 Sasha R. Howard
Sasha R. Howard- Centre for Endocrinology, William Harvey Research Institute, Barts and the London School of Medicine and Dentistry, Queen Mary, University of London, London, United Kingdom
Delayed pubertal onset has many etiologies, but on average two-thirds of patients presenting with late puberty have self-limited (or constitutional) delayed puberty. Self-limited delayed puberty often has a strong familial basis. Segregation analyses from previous studies show complex models of inheritance, most commonly autosomal dominant, but also including autosomal recessive, bilineal, and X-linked. Sporadic cases are also observed. Despite this, the neuroendocrine mechanisms and genetic regulation remain unclear in the majority of patients with self-limited delayed puberty. Only rarely have mutations in genes known to cause aberrations of the hypothalamic-pituitary-gonadal axis been identified in cases of delayed puberty, and the majority of these are in relatives of patients with congenital hypogonadotropic hypogonadism (CHH), for example in the FGFR1 and GNRHR genes. Using next generation sequencing in a large family with isolated self-limited delayed puberty, a pathogenic mutation in the CHH gene HS6ST1 was found as the likely cause for this phenotype. Additionally, a study comparing the frequency of mutations in genes that cause GnRH deficiency between probands with CHH and probands with isolated self-limited delayed puberty identified that a significantly higher proportion of mutations with a greater degree of oligogenicity were seen in the CHH group. Mutations in the gene IGSF10 have been implicated in the pathogenesis of familial late puberty in a large Finnish cohort. IGSF10 disruption represents a fetal origin of delayed puberty, with dysregulation of GnRH neuronal migration during embryonic development presenting for the first time in adolescence as late puberty. Some patients with self-limited delayed puberty have distinct constitutional features of growth and puberty. Deleterious variants in FTO have been found in families with delayed puberty with extremely low BMI and maturational delay in growth in early childhood. Recent exciting evidence highlights the importance of epigenetic up-regulation of GnRH transcription by a network of miRNAs and transcription factors, including EAP1, during puberty. Whilst a fascinating heterogeneity of genetic defects have been shown to result in delayed and disordered puberty, and many are yet to be discovered, genetic testing may become a realistic diagnostic tool for the differentiation of conditions of delayed puberty.
Introduction
The timing of puberty in humans and other mammals is strongly influenced by genetic regulation. Studies using epidemiological and intra-familial tools give an estimate of 50–80% of the variation in timing of pubertal onset being under genetic control (1, 2). Another illustration of this is the high correlation of the timing of sexual maturation observed between twins (3). Although the precise age of onset of puberty varies within and between different populations, it is a highly heritable phenotypic feature (4). Despite this strong genetic component, there is much that we still do not understand about the physiological control of the timing of onset of, or progression through, puberty (5).
The clinical phenotype of delayed puberty can be a feature of several different conditions (6). However, the most common presentation is with isolated and self-limited delayed puberty (also known as constitutional delay of growth and puberty, or CDGP). Self-limited delayed puberty has been shown in several observational studies to be the commonest cause of delayed puberty in males and females (7). More than 80% of boys and around one-third of girls presenting with late pubertal onset have this disorder of pubertal timing. The term “self-limited” has been coined as in these patients puberty will have commenced by the age of 18 years. Notably, constitutional features involving short stature or slow growth in early childhood are not seen in all patients with “simple” delayed puberty. In a patient presenting with delayed puberty in adolescence there are three main differential diagnoses: (1) central hypogonadism which is functional or temporary, where inhibition of the hypothalamic-pituitary-gonadal (HPG) axis is secondary to chronic disease (in one-fifth of those with late pubertal onset), under-nutrition, excessive exercise, or psychological distress; (2) permanent (central) hypogonadotropic hypogonadism, either congenital hypogonadotropic hypogonadism (CHH) or acquired, with classically low or normal LH and FSH levels (seen in 9% of males and up to one-fifth% of females); and (3) primary hypogonadism, with elevated gonadotropin levels secondary to gonadal failure, low sex steroid concentrations, and failure of negative feedback (in ~7% of males and one-quarter of females with late pubertal onset) (8).
Self-limited delayed puberty represents a timing of puberty onset at the extreme end of normal. Thus, those patients with this condition have a lack of testicular enlargement in males or breast development in females at an age that is 2 to 2.5 standard deviations (SD) later than the population mean (Figure 1) (6). Moreover, children with slow or stuttering progression through puberty, as diagnosed through the use of puberty normograms, can also fall within this diagnostic category (9) (Figure 1). Delay of pubertal development has now been recognized to be associated with several long-term sequelae and is no longer seen as a benign developmental variant (10). These adverse consequences include a higher risk for early natural menopause and poor overall health (11) and negatively affected psychosocial well-being and peer relationships (12). There is some evidence that delayed puberty is associated with lower bone density (13). Adult height can be affected by late pubertal timing but on average it is only slightly below the genetic target (12).
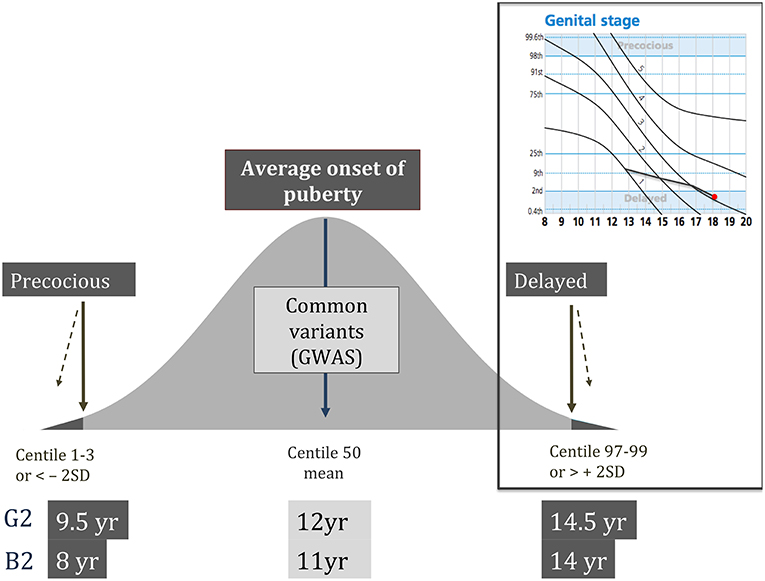
Figure 1. Schematic showing the normal distribution of timing of pubertal onset in the general population, with definitions of precocious and delayed being < or > 2 standard deviations from the mean age, respectively. Top right panel shows an example of a male puberty normogram demonstrating arrested puberty at G3. G–genital stage (Tanner); B–breast stage (Tanner); GWAS–genome wide association studies; SD–standard deviation.
Between half and two-thirds of those patients with self-limited delayed puberty have a family history of late puberty (14). Observational studies have demonstrated that self-limited delayed puberty is inherited with several different inheritance patterns including autosomal dominant or recessive, bilineal (both parents affected by delayed puberty), and X-linked. Sporadic cases are also observed (Figure 2). However, the majority of families display an autosomal dominant pattern of inheritance (with or without complete penetrance) (14–16). Whilst previously considered to be more common in males, evidence suggests that self-limited delayed puberty is not sex-specific, as within families there are near equal ratios of males and females affected with the trait (16). Indeed, in a cohort review by Winter et al., there were a higher number of female than male relatives affected with delayed puberty (47 females vs. 34 males) (17). The higher number of males that present to a medical team may well be a consequence of referral bias.
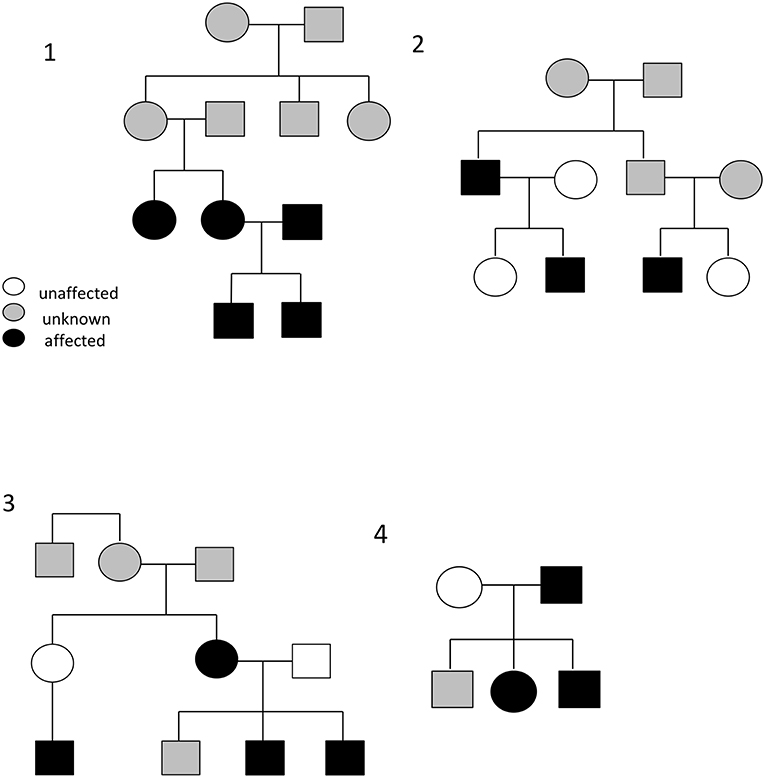
Figure 2. Example pedigrees demonstrating the typical autosomal dominance inheritance pattern seen in self-limited delayed puberty (pedigrees 2 and 4), including bilineal inheritance (shown in pedigree 1), and incomplete penetrance (pedigree 3). Black circles/squares–delayed puberty; clear circles/squares–normal timing of puberty; gray circles/squares–timing of puberty not known.
The etiology is unknown in the majority of patients with delayed puberty (18, 19). Identification of causal genetic defects in familial delayed puberty is complex for a numbers of reasons. Firstly, delayed puberty is not a rare condition, occurring (by statistical definition) in ~2% of the population. Secondly, whilst some pedigrees display clear Mendelian inheritance patterns it is likely that patients may have a di- or oligogenic (where variants in more two genes contribute to the phenotype) genetic basis for their phenotype in many cases. Thirdly, as noted above, self-limited delayed puberty represents a timing of puberty onset at the extreme end of a near-normally distributed trait in the general population, so there may be a low level of causal variants for this condition seen in population databases. Therefore, we cannot, as is often applied for rare diseases, filter out all non-novel variants from our sequencing datasets when searching for causal variants. Instead, we need to compare the prevalence of all rare and predicted damaging variants in a certain gene between cases and controls, in order to identify those genes that are enriched for deleterious variants in patients compared to the general population (20). Finally, the impact of environmental factors such as nutrition and endocrine disruptors superimposed on genetic regulation can “muddy the waters” for those attempting to isolate definitive genetic causes of delayed puberty.
