 María Inés Diaz Bessone
María Inés Diaz Bessone María José Gattas1
María José Gattas1 Marina Simian
Marina Simian- 1Laboratory of NanoBiology, Instituto de Nanosistemas, Universidad Nacional de San Martín, Buenos Aires, Argentina
- 2Amsterdam UMC, VUmc School of Medical Sciences, University of Vrije, Amsterdam, Netherlands
Estrogen receptor positive breast neoplasias represent over 70% of diagnosed breast cancers. Depending on the stage at which the tumor is detected, HER2 status and genomic risk, endocrine therapy is combined with either radio, chemo and/or targeted therapy. A growing amount of evidence supports the notion that components of the tumor microenvironment play specific roles in response to treatment and that strategies targeting these key interactions with tumor cells could pave the way to a new generation of therapies. In this review, we analyze the evidence suggesting different components of the tumor microenvironment play a role in hormone receptor positive breast cancer progression. In particular we focus on the immune system, carcinoma associated fibroblasts and the extracellular matrix. Further insight into the cross talk between these constituents of the microenvironment and the tumor cells may lead to therapies that eliminate disseminated metastatic cells early on, and thus reduce distant disease relapse which is the leading cause of death for patients who are diagnosed with this illness.
Introduction
Breast cancer is the most frequent cancer in women in the western world; one in eight women will have breast cancer at some point in their life (1). Seventy-five percent of diagnosed breast tumors express estrogen receptor-alpha (ERα) and endocrine therapy is the treatment of choice for patients with tumors of these characteristics. Within the scope of endocrine therapies, tamoxifen, a selective estrogen receptor modulator (SERM), has been the most widely used over the last 30 years (2). However, today other options are available such as aromatase inhibitors (AI) and selective estrogen receptor downregulators (SERDs) such as Fulvestrant (3). Standard-of-care regimens have not been established for ER+ tumors. Depending on the stage at which the tumor is detected, HER2 status and genomic risk, endocrine therapy is combined with either radio, chemo and/or targeted therapy (4). Patients with ER+ early stage breast cancer are susceptible to late recurrence that can take place even after 15 years of treatment interruption. Several strategies have been studied to prolong adjuvant endocrine therapy from 5 to 10 years, including 10 years of therapy with tamoxifen, 10 years of tamoxifen with an AI, or a 5-year period of tamoxifen followed by 2–5 years of treatment with an AI. Overall, the best outcomes with extended therapy in patients with a high risk of relapse are found in those who have received 5 years of tamoxifen and up to 3 years of an AI (5, 6).
Hormones and ER-α
ERα is a transcription factor present in different adult tissues such as mammary gland, ovaries, uterus and brain (7, 8) regulating cell proliferation, migration and survival. In the breast ERα controls development and plays a key role in tumor growth as mentioned above (9). Full length ERα is a 66-kDa ligand-dependent transcription factor that is activated by 17-β-estradiol. ERα shares a common structural organization with other steroid hormone receptors consisting of two transcriptional activation domains, the AF-1 N-terminal ligand-independent activation function domain and the AF-2 C-terminal ligand-dependent domain. A ligand-binding domain (LBD) also resides in the C-terminal region, and the DNA-binding and hinge domains are located in the central region of the protein (Figure 1). A palmitoylation site is found at Cys447 within the AF-2 domain (8, 10). In breast cancer cells two shorter isoforms of ERα have been described, ERα46 that lacks the A/B domain (11) and ERα36 that lacks both AF-1 and AF-2 transactivation domains and has an extra 27aa c-terminal domain (12, 13). According to the accepted mechanism of action, unbound full length ERs are located as monomers mostly in the nucleus and dimerize upon steroid binging. A percentage of the receptor sits in the cytoplasm bound to HSP90, that is released upon estrogen binding, enabling dimerization and traslocation of the receptor to the nucleus. In the nucleus, dimerized ERs bind estrogen-response-elements in the promoter regions of target genes regulating their transcription. ERα can also act as a co-regulator of other transcription factors such as AP-1, SP-1, and NF-κB especially when increased tyrosine kinase activity leads to phosphorylation of the AF-1 N-terminal region. Palmitoylated ERα localizes at diverse extranuclear compartments, including the plasma membrane, and is proposed to mediate rapid, non-genomic actions of ERα in the context of a cross talk with tyrosine kinase membrane receptors such as EGFR (14). It is now well-accepted that it is the same nuclear ERα that is modified and localizes at the membrane. In depth reviews of ER signaling have been recently published elsewhere (10, 15).
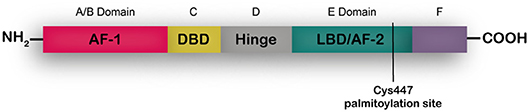
Figure 1. ERα structume. The functional domains ERα include the DNA binding domain (DBD), ligand binding domain (LBD), and two transcriptional activation functions (AF), the AF-1 ligand-independent activation function domain and the AF-2 ligand-dependent activation domain. The A/B domain, at the amino terminus of the protein, contains AF-1. The C domain binds to DNA motifs (EREs) at target genes. The D domain or hinge region contributes to DNA binding specificity and nuclear localization. The E domain or LBD interacts with estrogens or SERMs. At the C- terminus is the F domain.
Endocrine therapy for the treatment of breast cancer, in the form of tamoxifen, was the first targeted therapy to be developed (2). Tamoxifen is classified as a SERM because it modulates ER's activity by leading to the recruitment of co-inhibitors when the receptor binds 17-β-estradiol and thus impairs its transcriptional activity. Aromatase inhibitors inhibit the local synthesis of estrogens and are recommended for postmenopausal patients (16). SERD's, like Fulvestrant, lead to the degradation and dowregulation of the bound receptor (17).
While endocrine therapy is the most effective treatment for ER+ breast cancer, its effectiveness is limited by considerable rates of de novo (intrinsic) and acquired resistance. It is estimated that 50% of patients with metastatic disease will not respond to endocrine therapy and 30% of patients with early disease will eventually relapse having initially responded to therapy (18). Thus, understanding what leads to resistance is of great clinical importance considering that about 12% women in the U.S. are expected to have breast cancer at some point in their life (1).
Distant disease relapse is the main cause of death for patients who are diagnosed with early stage breast cancer. Breast cancer cells spread and settle in other tissues in small foci called micrometastasis. Evidence suggests that cells leave the primary tumor in the initial stages of tumor development (19). Genomic profiling of disseminated tumor cells in bone marrow shows that they are less genomically aberrant and have fewer copy number alterations than their corresponding primary tumors (20). Cells within micrometastasis can remain dormant for many years (21). The mechanisms involved in maintaining the dormant state and what actually triggers the onset of proliferation and the development of overt metastasis is still under debate. Interestingly, even though ER+ breast tumors are a perfect example of this clinical scenario, very few papers have actually addressed the mechanisms that are involved in recurrence in the context of endocrine therapy. Clinical data reveal that more than half of the recurrences of ER+ tumors take place 5 years or longer after diagnosis and surgery of the primary tumor; some patients experience recurrence after more than 20 years (22). The fact that prolonging adjuvant treatment from 5 to 10 years reduces recurrence and death between years 10 and 15 strongly suggests that blocking ER signaling maintains cell proliferation suppressed and impedes the exit from the dormant state. In this sense, Ogba et al. recently showed that breast cancer metastases and tumor arousal from dormancy are promoted by direct actions of estradiol and progesterone on the malignant cells (23). Through a series of elegant experiments using ovariectomized nude mice and four breast cancer cell lines that differed in their levels of expression of ER, PR, and CK5 they showed that ER+ PR+ luminal tumor cells can seed distant organs, where they remain dormant as micrometastases and sheltered from therapies but arousable by hormone repletion. Interestingly, as demonstrated in human clinical samples, the micromestastasis were composed of heterogeneous cell populations even though pure luminal cells were initially inoculated into the mice, to the best of the researchers' knowledge (23). In another study, the expression of HER2, ER, PR, Ki-67, and CK5 were studied in 72 primary breast cancers and their corresponding metastatic lesions (24). In accordance to previous studies, ER expression in the primary tumor was associated to late recurrence when the tissue was also positive for Ki67 (24). Hess and collaborators showed, when analyzing the distinct patterns of relapse of 558 patients, that rates of recurrence were significantly higher in patients with ER-negative status for the first 2 years of follow-up, but not thereafter (25). ER+ tumors showed increased recurrence in bone whereas ER- cancers were found to metastasize preferentially to viscera and soft tissues (25).
