driven Research project Molecular markers of pituitary tumor development, progression and therapy response (Project No. 1.1.1.1/16/A/066, 2017), and Roche Academy project Immunohistochemical analysis of pituitary adenoma tissue.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge the Latvian Biomedical Research and Study Center and the Genome Database of the Latvian Population for providing infrastructure, biological material, and data.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2019.00615/full#supplementary-material
Supplementary Figure 1. Size and concentration of two representative cfDNA samples that have been extracted from 2 ml of PA patients' blood plasma and eluted in 20 μl. Measurement was done using Agilent High Sensitivity DNA kit and Bioanalyzer 2100 (Agilent Technologies, USA). (1) indicates cfDNA sample, (2) indicates High Sensitivity DNA ladder lower marker, (4) indicates High Sensitivity DNA ladder upper marker. In both samples, cfDNA size was around 170 bp, but concentration warried from 13.96 to 224.40 pg/μl.
Supplementary Figure 2. Validation of LDLRAD2, SPEN, VPS13D, G6PC2, GPATCH4, CLCNKA, MTFMT, PDE3A, PRPF8, SMARCAD1, CCDC138, CLEC1B, FXR1, RYR1, ATF4, MPRIP gene variants with Sanger sequencing in pituitary adenoma somatic (PA) DNA and patients germline DNA from samples HA065, HA066, HA067, HA070, HA073, and HA090.
Supplementary Table 1. Used primers (Metabion international AG, Germany).
References
1. Fernandez A, Karavitaki N, Wass JAH. Prevalence of pituitary adenomas: a community-based, cross-sectional study in Banbury (Oxfordshire, UK). Clin Endocrinol. (2010) 72:377–82. doi: 10.1111/j.1365-2265.2009.03667.x
2. Tjörnstrand A, Gunnarsson K, Evert M, Holmberg E, Ragnarsson O, Rosén T, et al. The incidence rate of pituitary adenomas in western Sweden for the period 2001-2011. Eur J Endocrinol. (2014) 171:519–26. doi: 10.1530/EJE-14-0144
3. Agustsson TT, Baldvinsdottir T, Jonasson JG, Olafsdottir E, Steinthorsdottir V, Sigurdsson G, et al. The epidemiology of pituitary adenomas in Iceland, 1955-2012: a nationwide population-based study. Eur J Endocrinol. (2015) 173:655–64. doi: 10.1530/EJE-15-0189
4. Ezzat S, Asa SL, Couldwell WT, Barr CE, Dodge WE, Vance ML, et al. The prevalence of pituitary adenomas. Cancer. (2004) 101:613–9. doi: 10.1002/cncr.20412
5. Caimari F, Korbonits M. Novel genetic causes of pituitary adenomas. Clin Cancer Res. (2016) 22:5030–42. doi: 10.1158/1078-0432.CCR-16-0452
6. Newey PJ, Nesbit MA, Rimmer AJ, Head RA, Gorvin CM, Attar M, et al. Whole-exome sequencing studies of nonfunctioning pituitary adenomas. J Clin Endocrinol Metab. (2013) 98:E796–800. doi: 10.1210/jc.2012-4028
7. Ronchi CL, Peverelli E, Herterich S, Weigand I, Mantovani G, Schwarzmayr T, et al. Landscape of somatic mutations in sporadic GH-secreting pituitary adenomas. Eur J Endocrinol. (2016) 174:363–72. doi: 10.1530/EJE-15-1064
8. Song Z-J, Reitman ZJ, Ma Z-Y, Chen J-H, Zhang Q-L, Shou X-F, et al. The genome-wide mutational landscape of pituitary adenomas. Cell Res. (2016) 26:1255–9. doi: 10.1038/cr.2016.114
9. Leung F, Kulasingam V, Diamandis EP, Hoon DSB, Kinzler K, Pantel K, et al. Circulating tumor DNA as a cancer biomarker: fact or fiction? Clin Chem. (2016) 62:1054–60. doi: 10.1373/clinchem.2016.260331
10. Allin DM, Shaikh R, Carter P, Thway K, Sharabiani MTA, Gonzales-de-Castro D, et al. Circulating tumour DNA is a potential biomarker for disease progression and response to targeted therapy in advanced thyroid cancer. Eur J Cancer. (2018) 103:165–75. doi: 10.1016/j.ejca.2018.08.013
