 Wenting Dai1
Wenting Dai1 Lei Jiang1,2*
Lei Jiang1,2*- 1Department of Molecular and Cellular Endocrinology, Diabetes and Metabolism Research Institute, Duarte, CA, United States
- 2Comprehensive Cancer Center, City of Hope Medical Center, Duarte, CA, United States
Metabolism describes the life-sustaining chemical reactions in organisms that provide both energy and building blocks for cellular survival and proliferation. Dysregulated metabolism leads to many life-threatening diseases including obesity, diabetes, and cancer. Mitochondria, subcellular organelles, contain the central energy-producing metabolic pathway, the tricarboxylic acid (TCA) cycle. Also, mitochondria exist in a dynamic network orchestrated by extracellular nutrient levels and intracellular energy needs. Upon stimulation, mitochondria undergo consistent interchange through fusion (small to big) and fission (big to small) processes. Mitochondrial fusion is primarily controlled by three GTPases, mitofusin 1 (Mfn1), Mfn2, and optic atrophy 1 (Opa1), while mitochondrial fission is primarily regulated by GTPase dynamin-related protein 1 (Drp1). Dysregulated activity of these GTPases results in disrupted mitochondrial dynamics and cellular metabolism. This review will update the metabolic roles of these GTPases in obesity, diabetes, and cancer.
Introduction
The word mitochondrion comes from the Greek words mitos (thread in English) and chondrion (granule in English) (1). The field of mitochondrial dynamics has a history of over 100 years (1–3). In 1914, Mrs. and Mr. Lewis published “Mitochondria in Tissue Culture” as a special article in Science (2) and stated, “The mitochondria are almost never at rest, but are continually changing their position and also their shape.” The full picture of mitochondrial dynamics includes fusion, fission, transportation, and degradation, which have been covered in other reviews (1, 4–6). Mitochondria exist widely in most eukaryotic organisms and have a double membrane structure. The outer mitochondrial membrane contains large quantities of integral membrane proteins called porins, which form channels allowing for small molecules to freely diffuse from one side of the membrane to the other. In contrast, the inner mitochondrial membrane is highly impermeable, even to small metabolites. Three mitochondrial membrane–located GTPases regulate the mitochondrial fusion process, among which Mfn1 and Mfn2 control outer mitochondrial membrane fusion (7), while Opa1 regulates inner mitochondrial membrane fusion (8–10). In contrast, the cytosol-located GTPase Drp1 mediates the mitochondrial fission process (11). Mutations of these GTPases are closely related to human disease, such as Charcot–Marie–Tooth disease type 2A (12) and optic atrophy (13, 14). In this review, we primarily focus on these small GTPases and their influences on metabolic alterations in obesity, diabetes, and cancer.
SMALL GTPases Control Mitochondrial Fusion and Fission
Mfn1 and Mfn2, homologs of the drosophila protein fuzzy onion (Fzo) (15), are the first reported mammalian GTPases to mediate mitochondrial fusion (16, 17). Mice lacking Mfn1 or Mfn2 are embryonically lethal, which suggests their essential role in embryonic development (7, 18). In mouse embryonic fibroblasts (MEFs), Mfn1 or Mfn2 single-knockout results in fragmented mitochondria (7), while Mfn1 and Mfn2 double-knockout completely blocks mitochondrial fusion (18, 19). Conversely, overexpression of either Mfn1 or Mfn2 can rescue the mitochondrial fusion (7). These genetic studies show that Mfn1 and Mfn2 have redundant and collaborative roles in mitochondrial fusion.
Opa1 is named after a neurogenic disease optic atrophy. Patients with mutant Opa1 have degenerated retinal ganglion cells and atrophy of the optic nerve (13, 14). Opa1 is the mammalian ortholog of Mgm1, which is essential for mitochondrial fusion in yeast (20, 21). Opa1 is encoded by a complicated set of at least eight mRNA splice forms, and each long Opa1 forms (L-Opa1) peptides that can be further cleaved into short forms (S-Opa1) by proteases Yeast Mitochondrial Escape 1 Like 1 ATPase (YME1L1) and metalloendopeptidase Oma1 (22, 23). YME1L1 and Oma1 double-knockout MEFs only contain L-Opa1 (24). Importantly, YME1L1 and Oma1 establish a near-balance of L-Opa1 and S-Opa1 under basal conditions, and they regulate this balance in response to metabolic changes or mitochondrial dysfunction (24–26). Moreover, the distinction between L-Opa1 and S-Opa1 lies in their different locations within mitochondria, namely, L-Opa1 in the inner mitochondrial membrane and S-Opa1 within the mitochondrial matrix. Consistent with their respective locations, L-Opa1 promotes mitochondrial inner membrane fusion, while S-Opa1 enhances mitochondrial fission (27).
Drp1, a cytosolic protein, plays a crucial role in regulating mitochondrial fission (4). A newborn girl with a dominant-negative mutation in Drp1 had abnormal brain development and several other diseases, which were associated with a severe defect in the fission of both mitochondria and peroxisomes (28). Drp1 knockout mice also exhibit abnormal brain development and die around day 12 of embryonic development (29, 30). Upon activation, Drp1 relocates to the outer mitochondrial membrane and forms a ring structure to constrict mitochondrion. This translocation of Drp1 is regulated by several outer mitochondrial membrane proteins, including fission protein 1 (Fis1), mitochondrial fission factor (Mff), and mitochondrial dynamics proteins of 49 and 51 kDa (MiD49 and MiD51) (31, 32). In mammals, overexpression of Fis1 in mammalian cells promotes mitochondrial fission, while inhibition of Fis1 results in elongation (fusion) (33–35). Knockdown of Mff results in mitochondrial elongation in mammalian cells (35) and reduces the amount of Drp1 recruited to mitochondria (36). In 2011, Palmer et al. reported that a double knockdown of MiD49 and MiD51 leads to mitochondrial elongation and reduces recruitment of Drp1 to mitochondria (37). Subsequently, MiD49 and MiD51 proteins were found to promote mitochondrial fission independent of both Fis1 and Mff (31, 38). Furthermore, the activity of Drp1 could be controlled by its phosphorylation. The cyclin-dependent Drp1 phosphorylation at serine 585 (S585) is essential to promote mitochondrial fission in mitotic cells (39). The cyclic adenosine monophosphate (cAMP)-dependent protein kinase (PKA)–dependent Drp1 phosphorylation at S656 inhibits mitochondrial fission, and the calcineurin-dependent dephosphorylation of Drp1–S656 promotes mitochondrial fragmentation (40). Furthermore, mitogen-activated protein kinase 1 (MAPK1, also known as Erk2)–dependent Drp1 phosphorylation at S616 activates Drp1 and promotes mitochondrial fission (41, 42). In HeLa cells, Drp1 was found to be phosphorylated by protein kinase A (PKA) at S637, and this phosphorylation attenuated the Drp1 GTPase activity (43). Besides, calcineurin-dependent dephosphorylation of Drp1 at the S637 site drives its translocation to mitochondria and promotes mitochondrial fission (44). Genetical and chemical inhibition of Drp1 both result in elongated mitochondria (45).
Imbalanced Mitochondrial Metabolism Results in Obesity and Diabetes
Given the essential role of mitochondria in energy metabolism, mitochondrial dysfunction acts as a key regulator in the pathophysiology of obesity and diabetes. Mutations in some mitochondrial genes have been demonstrated to be the main causes of these metabolic diseases, such as Charcot–Marie–Tooth disease (Mfn2 mutation) and dominant optic atrophy (Opa1 mutation) (4). Interestingly, imbalanced mitochondrial metabolism induced by mitochondrial proteins (especially mitofusins, Opa1, and Drp1) in various tissues has been implicated in the pathology of metabolic diseases (Table 1).