Even though loss of ER expression may account for endocrine resistance, this phenomenon is observed in 15–20% of patients with progressed metastatic disease (26). Mutations in ERα are rare in primary tumors; only 0.5% of luminal breast cancers reveal mutations in ERα, and amplifications are observed in 2.6% of cases (27). In the metastatic setting however, a higher frequency of ERα mutations are observed, representing approximately 20% of cases (28, 29). Most mutations are found in the ligand binding domain of the receptor and are associated with the agonist conformation of the receptor as determined by biochemical and structural studies (29). The fact that mutations are found in progressed tumors suggests that they are the result of the expansion of rare clones in response to the selective pressure generated from targeted therapies against ER signaling (30).
Thus, evidence strongly supports the notion that ER signaling is critical even in later stages of tumor progression, years after initial diagnosis, surgery and prolonged treatment with endocrine therapies.
The Microenvironment as a Key Player in Endocrine Resistance
A growing body of evidence supports the notion that components of the tumor microenvironment play specific roles in response to therapy and that strategies targeting the interactions established with tumor cells could pave the way to a new generation of therapies. Carcinoma associated fibroblasts (CAFs), adipocytes, immune cells, endothelial cells, pericytes, the extracellular matrix (ECM) and soluble factors all contribute to tumor evolution (31–36). Thus, the resulting progression of a tumor is a consequence of the sum of interactions that are generated by all these players together with the tumor cell population. In ER+ breast cancer in particular, we are only now starting to understand the role played by the tumor stroma in response to therapy. The studies that will be reviewed in the following sections are, to our knowledge, those that have tackled how diverse microenvironmental players impact on the biology of ER+ tumors. However, it is important to understand that this is probably only the tip of the iceberg and that far more sophisticated interactions between these stromal players underlie the response to therapy.
The Immune System
Inflammation, one of the hallmarks of cancer, is associated to breast cancer development and progression. Studies show that regular use of non-steroidal anti-inflammatory drugs (NSAIDS), such as aspirin, significantly decrease the risk of ER-positive but not ER-negative breast cancers (37). Involution after pregnancy and obesity are two examples of risk factors associated to an inflammatory microenvironment and progression of breast cancer. In the case of pregnancy, involution, which follows pregnancy and takes place when lactation ends is considered to be responsible for the increase in breast cancer risk that has been described in the 10-years that follow parturition (38). The increased risk and poor prognosis of pregnancy associated breast cancer is thought to be associated to the inflammatory mediators that are present and active during involution. Extensive immune infiltration is present during involution, similar to what is observed during wound-healing (38). The ER status of pregnancy associated breast cancer is not clear (39). Although some studies suggest that there may be a reduced number of ER+ breast tumors amongst this population, others suggest that the high local and circulating levels of estrogens are responsible for the downregulation of ER and PR (39).
In postmenopausal women weight gain is associated to an increased risk of ER+ breast cancer (40, 41). Moreover, obese women with ER+ breast cancer have a higher rate of recurrence than lean women after treatment with endocrine therapy (42) and obesity on its own is related to tamoxifen resistance (43). Recruitment of macrophages into adipose tissue is a characteristic of obesity induced inflammation. Adipocytes and macrophages interact and have been shown to lead to activation of the proinflammatory transcription factor NF-κB. The degree of infiltration of macrophages is associated to the development of tamoxifen resistance (43).
Several studies suggest that tumor associated macrophages protect cancer cells from the anti-tumor immune responses. Macrophages isolated from mouse and human tumors can directly suppress T cell responses in vitro (44), and depletion of macrophages enhance CD8+ T-cells in a model of breast cancer under chemotherapy (45). A recent study analyzing circulating M2-like monocytes in breast cancer patients showed that they were increased in this population in comparison to healthy controls and patients with benign lesions (46). Another study evaluated the relationship between CD204 expression on tumor associated macrophages and clinicopathological factors in patients with invasive breast cancer. The authors found that in a sample of 108 luminal-like tumors high expression levels of CD204 was associated to decreased relapse-free survival and distant relapse-free survival (47).
Cytotoxic T cells, recognizable by the expression of CD8, play a major effector role in the adaptive immune system. Cells that present foreign antigens in association with the major histocompatibility complex class I molecule are recognized by cytotoxic T lymphocytes through a specific interaction between the T-cell receptor and the presented antigen. This interaction causes the activated T cell to release proteins such as perforin and granzyme that lead to cell death through lysis of the cell membrane [23]. These mechanisms can act on malignant cells which, unlike their normal counterparts, present atypical antigens [24, 25]. A recent paper analyzing 12,439 tumor samples, 8,775 of which were ER+ showed that for ER+ tumors that express HER-2, the presence of intratumor CD8+ T cells was associated with a 27% reduction in the hazard of dying from breast cancer (48). The analysis of the expression levels PD-L1 revealed that in the case of ER+ tumors, 20% of patients show detectable levels (49) compared to around 58% in the case of triple negative breast cancer (50). Response to anti PD-1/PD-L1 monotherapy in metastatic breast cancer showed durable clinical benefit for patients with triple negative breast cancer (50, 51). Regarding patients with ER+ tumors, few studies have specifically addressed this tumor type. Recently, Rugo et al. analyzed the antitumor activity of Pembrolizumab, an anti-PD-1 monoclonal antibody, in patients with ER+/Human Epidermal Growth Factor Receptor 2–negative advanced breast cancer (49). The objective response rate was 12% and a durable clinical benefit of more than 24 weeks was observed in 20% of patients (49). Another recent study was carried out analyzing 61 primary breast cancer tissues, 85% of which were ER+. Only eight of the samples were triple negative and one was ER-/HER2+ (52). The authors characterized the CD8+ tumor infiltrating lymphocytes and found that they retained robust capacity for production of effector cytokines and degranulation capacity even though they expressed PD-1, a hallmark of exhaustion (52). Additionally, they showed that CD8+ tumor infiltrating lymphocytes treated with CD3:CD19 bi-specific antibodies were able to kill breast cancer cells as efficiently as peripheral blood mononuclear cells from the same patients (52). The retention of polyfunctionality therefore implies the possibility that they are mostly composed of bystander T cells. This may explain the lack of impressive clinical responses to checkpoint blockade therapies in breast cancer.
