 Lap Hang Tse
Lap Hang Tse Yung Hou Wong
Yung Hou Wong- 1Division of Life Science, Biotechnology Research Institute, Hong Kong University of Science and Technology, Hong Kong, Hong Kong
- 2State Key Laboratory of Molecular Neuroscience, Molecular Neuroscience Center, Hong Kong University of Science and Technology, Hong Kong, Hong Kong
G protein-coupled receptors (GPCRs) constitute the largest superfamily of integral membrane protein receptors. As signal detectors, the several 100 known GPCRs are responsible for sensing the plethora of endogenous ligands that are critical for the functioning of our endocrine system. Although GPCRs are typically considered as detectors for first messengers in classical signal transduction pathways, they seldom operate in isolation in complex biological systems. Intercellular communication between identical or different cell types is often mediated by autocrine or paracrine signals that are generated upon activation of specific GPCRs. In the context of energy homeostasis, the distinct complement of GPCRs in each cell type bridges the autocrine and paracrine communication within an organ, and the various downstream signaling mechanisms regulated by GPCRs can be integrated in a cell to produce an ultimate output. GPCRs thus act as gatekeepers that coordinate and fine-tune a response. By examining the role of GPCRs in activating and receiving autocrine and paracrine signals, one may have a better understanding of endocrine diseases that are associated with GPCR mutations, thereby providing new insights for treatment regimes.
Introduction
To maintain homeostasis in humans, a wide array of extracellular factors is required to harmonize physiological activities between various organs and tissues. These signaling molecules in the form of hormones, peptides, neurotransmitters, proteins, ions, and lipids act via specific receptors to elicit cellular responses. Among the different receptor families, more than 700 G protein-coupled receptors (GPCRs) form the largest and the most diverse receptor superfamily that participate in virtually all aspects of human physiology. Most human GPCRs can be grouped into five families (Glutamate, Rhodopsin, Adhesion, Frizzled/Taste2, and Secretin) according to their structural similarity, which is known as the GRAFS classification system (1). The physiological relevance makes GPCRs one of the most popular drug targets (2), and their importance in the field of endocrinology is highlighted by the identification of naturally occurring GPCR mutations in patients with various endocrine diseases (3). In classical endocrine systems, hormones are released into the bloodstream and they modify target cells in a distant part of the body; it has become apparent that these processes are regulated by cellular communications encompassing autocrine, paracrine, intracrine, and juxtacrine interactions. In this review, we will focus on our current, yet evolving understanding of the autocrine and paracrine signals regulated by GPCRs in various physiological systems. Since signals mediated by GPCRs are regulated by a myriad of complex determinants, with crosstalk between different signaling pathways that culminate in the fine-tuning of cellular responses, it is important to examine the role of GPCRs in the context of signal processing within and between different cells and tissues.
GPCRs and the Hierarchy of Endocrine, Autocrine and Paracrine Signaling
Although the discovery of autocrine and paracrine interactions was initially overshadowed by the characterization of endocrine glands, the concept of cells being able to secrete regulatory factors was first appreciated more than 200 years ago by leading scientists of the time, including Brown-Séquard whom many regarded as the “father of endocrinology.” It is now firmly established that the endocrine glands are regulated by a plethora of internal and external signals via blood circulation, and that these input signals can further trigger the release of autocrine/paracrine messengers. Various autocrine/paracrine factors are known to contribute to the communications and intricate feedbacks between different types of cells within an endocrine gland, resulting in a coordinated hormonal output and the corresponding physiological outcome. Remarkably, the same chemical molecules can be used in multiple contexts of endocrine, paracrine or autocrine signaling, or even in synaptic signaling. The function of these signaling molecules can be considered in a hierarchical manner (Figure 1) for the majority of endocrine organs as: (1) a circulatory input that initiates the subsequent autocrine/paracrine interactions; (2) an autocrine/paracrine messenger that mediates the feedback networks among different cells within the endocrine gland; and (3) a hormonal output secreted by endocrine cells, which enter the circulation and further serve as a circulatory input for other organs. Among these signaling processes, GPCRs are ineluctable mediators in sensing both circulatory inputs and autocrine/paracrine factors. According to the “GRAFS” classification scheme, there are five classes of vertebrate GPCRs (1) whose differential distribution on different cell types helps to ensure specificity of the feedback network. Because optimal utilization of nutrient is a vital prerequisite for survival, this review will focus on examples of GPCRs in the Rhodopsin and Secretin families that regulate energy homeostasis. However, it should be noted that the concept of GPCRs mediating autocrine/paracrine responses is applicable to many other endocrine tissues/organs such as adipose tissue (4), the adrenal gland (5), and testis (6). Other classes of GPCRs are also functionally essential for various endocrine systems; a prime example is the calcium-sensing receptor of the Glutamate family for calcium homeostasis (7).
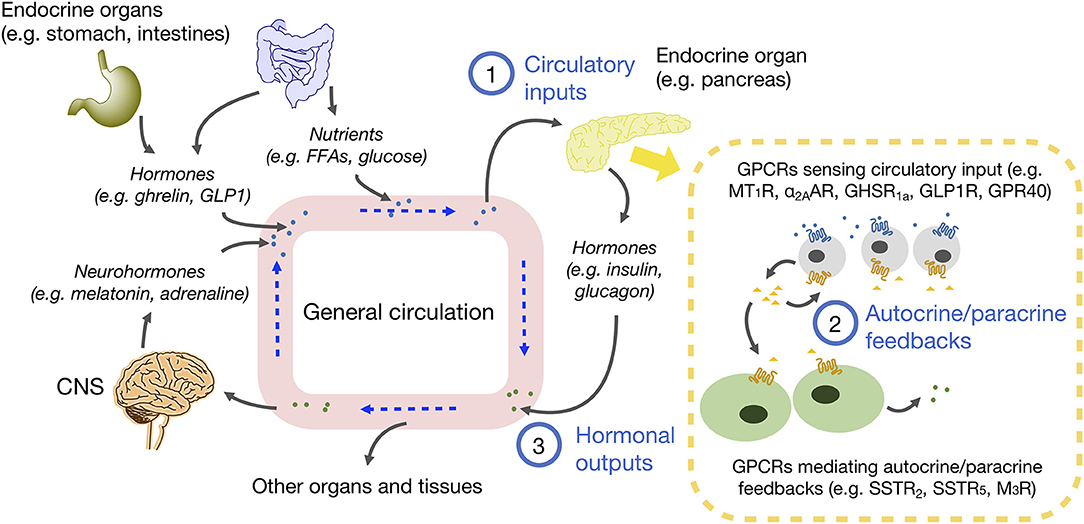
Figure 1. Schematic diagram of endocrine, autocrine, and paracrine communications in energy homeostasis. Circulating molecules such as neurohormones, hormones and nutrients are derived from CNS, endocrine organs and tissues, and small intestine. These molecules in blood are circulated to different parts of the body. The molecules diffused into endocrine organs act as circulatory inputs (1), which then bind to receptors on target cells and trigger the secretion of autocrine and paracrine factors, so as to foster communications between the same or different cell types (2). As a result of signal integration from various autocrine/paracrine factors, hormone secretions from endocrine cells are adjusted, and hormonal outputs are released into the circulation (3), which can further influence other organs and tissues to maintain homeostasis.
Pancreatic GPCRs in the Regulation of Energy Homeostasis
The human pancreas is composed of granular tissues embedded with a duct system. While exocrine cells constitute the major biomass of the pancreas, a small cluster of endocrine cells forms the pancreatic islet, including three key cell types: glucagon-secreting α-cells, insulin-secreting β-cells, somatostatin-secreting δ-cells (8) (Figure 2). The pancreatic islet is an important peripheral endocrine gland for maintaining blood glucose level and energy homeostasis. To adjust energy fluctuation caused by food intake, circadian rhythm or physical activities, the islet is sensitive to signals which are regulated by the hypothalamus as well as to other circulatory signals such as nutrients and hormones. Human islet GPCR mRNA profiling has identified 293 islet GPCRs that respond to 271 different endogenous ligands, of which at least 131 ligands are present in islet cells (9). However, the majority of islet GPCRs have unknown effects on pancreatic hormone secretion. Readers may refer to other reviews for the full list of islet GPCRs discovered in humans and their comparative analysis with mouse islet GPCRs (9, 10). Besides GPCRs, other receptor types such as tyrosine kinases are also able to regulate pancreatic responses. It should be noted that signal crosstalk and transactivation linkages between GPCR and non-PCR pathways constitute yet another layer of signaling complexity in energy homeostasis (11).