11. Takeshita T, Yamamoto Y, Yamamoto-Ibusuki M, Tomiguchi M, Sueta A, Murakami K, et al. Analysis of ESR1 and PIK3CA mutations in plasma cell-free DNA from ER-positive breast cancer patients. Oncotarget. (2017) 8:52142–55. doi: 10.18632/oncotarget.18479
12. Hirsch FR, Varella-Garcia M, Bunn PA, Franklin WA, Dziadziuszko R, Thatcher N, et al. Molecular predictors of outcome with gefitinib in a phase III placebo-controlled study in advanced non-small-cell lung cancer. J Clin Oncol. (2006) 24:5034–42. doi: 10.1200/JCO.2006.06.3958
13. Reinert T, Schøler L V, Thomsen R, Tobiasen H, Vang S, Nordentoft I, et al. Analysis of circulating tumour DNA to monitor disease burden following colorectal cancer surgery. Gut. (2016) 65:625–34. doi: 10.1136/gutjnl-2014-308859
14. Cangiano D, Ceolotto G, Caroccia B, Citton M, Seccia TH, Rossi GP. Detection of free-circulating dna in patients with aldosterone producing adenoma. J Hypertens. (2015) 33:e111–2. doi: 10.1097/01.hjh.0000467652.92919.31
15. Mead R, Duku M, Bhandari P, Cree IA. Circulating tumour markers can define patients with normal colons, benign polyps, and cancers. Br J Cancer. (2011) 105:239–45. doi: 10.1038/bjc.2011.230
16. Kopreski MS, Benko FA, Borys DJ, Khan A, McGarrity TJ, Gocke CD. Somatic mutation screening: identification of individuals harboring K-ras mutations with the use of plasma DNA. J Natl Cancer Inst. (2000) 92:918–23. doi: 10.1093/jnci/92.11.918
17. Perrone F, Lampis A, Bertan C, Verderio P, Ciniselli CM, Pizzamiglio S, et al. Circulating free DNA in a screening program for early colorectal cancer detection. Tumori. (2014) 100:115–21. doi: 10.1177/030089161410000201
18. Galanopoulos M, Papanikolaou IS, Zografos E, Viazis N, Papatheodoridis G, Karamanolis D, et al. Comparative study of mutations in single nucleotide polymorphism loci of KRAS and BRAF genes in patients who underwent screening colonoscopy, with and without premalignant intestinal polyps. Anticancer Res. (2017) 37:651–7. doi: 10.21873/anticanres.11360
19. Lupo M, Guttler R, Geck Z, Tonozzi TR, Kammesheidt A, Braunstein GD. Is measurement of circulating tumor dna of diagnostic use in patients with thyroid nodules? Endocr Pract. (2018) 24:453–9. doi: 10.4158/EP-2017-0213
20. Garcia J, Forestier J, Dusserre E, Wozny A-S, Geiguer F, Merle P, et al. Cross-platform comparison for the detection of RAS mutations in cfDNA (ddPCR Biorad detection assay, BEAMing assay, and NGS strategy). Oncotarget. (2018) 9:21122–31. doi: 10.18632/oncotarget.24950
21. Han X, Wang J, Sun Y. Circulating tumor DNA as biomarkers for cancer detection. Genomics Proteomics Bioinforma. (2017) 15:59–72. doi: 10.1016/j.gpb.2016.12.004
22. Plagnol V, Woodhouse S, Howarth K, Lensing S, Smith M, Epstein M, et al. Analytical validation of a next generation sequencing liquid biopsy assay for high sensitivity broad molecular profiling. PLoS ONE. (2018) 13:e0193802. doi: 10.1371/journal.pone.0193802
23. Landis CA, Harsh G, Lyons J, Davis RL, McCormick F, Bourne HR. Clinical characteristics of acromegalic patients whose pituitary tumors contain mutant Gs protein. J Clin Endocrinol Metab. (1990) 71:1416–20. doi: 10.1210/jcem-71-6-1416
24. Hayward BE, Barlier A, Korbonits M, Grossman AB, Jacquet P, Enjalbert A, et al. Imprinting of the G(s)alpha gene GNAS1 in the pathogenesis of acromegaly. J Clin Invest. (2001) 107:R31–6. doi: 10.1172/JCI11887
25. Efstathiadou ZA, Bargiota A, Chrisoulidou A, Kanakis G, Papanastasiou L, Theodoropoulou A, et al. Impact of gsp mutations in somatotroph pituitary adenomas on growth hormone response to somatostatin analogs: a meta-analysis. Pituitary. (2015) 18:861–7. doi: 10.1007/s11102-015-0662-5