In studies using breast cancer cell lines, ERα was shown to be a negative regulator of PD-L1 gene transcription as revealed by the mutually exclusive expression pattern of ERα and PD-L1 (53). Moreover, analysis of TCGA data derived from human breast cancer samples demonstrated that the average PD-L1 mRNA levels of ERα-positive tumors were significantly lower than those of ERα-negative tumors (53). Thus, immune response and ER signaling do not seem to be completely independent phenomenon. In this line of thought, one paper shows that antiestrogens induce immunosuppression in the tumor microenvironment, through a TGFβ-dependent mechanism contributing to the development of antiestrogen resistance in breast cancer (54). The impact of tamoxifen on the immune system has been reviewed in Behjati and Frank (55). Further studies analyzing the impact of anti-estrogen treatments on the interaction of CD8+ tumor infiltrating lymphocytes, macrophages and ER+ breast cancer cells may shed light on the development of future therapeutic strategies for breast cancer patients (Figure 2).
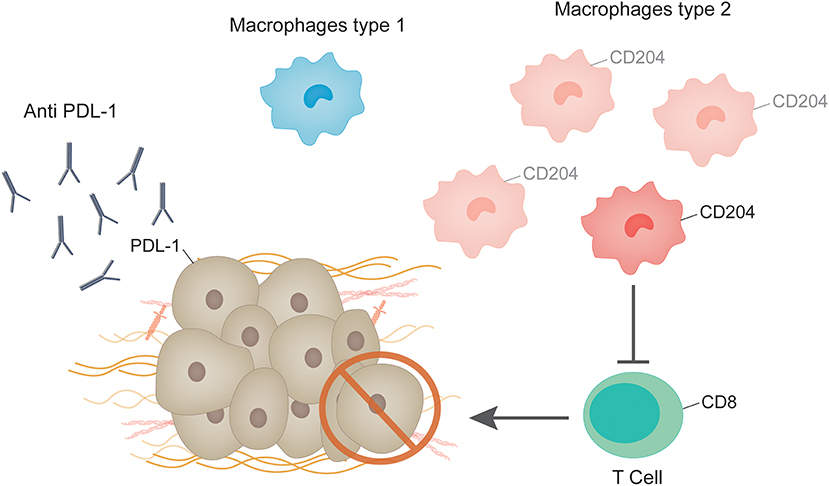
Figure 2. Immune system and ER+ breast tumors. Tumor associated macrophages protect cancer cells from the anti-tumor immune responses. M2 macrophages are increased in tumors in comparison to healthy controls and patients with benign lesions. CD204 expression in tumor associated macrophages is enhanced in patients with invasive breast cancer. Macrophages isolated from mouse and human tumors can directly suppress T cell responses in vitro, which are recognizable by the expression of CD8. Response to anti PD-1/PD-L1 monotherapy in metastatic ER+ breast cancer showed durable clinical benefit in a low percentage of patients, although very few studies have addressed this issue.
Carcinoma Associated Fibroblasts
In the normal mammary gland, fibroblasts in the stroma are located in close proximity to the epithelial ducts. They play a key role during puberty producing soluble factors, ECM components and proteases that are involved, amongst other things, as intermediaries in hormone signaling (56). Response to estrogens during development is the result of an intricate cross-talk established between ER+ sensor epithelial cells and stromal fibroblasts (56, 57). During ductal elongation, estrogens induce expression of amphiregulin that acts on EGFR positive stromal cells. In response, fibroblasts produce factors such as fibroblast growth factors and insulin-like growth factor-1 that signal back to the epithelium inducing proliferation (56). In ER+ breast cancer, in contrast to what is proposed for the normal mammary gland, estrogens impact directly on the proliferation of the ER+ tumor cells (15). Together with this, tumor progression is associated to changes in the stroma that contribute in the promotion of the malignant phenotype by secretion of additional growth stimulatory, angiogenic, immune-regulatory and pro-invasive soluble factors (58). A convincing link between tumor progression and the underlying stroma was elegantly demonstrated by Park's group where differential gene expression from the tumor stroma was shown to generate clusters linked to clinical outcome in breast cancer, independently of breast cancer subtypes (59). In the context of response to endocrine therapy we previously showed that soluble factors found in conditioned media derived from CAFs induce tamoxifen resistance in a murine model of ER+ breast cancer (60). Growth factors, proteases and signaling through β1 integrin were found to be involved in the protective effect the CAFs induced over the malignant cells exposed to endocrine therapy (60). In another study, using primary human CAFs co-cultured with MCF-7 cells, two CAF populations were identified through differential expression of CD146 in human breast tumors (61). CD146 (MCAM) is a stromal surface marker that defines fibroblast subtypes in the hematopoietic stem cell niche (62). These subtypes differentially influence the fate of peripheral blood monocytes (62). In the context of breast cancer, CD146− CAFs were shown to inhibit ER expression in MCF-7 cells, reduce sensitivity to estrogen, and increase resistance to tamoxifen. On the other hand, the presence of CD146+ CAFs stimulated ER expression and sustained estrogen-dependent proliferation and sensitivity to tamoxifen. Conditioned media from CD146+ CAFs reestablished tamoxifen sensitivity to tamoxifen-resistant breast cancer cells. Gene expression profiles of patient breast tumors with predominantly CD146− CAFs correlated with decreased clinical response to tamoxifen and worse patient outcomes (61). In the same line of thought, using a novel microfluidics-based organotypic model, Morgan et al. showed that when MCF-7 cells are co-cultured in 3-dimensions with immortalized human mammary fibroblasts, ER transactivation was increased in the presence of 17-β-estradiol. Moreover, the incorporation of fibroblasts increased the speed of development and size of estrogen-induced hyperplasias. This phenomenon was associated to reduced apoptosis in the co-culture model (63). Other publications have shown regulation of ER signaling in MCF-7 and T47D cells using either immortalized skin fibroblasts or marrow-derived stromal cells (64, 65). In first case the authors show that CAFs induce tamoxifen resistance by increasing mitochondrial activity in breast cancer cells. In the second case, paracrine stromal signaling leads ER downregulation in MCF-7 and T47D cells.
Another recently unraveled mechanisms of therapy resistance involves exosome transfer from stromal to breast cancer cells (66). In the context of ER positive breast cancer, transfer of OncomiR-221 containing microvesicles from CAFs to breast cancer cells has been shown to induce the expansion of cancer stem cells with increased self-renewing capacity, and resistance to endocrine therapy. Interestingly, tamoxifen has been previously shown to lead to the enrichment of breast cancer stem cells both in human and murine models, as well as in primary patient tissues (67–70).
Recent studies question the distinct origin of luminal and basal-like breast tumors and suggest a high degree of plasticity and heterogeneity between these two tumor types (71). Interconversion of luminal or basal-like tumor cells has been demonstrated to occur efficiently in vitro, establishing a common progenitor cell origin (72). PDGF receptors and ligands have recently been shown to actually modulate whether a tumor is luminal or basal and thus whether it may or not respond to endocrine therapy. Roswall et al. showed that CAFs act as determinants of the molecular subtype of breast cancer (73). Previous studies showed an association between PDGF-CC ligand expression in breast tumor cells and the triple negative subtype (74). Further insight revealed that PDGF-CC is an independent prognostic factor for poor survival in breast cancer (73). Experiments using triple-negative patient-derived xenografts (PDX), MDA-MB-231 triple negative breast cancer cells orthotopically inoculated in immunocompromised mice and FVB/N mice bearing orthotopically transplanted tumors from MMTV-PyMT; Pdgfc+/+ or MMTV-PyMT; Pdgfc−/− mice were shown to upregulate ERα expression when PDGF-CC was downregulated or inhibited and consequently respond to tamoxifen. PDGFRα and PDGFRβ were found to be expressed in the CAF compartment in all analyzed tumors (74) strongly indicating that the paracrine mode of signaling by PDGF-CC is from the epithelium to the stroma, and not autocrine within the stromal compartment. As a consequence of PDGF-CC signaling CAFs have been shown to produce molecules such as HGF, IGFBP3 and STC1 that are postulated as candidate mediators of the induction of the luminal phenotype (73) (Figure 3).