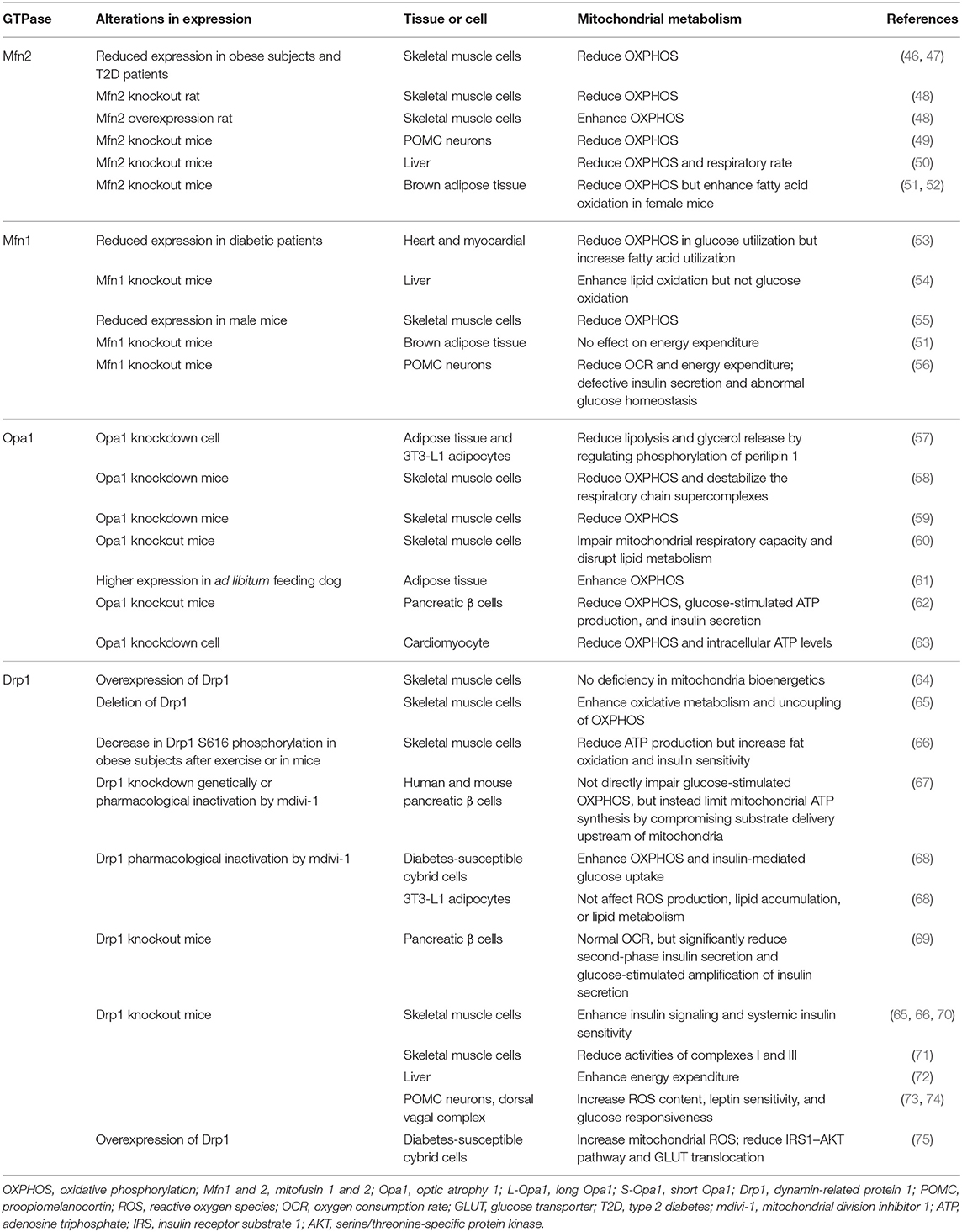
Table 1. The role of GTPases in obesity and diabetes.
In particular, type 2 diabetes (T2D) is related to the reduced expression of Mfn2, which depresses oxidative phosphorylation (OXPHOS) and impairs mitochondrial fusion in skeletal muscle (46, 76). Also, liver and skeletal muscle deletion of Mfn2 in mice both result in fragmented mitochondrial networks and numerous metabolic abnormalities, including glucose intolerance and enhanced hepatic gluconeogenesis (47, 54). These confirm the crucial regulatory role of Mfn2 in insulin signaling and glucose homeostasis associated with obesity and T2D. Besides, it was first reported that proopiomelanocortin (POMC)–specific ablation of Mfn2 results in endoplasmic reticulum (ER) stress–induced leptin resistance and decreased energy expenditure for protection against obesity (49). Moreover, Mfn1 deficiency leads to a highly fragmented mitochondrial network and enhanced mitochondrial respiration capacity in the myocardium (53) and liver (54) in diabetes. Interestingly, the liver from Mfn1 knockout mice demonstrates a preference for using lipids as the main energy source and a more active complex I to protect against insulin resistance (54). Also during the fast-to-fed transition, mice lacking Mfn1 in POMC neurons exhibit defective mitochondrial architecture and flexibility, which results in defective insulin secretion and abnormal glucose homeostasis by pancreatic β cells (56). Mfn2, but not Mfn1, deletion in brown adipose tissue (BAT) remodels the mitochondrial dysfunction, leading to an increase in insulin sensitivity and resistance to obesity (51, 52, 77). These observations have implicated a crucial role of mitofusins in the control of mitochondrial energy metabolism and insulin resistance. However, further studies should determine the distinct regulatory mechanisms of Mfn1 and Mfn2 targeting mitochondrial metabolism in diabetes and obesity.
The expanding evidence also displays the involvement of Opa1 in obesity and diabetes. With regard to adiposity, Opa1 has been reported to be involved in regulating lipolysis by anchoring a pool of PKA that phosphorylates perilipin and thereby triggers lipolysis in vitro (57) and in vivo (61). Further studies show the translocation of Opa1 from mitochondria to lipid droplets during human adipocyte differentiation (78). Proteases YME1L1 and Oma1 can cleave L-Opa1 into S-Opa1 (79). The deficiency of Oma1 perturbs the mitochondrial fusion–fission equilibrium, thereby reducing OXPHOS, enhancing fatty acid oxidation and decreasing energy expenditure, all of which collaboratively induce obesity in mice (79). Moreover, Opa1 deletion in pancreatic β cells impairs glucose-stimulated adenosine triphosphate (ATP) production and insulin secretion, which subsequently develops into hyperglycemia (62). Also, insulin increases Opa1 levels and promotes mitochondrial fusion in cardiomyocytes, which enhances OXPHOS, whereas Opa1 deletion suppresses the insulin-stimulated ATP synthesis (63). A recent study reported that sedentary but not active humans display an age-related decline in Opa1, which is associated with muscle loss (58). Acute muscle-specific deletion of Opa1 induces muscle inflammation, ER stress, and secretion of fibroblast growth factor 21 (FGF21) (58–60). Apart from these, a recent study addressed the physiological relevance of the concomitant impairment of mitochondrial fusion and fission machinery (80). These confirm the close relationship between Opa1 and mitochondrial insulin-stimulated energy metabolism, which provides a basis for its regulatory role in obesity and diabetes. Therefore, it is urgent to elucidate how Opa1 processing regulates mitochondrial metabolism in obesity and diabetes.