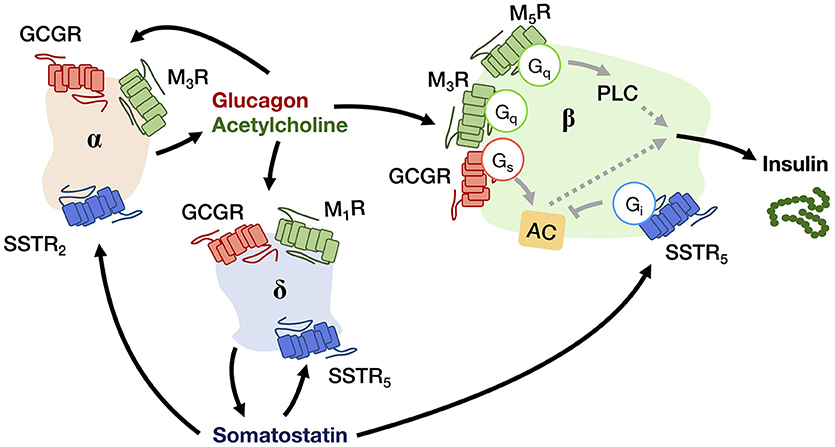
Figure 2. A schematic diagram showing the autocrine and paracrine interactions between pancreatic α-, β-, and δ-cells that regulate the insulin output. Glucagon and acetylcholine from α-cell (beige), and somatostatin from δ-cell (blue) are stimulatory and inhibitory signals for hormone secretion, respectively, that can act in both autocrine and paracrine manners via their receptors (GCGR for glucagon; M1R, M3R, and M5R for acetylcholine; SSTR2 and SSTR5 for somatostatin). The insulin output from β-cell (green) is adjusted by an integration of paracrine signals from both α- and δ-cells within the niche.
GPCRs for Sensing Circulatory Inputs
There are numerous GPCRs in the pancreas that can detect circulatory nutrients and related hormones, and the major examples are listed in Table 1. A few examples will be discussed to illustrate the role of GPCRs as circulatory signal sensors that induce subsequent autocrine/paracrine interactions in the pancreas. One of the best-known incretin hormones is glucagon-like peptide 1 (GLP1), which is secreted by the intestinal L-cells upon food consumption and circulates throughout the body, including the pancreatic islet which expresses GLP1 receptor (GLP1R). GLP1R is coupled to Gs proteins to control the secretion of insulin, glucagon, and somatostatin that facilitate glucose disposal. The activation of GLP1R on mouse β-cells induces a robust up-regulation of insulin-like growth factor 1 (IGF1) receptor, which triggers the IGF1/IGF2 receptor autocrine loop associated with an increase of Akt phosphorylation, with the Akt pathway bestowing an anti-apoptotic effect (25). GLP1R expressed on β- and δ-cells can also direct the paracrine regulations by activating the secretion of insulin and somatostatin, respectively, to inhibit glucagon secretion by α-cells. Gastric inhibitory polypeptide (GIP), another incretin hormone which acts on the Gs-coupled GIP receptor (GIPR), mediates similar responses as GLP1 but its mechanism of action is less understood (12). In addition, GIP can induce the production of interleukin-6 (IL6) by α cells, which in turn stimulates the production of GLP1 and insulin secretion by β-cells, forming another paracrine loop (24).
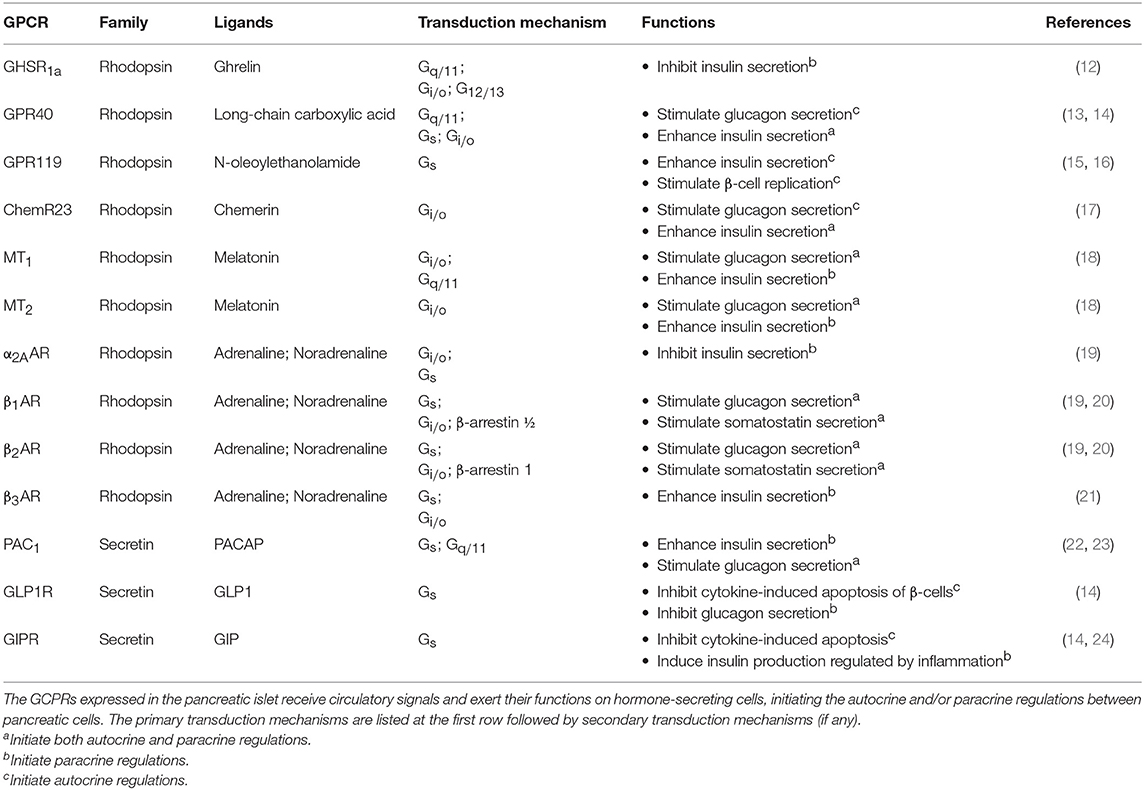
Table 1. Examples of GPCRs in the pancreatic islet for receiving circulatory inputs.
Interestingly, apart from circulatory hormones, energy sources like free fatty acids (FFAs) can also act as signaling molecules. FFAs are obtained from dietary fat. Depending on the length of the carbon chain, FFAs bind to a multitude of GPCRs which are known as FFA receptors (FFARs), including G protein-coupled receptor 41 (GPR41) and GPR43 that bind to short chain FFAs, GPR84 for medium chain FFAs, GPR40 and GPR120 for long-chain FFAs, and GPR119 for long-chain FFAs and cannabinoid (26). These FFARs are differentially distributed in tissues and they signal through different G proteins for energy homeostasis. For example, GPR40 (also known as FFAR1) is expressed in human islets at levels comparable to those of GLP1R. At fasting glucose level, palmitate can enhance the secretion of glucagon and insulin via GPR40 on α- and β-cells (27), and this positive regulation is primarily mediated via the Gq/11 signaling pathway (28). In contrast, GPR119 is expressed predominantly in β-cells. The binding of long-chain FFAs to GPR119 can increase the intracellular cAMP levels via Gs stimulation of adenylyl cyclase (AC) and promotes glucose-stimulated insulin secretion (29). Another example of organ crosstalk is between islet and the adipose tissues, which is mediated by adipose-derived signaling molecules (adipokines). Chemerin, one of the adipokines, was found to regulate glucose-stimulated insulin secretion and improve glucose tolerance via its receptor ChemR23 in mouse. The transduction mechanism of ChemR23 is coupled to the Gi/o pathway, while the functional relevance of the other two receptors of chemerin (GPR1 and CCRL2) remains unclear (17).