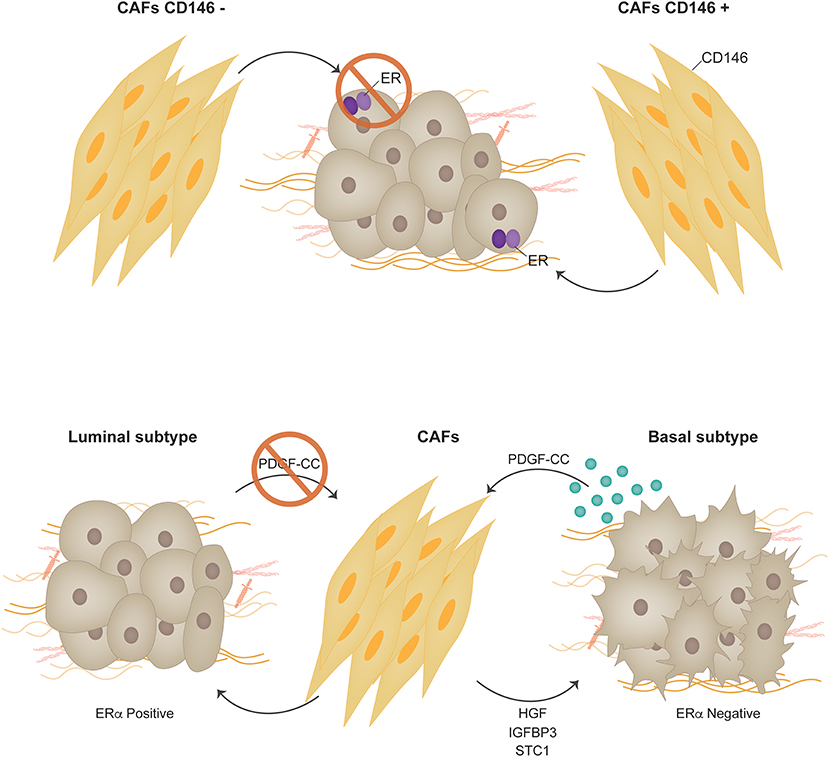
Figure 3. Carcinoma associated fibroblasts. Two CAF populations were identified through differential expression of CD146 in human breast tumors. CD146 is a stromal surface marker that defines fibroblast subtypes in the hematopoietic stem cell niche. In the context of breast cancer, CD146− CAFs were shown to suppress ERα expression in MCF-7 cells, decreased sensitivity to estrogen, and increase resistance to tamoxifen. On the other hand, the presence of CD146+ CAFs promoted ERα expression and sustained estrogen-dependent proliferation and sensitivity to tamoxifen. PDGF receptors and ligands have recently been shown to actually modulate whether a tumor is luminal or basal and thus whether it may or not respond to endocrine therapy [Permission obtained from Springer Nature (73)].
CAFs could be thus thought of as putative therapeutic targets to indirectly modulate the tumor epithelial compartment. Further insight in the understanding of the mechanisms implied in stromal epithelial interactions may produce a paradigm shift in the way we think about breast cancer classification and treatment.
The Extracellular Matrix
A central component of tumor tissue is the ECM that has in the last years been recognized as a key player in tumor progression and resistance to therapy in various malignancies, including breast cancer. It is well-accepted that tumors behave like wounds in the sense that the tumor host microenvironment is constantly in a fibrotic repair state. Changes in stromal composition and rigidity accompany breast cancer progression (75). In the normal mammary gland, the basement membrane clearly separates the epithelial compartment from the stroma. Laminin, collagen IV, fibronectin and entactin are the mayor constituents of the basement membrane that is produced jointly by epithelial, endothelial, and stromal cells. The interstitial ECM is composed of fibrillar collagens, fibronectin, glycoproteins and proteoglycans (75). The spacial arrangement together with the physical properties of the ECM determine tissue architecture and integrity. The biochemical characteristics of the matrix provide cues that modulate how the cells respond to different soluble factors such as hormones, polypeptide growth factors and chemokines.
The normal mammary gland is a soft and compliant tissue that upon malignant progression stiffens (76). This stiffening is accompanied by changes in the biochemical properties of the matrix. Remodeling of the tumor ECM involves continuous synthesis of matrix proteins, their assembly and crosslinking, as well as their turnover by proteases. This remodeling contributes to ECM stiffening, which is the consequence of increased collagen deposition, enhanced collagen crosslinking (as a result of lysyl oxidase (LOX) enzyme expression), and the reorientation of the collagen fibers to a parallel disposition (77–79). Significantly, increased collagen abundance and reorganization into thick, linearly oriented fibers correlates with tumor progression and clinical outcome (80). High ECM stiffness may also predispose individuals to develop certain types of cancer. Normal breast tissue clinically determined to have high mammographic density contains stiffer ECM, thicker collagen fibers and more linearized collagen than low mammographic-dense breast tissue (80), and was shown to increase the overall lifetime risk of breast cancer development (81). Tissue stiffness has been associated to breast cancer progression (82). Moreover, it is directly associated to response to chemotherapy (83, 84). Interestingly, reports analyzing tumor progression and stiffness suggest that tumors that have nodal metastasis usually have a stiffness >150 kpa (85).
An association between breast cancer progression and matrix composition analyzing human tumor samples was suggested as early as 2002 (86). However, the work of Els Burns's group was the first, to our knowledge, to focus on ER+ tumors. They showed a convincing association between an ECM gene cluster and disease progression in ER+ tumors derived from patients treated with tamoxifen (87). The authors examined 112 ER-positive primary breast carcinomas from patients with advanced disease and clearly defined therapy response types (i.e., 52 patients with objective response vs. 60 patients with progressive disease) from start of first-line treatment with tamoxifen. Eighty-one genes were found to be differentially expressed between the tamoxifen sensitive and the resistant tumors. From the 81 genes, 44 were extracted and validated on an independent set of 66 tumors. Within the group of identified genes, a cluster of ECM genes was identified: TIMP3, FN1 (fibronectin 1), LOX, COL1A1 (collagen type 1 alpha 1 chain), SPARC, and TNC (tenascin C). In all cases the overexpression of the genes was associated to disease progression. A second study by the same research group focused on these 6 genes and investigated in a sample of 1,286 tumors whether the mRNA expression levels were associated with the evolution of the disease, i.e., prognosis (independent of therapy response), clinical benefit from therapy with tamoxifen, or both (88). The results showed that high expression levels of FN1, LOX, and SPARC were associated with shorter metastasis free survival in lymph node negative patients who received no adjuvant systemic therapy. Other studies have analyzed the expression levels of FN in formalin fixed sections and also suggest that FN is associated to disease progression in breast cancer (86, 89–92).