26. Rovite V, Wolff-Sagi Y, Zaharenko L, Nikitina-Zake L, Grens E, Klovins J. Genome Database of the Latvian Population (LGDB): design, Goals, and Primary Results. J Epidemiol. (2018) 28:353–60. doi: 10.2188/jea.JE20170079
27. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. (2009) 25:1754–60. doi: 10.1093/bioinformatics/btp324
28. Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. (2011) 27:2987–93. doi: 10.1093/bioinformatics/btr509
29. Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. (2011) 29:24–6. doi: 10.1038/nbt.1754
30. Khatami F, Tavangar SM. Circulating tumor DNA (ctDNA) in the era of personalized cancer therapy. J Diabetes Metab Disord. (2018) 17:19–30. doi: 10.1007/s40200-018-0334-x
31. Zhang K, Lin G, Han Y, Xie J, Li J. Circulating unmethylated insulin DNA as a potential non-invasive biomarker of beta cell death in type 1 Diabetes: a review and future prospect. Clin Epigenetics. (2017) 9:44. doi: 10.1186/s13148-017-0343-5
32. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. (2015) 65:87–108. doi: 10.3322/caac.21262
33. Buurman H, Saeger W. Subclinical adenomas in postmortem pituitaries: classification and correlations to clinical data. Eur J Endocrinol. (2006) 154:753–8. doi: 10.1530/eje.1.02107
34. Sozzi G, Conte D, Leon M, Ciricione R, Roz L, Ratcliffe C, et al. Quantification of free circulating DNA as a diagnostic marker in lung cancer. J Clin Oncol. (2003) 21:3902–8. doi: 10.1200/JCO.2003.02.006
35. Kim K, Shin DG, Park MK, Baik SH, Kim TH, Kim S, et al. Circulating cell-free DNA as a promising biomarker in patients with gastric cancer: diagnostic validity and significant reduction of cfDNA after surgical resection. Ann Surg Treat Res. (2014) 86:136–42. doi: 10.4174/astr.2014.86.3.136
36. Shao X, He Y, Ji M, Chen X, Qi J, Shi W, et al. Quantitative analysis of cell-free DNA in ovarian cancer. Oncol Lett. (2015) 10:3478–82. doi: 10.3892/ol.2015.3771
37. Manier S, Park J, Capelletti M, Bustoros M, Freeman SS, Ha G, et al. Whole-exome sequencing of cell-free DNA and circulating tumor cells in multiple myeloma. Nat Commun. (2018) 9:1691. doi: 10.1038/s41467-018-04001-5
38. Song J, Yang Z. Case report: whole exome sequencing of circulating cell-free tumor DNA in a follicular thyroid carcinoma patient with lung and bone metastases. J Circ biomarkers. (2018) 7:1849454418763725. doi: 10.1177/1849454418763725
39. Wan JCM, Massie C, Garcia-Corbacho J, Mouliere F, Brenton JD, Caldas C, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. (2017) 17:223–38. doi: 10.1038/nrc.2017.7
40. Abbosh C, Birkbak NJ, Wilson GA, Jamal-Hanjani M, Constantin T, Salari R, et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature. (2017) 545:446–51. doi: 10.1038/nature22364
Keywords: pituitary adenoma, circulating tumor DNA, competitive allele-specific TaqMan, next-generation sequencing, GNAS
Citation: Megnis K, Peculis R, Rovite V, Laksa P, Niedra H, Balcere I, Caune O, Breiksa A, Nazarovs J, Stukens J, Konrade I, Pirags V and Klovins J (2019) Evaluation of the Possibility to Detect Circulating Tumor DNA From Pituitary Adenoma. Front. Endocrinol. 10:615. doi: 10.3389/fendo.2019.00615
Received: 03 June 2019; Accepted: 23 August 2019;
Published: 18 September 2019.
Edited by:
Mônica Gadelha, Federal University of Rio de Janeiro, BrazilReviewed by:
Odelia Cooper, Cedars-Sinai Medical Center, United StatesMurat Aydin Sav, Yeditepe University, Turkey
Giampaolo Trivellin, National Institutes of Health (NIH), United States
Copyright © 2019 Megnis, Peculis, Rovite, Laksa, Niedra, Balcere, Caune, Breiksa, Nazarovs, Stukens, Konrade, Pirags and Klovins. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Janis Klovins, a2xvdmluc0BiaW9tZWQubHUubHY=