The pancreatic islet is additionally regulated by signals from the central nervous system (CNS). Multiple studies have demonstrated the roles of circadian clocks in key metabolic tissues, including liver, pancreas, white adipose, and skeletal muscle (30). In mammals, the suprachiasmatic nuclei (SCN) express a robust rhythm of electrophysiological activity that controls the secretion of melatonin by the pineal gland, with the diurnal variation in melatonin being crucial for synchronizing the circadian rhythm (31). The expression of type 1 and type 2 melatonin receptors (MT1R and MT2R) in the human islets has been confirmed by molecular and immunocytochemical approaches (32). Upon ligand binding, MT1R and MT2R suppress intracellular cAMP production via Gi/o proteins and reduce insulin secretion. The MT2R can also inhibit insulin secretion by suppressing the guanylate cyclase/cyclic guanosine monophosphate (GC/cGMP) pathway (33). A strong functional link between MTNR1B (encode MT2R) and type 2 diabetes risk was further established by Bonnefond et al (34), and an inhibitory effect of melatonin on somatostatin secretion has recently been demonstrated in a human pancreatic δ-cell line (35). Like melatonin, other classical neurotransmitters can also act as neurohormones to modulate pancreatic responses, and these include noradrenaline and adrenaline that inhibit pancreatic hormone secretion. Several subtypes of α2-adrenoceptors (α2AAR, α2BAR, and α2CAR) and β adrenoceptors (β1AR, β2AR, and β3AR) are known to be expressed in the pancreatic islet (36). The α2AAR on β-cells is important for Gi/o-mediated inhibition of insulin secretion. Although less studied in humans, agonists of α2AAR can prevent excess insulin release (37) and variants of α2AAR are apparently associated with type 2 diabetes (38). Contrastingly, Gs-coupled β-adrenoceptors have opposing effects, resulting in enhanced insulin secretion (39). The reduced β2AR expression may contribute to the age-related decline of glucose tolerance in mice (40). It has also been suggested that β1/β2ARs can increase somatostatin levels in mice via a Gs-independent pathway composed of β-arrestin 1 and ERK1/2 (20). Overall, the available evidence supports the notion that Gs-coupled receptors facilitate insulin and somatostatin secretion, whereas Gi-coupled receptors tend to oppose these responses. However, co-activation of Gi- and Gq-coupled receptors was reported to have a synergistic stimulation on cytokine production (41). The mechanism of the observed synergism is presumably mediated via Gβγ-responsive isoforms of phospholipase Cβ (PLCβ2/3), which enable Gβγ dimers released from Gi-coupled receptors to further stimulate PLCβ2/3 (42). Thus, synergistic action in regulating pancreatic hormones by GPCRs represents a distinct possibility which should be examined.
GPCRs for Mediating Autocrine/Paracrine Regulations
Upon receiving various input signals, islet cells in turn secrete autocrine/paracrine molecules, a great number of which modulate the activity of neighboring cells through GPCRs (Table 2). Almost all endogenous GPCR ligands identified in the pancreas activate more than one type of receptor in the islet (9), suggesting that a ligand is able to trigger a variety of GPCRs present on multiple cell types, thereby diversifying the signaling event and inferring a robust paracrine regulatory mechanism. Among all the endocrine cell types within the islet, the paracrine interactions between α-, β-, and δ-cells have been proposed for a long time, with the somatostatin-secreting δ-cells providing essential negative feedback to both insulin and glucagon release, while the glucagon-secreting α-cells positively regulate insulin and somatostatin secretion (Figure 2) (53). Among the five human somatostatin receptor subtypes (SSTR1−5), only SSTR1, SSTR2, and SSTR5 show high expression levels in islet cells. SSTR1 and SSTR2 are selectively expressed on β-cells and α-cells, respectively. SSTR5 is highly expressed on both β- and δ-cells, and is moderately expressed on α-cells (54). Inhibition of glucagon and insulin secretion from the islet is primarily mediated by SSTR2 (43) and SSTR5 (44), respectively. Although all SSTRs are Gi/o-coupled, SSTR2 and SSTR5 can additionally signal through Gq/11 proteins. However, the Gq/11 transduction mechanism in regulating pancreatic hormone secretion remains to be fully elucidated. One potential pathway may involve the activation of nuclear factor κB (NFκB) which regulates inflammation and cell survival. This is in agreement with the demonstrated ability of SSTR2 to activate NFκB via Gq family proteins (55), and that NFκB has been implicated in fatty acid-induce β-cell dysfunction (56). In contrast, α-cells work as a positive regulator. Acetylcholine released by α-cells stimulates insulin secretion by β-cells via the muscarinic M3 and M5 receptors (M3R and M5R), as well as somatostatin secretion by δ-cells through M1R (46). Interestingly, M1R, M3R and M5R are all coupled to the Gq/11 pathway. Apart from the paracrine feedback system, α-cells have a glucagon autocrine feedback loop. Glucagon secreted by the α-cells can upregulate its own expression, the process of which is mediated by the glucagon receptor (GCGR) via Gs-dependent signal transduction (49). Since the release of glucagon is stimulated by a lack of glucose, this kind of positive feedback may help to optimize the hormonal output response under less favorable energy conditions. In general, pancreatic hormone secretion is modulated by intracellular cAMP level. Stimulation of cAMP production by Gs-coupled receptors leads to hormone secretion, while activation of Gi/o-coupled receptors oppose this effect.
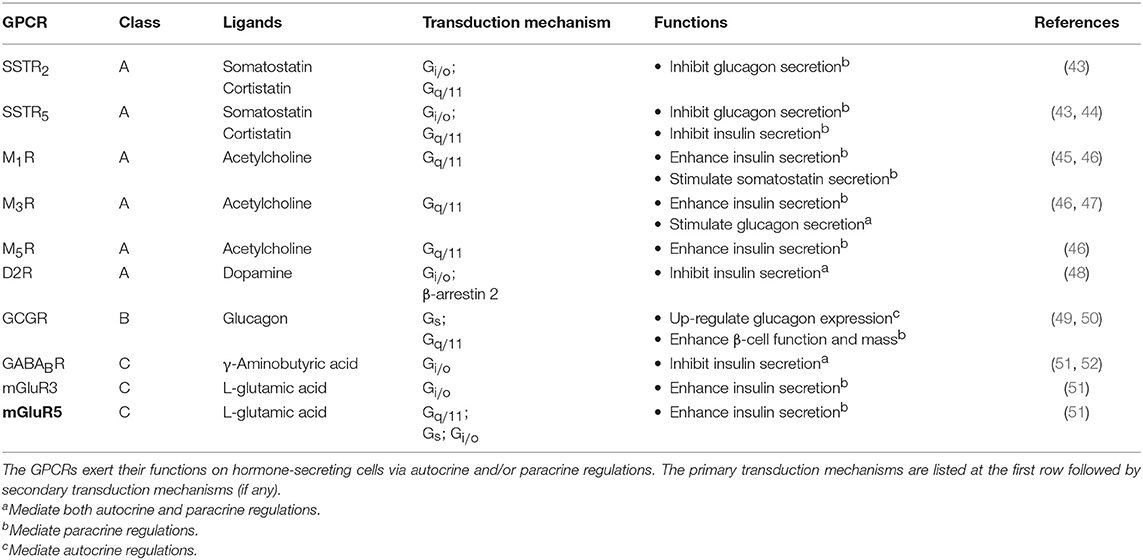
Table 2. Examples of GPCRs in the pancreatic islet for regulating pancreatic hormone secretion.
GPCRs for Regulating Energy Homeostasis in the Brain
The brain modulates the endocrine system in response to external environment. The effect of circulatory hormones, in turn, can regulate brain chemistry and function. Similar to peripheral endocrine glands, stimulation of the brain by circulatory inputs can trigger a sophisticated autocrine/paracrine feedback network to generate an integrated output signal that regulates the body. However, the feedback network is more complex in the brain, and thus more difficult to determine the particular type of GPCR that stimulates or suppresses the release of hormones or neurotransmitters, as synaptic signal transduction and membrane potential need to be taken into account, the latter of which can be modulated not only by Gα subunits, but also by Gβγ dimers. Examples of GPCRs for regulating energy homeostasis in the brain are listed in Table 3.