Overlap With Common Genetic Variants of Pubertal Timing
Leptin and Its Pathways
The noted secular trend toward an earlier age of pubertal onset in the developed world has been a subject of study for some time. The importance of energy balance and over- or under-nutrition is clear; a minimum level of energy availability is needed for puberty to ensue, but in contrast higher BMI is associated with earlier puberty (21). This latter statement has been seen especially in females (22, 23), but the underlying patho-biology is still not entirely clear. Leptin, a key metabolic hormone and modulator of BMI in humans, is produced from white adipose tissue (Figure 3). It is a major signal of energy sufficiency and mediates, at least in part, the influence of fat mass on pubertal timing. Leptin is a permissive signal for puberty and is necessary for normal reproduction. In females, serum leptin concentrations rise at the onset of puberty (26). Both humans and mice which lack leptin (Lep ob/ob) or its receptor (LepR db/db) show failure to complete puberty and are infertile (27). However, leptin is not the key coordinator in the up-regulation of GnRH signaling pathways at pubertal onset. Leptin alone does not stimulate pubertal onset and, whilst in females leptin concentrations rise during puberty, levels are lower in males and decrease during puberty (28). GnRH neurons do not express LepR therefore leptin cannot act directly to regulate GnRH neurons. Instead its acts indirectly via leptin-sensitive afferents which project to GnRH neurons (29). These afferents are likely to include LEPR-expressing GABA neurons from the arcuate nucleus, nitric oxide (which is required for its action) pathways, mTOR signaling, as well as kisspeptin/neuropeptide Y neurons (30, 31).
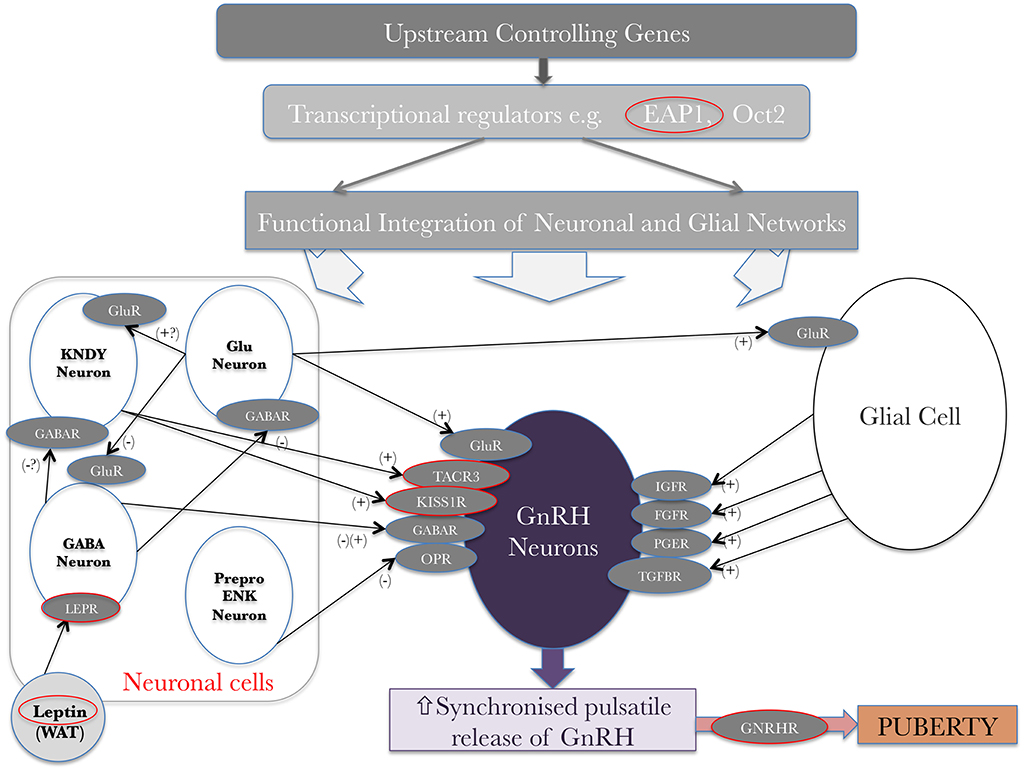
Figure 3. Genetic regulators in the trans-synaptic and glial control of GnRH neurons at the onset of puberty, original idea from Ojeda et al. (24) and adapted from Howard (25) under the CC-BY license. + represents an activating signal, – represents a repressing signal. Red circles highlight genes, mutations in which have been shown to affect pubertal timing. WAT–white adipose tissue; glu–glutamate; gluR–glutamate receptor; KNDY–see text.
Genome-Wide Association Studies
A key strategy in the attempt to uncover the key genetic regulators of pubertal timing in the general population has been genome-wide association studies (GWAS) of age at menarche and voice-breaking in healthy women and men, respectively. The first locus to be identified as associated with pubertal timing was the single nucleotide polymorphism (SNP) rs314276 in the gene LIN28B (32). The major allele of this SNP correlates with earlier breast development and menarche in girls (32). LIN28B is a human ortholog of a Caenorhabditis elegans gene important for developmental timing. The lin-28 family regulates, and is regulated by, the let-7 family of microRNAs (miRNAs). However, no human mutations in LIN28B have been identified, neither with delayed (33) nor with early puberty (34).
Since this initial discovery several increasingly large meta-analyses have been carried out on GWAS of timing of puberty. Whilst the first of these identified 42 (30 new, 2 previously confirmed and 10 possible) loci for age at menarche (35), an analysis of 182,416 European women encompassing 57 studies (36) identified 106 genomic loci. These meta-analyses are ongoing, but the largest to date which comprises 1000 Genomes Project-imputed genotype data in ~370,000 women has isolated 389 independent signals (P < 5 × 10−8) for age at menarche (37). The effect size of each of these alleles on the timing of menarche is between 1 week and 5 months. In total the loci identified in this study can explain ~7.4% of the variation in the timing of menarche in the general population, which corresponds to ~25% of the estimated heritability. Together this data suggests that individually many of these genetic variants have a low impact in the general population (37). Hence these huge studies suggest that there is a large degree of heterogeneity in the genetic determinants of normal pubertal timing. A large number of these signals show a significant association with Tanner staging in men and women, implying that the data is applicable to both genders. Additionally, many of these signals have been shown to have concordant effects on the age at voice breaking. However, in women the signals identified have stronger effects on early than on late age of menarche, but in contrast have larger effect estimates for relatively late than relatively early voice breaking in males (37).
Multiple signals in or near genes regulating the HPG axis function have been found by these studies including LEPR-LEPROT, GNRH1, and TACR3, mutations in which have been shown to be causal in CHH (38, 39). Loci in or near several further genes related to development of the pituitary and its function were also seen, including POU1F1, TENM2, and LGR4, the last of which acts as an enhancer for the pituitary development factor SOX2.
Energy Metabolism Genes Found by GWAS
In addition to leptin signaling, several other genes implicated in body mass index including FTO, SEC16B, TMEM18, and NEGR1 have been implicated by GWAS as having a role in the timing of puberty. FTO had already been identified via GWAS of susceptibility to obesity, and it remains the original and most impactful locus with respect to effect on BMI and risk of obesity (40). Subsequently, using next generation sequencing techniques rare heterozygous variants in FTO have been identified in pedigrees with self-limited delayed puberty associated with extreme low BMI and maturational delay in growth in early childhood (41). In a parallel murine experiment, mice that were heterozygous for FTO gene knockout were shown to have significantly delayed timing of puberty (41).
A further gene, IRX3, identified at the same GWAS locus as FTO, was later found also to be of importance in influencing BMI (42); however the evidence from animal models on the effect of FTO on food intake regulation remains robust (43), although its actions may be complex (44). FTO-knockout mice (45) and in vitro studies have demonstrated that essential amino acids act to modulate the expression of FTO and that FTO acts downstream to influence mTORC1 signaling (46). mTOR acts as a coupler of energy balance and the activity of the reproductive axis by regulation of the hypothalamic expression of the kisspeptin gene (47). Blockade of mTOR in a rodent model led to delayed vaginal opening with blunting of the positive effects of leptin on puberty onset in food-restricted females (48). However, it is still unknown if the effect of FTO on pubertal timing is facilitated via effects on BMI, via mTOR signaling, or by both.
Other Energy Metabolism Genes
Neuropeptide Y (NPY) is another protein implicated in the regulation of food intake and satiety, as well as the hypothalamic-pituitary axis. NPY increases the response of pituitary gonadotrope cells to GnRH (49), both by stimulating GnRH binding to pituitary GnRH receptors and by its action upstream at the median eminence to potentiate GnRH secretion from GnRH axon terminals (50). Studies with primate models imply that NPY may contribute to the brake that restrains the onset of puberty between infancy and mid-childhood (51). The link between energy homeostasis and reproductive development may also be mediated by ghrelin and other gut-derived peptides (52–54). α-MSH signaling via MC3/4 receptors, acting to increase Kiss1 expression and mediate the permissive effects of leptin on puberty, has also been implicated recently as an important element in the metabolic control of puberty (55). Lastly, mice lacking the insulin receptor in astrocytes have delayed puberty and irregular estrus cycles, with reduced astrocyte prostaglandin E synthase 2 levels (56). However, roles for the majority of these genes involved in fat mass and metabolic regulation have not been demonstrably shown in human delay of puberty. A small cohort of 31 patients was analyzed for mutations in the ghrelin receptor, or GHSR, and 5 patients were found to have point mutations in this gene (57).
Importance of GnRH Neuroendocrine Network in the Pathogenesis of Delayed Puberty
Overlap Between GnRH Deficiency and Delayed Puberty
It is biologically very plausible that the pathophysiology of delayed puberty and conditions of GnRH and gonadotropin deficiency share a common genetic basis. Therefore, investigations have been carried out into the role of genes known to cause CHH in the phenotype of isolated delayed puberty. Previous studies in CHH cohorts have found mutations in HS6ST1, FGFR1, and recently in KLB, not only in small numbers of patients with CHH but also in their relatives with delayed puberty (58–60). Last year, a study was completed that aimed to compare the frequency with which mutations in genes (n = 24) known to cause GnRH or gonadotropin deficiency were found in patients with CHH and individuals with self-limited delayed puberty. This comparison found a significantly higher proportion of mutations in the CHH group (51% of CHH probands vs. 7% of delayed puberty probands, p = 7.6 × 10−11). Whilst this is perhaps unsurprising, a greater degree of oligogenicity in these GnRH deficiency genes was also seen in the CHH group, suggesting a mostly distinct or as yet undiscovered genetic basis of these two conditions (61). Mutations in Kallmann Syndrome (KS) genes such as ANOS1 and NSMF, leading to hypogonadotropic hypogonadism with anosmia, have not been found in individuals with self-limited pubertal delay.