Our research group has been working on establishing a mechanistic link between ECM components and response to tamoxifen in ER+ breast cancer cells. We previously showed that culturing ER+ human and murine breast cancer cells on FN leads to endocrine resistance through binding to β1 integrin (60). We also showed that FN induces phosphorylation of ERα in serine-118, a site that has been associated previously to ligand-independent activation of ER transcriptional activity and tamoxifen resistance (93, 94). To unravel the mechanism behind the induction of tamoxifen resistance by FN we looked into the dynamics of ERα recycling for cells cultured on FN as compared to bovine serum albumin. Surprisingly, we found that when cells are in contact with FN, ERα is not downregulated after 1 h treatment with estradiol. Further studies led us to find that estradiol actually induces endocytosis in breast cancer cells and that it is ERα located at the cell membrane that travels in endosomes to the nucleus (95). Previously, others had shown an association of ERα to the endosomal compartment (96), but induction of endocytosis by estrogens had been only described for neurons (97), to our knowledge. Inhibition of endocytosis by both pharmacological and genetic approaches led to inhibition of ER's transcriptional activity. Importantly, what our work shows is that when FN is present, ERα is endocytosed and “dragged” by β1 integrin back to the cell surface, apart from traveling to the nucleus (95). Moreover, in these conditions there is no co-localization of ERα with the lysosomal compartment strongly supporting the notion that the β1 integrin/FN interaction directs the fate of ERα and thus the response to tamoxifen (95). The fact that membrane ERα is critical for the transcriptional activity in breast cancer cells had already been strongly suggested by previous work of Filipo Acconcia and collaborators (96, 98). Our work confirms their findings and unravels a key role for an ECM component in the regulation of ERα's half-life and transcriptional activity (95). Thus, the ECM appears to have a central role not only through the establishment of mechanical cues, but by directly regulating the impact of hormone-action in breast cancer cells. Further studies are needed to understand whether integrins such as β1 interact with other hormone receptor and impact on their fate and signaling capacity (Figure 4).
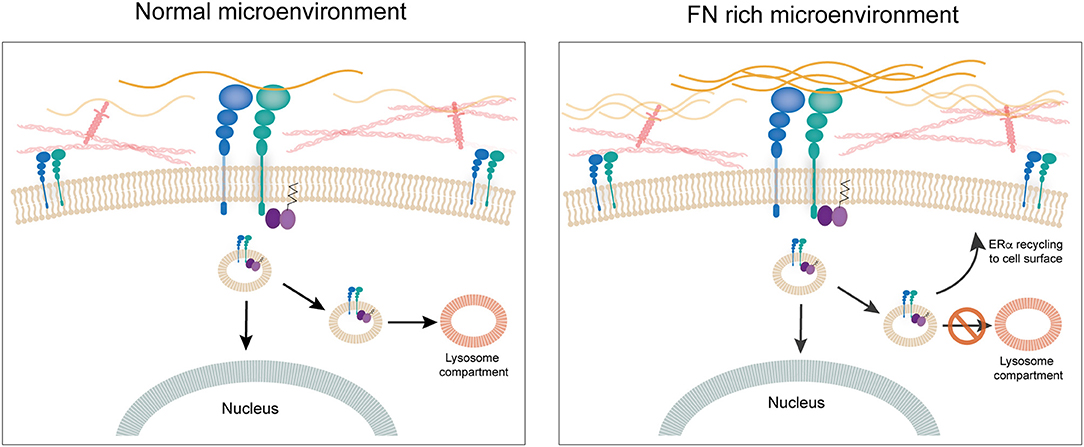
Figure 4. The extracellular matrix. FN is associated to disease progression in breast cancer. Culturing ERα+ human breast cancer cells on FN leads to endocrine resistance through binding to β1 integrin. When cells are in contact with FN, ERα is not downregulated after 1 h treatment with estradiol. Estradiol induces endocytosis in breast cancer cells and ERα located at the cell membrane travels in endosomes to the nucleus. When FN is present, ERα is endocytosed and “dragged” by β1 integrin back to the cell surface, apart from going to the nucleus. Also, there is no co-localization of ERα with the lysosomal compartment strongly supporting the notion that the β1 integrin/FN interaction directs the fate of ERα and thus response to tamoxifen.
Conclusion
The current management of ER+ breast cancer is based on how far the disease has progressed at the time of diagnosis, HER2 status and genomic risk leading to treatments that include chemotherapy, endocrine therapy, and targeted therapy (6). Women who are diagnosed with early stage breast cancer and can access adequate therapy have a 90% chance of being cured and multiple treatment options (6). However, for those who are diagnosed later the scenario is not that easy to manage. Targeted therapies are centered on the tumor cells. However, there is growing evidence that the tumor microenvironment plays a pivotal role in tumor progression and response to therapy. Immunotherapy is in its early years and is proving to revolutionize cancer treatment in general and impacting breast cancer management in particular. We propose that a deeper understanding of the role played by different components of the tumor microenvironment, especially focused on the niche where micrometastasis sit, may lead to the development of new therapies that could eliminate these residual cells early during treatment and thus reduce the late recurrences that characterize ER+ breast cancer.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This review was supported by a grant from the Agencia Nacional de Promoción Científica y Tecnológica, Préstamo BID- PICT 2016-0222 to MS. MG and TL are supported by a Conicet Doctoral Fellowship, MD by a Puente Fellowship from the Universidad Nacional de San Martín and MT by Student Fund from the VUmc Faculty.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Cronin KA, Lake AJ, Scott S, Sherman RL, Noone AM, Howlader N, et al. Annual report to the Nation on the status of cancer part I: National cancer statistics. Cancer. (2018) 124:2785–800. doi: 10.1002/cncr.31551
2. Jordan VC. Tamoxifen as the first targeted long-term adjuvant therapy for breast cancer. Endocr Relat Cancer. (2014) 21:R235–246. doi: 10.1530/ERC-14-0092
3. Waks AG, Winer EP. Breast cancer treatment: a review. JAMA. (2019) 321:288–300. doi: 10.1001/jama.2018.19323
4. Denduluri N, Chavez-MacGregor M, Telli ML, Eisen A, Graff SL, Hassett MJ, et al. Selection of optimal adjuvant chemotherapy and targeted therapy for early breast cancer: ASCO clinical practice guideline focused update. J Clin Oncol. (2018) 36:2433–43. doi: 10.1200/JCO.2018.78.8604
5. Burstein HJ, Lacchetti C, Anderson H, Buchholz TA, Davidson NE, Gelmon KA, et al. Adjuvant endocrine therapy for women with hormone receptor-positive breast cancer: ASCO clinical practice guideline focused update. J Clin Oncol. (2019) 37:423–38. doi: 10.1200/JCO.18.01160
6. Ponde NF, Zardavas D, Piccart M. Progress in adjuvant systemic therapy for breast cancer. Nat Rev Clin Oncol. (2019) 16:27–44. doi: 10.1038/s41571-018-0089-9
7. Couse JF, Lindzey J, Grandien K, Gustafsson JA, Korach KS. Tissue distribution and quantitative analysis of estrogen receptor-alpha (ERalpha) and estrogen receptor-beta (ERbeta) messenger ribonucleic acid in the wild-type and ERalpha-knockout mouse. Endocrinology. (1997) 138:4613–21. doi: 10.1210/endo.138.11.5496
8. Hewitt SC, Korach KS. Estrogen receptors: new directions in the new millennium. Endocr Rev. (2018) 39:664–67. doi: 10.1210/er.2018-00087