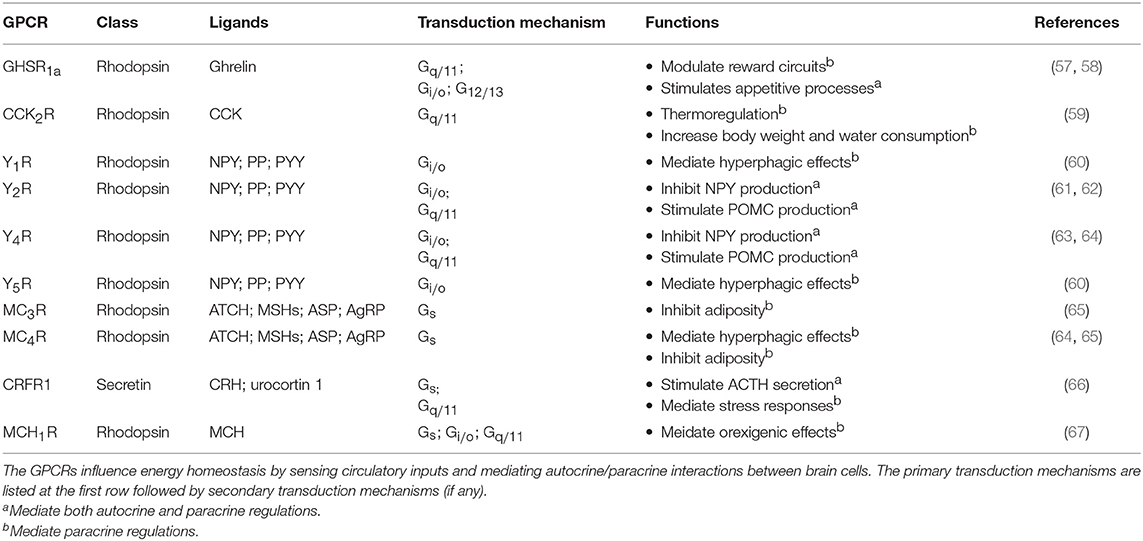
Table 3. Examples of GPCRs for regulating neuoendocrine functions in brain.
From Circulatory Inputs to Neuroendocrine Signals
In the case of energy homeostasis, it is important for the brain to sense the level of metabolic substances in order to regulate energy usage. In the CNS, the hypothalamus is considered as an essential area where the nervous system and the endocrine system meet. Metabolic signals such as glucose, insulin, cholecystokinin (CCK), pancreatic polypeptides (PP), and peptide YY (PYY), and ghrelin have all been found to modulate the activities of the hypothalamic arcuate nucleus (ARC), hence altering food intake and metabolism (68). Among these signals, ghrelin has received intense interest as it can upregulate food intake while the majority acts in the opposite manner (68). Circulatory ghrelin is mainly produced by the gastric X/A-like cells of oxyntic stomach mucosa under hunger situation (69). Ghrelin receptor type 1a (GHSR1a) is highly expressed in the ARC and ventromendial nucleus (VMN) of the hypothalamus (70). By activating phospholipase C (PLC) via Gq/11 protein, GHSR1a triggers the release of neuropeptide Y (NPY) that exerts paracrine effects (which will be discussed later). The heteromerization of GHSR1a with other GPCRs further broadens its downstream responses. Various studies have demonstrated that GHSR1a specifically forms dimers with the SSTR5 (71), dopamine D1 and D2 receptors (D1R and D2R) (72, 73), melanocortin-3 receptor (MC3R), and 5-hydroxytryptamine receptor 2C (5HT2cR) (74). The protomers within these dimers exhibit different signaling effects that range from facilitation, inhibition, and even modification of the pathways (75). Thus, heteromerization of GHSR1a represents a putative mechanism to regulate food intake and energy balance. Ghrelin treatment is also found to alter the dopamine and acetylcholine receptor gene expression in the mesolimbic reward circuitry (76). In contrast to ghrelin, many circulatory signals tend to down-regulate the energy intake. PYYs are stimulated during meal intake by the presence of nutrients (especially fat) in the small intestine. The Y2 receptor (Y2R), which is coupled to Gi/o and Gq/11 proteins, is critical in mediating the effects of PYY3−36 on reducing adiposity and feeding (77). The expression of Y2R can be found throughout the CNS, within the nodose ganglion and on vagal afferents, thus the feeding effects of PYY3−36 is possibly mediated through central, vagal activation, or a combination of both (77). The pattern of c-fos neuronal activation following peripheral administration of PYY3−36 further suggests the involvement of Y2R in the ARC (77).
GPCR-Mediated Autocrine and Paracrine Regulations in the Hypothalamus for Energy Homeostasis
Within the hypothalamus, ghrelin can be synthesized by different hypothalamic nuclei including dorsomedial, ventromedial, paraventricular nucleus (PVN), and the ARC. In the brain, ghrelin mainly acts on the presynaptic terminals of NPY neurons and stimulates the activity of arcuate NPY as demonstrated by electrophysiological recordings (78). NPY neuron is known to inhibit the release of pro-opiomelanocortin (POMC) from POMC neurons using neurotransmitter GABA, at the same time it can stimulate the secretion of melanin-concentrating hormone (MCH) and hypocretin/orexin from the lateral hypothalamus via NPY. The inhibition of POMC together with the secretion of MCH and hypocretin/orexin appears to increase food intake and reduce metabolic rate by acting on the PVN of hypothalamus. This network of paracrine regulation adjust metabolism through multiple output pathways that eventually enhance appetite (68). All NPY receptors in the human hypothalamus, including Y1R, Y2R, Y4R and Y5R, activate the Gi/o-signaling pathway. Depression of Ca2+ channel and increasing G protein coupled inwardly rectifying potassium channel (GIRK) currents are also observed upon G protein activation by NPY (79). The Y1R expressed in the ARC has been suggested to mediate the hyperphagic effect of NPY (60), while the ARC NPY expression is negatively regulated in an autocrine manner via presynaptic Y2R and Y4R present in NPY neurons (62, 63). The perifornical part of the lateral hypothalamus, which is considered as the feeding center, contains a high density of Y5R that mediates NPY-induced hyperphagia (60). Besides, NPY neurons produce another orexigenic peptide, the agouti-related peptide (AgRP), as an endogenous antagonist to the MC3R and MC4R (80). MC3R and MC4R are Gs-coupled receptors present in various hypothalamic nuclei which mediate the neuronal circuits to reduce food intake and increase energy expenditure (81, 82), and their endogenous agonist, α-melanocyte stimulating hormone (α-MSH), is synthesized by POMC neurons in the ARC using precursor POMC protein (81). An example of autocrine and paracrine interaction between the NPY neuron, the POMC neuron, and their downstream effector neuron is shown in Figure 3. Additionally, the POMC neurons can produce another anorexic peptide known as CART or cocaine and amphetamine related transcript (81). Although the receptor for CART remains to be characterized, the activation of Gi/o signaling pathway has been observed upon CART application (83). The paracrine communication between NPY/AgRP and POMC/CART neurons can further modulate downstream neuronal activity via GABAergic signaling (84). Overall, with the participation of GPCRs, the autocrine/paracrine communication between the orexigenic NPY/AgRP neuron and the anorexigenic POMC/CART neuron regulates a variety of physiological and behavioral events to maintain energy balance.
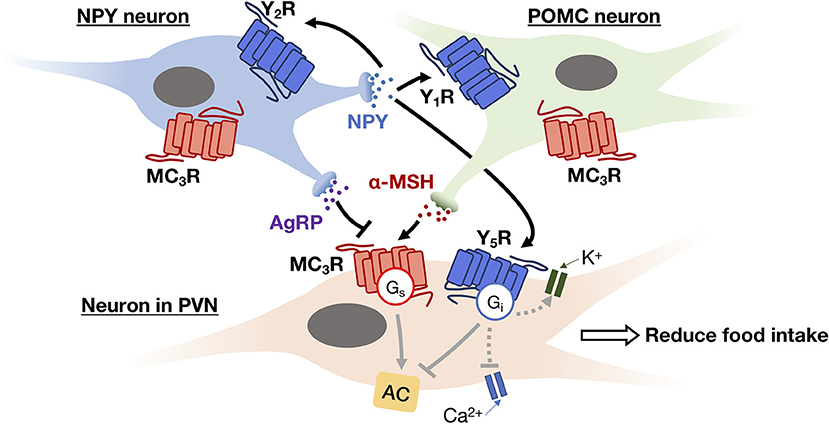
Figure 3. An example of autocrine and paracrine interactions between NPY neuron (blue) and POMC neuron (green) in the ARC and their downstream effector neuron (i.e., neuron in PVN; beige). NPY secreted by NPY neuron can trigger Gi signaling pathway to inhibit Ca2+ channel activity and activate GIRK current via its receptors (Y1R, Y2R, and Y5R) in both autocrine and paracrine manners, thereby inhibiting the release of neurotransmitters. While AgRP and α-MSH secreted by NPY neuron and POMC neuron, respectively, act as antagonist and agonist of MC3R. The activation of MC3R can reduce food intake via the feeding circuit in the hypothalamus.