Studies using next generation sequencing to examine cohorts of patients with delayed puberty have identified variants in several CHH genes, particularly GNRHR, TAC3 and its receptor TACR3, but also in IL17RD and SEMA3A (62). However, these variants have not been tested in vitro or in vivo for pathogenicity, or investigated for within pedigree segregation. Many syndromic conditions have delayed or absent puberty within the phenotypic spectrum of the condition, see Table 1.
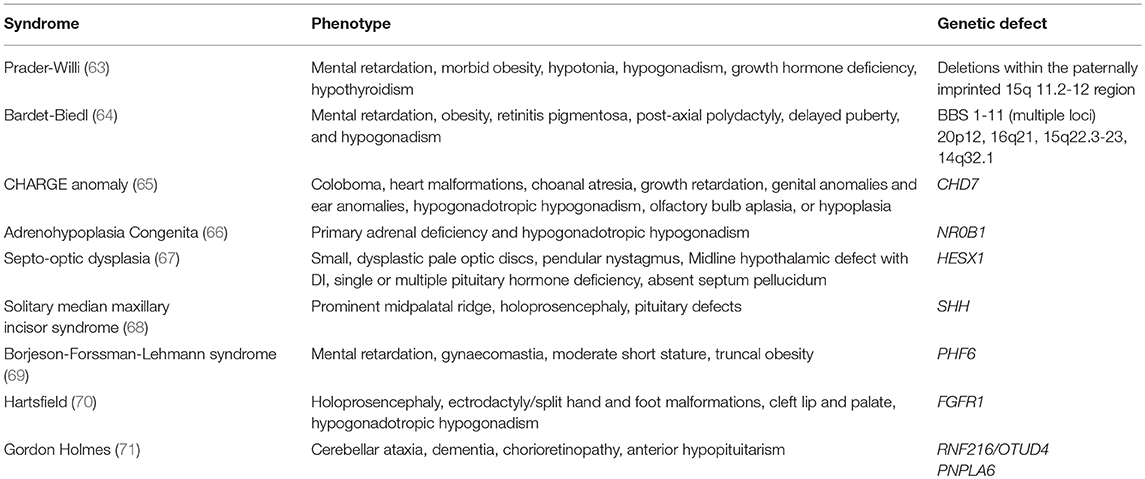
Table 1. Genetic syndromes associated with pubertal delay.
Heparin Sulfate 6O Sulphotransferase 1
Recently, using whole and targeted exome analysis a mutation in HS6ST1 was found in one extended pedigree from a large cohort of patients with isolated familial delayed puberty, for the first time without associated CHH in patient relatives (72). All of the six family members in three generations that carried the mutation had a classical self-limited delayed puberty phenotype, with no individuals displaying CHH. A spontaneous onset of puberty was seen in the proband at 14.3 years. A mouse heterozygous knockout model was also examined in parallel. This work substantiated that loss of one allele of Hs6st1 can provoke pubertal delay but with normal adult reproductive capacity. The Hs6st1+/− mice displayed no compromise in their fertility, GnRH neuron or testes development or spermatogenesis and were born at normal Mendelian ratios. However, female mice were seen to have a significant delay in the timing of vaginal opening, a surrogate for onset of puberty in female rodents.
Notably the Hs6st1+/− mice had no defects of olfactory bulb morphology and no significant reduction in the total number of GnRH neurons in the hypothalamus or extending to the median eminence to explain the pubertal delay. Instead, this might be mediated by changes in either GnRH neuron activity or other relevant downstream pathways, implied by the expression of Hs6st1 mRNA in both the arcuate nucleus and paraventricular nucleus (73, 74). These results indicate whilst, as above, many patients with familial self-limited delayed puberty do not carry mutations in CHH genes, perturbations in a single allele of a particular subset of genes that modulate the HPG axis may be enough to result in a phenotype of self-limited pubertal delay. In contrast, more deleterious alterations in these genes, mutations in both alleles of a gene or a heterozygous mutation in combination with mutations in further genes, are needed to produce the more severe phenotypes of CHH and KS (75).
Immunoglobulin Superfamily Member 10
A further study utilizing whole and targeted exome sequencing methods in the same large Finnish cohort of individuals with familial self-limited delayed puberty, identified deleterious mutations in the IGSF10 gene in six unrelated families (76). Mutations in this gene affect the migration of GnRH neurons from the vomeronasal organ in the nose to the forebrain during embryonic development (Figure 4). The patients with these mutations presented in adolescence with pubertal delay without features of constitutional delay in growth. Given that a functional GnRH neurosecretory network is required for the onset of puberty, the hypothesis produced from this work is that disruption of GnRH neuronal migration, as caused by aberrant IGSF10 signaling, could result in arrival of fewer (or delayed) GnRH neurons at the hypothalamus. This would then in turn lead to a functional defect in the GnRH neuroendocrine network and an increased “threshold” for the onset of puberty, with a resultant delay. In addition, loss-of-function mutations in IGSF10 were found in patients with a hypothalamic amenorrhea-like phenotype, implying a shared genetic basis of functional central hypogonadism with both CHH (77) and delayed puberty. However, although deleterious mutations were enriched in CHH patients, there was lack of complete segregation with trait in these permanent hypogonadotropic hypogonadism families, suggesting that haploinsufficiency of IGSF10 is not sufficient to cause this phenotype. Interesting, mutations in IGSF10 have very recently also been found in patients with both premature ovarian insufficiency and disorders of neuronal development, and in the same report in a further pedigree with a Kallmann-like phenotype (78). The results of the studies on HS6ST1 and IGSF10 in delayed puberty point to a mechanism by which developmental defects in the GnRH system during fetal life can modulate the timing of pubertal onset in adolescence, seemingly without other phenotypic features. It remains to be determined whether these patients have any deficiency of the their long-term reproductive capacity or sexual lifespan.
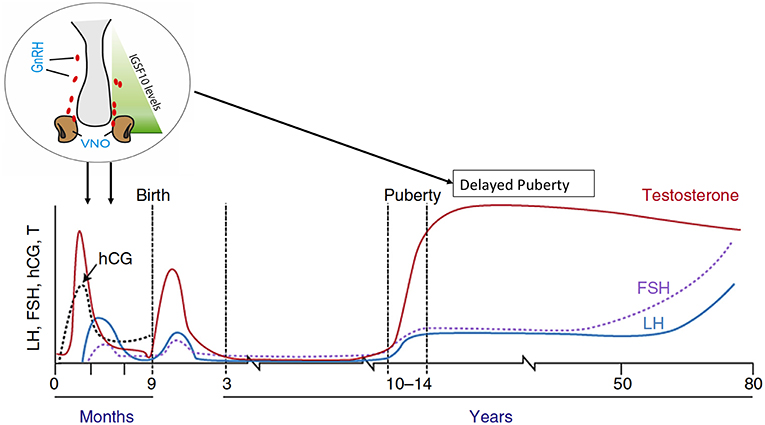
Figure 4. Schematic of the mechanism by which IGSF10 mutations lead to delayed puberty. Reduced levels of IGSF10 expression during embryogenesis in the corridor of nasal mesenchyme from the vomeronasal organ to the olfactory bulbs result in delayed migration of GnRH neurons to the hypothalamus. This leads to a phenotype of delayed puberty first evident in adolescence, due to abnormalities of the GnRH neuroendocrine network. Adapted from doi: 10.1210/er.2018-00248.
Genes Downstream of GnRH
Autosomal recessive CHH is most frequently caused by loss-of-function mutations within the GnRH receptor, accounting for 16–40% of this patient group. Mutations have been found within the extracellular, transmembrane, and intracellular domains of the receptor leading to impaired GnRH action (79). Sequencing studies that have analyzed the GNRHR gene in cohorts with self-limited delayed puberty (79), have found just a handful of deleterious mutations. A homozygous partial loss-of-function mutation in GNRHR was found in two brothers, one with self-limited delayed puberty and one with CHH (80), and a further heterozygous mutation found in one male with self-limited delayed puberty (81). Far more rarely defects of the glycoprotein hormones luteinizing hormone (LH) or follicle-stimulating hormone (FSH) can lead to CHH, in particular via mutations in the specific β-subunits (82, 83). In women, loss-of-function mutations of LHβ result in normal or late timing of menarche (following a normally timed onset of puberty) but with later infertility resulting from lack of ovulation (84). In men, similar defects lead to a presentation with absent pubertal development secondary to Leydig cell hypoplasia resulting in testosterone deficiency and failure of spermatogenesis. Women with inactivating FSHβ mutations display pubertal arrest and primary amenorrhea whilst men have a similar pattern of spontaneous entry into puberty followed by arrest with azoospermia (85). Defects in these two genes do not usually present with a classical picture of self-limited delayed puberty.
Overall, from the evidence we have from current published work we can conclude that although there are some shared gene defects, the genetic basis of CHH and delayed puberty is likely to be due to different, currently unrecognized, genes in many cases (Table 2) (81).
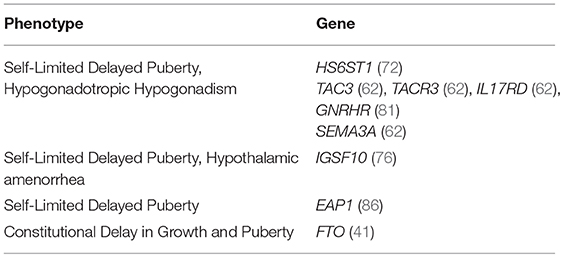
Table 2. Non-syndromic genetic defects associated with pubertal delay.
However, there is a wide spectrum of phenotypes in patients with central hypogonadism, ranging from complete hypogonadotropic hypogonadism, with failure of pubertal development, to partial hypogonadism with an arrest of pubertal development, and even reversible hypogonadotropic hypogonadism in some patients post treatment (87–90). It may be a prudent strategy for clinicians to focus the use of genetic testing of known CHH genes in delayed puberty patients on those patients with either extreme delayed puberty, clear familial inheritance or red flags (such as micropenis, cryptorchidism, anosmia, cleft lip or palate or renal agenesis) which would point to a syndromic or CHH phenotype.