The mitochondrial fission machinery protein Drp1 is also involved in obesity and T2D through its effect on the rate of mitochondrial oxidative metabolism. Drp1-mediated mitochondrial fission results in mitochondrial fragmentation along with decreased ATP content, which further leads to reduced insulin-mediated glucose uptake in human skeletal muscle (66) and significant loss of glucose-stimulated insulin secretion in mouse pancreatic β cells (69). Also in pancreatic β cells, Drp1 deletion largely impairs glucose-stimulated insulin secretion without affecting oxygen consumption and intracellular calcium mobilization (69). On the contrary, Drp1 regulates insulin secretion via glucose-fueled respiration, and pyruvate completely rescues the impaired insulin secretion of fission-deficient β cells (67). These data appear inconsistent, and the regulatory machinery of Drp1 affecting insulin secretion requires further clarification. When used to treat mice, mitochondrial division inhibitor 1 (mdivi-1), a Drp1 inhibitor, rescues tubular mitochondrial network and membrane potential, decreases oxidative stress, and increases insulin-mediated glucose uptake in diabetes-susceptible cybrid cells (68). However, the inhibition of Drp1 by mdivi-1 has no effect on reactive oxygen species (ROS) production, lipid accumulation, or the expression of adipogenic-related proteins in differentiated 3T3-L1 adipocytes (75). In contrast, overexpression of Drp1 decreases mitochondrial network formation and increases mitochondrial ROS, subsequently suppressing the insulin receptor substrate 1 (IRS1) – serine/threonine-specific protein kinase (AKT) pathway and glucose transporter (GLUT) translocation stimulated by insulin (68). Also, the depressed phosphorylation of Drp1 at S616 in skeletal muscle is negatively correlated with enhancement in fat oxidation and insulin sensitivity (66), which is further supported by the observation that Drp1 deletion induced aberrant mitochondrial fission, improving insulin signaling and systemic insulin sensitivity in skeletal muscle of obese mice (70). Moreover, Drp1 deficiency reduces fat mass, induces ER stress, and promotes energy expenditure through increasing the expression of FGF21 in the liver, which helps mice fed with a high-fat diet to be protected against obesity (72). Mice lacking Drp1 in skeletal muscle develop a lethal mitochondrial myopathy, and this muscle-specific Drp1 deletion impairs mitochondrial function, including the reduced activities of complexes I and III (71). In contrast, Drp1 overexpression in skeletal muscle displayed a drastic impairment in postnatal muscle growth, with reorganization of the mitochondrial network and reduction of mitochondrial DNA (mtDNA) quantity, without the deficiency of mitochondrial bioenergetics (64). Muscle-specific Drp1 heterozygote mice have impaired muscle endurance and running performance (65). The impaired mitochondrial remodeling induced by Drp1 in skeletal muscle is also associated with derangements in metabolism and insulin sensitivity (65, 66). Additionally, inducible deletion of Drp1 in POMC neurons (73) and the dorsal vagal complex (DVC) (74) increases ROS content, leptin sensitivity, and glucose responsiveness. Thus, Drp1-mediated mitochondrial fission may underlie the pathogenesis of insulin resistance in obesity and T2D. However, further studies are required to explore potential mechanisms for therapeutic interventions.
Mitochondrial Fusion and Fission Shape Cancer Metabolism
Most studies of the cancer-related mitochondrial dynamics focus on their functions in apoptosis, which have been well-reviewed (1, 6). Therefore, here we focus on energy metabolism in cancer, another important function of mitochondria (Table 2). Cancer metabolism also has a history of about 100 years. In 1925, Dr. Otto Warburg first described that cancer cells tend to favor metabolism via aerobic glycolysis rather than the much more efficient OXPHOS pathway, which is widely known as the “Warburg effect” (93–96). Although Dr. Warburg hypothesized that dysfunctional mitochondria were the source of anaerobic glycolysis, more recent studies found that cancer cells often have intact mitochondrial metabolism (97, 98).
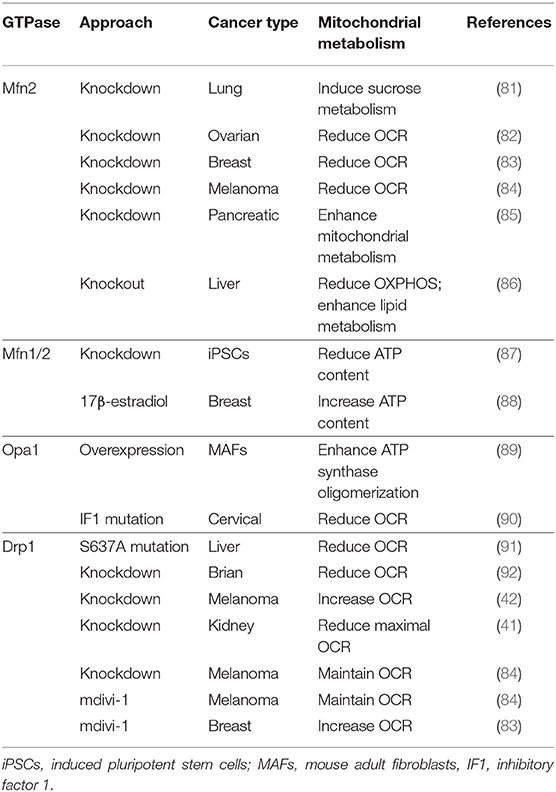
Table 2. The role of GTPases in cancer metabolism.
Mitochondrial morphology undergoes a dynamic balance between fusion and fission in response to the altered extracellular nutrient level (5). Meanwhile, cancer metabolism is also known for its flexibility to the surrounding nutrient composition (99), and cancer cells acquire different metabolic programs corresponding to their development stages. Rapid proliferating cancer cells have a high glycolytic activity and fatty acid synthesis rate, which provide the building blocks for cell growth (100). Metastatic cancer cells have high oxidative metabolism, which maintains the energy need for mobility (101, 102). In general, oxidative metabolism is less active in the fragmented mitochondria when compared to the tubular mitochondria. Limited mitochondrial oxidation preserves glycolytic intermediates, which can be used as the building blocks for cancer cell proliferation. The highly activated glycolysis has been linked to mitochondrial fission in many types of cancer, including lung cancer (103), metastatic breast cancer (104), ovarian cancer (105), colorectal cancer (106, 107), pancreatic cancers (41), and melanoma (42). In the meantime, active oxidative metabolism has also been connected to mitochondrial fusion in some metastatic cancer models, including pancreatic cancer (108).
Mfn1/2 shows altered expression in human tumors (109, 110). Lower Mfn2 expression is observed in breast cancer (111), lung cancer (103), urinary bladder cancers (112), hepatocellular carcinoma (113), colorectal cancer (106), and gastric cancer (114), compared to the normal tissue. In line with the lower Mfn2 expression in cancer, several studies demonstrate that Mfn2 overexpression inhibits cancer cell proliferation and colony formation and weakens the invasion and migratory ability (106, 114–116). It is worth noting that the higher expression of Mfn2 has also been reported in lung adenocarcinoma tissues as compared to adjacent normal tissues, and Mfn2 knockdown results in impaired cancer cell proliferation (81). In ovarian cancer (82), breast cancer (83), and melanoma (84), Mfn2 knockdown suppresses oxygen consumption rate (OCR). In pancreatic cancer cells (PANC-1), Mfn2 knockdown enhances mitochondria-dependent energy metabolism by promoting activity of electron transport chain complexes (85). Besides, Mfn2 knockdown induces mitochondrial biogenesis and elevates OXPHOS in Ras-transformed mice fibroblasts (117). A recent study reported that liver-specific ablation of Mfn2 in mice provoked inflammation, triglyceride accumulation, fibrosis, and liver cancer (86). As both knockdown and overexpression of Mfn2 impair cancer cell survival in certain cancer types, further studies need to further explore the different roles of Mfn2 in various tumor types.