GPCRs for Bridging the Autocrine/Paracrine Network and for Signal Integration
While an autocrine signal is an amplifier or a brake for message transmission, a paracrine ligand is a tool to disperse the message from one cell type to the others. It has been observed that many ligands can be recognized by multiple GPCR subtypes that are expressed in different cell types. Hence, once a paracrine signal is generated in an organ, various cell types could be differentially activated or inhibited by the same signal via different subtypes of GPCRs. Using pancreatic islet as an example again, somatostatin produced by δ-cells can simultaneously target subtypes of SSTRs on α-cells, β-cells and δ-cells, thereby inhibiting the secretion of both glucagon, insulin and somatostatin itself (53) (Figure 2). The biological setting of SSTR distribution indeed bridges the autocrine/paracrine feedback network. Since multiple cellular responses can be elicited by the same ligand, it results in a synchronized signal propagation. Apart from the pleiotropic regulation by a ligand, some GPCRs recognize more than one type of ligands so that enabling the integration of diverse signals. For example, MC3R in ARC can bind MSHs as well as AgRP with the former acting as agonists and the latter as an antagonist. Competition between these two ligands for the MC3R population on the cell surface will ultimately determine the downstream cellular response (Figure 3). Biased GPCR signaling, wherein an agonist preferentially triggers a specific downstream pathway, may offer additional avenues to modulate autocrine/paracrine signaling. GPR40 is a pharmacological target to increase insulin secretion in type 2 diabetes, and a synthetic ligand TAK-872 has been shown to induce a β-arrestin-biased pathway instead of G protein signals that are typically elicited by native ligands such as palmitate and oleate (85). It is generally accepted that biased agonists have great clinical potentials, but their importance in the regulation of autocrine/paracrine signaling remains to be fully appreciated. The ultimate hormonal output from an endocrine cell is influenced by multiple pathways that are mediated by different receptors. Pancreatic β-cells express Gs-coupled GLP1R and Gi/o-coupled SSTR5, and when both GLP1 and somatostatin are simultaneously presented to the β-cells, GLP1R would give activation signal while the SSTR5 sends inhibition signal to adenylyl cyclase (14, 44). The net effect of which represents an integrated outcome that depends on the relative strengths of the two signals. Furthermore, a single cell can express over a 100 types of GPCRs. The overlapping downstream pathways allow further integration of different messages. Since most cells express numerous GPCRs that receive ligands including circulatory hormones and other molecules including autocrine and paracrine factors, the final outputs from the cell are hence adjusted by signaling molecules in multiple contexts, producing more than one type of signal output, with specific cellular responses regulated by groups of receptors. Therefore, the cell-specific expression profile of GPCRs contributes to the incredibly complicated interactions: for cells to implement the autocrine/paracrine feedback networks, and to integrate various signals for fine-tuning the output. GPCRs such as GLP1R, GHSR, and MC4R are popular pharmaceutical targets for diseases related to energy homeostasis, especially for obesity and diabetes (86). With recent advancements in structural determination and computational techniques, it is envisioned that major strides in our understanding of ligand/receptor interactions may lead to the identification of novel compounds that act as orthosteric, allosteric, or biased ligands of GPCRs. However, it is tremendously important to fully elucidate the pharmacological capabilities of individual GPCRs in the context of signaling networks, as adverse drug effects are often associated with indirect modulations of physiological systems. Hence, a thorough understanding of the diversity and complexity of GPCR signaling is critical for successful therapeutic development of GPCR ligands.
GPCRs Mutations and Perturbation of Energy Homeostasis
Since GPCRs are intricately involved in the regulation of energy homeostasis in both the CNS and peripheral organs, it is not surprising that mutations of GPCRs are found to be associated with endocrine diseases (3). For example, the MT2R mutations in ligand binding and G protein activation are associated with type 2 diabetes (87), and MC4R deficiency is prevalently found in obesity (88) (please refer to sections GPCRs for Sensing Circulatory Inputs and GPCR-Mediated Autocrine and Paracrine Regulations in the Hypothalamus for Energy Homeostasis, respectively). The pathogenesis of which may be attributed to the dysregulation of GPCRs in intercellular communication, can impair cell-to-cell interactions and the autocrine/paracrine feedback loops that are critical for maintaining homeostasis. As illustrated in animal models, mice with GCGR null mutation (GCGR−/−) display supraphysiological glucagon levels, increased proglucagon expression and increased pancreatic and circulating GLP1 (89). The GCGR−/− mice also exhibit reduced adiposity and leptin levels while having normal body weight, food intake, and energy expenditure. These disease phenotypes are associated with postnatal enlargement of the pancreas and hyperplasia of islets, which is mainly due to α-cell, and to a lesser extent, δ-cell proliferation (89). Likewise, the ablation of GCGR delays β-cell differentiation and perturbs the proportion of β- to α-cells in embryonic islets, inhibits the progression of α-cells to maturity in adult mice, as well as affecting the expression of several β-cell-specific genes (90). It has been noted that an augmentation in both islet number and in the rate of proliferation of α- or β-cells led to increased cell mass of the islet (including the δ-cell mass) in those mutant mice (90). These findings in mice suggest that glucagon participates in an autocrine/paracrine feedback loop that regulates the proportion of the different endocrine cell types in islets, the number of islets per pancreas, and development of the mature α-cell phenotype. It was subsequently demonstrated that the overexpression of GCGR increases β-cell mass (89). In humans, a study reported that a homozygous P86S mutation of the human GCGR is associated with hyperglucagonemia, α-cell hyperplasia, and islet cell tumor α-cell hyperplasia (91). This receptor mutant displayed lower affinity to glucagon and decreased cAMP production of the cells at physiological glucagon level. It is likely that the insufficient glucagon signaling perturbs the negative feedback on α-cell proliferation by other islet cells. In this case, the disease is not simply due the alteration of their signaling functions, the influence of malfunction is stepped up by the disruption of autocrine/paracrine feedback loop, which eventually affect the endocrine organ and the greater biological system. Indeed, the engagement of various autocrine/paracrine regulations in human physiology and pathophysiology has been well-recognized over the years. The therapeutic implications of autocrine/paracrine modulators that act on GPCRs are being explored in not only endocrine diseases such as obesity and diabetes (92), but also in cancers (93) and heart failure (94). A number of studies suggested that drugs that can propagate autocrine/paracrine signals could further enhance the efficacy of the therapy. Nevertheless, although GPCRs are one of the most popular pharmacological targets, very little is known with regard to the pathophysiological mechanisms of GPCRs that regulate autocrine/paracrine communications. By better understanding the roles of GPCRs in autocrine/paracrine regulations, one might reveal new avenues for therapeutic interventions against various diseases.
Concluding Remarks
It is known that GPCRs participate in almost every process in the regulation of energy homeostasis as well as other physiological processes that are not mentioned in this review. In the process of maintaining homeostasis, an intricate network of autocrine/paracrine feedbacks is often involved. Due to the pleiotropic property of many GPCR ligands, the diversity of GPCRs and their subtypes, and the potential involvement of multiple intracellular signaling pathways and crosstalks, it is rather difficult to distinguish the physiological roles of autocrine and paracrine factors in a tissue. Tools like co-culture transwell system (95) and the more recently established microcavity platform (96) have been used to study the interactions of selected cell types, which help to unmask autocrine/paracrine communications between cells. Moreover, advances in single cell sequencing techniques enable the identification of GPCR expression profile on different cell types, thus providing a clear mapping of possible GPCR autocrine/paracrine pathways, and it may even be possible to discern between the two mechanisms. By profiling the expression of GPCRs in various cell types within a tissue or organ, we may begin to elucidate the complexity of autocrine/paracrine regulatory pathways under physiological as well as pathophysiological conditions. Subsequent knockdown or knockout experiments can then be conducted to confirm the importance of potential GPCR autocrine/paracrine pathways in healthy and disease states. Given that GPCRs represent highly amenable targets for drug development, deciphering their roles in autocrine/paracrine regulations of human physiology may provide new insights for treatment regimens against complex diseases such as diabetes.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported in part by grants from the Research Grants Council of Hong Kong (16102018 and T13-605/18-W), the University Grants Committee of Hong Kong (AoE/M-604/16), Innovation and Technology Commission of Hong Kong (ITCPD/17-9), and the Hong Kong Jockey Club.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Joy Chan for helpful discussions.