Transcriptional and Epigenetic Control of GnRH Signaling
The GnRH Pulse Generator
The central control of pubertal onset, after the mid-childhood period of HPG axis quiescence, is orchestrated by a resurgence of the GnRH pulse generator, with a reduction in central inhibition and a sharp upregulation in the activity of this axis. This activity is permitted by a change in the balance of GABA-glutamate signaling in the brain (91). Around this time morphological changes in GnRH neurons have been observed including increases in dendritic spine density and a simplification of their dendritic architecture. The intensification of kisspeptin signaling in the hypothalamus, one of the key hormonal players in puberty onset, at this time is a consequence of both an increase of kisspeptin synthesis and a rise in the responsiveness of GnRH neurons to kisspeptin stimulation. This mechanism has been well observed in murine models, but also in primates and is relatively conserved during evolution (92). However, what is far less well understood are what the triggers are for this upregulation of kisspeptin biosynthesis in the hypothalamus at the end of the juvenile period. Thus, whilst there is strong evidence that the secretion of kisspeptins from KNDy neurons in the arcuate nucleus is one of the vital stimulatory inputs on the GnRH pulse generator, it is not likely to be the ultimate controller of the release of the puberty brake. Rather, kisspeptin is the conductor of the orchestra of upstream stimulators and repressors influencing the system at this crucial developmental stage (51).
Transcriptional Control of the GnRH Network
It is likely, therefore, that there is no one single gene that is capable of the hypothalamic control of puberty onset. Instead, we can imagine a hierarchical network of genes acting together to lift the brake applied during the dormancy of the HPG axis in mid-childhood (Figure 3). Data to support this hypothesis have largely come from a systems biology approach (93) and animal models (51), with little data from human subjects. It is clear that transcriptional repression is fundamentally important to the regulation of gene expression in mammals. Transcriptional repressors containing zinc finger motifs, which recognize specific DNA sequences in regulatory regions of the genome, are particularly appealing candidates to have major roles in this governing network (94). Potential key regulators include Oct-2, Ttf-1, Yy1, and Eap1. Oct-2 is a transcriptional regulator of the POU-domain family of homeobox-containing genes. Oct-2 mRNA is upregulated in the hypothalamus in juvenile rodents; blockage of Oct-2 synthesis delays age at first ovulation and hypothalamic lesions which induce precocious puberty (e.g., hamartomas) activate Oct-2 expression (95). Ttf-1 is another homeobox gene that enhances GnRH expression (96). Ttf-1 expression is increased in pubertal rhesus monkeys (97). Yy1 is a zinc-finger transcription factor with crucial roles in normal development and malignancy (98). Eap1, or Enhanced at puberty 1, codes for a nuclear transcription factor, characterized by a dual transcriptional activity: it both trans-activates the GnRH promoter, which facilitates GnRH secretion, and inhibits the preproenkephalin promoter, which represses GnRH secretion. Eap1 mRNA levels increase in the hypothalamus of primates and rodents during puberty, and Eap1 knockdown with siRNA causes delayed puberty and disrupted estrous cyclicity in a rodent model (99–103). Therefore, Eap1 transcriptional activity facilitates the initiation of female puberty, in a manner that is independent of hypothalamic Kiss1 expression (101). Eap1 gene expression is itself regulated by both activation by Ttf-1, and repression by Yy1 and a further transcriptional regulator Cux1 (104).
Enhanced at Puberty 1
A very recent discovery is of the first human EAP1 mutations that appear to be causal for self-limited delayed puberty in two families (86). The affected individuals from these two families had classical clinical and biochemical features of self-limited delayed puberty, with presentation at more than 15.5 years with delayed onset of Tanner stage 2 and delayed peak height velocity. Both probands had spontaneous pubertal development by the age of 18 years without testosterone therapy, thus excluding CHH. By whole exome sequencing of probands with familial delayed puberty two highly conserved variants—one in-frame deletion and one rare missense variant in EAP1—were identified. Using a luciferase reporter assay, EAP1 mutants showed a reduced ability to trans-activate the GnRH promoter compared to wild-type EAP1, due to reduced protein levels caused by the in-frame deletion and sub-cellular mis-location caused by the missense mutation. This study also demonstrated by chromatin immunoprecipitation that EAP1 binding to the GnRH1 promoter increases in monkey hypothalamus at the onset of puberty.
Polycomb Complex Genes
Furthermore, evidence from a recent study has emphasized the importance of the transcriptional control of the Kisspeptin gene Kiss1. This regulation by the polycomb complex proteins EED and Cbx7 is thought to be an important transcriptional repressive mechanism to prevent the premature onset of puberty (105). In the latter stages of mid-childhood there is an increase in the methylation of the promoters of these genes, resulting in a reduction in expression, as well as a decrease in the binding of EED on the Kiss1 promoter. This inhibition of Kiss1 repression also correlates with reduced expression of transcription factors containing certain zinc finger motifs. Moreover, there is also reorganization of the chromatin status and changes in histone methylation to accompany the loss of these polycomb complex proteins from the Kiss1 promoter (106). Studies on both rats and goats also provide data on changes in histone acetylation and gene methylation resulting in alterations in gene expression during puberty (107, 108).
Epigenetic Mechanisms in the Timing of Puberty
There are a number of different epigenetic mechanisms that may have importance for the regulation of the pubertal timing, including imprinting. Imprinted genes are known to influence the timing of several key developmental stages in humans including weaning and adrenarche. In general, paternally expressed genes promote later childhood maturation and maternally expressed genes promote a more premature maturation (109). This holds true for two paternally inherited genes, MKRN3 and DLK1, which are associated with age at menarche in girls and voice-breaking in boys from the GWAS discussed above (37). Variants in both of these genes have been discovered in patients with familial central precocious pubertal timing, with paternally-inherited mutations leading to the expression of the phenotype (110, 111). MKRN3 is thought to contribute to the puberty brake restraining the HPG axis via inhibition of GnRH release. This gene encodes Makorin Ring finger protein 3, a zinc finger protein containing a C3HC4 motif (known as a RING domain) associated with E3 ubiquitin ligase activity (112, 113). Since MKRN3 expression in the arcuate nucleus falls in murine models between birth and weaning, and in humans serum concentrations decline at puberty onset, it is thought to have an inhibitory effect on the GnRH network (114, 115). This supports the hypothesis that the onset of puberty is a consequence of the removal of gonadotropic axis repression. However, what is still unclear is where MKRN3 is placed in this hierarchy of gene regulators controlling kisspeptin levels. Very new data has demonstrated that knock-out of MKRN3 in pluripotent stem cells does not affect GNRH1-expression when these cells are later differentiated into neurons (116). In terms of delayed puberty, mutations in neither MKRN3 nor DLK1 genes have been described in human patients with these conditions.
Imprinting and Pubertal Timing
Prader-Willi syndrome (PWS) is frequently caused by disorders of imprinting and is often associated with either absent or delayed puberty (117). In most patients with PWS the syndrome is due to a deletion of a cluster of imprinted genes (including MKRN3) on the paternally inherited copy of chromosome 15 (paternal deletion), or by inheritance of both copies of this cluster from the mother (maternal uniparental disomy) (118). Precocious puberty is relatively uncommon in PWS (119), but most individuals show some degree of pubertal failure, with one or a combination of an absent pubertal growth spurt, hypogonadotropic hypogonadism, cryptorchidism, underdeveloped genitalia, or primary amenorrhea (120). The probable explanation for the rarity of precocious puberty in individuals with PWS, despite the lack of MKRN3 expression, is the effects of other genes inactivated by the imprinting defect, in particular MAGEL2 (121, 122). This points to a complex role for imprinted genes in the pubertal timing, with tissue type and developmental stage specific gene expression (109, 118).
Non-coding RNAs
Evidence from murine models has demonstrated that non-coding RNAs can act as epigenetic modulators of the timing of puberty. Specific microRNAs play a role in the epigenetic up-regulation of GnRH transcription during what is known in mice as “the critical period,” or infantile mini-puberty in humans (123). A key pair of microRNAs (miR-200 and mIR-155) are thought to regulate Gnrh1 expression, and to control the expression of two important transcriptional repressors of Gnrh1, Zeb-1, and Cebpb. There is an associated increase in the transcriptional activation of GnRH1 with a reduction in Zeb-1 and Cebpb, the latter a nitric oxide-mediated repressor of Gnrh1 that acts both directly and through Zeb1. These changes lead to the up-regulation of Gnrh1 synthesis in GnRH neurons (123). Moreover, miR-7a2 has been demonstrated to be essential for normal murine pituitary development and HPG function, with deletion in mice leading to hypogonadotropic infertility (124).
Endocrine-Disrupting Chemicals
The increase of kisspeptin and GnRH expression in the hypothalamus at puberty is therefore the result of the actions of an intricate arrangement of repressing and activating transcription factors controlling Kiss1 and GnRH1 transcription, with these being themselves influenced by several epigenetic mechanisms including DNA methylation, histone modification and non-coding RNAs (106, 123, 125, 126). Moreover, these epigenetic mechanisms are possible facilitators of gene-environment interactions that also have influence on the hypothalamic regulation of puberty. A number of different sources of evidence have demonstrated that the brain epigenome at puberty is affected by environmental disturbances (107). Endocrine-disrupting chemicals (EDCs), often found in products commonly used in the developed world, have been considered as a potential cause for pubertal timing disturbance for many years, with increasing concern among the lay population (127). Many and varied substances have been identified as possible EDCs, such as polybrominated biphenyls, bisphenol A, atrazine (herbicides), and glyphosate, but also common medicines including paracetamol and betamethasone (128–131). It has been observed that adolescents who have been exposed to the estrogenic insecticide DTT and then adopted internationally display early or precocious pubertal timing (132).
The most important timing of EDC exposure for impact on pubertal timing was historically considered to be in late childhood, but there is now clear data that there may be prenatal and infantile origin of alterations in the timing of puberty. In utero exposure in males to EDCs, in particular to phthalates, can result in under-masculinization of genitalia (133). Moreover, exposure of pregnant rodents to EDCs has been associated with epigenetic alterations in testis as well as other systemic effects. This together suggests that epigenetic changes in the fetal period are a potential mechanism for the hypothalamic effects of prenatal exposure to EDCs (131). These effects may manifest in pregnant rodents, their unborn fetus but also into the next two or more generations as well (134).
However, it is difficult to definitively demonstrate a mechanism of action for EDCs through the premature activation of the hypothalamic GnRH pulse generator. Recently, exposure of female mice to arsenic in utero was shown to alter the hypothalamic expression not only of GnRH and LH but also of their upstream transcriptional regulators, in particular Oct-2 and Ttf-1 (135). Mice exposed to arsenic demonstrated precocious puberty with premature vaginal opening, a marker of the onset of puberty rodents. However, in most datasets it has been difficult to unpick the most likely differing, and possibly conflicting, influence of varying doses and combination of EDCs affecting estrogenic, androgenic or other pathways, and changes in effects depending on age and length of exposure (136).