9. Brisken C, Ataca D. Endocrine hormones and local signals during the development of the mouse mammary gland. Wiley Interdiscip Rev Dev Biol. (2015) 4:181–95. doi: 10.1002/wdev.172
10. Arnal JF, Lenfant F, Metivier R, Flouriot G, Henrion D, Adlanmerini M, et al. Membrane and nuclear estrogen receptor alpha actions: from tissue specificity to medical implications. Physiol Rev. (2017) 97:1045–87. doi: 10.1152/physrev.00024.2016
11. Chantalat E, Boudou F, Laurell H, Palierne G, Houtman R, Melchers D, et al. The AF-1-deficient estrogen receptor ERalpha46 isoform is frequently expressed in human breast tumors. Breast Cancer Res. (2016) 18:123. doi: 10.1186/s13058-016-0780-7
12. Wang Z, Zhang X, Shen P, Loggie BW, Chang Y, Deuel TF. Identification, cloning, and expression of human estrogen receptor-alpha36, a novel variant of human estrogen receptor-alpha66. Biochem Biophys Res Commun. (2005) 336:1023–7. doi: 10.1016/j.bbrc.2005.08.226
13. Wang Z, Zhang X, Shen P, Loggie BW, Chang Y, Deuel TF. A variant of estrogen receptor-{alpha}, hER-{alpha}36: transduction of estrogen- and antiestrogen-dependent membrane-initiated mitogenic signaling. ProcNatlAcadSci USA. (2006) 103:9063–8. doi: 10.1073/pnas.0603339103
14. Levin ER, Hammes SR. Nuclear receptors outside the nucleus: extranuclear signalling by steroid receptors. Nat Rev Mol Cell Biol. (2016) 17:783–97. doi: 10.1038/nrm.2016.122
15. Siersbaek R, Kumar S, Carroll JS. Signaling pathways and steroid receptors modulating estrogen receptor alpha function in breast cancer. Genes Dev. (2018) 32:1141–54. doi: 10.1101/gad.316646.118
16. Chumsri S, Howes T, Bao T, Sabnis G, Brodie A. Aromatase, aromatase inhibitors, and breast cancer. J Steroid Biochem Mol Biol. (2011) 125:13–22. doi: 10.1016/j.jsbmb.2011.02.001
17. Traboulsi T, El Ezzy M, Gleason JL, Mader S. Antiestrogens: structure-activity relationships and use in breast cancer treatment. J Mol Endocrinol. (2017) 58:R15–R31. doi: 10.1530/JME-16-0024
18. Reinert T, Barrios CH. Optimal management of hormone receptor positive metastatic breast cancer in 2016. Ther Adv Med Oncol. (2015) 7:304–20. doi: 10.1177/1758834015608993
19. Hosseini H, Obradovic MMS, Hoffmann M, Harper KL, Sosa MS, Werner-Klein M, et al. Early dissemination seeds metastasis in breast cancer. Nature. (2016) 540:552–8. doi: 10.1038/nature20785
20. Magbanua MJM, Rugo HS, Hauranieh L, Roy R, Scott JH, Lee JC, et al. Genomic and expression profiling reveal molecular heterogeneity of disseminated tumor cells in bone marrow of early breast cancer. NPJ Breast Cancer. (2018) 4:31. doi: 10.1038/s41523-018-0083-5
21. Demicheli R, Desmedt C, Piccart M, Biganzoli E. Tumor dormancy at bedside: a late awakening. Breast. (2019) 45:61–3. doi: 10.1016/j.breast.2019.03.001
22. Early Breast Cancer Trialists' Collaborative G. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. (2005) 365:1687–717. doi: 10.1016/S0140-6736(05)66544-0
23. Ogba N, Manning NG, Bliesner BS, Ambler SK, Haughian JM, Pinto MP, et al. Luminal breast cancer metastases and tumor arousal from dormancy are promoted by direct actions of estradiol and progesterone on the malignant cells. Breast Cancer Res. (2014) 16:489. doi: 10.1186/s13058-014-0489-4
24. Joensuu K, Leidenius M, Kero M, Andersson LC, Horwitz KB, Heikkila P. ER, PR, HER2, Ki-67 and CK5 in early and late relapsing breast cancer-reduced CK5 expression in metastases. Breast Cancer. (2013) 7:23–34. doi: 10.4137/BCBCR.S10701
25. Hess KR, Pusztai L, Buzdar AU, Hortobagyi GN. Estrogen receptors and distinct patterns of breast cancer relapse. Breast Cancer Res Treat. (2003) 78:105–18. doi: 10.1023/A:1022166517963
26. Hoefnagel LD, Moelans CB, Meijer SL, van Slooten HJ, Wesseling P, Wesseling J, et al. Prognostic value of estrogen receptor alpha and progesterone receptor conversion in distant breast cancer metastases. Cancer. (2012) 118:4929–35. doi: 10.1002/cncr.27518
27. Jeselsohn R, De Angelis C, Brown M, Schiff R. The evolving role of the estrogen receptor mutations in endocrine therapy-resistant breast cancer. Curr Oncol Rep. (2017) 19:35. doi: 10.1007/s11912-017-0591-8
28. Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. (2013) 45:1446–51. doi: 10.1038/ng.2823
29. Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet. (2013) 45:1439–45. doi: 10.1038/ng.2822
30. Turner NC, Reis-Filho JS. Genetic heterogeneity and cancer drug resistance. Lancet Oncol. (2012) 13:e178–185. doi: 10.1016/S1470-2045(11)70335-7
31. Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer. (2001) 1:46–54. doi: 10.1038/35094059
32. Hui L, Chen Y. Tumor microenvironment: sanctuary of the devil. Cancer Lett. (2015) 368:7–13. doi: 10.1016/j.canlet.2015.07.039
33. Bussard KM, Mutkus L, Stumpf K, Gomez-Manzano C, Marini FC. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. (2016) 18:84. doi: 10.1186/s13058-016-0740-2
34. Wu T, Dai Y. Tumor microenvironment and therapeutic response. Cancer Lett. (2017) 387:61–8. doi: 10.1016/j.canlet.2016.01.043
35. Maman S, Witz IP. A history of exploring cancer in context. Nat Rev Cancer. (2018) 18:359–76. doi: 10.1038/s41568-018-0006-7
36. Kim J. Pericytes in breast cancer. Adv Exp Med Biol. (2019) 1147:93–107. doi: 10.1007/978-3-030-16908-4_3
37. Terry MB, Gammon MD, Zhang FF, Tawfik H, Teitelbaum SL, Britton JA, et al. Association of frequency and duration of aspirin use and hormone receptor status with breast cancer risk. JAMA. (2004) 291:2433–40. doi: 10.1001/jama.291.20.2433
38. Schedin P. Pregnancy-associated breast cancer and metastasis. Nat Rev Cancer. (2006) 6:281–91. doi: 10.1038/nrc1839
39. Bure LA, Azoulay L, Benjamin A, Abenhaim HA. Pregnancy-associated breast cancer: a review for the obstetrical care provider. J Obstet Gynaecol Can. (2011) 33:330–7. doi: 10.1016/S1701-2163(16)34850-2
40. Feigelson HS, Patel AV, Teras LR, Gansler T, Thun MJ, Calle EE. Adult weight gain and histopathologic characteristics of breast cancer among postmenopausal women. Cancer. (2006) 107:12–21. doi: 10.1002/cncr.21965
41. Reeves GK, Pirie K, Beral V, Green J, Spencer E, Bull D, et al. Cancer incidence and mortality in relation to body mass index in the million women study: cohort study. BMJ. (2007) 335:1134. doi: 10.1136/bmj.39367.495995.AE
42. Sestak I, Distler W, Forbes JF, Dowsett M, Howell A, Cuzick J. Effect of body mass index on recurrences in tamoxifen and anastrozole treated women: an exploratory analysis from the ATAC trial. J Clin Oncol. (2010) 28:3411–5. doi: 10.1200/JCO.2009.27.2021