References
1. Schiöth HB, Fredriksson R. The GRAFS classification system of G protein-coupled receptors in comparative perspective. Gen Comp Endocrinol. (2005) 142:94–101. doi: 10.1016/J.YGCEN.2004.12.018
2. Hauser AS, Attwood MM, Rask-Andersen M, Schiöth HB, Gloriam DE. Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov. (2017) 16:829. doi: 10.1038/nrd.2017.178
3. Vassart G, Costagliola S. G protein-coupled receptors: mutations and endocrine diseases. Nat Rev Endocrinol. (2011) 7:362–72. doi: 10.1038/nrendo.2011.20
4. Mohamed-Ali V, Pinkney J, Coppack S. Adipose tissue as an endocrine and paracrine organ. Int J Obes. (1998) 22:1145–58. doi: 10.1038/sj.ijo.0800770
5. Conconi MT, Spinazzi R, Nussdorfer GG. Endogenous ligands of PACAP/VIP receptors in the autocrine–paracrine regulation of the adrenal gland. Int Rev Cytol. (2006) 249:1–51. doi: 10.1016/S0074-7696(06)49001-X
6. Roser JF. Regulation of testicular function in the Stallion: an intricate network of endocrine, paracrine and autocrine systems. Anim Reprod Sci. (2008) 107:179–96. doi: 10.1016/J.ANIREPROSCI.2008.05.004
7. Kos CH, Karaplis AC, Peng J-B, Hediger MA, Goltzman D, Mohammad KS, et al. The calcium-sensing receptor is required for normal calcium homeostasis independent of parathyroid hormone. J Clin Invest. (2003) 111:1021–8. doi: 10.1172/JCI17416
8. In't Veld P, Marichal M. Microscopic anatomy of the human islet of Langerhans. Adv Exp Med Biol. (2010) 654:1–19. doi: 10.1007/978-90-481-3271-3_1
9. Amisten S, Salehi A, Rorsman P, Jones PM, Persaud SJ. An atlas and functional analysis of G protein-coupled receptors in human islets of langerhans. Pharmacol Ther. (2013) 139:359–91. doi: 10.1016/j.pharmthera.2013.05.004
10. Amisten S, Atanes P, Hawkes R, Ruz-Maldonado I, Liu B, Parandeh F, et al. A comparative analysis of human and mouse islet G protein-coupled receptor expression. Sci Rep. (2017) 7:46600. doi: 10.1038/srep46600
11. Delcourt N, Bockaert J, Marin P. GPCR-jacking: from a new route in RTK signalling to a new concept in GPCR activation. Trends Pharmacol Sci. (2007) 28:602–7. doi: 10.1016/j.tips.2007.09.007
12. Ahrén B. Islet G protein-coupled receptors as potential targets for treatment of type 2 diabetes. Nat Rev Drug Discov. (2009) 8:369–85. doi: 10.1038/nrd2782
13. Flodgren E, Olde B, Meidute-Abaraviciene S, Winzell MS, Ahrén B, Salehi A. GPR40 is expressed in glucagon producing cells and affects glucagon secretion. Biochem Biophys Res Commun. (2007) 354:240–5. doi: 10.1016/j.bbrc.2006.12.193
14. Cornu M, Yang J-Y, Jaccard E, Poussin C, Widmann C, Thorens B. Glucagon-like peptide-1 protects β-cells against apoptosis by increasing the activity of an IGF-2/IGF-1 receptor autocrine loop. Diabetes. (2009) 58:1816–25. doi: 10.2337/db09-0063
15. Nolan CJ, Madiraju MSR, Delghingaro-Augusto V, Peyot M-L, Prentki M. Fatty acid signaling in the β-cell and insulin secretion. Diabetes. (2006) 55:S16–23. doi: 10.2337/db06-S003
16. Gao J, Tian L, Weng G, Bhagroo NV, Sorenson RL, O'Brien TD, et al. Stimulating β-cell replication and improving islet graft function by GPR119 agonists. Transpl Int. (2011) 24:1124–34. doi: 10.1111/j.1432-2277.2011.01332.x
17. Takahashi M, Okimura Y, Iguchi G, Nishizawa H, Yamamoto M, Suda K, et al. Chemerin regulates β-cell function in mice. Sci Rep. (2011) 1:123. doi: 10.1038/srep00123
18. Ramracheya RD, Muller DS, Squires PE, Brereton H, Sugden D, Huang GC, et al. Function and expression of melatonin receptors on human pancreatic islets. J Pineal Res. (2008) 44:273–9. doi: 10.1111/j.1600-079X.2007.00523.x
19. Vieira E, Liu Y-J, Gylfe E. Involvement of α1 and β-adrenoceptors in adrenaline stimulation of the glucagon-secreting mouse α-cell. Naunyn Schmiedebergs Arch Pharmacol. (2004) 369:179–83. doi: 10.1007/s00210-003-0858-5
20. Wang H-M, Dong J-H, Li Q, Hu Q, Ning S-L, Zheng W, et al. A stress response pathway in mice upregulates somatostatin level and transcription in pancreatic delta cells through Gs and β-arrestin 1. Diabetologia. (2014) 57:1899–910. doi: 10.1007/s00125-014-3290-0
21. Perfetti R, Hui H, Chamie K, Binder S, Seibert M, McLenithan J, et al. Pancreatic β-cells expressing the Arg64 variant of the β3-adrenergic receptor exhibit abnormal insulin secretory activity. J Mol Endocrinol. (2001) 27:133–44. doi: 10.1677/jme.0.0270133
22. Yada T, Sakurada M, Ishihara H, Nakata M, Shioda S, Yaekura K, et al. Pituitary adenylate cyclase-activating polypeptide. (PACAP) is an islet substance serving as an intra-islet amplifier of glucose-induced insulin secretion in rats. J Physiol. (2004) 505:319–28. doi: 10.1111/j.1469-7793.1997.319bb.x
23. Winzell MS, Ahrén B. Role of VIP and PACAP in islet function. Peptides. (2007) 28:1805–13. doi: 10.1016/j.peptides.2007.04.024
24. Timper K, Dalmas E, Dror E, Rutti S, Thienel C, Sauter NS, et al. Glucose-dependent insulinotropic peptide stimulates glucagon-like peptide 1 production by pancreatic islets via interleukin 6, produced by α cells. Gastroenterology. (2016) 151:165–79. doi: 10.1053/j.gastro.2016.03.003
25. Cornu M, Modi H, Kawamori D, Kulkarni RN, Joffraud M, Thorens B. Glucagon-like peptide-1 increases β-cell glucose competence and proliferation by translational induction of insulin-like growth factor-1 receptor expression. J Biol Chem. (2010) 285:10538–45. doi: 10.1074/jbc.M109.091116
26. Ichimura A, Hirasawa A, Hara T, Tsujimoto G. Free fatty acid receptors act as nutrient sensors to regulate energy homeostasis. Prostag Oth Lipid M. (2009) 89:82–8. doi: 10.1016/j.prostaglandins.2009.05.003
27. Kristinsson H, Sargsyan E, Manell H, Smith DM, Göpel SO, Bergsten P. Basal hypersecretion of glucagon and insulin from palmitate-exposed human islets depends on FFAR1 but not decreased somatostatin secretion. Sci Rep. (2017) 7:1–11. doi: 10.1038/s41598-017-04730-5
28. Briscoe CP, Tadayyon M, Andrews JL, Benson WG, Chambers JK, Eilert MM, et al. The orphan G protein-coupled receptor GPR40 is activated by medium and long-chain fatty acids. J Biol Chem. (2003) 278:11303–11. doi: 10.1074/jbc.M211495200
29. Overton HA, Fyfe MCT, Reynet C. GPR119, a novel novel G protein-coupled receptor target for the treatment of type 2 diabetes and obesity. Br J Pharmacol. (2008) 153:S76–81. doi: 10.1038/sj.bjp.0707529
30. Huang W, Ramsey KM, Marcheva B, Bass J. Circadian rhythms, sleep, and metabolism. J Clin Invest. (2011) 121:2133–41. doi: 10.1172/JCI46043
31. Takahashi JS. Transcriptional architecture of the mammalian circadian clock. Nat Rev Genet. (2016) 18:164. doi: 10.1038/nrg.2016.150
32. Peschke E, Stumpf I, Bazwinsky I, Litvak L, Dralle H, Mühlbauer E. Melatonin and type 2 diabetes - A possible link? J Pineal Res. (2007) 42:350–8. doi: 10.1111/j.1600-079X.2007.00426.x
33. Stumpf I, Mühlbauer E, Peschke E. Involvement of the cGMP pathway in mediating the insulin-inhibitory effect of melatonin in pancreatic β-cells. J Pineal Res. (2008) 45:318–27. doi: 10.1111/j.1600-079X.2008.00593.x