Future Directions
Over the last 2 years there have been very exciting developments in the understanding of the genetic basis of delayed puberty, particularly with respect to the transcriptional and epigenetic control of the GnRH “master switch.” We anticipate further discoveries in the near future that will help to elucidate these control mechanisms and better understand the genetic predisposition to familial delayed puberty and to conditions of functional hypogonadism. It is, of course, hoped that this knowledge can be rapidly translated into more efficient clinical diagnosis and management.
Conclusion
Puberty represents the remarkable transition from childhood to adult life with the attainment of reproduction and adult stature. The onset of puberty is elicited by the re-activation of the HPG axis, which is first functional in fetal life, through a rise in the pulsatile release of hypothalamic GnRH. Puberty can be deemed the consequence of a neurodevelopmental program, that begins prenatally but has many features in common with the postnatal development of other neuronal processes. However, its unique feature is as a functional system that lies dormant for most of childhood and then reactivates in the majority of the population within a short time window. This timing is controlled by genetic factors, relies upon an intact hormonal axis and influenced by the environment. It is thus not so surprising that pubertal delay and even aberrant pubertal development are not infrequent human pathologies.
The genetic regulators that determine timing of puberty in the general population, a trait that follows a skewed near-normal distribution, have relevance to conditions of delayed and even aberrant pubertal onset (Figure 5). There is also overlap between those pathways found to be defective in self-limited delayed, precocious, and absent puberty conditions, with the phenotype varying dependent on the impact of the gene defect and mutational burden. So whilst there are shared pathogenic mechanisms between these conditions, there is also much heterogeneity in the genetic changes responsible for delayed puberty. Defects in GnRH neuronal development and function, transcriptional regulation of the HPG axis, epigenetic mechanisms including DNA methylation, histone modification and non-coding RNAs, and metabolic and energy homeostatic derangements, can all lead to the final common pathway of delayed puberty. Moreover, these genomic regulators can exert their influence in fetal life, during postnatal development and in mid-childhood, all having an effect in adolescence on pubertal timing.
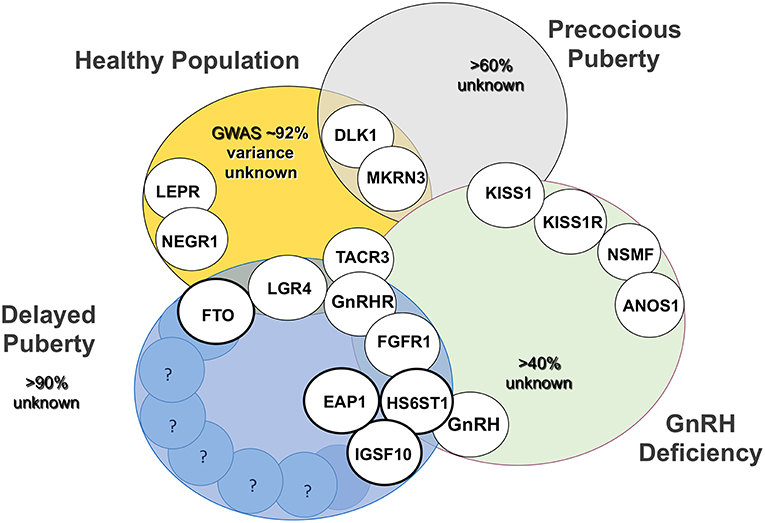
Figure 5. Established genetic basis of common genetic variants of pubertal timing from genome wide association studies (GWAS), conditions of GnRH deficiency (CHH and KS), precocious puberty and delayed puberty and their overlap. Activating and inactivating mutations in KISS1 and KISS1R cause the opposite phenotypes, precocious puberty and CHH, respectively. Bold circles highlight those genes, mutations in which have been identified in familial delayed puberty. Adapted from Howard (25) under the CC-BY license.
Genetic testing may allow the translation of this understanding to benefit patient care in the future: as a diagnostic tool for the investigation of delayed puberty, by informing the natural history of the condition, possible inheritance in the individual's family and optimization of treatment. Rapid and accurate diagnostic testing in clinic would greatly improve patient care and most likely represent a significant advantage in terms of health economics.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
SH is funded by the NIHR (CL-2017-19-002), The Rosetrees Trust (M222-F1), and supported by the Academy of Medical sciences, Wellcome Trust, Medical Research Council, British Heart Foundation, Arthritis Research UK and Diabetes UK through the clinical lecturers scheme (SGL019\1043).
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Morris DH, Jones ME, Schoemaker MJ, Ashworth A, Swerdlow AJ. Familial concordance for age at menarche: analyses from the Breakthrough Generations Study. Paediatr Perinat Epidemiol. (2011) 25:306–11. doi: 10.1111/j.1365-3016.2010.01183.x
2. Eaves L, Silberg J, Foley D, Bulik C, Maes H, Erkanli A, et al. Genetic and environmental influences on the relative timing of pubertal change. Twin Res. (2004) 7:471–81. doi: 10.1375/1369052042335278
3. Van Den Berg SM, Setiawan A, Bartels M, Polderman TJ, Van Der Vaart AW, Boomsma DI. Individual differences in puberty onset in girls: bayesian estimation of heritabilities and genetic correlations. Behav Genet. (2006) 36:261–70. doi: 10.1007/s10519-005-9022-y
4. Palmert MR, Boepple PA. Variation in the timing of puberty: clinical spectrum and genetic investigation. J Clin Endocrinol Metab. (2001) 86:2364–8. doi: 10.1210/jcem.86.6.7603
5. Divall SA, Radovick S. Pubertal development and menarche. Ann N Y Acad Sci. (2008) 1135:19–28. doi: 10.1196/annals.1429.026
6. Palmert MR, Dunkel L. Clinical practice. Delayed puberty. N Engl J Med. (2012) 366:443–53. doi: 10.1056/NEJMcp1109290
7. Abitbol L, Zborovski S, Palmert MR. Evaluation of delayed puberty: what diagnostic tests should be performed in the seemingly otherwise well adolescent? Arch Dis Child. (2016) 101:767–71. doi: 10.1136/archdischild-2015-310375
8. Sedlmeyer IL. Delayed puberty: analysis of a large case series from an academic center. J Clin Endocrinol Metabol. (2002) 87:1613–20. doi: 10.1210/jc.87.4.1613
9. Lawaetz JG, Hagen CP, Mieritz MG, Blomberg Jensen M, Petersen JH, Juul A. Evaluation of 451 Danish boys with delayed puberty: diagnostic use of a new puberty nomogram and effects of oral testosterone therapy. J Clin Endocrinol Metab. (2015) 100:1376–85. doi: 10.1210/jc.2014-3631
10. Zhu J, Chan YM. Adult consequences of self-limited delayed puberty. Pediatrics. (2017) 2017:3177. doi: 10.1542/peds.2016-3177
11. Day FR, Elks CE, Murray A, Ong KK, Perry JR. Puberty timing associated with diabetes, cardiovascular disease and also diverse health outcomes in men and women: the UK Biobank study. Sci Rep. (2015) 5:11208. doi: 10.1038/srep11208
12. Albanese A, Stanhope R. Predictive factors in the determination of final height in boys with constitutional delay of growth and puberty. J Pediatr. (1995) 126:545–50. doi: 10.1016/S0022-3476(95)70347-0
13. Parker SE, Troisi R, Wise LA, Palmer JR, Titus-Ernstoff L, Strohsnitter WC, et al. Menarche, menopause, years of menstruation, and the incidence of osteoporosis: the influence of prenatal exposure to diethylstilbestrol. J Clin Endocrinol Metab. (2014) 99:594–601. doi: 10.1210/jc.2013-2954
14. Sedlmeyer IL. Pedigree analysis of constitutional delay of growth and maturation: determination of familial aggregation and inheritance patterns. J Clin Endocrinol Metabol. (2002) 87:5581–6. doi: 10.1210/jc.2002-020862
15. Howard SR, Dunkel L. The genetic basis of delayed puberty. Neuroendocrinology. (2017) 2017:481569. doi: 10.1159/000481569
16. Wehkalampi K, Widen E, Laine T, Palotie A, Dunkel L. Patterns of inheritance of constitutional delay of growth and puberty in families of adolescent girls and boys referred to specialist pediatric care. J Clin Endocrinol Metab. (2008) 93:723–8. doi: 10.1210/jc.2007-1786
17. Winter S, Ousidhoum A, Mcelreavey K, Brauner R. Constitutional delay of puberty: presentation and inheritance pattern in 48 familial cases. BMC Pediatr. (2016) 16:37. doi: 10.1186/s12887-016-0580-3
18. Cousminer DL, Leinonen JT, Sarin AP, Chheda H, Surakka I, Wehkalampi K, et al. Targeted resequencing of the pericentromere of chromosome 2 linked to constitutional delay of growth and puberty. PLoS ONE. (2015) 10:e0128524. doi: 10.1371/journal.pone.0128524
19. Wehkalampi K, Widen E, Laine T, Palotie A, Dunkel L. Association of the timing of puberty with a chromosome 2 locus. J Clin Endocrinol Metab. (2008) 93:4833–9. doi: 10.1210/jc.2008-0882
20. Guo MH, Plummer L, Chan YM, Hirschhorn JN, Lippincott MF. Burden testing of rare variants identified through exome sequencing via publicly available control data. Am J Hum Genet. (2018) 103:522–34. doi: 10.1016/j.ajhg.2018.08.016
21. Wang Y. Is obesity associated with early sexual maturation? A comparison of the association in American boys versus girls. Pediatrics. (2002) 110:903–10. doi: 10.1542/peds.110.5.903
22. Sorensen K, Aksglaede L, Petersen JH, Juul A. Recent changes in pubertal timing in healthy Danish boys: associations with body mass index. J Clin Endocrinol Metab. (2010) 95:263–70. doi: 10.1210/jc.2009-1478
23. He Q, Karlberg J. Bmi in childhood and its association with height gain, timing of puberty, and final height. Pediatr Res. (2001) 49:244–51. doi: 10.1203/00006450-200102000-00019