43. Xuan QJ, Wang JX, Nanding A, Wang ZP, Liu H, Lian X, et al. Tumor-associated macrophages are correlated with tamoxifen resistance in the postmenopausal breast cancer patients. Pathol Oncol Res. (2014) 20:619–24. doi: 10.1007/s12253-013-9740-z
44. Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell. (2015) 27:462–72. doi: 10.1016/j.ccell.2015.02.015
45. DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. (2011) 1:54–67. doi: 10.1158/2159-8274.CD-10-0028
46. Zhang B, Cao M, He Y, Liu Y, Zhang G, Yang C, et al. Increased circulating M2-like monocytes in patients with breast cancer. Tumour Biol. (2017) 39:1010428317711571. doi: 10.1177/1010428317711571
47. Miyasato Y, Shiota T, Ohnishi K, Pan C, Yano H, Horlad H, et al. High density of CD204-positive macrophages predicts worse clinical prognosis in patients with breast cancer. Cancer Sci. (2017) 108:1693–700. doi: 10.1111/cas.13287
48. Ali HR, Provenzano E, Dawson SJ, Blows FM, Liu B, Shah M, et al. Association between CD8+ T-cell infiltration and breast cancer survival in 12,439 patients. Ann Oncol. (2014) 25:1536–43. doi: 10.1093/annonc/mdu191
49. Rugo HS, Delord JP, Im SA, Ott PA, Piha-Paul SA, Bedard PL, et al. Safety and antitumor activity of pembrolizumab in patients with estrogen receptor-positive/human epidermal growth factor receptor 2-negative advanced breast cancer. Clin Cancer Res. (2018) 24:2804–11. doi: 10.1158/1078-0432.CCR-17-3452
50. Nanda R, Chow LQ, Dees EC, Berger R, Gupta S, Geva R, et al. Pembrolizumab in patients with advanced triple-negative breast cancer: phase Ib KEYNOTE-012 study. J Clin Oncol. (2016) 34:2460–7. doi: 10.1200/JCO.2015.64.8931
51. Dirix LY, Takacs I, Jerusalem G, Nikolinakos P, Arkenau HT, Forero-Torres A, et al. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: a phase 1b JAVELIN Solid Tumor study. Breast Cancer Res Treat. (2018) 167:671–86. doi: 10.1007/s10549-017-4537-5
52. Egelston CA, Avalos C, Tu TY, Simons DL, Jimenez G, Jung JY, et al. Human breast tumor-infiltrating CD8(+) T cells retain polyfunctionality despite PD-1 expression. Nat Commun. (2018) 9:4297. doi: 10.1038/s41467-018-06653-9
53. Liu L, Shen Y, Zhu X, Lv R, Li S, Zhang Z, et al. ERα is a negative regulator of PD-L1 gene transcription in breast cancer. Biochem Biophys Res Commun. (2018) 505:157–61. doi: 10.1016/j.bbrc.2018.09.005
54. Joffroy CM, Buck MB, Stope MB, Popp SL, Pfizenmaier K, Knabbe C. Antiestrogens induce transforming growth factor beta-mediated immunosuppression in breast cancer. Cancer Res. (2010) 70:1314–22. doi: 10.1158/0008-5472.CAN-09-3292
55. Behjati S, Frank MH. The effects of tamoxifen on immunity. Curr Med Chem. (2009) 16:3076–80. doi: 10.2174/092986709788803042
56. Tanos T, Rojo L, Echeverria P, Brisken C. ER and PR signaling nodes during mammary gland development. Breast Cancer Res. (2012) 14:210. doi: 10.1186/bcr3166
57. Haslam SZ. Mammary fibroblast influence on normal mouse mammary epithelial cell responses to estrogen in vitro. Cancer Res. (1986) 46:310.
58. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. (2006) 6:392–401. doi: 10.1038/nrc1877
59. Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. (2008) 14:518–27. doi: 10.1038/nm1764
60. Pontiggia O, Sampayo R, Raffo D, Motter A, Xu R, Bissell MJ, et al. The tumor microenvironment modulates tamoxifen resistance in breast cancer: a role for soluble stromal factors and fibronectin through beta1 integrin. Breast Cancer Res Treat. (2012) 133:459–71. doi: 10.1007/s10549-011-1766-x
61. Brechbuhl HM, Finlay-Schultz J, Yamamoto TM, Gillen AE, Cittelly DM, Tan AC, et al. Fibroblast subtypes regulate responsiveness of luminal breast cancer to estrogen. Clin Cancer Res. (2017) 23:1710–21. doi: 10.1158/1078-0432.CCR-15-2851
62. Iwata M, Sandstrom RS, Delrow JJ, Stamatoyannopoulos JA, Torok-Storb B. Functionally and phenotypically distinct subpopulations of marrow stromal cells are fibroblast in origin and induce different fates in peripheral blood monocytes. Stem Cells Dev. (2014) 23:729–40. doi: 10.1089/scd.2013.0300
63. Morgan MM, Livingston MK, Warrick JW, Stanek EM, Alarid ET, Beebe DJ, et al. Mammary fibroblasts reduce apoptosis and speed estrogen-induced hyperplasia in an organotypic MCF7-derived duct model. Sci Rep. (2018) 8:7139. doi: 10.1038/s41598-018-25461-1
64. Martinez-Outschoorn UE, Goldberg A, Lin Z, Ko YH, Flomenberg N, Wang C, et al. Anti-estrogen resistance in breast cancer is induced by the tumor microenvironment and can be overcome by inhibiting mitochondrial function in epithelial cancer cells. Cancer Biol Ther. (2011) 12:924–38. doi: 10.4161/cbt.12.10.17780
65. Huang J, Woods P, Normolle D, Goff JP, Benos PV, Stehle CJ, et al. Downregulation of estrogen receptor and modulation of growth of breast cancer cell lines mediated by paracrine stromal cell signals. Breast Cancer Res Treat. (2017) 161:229–43. doi: 10.1007/s10549-016-4052-0
66. Boelens MC, Wu TJ, Nabet BY, Xu B, Qiu Y, Yoon T, et al. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell. (2014) 159:499–513. doi: 10.1016/j.cell.2014.09.051
67. Ao A, Morrison BJ, Wang H, Lopez JA, Reynolds BA, Lu J. Response of estrogen receptor-positive breast cancer tumorspheres to antiestrogen treatments. PLoS ONE. (2011) 6:e18810. doi: 10.1371/journal.pone.0018810
68. Simoes BM, Piva M, Iriondo O, Comaills V, Lopez-Ruiz JA, Zabalza I, et al. Effects of estrogen on the proportion of stem cells in the breast. Breast Cancer Res Treat. (2011) 129:23–35. doi: 10.1007/s10549-010-1169-4
69. Simoes BM, Vivanco MD. Cancer stem cells in the human mammary gland and regulation of their differentiation by estrogen. Future Oncol. (2011) 7:995–1006. doi: 10.2217/fon.11.80
70. Raffo D, Berardi DE, Pontiggia O, Todaro L, de Kier Joffe EB, Simian M. Tamoxifen selects for breast cancer cells with mammosphere forming capacity and increased growth rate. Breast Cancer Res Treat. (2013) 142:537–48. doi: 10.1007/s10549-013-2760-2
71. Sflomos G, Dormoy V, Metsalu T, Jeitziner R, Battista L, Scabia V, et al. A preclinical model for eralpha-positive breast cancer points to the epithelial microenvironment as determinant of luminal phenotype and hormone response. Cancer Cell. (2016) 29:407–22. doi: 10.1016/j.ccell.2016.02.002
72. Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. (2011) 146:633–44. doi: 10.1016/j.cell.2011.07.026