34. Bonnefond A, Clément N, Fawcett K, Yengo L, Vaillant E, Guillaume J-L, et al. Rare MTNR1B variants impairing melatonin receptor 1B function contribute to type 2 diabetes. Nat Genet. (2012) 44:297–301. doi: 10.1038/ng.1053
35. Zibolka J, Mühlbauer E, Peschke E. Melatonin influences somatostatin secretion from human pancreatic δ-cells via MT1 and MT2 receptors. J Pineal Res. (2015) 58:198–209. doi: 10.1111/jpi.12206
36. Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion N V, Peters JA, et al. The concise guide to pharmacology 2017/18: G protein-coupled receptors. Br J Pharmacol. (2017) 174:S17–29. doi: 10.1111/bph.13878
37. Fagerholm V, Haaparanta M, Scheinin M. α2-adrenoceptor regulation of blood glucose homeostasis. Basic Clin Pharmacol Toxicol. (2011) 108:365–70. doi: 10.1111/j.1742-7843.2011.00699.x
38. Talmud PJ, Cooper JA, Gaunt T, Holmes MV, Shah S, Palmen J, et al. Variants of ADRA2A are associated with fasting glucose, blood pressure, body mass index and type 2 diabetes risk: meta-analysis of four prospective studies. Diabetologia. (2011) 54:1710–9. doi: 10.1007/s00125-011-2108-6
39. Porte D. β-adrenergic stimulation of insulin release in man. Diabetes. (1967) 16:150–5. doi: 10.2337/diab.16.3.150
40. Santulli G, Lombardi A, Sorriento D, Anastasio A, Del Giudice C, Formisano P, et al. Age-related impairment in insulin release: the essential role of β2-adrenergic receptor. Diabetes. (2012) 61:692–701. doi: 10.2337/db11-1027
41. Chan ASL, Lau WWI, Szeto ACH, Wang J, Wong YH. Differential regulation of CXCL8 production by different G protein subunits with synergistic stimulation by Gi- and Gq-regulated pathways. J Mol Biol. (2016) 428:3869–84. doi: 10.1016/j.jmb.2016.03.026
42. Chan JSC, Lee JWM, Ho MKC, Wong YH. Preactivation permits subsequent stimulation of phospholipase C by Gi-coupled receptors. Mol. Pharmacol. (2000) 57:700–8. doi: 10.1124/mol.57.4.700
43. Strowski MZ, Parmar RM, Blake AD, Schaeffer JM. Somatostatin inhibits insulin and glucagon secretion via two receptor subtypes: an in vitro study of pancreatic islets from somatostatin receptor 2 knockout mice. Endocrinology. (2000) 141:111–7. doi: 10.1210/endo.141.1.7263
44. Strowski MZ, Kohler M, Chen HY, Trumbauer ME, Li Z, Szalkowski D, et al. Somatostatin receptor subtype 5 regulates insulin secretion and glucose homeostasis. Mol Endocrinol. (2003) 17:93–106. doi: 10.1210/me.2001-0035
45. Renuka TR, Robinson R, Paulose CS. Increased insulin secretion by muscarinic M1 and M3 receptor function from rat pancreatic islets in vitro. Neurochem Res. (2006) 31:313–20. doi: 10.1007/s11064-005-9022-6
46. Molina J, Rodriguez-Diaz R, Fachado A, Jacques-Silva MC, Berggren PO, Caicedo A. Control of insulin secretion by cholinergic signaling in the human pancreatic islet. Diabetes. (2014) 63:2714–26. doi: 10.2337/db13-1371
47. Duttaroy A, Zimliki CL, Gautam D, Cui Y, Mears D, Wess J. Muscarinic stimulation of pancreatic insulin and glucagon release is abolished in M3 muscarinic acetylcholine receptor-deficient mice. Diabetes. (2004) 53:1714–20. doi: 10.2337/diabetes.53.7.1714
48. Ustione A, Piston DW, Harris PE. Minireview: dopaminergic regulation of insulin secretion from the pancreatic islet. Mol Endocrinol. (2013) 27:1198–207. doi: 10.1210/me.2013-1083
49. Leibiger B, Moede T, Muhandiramlage TP, Kaiser D, Sanchez PV, Leibiger IB, et al. Glucagon regulates its own synthesis by autocrine signaling. Proc Natl Acad Sci USA. (2012) 109:20925–30. doi: 10.1073/pnas.1212870110
50. Gelling RW, Vuguin PM, Du XQ, Cui L, Rømer J, Pederson RA, et al. Pancreatic β-cell overexpression of the glucagon receptor gene results in enhanced β-cell function and mass. Am J Physiol Endocrinol Metab. (2009) 297:E695–707. doi: 10.1152/ajpendo.00082.2009
51. Brice NL, Varadi A, Ashcroft SJH, Molnar E. Metabotropic glutamate and GABAB receptors contribute to the modulation of glucose-stimulated insulin secretion in pancreatic β-cells. Diabetologia. (2002) 45:242–52. doi: 10.1007/s00125-001-0750-0
52. Taneera J, Jin Z, Jin Y, Muhammed SJ, Zhang E, Lang S, et al. γ-aminobutyric acid. (GABA) signalling in human pancreatic islets is altered in type 2 diabetes. Diabetologia. (2012) 55:1985–94. doi: 10.1007/s00125-012-2548-7
53. Caicedo A. Paracrine and autocrine interactions in the human islet: more than meets the eye. Semin Cell Dev Biol. (2013) 24:11–21. doi: 10.1016/j.semcdb.2012.09.007
54. Kumar U, Sasi R, Suresh S, Patel A, Thangaraju M, Metrakos P, et al. Subtype-selective expression of the five somatostatin receptors. (hSSTR1−5) in human pancreatic islet cells: a quantitative double-label immunohistochemical analysis. Diabetes. (1999) 48:77–85. doi: 10.2337/diabetes.48.1.77
55. Liu AMF, Wong YH. Activation of nuclear factor κb by somatostatin type 2 receptor in pancreatic acinar AR42J cells involves Gα14 and multiple signaling components: a mechanism requiring protein kinase C, calmodulin-dependent kinase II, ERK, and c-Src. J Biol Chem. (2005) 280:34617–25. doi: 10.1074/jbc.M504264200
56. Amyot J, Semache M, Ferdaoussi M, Fontés G, Poitout V. Lipopolysaccharides impair insulin gene expression in isolated islets of Langerhans via toll-like receptor-4 and NF-κB signalling. PLoS ONE. (2012) 7:e36200. doi: 10.1371/journal.pone.0036200
57. Dickson SL, Egecioglu E, Landgren S, Skibicka KP, Engel JA, Jerlhag E. The role of the central ghrelin system in reward from food and chemical drugs. Mol Cell Endocrinol. (2011) 340:80–7. doi: 10.1016/j.mce.2011.02.017
58. Kirsz K, Zieba DA. Ghrelin-mediated appetite regulation in the central nervous system. Peptides. (2011) 32:2256–64. doi: 10.1016/j.peptides.2011.04.010
59. Weiland TJ, Voudouris NJ, Kent S. The role of CCK2 receptors in energy homeostasis: Insights from the CCK2 receptor deficient mouse. Physiol Behav. (2004) 82:471–6. doi: 10.1016/j.physbeh.2004.04.065
60. Williams G, Bing C, Cai XJ, Harrold JA, King PJ, Liu XH. The hypothalamus and the control of energy homeostasis: different circuits, different purposes. Physiol Behav. (2001) 74:683–701. doi: 10.1016/S0031-9384(01)00612-6
61. Ellacott KLJ, Cone RD. The central melanocortin system and the integration of short- and long-term regulators of energy homeostasis. Recent Prog Horm Res. (2004) 59:395–408. doi: 10.1210/rp.59.1.395
62. Acuna-Goycolea C, van den Pol AN. Peptide YY(3−36) inhibits both anorexigenic proopiomelanocortin and orexigenic neuropeptide Y neurons: implications for hypothalamic regulation of energy homeostasis. J Neurosci. (2005) 25:10510–9. doi: 10.1523/JNEUROSCI.2552-05.2005
63. King PJ, Widdowson PS, Doods HN, Williams G. Regulation of neuropeptide Y release by neuropeptide Y receptor ligands and calcium channel antagonists in hypothalamic slices. J Neurochem. (1999) 73:641–6.