24. Ojeda SR, Lomniczi A, Mastronardi C, Heger S, Roth C, Parent AS, et al. Minireview: the neuroendocrine regulation of puberty: is the time ripe for a systems biology approach? Endocrinology. (2006) 147:1166–74. doi: 10.1210/en.2005-1136
25. Howard SR. Genes underlying delayed puberty. Mol Cell Endocrinol. (2018) 2018:001. doi: 10.1016/j.mce.2018.05.001
26. Ahmed ML, Ong KK, Morrell DJ, Cox L, Drayer N, Perry L, et al. Longitudinal study of leptin concentrations during puberty: sex differences and relationship to changes in body composition. J Clin Endocrinol Metab. (1999) 84:899–905. doi: 10.1210/jc.84.3.899
27. Farooqi IS. Leptin and the onset of puberty: insights from rodent and human genetics. Semin Reprod Med. (2002) 20:139–44. doi: 10.1055/s-2002-32505
28. Garcia-Mayor RV, Andrade MA, Rios M, Lage M, Dieguez C, Casanueva FF. Serum leptin levels in normal children: relationship to age, gender, body mass index, pituitary-gonadal hormones, and pubertal stage. J Clin Endocrinol Metab. (1997) 82:2849–55. doi: 10.1210/jc.82.9.2849
29. Elias CF, Purohit D. Leptin signaling and circuits in puberty and fertility. Cell Mol Life Sci. (2013) 70:841–62. doi: 10.1007/s00018-012-1095-1
30. Roa J, Tena-Sempere M. Energy balance and puberty onset: emerging role of central mTOR signaling. Trends Endocrinol Metab. (2010) 21:519–28. doi: 10.1016/j.tem.2010.05.003
31. Bellefontaine N, Chachlaki K, Parkash J, Vanacker C, Colledge W, D'anglemont De Tassigny X, et al. Leptin-dependent neuronal NO signaling in the preoptic hypothalamus facilitates reproduction. J Clin Invest. (2014) 124:2550–9. doi: 10.1172/JCI65928
32. Ong KK, Elks CE, Li S, Zhao JH, Luan J, Andersen LB, et al. Genetic variation in LIN28B is associated with the timing of puberty. Nat Genet. (2009) 41:729–33. doi: 10.1038/ng.382
33. Tommiska J, Wehkalampi K, Vaaralahti K, Laitinen EM, Raivio T, Dunkel L. LIN28B in constitutional delay of growth and puberty. J Clin Endocrinol Metab. (2010) 95:3063–6. doi: 10.1210/jc.2009-2344
34. Silveira-Neto AP, Leal LF, Emerman AB, Henderson KD, Piskounova E, Henderson BE, et al. Absence of functional LIN28B mutations in a large cohort of patients with idiopathic central precocious puberty. Horm Res Paediatr. (2012) 78:144–50. doi: 10.1159/000342212
35. Elks CE, Perry JR, Sulem P, Chasman DI, Franceschini N, He C, et al. Thirty new loci for age at menarche identified by a meta-analysis of genome-wide association studies. Nat Genet. (2010) 42:1077–85. doi: 10.1038/ng.714
36. Perry JR, Day F, Elks CE, Sulem P, Thompson DJ, Ferreira T, et al. Parent-of-origin-specific allelic associations among 106 genomic loci for age at menarche. Nature. (2014) 514:92–7. doi: 10.1038/nature13545
37. Day FR, Thompson DJ, Helgason H, Chasman DI, Finucane H, Sulem P, et al. Genomic analyses identify hundreds of variants associated with age at menarche and support a role for puberty timing in cancer risk. Nat Genet. (2017) 49:834–41. doi: 10.1038/ng.3841
38. Farooqi IS, Wangensteen T, Collins S, Kimber W, Matarese G, Keogh JM, et al. Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor. N Engl J Med. (2007) 356:237–47. doi: 10.1056/NEJMoa063988
39. Silveira LF, Trarbach EB, Latronico AC. Genetics basis for GnRH-dependent pubertal disorders in humans. Mol Cell Endocrinol. (2010) 324:30–8. doi: 10.1016/j.mce.2010.02.023
40. Yeo GS. The role of the FTO (Fat Mass and Obesity Related) locus in regulating body size and composition. Mol Cell Endocrinol. (2014) 397:34–41. doi: 10.1016/j.mce.2014.09.012
41. Howard SR, Guasti L, Poliandri A, David A, Cabrera CP, Barnes MR, et al. Contributions of function-altering variants in genes implicated in pubertal timing and body mass for self-limited delayed puberty. J Clin Endocrinol Metab. (2018) 103:649–59. doi: 10.1210/jc.2017-02147
42. Smemo S, Tena JJ, Kim KH, Gamazon ER, Sakabe NJ, Gomez-Marin C, et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature. (2014) 507:371–5. doi: 10.1038/nature13138
43. Mcmurray F, Church CD, Larder R, Nicholson G, Wells S, Teboul L, et al. Adult onset global loss of the fto gene alters body composition and metabolism in the mouse. PLoS Genet. (2013) 9:e1003166. doi: 10.1371/journal.pgen.1003166
44. Merkestein M, Laber S, Mcmurray F, Andrew D, Sachse G, Sanderson J, et al. FTO influences adipogenesis by regulating mitotic clonal expansion. Nat Commun. (2015) 6:6792. doi: 10.1038/ncomms7792
45. Fischer J, Koch L, Emmerling C, Vierkotten J, Peters T, Bruning JC, et al. Inactivation of the Fto gene protects from obesity. Nature. (2009) 458:894–8. doi: 10.1038/nature07848
46. Speakman JR. The 'fat mass and obesity related' (FTO) gene: mechanisms of impact on obesity and energy balance. Curr Obes Rep. (2015) 4:73–91. doi: 10.1007/s13679-015-0143-1
47. Martinez De Morentin PB, Martinez-Sanchez N, Roa J, Ferno J, Nogueiras R, Tena-Sempere M, et al. Hypothalamic mTOR: the rookie energy sensor. Curr Mol Med. (2014) 14:3–21. doi: 10.2174/1566524013666131118103706
48. Roa J, Garcia-Galiano D, Varela L, Sanchez-Garrido MA, Pineda R, Castellano JM, et al. The mammalian target of rapamycin as novel central regulator of puberty onset via modulation of hypothalamic Kiss1 system. Endocrinology. (2009) 150:5016–26. doi: 10.1210/en.2009-0096
49. Parker SL, Kalra SP, Crowley WR. Neuropeptide Y modulates the binding of a gonadotropin-releasing hormone (GnRH) analog to anterior pituitary GnRH receptor sites. Endocrinology. (1991) 128:2309–16. doi: 10.1210/endo-128-5-2309
50. Crowley WR, Kalra SP. Neuropeptide Y stimulates the release of luteinizing hormone-releasing hormone from medial basal hypothalamus in vitro: modulation by ovarian hormones. Neuroendocrinology. (1987) 46:97–103. doi: 10.1159/000124804
51. Plant TM. Neuroendocrine control of the onset of puberty. Front Neuroendocrinol. (2015) 38:73–88. doi: 10.1016/j.yfrne.2015.04.002
52. Couce ME, Cottam D, Esplen J, Teijeiro R, Schauer P, Burguera B. Potential role of hypothalamic ghrelin in the pathogenesis of human obesity. J Endocrinol Invest. (2006) 29:599–605. doi: 10.1007/BF03344158
53. Pomerants T, Tillmann V, Jurimae J, Jurimae T. Relationship between ghrelin and anthropometrical, body composition parameters and testosterone levels in boys at different stages of puberty. J Endocrinol Invest. (2006) 29:962–7. doi: 10.1007/BF03349208
54. Pomerants T, Tillmann V, Karelson K, Jurimae J, Jurimae T. Ghrelin response to acute aerobic exercise in boys at different stages of puberty. Horm Metab Res. (2006) 38:752–7. doi: 10.1055/s-2006-955087
55. Manfredi-Lozano M, Roa J, Ruiz-Pino F, Piet R, Garcia-Galiano D, Pineda R, et al. Defining a novel leptin-melanocortin-kisspeptin pathway involved in the metabolic control of puberty. Mol Metab. (2016) 5:844–57. doi: 10.1016/j.molmet.2016.08.003
56. Manaserh IH, Chikkamenahalli L, Ravi S, Dube PR, Park JJ, Hill JW. Ablating astrocyte insulin receptors leads to delayed puberty and hypogonadism in mice. PLoS Biol. (2019) 17:e3000189. doi: 10.1371/journal.pbio.3000189
57. Pugliese-Pires PN, Fortin JP, Arthur T, Latronico AC, Mendonca BB, Villares SM, et al. Novel inactivating mutations in the GH secretagogue receptor gene in patients with constitutional delay of growth and puberty. Eur J Endocrinol. (2011) 165:233–41. doi: 10.1530/EJE-11-0168
58. Tornberg J, Sykiotis GP, Keefe K, Plummer L, Hoang X, Hall JE, et al. Heparan sulfate 6-O-sulfotransferase 1, a gene involved in extracellular sugar modifications, is mutated in patients with idiopathic hypogonadotrophic hypogonadism. Proc Natl Acad Sci USA. (2011) 108:11524–9. doi: 10.1073/pnas.1102284108
59. Pitteloud N, Meysing A, Quinton R, Acierno JS Jr Dwyer AA, Plummer L, et al. Mutations in fibroblast growth factor receptor 1 cause Kallmann syndrome with a wide spectrum of reproductive phenotypes. Mol Cell Endocrinol. (2006) 254–255:60–9. doi: 10.1016/j.mce.2006.04.021
60. Xu C, Messina A, Somm E, Miraoui H, Kinnunen T, Acierno J Jr, et al. KLB, encoding beta-Klotho, is mutated in patients with congenital hypogonadotropic hypogonadism. EMBO Mol Med. (2017) 2017:7376. doi: 10.15252/emmm.201607376
61. Cassatella D, Howard SR, Acierno JS, Xu C, Papadakis GE, Santoni FA, et al. Congenital hypogonadotropic hypogonadism and constitutional delay of growth and puberty have distinct genetic architectures. Eur J Endocrinol. (2018) 178:377–88. doi: 10.1530/EJE-17-0568
62. Zhu J, Choa RE, Guo MH, Plummer L, Buck C, Palmert MR, et al. A shared genetic basis for self-limited delayed puberty and idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. (2015) 2015:jc20151080. doi: 10.1210/jc.2015-1080