73. Roswall P, Bocci M, Bartoschek M, Li H, Kristiansen G, Jansson S, et al. Microenvironmental control of breast cancer subtype elicited through paracrine platelet-derived growth factor-CC signaling. Nat Med. (2018) 24:463–73. doi: 10.1038/nm.4494
74. Jansson S, Aaltonen K, Bendahl PO, Falck AK, Karlsson M, Pietras K, et al. The PDGF pathway in breast cancer is linked to tumour aggressiveness, triple-negative subtype and early recurrence. Breast Cancer Res Treat. (2018) 169:231–41. doi: 10.1007/s10549-018-4664-7
75. Kaushik S, Pickup MW, Weaver VM. From transformation to metastasis: deconstructing the extracellular matrix in breast cancer. Cancer Metastasis Rev. (2016) 35:655–67. doi: 10.1007/s10555-016-9650-0
76. Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. (2005) 8:241–54. doi: 10.1016/j.ccr.2005.08.010
77. Butcher DT, Alliston T, Weaver VM. A tense situation: forcing tumour progression. Nat Rev Cancer. (2009) 9:108–22. doi: 10.1038/nrc2544
78. Kumar S, Weaver VM. Mechanics, malignancy, and metastasis: the force journey of a tumor cell. Cancer Metastasis Rev. (2009) 28:113–27. doi: 10.1007/s10555-008-9173-4
79. Yu H, Mouw JK, Weaver VM. Forcing form and function: biomechanical regulation of tumor evolution. Trends Cell Biol. (2011) 21:47–56. doi: 10.1016/j.tcb.2010.08.015
80. Acerbi I, Cassereau L, Dean I, Shi Q, Au A, Park C, et al. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr Biol. (2015) 7:1120–34. doi: 10.1039/c5ib00040h
81. Razzaghi H, Troester MA, Gierach GL, Olshan AF, Yankaskas BC, Millikan RC. Mammographic density and breast cancer risk in white and African American women. Breast Cancer Res Treat. (2012) 135:571–80. doi: 10.1007/s10549-012-2185-3
82. Northcott JM, Dean IS, Mouw JK, Weaver VM. Feeling stress: the mechanics of cancer progression and aggression. Front Cell Dev Biol. (2018) 6:17. doi: 10.3389/fcell.2018.00017
83. Evans A, Armstrong S, Whelehan P, Thomson K, Rauchhaus P, Purdie C, et al. Can shear-wave elastography predict response to neoadjuvant chemotherapy in women with invasive breast cancer? Br J Cancer. (2013) 109:2798–802. doi: 10.1038/bjc.2013.660
84. Hayashi M, Yamamoto Y, Sueta A, Tomiguchi M, Fujiwara S, Yamamoto S, et al. Correlation between clinical tumor stiffness by elastography and response to neoadjuvant chemotherapy in patients with breast cancer. J Clin Oncol. (2014) 32(15Suppl.):1061. doi: 10.1200/jco.2014.32.15_suppl.1061
85. Evans A, Rauchhaus P, Whelehan P, Thomson K, Purdie CA, Jordan LB, et al. Does shear wave ultrasound independently predict axillary lymph node metastasis in women with invasive breast cancer? Breast Cancer Res Treat. (2014) 143:153–7. doi: 10.1007/s10549-013-2747-z
86. Ioachim E, Charchanti A, Briasoulis E, Karavasilis V, Tsanou H, Arvanitis DL, et al. Immunohistochemical expression of extracellular matrix components tenascin, fibronectin, collagen type IV and laminin in breast cancer: their prognostic value and role in tumour invasion and progression. Eur J Cancer. (2002) 38:2362–70. doi: 10.1016/S0959-8049(02)00210-1
87. Jansen MP, Foekens JA, van Staveren IL, Dirkzwager-Kiel MM, Ritstier K, Look MP, et al. (2005). Molecular classification of tamoxifen-resistant breast carcinomas by gene expression profiling. J Clin.Oncol. 23, 732–740. doi: 10.1200/JCO.2005.05.145
88. Helleman J, Jansen MP, Ruigrok-Ritstier K, van Staveren IL, Look MP, Meijer-van Gelder ME, et al. Association of an extracellular matrix gene cluster with breast cancer prognosis and endocrine therapy response. Clin Cancer Res. (2008) 14:5555–64. doi: 10.1158/1078-0432.CCR-08-0555
89. Yao ES, Zhang H, Chen YY, Lee B, Chew K, Moore D, et al. Increased beta1 integrin is associated with decreased survival in invasive breast cancer. Cancer Res. (2007) 67:659–64. doi: 10.1158/0008-5472.CAN-06-2768
90. Loi S, Haibe-Kains B, Desmedt C, Wirapati P, Lallemand F, Tutt AM, et al. Predicting prognosis using molecular profiling in estrogen receptor-positive breast cancer treated with tamoxifen. BMC Genomics. (2008) 9:239. doi: 10.1186/1471-2164-9-239
91. Fernandez-Garcia B, Eiro N, Marin L, Gonzalez-Reyes S, Gonzalez LO, Lamelas ML, et al. Expression and prognostic significance of fibronectin and matrix metalloproteases in breast cancer metastasis. Histopathology. (2014) 64:512–22. doi: 10.1111/his.12300
92. Bae YK, Kim A, Kim MK, Choi JE, Kang SH, Lee SJ. Fibronectin expression in carcinoma cells correlates with tumor aggressiveness and poor clinical outcome in patients with invasive breast cancer. Hum Pathol. (2013) 44:2028–37. doi: 10.1016/j.humpath.2013.03.006
93. Murphy LC, Weitsman GE, Skliris GP, Teh EM, Li L, Peng B, et al. Potential role of estrogen receptor alpha (ERalpha) phosphorylated at serine118 in human breast cancer in vivo. J Steroid BiochemMolBiol. (2006) 102:139–46. doi: 10.1016/j.jsbmb.2006.09.021
94. Chen M, Cui YK, Huang WH, Man K, Zhang GJ. Phosphorylation of estrogen receptor alpha at serine 118 is correlated with breast cancer resistance to tamoxifen. Oncol Lett. (2013) 6:118–24. doi: 10.3892/ol.2013.1324
95. Sampayo RG, Toscani AM, Rubashkin MG, Thi K, Masullo LA, Violi IL, et al. Fibronectin rescues estrogen receptor alpha from lysosomal degradation in breast cancer cells. J Cell Biol. (2018) 217:2777–98. doi: 10.1083/jcb.201703037
96. Totta P, Pesiri V, Marino M, Acconcia F. Lysosomal function is involved in 17beta-estradiol-induced estrogen receptor alpha degradation and cell proliferation. PLoS ONE. (2014) 9:e94880. doi: 10.1371/journal.pone.0094880
97. Kisler K, Chow RH, Dominguez R. Fluorescently-labeled estradiol internalization and membrane trafficking in Live N-38 neuronal cells visualized with total internal reflection fluorescence microscopy. J Steroids Horm Sci. (2013) (Suppl. 12). doi: 10.4172/2157-7536.S12-002
Keywords: breast cancer, endocrine resistance, microenvironment, immune system, extracellular matrix, carcinoma associated fibroblasts, estrogen receptor
Citation: Diaz Bessone MI, Gattas MJ, Laporte T, Tanaka M and Simian M (2019) The Tumor Microenvironment as a Regulator of Endocrine Resistance in Breast Cancer. Front. Endocrinol. 10:547. doi: 10.3389/fendo.2019.00547
Received: 07 May 2019; Accepted: 22 July 2019;
Published: 08 August 2019.
Edited by:
Stephen Hiscox, Cardiff University, United KingdomReviewed by:
Dragana Nikitovic, University of Crete, GreeceYun Zhu, University of Texas MD Anderson Cancer Center, United States
Copyright © 2019 Diaz Bessone, Gattas, Laporte, Tanaka and Simian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marina Simian, bXNpbWlhbkB1bnNhbS5lZHUuYXI=