Opa1 and its proteolytic processing are important to maintain mitochondrial fusion. Studies have displayed a significant decrease in the expression of Opa1 along with high mitochondrial fragmentation in hepatocellular carcinoma (118). Tumor suppressor p53 is involved in Oma1-mediated L-Opa1 processing and mitochondrial fragmentation in ovarian and cervical cancer cells (119), the role of which is further supported by the close relationship between S-Opa1 and the p53 signaling pathway (120). Interestingly, in mouse adult fibroblasts (MAFs), Opa1 interacts with mitochondrial F1F0-ATP synthase, favors ATP synthase oligomerization, and finally, protects mitochondria from respiratory chain inhibition by modulating crista shape (89). Subsequent studies have discovered that the ATPase inhibitory factor 1 (IF1), a mitochondrial protein, prevents apoptotic remodeling of mitochondria by inhibiting Oma1 activation and Opa1 processing in HeLa cells, which further decreases OCR and intracellular ATP synthesis (90). Furthermore, Oma1 is dependent on the IF1 protective activity against apoptotic processing of Opa1 in HeLa cells (90). These observations display an essential role of ATP synthase in Opa1 processing to ensure normal mitochondrial bioenergetics. Since Opa1 processing complicates its function in maintenance of mitochondrial metabolism, future studies are required to focus more on the distinct regulatory machinery of Opa1 and its processing involved in mitochondrial metabolism within various cancer cells. This will provide insights into anti-tumor pharmaceutical therapy targeting Opa1-regulating mitochondrial metabolism.
Drp1 expression and activity have been associated with mitochondria fragmentation, which facilitates glycolysis in cancer cells. High Drp1 expression has been detected in lung cancer (103), breast cancer (104), thyroid cancer (121), glioblastoma (92), and hepatocellular carcinoma (91). In 2015, two back-to-back studies demonstrated the essential role of Drp1 activation in MAPK-driven tumor growth (41, 42). Moreover, Drp1 can be activated and phosphorylated by extracellular signal-regulated kinase (ERK1/2) on S616, which is required for Ras-induced transformation in MEFs (42). This S616 phosphorylation and mitochondrial fragmentation is enhanced in human pancreatic cancer (41) and brain tumor initiating cells (BTICs) (92). Similarly, the inhibition of Erk signaling promotes mitochondrial fusion and increases mitochondrial metabolism but reduces the Drp1 activity in A375, SK-MEL-28, and BT-474 cells (42). On the contrary, the reduced expression of Drp1 exerts different effects on oxidative metabolism in different cells. For instance, Drp1 knockdown increases OCR and promotes ATP production in Ras-transformed MEFs and the SK-MEL-28 cells (42). However, Drp1 knockdown decreases the maximal OCR in the HRas-transformed HEK-TtH cells (41) and in the T387 BTICs (92), which further prevents cell growth and tumor formation. A recent study shows that in lung cancer cells, sirtuin 4 (SIRT4, an nicotinamide-adenine dinucleotide+ (NAD+)-dependent protein deacetylase) inhibits Drp1 phosphorylation at the S616 site and weakens Drp1 recruitment to the mitochondrial membrane via an interaction with Fis1 (122). Besides, Drp1 can also be phosphorylated on S637, which subsequently represses its activity and mitochondrial translocation. Particularly, S637-mediated mitochondrial elongation under energy stress is essential for hepatocellular carcinoma cell survival both in vitro and in vivo (91). Interestingly, lung cancer cell lines have higher S616 and lower S637 phosphorylation, when compared to human bronchial epithelial cell (hBEC) (103).
mdivi-1 is widely used as a Drp1 inhibitor (30, 123). In 2008, Dr. Nunnari's group first utilized mdivi-1 to study mitochondrial dynamics (45), and most of the studies have followed this application of mdivi-1 to examine the metabolic role of Drp1. mdivi-1 treatments result in both increased and decreased OCR, depending on different cell types. In most conditions, mdivi-1 treatment induces mitochondrial fusion and increases mitochondrial activity, which further enhances OCR (124). In MDA-MB-231 cells and H1299 cells, mdivi-1 treatment significantly depresses OCR, without changing the cellular ATP level (125). Interestingly, a study shows that mdivi-1 represses OCR to a similar degree in both wild-type and Drp1 knockout MEF cells (126). This Drp1-independent role of mdivi-1 is further supported by the finding that mdivi-1 works as mitochondrial complex I inhibitor, to induce ROS and repress OCR (126). In the highly oxidative estrogen receptor–positive breast cancer cells, mdivi-1 reduces cellular mitochondrial bioenergetics and increases glycolysis without any influence on mitochondrial morphology (127). Overall, mdivi-1 has potential therapeutic avenues in suppressing tumor progression. Further studies are required to explore its effects on the oxidative metabolic alterations in cancer.
Summary
Dysregulated metabolism is a common feature of the metabolic diseases, including obesity, diabetes, and cancer, which can be regulated by mitochondrial dynamics (Figure 1). Obese and diabetic patients have pathology in multiple tissues, such as fat, muscle, liver, kidney, and pancreas. Cancer patients also have tumors from different origins, and all cancer types undergo different stages. Moreover, the specific mitochondrial dynamics under these pathology conditions are in response to different metabolic states, and thus, it is important to discuss the mitochondrial dynamics and metabolism in a context-dependent manner. For example, mild nutrient deprivation induces mitochondrial fusion and increases energy-producing efficiency to compensate the energy demand for growth, while severe nutrient deprivation drives mitochondrial fission and promotes cell apoptosis to preserve energy for neighbor cells. As demonstrated here, the GTPases (Mfn1, Mfn2, Opa1, and Drp1) have strong regulatory effects in balancing mitochondrial fusion and fission, and these GTPases control the aforementioned metabolic diseases predominantly through orchestrating oxidative metabolism. It is essential to explore how the GTPases orchestrate metabolic alterations under certain nutrient environments. Since mitochondrial morphology undergoes a dynamic process balanced by the above GTPases, knockdown and knockout of these result in abnormal chronical inhibition and can potentially cause indirect metabolic effects. Therefore, it is important to develop new chemical inhibitors to acutely block the function of these GTPases. Oxygen consumption is only a small fraction in metabolism, and more direct metabolomics and metabolic flux analyses will provide a clearer picture of the mitochondrial dynamics and metabolism in obesity, diabetes, and cancer.
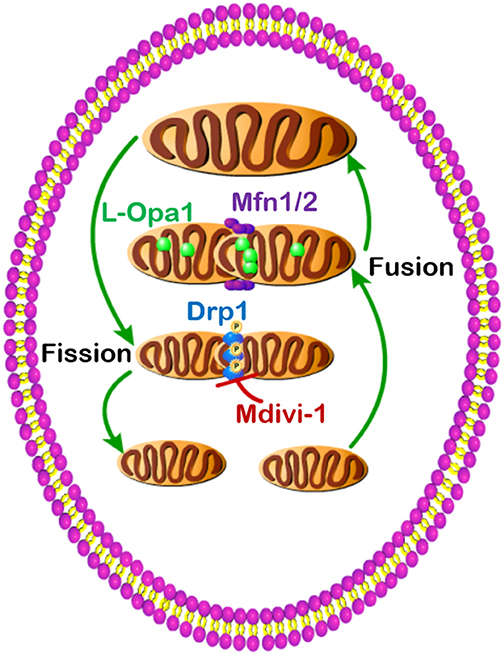
Figure 1. Dysregulated mitochondrial dynamics and metabolism in obesity, diabetes, and cancer.