63. Emerick JE, Vogt KS. Endocrine manifestations and management of Prader-Willi syndrome. Int J Pediatr Endocrinol. (2013) 2013:14. doi: 10.1186/1687-9856-2013-14
64. Forsythe E, Beales PL. Bardet-Biedl syndrome. Eur J Hum Genet. (2013) 21:8–13. doi: 10.1038/ejhg.2012.115
65. Dauber A, Hirschhorn JN, Picker J, Maher TA, Milunsky A. Delayed puberty due to a novel mutation in CHD7 causing CHARGE syndrome. Pediatrics. (2010) 126:e1594–e1598. doi: 10.1542/peds.2010-0164
66. Loureiro M, Reis F, Robalo B, Pereira C, Sampaio L. Adrenal hypoplasia congenita: a rare cause of primary adrenal insufficiency and hypogonadotropic hypogonadism. Pediatr Rep. (2015) 7:5936. doi: 10.4081/pr.2015.5936
67. Nagasaki K, Kubota T, Kobayashi H, Sawada H, Numakura C, Harada S, et al. Clinical characteristics of septo-optic dysplasia accompanied by congenital central hypothyroidism in Japan. Clin Pediatr Endocrinol. (2017) 26:207–13. doi: 10.1297/cpe.26.207
68. Szakszon K, Felszeghy E, Csizy I, Jozsa T, Kaposzta R, Balogh E, et al. Endocrine and anatomical findings in a case of Solitary Median Maxillary Central Incisor Syndrome. Eur J Med Genet. (2012) 55:109–11. doi: 10.1016/j.ejmg.2011.11.002
69. Turner G, Lower KM, White SM, Delatycki M, Lampe AK, Wright M, et al. The clinical picture of the Borjeson-Forssman-Lehmann syndrome in males and heterozygous females with PHF6 mutations. Clin Genet. (2004) 65:226–32. doi: 10.1111/j.0009-9163.2004.00215.x
70. Simonis N, Migeotte I, Lambert N, Perazzolo C, De Silva DC, Dimitrov B, et al. FGFR1 mutations cause Hartsfield syndrome, the unique association of holoprosencephaly and ectrodactyly. J Med Genet. (2013) 50:585–92. doi: 10.1136/jmedgenet-2013-101603
71. Wortmann SB, Espeel M, Almeida L, Reimer A, Bosboom D, Roels F, et al. Inborn errors of metabolism in the biosynthesis and remodelling of phospholipids. J Inherit Metab Dis. (2015) 38:99–110. doi: 10.1007/s10545-014-9759-7
72. Howard SR, Oleari R, Poliandri A, Chantzara V, Fantin A, Ruiz-Babot G, et al. HS6ST1 insufficiency causes self-limited delayed puberty in contrast with other GnRH deficiency genes. J Clin Endocrinol Metab. (2018) 2018:646. doi: 10.1210/jc.2018-00646
73. Parkash J, Messina A, Langlet F, Cimino I, Loyens A, Mazur D, et al. Semaphorin7A regulates neuroglial plasticity in the adult hypothalamic median eminence. Nat Commun. (2015) 6:6385. doi: 10.1038/ncomms7385
74. Pielecka-Fortuna J, Chu Z, Moenter SM. Kisspeptin acts directly and indirectly to increase gonadotropin-releasing hormone neuron activity and its effects are modulated by estradiol. Endocrinology. (2008) 149:1979–86. doi: 10.1210/en.2007-1365
75. Pitteloud N, Quinton R, Pearce S, Raivio T, Acierno J, Dwyer A, et al. Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. J Clin Invest. (2007) 117:457–63. doi: 10.1172/JCI29884
76. Howard SR, Guasti L, Ruiz-Babot G, Mancini A, David A, Storr HL, et al. IGSF10 mutations dysregulate gonadotropin-releasing hormone neuronal migration resulting in delayed puberty. EMBO Mol Med. (2016) 2016:6250. doi: 10.15252/emmm.201606250
77. Caronia LM, Martin C, Welt CK, Sykiotis GP, Quinton R, Thambundit A, et al. A genetic basis for functional hypothalamic amenorrhea. N Engl J Med. (2011) 364:215–25. doi: 10.1056/NEJMoa0911064
78. Jolly A, Bayram Y, Turan S, Aycan Z, Tos T, Abali ZY, et al. Exome sequencing of a primary ovarian insufficiency cohort reveals common molecular etiologies for a spectrum of disease. J Clin Endocrinol Metab. (2019) 2019:248. doi: 10.1210/jc.2019-00248
79. Chevrier L, Guimiot F, De Roux N. GnRH receptor mutations in isolated gonadotropic deficiency. Mol Cell Endocrinol. (2011) 346:21–8. doi: 10.1016/j.mce.2011.04.018
80. Lin L, Conway GS, Hill NR, Dattani MT, Hindmarsh PC, Achermann JC. A homozygous R262Q mutation in the gonadotropin-releasing hormone receptor presenting as constitutional delay of growth and puberty with subsequent borderline oligospermia. J Clin Endocrinol Metab. (2006) 91:5117–21. doi: 10.1210/jc.2006-0807
81. Vaaralahti K, Wehkalampi K, Tommiska J, Laitinen EM, Dunkel L, Raivio T. The role of gene defects underlying isolated hypogonadotropic hypogonadism in patients with constitutional delay of growth and puberty. Fertil Steril. (2011) 95:2756–8. doi: 10.1016/j.fertnstert.2010.12.059
82. Themmen APN, Huhtaniemi IT. Mutations of gonadotropins and gonadotropin receptors: elucidating the physiology and pathophysiology of pituitary-gonadal function. Endocr Rev. (2000) 21:551–83. doi: 10.1210/edrv.21.5.0409
83. Potorac I, Rivero-Muller A, Trehan A, Kielbus M, Jozwiak K, Pralong F, et al. A vital region for human glycoprotein hormone trafficking revealed by an LHB mutation. J Endocrinol. (2016) 231:197–207. doi: 10.1530/JOE-16-0384
84. Lofrano-Porto A, Barra GB, Giacomini LA, Nascimento PP, Latronico AC, Casulari LA, et al. Luteinizing hormone beta mutation and hypogonadism in men and women. N Engl J Med. (2007) 357:897–904. doi: 10.1056/NEJMoa071999
85. Layman LC, Lee EJ, Peak DB, Namnoum AB, Vu KV, Van Lingen BL, et al. Delayed puberty and hypogonadism caused by mutations in the follicle-stimulating hormone beta-subunit gene. N Engl J Med. (1997) 337:607–11. doi: 10.1056/NEJM199708283370905
86. Mancini A, Howard SR, Cabrera CP, Barnes MR, David A, Wehkalampi K, et al. EAP1 regulation of GnRH promoter activity is important for human pubertal timing. Hum Mol Genet. (2019) 2019:451. doi: 10.1093/hmg/ddy451
87. Hutchins BI, Kotan LD, Taylor-Burds C, Ozkan Y, Cheng PJ, Gurbuz F, et al. CCDC141 mutation identified in anosmic hypogonadotropic hypogonadism (Kallmann Syndrome) alters GnRH neuronal migration. Endocrinology. (2016) 157:1956–66. doi: 10.1210/en.2015-1846
88. Sarfati J, Bouvattier C, Bry-Gauillard H, Cartes A, Bouligand J, Young J. Kallmann syndrome with FGFR1 and KAL1 mutations detected during fetal life. Orphanet J Rare Dis. (2015) 10:71. doi: 10.1186/s13023-015-0287-9
89. Sarfati J, Guiochon-Mantel A, Rondard P, Arnulf I, Garcia-Pinero A, Wolczynski S, et al. A comparative phenotypic study of kallmann syndrome patients carrying monoallelic and biallelic mutations in the prokineticin 2 or prokineticin receptor 2 genes. J Clin Endocrinol Metab. (2010) 95:659–69. doi: 10.1210/jc.2009-0843
90. Raivio T, Falardeau J, Dwyer A, Quinton R, Hayes FJ, Hughes VA, et al. Reversal of idiopathic hypogonadotropic hypogonadism. N Engl J Med. (2007) 357:863–73. doi: 10.1056/NEJMoa066494
91. Bourguignon JP, Gerard A, Purnelle G, Czajkowski V, Yamanaka C, Lemaitre M, et al. Duality of glutamatergic and GABAergic control of pulsatile GnRH secretion by rat hypothalamic explants: II. Reduced NR2C- and GABAA-receptor-mediated inhibition at initiation of sexual maturation. J Neuroendocrinol. (1997) 9:193–9. doi: 10.1046/j.1365-2826.1997.00568.x
92. Plant TM, Barker-Gibb ML. Neurobiological mechanisms of puberty in higher primates. Hum Reprod Update. (2004) 10:67–77. doi: 10.1093/humupd/dmh001
93. Ojeda SR, Dubay C, Lomniczi A, Kaidar G, Matagne V, Sandau US, et al. Gene networks and the neuroendocrine regulation of puberty. Mol Cell Endocrinol. (2010) 324:3–11. doi: 10.1016/j.mce.2009.12.003
94. Lomniczi A, Wright H, Castellano JM, Matagne V, Toro CA, Ramaswamy S, et al. Epigenetic regulation of puberty via Zinc finger protein-mediated transcriptional repression. Nat Commun. (2015) 6:10195. doi: 10.1038/ncomms10195
95. Ojeda SR, Hill J, Hill DF, Costa ME, Tapia V, Cornea A, et al. The Oct-2 POU domain gene in the neuroendocrine brain: a transcriptional regulator of mammalian puberty. Endocrinology. (1999) 140:3774–89. doi: 10.1210/endo.140.8.6941
96. Mastronardi C, Smiley GG, Raber J, Kusakabe T, Kawaguchi A, Matagne V, et al. Deletion of the Ttf1 gene in differentiated neurons disrupts female reproduction without impairing basal ganglia function. J Neurosci. (2006) 26:13167–79. doi: 10.1523/JNEUROSCI.4238-06.2006
97. Lee BJ, Cho GJ, Norgren RB Jr, Junier MP, Hill DF, Tapia V, et al. TTF-1, a homeodomain gene required for diencephalic morphogenesis, is postnatally expressed in the neuroendocrine brain in a developmentally regulated and cell-specific fashion. Mol Cell Neurosci. (2001) 17:107–26. doi: 10.1006/mcne.2000.0933
98. Gabriele M, Vulto-Van Silfhout AT, Germain PL, Vitriolo A, Kumar R, Douglas E, et al. YY1 haploinsufficiency causes an intellectual disability syndrome featuring transcriptional and chromatin dysfunction. Am J Hum Genet. (2017) 100:907–25. doi: 10.1016/j.ajhg.2017.05.006
99. Heger S, Mastronardi C, Dissen GA, Lomniczi A, Cabrera R, Roth CL, et al. Enhanced at puberty 1 (EAP1) is a new transcriptional regulator of the female neuroendocrine reproductive axis. J Clin Invest. (2007) 117:2145–54. doi: 10.1172/JCI31752