64. Lin S, Shi Y-C, Yulyaningsih E, Aljanova A, Zhang L, Macia L, et al. Critical role of arcuate Y4 receptors and the melanocortin system in pancreatic polypeptide-induced reduction in food intake in mice. PLoS ONE. (2010) 4:e8488. doi: 10.1371/journal.pone.0008488
65. Butler AA, Cone RD. The melanocortin receptors: lessons from knockout models. Neuropeptides. (2002) 36:77–84. doi: 10.1054/npep.2002.0890
66. Dautzenberg FM, Hauger RL. The CRF peptide family and their receptors: yet more partners discovered. Trends Pharmacol Sci. (2018) 23:71–7. doi: 10.1016/S0165-6147(02)01946-6
67. MacNeil DJ. The role of melanin-concentrating hormone and its receptors in energy homeostasis. Front Endocrinol. (2013) 4:49. doi: 10.3389/fendo.2013.00049
68. Abizaid A, Horvath TL. Brain circuits regulating energy homeostasis. Regul Pept. (2008) 149:3–10. doi: 10.1016/j.regpep.2007.10.006
69. Date Y, Kojima M, Hosoda H, Sawaguchi A, Mondal MS, Suganuma T, et al. Ghrelin, a novel growth hormone-releasing acylated peptide, is synthesized in a distinct endocrine cell type in the gastrointestinal tracts of rats and humans. Endocrinology. (2000) 141:4255–61. doi: 10.1210/endo.141.11.7757
70. Ferrini F, Salio C, Lossi L, Merighi A. Ghrelin in central neurons. Curr Neuropharmacol. (2009) 7:37–49. doi: 10.2174/157015909787602779
71. Park S, Jiang H, Zhang H, Smith RG. Modification of ghrelin receptor signaling by somatostatin receptor-5 regulates insulin release. Proc Natl Acad Sci USA. (2012) 109:19003–8. doi: 10.1073/pnas.1209590109
72. Jiang H, Betancourt L, Smith RG. Ghrelin amplifies dopamine signaling by cross talk involving formation of growth hormone secretagogue receptor/dopamine receptor subtype 1 heterodimers. Mol Endocrinol. (2006) 20:1772–85. doi: 10.1210/me.2005-0084
73. Kern A, Albarran-Zeckler R, Walsh HE, Smith RG. Apo-ghrelin receptor forms heteromers with DRD2 in hypothalamic neurons and is essential for anorexigenic effects of DRD2 agonism. Neuron. (2012) 73:317–32. doi: 10.1016/j.neuron.2011.10.038
74. Schellekens H, van Oeffelen WEPA, Dinan TG, Cryan JF. Promiscuous dimerization of the growth hormone secretagogue receptor. (GHS-R1a) attenuates ghrelin-mediated signaling. J Biol Chem. (2013) 288:181–91. doi: 10.1074/jbc.M112.382473
75. Schellekens H, De Francesco PN, Kandil D, Theeuwes WF, McCarthy T, van Oeffelen WEPA, et al. Ghrelin's orexigenic effect is modulated via a serotonin 2C receptor interaction. ACS Chem Neurosci. (2015) 6:1186–97. doi: 10.1021/cn500318q
76. Skibicka KP, Hansson C, Egecioglu E, Dickson SL. Role of ghrelin in food reward: impact of ghrelin on sucrose self-administration and mesolimbic dopamine and acetylcholine receptor gene expression. Addict Biol. (2012) 17:95–107. doi: 10.1111/j.1369-1600.2010.00294.x
77. Karra E, Chandarana K, Batterham RL. The role of peptide YY in appetite regulation and obesity. J Physiol. (2009) 587:19–25. doi: 10.1113/jphysiol.2008.164269
78. Cowley MA, Cone RD, Enriori P, Louiselle I, Williams SM, Evans AE. Electrophysiological actions of peripheral hormones on melanocortin neurons. Ann N Y Acad Sci. (2003) 994:175–86. doi: 10.1111/j.1749-6632.2003.tb03178.x
79. Brothers SP, Wahlestedt C. Therapeutic potential of neuropeptide Y. (NPY) receptor ligands. EMBO Mol Med. (2010) 2:429–439. doi: 10.1002/emmm.201000100
80. Ollmann MM, Wilson BD, Yang Y-K, Kerns JA, Chen Y, Gantz I, et al. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science. (1997) 278:135–8. doi: 10.1126/science.278.5335.135
81. Millington GWM. The role of proopiomelanocortin. (POMC) neurones in feeding behaviour. Nutr Metab. (2007) 4:18. doi: 10.1186/1743-7075-4-18
82. Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. (2006) 443:289–95. doi: 10.1038/nature05026
83. Rogge G, Jones D, Hubert GW, Lin Y, Kuhar MJ. CART peptides: regulators of body weight, reward and other functions. Nat Rev Neurosci. (2008) 9:747–58. doi: 10.1038/nrn2493
84. Tong Q, Ye CP, Jones JE, Elmquist JK, Lowell BB. Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat Neurosci. (2008) 11:998–1000. doi: 10.1038/nn.2167
85. Mancini AD, Bertrand G, Vivot K, Carpentier É, Tremblay C, Ghislain J, et al. β-arrestin recruitment and biased agonism at free fatty acid receptor 1. J Biol Chem. (2015) 290:21131–40. doi: 10.1074/jbc.M115.644450
86. Cooke D, Bloom S. The obesity pipeline: current strategies in the development of anti-obesity drugs. Nat Rev Drug Discov. (2006) 5 919–31. doi: 10.1038/nrd2136
87. Chambers JC, Zhang W, Zabaneh D, Sehmi J, Jain P, McCarthy MI, et al. Common genetic variation near melatonin receptor MTNR1B contributes to raised plasma glucose and increased risk of type-2 diabetes amongst indian asians and european whites. Diabetes. (2009) 67:8. doi: 10.2337/db08-1805
88. Farooqi IS, Keogh JM, Yeo GSH, Lank EJ, Cheetham T, O'Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med. (2003) 348:1085–95. doi: 10.1056/NEJMoa022050
89. Gelling RW, Du XQ, Dichmann DS, Rømer J, Huang H, Cui L, et al. Lower blood glucose, hyperglucagonemia, and pancreatic α cell hyperplasia in glucagon receptor knockout mice. Proc Natl Acad Sci USA. (2003) 100:1438–43. doi: 10.1073/pnas.0237106100
90. Vuguin PM, Kedees MH, Cui L, Guz Y, Gelling RW, Nejathaim M, et al. Ablation of the glucagon receptor gene increases fetal lethality and produces alterations in islet development and maturation. Endocrinology. (2006) 147:3995–4006. doi: 10.1210/en.2005-1410
91. Zhou C, Dhall D, Nissen NN, Chen C-R, Yu R. A homozygous P86S mutation of the human glucagon receptor is associated with hyperglucagonemia, α-cell hyperplasia, and islet cell tumor. Pancreas. (2009) 38:941–6. doi: 10.1097/MPA.0b013e3181b2bb03
92. Müller TD, Finan B, Clemmensen C, DiMarchi RD, Tschöp MH. The new biology and pharmacology of glucagon. Physiol Rev. (2017) 97:721–66. doi: 10.1152/physrev.00025.2016
93. Balkwill F, Mantovani A. Cancer and inflammation: implications for pharmacology and therapeutics. Clin Pharmacol Ther. (2010) 87:401–6. doi: 10.1038/clpt.2009.312
94. Lionetti V, Bianchi G, Recchia FA, Ventura C. Control of autocrine and paracrine myocardial signals: an emerging therapeutic strategy in heart failure. Heart Fail Rev. (2010) 15:531–42. doi: 10.1007/s10741-010-9165-7
95. Domenech M, Yu H, Warrick J, Badders NM, Meyvantsson I, Alexander CM, et al. Cellular observations enabled by microculture: paracrine signaling and population demographics. Integr Biol. (2009) 1:267–74. doi: 10.1039/b823059e
Keywords: G protein, receptor, autocrine, paracrine, signaling
Citation: Tse LH and Wong YH (2019) GPCRs in Autocrine and Paracrine Regulations. Front. Endocrinol. 10:428. doi: 10.3389/fendo.2019.00428
Received: 17 April 2019; Accepted: 14 June 2019;
Published: 12 July 2019.
Edited by:
Eric Reiter, Institut National de la Recherche Agronomique (INRA), FranceReviewed by:
Mohammed Akli Ayoub, United Arab Emirates University, United Arab EmiratesLivio Casarini, University of Modena and Reggio Emilia, Italy
Aylin Carla Hanyaloglu, Imperial College London, United Kingdom
Copyright © 2019 Tse and Wong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yung Hou Wong, Ym95dW5nQHVzdC5oaw==