Author Contributions
LJ designed the review. WD made the figure and tables. WD and LJ wrote the review.
Funding
Research reported in this publication was also supported by the National Cancer Institute of the National Institutes of Health under award number P30CA33572.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to apologize to those colleagues whose work could not be cited or discussed in sufficient detail due to space limitations.
References
1. Archer SL. Mitochondrial dynamics—mitochondrial fission and fusion in human diseases. N Engl J Med. (2013) 369:2236–51. doi: 10.1056/NEJMra1215233
2. Lewis MR, Lewis WH. Mitochondria in tissue culture. Science. (1914) 39:330–3. doi: 10.1126/science.39.1000.330
3. Lewis MR, Lewis WH. Mitochondria (and other cytoplasmic structures) in tissue cultures. Am J Anat. (1915) 17:339–401. doi: 10.1002/aja.1000170304
4. Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet. (2012) 46:265–87. doi: 10.1146/annurev-genet-110410-132529
5. Chen H, Chan DC. Mitochondrial dynamics in regulating the unique phenotypes of cancer and stem cells. Cell Metabol. (2017) 26:39–48. doi: 10.1016/j.cmet.2017.05.016
6. Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. (2010) 11:872–84. doi: 10.1038/nrm3013
7. Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. (2003) 160:189–200. doi: 10.1083/jcb.200211046
8. Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci USA. (2004) 101:15927–32. doi: 10.1073/pnas.0407043101
9. Song Z, Ghochani M, McCaffery JM, Frey TG, Chan DC. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol Biol Cell. (2009) 20:3525–32. doi: 10.1091/mbc.E09-03-0252
10. Mishra P, Carelli V, Manfredi G, Chan DC. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metabol. (2014) 19:630–41. doi: 10.1016/j.cmet.2014.03.011
11. Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, et al. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. (2001) 1:515–25. doi: 10.1016/s1534-5807(01)00055-7
12. Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot–Marie–Tooth neuropathy type 2A. Nat Genet. (2004) 36:449–51. doi: 10.1038/ng1341
13. Alexander C, Votruba M, Pesch UEA, Thiselton DL, Mayer S, Moore A, et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. (2000) 26:211. doi: 10.1038/79944
14. Delettre C, Lenaers G, Griffoin J-M, Gigarel N, Lorenzo C, Belenguer P, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. (2000) 26:207. doi: 10.1038/79936
15. Hales KG, Fuller MT. Developmentally regulated mitochondrial fusion mediated by a conserved, novel, predicted GTPase. Cell. (1997) 90:121–9. doi: 10.1016/S0092-8674(00)80319-0
16. Santel A, Fuller MT. Control of mitochondrial morphology by a human mitofusin. J Cell Sci. (2001) 114:867–74.
17. Legros F, Lombès A, Frachon P, Rojo M. Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol Biol Cell. (2002) 13:4343–54. doi: 10.1091/mbc.e02-06-0330
18. Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. (2005) 280:26185–92. doi: 10.1074/jbc.M503062200
19. Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science. (2004) 305:858–62. doi: 10.1126/science.1099793
20. Wong ED, Wagner JA, Gorsich SW, McCaffery JM, Shaw JM, Nunnari J. The dynamin-related GTPase, Mgm1p, is an intermembrane space protein required for maintenance of fusion competent mitochondria. J Cell Biol. (2000) 151:341–52. doi: 10.1083/jcb.151.2.341
21. Sesaki H, Southard SM, Yaffe MP, Jensen RE. Mgm1p, a Dynamin-related GTPase, is essential for fusion of the mitochondrial outer membrane. Mol Biol Cell. (2003) 14:2342–56. doi: 10.1091/mbc.e02-12-0788
22. Olichon A, Elachouri G, Baricault L, Delettre C, Belenguer P, Lenaers G. OPA1 alternate splicing uncouples an evolutionary conserved function in mitochondrial fusion from a vertebrate restricted function in apoptosis. Cell Death Differ. (2007) 14:682–92. doi: 10.1038/sj.cdd.4402048
23. Song Z, Chen H, Fiket M, Alexander C, Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol. (2007) 178:749–55. doi: 10.1083/jcb.200704110
24. Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, et al. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol. (2014) 204:919–29. doi: 10.1083/jcb.201308006
25. Ehses S, Raschke I, Mancuso G, Bernacchia A, Geimer S, Tondera D, et al. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J Cell Biol. (2009) 187:1023–36. doi: 10.1083/jcb.200906084
26. MacVicar T, Langer T. OPA1 processing in cell death and disease—the long and short of it. J Cell Sci. (2016) 129:2297–306. doi: 10.1242/jcs.159186
27. Lee H, Smith SB, Yoon Y. The short variant of the mitochondrial dynamin OPA1 maintains mitochondrial energetics and cristae structure. J Biol Chem. (2017) 292:7115–30. doi: 10.1074/jbc.M116.762567
28. Waterham HR, Koster J, van Roermund CW, Mooyer PA, Wanders RJ, Leonard JV. A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med. (2007) 356:1736–41. doi: 10.1056/NEJMoa064436
29. Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol. (2009) 11:958–66. doi: 10.1038/ncb1907
30. Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, et al. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol. (2009) 186:805–16. doi: 10.1083/jcb.200903065
31. Loson OC, Song ZY, Chen HC, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell. (2013) 24:659–67. doi: 10.1091/mbc.E12-10-0721
32. Samangouei P, Crespo-Avilan GE, Cabrera-Fuentes H, Hernandez-Resendiz S, Ismail NI, Katwadi KB, et al. MiD49 and MiD51: new mediators of mitochondrial fission and novel targets for cardioprotection. Cond Med. (2018) 1:239–46.