100. Dissen GA, Lomniczi A, Heger S, Neff TL, Ojeda SR. Hypothalamic EAP1 (enhanced at puberty 1) is required for menstrual cyclicity in nonhuman primates. Endocrinology. (2012) 153:350–61. doi: 10.1210/en.2011-1541
101. Li C, Li P. Enhanced at puberty-1 (Eap1) expression critically regulates the onset of puberty independent of hypothalamic kiss1 expression. Cell Physiol Biochem. (2017) 43:1402–12. doi: 10.1159/000481872
102. Lomniczi A, Garcia-Rudaz C, Ramakrishnan R, Wilmot B, Khouangsathiene S, Ferguson B, et al. A single-nucleotide polymorphism in the EAP1 gene is associated with amenorrhea/oligomenorrhea in nonhuman primates. Endocrinology. (2012) 153:339–49. doi: 10.1210/en.2011-1540
103. Xu J, Li P. Expression of EAP1 and CUX1 in the hypothalamus of female rats and relationship with KISS1 and GnRH. Endocr J. (2016) 63:681–90. doi: 10.1507/endocrj.EJ16-0123
104. Mueller JK, Koch I, Lomniczi A, Loche A, Rulfs T, Castellano JM, et al. Transcription of the human EAP1 gene is regulated by upstream components of a puberty-controlling Tumor Suppressor Gene network. Mol Cell Endocrinol. (2012) 351:184–98. doi: 10.1016/j.mce.2011.12.004
105. Lomniczi A, Ojeda SR. The emerging role of epigenetics in the regulation of female puberty. Endocr Dev. (2016) 29:1–16. doi: 10.1159/000438840
106. Lomniczi A, Loche A, Castellano JM, Ronnekleiv OK, Bosch M, Kaidar G, et al. Epigenetic control of female puberty. Nat Neurosci. (2013) 16:281–9. doi: 10.1038/nn.3319
107. Morrison KE, Rodgers AB, Morgan CP, Bale TL. Epigenetic mechanisms in pubertal brain maturation. Neuroscience. (2014) 264:17–24. doi: 10.1016/j.neuroscience.2013.11.014
108. Yang C, Ye J, Li X, Gao X, Zhang K, Luo L, et al. DNA methylation patterns in the hypothalamus of female pubertal goats. PLoS ONE. (2016) 11:e0165327. doi: 10.1371/journal.pone.0165327
109. Peters J. The role of genomic imprinting in biology and disease: an expanding view. Nat Rev Genet. (2014) 15:517–30. doi: 10.1038/nrg3766
110. Abreu AP, Dauber A, Macedo DB, Noel SD, Brito VN, Gill JC, et al. Central precocious puberty caused by mutations in the imprinted gene MKRN3. N Engl J Med. (2013) 368:2467–75. doi: 10.1056/NEJMoa1302160
111. Dauber A, Cunha-Silva M, Macedo DB, Brito VN, Abreu AP, Roberts SA, et al. Paternally inherited DLK1 deletion associated with familial central precocious puberty. J Clin Endocrinol Metab. (2017) 102:1557–67. doi: 10.1210/jc.2016-3677
112. Simon D, Ba I, Mekhail N, Ecosse E, Paulsen A, Zenaty D, et al. Mutations in the maternally imprinted gene MKRN3 are common in familial central precocious puberty. Eur J Endocrinol. (2016) 174:1–8. doi: 10.1530/EJE-15-0488
113. Jong MT, Carey AH, Caldwell KA, Lau MH, Handel MA, Driscoll DJ, et al. Imprinting of a RING zinc-finger encoding gene in the mouse chromosome region homologous to the Prader-Willi syndrome genetic region. Hum Mol Genet. (1999) 8:795–803. doi: 10.1093/hmg/8.5.795
114. Hagen CP, Sorensen K, Mieritz MG, Johannsen TH, Almstrup K, Juul A. Circulating MKRN3 levels decline prior to pubertal onset and through puberty: a longitudinal study of healthy girls. J Clin Endocrinol Metab. (2015) 100:1920–6. doi: 10.1210/jc.2014-4462
115. Busch AS, Hagen CP, Almstrup K, Juul A. Circulating MKRN3 levels decline during puberty in healthy boys. J Clin Endocrinol Metab. (2016) 101:2588–93. doi: 10.1210/jc.2016-1488
116. Yellapragada V, Liu X, Lund C, Kansakoski J, Pulli K, Vuoristo S, et al. MKRN3 interacts with several proteins implicated in puberty timing but does not influence GNRH1 expression. Front Endocrinol. (2019) 10:48. doi: 10.3389/fendo.2019.00048
117. Hirsch HJ, Eldar-Geva T, Bennaroch F, Pollak Y, Gross-Tsur V. Sexual dichotomy of gonadal function in Prader-Willi syndrome from early infancy through the fourth decade. Hum Reprod. (2015) 30:2587–96. doi: 10.1093/humrep/dev213
118. Butler MG. Genomic imprinting disorders in humans: a mini-review. J Assist Reprod Genet. (2009) 26:477–86. doi: 10.1007/s10815-009-9353-3
119. Lee HS, Hwang JS. Central precocious puberty in a girl with Prader-Willi syndrome. J Pediatr Endocrinol Metab. (2013) 26:1201–4. doi: 10.1515/jpem-2013-0040
120. Crino A, Schiaffini R, Ciampalini P, Spera S, Beccaria L, Benzi F, et al. Hypogonadism and pubertal development in Prader-Willi syndrome. Eur J Pediatr. (2003) 162:327–33. doi: 10.1007/s00431-002-1132-4
121. Kanber D, Giltay J, Wieczorek D, Zogel C, Hochstenbach R, Caliebe A, et al. A paternal deletion of MKRN3, MAGEL2 and NDN does not result in Prader-Willi syndrome. Eur J Hum Genet. (2009) 17:582–90. doi: 10.1038/ejhg.2008.232
122. de Smith AJ, Purmann C, Walters RG, Ellis RJ, Holder SE, Van Haelst MM, et al. A deletion of the HBII-85 class of small nucleolar RNAs (snoRNAs) is associated with hyperphagia, obesity and hypogonadism. Hum Mol Genet. (2009) 18:3257–65. doi: 10.1093/hmg/ddp263
123. Messina A, Langlet F, Chachlaki K, Roa J, Rasika S, Jouy N, et al. A microRNA switch regulates the rise in hypothalamic GnRH production before puberty. Nat Neurosci. (2016) 19:835–44. doi: 10.1038/nn.4298
124. Ahmed K, Lapierre MP, Gasser E, Denzler R, Yang Y, Rulicke T, et al. Loss of microRNA-7a2 induces hypogonadotropic hypogonadism and infertility. J Clin Invest. (2017) 127:1061–74. doi: 10.1172/JCI90031
125. Kurian JR, Olesen KM, Auger AP. Sex differences in epigenetic regulation of the estrogen receptor-alpha promoter within the developing preoptic area. Endocrinology. (2010) 151:2297–305. doi: 10.1210/en.2009-0649
126. Toro CA, Wright H, Aylwin CF, Ojeda SR, Lomniczi A. Trithorax dependent changes in chromatin landscape at enhancer and promoter regions drive female puberty. Nat Commun. (2018) 9:57. doi: 10.1038/s41467-017-02512-1
127. Mouritsen A, Aksglaede L, Sorensen K, Mogensen SS, Leffers H, Main KM, et al. Hypothesis: exposure to endocrine-disrupting chemicals may interfere with timing of puberty. Int J Androl. (2010) 33:346–59. doi: 10.1111/j.1365-2605.2010.01051.x
128. Pinson A, Franssen D, Gerard A, Parent AS, Bourguignon JP. Neuroendocrine disruption without direct endocrine mode of action: polychloro-biphenyls (PCBs) and bisphenol A (BPA) as case studies. C R Biol. (2017) 340:432–8. doi: 10.1016/j.crvi.2017.07.006
129. Drobna Z, Henriksen AD, Wolstenholme JT, Montiel C, Lambeth PS, Shang S, et al. Transgenerational Effects of Bisphenol A on Gene Expression and DNA Methylation of Imprinted Genes in Brain. Endocrinology. (2018) 159:132–44. doi: 10.1210/en.2017-00730
130. Milesi MM, Lorenz V, Pacini G, Repetti MR, Demonte LD, Varayoud J, et al. Perinatal exposure to a glyphosate-based herbicide impairs female reproductive outcomes and induces second-generation adverse effects in Wistar rats. Arch Toxicol. (2018) 92:2629–43. doi: 10.1007/s00204-018-2236-6
131. Parent AS, Franssen D, Fudvoye J, Gerard A, Bourguignon JP. Developmental variations in environmental influences including endocrine disruptors on pubertal timing and neuroendocrine control: revision of human observations and mechanistic insight from rodents. Front Neuroendocrinol. (2015) 38:12–36. doi: 10.1016/j.yfrne.2014.12.004
132. Krstevska-Konstantinova M, Charlier C, Craen M, Du Caju M, Heinrichs C, De Beaufort C, et al. Sexual precocity after immigration from developing countries to Belgium: evidence of previous exposure to organochlorine pesticides. Hum Reprod. (2001) 16:1020–6. doi: 10.1093/humrep/16.5.1020
133. Swan SH, Main KM, Liu F, Stewart SL, Kruse RL, Calafat AM, et al. Decrease in anogenital distance among male infants with prenatal phthalate exposure. Environ Health Perspect. (2005) 113:1056–61. doi: 10.1289/ehp.8100
134. Rissman EF, Adli M. Minireview: transgenerational epigenetic inheritance: focus on endocrine disrupting compounds. Endocrinology. (2014) 155:2770–80. doi: 10.1210/en.2014-1123
135. Li X, Sun Z, Manthari RK, Li M, Guo Q, Wang J. Effect of gestational exposure to arsenic on puberty in offspring female mice. Chemosphere. (2018) 202:119–26. doi: 10.1016/j.chemosphere.2018.03.095
Keywords: puberty, constitutional delay of growth and puberty (CDGP), endocrine genetics, self-limited delayed puberty, EAP1, IGSF10, HS6ST1, FTO
Citation: Howard SR (2019) The Genetic Basis of Delayed Puberty. Front. Endocrinol. 10:423. doi: 10.3389/fendo.2019.00423
Received: 15 April 2019; Accepted: 12 June 2019;
Published: 26 June 2019.
Edited by:
Vassos Neocleous, The Cyprus Institute of Neurology and Genetics, CyprusReviewed by:
Andrew A. Dwyer, Boston College, United StatesAna Paula Abreu, Brigham and Women's Hospital and Harvard Medical School, United States
Copyright © 2019 Howard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sasha R. Howard, cy5ob3dhcmRAcW11bC5hYy51aw==