33. Yoon Y, Krueger EW, Oswald BJ, McNiven MA. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol. (2003) 23:5409–20. doi: 10.1128/mcb.23.15.5409-5420.2003
34. Stojanovski D, Koutsopoulos OS, Okamoto K, Ryan MT. Levels of human Fis1 at the mitochondrial outer membrane regulate mitochondrial morphology. J Cell Sci. (2004) 117(Pt 7):1201–10. doi: 10.1242/jcs.01058
35. Gandre-Babbe S, van der Bliek AM. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell. (2008) 19:2402–12. doi: 10.1091/mbc.E07-12-1287
36. Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, et al. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. (2010) 191:1141–58. doi: 10.1083/jcb.201007152
37. Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. (2011) 12:565–73. doi: 10.1038/embor.2011.54
38. Palmer CS, Elgass KD, Parton RG, Osellame LD, Stojanovski D, Ryan MT. Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. J Biol Chem. (2013) 288:27584–93. doi: 10.1074/jbc.M113.479873
39. Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. (2007) 282:11521–9. doi: 10.1074/jbc.M607279200
40. Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP–dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. (2007) 8:939–44. doi: 10.1038/sj.embor.7401062
41. Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, et al. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol Cell. (2015) 57:537–51. doi: 10.1016/j.molcel.2015.01.002
42. Serasinghe MN, Wieder SY, Renault TT, Elkholi R, Asciolla JJ, Yao JL, et al. Mitochondrial division is requisite to RAS-induced transformation and targeted by oncogenic MAPK pathway inhibitors. Mol Cell. (2015) 57:521–36. doi: 10.1016/j.molcel.2015.01.003
43. Chang CR, Blackstone C. Cyclic AMP–dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem. (2007) 282:21583–7. doi: 10.1074/jbc.C700083200
44. Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, et al. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci USA. (2008) 105:15803–8. doi: 10.1073/pnas.0808249105
45. Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. (2008) 14:193–204. doi: 10.1016/j.devcel.2007.11.019
46. Zorzano A, Liesa M, Palacin M. Mitochondrial dynamics as a bridge between mitochondrial dysfunction and insulin resistance. Arch Physiol Biochem. (2009) 115:1–12. doi: 10.1080/13813450802676335
47. Bach D, Naon D, Pich S, Soriano FX, Vega N, Rieusset J, et al. Expression of Mfn2, the Charcot–Marie–Tooth neuropathy type 2A gene, in human skeletal muscle: effects of type 2 diabetes, obesity, weight loss, and the regulatory role of tumor necrosis factor alpha and interleukin-6. Diabetes. (2005) 54:2685–93. doi: 10.2337/diabetes.54.9.2685
48. Pich S, Bach D, Briones P, Liesa M, Camps M, Testar X, et al. The Charcot–Marie–Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum Mole Genet. (2005) 14:1405–15. doi: 10.1093/hmg/ddi149
49. Schneeberger M, Dietrich MO, Sebastian D, Imbernon M, Castano C, Garcia A, et al. Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell. (2013) 155:172–87. doi: 10.1016/j.cell.2013.09.003
50. Naon D, Zaninello M, Giacomello M, Varanita T, Grespi F, Lakshminaranayan S, et al. Critical reappraisal confirms that mitofusin 2 is an endoplasmic reticulum–mitochondria tether. Proc Natl Acad Sci USA. (2016) 113:11249–54. doi: 10.1073/pnas.1606786113
51. Boutant M, Kulkarni SS, Joffraud M, Ratajczak J, Valera-Alberni M, Combe R, et al. Mfn2 is critical for brown adipose tissue thermogenic function. EMBO J. (2017) 36:1543–58. doi: 10.15252/embj.201694914
52. Mahdaviani K, Benador IY, Su S, Gharakhanian RA, Stiles L, Trudeau KM, et al. Mfn2 deletion in brown adipose tissue protects from insulin resistance and impairs thermogenesis. EMBO Rep. (2017) 18:1123–38. doi: 10.15252/embr.201643827
53. Montaigne D, Marechal X, Coisne A, Debry N, Modine T, Fayad G, et al. Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation. (2014) 130:554–64. doi: 10.1161/circulationaha.113.008476
54. Kulkarni SS, Joffraud M, Boutant M, Ratajczak J, Gao AW, Maclachlan C, et al. Mfn1 deficiency in the liver protects against diet-induced insulin resistance and enhances the hypoglycemic effect of metformin. Diabetes. (2016) 65:3552–60. doi: 10.2337/db15-1725
55. Liu R, Jin P, Yu L, Wang Y, Han L, Shi T, et al. Impaired mitochondrial dynamics and bioenergetics in diabetic skeletal muscle. PLoS ONE. (2014) 9:e92810. doi: 10.1371/journal.pone.0092810
56. Ramirez S, Gomez-Valades AG, Schneeberger M, Varela L, Haddad-Tovolli R, Altirriba J, et al. Mitochondrial dynamics mediated by mitofusin 1 is required for POMC neuron glucose-sensing and insulin release control. Cell Metabol. (2017) 25:1390–9.e6. doi: 10.1016/j.cmet.2017.05.010
57. Pidoux G, Witczak O, Jarnaess E, Myrvold L, Urlaub H, Stokka AJ, et al. Optic atrophy 1 is an A-kinase anchoring protein on lipid droplets that mediates adrenergic control of lipolysis. EMBO J. (2011) 30:4371–86. doi: 10.1038/emboj.2011.365
58. Tezze C, Romanello V, Desbats MA, Fadini GP, Albiero M, Favaro G, et al. Age-associated loss of OPA1 in muscle impacts muscle mass, metabolic homeostasis, systemic inflammation, and epithelial senescence. Cell Metabol. (2017) 25:1374–89.e6. doi: 10.1016/j.cmet.2017.04.021
59. Rodriguez-Nuevo A, Diaz-Ramos A, Noguera E, Diaz-Saez F, Duran X, Munoz JP, et al. Mitochondrial DNA and TLR9 drive muscle inflammation upon Opa1 deficiency. EMBO J. (2018) 37:e96553. doi: 10.15252/embj.201796553
60. Pereira RO, Tadinada SM, Zasadny FM, Oliveira KJ, Pires KMP, Olvera A, et al. OPA1 deficiency promotes secretion of FGF21 from muscle that prevents obesity and insulin resistance. EMBO J. (2017) 36:2126–45. doi: 10.15252/embj.201696179
61. Grant RW, Vester Boler BM, Ridge TK, Graves TK, Swanson KS. Adipose tissue transcriptome changes during obesity development in female dogs. Physiol Genomics. (2011) 43:295–307. doi: 10.1152/physiolgenomics.00190.2010
62. Zhang Z, Wakabayashi N, Wakabayashi J, Tamura Y, Song WJ, Sereda S, et al. The dynamin-related GTPase Opa1 is required for glucose-stimulated ATP production in pancreatic beta cells. Mol Biol Cell. (2011) 22:2235–45. doi: 10.1091/mbc.E10-12-0933
63. Parra V, Verdejo HE, Iglewski M, Del Campo A, Troncoso R, Jones D, et al. Insulin stimulates mitochondrial fusion and function in cardiomyocytes via the Akt–mTOR–NFkappaB–Opa-1 signaling pathway. Diabetes. (2014) 63:75–88. doi: 10.2337/db13-0340
64. Touvier T, De Palma C, Rigamonti E, Scagliola A, Incerti E, Mazelin L, et al. Muscle-specific Drp1 overexpression impairs skeletal muscle growth via translational attenuation. Cell Death Dis. (2015) 6:e1663. doi: 10.1038/cddis.2014.595
65. Moore TM, Zhou Z, Cohn W, Norheim F, Lin AJ, Kalajian N, et al. The impact of exercise on mitochondrial dynamics and the role of Drp1 in exercise performance and training adaptations in skeletal muscle. Mol Metabol. (2019) 21:51–67. doi: 10.1016/j.molmet.2018.11.012
66. Fealy CE, Mulya A, Lai N, Kirwan JP. Exercise training decreases activation of the mitochondrial fission protein dynamin-related protein-1 in insulin-resistant human skeletal muscle. J Appl Physiol. (2014) 117:239–45. doi: 10.1152/japplphysiol.01064.2013
67. Kabra UD, Pfuhlmann K, Migliorini A, Keipert S, Lamp D, Korsgren O, et al. Direct substrate delivery into mitochondrial fission-deficient pancreatic islets rescues insulin secretion. Diabetes. (2017) 66:1247–57. doi: 10.2337/db16-1088
68. Lin HY, Weng SW, Chang YH, Su YJ, Chang CM, Tsai CJ, et al. The causal role of mitochondrial dynamics in regulating insulin resistance in diabetes: link through mitochondrial reactive oxygen species. Oxid Med Cell Longev. (2018) 2018:7514383. doi: 10.1155/2018/7514383
69. Hennings TG, Chopra DG, DeLeon ER, VanDeusen HR, Sesaki H, Merrins MJ, et al. In vivo deletion of beta-cell Drp1 impairs insulin secretion without affecting islet oxygen consumption. Endocrinology. (2018) 159:3245–56. doi: 10.1210/en.2018-00445
70. Jheng HF, Tsai PJ, Guo SM, Kuo LH, Chang CS, Su IJ, et al. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol Cell Biol. (2012) 32:309–19. doi: 10.1128/mcb.05603-11
71. Favaro G, Romanello V, Varanita T, Andrea Desbats M, Morbidoni V, Tezze C, et al. DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat Commun. (2019) 10:2576. doi: 10.1038/s41467-019-10226-9
72. Wang L, Ishihara T, Ibayashi Y, Tatsushima K, Setoyama D, Hanada Y, et al. Disruption of mitochondrial fission in the liver protects mice from diet-induced obesity and metabolic deterioration. Diabetologia. (2015) 58:2371–80. doi: 10.1007/s00125-015-3704-7
73. Santoro A, Campolo M, Liu C, Sesaki H, Meli R, Liu ZW, et al. DRP1 suppresses leptin and glucose sensing of POMC neurons. Cell Metabol. (2017) 25:647–60. doi: 10.1016/j.cmet.2017.01.003
74. Filippi BM, Abraham MA, Silva PN, Rasti M, LaPierre MP, Bauer PV, et al. Dynamin-related protein 1–dependent mitochondrial fission changes in the dorsal vagal complex regulate insulin action. Cell Rep. (2017) 18:2301–9. doi: 10.1016/j.celrep.2017.02.035
75. Kim JH, Park SJ, Kim B, Choe YG, Lee DS. Insulin-stimulated lipid accumulation is inhibited by ROS-scavenging chemicals, but not by the Drp1 inhibitor mdivi-1. PLoS ONE. (2017) 12:e0185764. doi: 10.1371/journal.pone.0185764
76. Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. (2002) 51:2944–50. doi: 10.2337/diabetes.51.10.2944
77. Scheideler M, Herzig S. Let's burn whatever you have: mitofusin 2 metabolically re-wires brown adipose tissue. EMBO Rep. (2017) 18:1039–40. doi: 10.15252/embr.201744341
78. Rogne M, Chu DT, Kuntziger TM, Mylonakou MN, Collas P, Tasken K. OPA1-anchored PKA phosphorylates perilipin 1 on S522 and S497 in adipocytes differentiated from human adipose stem cells. Mol Biol Cell. (2018) 29:1487–501. doi: 10.1091/mbc.E17-09-0538
79. Quiros PM, Ramsay AJ, Sala D, Fernandez-Vizarra E, Rodriguez F, Peinado JR, et al. Loss of mitochondrial protease OMA1 alters processing of the GTPase OPA1 and causes obesity and defective thermogenesis in mice. EMBO J. (2012) 31:2117–33. doi: 10.1038/emboj.2012.70
80. Romanello V, Scalabrin M, Albiero M, Blaauw B, Scorrano L, Sandri M. Inhibition of the fission machinery mitigates OPA1 Impairment in adult skeletal muscles. Cells. (2019) 8:E597. doi: 10.3390/cells8060597
81. Lou Y, Li R, Liu J, Zhang Y, Zhang X, Jin B, et al. Mitofusin-2 over-expresses and leads to dysregulation of cell cycle and cell invasion in lung adenocarcinoma. Med Oncol. (2015) 32:132. doi: 10.1007/s12032-015-0515-0
82. Chakraborty PK, Murphy B, Mustafi SB, Dey A, Xiong X, Rao G, et al. Cystathionine beta-synthase regulates mitochondrial morphogenesis in ovarian cancer. FASEB J. (2018) 32:4145–57. doi: 10.1096/fj.201701095R
83. Cheng CT, Kuo CY, Ouyang C, Li CF, Chung Y, Chan DC, et al. Metabolic stress–induced phosphorylation of KAP1 Ser473 blocks mitochondrial fusion in breast cancer cells. Cancer Res. (2016) 76:5006–18. doi: 10.1158/0008-5472.CAN-15-2921
84. Dal Yontem F, Kim SH, Ding Z, Grimm E, Ekmekcioglu S, Akcakaya H. Mitochondrial dynamic alterations regulate melanoma cell progression. J Cell Biochem. (2018) 120:2098–108. doi: 10.1002/jcb.27518
85. Pan L, Zhou L, Yin W, Bai J, Liu R. miR-125a induces apoptosis, metabolism disorder and migration impairment in pancreatic cancer cells by targeting Mfn2-related mitochondrial fission. Int J Oncol. (2018) 53:124–36. doi: 10.3892/ijo.2018.4380
86. Hernandez-Alvarez MI, Sebastian D, Vives S, Ivanova S, Bartoccioni P, Kakimoto P, et al. Deficient endoplasmic reticulum–mitochondrial phosphatidylserine transfer causes liver disease. Cell. (2019) 177:881–95.e17. doi: 10.1016/j.cell.2019.04.010
87. Yamada S, Yamazaki D, Kanda Y. 5-Fluorouracil inhibits neural differentiation via Mfn1/2 reduction in human induced pluripotent stem cells. J Toxicol Sci. (2018) 43:727–34. doi: 10.2131/jts.43.727
88. Sastre-Serra J, Nadal-Serrano M, Pons DG, Roca P, Oliver J. Mitochondrial dynamics is affected by 17beta-estradiol in the MCF-7 breast cancer cell line. Effects on fusion and fission related genes. Int J Biochem Cell Biol. (2012) 44:1901–5. doi: 10.1016/j.biocel.2012.07.012
89. Quintana-Cabrera R, Quirin C, Glytsou C, Corrado M, Urbani A, Pellattiero A, et al. The cristae modulator Optic atrophy 1 requires mitochondrial ATP synthase oligomers to safeguard mitochondrial function. Nat Commun. (2018) 9:3399. doi: 10.1038/s41467-018-05655-x
90. Faccenda D, Nakamura J, Gorini G, Dhoot GK, Piacentini M, Yoshida M, et al. Control of mitochondrial remodeling by the ATPase inhibitory factor 1 unveils a pro-survival relay via OPA1. Cell Rep. (2017) 18:1869–83. doi: 10.1016/j.celrep.2017.01.070
91. Li J, Huang Q, Long X, Guo X, Sun X, Jin X, et al. Mitochondrial elongation–mediated glucose metabolism reprogramming is essential for tumour cell survival during energy stress. Oncogene. (2017) 36:4901–12. doi: 10.1038/onc.2017.98
92. Xie Q, Wu Q, Horbinski CM, Flavahan WA, Yang K, Zhou W, et al. Mitochondrial control by DRP1 in brain tumor initiating cells. Nat Neurosci. (2015) 18:501–10. doi: 10.1038/nn.3960
93. Warburg O. Iron, the oxygen-carrier of respiration-ferment. Science. (1925) 61:575–82. doi: 10.1126/science.61.1588.575
94. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. (1927) 8:519–30.
96. Otto AM. Warburg effect(s)—a biographical sketch of Otto Warburg and his impacts on tumor metabolism. Cancer Metabol. (2016) 4:5. doi: 10.1186/s40170-016-0145-9
97. DeBerardinis RJ, Thompson CB. Cellular metabolism and disease: what do metabolic outliers teach us? Cell. (2012) 148:1132–44. doi: 10.1016/j.cell.2012.02.032
98. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. (2011) 11:85–95. doi: 10.1038/nrc2981
99. Palm W, Thompson CB. Nutrient acquisition strategies of mammalian cells. Nature. (2017) 546:234–42. doi: 10.1038/nature22379
101. Jiang L, Deberardinis R, Boothman DA. The cancer cell ‘energy grid’: TGF-beta1 signaling coordinates metabolism for migration. Mol Cell Oncol. (2015) 2:e981994. doi: 10.4161/23723556.2014.981994
102. Jiang L, Xiao L, Sugiura H, Huang X, Ali A, Kuro-o M, et al. Metabolic reprogramming during TGFbeta1-induced epithelial-to-mesenchymal transition. Oncogene. (2015) 34:3908–16. doi: 10.1038/onc.2014.321
103. Rehman J, Zhang HJ, Toth PT, Zhang Y, Marsboom G, Hong Z, et al. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. (2012) 26:2175–86. doi: 10.1096/fj.11-196543
104. Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, et al. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene. (2013) 32:4814–24. doi: 10.1038/onc.2012.494
105. Tanwar DK, Parker DJ, Gupta P, Spurlock B, Alvarez RD, Basu MK, et al. Crosstalk between the mitochondrial fission protein, Drp1, and the cell cycle is identified across various cancer types and can impact survival of epithelial ovarian cancer patients. Oncotarget. (2016) 7:60021–37. doi: 10.18632/oncotarget.11047
106. Cheng X, Zhou D, Wei J, Lin J. Cell-cycle arrest at G2/M and proliferation inhibition by adenovirus-expressed mitofusin-2 gene in human colorectal cancer cell lines. Neoplasma. (2013) 60:620–6. doi: 10.4149/neo_2013_080
107. Inoue-Yamauchi A, Oda H. Depletion of mitochondrial fission factor DRP1 causes increased apoptosis in human colon cancer cells. Biochem Biophys Res Commun. (2012) 421:81–5. doi: 10.1016/j.bbrc.2012.03.118
108. Yu M, Huang Y, Deorukhkar A, Fujimoto T, Govindaraju S, Molkentine J, et al. Mitochondrial fusion suppresses pancreatic cancer growth via reduced oxidative metabolism. bioRxiv. (2018) 2018:279745. doi: 10.1101/279745
109. Senft D, Ronai ZA. Regulators of mitochondrial dynamics in cancer. Curr Opin Cell Biol. (2016) 39:43–52. doi: 10.1016/j.ceb.2016.02.001
110. Schrepfer E, Scorrano L. Mitofusins, from mitochondria to metabolism. Mol Cell. (2016) 61:683–94. doi: 10.1016/j.molcel.2016.02.022
111. Xu K, Chen G, Li X, Wu X, Chang Z, Xu J, et al. MFN2 suppresses cancer progression through inhibition of mTORC2/Akt signaling. Sci Rep. (2017) 7:41718. doi: 10.1038/srep41718
112. Jin B, Fu G, Pan H, Cheng X, Zhou L, Lv J, et al. Anti-tumour efficacy of mitofusin-2 in urinary bladder carcinoma. Med Oncol. (2011) 28(Suppl. 1):S373–80. doi: 10.1007/s12032-010-9662-5
113. Wu Y, Zhou D, Xu X, Zhao X, Huang P, Zhou X, et al. Clinical significance of mitofusin-2 and its signaling pathways in hepatocellular carcinoma. World J Surg Oncol. (2016) 14:179. doi: 10.1186/s12957-016-0922-5
114. Zhang GE, Jin HL, Lin XK, Chen C, Liu XS, Zhang Q, et al. Anti-tumor effects of Mfn2 in gastric cancer. Int J Mol Sci. (2013) 14:13005–21. doi: 10.3390/ijms140713005
115. Xue R, Meng Q, Lu D, Liu X, Wang Y, Hao J. Mitofusin2 induces cell autophagy of pancreatic cancer through inhibiting the PI3K/Akt/mTOR signaling pathway. Oxid Med Cell Longev. (2018) 2018:2798070. doi: 10.1155/2018/2798070
116. Wang W, Liu X, Guo X, Quan H. Mitofusin-2 triggers cervical carcinoma cell Hela apoptosis via mitochondrial pathway in mouse model. Cell Physiol Biochem. (2018) 46:69–81. doi: 10.1159/000488410
117. Yao CH, Wang R, Wang Y, Kung CP, Weber JD, Patti GJ. Mitochondrial fusion supports increased oxidative phosphorylation during cell proliferation. eLife. (2019) 8:41351. doi: 10.7554/eLife.41351
118. Zhao X, Tian C, Puszyk WM, Ogunwobi OO, Cao M, Wang T, et al. OPA1 downregulation is involved in sorafenib-induced apoptosis in hepatocellular carcinoma. Lab Invest. (2013) 93:8–19. doi: 10.1038/labinvest.2012.144
119. Kong B, Wang Q, Fung E, Xue K, Tsang BK. p53 is required for cisplatin-induced processing of the mitochondrial fusion protein L-Opa1 that is mediated by the mitochondrial metallopeptidase Oma1 in gynecologic cancers. J Biol Chem. (2014) 289:27134–45. doi: 10.1074/jbc.M114.594812
120. Alavi MV. Targeted OMA1 therapies for cancer. Int J Cancer. (2019) 2019:32177. doi: 10.1002/ijc.32177
121. Ferreira-da-Silva A, Valacca C, Rios E, Populo H, Soares P, Sobrinho-Simoes M, et al. Mitochondrial dynamics protein Drp1 is overexpressed in oncocytic thyroid tumors and regulates cancer cell migration. PLoS ONE. (2015) 10:e0122308. doi: 10.1371/journal.pone.0122308
122. Fu L, Dong Q, He J, Wang X, Xing J, Wang E, et al. SIRT4 inhibits malignancy progression of NSCLCs, through mitochondrial dynamics mediated by the ERK–Drp1 pathway. Oncogene. (2017) 36:2724–36. doi: 10.1038/onc.2016.425
123. Smith G, Gallo G. To mdivi-1 or not to mdivi-1: is that the question? Dev Neurobiol. (2017) 77:1260–8. doi: 10.1002/dneu.22519
124. Sharp WW, Fang YH, Han M, Zhang HJ, Hong Z, Banathy A, et al. Dynamin-related protein 1 (Drp1)–mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J. (2014) 28:316–26. doi: 10.1096/fj.12-226225
125. Qian W, Wang J, Roginskaya V, McDermott LA, Edwards RP, Stolz DB, et al. Novel combination of mitochondrial division inhibitor 1 (mdivi-1) and platinum agents produces synergistic pro-apoptotic effect in drug resistant tumor cells. Oncotarget. (2014) 5:4180–94. doi: 10.18632/oncotarget.1944
126. Bordt EA, Clerc P, Roelofs BA, Saladino AJ, Tretter L, Adam-Vizi V, et al. The putative Drp1 inhibitor mdivi-1 is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Dev Cell. (2017) 40:583–94.e6. doi: 10.1016/j.devcel.2017.02.020
Keywords: energy metabolism, TCA cycle, mitochondrial dynamics, GTPase, mdivi-1
Citation: Dai W and Jiang L (2019) Dysregulated Mitochondrial Dynamics and Metabolism in Obesity, Diabetes, and Cancer. Front. Endocrinol. 10:570. doi: 10.3389/fendo.2019.00570
Received: 13 March 2019; Accepted: 05 August 2019;
Published: 03 September 2019.
Edited by:
Mei Kong, University of California, Irvine, United StatesReviewed by:
Jiangbin Ye, Stanford University, United StatesVanina Romanello, Veneto Institute of Molecular Medicine (VIMM), Italy
Copyright © 2019 Dai and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lei Jiang, bGppYW5nQGNvaC5vcmc=